

Abstract
Previously, we showed that expression of a dominant-negative form of the transforming growth factor β (TGF-β) type II receptor in skeletal tissue resulted in increased hypertrophic differentiation in growth plate and articular chondrocytes, suggesting a role for TGF-β in limiting terminal differentiation in vivo. Parathyroid hormone–related peptide (PTHrP) has also been demonstrated to regulate chondrocyte differentiation in vivo. Mice with targeted deletion of the PTHrP gene demonstrate increased endochondral bone formation, and misexpression of PTHrP in cartilage results in delayed bone formation due to slowed conversion of proliferative chondrocytes into hypertrophic chondrocytes. Since the development of skeletal elements requires the coordination of signals from several sources, this report tests the hypothesis that TGF-β and PTHrP act in a common signal cascade to regulate endochondral bone formation. Mouse embryonic metatarsal bone rudiments grown in organ culture were used to demonstrate that TGF-β inhibits several stages of endochondral bone formation, including chondrocyte proliferation, hypertrophic differentiation, and matrix mineralization. Treatment with TGF-β1 also stimulated the expression of PTHrP mRNA. PTHrP added to cultures inhibited hypertrophic differentiation and matrix mineralization but did not affect cell proliferation. Furthermore, terminal differentiation was not inhibited by TGF-β in metatarsal rudiments from PTHrP-null embryos; however, growth and matrix mineralization were still inhibited. The data support the model that TGF-β acts upstream of PTHrP to regulate the rate of hypertrophic differentiation and suggest that TGF-β has both PTHrP-dependent and PTHrP-independent effects on endochondral bone formation.
Keywords: chondrocyte differentiation, skeletal development, perichondrium, organ culture, transforming growth factor β receptors
The rate of cartilage differentiation must be carefully regulated so that bones attain the proper shape and length. Early in skeletal development, mesenchymal cells condense and differentiate into chondroblasts, which form the initial shape of the bone rudiment. Chondroblasts then undergo a complex program of proliferation, maturation, and hypertrophy. Hypertrophic cartilage is then replaced with bone. Endochondral bone formation is complex and requires the coordination of signals from several factors and multiple cell types (reviewed in Cancedda et al., 1995; Erlebacher et al., 1995). Chondrocyte differentiation is regulated by factors synthesized by both chondrocytes and cells in the perichondrium, the layer of mesenchyme that surrounds the cartilage rudiment.
Parathyroid hormone–related peptide (PTHrP)1 was first identified as a factor associated with humoral hypercalcemia of malignancy (Broadus and Stewart, 1994). PTHrP is expressed in a wide variety of adult and embryonic cell types, including osteoblasts and chondrocytes (Broadus and Stewart, 1994; Suva et al., 1987; Lee et al., 1995). During the development of endochondral bones, PTHrP is expressed in periarticular chondrocytes and resting and maturing chondrocytes of the growth plate. PTHrP signals through the G protein–coupled parathyroid hormone (PTH) receptor, which is also expressed in a wide range of adult and embryonic cells, including chondrocytes at the transition between the zones of proliferation and hypertrophy (Lee et al., 1993, 1995). The importance of PTHrP in endochondral bone formation is demonstrated in mice homozygous for a targeted disruption of the PTHrP gene (Karaplis et al., 1994; Amizuka et al., 1994, 1996). PTHrP-null mice demonstrate increased endochondral bone formation. The epiphyses demonstrate diminished resting and proliferating zones and accelerated chondrocyte maturation, apoptosis, and replacement with bone. Mice with targeted deletion of the PTH receptor demonstrate a similar phenotype (Lanske et al., 1996). Misexpression of PTHrP in cartilage with the promoter from the type II collagen gene results in accumulation of prehypertrophic chondrocytes and inhibition of apoptosis in late stage hypertrophic chondrocytes (Weir et al., 1996; Amling et al., 1997). The Jansen type chondrodysplasia in humans, which is also characterized by a delay in endochondral maturation, has been attributed to a mutation in the PTH receptor gene that results in a constitutively active protein (Schipani et al., 1995). Transgenic mice that express this mutation demonstrate slowed conversion of proliferating chondrocytes into hypertrophic chondrocytes (Schipani et al., 1997). Recently, it was proposed that Indian hedgehog (Ihh) and PTHrP form a negative feedback loop that provides a mechanism for chondrocytes to sense and regulate their rate of differentiation (Vortkamp et al., 1996; Lanske et al., 1996; Wallis, 1996). Vertebrate hedgehog (Hh) proteins are a family of secreted molecules that are important regulators of embryonic patterning (Hammerschmidt et al., 1997) related to the Drosophila segment polarity gene, Hh (Nusslein-Volhard and Wieschaus, 1980). Ihh is expressed in the developing cartilage rudiments, in a population of cells that are committed to become hypertrophic (Bitgood and McMahon, 1995; Vortkamp et al., 1996). Misexpression of Ihh in chick limb cartilage rudiments resulted in inhibition of chondrocyte differentiation and stimulation of PTHrP mRNA expression. Furthermore, PTHrP was required for the inhibitory activities of Hh (Vortkamp et al., 1996; Lanske et al., 1996).
Members of the TGF-β superfamily are secreted signaling molecules that regulate many aspects of growth and differentiation (reviewed in Massague et al., 1990; Moses, 1990; Moses and Serra, 1996). This family includes several TGF-β isoforms, the activin and inhibins, growth and differentiation factors (GDFs), and the bone morphogenetic proteins (BMPs), which were first identified by their ability to induce ectopic bone formation when injected into intramuscular sites. TGF-β1, 2, and 3 mRNA are synthesized in the mouse perichondrium and periosteum from 13.5 d post coitum (p.c.) until after birth (Sandberg et al., 1988; Pelton et al., 1990a; Gatherer et al., 1990; Millan et al., 1991). TGF-β alters chondrocyte differentiation in vitro and has varying effects depending on the status of the cells (reviewed in Moses and Serra, 1996). TGF-β promotes chondrogenesis in cultures of early undifferentiated mesenchyme (Kulyk et al., 1989; Leonard et al., 1991; Denker et al., 1994) but inhibits terminal chondrocyte differentiation in high density chondrocyte pellet cultures or organ cultures (Kato et al., 1988; Ballock et al., 1993; Tschan et al., 1993; Bohme et al., 1995; Dieudonne et al., 1994).
Previously, we generated transgenic mice that express a dominant-negative mutation of the TGF-β type II receptor (DNIIR) in the periosteum, perichondrium, articular cartilage, synovium, and the lower hypertrophic zone of the growth plate (Serra et al., 1997). Transgenic mice demonstrated increased terminal differentiation and persistent expression of Ihh in growth plate chondrocytes, suggesting that TGF-β regulates hypertrophic differentiation in vivo. Since Ihh normally acts as an inhibitor of chondrocyte differentiation, it was proposed that TGF-β was required for Ihh-mediated inhibition. This report tests the hypothesis that TGF-β and PTHrP act in a common signaling cascade to regulate chondrocyte differentiation and demonstrates that TGF-β has both PTHrP-dependent and PTHrP-independent effects on endochondral bone formation.
Materials and Methods
Embryonic Metatarsal Rudiment Organ Cultures
Metatarsal rudiments were isolated from 15.5 d p.c. ICR/B6D2 mouse embryos or embryos from crosses of PTHrP+/− mice (Karaplis et al., 1994) and were used as noted. Noon on the day of the vaginal plug is 0.5 d p.c. The three central metatarsal rudiments were cultured in each well of a 24-well plate in 1 ml of chemically defined medium containing α-MEM (cat. no. 12000-063; GIBCO BRL) supplemented with 0.05 mg/ml ascorbic acid (Sigma Chemical Co.), 0.3 mg/ml l-glutamine (Sigma Chemical Co.), 0.05 mg/ml gentamicin 90, 1 mM β-glycerophosphate (Sigma Chemical Co.), and 0.2% endotoxin-free fraction V BSA (Sigma Chemical Co.) as previously described (Dieudonne et al., 1994). Explants were grown at 37°C in a humidified 5% CO2 incubator. TGF-β1 (1 or 10 ng/ml) in 4 mM HCl (R&D Systems) or PTHrP (1–34) in 10 mM acetic acid containing 1% BSA (Bachem) at varying concentration was added to cultures 12–16 h after dissection. Medium was changed on the third day of culture. Cultures were observed and photographed with an Olympus SZH10 dissecting microscope at 24 h, 3 d, and 5 d of treatment. The length of metatarsals was calculated by measuring the length of rudiment from photographs taken on the dissecting microscope and dividing by the magnification factor (usually ×5). The length of several bone rudiments (at least three for each condition) was calculated, and the data are shown as the mean ± SD.
Histology
Metatarsal rudiments were fixed overnight at 4°C in fresh 4% paraformaldehyde and then decalcified 2 h to overnight at 4°C in 1 mM Tris, pH 7.5, 10% EDTA tetrasodium salt, 7.5% polyvinyl pyrolidone, and 1 μl/ml diethyl pyrocarbonate (DEPC). The explants were dehydrated through a series of ethanols and xylene and then embedded in paraffin and cut into 5-μm sections. Sections were stained with hematoxylin and eosin as noted using standard procedures.
Bromo Deoxyuridine Labeling
Metatarsal rudiments were treated with 10 μM bromo deoxyuridine (BrdU; Boehringer Mannheim) for 2.5 h. Metatarsals were then washed twice in PBS at 37°C, fixed in paraformaldehyde at 4°C overnight, embedded in paraffin, and cut into 5-μm sections. Sections were deparaffinized, denatured in 2 N HCl for 20 min at 37°C, and neutralized in 1% boric acid/ 0.285% sodium borate, pH 7.6. Next, the sections were treated with 0.005 mg trypsin/ml 0.05 M Tris, pH 7.6, for 3 min at 37°C and washed three times in PBS. Immunostaining was then performed using components and directions from the Vectastain Elite staining kit (Vector Laboratories). A rat mAb directed to BrdU (Harlan) was used as the primary antibody at a 1:200 dilution. Cy3-conjugated avidin (Vector Laboratories) was substituted for the avidin-biotin-peroxidase complex. Excess Cy3-conjugated avidin was removed from the sections by washing three times for 10 min each in PBS at room temperature, and the sections were immediately mounted with Aquapoly mount (Poly Sciences). Fluorescence was observed and imaged using a Zeiss Axiophot microscope and a Princeton Instruments CCD camera with Sellomics imaging software.
Immunohistochemistry
Immunohistochemical staining of type X collagen was performed using polyclonal antibodies to mouse type X collagen (a generous gift from Bjorn Olsen, Harvard Medical School, Boston, MA). Sections were dewaxed, rehydrated, and incubated with 1 mg/ml hyaluronidase (Sigma Chemical Co.) in PBS at 37°C for 30 min. Immunohistochemistry was then performed following the directions supplied with the Vectastain Elite immunoperoxidase staining kit (Vector Laboratories). The color reaction was performed using the DAB substrate kit, also from Vector Laboratories. Sections were very lightly counterstained with toluidine blue, and photographs were taken under bright field illumination with a Zeiss Axiophot microscope. The percentage of the bone rudiment stained for type X collagen was calculated as follows:
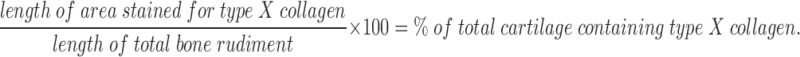 |
This measurement takes into account changes in the total length of the bone rudiment.
Immunofluorescent staining for the TGF-β type I and type II receptors was performed using polyclonal antibodies obtained from Santa Cruz Biotechnology (type I, cat. no. sc 398; type II, cat. no. sc 220). Sections were dewaxed, rehydrated, and treated with 0.05% Saponin in water for 30 min at room temperature. Saponin was removed by washing three times for 5 min each in TBS with 0.1% Tween 20 at room temperature. Immunostaining was then performed using components and directions from the Vectastain Elite staining kit; however, Cy3-conjugated avidin (Vector Laboratories) was substituted for the avidin-biotin-peroxidase complex. Excess Cy3-conjugated avidin was removed from the sections by washing three times for 10 min each in TBS with 0.1% Tween 20 at room temperature, and the sections were immediately mounted with Aquapoly mount (Poly Sciences). Fluorescence was observed and imaged using a Zeiss Axiophot microscope and a Princeton Instruments CCD camera with Sellomics imaging software.
In Situ Hybridization
In situ hybridization was performed as described (Pelton et al., 1990b). Metatarsal rudiments were fixed overnight in paraformaldehyde at 4°C, then decalcified in 1 mM Tris, pH 7.5, 10% EDTA tetrasodium salt, 7.5% polyvinyl pyrolidone, and 1 μl/ml DEPC at 4°C for 2 h to overnight. The metatarsals were then dehydrated in alcohol and embedded in paraffin. Sections (5 μm) were hybridized to 35S-labeled antisense riboprobes. The Ihh plasmid (a kind gift from Andy McMahon, Harvard University, Cambridge, MA) was linearized with XbaI, and riboprobe was synthesized using T7 polymerase. The PTHrP plasmid (a generous gift from Henry Kronenberg, Harvard Medical School, Boston, MA) was linearized with EcoRI, and riboprobe was synthesized using T3 polymerase. Slides were exposed to photographic emulsion at 4°C for 2 wk, then developed, fixed, and cleared. Sections were counterstained with 0.02% toluidine blue. Bright field and dark field images were captured with a Princeton Instruments CCD camera. Bright field and dark field images were merged using the electronic photography imaging program from Sellomics.
Reverse Transcription PCR Analysis
RNA was extracted from cartilage rudiment cultures by lysis in guanidine thiocyanate using the Ambion RNaqueous kit (cat. no. 1912) and the manufacturer's instructions with the modification that the cartilage rudiments were first homogenized in the guanidine solution in a microcentrifuge tube with a small pestle. RNA was treated with RNase-free DNase (Promega Corp.) for 1 h at 37°C, phenol/chloroform extracted, and ethanol precipitated. Precipitation of RNA was facilitated with 20 μg glycogen per sample (Boehringer Mannheim). RNA concentration was determined spectrophotometrically. For reverse transcription (RT)-PCR analysis, cDNA was synthesized from 1 μg of total RNA as described in the GeneAmp RNA PCR kit (Perkin-Elmer) using the oligo dT primers. For each sample, 5 μl cDNA was amplified with 0.2 μM primers and 0.2 mM nucleotides for 20 cycles. These conditions were determined to fall within the linear range of PCR product formation for both PTHrP and glyceraldehyde-6-phosphate dehydrogenase (GAPDH). Samples incubated without reverse transcriptase were used to determine if there was DNA contamination in the RNA samples. DNA contamination was not detected in the RNA from the three separate experiments performed. PCR products were blotted to nylon membrane as described (Southern, 1975) and probed with 32P-labeled cDNA probes for PTHrP (obtained from H. Kronenberg, Harvard Medical School, Boston, MA) and GAPDH. GAPDH was used as an internal control for the amount of cDNA used in each reaction. Relative levels of mRNA were quantified using a Molecular Dynamics PhosphorImager. GAPDH primers were purchased from Clontech. The PTHrP primers used were as follows (Suda et al., 1996): PTHrP5′, TGG TGT TCC TGC TCA GCT A, and PTHrP3′, CCT CGT CGT CTG ACC CAA A.
Results
TGF-β1 Alters Development of Embryonic Metatarsal Rudiments Grown in Organ Culture
Previously, we showed that expression of a dominant-negative mutation of the TGF-β type II receptor in skeletal tissue in transgenic mice resulted in increased hypertrophic differentiation (Serra et al., 1997), suggesting that TGF-β regulates terminal differentiation in vivo. To determine the effects of exogenously added TGF-β1 on endochondral bone development, mouse metatarsal rudiments isolated from embryos at 15.5 d of gestation and grown in a chemically defined medium (Dieudonne et al., 1994) were used in the following experiments (Fig. 1). Organ culture allows for the study of complex biological processes in a three-dimensional structure in the context of native cell–cell and cell–extracellular matrix interactions. Over 5 d, cartilage organ cultures grew longitudinally and several stages of endochondral bone formation were detected (Fig. 1, a–d). The zone of hypertrophic cartilage (clear area plus dark area in the center of the rudiment), calcified zone (dark area), and proximal and distal bone ends representing resting and proliferating zones of cartilage have been described previously (Dieudonne et al., 1994; and see Fig. 1 d and Fig. 2 A). Treatment with 1 ng/ ml TGF-β1 (data not shown) and 10 ng/ml TGF-β1 (Fig. 1 e) for 5 d inhibited longitudinal growth of the cartilage rudiment (Fig. 1, d and e; control length = 3.5 ± 0.12 mm, n = 4; 10 ng/ml TGF-β1 = 2.46 ± 0.12 mm, n = 3). In addition, mineralized matrix was not detected in TGF-β1–treated cultures (compare Fig. 1, d and e).
Figure 1.

Embryonic mouse metatarsal bone rudiments grown in organ culture. Mouse metatarsals were isolated at 15.5 d p.c. (a) and placed in a chemically defined medium. By 1 (b) and 3 d in culture (c), the explants had grown longitudinally and a clear area of hypertrophic cartilage was observed in the center of the bone rudiments. After 5 d in culture (d), matrix mineralization was observed in the center of the explants. Treatment with 10 ng/ml TGF-β1 (e) inhibited longitudinal growth and matrix mineralization.
Figure 2.
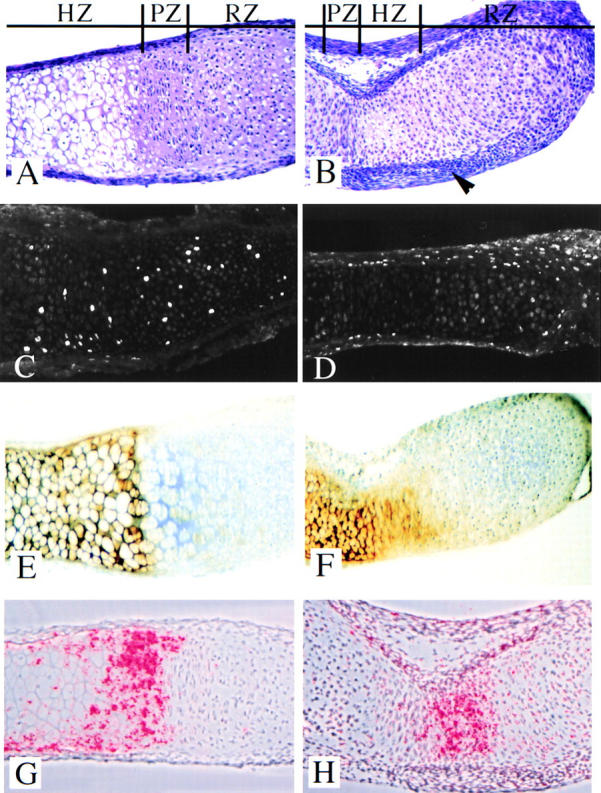
Metatarsal bone rudiment histology. Sections from embryonic metatarsal rudiments untreated (A) or treated with 10 ng/ml TGF-β1 (B) for 5 d were stained with hematoxylin and eosin. Zones of histologically defined resting (RZ), proliferating (PZ), and hypertrophic (HZ) cartilage were easily detected in sections from untreated cultures (A). Cartilage zones were not clearly demarcated in the TGF-β1–treated cultures, and the perichondrium (arrowhead) was thicker relative to the untreated controls (B). BrdU incorporation. Metatarsals in organ culture that were either untreated (C) or treated (D) with 10 ng/ml TGF-β1 for 24 h were labeled with BrdU for an additional 2.5 h. BrdU incorporation was detected using immunofluorescence. Labeled cells were detected throughout the prehypertrophic cartilage in untreated cultures (C). Labeled cells were not detected in the cartilage of TGF-β1–treated metatarsal cultures; however, many cells were labeled throughout the perichondrium (D). Differentiation markers. Type X collagen, a marker of hypertrophic cartilage, was immunolocalized to the histologically hypertrophic cartilage in untreated (E) and TGF-β1– treated (F) cultures. The fraction of cartilage that stained for type X collagen was reduced in TGF-β1–treated cultures (F) relative to the untreated control (E). No staining was detected in the absence of primary antibody (data not shown). Ihh mRNA, a marker for cells committed to become hypertrophic, was localized by in situ hybridization to sections from control (G) and TGF-β1– treated (H) metatarsal cultures. In untreated rudiments, Ihh mRNA was localized to a population of flat cells closest to the zone of hypertrophy, and expression continued into the hypertrophic layer (G). Ihh mRNA was localized to a very small population of cells in the center of the TGF-β1–treated cultures (H).
To further characterize the effects of TGF-β1 on bone development, the histology of untreated and TGF-β1– treated cartilage rudiments was examined. In untreated cartilage rudiments grown in culture for 5 d, histologically defined resting, proliferating, and hypertrophic zones were clearly demarcated in hematoxylin and eosin–stained sections (Fig. 2 A). These zones of cartilage were not easily recognized in cultures treated with 10 ng/ml TGF-β1, and treatment appeared to result in a decreased fraction of histologically hypertrophic cartilage (Fig. 2 B). In addition, the perichondrium in TGF-β1–treated cultures contained up to eight layers of cells, while the perichondrium in control cultures was only two to three cell layers thick (Fig. 2, A and B). The morphology and histology of TGF-β1–treated metatarsal bone rudiments suggested that TGF-β could have effects on both cell proliferation and differentiation.
The effect of TGF-β1 on bone length could be in part due to effects on cell proliferation. To test this hypothesis, bone rudiments were either treated with the TGF-β1 vehicle or were treated with 10 ng/ml TGF-β1 for 24 h followed by treatment with BrdU for 2.5 h. BrdU incorporation was assayed using immunofluorescence (Fig. 2, C and D). In control cultures, BrdU-labeled cells were detected throughout the histologically defined zones of resting and proliferating cartilage (Fig. 2 C). Cell proliferation in the histologically defined resting zone is not unusual in fetal bones. Treatment with TGF-β1 dramatically inhibited chondrocyte proliferation as measured by the lack of BrdU-labeled cells in the cartilage (Fig. 2 D), suggesting that inhibition of chondrocyte growth participates in the decrease in bone length observed after treatment with TGF-β1. In contrast, TGF-β1–treated rudiments had an increase in the number of BrdU-labeled cells in the perichondrium compared with the untreated cultures (Fig. 2, C and D), suggesting that TGF-β1 stimulated growth of perichondrial cells.
Immunolocalization of type X collagen was used to determine the effects of TGF-β1 on hypertrophic differentiation in the organ culture model. Type X collagen is a well-documented marker for hypertrophic cartilage (Schmid and Linsenmayer, 1987). Expression of Ihh, a marker for cells committed to become hypertrophic (Bitgood and McMahon, 1995; Vortkamp et al., 1996), was also used to determine the effects of TGF-β1 on hypertrophic differentiation. In untreated metatarsal rudiments grown in culture for 5 d, the matrix of the histologically hypertrophic cartilage contained type X collagen (Fig. 2 E). Ihh expression was localized to prehypertrophic chondrocytes in the lower zone of proliferation (transition cells) and continued into the zone of hypertrophy (Fig. 2 G). Treatment with TGF-β1 resulted in a decrease in the amount of histologically hypertrophic cartilage as well as a decrease in the fraction of the cartilage area stained for type X collagen (Fig. 2, E and F; control = 30 ± 3.7%, n = 11; TGF-β1 = 19.3 ± 3.9%, n = 10). This measurement takes into account changes in the total length of the bone rudiment and suggests that inhibition of hypertrophic differentiation occurs independent of the inhibition in longitudinal growth. Ihh mRNA was restricted to a small population of cells in the center of the TGF-β1–treated rudiment (Fig. 2 H), suggesting that treatment with TGF-β1 resulted in a decrease in the number of cells committed to hypertrophic differentiation in the metatarsal organ cultures. The data taken together suggest that TGF-β1 acts to inhibit several points of endochondral bone formation, including cell growth, hypertrophic differentiation, and matrix mineralization.
Embryonic Metatarsal Rudiments in Organ Culture Express the TGF-β Type I and Type II Receptors
TGF-βs signal through heteromeric serine/threonine kinase receptors (reviewed in Derynck, 1994; Massague et al., 1994; Ten Dijke et al., 1996; Alevizopoulos and Mermod, 1997). Both type I and type II receptors are required to generate a response to TGF-β (Laiho et al., 1990; Wrana et al., 1992; Ruberte et al., 1995). The current model is that TGF-β ligand binds to the TGF-β type II receptor on the cell surface (Wrana et al., 1994). The type II receptor is then able to recruit the type I receptor to form a heterotetrameric complex of two type I and two type II receptors. The type II receptor, which is a constitutively active kinase, phosphorylates the type I receptor, activating the type I serine/threonine kinase. Downstream targets of the type I receptor then transduce the signal to the nucleus. To determine which cell types in the cartilage rudiment cultures potentially respond to the TGF-β1 signal, expression of the TGF-β type I and type II receptors in embryonic mouse metatarsal rudiments isolated at 15.5 d of gestation and kept in culture for 24 h (data not shown) or 5 d (Fig. 3) was examined by immunofluorescence. The receptor expression pattern was similar at 24 h and 5 d of culture. Staining for the TGF-β type II receptor was detected at varying levels in all the cell types in the cartilage rudiment (Fig. 3). The highest levels of staining were detected in the perichondrium, the resting cartilage at the most distal ends of the metatarsal, the cells in the portion of proliferating zone closest to the hypertrophic zone (transition zone), and hypertrophic cells in the center of the rudiment. Strong staining was also observed in small round cells within the osteoid seam between the zone of hypertrophy and the perichondrium/periosteum. These cells are presumably osteoblasts. Immunofluorescent staining with an antibody directed to the TGF-β type I receptor demonstrated an expression pattern for the type I receptor similar to that observed for the type II receptor (Fig. 3). The pattern of expression for the receptor proteins suggests that all cells are potentially capable of responding to TGF-β1, but also suggests that cells in the perichondrium, distal tips of the rudiment, lower proliferating, and hypertrophic zones could be more sensitive to treatment with TGF-β1.
Figure 3.

Immunofluorescent localization of TGF-β type I and type II receptors. TGF-β type II (A and C) and type I (B and D) receptor proteins were localized by immunofluorescence in sections of metatarsal bone rudiments grown in culture for 5 d. Staining was observed in all cells in the metatarsal explants but was detected at higher levels in the perichondrium (white arrow, C and D), in flat cells in the lower portion of the zone of proliferation (dashed arrows, A and B), and in resting chondrocytes at the most distal tips of the metatarsal (A). Round cells, presumably osteoblasts, located between the perichondrium and zone of hypertrophy were also stained for both type I and type II receptors (arrowheads, C and D). Bar: (A and B) 100 microns; (C and D) 18 microns. No staining was detected in the absence of primary antibody (data not shown).
Treatment with TGF-β1 Stimulates Expression of PTHrP mRNA, and PTHrP Inhibits Hypertrophic Differentiation
To provide evidence for the model that TGF-β and PTHrP act in a common signal cascade to regulate the rate of chondrocyte differentiation, the hypothesis that TGF-β1 regulates expression of PTHrP mRNA in metatarsal cultures was tested. RNA extracted from cartilage rudiments either untreated or treated with 10 ng/ml TGF-β1 for 8 h or 5 d was analyzed using RT-PCR analysis (Fig. 4, A and B). Conditions were determined such that PCR product formation would be in the linear range. PTHrP mRNA levels were normalized to the expression of GAPDH, a constitutively active housekeeping gene. In three separate experiments, treatment with TGF-β1 resulted in an approximately threefold increase in PTHrP mRNA levels after 8 h, and the increased levels of PTHrP mRNA persisted for up to 5 d of treatment (Fig. 4 B). In situ hybridization was used to localize expression of PTHrP in cartilage rudiments untreated or treated with TGF-β1 for 6 h or 5 d (Fig. 4 C). PTHrP mRNA was localized to the periarticular region in both untreated and TGF-β1– treated cultures at 6 h and 5 d (Fig. 4 C). Cultures treated with TGF-β1 also demonstrated ectopic PTHrP mRNA expression in the perichondrium (Fig. 4 C). These data support the model that TGF-β1 acts to positively regulate PTHrP expression.
Figure 4.



PTHrP mRNA expression. (A) Metatarsal organ cultures were grown in the absence (−) or presence (+) or 10 ng/ml TGF-β1, and RNA was extracted after 8 h or 5 d of treatment. RNA was used in RT-PCR assays to determine PTHrP mRNA expression levels. PCR was performed under conditions determined to be in the linear range of product formation. PCR fragments were blotted to nylon membranes and hybridized to 32P-labeled PTHrP and GAPDH probes. (B) PTHrP levels were quantified using a Molecular Dynamics PhosphorImager. PTHrP expression was normalized to expression of GAPDH, a constitutive housekeeping gene. Data are shown as the fold induction relative to the untreated control in three separate experiments. N.D., not determined. (C) PTHrP mRNA was localized by in situ hybridization to sections from metatarsals either untreated (0β) or treated (+β) with 10 ng/ml TGF-β1 for 6 h or 5 d. In untreated cultures, PTHrP was localized to the periarticular region (arrow). TGF-β1–treated cultures demonstrated that PTHrP mRNA localized to the perichondrium (arrow) as well as to the periarticular region.
If TGF-β1 functions via increasing PTHrP expression, we would predict that PTHrP would have some of the same effects as TGF-β1 on bone development. Previously, it was shown that transgenic mice that misexpress PTHrP from the type II collagen promoter and mice that express a constitutively active PTH/PTHrP receptor show a delay in hypertrophic differentiation (Weir et al., 1996; Amling et al., 1997; Schipani et al., 1997). The hypothesis that PTHrP inhibits development of metatarsal rudiments grown in organ culture was tested, and the effects of PTHrP were compared with those of TGF-β1 (Figs. 5 and 6). Similar to treatment with TGF-β1 (Fig. 5 e), treatment with as little as 10−8 M PTHrP inhibited mineralization of the cartilage matrix in the explant cultures (Fig. 5 b) relative to the untreated control (Fig. 5 a). In contrast to TGF-β1, PTHrP at a concentration as high as 10−5 M did not result in significant inhibition of longitudinal growth in the explants when compared with the untreated cultures (Fig. 5, a, d, and e; control length = 3.64 ± 0.16 mm, n = 5; PTHrP = 3.86 ± 0.3 mm, n = 3; TGF-β1 = 2.52 ± 0.22 mm, n = 5). The hypothesis that PTHrP did not affect chondrocyte proliferation was confirmed using the BrdU incorporation assay (Fig. 6, D and E). Cultures were untreated or treated with 10−6 M PTHrP for 24 h followed by treatment with BrdU for an additional 2.5 h. BrdU was detected using immunofluorescence. There were no detectable differences in BrdU labeling in control (Fig. 6 D) and PTHrP-treated (Fig. 6 E) cultures. The effects of PTHrP on metatarsal organ cultures were further characterized by staining with hematoxylin and eosin (Fig. 6, A–C) and immunostaining for type X collagen (data not shown). Histological examination of organ cultures revealed a decrease, relative to the untreated control (Fig. 6 A), in the fraction of the cartilage area that contained histologically hypertrophic cartilage and stained with type X collagen in both PTHrP- (Fig. 6 B) and TGF-β1– (Fig. 6 C) treated cultures (control = 30 ± 3.7%, n = 11; TGF-β1 = 19.3 ± 3.9%, n = 10; PTHrP = 16.7 ± 3.2%, n = 3). The histology and BrdU labeling of perichondrium were not affected by PTHrP (Fig. 6, B and E). These data indicate that PTHrP and TGF-β have some distinct and some overlapping functions. Both PTHrP and TGF-β1 inhibit hypertrophic differentiation and matrix mineralization in the assays described above; however, in contrast to TGF-β1, PTHrP did not affect longitudinal growth, chondrocyte proliferation, or the perichondrium. A model whereby TGF-β could act through PTHrP to regulate hypertrophic differentiation and/or matrix mineralization is suggested.
Figure 5.

Effects of PTHrP on metatarsal bone rudiments grown in organ culture. Metatarsal explants were either untreated (a) or treated with 10−8 (b), 10−7 (c), or 10−5 M (d) PTHrP, or with 10 ng/ml TGF-β1 for 5 d (e). Matrix mineralization was inhibited at all concentrations of PTHrP. Longitudinal growth was not affected by treatment with PTHrP.
Figure 6.


Effects of PTHrP on metatarsal histology. Hematoxylin and eosin–stained sections of metatarsal bone rudiments grown in the absence of growth factors (A) or in the presence of 10−5 M PTHrP (B) or 10 ng/ml TGF-β1 (C) are shown. PTHrP (B) and TGF-β1 (C) treated rudiments demonstrate a smaller zone of hypertrophy (HZ) relative to the untreated control (A). Cells that were not hypertrophic (arrow) were scattered throughout the zone of hypertrophy in both PTHrP (B) and TGF-β1 (C) treated cultures. PTHrP-treated rudiments (B) had an extended histologically defined zone of proliferation (PZ) and resting cartilage (RZ), whereas TGF-β1– treated cultures (C) demonstrated a decrease in the length of the zone of proliferation. Thickened perichondrium was observed in the TGF-β1–treated explants (arrowhead, C) but not in the PTHrP-treated explants (B). BrdU labeling. Metatarsals either untreated (D) or treated with 10−5 M PTHrP (E) for 24 h were labeled with BrdU for an additional 2.5 h. BrdU incorporation was detected using immunofluorescence. There were no detectable differences in labeled cells in PTHrP-treated (E) and control (D) cultures.
TGF-β1 Has PTHrP-dependent and PTHrP-independent Effects on the Development of Embryonic Metatarsal Rudiments Grown in Organ Culture
Metatarsal organ cultures from embryos with targeted deletion of the PTHrP gene (Karaplis et al., 1994) were used to test the hypothesis that PTHrP is required for TGF-β–mediated effects on endochondral bone development. Two separate experiments were performed to compare the effects of 0 (Fig. 7, A and D), 1 (Fig. 7, B and E), and 10 ng/ml TGF-β1 (Fig. 7, C and F) on rudiments from PTHrP-positive (+/+ or +/−; Fig. 7, A–C) and PTHrP-null (−/−; Fig. 7, D–F) embryos. There were no detectable differences in cartilage rudiments from PTHrP-positive and PTHrP-null cultures at the time of dissection (15.5 d p.c.) or at the time TGF-β was added (16 h after dissection; data not shown). By 5 d of culture, rudiments from mice homozygous for the PTHrP deletion demonstrated increased matrix mineralization relative to PTHrP-positive controls (Fig. 7, A and D). This observation is consistent with the reported phenotype of PTHrP-null mice (Karaplis et al., 1994; Amizuka et al., 1994, 1996). PTHrP-positive cultures treated with TGF-β1 for 5 d demonstrated a dose-dependent decrease in length and mineralized matrix (Fig. 7, A–C; Table I). Cultures from PTHrP-null embryos demonstrated similar effects (Fig. 7, D–F; Table I), suggesting that PTHrP is not required for TGF-β1–mediated effects on growth or matrix mineralization. The effects on longitudinal growth were predicted in the previous experiment where addition of PTHrP to explant cultures did not alter growth.
Figure 7.

Effects of TGF-β1 on metatarsal explants from PTHrP-null mice. Metatarsal bone rudiments from PTHrP-positive (A–C) and PTHrP-null (D–F) mice were left untreated (A and D) or were treated with 1 (B and E) or 10 ng/ml TGF-β1 (C and F) for 5 d. Treatment with TGF-β1 resulted in decreased length and matrix mineralization in both PTHrP-positive and PTHrP-null cultures. Representative explants from one of the two separate experiments are shown.
Table I.
Metatarsal Bone Rudiment Length
| 0 ng TGF-β1 | 1 ng TGF-β1 | 10 ng TGF-β1 | ||||
|---|---|---|---|---|---|---|
| PTHrP+ | 3.44 ± 0.16 mm | 2.72 ± 0.10 mm | 2.14 ± 0.16 mm | |||
| n = 13 | n = 8 | n = 6 | ||||
| PTHrP− | 3.14 ± 0.13 mm | 2.56 ± 0.22 mm | 2.20 ± 0.22 mm | |||
| n = 11 | n = 5 | n = 6 |
To determine if PTHrP is required for the effects of TGF-β1 on metatarsal histology, hematoxylin and eosin– stained sections of organ cultures from PTHrP-positive (Fig. 8, A–C) and PTHrP-null embryos (Fig. 8, D–F) either untreated or treated with TGF-β1 were examined. Consistent with the assertion that PTHrP normally inhibits hypertrophic differentiation (Karaplis et al., 1994; Amizuka et al., 1994, 1996), all of the chondrocytes in untreated PTHrP-null rudiments were histologically hypertrophic (Fig. 8 D), whereas only a fraction of the cartilage in PTHrP-positive cultures is hypertrophic (Fig. 8 A). In PTHrP-positive cultures, TGF-β1 treatment resulted in a dose-dependent decrease in the fraction of chondrocytes in the histologically hypertrophic and proliferating zones (Fig. 8, A–C). In contrast, most of the chondrocytes in rudiments from PTHrP-null mice remained histologically hypertrophic despite treatment with 1 or 10 ng/ml TGF-β1 (Fig. 8, D–F). Treatment with 1 (Fig. 8, B and E) and 10 ng/ml TGF-β1 (Fig. 8, C and F) resulted in an increase in the thickness of the perichondrium relative to control cultures (Fig. 8, A and D) in both PTHrP-positive and PTHrP-null cultures. The data suggest PTHrP is not required for the effects of TGF-β on the perichondrium but indicate that PTHrP may be required for TGF-β–mediated effects on hypertrophic differentiation. To specifically determine the role of PTHrP in TGF-β–mediated effects on hypertrophic differentiation, sections from PTHrP-positive and PTHrP-null organ cultures untreated or treated with TGF-β1 were immunostained for type X collagen, a marker of hypertrophic differentiation (Fig. 9). Treatment of PTHrP-positive cultures with 1 ng (Fig. 9 B) and 10 ng/ml TGF-β1 (Fig. 9 C) resulted in a decrease in the fraction of cartilage stained for type X collagen relative to untreated cultures (Fig. 9 A). In contrast, TGF-β1 had no effect on the fraction of cartilage stained for type X collagen in PTHrP-null cultures (Fig. 9, D–F). Since treatment with TGF-β1 did not inhibit hypertrophic differentiation in PTHrP-null cultures, the data indicate that PTHrP is required for the effects of TGF-β1 on hypertrophic differentiation and suggest TGF-β acts upstream of PTHrP in a common signaling cascade to regulate differentiation. The data also suggest that TGF-β1 has both PTHrP-dependent and PTHrP-independent effects on endochondral bone formation.
Figure 8.
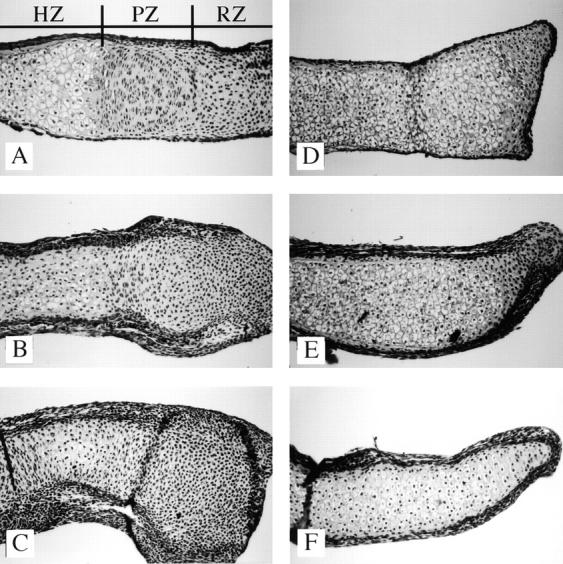
Effects of TGF-β1 on the histology of metatarsal explants from PTHrP-null mice. Sections of metatarsals from PTHrP-positive (A–C) and PTHrP-null (D–F) mice untreated (A and D) or treated with 1 (B and E) or 10 ng/ml TGF-β1 (C and F) were stained with hematoxylin and eosin. Zones of histologically defined hypertrophic (HZ), proliferating (PZ), and resting (RZ) cartilage were clearly visible in untreated cultures from PTHrP-positive mice (A). All of the cartilage in the untreated PTHrP-null cultures appeared hypertrophic (B). Treatment with TGF-β1 inhibited the fraction of histologically hypertrophic cartilage in the PTHrP-positive cultures (A–C) but not in the PTHrP-null cultures (D–F).
Figure 9.

Effects of TGF-β1 on type X collagen expression in PTHrP-null cultures. Sections of metatarsals from PTHrP-positive (A–C) and PTHrP-null (D–F) mice that were untreated (A and D) or treated with 1 (B and E) or 10 ng/ml TGF-β1 (C and F) were stained with an antibody to type X collagen and a peroxidase DAB substrate. Type X collagen protein is seen as the brown stain and is a marker for hypertrophic cartilage. All of the cartilage in the untreated PTHrP-null cultures stained positive for type X collagen (D). Treatment with TGF-β1 resulted in a decrease in the fraction of cartilage stained for type X collagen in the PTHrP-positive cultures (A–C) but did not affect the fraction of cartilage stained for type X collagen in the PTHrP-null cultures (D–F).
Discussion
Embryonic metatarsal bone rudiments grown in organ culture were used to test the hypothesis that TGF-β1 acts upstream of PTHrP to regulate chondrocyte differentiation. TGF-β1 acted at several check points during endochondral bone formation, inhibiting longitudinal growth, hypertrophic differentiation, and matrix mineralization. TGF-β drastically reduced BrdU labeling in chondrocytes of the histologically defined resting/proliferating zone by 24 h of treatment, suggesting that TGF-β affects longitudinal growth by regulating chondrocyte proliferation. However, additional effects of TGF-β on apoptosis or extracellular matrix production cannot be ruled out by the experiments presented here. PTHrP also inhibited chondrocyte hypertrophy and matrix mineralization but did not affect cell proliferation. TGF-β1 stimulated expression of PTHrP mRNA, suggesting that TGF-β1 and PTHrP could be part of the same signaling cascade to regulate hypertrophic differentiation and/or matrix mineralization. The hypothesis that PTHrP is required for TGF-β1–mediated effects on endochondral bone formation was tested using organ cultures from PTHrP-null mice. TGF-β1 did not inhibit hypertrophic differentiation in PTHrP-null cultures, suggesting that inhibition of hypertrophic differentiation occurred through a PTHrP-dependent mechanism. In contrast, longitudinal growth and matrix mineralization were inhibited by TGF-β1 in both PTHrP-expressing and PTHrP-null cultures, suggesting TGF-β has PTHrP-independent effects as well.
A model where longitudinal growth, hypertrophic differentiation, and matrix mineralization can be regulated independently is supported by data presented here and by others (Dieudonne et al., 1994; Long and Linsenmayer, 1998). Inhibition of hypertrophic differentiation by TGF-β1 is not solely a consequence of reduced proliferation. Treatment with TGF-β resulted in a decrease in the fraction of the bone area that stained for type X collagen. This measurement took into account differences in the total length of the bone rudiment and suggest that TGF-β inhibits hypertrophic differentiation in addition to inhibiting longitudinal growth. TGF-β was previously shown to be sufficient to inhibit hypertrophic differentiation in chondrocyte cultures (Kato et al., 1988; Ballock et al., 1993; Tschan et al., 1993; Bohme et al., 1995; Dieudonne et al., 1994). In addition, expression of a dominant-negative form of the TGF-β type II receptor in transgenic mice resulted in increased hypertrophic differentiation (Serra et al., 1997), suggesting that TGF-β is necessary to prevent hypertrophy in vivo. Likewise, inhibition of matrix mineralization by TGF-β1 is most likely not a consequence of reduced hypertrophic differentiation. Previously, it was shown that treatment with TGF-β1 resulted in only a 40% inhibition in hypertrophy but a complete inhibition of matrix mineralization, suggesting that TGF-β inhibited matrix mineralization independently of cellular differentiation (Dieudonne et al., 1994). In the present report, we demonstrate that TGF-β1 completely inhibited matrix mineralization in the absence of PTHrP even though these cultures were completely hypertrophic.
Immunolocalization of TGF-β type I and type II receptors suggests that all cells in the cartilage rudiment are potentially able to respond to TGF-β1; however, the data also suggest that some cell types may be more sensitive to treatment. High levels of receptor protein were localized to perichondrial cells, a subset of resting chondrocytes at the ends of the skeletal element, cells in the proliferating zone closest to the hypertrophic zone, and the hypertrophic cells in the center of the long bone rudiment. High levels of the TGF-β receptors in the perichondrium suggest that these cells are able to respond to TGF-β directly. Treatment with TGF-β was shown to increase the number of BrdU-labeled cells in the perichondrium, and TGF-β1 was able to stimulate perichondrial growth in the absence of PTHrP. Localization of the TGF-β receptors in chondrocytes suggests that TGF-β could act directly on chondrocytes to regulate growth or hypertrophic differentiation; alternatively, TGF-β1 could act indirectly through the perichondrium. Recently it was demonstrated that growth and differentiation of long bone cartilage are mediated by factors from the perichondrium (Long and Linsenmayer, 1998). Removal of the perichondrium from chick long bone rudiments resulted in an increase in the length of the explants, an increase in DNA synthesis in the zone of proliferation, and increased hypertrophic differentiation (Long and Linsenmayer, 1998). Long growth continued in the perichondrium-free chick cultures in the presence of exogenously added PTHrP even though hypertrophic differentiation was inhibited (Long and Linsenmayer, 1998). This is in agreement with our results in mouse metatarsal organ cultures where PTHrP inhibited hypertrophic differentiation but did not affect incorporation of BrdU. In addition, we observed that inhibition of longitudinal growth by TGF-β1 occurred independently of PTHrP, suggesting that, if the effect of TGF-β1 on growth is mediated through the perichondrium, another factor is involved. Fibroblast growth factor receptor 3 (FGFR3) is expressed in chondrocytes in the proliferating zone (Peters et al., 1993; Deng et al., 1996), and FGFR3-null mice demonstrate enhanced and prolonged longitudinal bone growth (Deng et al., 1996; Colvin et al., 1996). The ligand for the FGFR3 that regulates endochondral bone formation is not known. TGF-β may modulate the expression or activity of FGFs from the perichondrium and thereby indirectly regulate longitudinal growth.
This suggests that TGF-β1 mediates hypertrophic differentiation indirectly through PTHrP which is most likely secreted from the perichondrium. TGF-β1 stimulated expression of PTHrP mRNA in the cartilage rudiment cultures, and expression was localized to the perichondrium and periarticular area. TGF-β1 has been shown to stimulate expression of PTHrP mRNA and protein in several cultured cell types, including carcinoma cell lines, uterine and ovarian epithelial cells, and articular chondrocytes (Gillespie and Martin, 1994; Kiriyama et al., 1993; Merryman et al., 1994; Southby et al., 1996; Tsukazaki et al., 1995). Additional support of an indirect effect of TGF-β mediated through the perichondrium comes from transgenic mice that express a truncated, dominant-negative TGF-β type II receptor in the perichondrium. These mice demonstrate increased hypertrophic differentiation in the growth plate and suggest that TGF-β signaling in the perichondrium is required to regulate hypertrophy (Serra et al., 1997).
Development of skeletal elements requires the coordination of signals from several sources. It was recently shown that Ihh and PTHrP form a negative feedback loop that provides a mechanism for chondrocytes to sense and downregulate their rate of differentiation (Vortkamp et al., 1996; Lanske et al., 1996; Wallis, 1996). Misexpression of Ihh in chick limb cartilage rudiments resulted in inhibition of chondrocyte differentiation and increased expression of Ptc and Gli in the perichondrium, suggesting that the perichondrium contained the Ihh responding cells (Vortkamp et al., 1996). Misexpression of Ihh also resulted in increased expression of PTHrP, and it was shown that PTHrP was required for the inhibitory activities of Hh on chondrocyte differentiation (Vortkamp et al., 1996; Lanske et al., 1996). Expression of a dominant-negative form of the TGF-β type II receptor in mouse perichondrium results in increased terminal chondrocyte differentiation and increased and persistent expression of Ihh (Serra et al., 1997). Since Ihh normally acts as a negative regulator of differentiation, it was proposed that TGF-β signaling is required for Ihh-mediated inhibition of chondrocyte differentiation. There is precedent for such a model. The Drosophila protein, Dpp, a member of the TGF-β superfamily, can act as a secondary signal downstream of Hh to regulate the patterning of the imaginal disks (Heberlein et al., 1993; Ingham and Fietz, 1995), and BMP-2 and BMP-4 are induced by sonic Hh in the chick limb and hind gut (Laufer et al., 1994; Roberts et al., 1995). Furthermore, Hh- and TGF-β–related genes are coexpressed at many sites of cell–cell communication in the mouse embryo (Bitgood and McMahon, 1995). The present study suggests that TGF-β acts upstream of PTHrP to negatively regulate chondrocyte differentiation; however, whether or not TGF-β mediates the effects of Ihh on PTHrP has not yet been addressed. Future experiments will provide further evidence of how signaling from several factors is coordinated to build and maintain a functional skeletal system.
Acknowledgments
We wish to thank Dr. Bjorn Olsen for providing the antibody to mouse type X collagen, and Drs. McMahon and Kronenberg for providing Ihh and PTHrP cDNA probes. We would also like to thank Drs. Chin Chiang and Justin Grindley for suggestions during the preparation of the manuscript, and Rachael Evans for excellent technical assistance.
Abbreviations used in this paper
- BMP
bone morphogenic protein
- BrdU
bromo deoxyuridine
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- Hh
hedgehog
- Ihh
Indian hedgehog
- p.c.
post coitum
- PTH
parathyroid hormone
- PTHrP
PTH-related peptide
- RT
reverse transcription
Footnotes
Imaging work and analysis were performed in cooperation with the Vanderbilt University Medical Center Cell Imaging Resource supported by National Institutes of Health grants CA68485 and DK20593. This work was supported by grant AR45605 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R. Serra). We would also like to acknowledge Dr. H.L. Moses for his support (National Institutes of Health grants CA42572 and CA48799).
Address correspondence before June 1, 1999, to Rosa Serra, Department of Cell Biology, 649 MRBII, Vanderbilt Cancer Center, Nashville, TN 37232-3868. Tel.: (615) 936-1507. Fax: (615) 936-1790. After June 1, 1999, R. Serra's address will be Department of Cellular and Molecular Physiology, University of Cincinnati School of Medicine, 231 Bethesda, Rm. 4251, Cincinnati, OH 45267. Fax: (513) 558-5738.
References
- Alevizopoulos A, Mermod N. Transforming growth factor-β: the breaking open of a black box. BioEssays. 1997;19:581–591. doi: 10.1002/bies.950190709. [DOI] [PubMed] [Google Scholar]
- Amizuka N, Warshawsky H, Henderson JE, Goltzman D, Karaplis AC. Parathyroid hormone–related peptide-depleted mice show abnormal epiphyseal cartilage development and altered endochondral bone formation. J Cell Biol. 1994;126:1611–1623. doi: 10.1083/jcb.126.6.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amizuka N, Henderson JE, Hoshi K, Warshawsky H, Ozawa H, Goltzman D, Karaplis AC. Programmed cell death of chondrocytes and aberrant chondrogenesis in mice homozygous for parathyroid hormone-related peptide gene deletion. Endocrinology. 1996;137:5055–5067. doi: 10.1210/endo.137.11.8895380. [DOI] [PubMed] [Google Scholar]
- Amling M, Neff L, Tanaka S, Inoue D, Kuida K, Weir E, Philbrick WM, Broadus AE, Baron R. Bcl-2 lies downstream of parathyroid hormone–related peptide in a signaling pathway that regulates chondrocyte maturation during skeletal development. J Cell Biol. 1997;136:205–213. doi: 10.1083/jcb.136.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballock RT, Heydemann A, Wakefield LM, Flanders KC, Roberts AB, Sporn MB. TGF-β1 prevents hypertrophy of epiphyseal chondrocytes: regulation of gene expression for cartilage matrix proteins and metalloproteases. Dev Biol. 1993;158:414–429. doi: 10.1006/dbio.1993.1200. [DOI] [PubMed] [Google Scholar]
- Bitgood MJ, McMahon AP. Hedgehog and Bmpgenes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev Biol. 1995;172:126–138. doi: 10.1006/dbio.1995.0010. [DOI] [PubMed] [Google Scholar]
- Bohme K, Winterhalter KH, Bruckner P. Terminal differentiation of chondrocytes in culture is a spontaneous process and is arrested by transforming growth factor-β2 and basic fibroblast growth factor in synergy. Exp Cell Res. 1995;216:191–198. doi: 10.1006/excr.1995.1024. [DOI] [PubMed] [Google Scholar]
- Broadus, A.E., and A.F. Stewart. 1994. Parathyroid hormone-related protein. In The Parathyroids. J.P. Bilezikian, M.A. Levine, and R. Marcus, editors. Raven Press, Inc., New York. 259–294.
- Cancedda R, Cancedda FD, Castagnola P. Chondrocyte differentiation. Int Rev Cytol. 1995;159:265–358. doi: 10.1016/s0074-7696(08)62109-9. [DOI] [PubMed] [Google Scholar]
- Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390–397. doi: 10.1038/ng0496-390. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Denker AE, Nicoll SB, Tuan RS. Formation of cartilage-like spheroids by micromass cultures of murine C3H10T1/2 cells upon treatment with TGF-β1. Differentiation. 1994;59:25–34. doi: 10.1046/j.1432-0436.1995.5910025.x. [DOI] [PubMed] [Google Scholar]
- Derynck R. TGF-β-receptor-mediated signaling. Trends Biochem Sci. 1994;19:548–553. doi: 10.1016/0968-0004(94)90059-0. [DOI] [PubMed] [Google Scholar]
- Dieudonne SC, Semeins CM, Goei SW, Vukicevic S, Nulend JK, Sampath TK, Helder M, Burger EH. Opposite effects of osteogenic protein and TGF-β on chondrogenesis in cultured long bone rudiments. J Bone Miner Res. 1994;9:771–780. doi: 10.1002/jbmr.5650090603. [DOI] [PubMed] [Google Scholar]
- Erlebacher A, Filvaroff EH, Gitelman SE, Derynck R. Toward a molecular understanding of skeletal development. Cell. 1995;80:371–378. doi: 10.1016/0092-8674(95)90487-5. [DOI] [PubMed] [Google Scholar]
- Gatherer D, Ten P, Dijke, Baird DT, Akhurst RJ. Expression of TGF-β isoforms during first trimester human embryogenesis. Development. 1990;110:445–460. doi: 10.1242/dev.110.2.445. [DOI] [PubMed] [Google Scholar]
- Gillespie MT, Martin TJ. The parathyroid hormone-related protein gene and its expression. Mol Cell Endocrinol. 1994;100:143–147. doi: 10.1016/0303-7207(94)90293-3. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt M, Brook A, McMahon AP. The world according to hedgehog. TIG (Trends Genet) 1997;13:14–21. doi: 10.1016/s0168-9525(96)10051-2. [DOI] [PubMed] [Google Scholar]
- Heberlein U, Wolff T, Rubin GM. The TGFβ homolog dpp and the segment polarity gene hedgehog are required for propagation of a morphogenetic wave in the Drosophilaretina. Cell. 1993;75:913–926. doi: 10.1016/0092-8674(93)90535-x. [DOI] [PubMed] [Google Scholar]
- Ingham PW, Fietz MJ. Quantitative effects of hedgehog and decapentaplegic activity on the patterning of the Drosophilawing. Curr Biol. 1995;5:432–440. doi: 10.1016/s0960-9822(95)00084-4. [DOI] [PubMed] [Google Scholar]
- Karaplis AC, Luz A, Glowacki J, Bronson RT, Tybulewicz VLJ, Kronenberg HM, Mulligan RC. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994;8:277–289. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]
- Kato Y, Iwamoto M, Koike T, Suzuki F, Takano Y. Terminal differentiation and calcification in rabbit chondrocyte cultures grown in centrifuge tubes: regulation by transforming growth factor beta and serum factors. Proc Natl Acad Sci USA. 1988;85:9552–9556. doi: 10.1073/pnas.85.24.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiriyama T, Gillespie MT, Glatz JA, Fukumoto S, Moseley JM, Martin TJ. Transforming growth factor-β stimulation of parathyroid hormone-related protein (PTHrP): a paracrine regulator? . Mol Cell Endocrinol. 1993;92:55–62. doi: 10.1016/0303-7207(93)90074-t. [DOI] [PubMed] [Google Scholar]
- Kulyk WM, Rodgers BJ, Greer K, Kosher RA. Promotion of embryonic chick limb cartilage differentiation by transforming growth factor-beta. Dev Biol. 1989;135:424–430. doi: 10.1016/0012-1606(89)90191-7. [DOI] [PubMed] [Google Scholar]
- Laiho M, Weis MB, Massague J. Concomitant loss of transforming growth factor (TGF)-β receptor types I and II in TGF-β-resistant cell mutants implicates both receptor types in signal transduction. J Biol Chem. 1990;265:18518–18524. [PubMed] [Google Scholar]
- Lanske B, Karaplis AC, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize LHK, Ho C, Mulligan RC, et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273:663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- Laufer E, Nelson CE, Johnson RL, Morgan BA, Tabin C. Sonic hedgehog and FGF-4 act through a signal cascade and feedback loop to integrate growth and patterning of the developing limb bud. Cell. 1994;79:993–1003. doi: 10.1016/0092-8674(94)90030-2. [DOI] [PubMed] [Google Scholar]
- Lee K, Deeds JD, Bond AT, Juppner H, Abou-Samara AB, Segre GV. In situ localization of PTH/PTHrP receptor mRNA in the bone of fetal and young rats. Bone. 1993;14:341–345. doi: 10.1016/8756-3282(93)90162-4. [DOI] [PubMed] [Google Scholar]
- Lee K, Deeds JD, Segre GV. Expression of parathyroid hormone-related peptide and its receptor mRNA during fetal development of rats. Endocrinology. 1995;136:453–463. doi: 10.1210/endo.136.2.7835276. [DOI] [PubMed] [Google Scholar]
- Leonard CM, Fuld HM, Frenz DA, Downie SA, Massague J, Newman SA. Role of transforming growth factor-β in chondrogenic pattern formation in the embryonic limb: stimulation of mesenchymal condensation and fibronectin gene expression by exogenous TGF-β and evidence for endogenous TGF-β-like activity. Dev Biol. 1991;145:99–109. doi: 10.1016/0012-1606(91)90216-p. [DOI] [PubMed] [Google Scholar]
- Long F, Linsenmayer TF. Regulation of growth region cartilage proliferation and differentiation by perichondrium. Development. 1998;125:1067–1073. doi: 10.1242/dev.125.6.1067. [DOI] [PubMed] [Google Scholar]
- Massague J, Cheifetz S, Boyd FT, Andres JL. TGF-beta receptors and TGF-beta binding proteoglycans: recent progress in identifying their functional properties. Ann NY Acad Sci. 1990;593:59–72. doi: 10.1111/j.1749-6632.1990.tb16100.x. [DOI] [PubMed] [Google Scholar]
- Massague J, Attisano L, Wrana JL. The TGF-β family and its composite receptors. Trends Cell Biol. 1994;4:172–178. doi: 10.1016/0962-8924(94)90202-x. [DOI] [PubMed] [Google Scholar]
- Merryman JI, DeWille JW, Werkmeister JR, Capen CC, Rosol TJ. Effects of transforming growth factor-β on parathyroid hormone-related protein production and ribonucleic acid expression by a squamous carcinoma cell line in vitro. Endocrinology. 1994;134:2424–2430. doi: 10.1210/endo.134.6.8194469. [DOI] [PubMed] [Google Scholar]
- Millan FA, Denhez F, Kondaiah P, Akhurst R. Embryonic gene expression patterns of TGF beta 1, beta 2, and beta 3 suggest different developmental functions in vivo. Development. 1991;111:131–143. doi: 10.1242/dev.111.1.131. [DOI] [PubMed] [Google Scholar]
- Moses, H.L. 1990. The biological actions of transforming growth factor beta. In Growth Factors from Genes to Clinical Application. V. Sara, K. Hall, and H. Low, editors. Raven Press, Inc., New York. 141–155.
- Moses HL, Serra R. Regulation of differentiation by TGF-β. Curr Opin Genet Dev. 1996;6:581–586. doi: 10.1016/s0959-437x(96)80087-6. [DOI] [PubMed] [Google Scholar]
- Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. . Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- Pelton RW, Dickinson ME, Moses HL, Hogan BLM. In situ hybridization analysis of TGF-β3 RNA expression during mouse development: comparative studies with TGF-β1 and -β2. Development. 1990a;110:600–620. doi: 10.1242/dev.110.2.609. [DOI] [PubMed] [Google Scholar]
- Pelton RW, Hogan BLM, Miller DA, Moses HL. Differential expression of genes encoding TGFs β-1, β-2, and β-3 during murine palate formation. Dev Biol. 1990b;141:456–460. doi: 10.1016/0012-1606(90)90401-4. [DOI] [PubMed] [Google Scholar]
- Peters K, Ornitz D, Werner S, Williams L. Unique expression pattern of the FGF receptor 3 gene during mouse organogenesis. Dev Biol. 1993;155:423–430. doi: 10.1006/dbio.1993.1040. [DOI] [PubMed] [Google Scholar]
- Roberts DJ, Johnson RL, Burke AC, Nelson CE, Morgan BA, Tabin C. Sonic hedgehog is an endodermal signal inducing Bmp-4 and Hoxgenes during induction and regionalization of the chick hindgut. Development. 1995;121:3163–3174. doi: 10.1242/dev.121.10.3163. [DOI] [PubMed] [Google Scholar]
- Ruberte E, Marty T, Nellen D, Affolter M, Basler K. An absolute requirement for both the type II and type I receptors, punt and thick veins, for dpp signaling in vivo. Cell. 1995;80:889–897. doi: 10.1016/0092-8674(95)90292-9. [DOI] [PubMed] [Google Scholar]
- Sandberg M, Vurio T, Hirrovan H, Alitalo K, Vuorio E. Enhanced expression of TGF-beta and c-fos mRNAs in the growth plates of developing human long bones. Development. 1988;102:461–470. doi: 10.1242/dev.102.3.461. [DOI] [PubMed] [Google Scholar]
- Schipani E, Kruse K, Juppner H. A constitutively active mutant PTH/PTHrP receptor in Jansen-type metaphyseal chondrodysplasia. Science. 1995;268:98–100. doi: 10.1126/science.7701349. [DOI] [PubMed] [Google Scholar]
- Schipani E, Lanske B, Hunzelman J, Luz A, Kovacs CS, Lee K, Pirro A, Kronenberg HM, Juppner H. Targeted expression of constitutively active receptors for parathyroid hormone and parathyroid hormone-related peptide delays endochondral bone formation and rescues mice that lack parathyroid hormone-related peptide. Proc Natl Acad Sci USA. 1997;94:13689–13694. doi: 10.1073/pnas.94.25.13689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, T.M., and T.F. Linsenmayer. 1987. Type X collagen. In Structure and function of collagen types. R. Mayne and R.E. Bergeson, editors. Academic Press, Inc., Orlando. 223–259.
- Serra R, Johnson M, Filvaroff EH, LaBorde J, Sheehan DM, Derynck R, Moses HL. Expression of a truncated, kinase-defective TGF-β type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J Cell Biol. 1997;139:541–552. doi: 10.1083/jcb.139.2.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southby J, Murphy LM, Martin TJ, Gillespie MT. Cell-specific and regulator-induced promoter usage and mRNA splicing for parathyroid hormone-related protein. Endocrinology. 1996;137:1349–1357. doi: 10.1210/endo.137.4.8625910. [DOI] [PubMed] [Google Scholar]
- Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975;98:503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- Suda N, Gillespie MT, Traianedes K, Zhou H, Ho PWM, Hards DK, Allan EH, Martin TJ, Moseley JM. Expression of parathyroid hormone related protein in cells of osteoblastic lineage. J Cell Physiol. 1996;116:94–104. doi: 10.1002/(SICI)1097-4652(199601)166:1<94::AID-JCP11>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Suva LJ, Winslow GA, Wettenhall REH, Hammonds RG, Moseley JM, Diefenbach-Jagger H, Rodda CP, Kemp BE, Rodrigues H, Chen EY. A parathyroid hormone-related protein implicated in malignant hypercalcemia: cloning and expression. Science. 1987;237:893–896. doi: 10.1126/science.3616618. [DOI] [PubMed] [Google Scholar]
- Ten Dijke, P., K. Miyazono, and C.-H. Heldin. Signaling via hetero-oligomeric complexes of type I and type II serine threonine kinase receptors. Curr Opin Cell Biol. 1996;8:139–145. doi: 10.1016/s0955-0674(96)80058-5. [DOI] [PubMed] [Google Scholar]
- Tschan T, Bohme K, Conscience EM, Zenke G, Winterhalter KH, Bruckner P. Autocrine or paracrine TGF-β modulates the phenotype of chick embryo sternal chondrocytes in serum-free agarose culture. J Biol Chem. 1993;5:5156–5161. [PubMed] [Google Scholar]
- Tsukazaki T, Ohtsuru A, Enomoto H, Yano H, Motomura K, Ito M, Namba H, Iwasaki K, Yamashita S. Expression of parathyroid hormone related protein in rat articular cartilage. Calcif Tissue Int. 1995;57:196–200. doi: 10.1007/BF00310258. [DOI] [PubMed] [Google Scholar]
- Vortkamp A, Lee K, Lanske B, Segre GV, Kroneberg HM, Tabin CJ. Regulation of rate of chondrocyte differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–621. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- Wallis GA. Bone growth: coordinating chondrocyte differentiation. Curr Biol. 1996;6:1577–1580. doi: 10.1016/s0960-9822(02)70776-8. [DOI] [PubMed] [Google Scholar]
- Weir EC, Philbrick WM, Amling M, Neff L, Baron R, Broadus AE. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc Natl Acad Sci USA. 1996;93:10240–10245. doi: 10.1073/pnas.93.19.10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana JL, Carcamo J, Attisano L, Cheifetz S, Zentella A, Lopez-Casillas F, Massagué J. The type II TGF-β receptor signals diverse responses in cooperation with the type I receptor. Cold Spring Harbor Symp Quant Biol. 1992;57:81–86. doi: 10.1101/sqb.1992.057.01.011. [DOI] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Wieser R, Ventura F, Massagué J. Mechanism of activation of the TGF-β receptor. Nature. 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]