

Abstract
Tyrosine phosphorylation of the high affinity immunoglobulin (Ig)E receptor (FcεRI) by the Src family kinase Lyn is the first known biochemical step that occurs during activation of mast cells and basophils after cross-linking of FcεRI by antigen. The hypothesis that specialized regions in the plasma membrane, enriched in sphingolipids and cholesterol, facilitate the coupling of Lyn and FcεRI was tested by investigating functional and structural effects of cholesterol depletion on Lyn/FcεRI interactions. We find that cholesterol depletion with methyl-β-cyclodextrin substantially reduces stimulated tyrosine phosphorylation of FcεRI and other proteins while enhancing more downstream events that lead to stimulated exocytosis. In parallel to its inhibition of tyrosine phosphorylation, cholesterol depletion disrupts the interactions of aggregated FcεRI and Lyn on intact cells and also disrupts those interactions with detergent-resistant membranes that are isolated by sucrose gradient ultracentrifugation of lysed cells. Importantly, cholesterol repletion restores receptor phosphorylation together with the structural interactions. These results provide strong evidence that membrane structure, maintained by cholesterol, plays a critical role in the initiation of FcεRI signaling.
Keywords: methyl-β-cyclodextrin, immunoglobulin E receptor, plasma membrane structure, lipid domains, signal transduction
T he first known biochemical event in antigen stimulation of mast cells and basophils is tyrosine phosphorylation of the β and γ subunits of the high affinity IgE receptor (FcεRI) by the Src family kinase Lyn; however, the mechanism by which these proteins interact is not fully understood. A common view is that this receptor–kinase coupling occurs strictly via protein–protein interactions. For example, in the transphosphorylation model, antigen-induced cross-linking causes Lyn, which is bound weakly to one receptor, to phosphorylate immune tyrosine activation motifs on a juxtaposed receptor, thereby initiating signal transduction (Jouvin et al., 1994; Pribluda et al., 1994). We recently presented a different view that the plasma membrane structure plays an integral role in facilitating coupling between Lyn and FcεRI. In particular, we proposed that FcεRI tyrosine phosphorylation occurs within specialized regions in the plasma membrane, enriched in sphingolipids and cholesterol (Field et al., 1997; Sheets et al., 1999). Membrane structures of this composition have been isolated from many cell types and primarily characterized on the basis of their resistance to solubilization by nonionic detergents such as Triton X-100 (TX-100)1 and their consequent buoyancy in sucrose density gradients. These isolated detergent-resistant membranes (DRMs) (also referred to in the literature as DIGs [detergent insoluble glycolipid domains], GEMs [glycolipid-enriched membranes], and membrane rafts) are enriched in cholesterol, sphingomyelin, glycosphingolipids, and saturated glycerophospholipids, as well as dually acylated Src family kinases (e.g., Lyn, Fyn, Yes) and glycosylphosphatidylinositol (GPI)-anchored proteins (for review see Brown and London, 1998a; Simons and Ikonen, 1997). They are postulated to represent plasma membrane domains that may function as centers for signal transduction and membrane trafficking, although their nature on the cell surface is controversial (Brown and London, 1998a; Edidin, 1997).
In initial studies we found that cross-linking of FcεRI increases the percentage of cellular Lyn recovered in DRMs from TX-100–lysed RBL-2H3 mast cells, suggesting that FcεRI aggregation causes an alteration of DRMs that may be involved in FcεRI-mediated signaling (Field et al., 1995). With sufficiently low concentrations of TX-100 to lyse the cells, aggregated (but not monomeric) FcεRI also associate with DRM vesicles, and only this population of receptors is phosphorylated upon cross-linking (Field et al., 1997). Furthermore, fluorescence microscopy on intact cells revealed that cross-linking of FcεRI induces co-redistribution with DRM components, including a GD1b ganglioside (Pierini et al., 1996), as well as Lyn and the GPI-anchored protein Thy-1 (Holowka, D., E.D. Sheets, and B. Baird, manuscript in preparation), and with saturated phospholipid analogues (Thomas et al., 1994). Together, these results are consistent with the view that specialized membrane domains function in cells to facilitate coupling between aggregated FcεRI and Lyn.
The detergent resistance of these membrane structures has been hypothesized to depend upon their lipid phase (Brown and London, 1998b). Cholesterol, which profoundly affects phase behavior of lipids, was found to be a major lipid component of isolated DRMs derived from MDCK cells (Brown and Rose, 1992). In subsequent studies, Brown and colleagues found that model membranes with compositions similar to DRMs are not solubilized by TX-100, and this detergent resistance was observed to correlate with cholesterol concentrations that induce formation of the liquid-ordered (L o) phase (Schroeder et al., 1994; Ahmed et al., 1997; Schroeder et al., 1998). This phase results from cholesterol having a gel phase–like ordering effect on the saturated and near-saturated acyl chains of glycerophospholipids and sphingolipids; yet the lipids retain a high degree of lateral and rotational mobility, similar to lipids in the fluid liquid crystalline phase (Brown and London, 1998a,b). The relevance of this structure for biological membranes is supported by recent electron spin resonance measurements that showed parameters characteristic of the L o phase for DRM vesicles isolated from RBL cells (Ge et al., 1999). Overall, these results support the hypothesis that ordered lipid domains coalescing on the plasma membrane after FcεRI aggregation serve to co-localize FcεRI and Lyn and thereby initiate receptor phosphorylation and signaling.
As reported here, we tested this hypothesis by using methyl-β-cyclodextrin (MβCD) to selectively deplete cholesterol from RBL-2H3 cells, and we investigated the functional and structural effects of the MβCD treatment on FcεRI/Lyn interactions. This reagent has been used recently for efficient removal of cholesterol from a variety of cell types (Kilsdonk et al., 1995; Yancey et al., 1996; Christian et al., 1997; Gimpl et al., 1997; Scheiffele et al., 1997; Friedrichson and Kurzchalia, 1998; Keller and Simons, 1998; Varma and Mayor, 1998). We find that cholesterol depletion substantially reduces stimulated tyrosine phosphorylation of FcεRI and other substrates in RBL cells. Furthermore, we find that cholesterol depletion selectively disrupts the structural interactions between aggregated FcεRI and Lyn both for DRM vesicles from lysed cells that are isolated on sucrose gradients and for intact cells as assessed by confocal fluorescence microscopy. When cholesterol levels are repleted, these functional effects and molecular associations are restored. These results provide strong evidence that cholesterol is required for effective functional coupling between aggregated FcεRI and Lyn, and they are consistent with an important structural role for liquid-ordered membrane domains in this coupling.
Materials and Methods
Cholesterol Depletion and Repletion
RBL-2H3 cells were maintained and harvested as previously described (Pierini et al., 1996). Mouse monoclonal IgE specific for 2,4-dinitrophenyl (DNP) (Liu et al., 1980) was purified as previously described (Subramanian et al., 1996); biotinylated and iodinated (Field et al., 1995) or FITC-labeled (Pierini et al., 1996) IgE was used to sensitize cells in some experiments. Mouse monoclonal anti-1,5-dansyl IgE was affinity purified as previously described (Weetall et al., 1993). Other RBL cell membrane components were labeled with AA4 mAb (a gift from Dr. Reuben Siraganian, National Institutes of Health, Bethesda, MD), specific for the α-galactosyl GD1b ganglioside derivative; OX-7 (PharMingen), specific for the GPI-anchored protein Thy-1; and anti-Lyn (Upstate Biotechnology, Inc., and Santa Cruz Biotechnology) as previously described (Field et al., 1995; Pierini et al., 1996). Transferrin receptors (TfRs; CD71) were labeled with a monoclonal antibody from PharMingen, followed by Cy3-goat anti–mouse γ chain (Southern Biotechnology Associates).
To remove cholesterol, suspended cells (2–4 × 106 cells/ml) were incubated for 1 h at 37°C in the presence or absence of 10 mM MβCD (Sigma Chemical Co.) in BSA-containing buffered saline solution (BSA/BSS: 20 mM Hepes, pH 7.4, 135 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5.6 mM glucose, and 1 mg/ml BSA), then washed with BSA/BSS before stimulation. For some experiments, cholesterol (Avanti Polar Lipids) was added back to cholesterol-depleted cells (2 × 106 cells/ml) in BSA/BSS by incubation for 2 h at 37°C with indicated concentrations of MβCD/cholesterol (8:1, mol/mol) complexes. These complexes were prepared similarly to a previously described procedure (Racchi et al., 1997). In brief, cholesterol in a chloroform solution was dried under nitrogen in a glass culture tube precleaned with ethanolic KOH. An appropriate volume of sterile-filtered 300 mM MβCD in BSA/BSS was added to the tube, and the resulting suspension was vortexed and bath sonicated until the suspension clarified. The complex was then incubated in a rocking water bath overnight at 37°C to maximize formation of soluble complexes.
In Vivo Tyrosine Phosphorylation Assays
Suspended RBL-2H3 cells that had been sensitized with anti-DNP IgE (Chang et al., 1995) and cholesterol depleted/repleted or not were stimulated at a density of 106 cell/ml with multivalent DNP-BSA (1 μg/ml; Xu et al., 1998a) at 37°C for indicated times, lysed by addition of 5× SDS sample buffer (50% glycerol, 0.25 M Tris, pH 6.8, 5% SDS, 0.5% bromphenol blue) and boiled for 5 min and centrifuged for 5 min at 13,000 g. Equal numbers of cell equivalents of lysates (typically, 8 × 103 cell equivalents) were electrophoresed on 12% nonreduced SDS polyacrylamide gels, transferred to Immobilon-P membranes (Millipore Corp.), and probed with horseradish peroxidase–conjugated antiphosphotyrosine (4G10-HRP; Upstate Biotechnology, Inc.). Enhanced chemiluminescence (Pierce) was used for detection. Phosphorylation as a function of stimulation time was quantified after scanning blots and analyzing with Un-Scan-It (Silk Scientific) and Igor Pro (WaveMetrics).
Quantitative Measurements of FcεRI
We determined the effect of MβCD on the amount of FcεRI associated with RBL-2H3 cells by two different methods. For some experiments, biotinylated 125I-IgE was bound to FcεRI under saturating conditions, and these labeled cells were used to monitor the loss of IgE–FcεRI complexes after treatment with or without MβCD as described above. The state of 125I-IgE released from the cells during MβCD treatment was assessed by collecting the supernatants after cell pelleting (200 g, 5 min) and subjecting these to a high speed centrifugation (250,000 g, 45 min, 4°C). Gamma counting indicated that 54% of the 125I-IgE was pelleted during the second centrifugation, in contrast to only 6% of the 125I-IgE that could be pelleted under these conditions for untreated cells. In another set of experiments, FcεRI were saturated with FITC-IgE, and cells were treated with or without MβCD. Receptor-bound FITC-IgE was measured on washed cells with steady-state fluorimetry as previously described (Xu et al., 1998b). Examination of these cells by fluorescence microscopy in the presence or absence of 15 mM NH4Cl to neutralize endosomes (Xu et al., 1998b) showed no evidence for internalization of FITC-IgE after MβCD treatment.
To examine the relationship between the density of anti-DNP IgE on the cells and antigen-stimulated tyrosine phosphorylation, FcεRI were saturated with mixtures of anti-DNP IgE and antidansyl IgE in percentage mixtures of 30:70, 50:50, and 100:0. Washed cells were stimulated with 1 μg/ml DNP-BSA at 37°C for various times and tyrosine phosphorylation was analyzed. Competition binding between each of these unlabeled antibodies and FITC-labeled anti-DNP IgE was carried out under identical conditions. With steady-state fluorimetry to quantify the amount of cell-bound FITC-IgE, we confirmed that the percentages of anti-DNP IgE and antidansyl IgE bound to FcεRI in the tyrosine phosphorylation experiments were identical to the percentages added with an uncertainty of ± 8%. Furthermore, fluorimetry experiments with mixtures of FITC anti-DNP IgE and unlabeled antidansyl IgE showed that DNP-BSA binding and cross-linking induced internalization of FITC-IgE–FcεRI proportionally to the amount of FITC-IgE bound in the range of 30–100% occupancy by this IgE (data not shown; Xu et al., 1998a,b).
Sucrose Gradients
Cholesterol-depleted/repleted or untreated cells were fractionated on sucrose step gradients as previously described (Field et al., 1997), except that the concentration of TX-100 during lysis was 0.04% instead of 0.05%. Aliquots (200 μl) were removed from the top of the gradient, and γ-radiation of biotinylated 125I-IgE was counted. The gradient fractions were then pooled as indicated, boiled with SDS sample buffer, and blotted as described above, except that the primary antibody was rabbit anti-Lyn antibody (Upstate Biotechnology, Inc.) and the secondary antibody was HRP-conjugated donkey anti–rabbit Ig (Amersham Pharmacia Biotech). To determine the location of GD1b in the gradients, cells labeled with 125I-AA4 and biotinylated IgE were analyzed as previously described (Field et al., 1995). In some experiments, the pooled gradient fractions were electrophoresed on 12% nonreduced SDS-acrylamide gels and subsequently were silver stained (Daiichi Silver Stain II; Owl Separation Systems).
Immunodepletion of FcεRI
Anti-DNP IgE–sensitized cells were stimulated for the indicated times with DNP-BSA (1 μg/ml) and lysed on ice with TX-100 lysis buffer [10 mM Tris, pH 8.0, 50 mM NaCl, 1 mM Na3VO4, 30 mM sodium pyrophosphate, 10 mM sodium glycerophosphate, 0.02 U/ml aprotinin, 0.01% NaN3, 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride, and 0.2% TX-100] followed by addition of 10 μM DNP-aminocaproyl-l-tyrosine, as previously described (Harris et al., 1997). After centrifugation at 13,000 g for 5 min to remove insoluble material, lysates were incubated with or without 20 μg/ml rabbit anti-IgE (Menon et al., 1984). After incubation with protein A–agarose beads (Pierce), samples were centrifuged to pellet the beads and supernatants were removed, boiled in SDS sample buffer, electrophoresed, and blotted with 4G10-HRP as described above. Rabbit anti-IgE/IgE–FcεRI complexes are bound to the protein A–agarose, which result in selective depletion of the FcεRI β and γ bands.
Degranulation Assays
The degranulation response that occurs after stimulation with the antigen DNP-BSA or the calcium ionophore A23187 (Calbiochem-Novabiochem) for 1 h at 37°C was carried out as described previously (Harris et al., 1997), except that cells were treated with or without MβCD immediately before stimulation.
Confocal Immunofluorescence Microscopy
Cells sensitized with FITC-IgE were treated with or without MβCD, then washed and incubated with cytochalasin D (1 μg/ml) for 5 min at room temperature to prevent antigen-stimulated IgE–FcεRI internalization and to sustain Lyn co-redistribution with patched IgE–FcεRI at the cell surface. The cells were then stimulated with 1.7 μg/ml DNP-BSA at room temperature for 20 min and subsequently fixed with cold methanol for Lyn labeling or with formaldehyde for GD1b or TfR labeling as previously described (Pierini et al., 1996). Confocal fluorescence microscopy was performed as previously described (Pierini et al., 1996). Cross correlation analysis of the co-redistribution of Lyn or TfR with antigen–cross-linked FITC-IgE–FcεRI were carried out on equatorial images of individual cells using a computational procedure similar to that previously described (Stauffer and Meyer, 1997). Peak values can be calculated from this analysis to a correlation coefficient (ρ) (Eq. 1; Barlow, 1989), and these values were averaged for 7–13 cells from each sample for numerical comparison of the degree of co-redistribution.
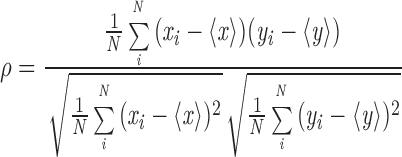 |
1 |
In Eq. 1, xi and yi are the intensities at each equatorial point of the FITC and Cy3 fluorescence, respectively, and < x > and < y > are the corresponding average values.
Lipid Extractions and Analyses
RBL-2H3 cells were depleted of cholesterol (or not) and repleted (or not) as described above. After treatment, the cells were washed with BSA/ BSS, and then resuspended in methanol. The cell lysate was homogenized with a Duall ground glass tissue grinder, transferred to an ethanolic KOH-cleaned glass vial, and an equal volume of chloroform was added, followed by vigorous vortexing and probe sonication. The samples were rocked overnight at room temperature, then centrifuged at 800 g for 5 min, and the supernatants were transferred to a new glass vial. Chloroform/methanol (1:1, vol/vol) was added to the pellets and the suspension was vortexed vigorously and centrifuged as described above. Supernatants consisting of the total lipid extracts were combined and stored under nitrogen at −20°C.
The extent of cholesterol depletion or repletion was measured using a colorimetric cholesterol oxidase assay (Boehringer Mannheim) that quantifies total free cholesterol. Total lipid extracts from paired samples of MβCD-treated and untreated cells were assayed simultaneously in these determinations. Thin layer chromatography of the total lipid extracts was carried out on silica gel 60 plates (EM Science) as previously described (Cartwright, 1993), with iodine detection. The developing solvent for polar lipids was chloroform/methanol/glacial acetic acid/water (60:50:1:4, vol/vol), and for neutral lipids, hexane/diethyl ether/glacial acetic acid (90: 10:1, vol/vol) (Cartwright, 1993).
Results
Cholesterol Depletion Inhibits Antigen-stimulated Tyrosine Phosphorylation of FcεRI
Because cholesterol is a critical component of liquid-ordered DRMs, we investigated whether reduction of the cholesterol content of the RBL-2H3 mast cells affects the tyrosine phosphorylation of FcεRI β and γ subunits. For this purpose, we incubated the cells with 10 mM MβCD for 1 h at 37°C to deplete cellular cholesterol before stimulation with multivalent antigen. Fig. 1 a shows a representative antiphosphotyrosine Western blot of lysates from control and MβCD-treated cells that have been stimulated with an optimal dose of antigen, DNP-BSA, for 0–30 min at 37°C. For the control cells, stimulated tyrosine phosphorylation is maximal after 2 min of stimulation with 1 μg/ml DNP-BSA, and then declines over time as previously reported (Pribluda and Metzger, 1992; Xu et al., 1998a). For the MβCD-treated cells, there is a substantial reduction in stimulated tyrosine phosphorylation in all bands detected, including the β and γ subunits of FcεRI that are readily observed under these conditions when the 4G10 mAb is used to detect phosphotyrosine. Fig. 1 b quantifies the time course of tyrosine phosphorylation of FcεRI β in this experiment. By this method of analysis, we found that maximal FcεRI β tyrosine phosphorylation is inhibited 95 ± 4% by pretreatment with MβCD in six separate experiments under these optimal conditions. The identity of FcεRI β and γ in the antiphosphotyrosine blots of whole cell lysates was confirmed by specific immunodepletion of these bands with anti-IgE (Fig. 1 c).
Figure 1.

Effects of cholesterol depletion on antigen-stimulated tyrosine phosphorylation. (a) Tyrosine phosphorylation of whole cell lysates. IgE-sensitized RBL-2H3 cells treated with (+) or without (−) 10 mM MβCD for 1 h at 37°C were washed and stimulated with 1 μg/ml DNP-BSA at 37°C for the indicated times, then lysed with SDS sample buffer. Equal numbers of cell equivalents (8,000/lane) were Western blot analyzed and probed with antiphosphotyrosine. (b) Quantitation of FcεRI β tyrosine phosphorylation. The blot shown in a was scanned and absorbances of the β band were measured. Open and closed circles denote untreated and MβCD-treated cells, respectively. (c) Immunodepletion of IgE–FcεRI. RBL-2H3 cells were stimulated as in a and lysed with TX-100 lysis buffer. Soluble lysates were incubated in the presence or absence of rabbit anti-IgE followed by protein A–agarose and centrifugation to immunodeplete IgE–FcεRI. Protein A–agarose supernatants (equal cell equivalents) were analyzed by Western blot as in a.
To further characterize the cells under conditions of MβCD treatment, we determined that untreated cells contain 6.7 ± 0.7 nmol free cholesterol/106 cells (n = 5), and, after incubation with 10 mM MβCD at 37°C for 1 h, the amount of free cholesterol was determined to be 2.7 ± 1.0 nmol/106 cells (n = 5). Thus, the fraction of free cholesterol remaining after treatment is 0.40 ± 0.15 as compared with untreated cells, consistent with previously described levels of cholesterol depletion for a variety of cell types under similar conditions (Kilsdonk et al., 1995; Yancey et al., 1996; Christian et al., 1997; Gimpl et al., 1997; Scheiffele et al., 1997; Friedrichson and Kurzchalia, 1998; Keller and Simons, 1998; Varma and Mayor, 1998). TLC analysis of cellular lipid extracts showed that there was no detectable difference in phospholipid composition before and after MβCD treatment, and this is also consistent with previous reports from other laboratories for other cell types (Kilsdonk et al., 1995; Yancey et al., 1996; Christian et al., 1997; Gimpl et al., 1997). In particular, we found that the amounts of phosphatidylethanolamine, phosphatidylcholine, and sphingomyelin present in the total lipid extracts from MβCD-treated cells were not obviously different from untreated cell lipids (data not shown). The amounts of two other unidentified lipid species (one polar lipid species and one neutral lipid species) observed with TLC also were unaffected by MβCD treatment.
To further investigate the basis for the dramatic reduction in tyrosine phosphorylation of FcεRI β, we measured the amount of FcεRI before and after MβCD treatment using 125I-IgE. Under our optimal conditions for inhibition of tyrosine phosphorylation (10 mM MβCD for 1 h at 37°C), we observed a 70 ± 6% (n = 6) loss of receptor-bound IgE from the cells that could be recovered in the supernatants of the cell washes after MβCD treatment. Similarly, 64 ± 7% (n = 2) of the ganglioside GD1b was lost from the cells due to MβCD treatment as detected using 125I-AA4 mAb. The loss of FcεRI is probably due to vesicle shedding caused by the MβCD treatment because a substantial fraction of 125I-IgE in the post-wash supernatant after MβCD treatment can be pelleted by high speed centrifugation as membrane vesicles that are detectable by phase contrast microscopy (data not shown).
In steady-state fluorescence measurements of FITC-IgE bound to MβCD-treated cells, 65 ± 10% (n = 3) IgE– FcεRI loss was observed. Qualitatively consistent results were visualized with FITC-IgE and Cy3-AA4 in fluorescence microscopy of labeled cells, and a similar reduction in the cell surface expression of the GPI-anchored protein Thy-1 was also observed (data not shown). Despite these substantial reductions in FcεRI and the outer leaflet markers for DRMs in these cells after MβCD treatment, the plasma membrane expression of Lyn was only modestly reduced as assessed from fluorescence microscopy and Western blot analysis of sucrose gradient fractions (see below). Silver-stained polyacrylamide gels of RBL cell lysates showed no significant alterations in the amounts or composition of proteins detected in this manner (data not shown).
These results suggest that the inhibition of antigen-stimulated tyrosine phosphorylation is due in part to a reduction in the amount of FcεRI available for phosphorylation in the MβCD-treated cells. To assess the magnitude of this effect, we determined the relationship between FcεRI tyrosine phosphorylation and the effective concentration of this receptor in the plasma membrane by comparing the amount of FcεRI β tyrosine phosphorylation for cells in which 30% of the receptors were occupied by anti-DNP IgE, and 70% were occupied by anti-1,5-dansyl IgE, which does not bind DNP ligands (Weetall et al., 1993; see Material and Methods). Using the same conditions for stimulation and analysis as was used for the MβCD-treated cells, we determined that at the time point for maximal stimulation DNP-BSA caused 66% (n = 2) less tyrosine phosphorylation of FcεRI β for the cells occupied with 30% anti-DNP IgE compared with those occupied with 100% anti-DNP IgE. Likewise, when FcεRI on cells were occupied by 50% anti-DNP IgE, DNP-BSA caused 41% (n = 2) less tyrosine phosphorylation of FcεRI β than those occupied with 100% anti-DNP IgE. Assuming that this approximately proportional relationship between receptor number and β phosphorylation is valid for the MβCD-treated cells over this range of anti-DNP IgE densities, then the expected reduction in tyrosine phosphorylation of FcεRI β due to 70% loss of FcεRI should be ∼70%. Thus, the actual reduction in this value (95 ± 4%) represents an 83% inhibition of the stimulated FcεRI β tyrosine phosphorylation expected for the amount of cross-linked receptors present.
Because of the nearly complete inhibition of stimulated FcεRI tyrosine phosphorylation caused by MβCD-mediated cholesterol depletion, we examined the effects of this treatment on cellular degranulation as measured by release of β-hexosaminidase. Fig. 2 summarizes the results from three separate experiments and shows that cholesterol depletion does not significantly inhibit degranulation stimulated by an optimal dose of antigen. Furthermore, cholesterol depletion actually enhances the amount of degranulation observed in response to stimulation by the Ca2+ ionophore, A23187, without altering the amount of β-hexosaminidase released in unstimulated cells. These results indicate that reduction in cellular cholesterol enhances one or more of the downstream events that follow Ca2+ elevation and lead to degranulation. They also demonstrate that cholesterol depletion under these conditions is not cytotoxic. As seen in Fig. 1, tyrosine phosphorylation of FcεRI is usually inhibited more strongly than stimulated phosphorylation of some other substrates (e.g., the stimulated bands in the range of 70–100 kD) that are known to be dependent on activation of the tyrosine kinase Syk (Zhang et al., 1996). These results indicate that relatively small amounts of stimulated tyrosine phosphorylation can result in substantial degranulation responses in the MβCD-treated cells. Consistent with this, stimulation of maximal degranulation requires effective cross-linking of only ∼10% of the IgE receptors on untreated RBL-2H3 cells (Fewtrell, 1985).
Figure 2.

Effects of MβCD treatment on degranulation of IgE-sensitized RBL-2H3. Cells pretreated with MβCD as in Fig. 1 a (hatched bars) or untreated controls (white bars) were assessed by the release of β-hexosaminidase. Cells were incubated with buffer alone (spontaneous), 100 ng/ml DNP-BSA, or 0.7 μM A23187 for 1 h at 37°C; β-hexosaminidase in the supernatant was measured as a percentage of total β-hexosaminidase. Bars represent mean ± SD (n = duplicate samples in three independent experiments).
Cholesterol Depletion Disrupts the Interactions of Cross-linked IgE–FcεRI and Lyn with DRM Vesicles
To determine the importance of cholesterol in DRM interactions, we examined the distributions of IgE–FcεRI and Lyn across sucrose gradients of cholesterol-depleted or untreated RBL cells. As previously demonstrated for untreated cells (Field et al., 1997), monomeric IgE–FcεRI (Fig. 3 a, •) is found predominantly in the 40% sucrose region of the gradient where cytoplasmic and detergent-solubilized membrane proteins are characteristically observed, and a large percentage of cross-linked IgE–FcεRI (Fig. 3 a, ○) is located in the low density region of the gradient where DRM vesicles are found (Field et al., 1997; Scheiffele et al., 1997; Wolf et al., 1998). After MβCD treatment, monomeric IgE–FcεRI (Fig. 3 a, ▪) is located in the 40% sucrose region, similar to those in the untreated cells. However, cross-linked IgE–FcεRI (Fig. 3 a, □) no longer float to the DRM region, but rather appear in the 50–60% sucrose region where aggregates of IgE– FcεRI characteristically locate in the absence of interactions with DRMs (Field et al., 1997). These results are representative of four experiments, and they show that ∼60% reduction in cholesterol almost completely prevents the association of cross-linked IgE–FcεRI with DRMs.
Figure 3.
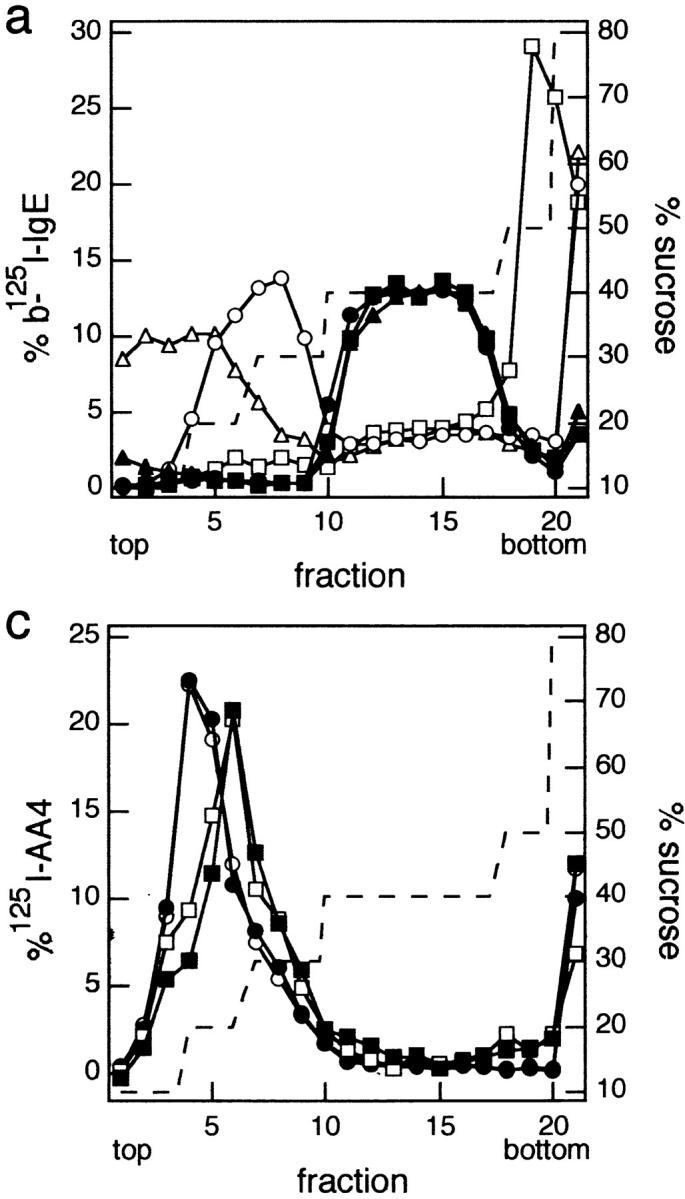

Molecular distributions of IgE–FcεRI, Lyn, and GD1b on sucrose density gradients. (a) IgE–FcεRI distributions. Shown are representative sucrose gradient results in which cells were sensitized with biotinylated 125I-IgE (b-125I-IgE) and either cross-linked with 10 nM streptavidin (sAv, open symbols) for 5 min at 37°C or not cross-linked (closed symbols). Cells were then lysed with cold TX-100 lysis buffer containing 0.040% TX-100, loaded onto sucrose step gradients, and analyzed. Before cross-linking with sAv (or not), cells were incubated: (1) without MβCD for 1 h at 37°C, then subsequently for 2 h at 37°C in the absence of MβCD or cholesterol– MβCD (untreated; circles); (2) with 10 mM MβCD for 1 h at 37°C, followed by a subsequent treatment with 3 mM MβCD for 2 h at 37°C (depleted; squares); or (3) with 10 mM MβCD for 1 h at 37°C, followed by cholesterol/MβCD (3 mM MβCD; MβCD/ cholesterol, 8:1, mol/mol) for 2 h at 37°C (repleted; triangles). (b) Lyn distributions. Equal amounts of sucrose gradient fractions pooled as indicated were loaded onto nonreducing SDS polyacrylamide gels and immunoblotted for Lyn. Top, untreated cells; middle, cholesterol-depleted cells; and bottom, cholesterol-repleted cells as described in a. Note that the fraction denoted bottom includes the pellet. (c) GD1b distributions. RBL cells sensitized with biotinylated IgE were labeled with 125I-AA4 (anti-GD1b), cross-linked or not with sAv, lysed, and subjected to sucrose density gradient analysis as in a. Distributions of GD1b from cells stimulated with sAv as in a are denoted by open symbols and those from unstimulated cells are denoted by closed symbols. Circles indicate untreated cells, and squares indicate cells that had been treated with 10 mM MβCD for 1 h.
As shown in Fig. 3 b (top), the distribution of Lyn in the gradient fractions of stimulated (+ sAv) and unstimulated (− sAv) cells that were not treated with MβCD is qualitatively similar to that observed by Field et al. (1995), who used higher concentrations of TX-100. In the present experiments, it is notable that the p56 isoform of Lyn is selectively enriched in the low density, DRM region of the gradient (fractions 4–9), whereas the p53 isoform is located predominately in the 40% sucrose region (fractions 10–18). After cholesterol depletion, Lyn no longer localizes in the low density region of the gradient for both stimulated and unstimulated cells, whereas the distribution of p53 Lyn in the 40% sucrose region remains essentially unchanged from the untreated cells (Fig. 3 b, middle). In the cholesterol-depleted cells, it appears that p56 Lyn is relatively enriched in the gradient pellet (bottom fraction), both for the stimulated and the unstimulated cells. Overall, the total amount of Lyn detected in the gradients is only moderately reduced in the MβCD-treated cells compared with untreated control cells.
The loss of both aggregated FcεRI and Lyn from the low density region of the gradient raises the question of whether DRMs are disrupted entirely in the cholesterol-depleted cells. To address this question, we investigated the distribution of other DRM markers in sucrose gradients after lysis of cholesterol-depleted cells. As shown in Fig. 3 c, AA4-labeled GD1b from cholesterol-depleted cells is found almost completely in the low density region of the gradients, indicating that DRMs still exist in some form after ∼60% cholesterol depletion. The shift in GD1b distribution to a slightly higher density after cholesterol depletion suggests an increase in the protein/lipid ratio, consistent with the substantial loss of cholesterol, a major lipid component of the DRM (Brown and Rose, 1992). These same trends are observed for both stimulated (Fig. 3 c, open symbols) and unstimulated (Fig. 3 c, closed symbols) cells. Another DRM marker, the GPI-anchored protein Thy-1 (Dráberová and Dráber, 1993; Field et al., 1995; Surviladze et al., 1998), localizes similarly to GD1b in the sucrose gradients before and after cholesterol depletion (data not shown). These results indicate that 60% cholesterol depletion can prevent the interactions of some proteins (FcεRI and Lyn) with DRMs without eliminating DRM structure.
Cholesterol Depletion Prevents the Redistribution of Lyn with Cross-linked IgE–FcεRI on Intact Cells
To evaluate how cholesterol depletion affects FcεRI on intact cells, we used confocal fluorescence microscopy to examine the redistributions of Lyn and GD1b with cross-linked IgE–FcεRI. Representative images in Fig. 4, a and b, show that both monomeric IgE–FcεRI (left panels) and Lyn (right panels) are uniformly distributed in the plasma membrane in the absence and presence of MβCD, respectively. When IgE–FcεRI is aggregated by antigen at 22°C for 20 min, small patches of these are formed, and these patches often cluster together on one side of the cell. As see in Fig. 4 c, the concentration of Lyn is enhanced in these regions of patched receptors in the absence of MβCD treatment. For MβCD-treated cells, IgE–FcεRI also redistributes into patches after aggregation by antigen (Fig. 4 d, left), indicating that lateral mobility is not impeded by cholesterol depletion; however, Lyn does not redistribute with IgE–FcεRI under these conditions (Fig. 4 d). As indicated in the first line of Table I, these differences are statistically significant when quantified by cross correlation analysis of multiple cells. Thus, cross-link– dependent interactions between FcεRI and Lyn on the cell surface are largely prevented by cholesterol depletion, consistent with the loss of interactions of these proteins with DRMs in the sucrose gradient analyses of lysed cells described above.
Figure 4.

Immunofluorescence of the cellular distributions of FcεRI, Lyn, GD1b, and TfRs. Each panel consists of paired confocal fluorescence images of FITC-anti-DNP IgE– FcεRI on the left side and the indicated Cy3-labeled proteins on the right side. Panels a, c, e, and g show cells not pretreated with MβCD, and panels b, d, f, and h show cells pretreated with 10 mM MβCD before IgE–FcεRI cross-linking by antigen (c–h) or uncross-linked (a and b). a–d are paired images of IgE– FcεRI and Lyn, e–f are of IgE–FcεRI and GD1b, and g–h are of IgE–FcεRI and TfRs. Arrows indicate regions of co-redistribution of Lyn (c) or GD1b (e and f) with patched IgE–FcεRI. Bars, 10 μm.
Table I.
Cross Correlation Analysis of the Co-redistribution of Lyn or TfR with Antigen–cross-linked FITC-IgE–FcεRI
| −MβCD ρ | n | +MβCD ρ | n | |||||
|---|---|---|---|---|---|---|---|---|
| IgE/Lyn | 0.67 ± 0.09 | 12 | 0.29 ± 0.16 | 13 | ||||
| IgE/TfR | 0.25 ± 0.16 | 7 | 0.15 ± 0.24 | 9 |
Quantitative analyses were carried out on fluorescent intensity profiles of equatorial images of individual cells as described in Materials and Methods. The peak values for each cross correlation plot (Eq.1) were averaged to yield a correlation coefficient (ρ) for numerical comparison of the degree of co-redistribution; 1 corresponds to complete co-localization. The correlation coefficients are reported as mean ± SD, and n is the number of individual cells analyzed.
Fig. 4 e shows that, as previously observed (Pierini et al., 1996), cross-linking of IgE–FcεRI at the cell surface results in co-redistribution of the GD1b ganglioside that is labeled by Cy3-AA4 mAb. For MβCD-treated cells, we find that co-redistribution of this outer leaflet DRM marker with cross-linked IgE–FcεRI is reduced compared with control cells but not completely disrupted as was the case for Lyn. Fig. 4 f shows an example of this variability, in which one cell exhibits partial co-redistribution of the labeled ganglioside, and the other shows a complete lack of co-redistribution with patched IgE–FcεRI. Under these conditions, ≤20% of the cells exhibited detectable co-redistribution of labeled ganglioside, whereas no detectable co-redistribution of Lyn with IgE–FcεRI patches was observed. From these results, it appears that the interaction between Lyn and FcεRI is more sensitive to cholesterol depletion than is the interaction between the ganglioside and FcεRI in the intact cells, although both are substantially prevented. The difference observed may be related to the greater sensitivity of the Lyn–DRM interactions than GD1b–DRM interactions, as shown in Fig. 3 (see Discussion).
As a further control, we compared the distribution of the transferrin receptor (CD71) to FITC-IgE–FcεRI cross-linked under the same conditions as above. Previous studies showed that this transmembrane protein does not associate with isolated DRMs (Melkonian et al., 1999), nor does it co-redistribute with other DRM-associated proteins when simultaneously but separately cross-linked on BHK and Jurkat T cells (Harder et al., 1998). As seen in Fig. 4, g and h, TfRs do not co-redistribute with cross-linked IgE–FcεRI, and they remain evenly distributed around the periphery of the cell, often in tiny clusters that may reflect interactions with coated pits. Quantitative analysis of the cross correlation of TfR with cross-linked IgE–FcεRI show no appreciable colocalization between these molecules whether or not the cells have been depleted of cholesterol (see Table I). These results support the significance of the co-redistribution of Lyn with cross-linked IgE–FcεRI described above, as well as its inhibition by cholesterol depletion with MβCD.
Cholesterol Repletion Restores FcεRI Tyrosine Phosphorylation Together with the Association of Cross-linked FcεRI and Lyn with DRMs
To investigate the reversibility of cholesterol effects on the functional and structural interactions of FcεRI with Lyn, we restored cholesterol levels in MβCD-treated cells by incubating them with cholesterol–MβCD complexes. The efficiency of repletion is dependent upon the incubation period of the cells with the complex, the molar ratio of cholesterol to MβCD, and the final concentration of MβCD (Christian et al., 1997; Sheets, E.D., unpublished results). To optimize repletion, we used several dilutions of cholesterol–MβCD complexes prepared as described in Materials and Methods. In our sequential depletion/repletion experiment, cells were initially left untreated (control samples) or incubated with MβCD to lower cholesterol levels as described above. During the second step, the cells were incubated for 2 h at 37°C at the indicated dilution of cholesterol/MβCD, 3 mM MβCD only, or buffer only. We found that (a) the cholesterol levels of cells depleted by exposure to MβCD in the first step did not change during the subsequent incubation in the absence of MβCD; (b) the presence of 3 mM of MβCD during the second step also did not cause additional cholesterol depletion, nor did it alter the distribution of IgE–FcεRI in the sucrose gradients; and (c) under optimal conditions of cholesterol repletion used (3–6 mM MβCD; 8:1, mol/mol MβCD/cholesterol), the cholesterol content of the repleted cells was 3.0–3.5-fold higher than that in the untreated control cells. Furthermore, TLC analyses of total lipid extracts indicated that cholesterol was the only lipid that changed detectably during the depletion/repletion treatments (data not shown).
As shown in Fig. 5, repletion of cholesterol in MβCD-treated cells results in partial restoration of antigen-stimulated tyrosine phosphorylation. In the experiment shown, maximal recovery of stimulated tyrosine phosphorylation of FcεRI β and other bands was achieved when 1:50 dilution of the preformed 8:1 MβCD/cholesterol complexes was used to give a final concentration of 6 mM MβCD during the repletion step (Fig. 5, lane 10). As seen in Fig. 5, lane 6, cells that had been treated with MβCD alone during both the depletion and repletion steps have no detectable β phosphorylation, and only a very small amount of stimulated tyrosine phosphorylation is seen in the higher molecular weight bands. Similar results to these were obtained in three separate experiments. Under the conditions of cholesterol depletion/repletion, loss of IgE– FcεRI was determined to be 77 ± 4% (n = 5), and, based upon the proportional relationship between receptor number and β phosphorylation (above), this leads us to expect that optimal restoration of FcεRI β phosphorylation should be ∼23% of the stimulated control in Fig. 5, lane 2. The somewhat smaller restoration that is apparent (Fig. 5, lanes 10 and 12) suggests that other factors, such as the loss of other outer-leaflet DRM components during cholesterol depletion noted above, may reduce the maximum restoration achievable (see Discussion).
Figure 5.

Tyrosine phosphorylation of FcεRI before and after cholesterol depletion/repletion. IgE-sensitized cells were sequentially treated with (+) or without (–) 10 mM MβCD for 1 h at 37°C, then for 2 h at 37°C with 3 mM MβCD (*) (lanes 5 and 6), with 300 mM MβCD/cholesterol (8:1, mol/mol) diluted as indicated (†) (lanes 7–12), or with buffer alone (lanes 1–4). Washed cells were stimulated for 2 min with (+) or without (−) 1 μg/ml DNP-BSA at 37°C, then lysed and analyzed by antiphosphotyrosine Western blot analysis as described for Fig. 1 a.
Repletion of cholesterol also results in restoration of cross-link–dependent association of IgE–FcεRI with isolated DRMs. When lysates of cholesterol-repleted cells are analyzed on sucrose gradients, cross-linked IgE–FcεRI (Fig. 3 a, ▵) migrates to the low density sucrose region, whereas uncross-linked IgE–FcεRI (Fig. 3 a, ▴) is found in the 40% sucrose region, similar to the gradient distributions from control cells (Fig. 3 a, ○ and •). Cross- linked IgE–FcεRI from cholesterol-repleted cells migrate at slightly lower densities in the sucrose gradients than this complex in the control cells, suggesting that the average density of DRMs in repleted cells is lower than in control cells, possibly due to a decrease in the protein/lipid ratio resulting from an increased cholesterol content. Furthermore, as shown in Fig. 3 b (bottom), p56 Lyn also migrates to the low density region of the gradient (fractions 1–6) after cholesterol repletion in cells with both cross-linked and uncross-linked FcεRI. These results, in parallel with the restoration of stimulated FcεRI tyrosine phosphorylation (Fig. 5), provide strong evidence that cholesterol is important for functional coupling of FcεRI with Lyn and for their mutual association with DRMs.
Discussion
Our results demonstrate that cholesterol plays a critical role in the initial step of FcεRI signaling: antigen-stimulated tyrosine phosphorylation of this receptor by the Src family tyrosine kinase Lyn. In parallel with loss of this stimulated phosphorylation (Fig. 1), reduction of cellular cholesterol by MβCD causes the loss of association of both Lyn and cross-linked FcεRI with DRMs isolated after cell lysis by TX-100 (Fig. 3). Restoration of the cholesterol content of the depleted cells using preformed cholesterol–MβCD complexes restores the association of Lyn and cross-linked IgE–FcεRI with DRMs (Fig. 3) and also causes partial restoration of antigen-stimulated tyrosine phosphorylation in the cells (Fig. 5). These results support the hypothesis that interactions of cross-linked IgE–FcεRI with DRMs are important for the initial coupling of FcεRI and Lyn that results in receptor phosphorylation. On the cell surface, the association of Lyn with aggregated IgE– FcεRI is lost as the result of cholesterol depletion (Fig. 4), indicating that the interactions detected in isolated DRMs are relevant to those occurring in intact cells. Furthermore, these microscopy results argue against a direct interaction of cross-linked FcεRI with Lyn as the basis for association of receptors with DRMs, and they support the view that the L o structure of the plasma membrane is important for FcεRI–Lyn interactions.
An initially surprising finding in our studies is the apparently selective loss of FcεRI and outer leaflet plasma membrane components of DRMs due to cholesterol depletion by MβCD. As indicated by Western blot analysis and fluorescence microscopy, there is a smaller loss of Lyn due to cholesterol depletion, and there is no detectable loss of other cellular proteins by silver stain analysis of whole cell lysates (data not shown). The mechanism by which cholesterol depletion causes this selective loss is not yet known, but it is interesting to speculate that vesicles containing these components may pinch off from the cells in a mechanism that depends on their local structural environment in the plasma membrane. In a recent study by Ilangumaran and Hoessli (1998), a similar preferential release of DRM components by MβCD treatment was characterized in lymphocytes and endothelial cells, and evidence for release of these components in membrane vesicles was described. Our results suggest that FcεRI may preferentially associate with DRM components on intact cells even in the absence of receptor cross-linking. Consistent with this, Basciano et al. showed that pre-binding of AA4 mAb or its Fab fragment to the α-galactosyl GD1b antigen on RBL-2H3 cells can effectively inhibit the subsequent binding of IgE to FcεRI (Basciano et al., 1986). By varying the cell surface density of antigen-specific IgE in the range of 30–100%, we show that the loss of FcεRI due to cholesterol depletion cannot account for the nearly complete inhibition of FcεRI tyrosine phosphorylation that is observed in these cells. Furthermore, the partial restoration of antigen-stimulated tyrosine phosphorylation without an increase in FcεRI expression after cholesterol repletion strengthens the evidence that cellular cholesterol critically regulates the coupling of the remaining FcεRI and Lyn.
An important observation by fluorescence microscopy is that cholesterol depletion does not prevent antigen-dependent aggregation of IgE–FcεRI on the cell surface, even though it prevents the co-redistribution of Lyn and inhibits stimulated tyrosine phosphorylation. We previously showed that the cholesterol-binding polyene antibiotic, filipin, prevents anti-IgE–mediated patching of IgE– FcεRI (Feder et al., 1994), indicating that it may prevent aggregation of the receptor necessary to initiate signaling. Unlike MβCD, which extracts cholesterol into a water-soluble complex that can be washed away, filipin forms complexes with cholesterol in the membrane that appear to restrict lateral diffusion of at least some membrane proteins, making this reagent less useful for studying the role of cholesterol in signaling by receptors that must aggregate in response to their ligands to be effective. The rapidity with which MβCD can reduce cell cholesterol by substantial amounts without significantly compromising cell integrity, as evidenced by our degranulation results, and the capacity to restore stimulated tyrosine phosphorylation by reintroduction of cholesterol via MβCD complexes, make this an extremely valuable tool for investigating the role of cholesterol in a wide variety of receptor systems.
Recent studies used MβCD to investigate the role of cholesterol in signaling by other receptors. Pike and Miller (1998) showed that cholesterol depletion by MβCD inhibits EGF- and bradykinin-stimulated phosphatidylinositol turnover, which can be restored by cholesterol repletion with MβCD–cholesterol complexes. These receptors belong to the families of intrinsic tyrosine kinase receptors and G protein–coupled receptors, respectively, suggesting the potentially general importance of cholesterol and the L o structure it confers on the plasma membrane in mediating receptor signaling. Interestingly, cholesterol depletion by MβCD does not inhibit EGF-stimulated tyrosine phosphorylation of its receptor (Pike, L.J., Y. Liu, K.N. Chung, and J.A. Heuser. 1998. FASEB J. 12:A1278 [abstr.]), which probably occurs via a transphosphorylation mechanism (Weiss and Schlessinger, 1998). Rather, cholesterol depletion appears to affect the compartmentalization of phosphatidylinositol 4,5-bisphosphate, the primary phospholipase C substrate in the plasma membrane, as revealed by its reduced localization with DRMs in sucrose gradients (Pike and Miller, 1998). In contrast, our results with IgE receptors indicate a role for cholesterol in the initial signaling step in which these receptors are phosphorylated by Lyn, and this finding may have general relevance for other receptors that function by interacting with Src family kinases. Indeed, Xavier et al. (1998) and Moran and Miceli (1998) showed that pretreatment of T cells with MβCD inhibits T cell receptor for antigen-mediated Ca2+ mobilization and tyrosine phosphorylation, respectively, providing evidence for an important role for cholesterol in the function of this related multichain immune recognition receptor family member.
Our degranulation results (Fig. 2) suggest that normal levels of cholesterol may negatively regulate downstream signaling or the exocytotic process in the RBL-2H3 cells. The enhancement of degranulation stimulated by Ca2+ ionophore, as well as the lack of inhibition of antigen-stimulated degranulation, despite the dramatic inhibition of tyrosine phosphorylation by cholesterol depletion, are consistent with this explanation. In addition, preliminary experiments on the effects of cholesterol depletion on Ca2+ mobilization by antigen indicate that this activity is inhibited less than is stimulated tyrosine phosphorylation of FcεRI, consistent with differential effects of cholesterol depletion on different signaling steps (Holowka, D., and E.D. Sheets, unpublished results). Membrane structural changes involved in exocytosis may also be affected. As described above, the proportionality of FcεRI tyrosine phosphorylation with the number of receptors cross-linked in the range of 30–100% does not hold for more downstream signaling events, as only a small fraction of FcεRI needs to be cross-linked to achieve maximal degranulation. In future experiments, it will be interesting to explore the effects of cholesterol depletion on specific downstream signaling pathways and exocytic membrane events through bypassing receptor-mediated signaling with alternate means of activation.
The results described here are consistent with the hypothesis that cross-linked IgE–FcεRI interact with Lyn-containing DRM domains on the cell surface and that these structural interactions are integral to the initiation of signal transduction. Monomeric FcεRI probably interact dynamically with DRM components, and these transient complexes may exist in the plasma membrane of unstimulated cells as small clusters similar to those recently described for certain GPI-anchored proteins (Friedrichson and Kurzchalia, 1998; Varma and Mayor, 1998). In this hypothesis, cross-linking of IgE–FcεRI on the cell surface causes them to cluster with DRM components, thereby creating larger L o regions containing FcεRI and Lyn that are segregated from more fluid regions of the plasma membrane. It is likely that most transmembrane proteins are more readily accommodated by phospholipids in the more fluid liquid crystalline phase, and these would then segregate from the L o regions containing FcεRI and DRM components. For example, the tyrosine phosphatase CD45 is a transmembrane protein that was shown to be largely excluded from DRMs on T cells (Rodgers and Rose, 1996) and has been recently shown to negatively regulate the Src family kinase Lck (D'Oro and Ashwell, 1999). Our microscopy results (Fig. 4 and Holowka, D., E.D. Sheets, and B. Baird, manuscript in preparation) indicate that segregated DRM domains occupy a large percentage of the cell surface (20–50%) as detectable within the limits of optical resolution, and earlier studies indicated that DRM phospholipids represent a similarly large percentage of plasma membrane phospholipids (Mescher and Apgar, 1985). Thus, segregation of certain proteins from others may be more important for signaling promoted by DRM interactions of cross-linked FcεRI and Lyn than an increased localized concentration of these DRM-associated proteins within domains.
In our model, cholesterol is an essential component for the L o phase, and its 60% reduction, as in the studies presented here, appears to most greatly affect the association of the transmembrane protein FcεRI and the inner leaflet component Lyn with DRMs (Figs. 3 and 4). Outer leaflet DRM components are retained in the low-density, TX-100–insoluble membrane vesicles (Fig. 3), and they still maintain a small but detectable association with cross-linked IgE–FcεRI on intact cells (Fig. 4 f and data not shown), which may reflect the continued presence of an L o environment in the outer leaflet under conditions of diminished cholesterol. It is possible that sphingomyelin and other sphingolipids enriched in the outer leaflet of the plasma membrane (Devaux, 1991) cause a preferential retention of cholesterol in this leaflet of the bilayer under conditions of limiting cholesterol, since this particular class of phospholipids may interact preferentially with cholesterol (Brown, 1998). Thus, cholesterol-dependent associations at the inner leaflet of the plasma membrane may be more sensitive to cholesterol depletion than are such interactions in the outer leaflet. As an alternative explanation for our results, it is possible that cholesterol serves as a critical boundary lipid for FcεRI that facilitates a direct interaction with Lyn. However, this explanation would not account for the association of these components with DRMs and the correlation between loss of this structural association and loss of functional coupling. The involvement of DRMs in functional coupling between FcεRI and Lyn as a means of promoting the proximity of these proteins while excluding transmembrane tyrosine phosphatases such as CD45 is an attractive hypothesis that warrants further examination.
Acknowledgments
We thank Eric Holowka for carrying out the cross correlation analysis, using software developed in our laboratory by Paul Pyenta.
Supported by National Institutes of Health grants AI09838 (E.D. Sheets) and AI22449.
Abbreviations used in this paper
- BSA/BSS
BSA-containing buffered saline solution
- DRM
detergent-resistant membrane
- GPI
glycosylphosphatidylinositol
- HRP
horseradish peroxidase
- Lo
liquid-ordered
- MβCD
methyl-β-cyclodextrin
- TfR
transferrin receptor
- TX-100
Triton X-100
Footnotes
Address correspondence to Barbara Baird or David Holowka, Department of Chemistry & Chemical Biology, Baker Laboratory, Cornell University, Ithaca, NY 14853-1301. Tel.: (607) 255-4095 or (607) 255-6140. Fax: (607) 255-4137. E-mail: bab13@cornell.edu or dah24@cornell.edu
References
- Ahmed SN, Brown DA, London E. On the origin of sphingolipid/ cholesterol-rich detergent-insoluble cell membranes: physiological concentrations of cholesterol and sphingolipid induce formation of a detergent-insoluble, liquid-ordered lipid phase in model membranes. Biochemistry. 1997;36:10944–10953. doi: 10.1021/bi971167g. [DOI] [PubMed] [Google Scholar]
- Barlow, R.J. 1989. Statistics: A Guide to the Use of Statistical Methods in the Physical Sciences. John Wiley and Sons, Chichester, UK. 204 pp.
- Basciano LK, Berenstein EH, Kmak L, Siraganian RP. Monoclonal antibodies that inhibit IgE binding. J Biol Chem. 1986;261:11823–11831. [PubMed] [Google Scholar]
- Brown DA, London E. Functions of lipid rafts in biological membranes. Annu Rev Cell Biol. 1998a;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- Brown DA, London E. Structure and origin of ordered lipid domains in biological membranes. J Membr Biol. 1998b;164:103–114. doi: 10.1007/s002329900397. [DOI] [PubMed] [Google Scholar]
- Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- Brown RE. Sphingolipid organization in biomembranes: what physical studies of model membranes reveal. J Cell Sci. 1998;111:1–9. doi: 10.1242/jcs.111.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright, I.J. 1993. Separation and analysis of phospholipids by thin layer chromatography. In Methods in Molecular Biology. Vol. 19. J.M. Graham and J.A. Higgins, editors. Humana Press, Totowa, NJ. 153–167. [DOI] [PubMed]
- Chang EY, Zheng Y, Holowka D, Baird B. Alteration of lipid composition modulates FcεRI signaling in RBL-2H3 cells. Biochemistry. 1995;34:4376–4384. doi: 10.1021/bi00013a029. [DOI] [PubMed] [Google Scholar]
- Christian AE, Haynes MP, Phillips MC, Rothblat GH. Use of cyclodextrins for manipulating cellular cholesterol content. J Lipid Res. 1997;38:2264–2272. [PubMed] [Google Scholar]
- Devaux PF. Static and dynamic lipid asymmetry in cell membranes. Biochemistry. 1991;30:1163–1173. doi: 10.1021/bi00219a001. [DOI] [PubMed] [Google Scholar]
- D'Oro U, Ashwell JD. Cutting edge: the CD45 tyrosine phosphatase is an inhibitor of Lck activity in thymocytes. J Immunol. 1999;162:1879–1883. [PubMed] [Google Scholar]
- Dráberová L, Dráber P. Thy-1 glycoprotein and src-like protein-tyrosine kinase p53/56lynare associated in large detergent-resistant complexes in rat basophilic leukemia cells. Proc Natl Acad Sci USA. 1993;90:3611–3615. doi: 10.1073/pnas.90.8.3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edidin M. Lipid microdomains in cell surface membranes. Curr Opin Struct Biol. 1997;7:528–532. doi: 10.1016/s0959-440x(97)80117-0. [DOI] [PubMed] [Google Scholar]
- Feder TJ, Chang E-Y, Holowka D, Webb WW. Disparate modulation of plasma membrane protein lateral mobility by various cell permeabilizing agents. J Cell Physiol. 1994;158:7–16. doi: 10.1002/jcp.1041580103. [DOI] [PubMed] [Google Scholar]
- Fewtrell, C. 1985. Activation and desensitization of receptors for IgE on tumor basophils. In Calcium in Biological Systems. R.P. Ruben, G.B. Weiss, and J.W. Putney, editors. Plenum, New York. 129–136.
- Field KA, Holowka D, Baird B. FcεRI-mediated recruitment of p53/56lynto detergent-resistant membrane domains accompanies cellular signaling. Proc Natl Acad Sci USA. 1995;92:9201–9205. doi: 10.1073/pnas.92.20.9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field KA, Holowka D, Baird B. Compartmentalized activation of the high affinity immunoglobulin E receptor within membrane domains. J Biol Chem. 1997;272:4276–4280. doi: 10.1074/jbc.272.7.4276. [DOI] [PubMed] [Google Scholar]
- Friedrichson T, Kurzchalia TV. Microdomains of GPI-anchored proteins in living cells revealed by crosslinking. Nature. 1998;394:802–805. doi: 10.1038/29570. [DOI] [PubMed] [Google Scholar]
- Ge, M., K.A. Field, R. Aneja, D. Holowka, B. Baird, and J. Freed. 1999. ESR characterization of liquid ordered phase of detergent resistant membranes from RBL-2H3 cells. Biophys. J. In press. [DOI] [PMC free article] [PubMed]
- Gimpl G, Burger K, Fahrenholz F. Cholesterol as modulator of receptor function. Biochemistry. 1997;36:10959–10974. doi: 10.1021/bi963138w. [DOI] [PubMed] [Google Scholar]
- Harder T, Scheiffele P, Verkade P, Simons K. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J Cell Biol. 1998;141:929–942. doi: 10.1083/jcb.141.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris NT, Goldstein B, Holowka D, Baird B. Altered patterns of tyrosine phosphorylation and Syk activation for sterically restricted cyclic dimers of IgE–FcεRI. Biochemistry. 1997;36:2237–2242. doi: 10.1021/bi9619839. [DOI] [PubMed] [Google Scholar]
- Ilangumaran S, Hoessli DC. Effects of cholesterol depletion by cyclodextrin on the sphingolipid microdomains of the plasma membrane. Biochem J. 1998;335:433–440. doi: 10.1042/bj3350433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvin M-HE, Adamczewski M, Numerof R, Letourneur O, Vallé A, Kinet J-P. Differential control of the tyrosine kinases Lyn and Syk by the two signaling chains of the high affinity immunoglobulin E receptor. J Biol Chem. 1994;269:5918–5925. [PubMed] [Google Scholar]
- Keller P, Simons K. Cholesterol is required for surface transport on influenza virus hemagglutinin. J Cell Biol. 1998;140:1357–1367. doi: 10.1083/jcb.140.6.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilsdonk EPC, Yancey PG, Stoudt GW, Bangerter FW, Johnson WJ, Phillips MC, Rothblat GH. Cellular cholesterol efflux mediated by cyclodextrins. J Biol Chem. 1995;270:17250–17256. doi: 10.1074/jbc.270.29.17250. [DOI] [PubMed] [Google Scholar]
- Liu F-T, Bohn JW, Ferry EL, Yamamoto H, Molinaro CA, Sherman LA, Klinman NR, Katz DH. Monoclonal dinitrophenyl-specific murine IgE antibody: preparation, isolation, and characterization. J Immunol. 1980;124:2728–2737. [PubMed] [Google Scholar]
- Melkonian KA, Ostermeyer AG, Chen JZ, Roth MG, Brown DA. Role of lipid modifications in targeting proteins to detergent-resistant membrane rafts. Many raft proteins are acylated, while few are prenylated. J Biol Chem. 1999;274:3910–3917. doi: 10.1074/jbc.274.6.3910. [DOI] [PubMed] [Google Scholar]
- Menon AK, Holowka D, Baird B. Small oligomers of immunoglobulin E (IgE) cause large-scale clustering of IgE receptors on the surface of rat basophilic leukemia cells. J Cell Biol. 1984;98:577–583. doi: 10.1083/jcb.98.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mescher MF, Apgar JR. The plasma membrane ‘skeleton' of tumor and lymphoid cells: a role in cell lysis? . Adv Exp Med Biol. 1985;184:387–400. doi: 10.1007/978-1-4684-8326-0_26. [DOI] [PubMed] [Google Scholar]
- Moran M, Miceli MC. Engagement of GPI-linked CD48 contributes to TCR signals and cytoskeletal reorganization: a role for lipid rafts in T cell activation. Immunity. 1998;9:787–796. doi: 10.1016/s1074-7613(00)80644-5. [DOI] [PubMed] [Google Scholar]
- Pierini L, Holowka D, Baird B. FcεRI-mediated association of 6-μm beads with RBL-2H3 mast cells results in exclusion of signaling proteins from the forming phagosome and abrogation of normal downstream signaling. J Cell Biol. 1996;134:1427–1439. doi: 10.1083/jcb.134.6.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LJ, Miller JM. Cholesterol depletion delocalizes phosphatidylinositol bisphosphate and inhibits hormone-stimulated phosphatidylinositol turnover. J Biol Chem. 1998;273:22298–22304. doi: 10.1074/jbc.273.35.22298. [DOI] [PubMed] [Google Scholar]
- Pribluda VS, Metzger H. Transmembrane signaling by the high-affinity IgE receptor on membrane preparations. Proc Natl Acad Sci USA. 1992;89:11446–11450. doi: 10.1073/pnas.89.23.11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribluda VS, Pribluda C, Metzger H. Transphosphorylation as the mechanism by which the high-affinity receptor for IgE is phosphorylated upon aggregation. Proc Natl Acad Sci USA. 1994;91:11246–11250. doi: 10.1073/pnas.91.23.11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racchi M, Baetta R, Salvietti N, Ianna P, Franceschini G, Paoletti R, Fumagalli R, Govoni S, Trabucci M, Soma M. Secretory processing of amyloid precursor protein is inhibited by increase in cellular cholesterol content. Biochem J. 1997;322:893–898. doi: 10.1042/bj3220893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers W, Rose JK. Exclusion of CD45 inhibits activity of p56lckassociated with glycolipid-enriched membrane domains. J Cell Biol. 1996;135:1515–1523. doi: 10.1083/jcb.135.6.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P, Roth MG, Simons K. Interaction of influenza virus haemagglutinin with sphingolipid–cholesterol membrane domains via its transmembrane domain. EMBO (Eur Mol Biol Organ) J. 1997;16:5501–5508. doi: 10.1093/emboj/16.18.5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder R, London E, Brown D. Interactions between saturated acyl chains confer detergent resistance on lipids and glycosylphosphatidylinositol (GPI)-anchored proteins: GPI-anchored proteins in liposomes and cells show similar behavior. Proc Natl Acad Sci USA. 1994;91:12130–12134. doi: 10.1073/pnas.91.25.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder RJ, Ahmed SN, Zhu Y, London E, Brown DA. Cholesterol and sphingolipid enhance the Triton X-100 insolubility of glycosylphosphatidylinositol-anchored proteins by promoting the formation of detergent-insoluble ordered membrane domains. J Biol Chem. 1998;273:1150–1157. doi: 10.1074/jbc.273.2.1150. [DOI] [PubMed] [Google Scholar]
- Sheets ED, Holowka D, Baird B. Membrane organization in immunoglobulin E receptor signaling. Curr Opin Chem Biol. 1999;3:95–99. doi: 10.1016/s1367-5931(99)80017-9. [DOI] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Stauffer TP, Meyer T. Compartmentalized IgE receptor–mediated signal transduction in living cells. J Cell Biol. 1997;139:1447–1454. doi: 10.1083/jcb.139.6.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian K, Holowka D, Baird B, Goldstein B. The Fc segment of IgE influences the kinetics of dissociation of a symmetrical bivalent ligand from cyclic dimeric complexes. Biochemistry. 1996;35:5518–5527. doi: 10.1021/bi9523522. [DOI] [PubMed] [Google Scholar]
- Surviladze Z, Dráberová L, Kubínová L, Dráber P. Functional heterogeneity of Thy-1 membrane microdomains in rat basophilic leukemia cells. Eur J Immunol. 1998;28:1847–1858. doi: 10.1002/(SICI)1521-4141(199806)28:06<1847::AID-IMMU1847>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Thomas JL, Holowka D, Baird B, Webb WW. Large-scale co-aggregation of fluorescent lipid probes with cell surface proteins. J Cell Biol. 1994;125:795–802. doi: 10.1083/jcb.125.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma R, Mayor S. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature. 1998;394:798–801. doi: 10.1038/29563. [DOI] [PubMed] [Google Scholar]
- Weetall M, Holowka D, Baird B. Heterologous desensitization of the high affinity receptor for IgE (FcεR1) on RBL cells. J Immunol. 1993;150:4072–4083. [PubMed] [Google Scholar]
- Weiss A, Schlessinger J. Switching signals on or off by receptor dimerization. Cell. 1998;94:277–280. doi: 10.1016/s0092-8674(00)81469-5. [DOI] [PubMed] [Google Scholar]
- Wolf AA, Jobling MG, Wimer-Macklin S, Ferguson-Maltzman M, Madera JL, Holmes RK, Lencer WI. Ganglioside structure dictates signal transduction by cholera toxin and association with caveolae-like membrane domains in polarized epithelia. J Cell Biol. 1998;141:917–927. doi: 10.1083/jcb.141.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier R, Brennan T, Li Q, McCormack C, Seed B. Membrane compartmentation is required for efficient T cell activation. Immunity. 1998;8:723–732. doi: 10.1016/s1074-7613(00)80577-4. [DOI] [PubMed] [Google Scholar]
- Xu K, Goldstein B, Holowka D, Baird B. Kinetics of multivalent antigen DNP-BSA binding to IgE–FcεRI in relationship to the stimulated tyrosine phosphorylation of FcεRI. J Immunol. 1998a;160:3225–3235. [PubMed] [Google Scholar]
- Xu K, Williams RM, Holowka D, Baird B. Stimulated release of fluorescently labeled IgE fragments that efficiently accumulate in secretory granules after endocytosis in RBL-2H3 mast cells. J Cell Sci. 1998b;111:2385–2396. doi: 10.1242/jcs.111.16.2385. [DOI] [PubMed] [Google Scholar]
- Yancey PG, Rodrigueza WV, Kilsdonk EPC, Stoudt GW, Johnson WJ, Phillips MC, Rothblat GH. Cellular cholesterol efflux mediated by cyclodextrins. Demonstration of kinetic pools and mechanism of efflux. J Biol Chem. 1996;271:16026–16034. doi: 10.1074/jbc.271.27.16026. [DOI] [PubMed] [Google Scholar]
- Zhang J, Berenstein EH, Evans RL, Siraganian RP. Transfection of Syk protein tyrosine kinase reconstitutes high affinity IgE receptor–mediated degranulation in a Syk-negative variant of rat basophilic leukemia RBL-2H3 cells. J Exp Med. 1996;184:71–79. doi: 10.1084/jem.184.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]