

Abstract
We investigated the requirements for targeting the centromeric histone H3 homologue CENP-A for assembly at centromeres in human cells by transfection of epitope-tagged CENP-A derivatives into HeLa cells. Centromeric targeting is driven solely by the conserved histone fold domain of CENP-A. Using the crystal structure of histone H3 as a guide, a series of CENPA/histone H3 chimeras was constructed to test the role of discrete structural elements of the histone fold domain. Three elements were identified that are necessary for efficient targeting to centromeres. Two correspond to contact sites between histone H3 and nucleosomal DNA. The third maps to a homotypic H3–H3 interaction site important for assembly of the (H3/H4)2 heterotetramer. Immunoprecipitation confirms that CENP-A self-associates in vivo. In addition, targeting requires that CENP-A expression is uncoupled from histone H3 synthesis during S phase. CENP-A mRNA accumulates later in the cell cycle than histone H3, peaking in G2. Isolation of the gene for human CENP-A revealed a regulatory motif in the promoter region that directs the late S/G2 expression of other cell cycle–dependent transcripts such as cdc2, cdc25C, and cyclin A. Our data suggest a mechanism for molecular recognition of centromeric DNA at the nucleosomal level mediated by a cooperative series of differentiated CENP-A–DNA contact sites arrayed across the surface of a CENP-A nucleosome and a distinctive assembly pathway occurring late in the cell cycle.
The accurate transmission of replicated eukaryotic chromosomes is mediated by centromeres. Structurally distinct loci present once per chromosome, centromeres provide the essential functions of chromosome segregation. These include specifying the assembly of the kinetochore, a microtubule-dependent motor complex at the surface of the chromosome, and the maintenance of sister chromatid cohesion until their separation at the onset of anaphase (Bloom, 1993; Miyazaki and Orr-Weaver, 1994; Pluta et al., 1995). In addition to these primarily mechanical functions, centromeres act as important regulators of mitosis and meiosis through a mechanism that monitors attachment of chromosomes to the spindle and reports to a spindle assembly checkpoint that regulates progression into anaphase (McIntosh, 1991; Li and Nicklas, 1995; Nicklas et al., 1995; Rieder et al., 1995). Understanding how these functions are specified at a molecular level begins with identification of the molecular recognition events that initiate centromere assembly on the chromosome.
By elegant molecular genetic approaches, it has been possible to identify discrete cis-acting DNA sequences from Saccharomyces cerevisiae (Clarke and Carbon, 1980; Hieter et al., 1985) and Saccharomyces pombe (Hahnenberger et al., 1989; Niwa et al., 1989) that are sufficient to establish centromere function on artificial chromosomes. Dissection of these sequences has revealed that centromere function is established at both the primary structural level of DNA sequence as well as at higher levels of DNA structure within chromatin. DNA sequence recognition is driven by sequence-specific DNA–protein interactions, exemplified by the essential CDE III element of the S. cerevisiae centromere; single point mutations in this 25-bp DNA sequence can completely abolish centromere activity (McGrew et al., 1986; Hegemann et al., 1988). CDE III plays a primary role in kinetochore assembly on the S. cerevisiae centromere by binding to a 240-kD multiprotein complex, CBF3, that mediates the association of a microtubule- dependent motor activity with the chromosome (Lechner and Carbon, 1991; Hyman et al., 1992; Middleton and Carbon, 1994). Cbf3b, a 60-kD subunit of CBF3, is an essential zinc finger protein that is thought to provide the DNA binding function of CBF3 (Lechner, 1994). Other examples of centromere proteins that directly recognize DNA sequence are the yeast helix-loop-helix protein CBF1 (Cai and Davis, 1990) and the mammalian protein CENP-B, which recognizes a discrete sequence element found in centromeric satellite DNA (Earnshaw et al., 1987; Masumoto et al., 1989; Sullivan and Glass, 1991). Thus, molecular recognition of centromeric loci occurs, at least in part, through direct DNA sequence recognition by proteins, interactions similar to the familiar DNA binding activities observed for transcription factors (Mitchell and Tjian, 1989; Harrison, 1991).
Centromere function is also established through essential interactions that take place at the level of DNA structure within chromatin. From the earliest cytological observations of centromeres as the primary constriction of mitotic chromosomes, it has been understood that centromeres are packaged distinctly as constitutive heterochromatin. In the point centromeres of budding yeast, 150–200 bp of cen DNA sequences are packaged in a core particle flanked on both sides by arrays of highly phased nucleosomes (Bloom and Carbon, 1982), and this specialized chromatin structure is necessary for centromere function (Saunders et al., 1988; Bloom et al., 1989). Sequence element CDE II, which comprises a 78–86-bp AT-rich segment conserved in composition but not in sequence among yeast centromeres, appears to adopt a uniquely folded conformation that plays an important role in providing complete centromere function (Sorger et al., 1994; Tal et al., 1994; Sears et al., 1995). The more complex centromeres of fission yeast exhibit a different type of chromatin structure, with several kilobases of DNA in the central core domain packaged in a highly irregular nucleosomal array that is assembled only in conjunction with functional centromere sequences in S. pombe (Polizzi and Clarke, 1991). The dependence of this structure on sequences distal to the central core DNA suggests that large scale folding of the centromere locus is required for the segregation function (Polizzi and Clarke, 1991; Marschall and Clarke, 1995).
Understanding what constitutes a functional centromere sequence in animal cells has been confounded by their large size, ranging from 500–5,000 kb in human chromosomes (Tyler-Smith and Willard, 1993). Nevertheless, it has been possible to map a Drosophila centromere to a 420-kb segment, revealing that both simple satellite sequences as well as islands of complex sequence are required for complete centromere function (Le et al., 1995; Murphy and Karpen, 1995). In mammalian cells, centromere function is also associated with large blocks of heterochromatin comprised of highly repetitive satellite DNA typified by the α satellite of primate chromosomes: extensive tandemly repeated arrays of a 171-bp monomer sequence (Willard, 1991). One of the abiding mysteries of animal centromeres, however, is the lack of sequence conservation of centromeric satellite DNA (Beridze, 1982). With the exception of a small sequence that functions as the binding site for CENP-B, the CENP-B box (Masumoto et al., 1989), no homology is seen in satellite DNA across different classes of vertebrates, and, indeed, satellite DNA is one of the most rapidly evolving compartments of the genome in vertebrates. An important role for CENP-B and the CENP-B box in centromere function is in doubt, however, since its presence is not correlated with centromere function (Earnshaw et al., 1989). Two hypotheses have been suggested to explain this lack of conservation in centromere sequences: either animal cell centromere DNA contains small, as yet unidentified sequence elements similar to yeast centromeres that possess kinetochore-nucleating capabilities, or centromere function is not specified directly by DNA sequence, but rather by higher order DNA or chromatin structure.
One protein situated to play a role in specifying the properties of centromeric chromatin is CENP-A, a centromere-specific homologue of the core nucleosomal protein histone H3 (Palmer et al., 1991; Sullivan et al., 1994). CENP-A was originally identified as a centromere-specific autoantigen that copurified with nucleosomal core particles (Earnshaw and Rothfield, 1985; Palmer and Margolis, 1985; Palmer et al., 1987). A potentially homologous protein has recently been identified in yeast as the product of a gene, CSE4, that is essential for mitotic chromosome segregation (Stoler et al., 1995). Together, CENP-A, CSE4, and histone H3 form a roughly equidistant triangle of homologous proteins linked at the level of ∼60% sequence identity, limited to a COOH-terminal domain of ∼90 amino acids (Sullivan et al., 1994; Stoler et al., 1995). This region corresponds to the domain in histone H3 that is sufficient for nucleosome assembly in vitro (for review see van Holde, 1989) and in vivo (Mann and Grunstein, 1992), and that is part of the highly ordered core of the histone octamer (Arents et al., 1991). Surprisingly, this conserved histone fold domain of CENP-A is required for targeting to human centromeres, rather than the unique sequences of the NH2 terminus (Sullivan et al., 1994).
In this work we have dissected the molecular features of CENP-A that are required for its assembly at human centromeres. By systematic replacement of structural elements of the CENP-A histone fold domain with the corresponding sequences of histone H3, we have identified three regions of the molecule that are required for targeting CENP-A to centromeres. These correspond to two nucleosomal DNA contact sites of histone H3 and a region that mediates self-association between the two copies of histone H3 within the nucleosome. In addition to these structural features, we show that CENP-A expression is uncoupled from normal histone H3 expression, occurring later in the cell cycle, and that this synthetic timing is important for appropriate targeting of CENP-A. Taken together, these data suggest a mechanism for specific molecular recognition of centromeric DNA at the level of the nucleosome.
Materials and Methods
Cell Culture and Transfection
HeLa (ATCC CCL3) and tTA-HeLa cells (Gossen and Bujard, 1992) were maintained in DME with 10% FCS (GIBCO BRL, Gaithersberg, MD) at 37°C in a 5% CO2 atmosphere. tTA-HeLa cultures were supplemented with 400 μg/ml G418. Stably transformed tTA cell lines (see below) were cultured in the presence of 400 μg/ml G418, 330 ng/ml puromycin, and 1 μg/ml tetracycline. For immunofluorescence experiments, cells were plated on glass coverslips at a density of 2–2.5 × 104 cells per cm2 the night before transfection. Transfection was performed in serum-free medium using Lipofectamine (GIBCO BRL) as previously described (Sullivan et al., 1994).
To establish a stable, inducible cell line expressing CENP-A–HA1, the CENP-A insert from pcDL-CAepi was subcloned into plasmid pUHD10.3 (Gossen and Bujard, 1992), forming plasmid pUHD10.3CAepi. tTA HeLa cells expressing the tetracycline transactivator were cotransfected with pUHD10.3-CAepi and pBS-PAC, a puromycin resistance marker (de la Luna et al., 1988) at a 10:1 ratio using Lipofectamine. Transformants were selected using 330 ng/ml puromycin in the presence of 1 μg/ml tetracycline, and individual clones were assayed by induction in tetracycline-free medium followed by Western blot analysis.
DNA Constructs
Construction of Segmental Mutants.
General methods were essentially as described by Ausubel et al. (1995) unless specified. Mutations were constructed in plasmid pcDL-CAepi, which is identical to pcDL-CAHA1 (Sullivan et al., 1994) except that three copies of the hemagglutinin (HA)1 1 epitope are present at the COOH terminus of the coding region. Histone H3 sequences were obtained from plasmid pMH3.2-614 (Taylor et al., 1986). Segments of CENP-A were replaced with the corresponding H3 sequence using a bimolecular recombinant PCR strategy. A pair of standard 5′ and 3′ oligonucleotide primers (GIBCO BRL) flanking the CENP-A coding region of pcDL-CAepi was prepared and used in all experiments. For each mutant, two divergent overlapping primers were constructed, each containing at least 12–15 bp of CENP-A sequence at their 3′ ends and a segment encoding the desired mutations at their 5′ ends. Each mutagenic oligonucleotide pair was designed with a 15–17-bp overlap. PCR reactions were performed using each mutagenic primer in conjunction with the appropriate flanking primers, synthesizing two DNA fragments that overlapped by 15–17 bp within the mutated region. PCR reactions (95°C × 90 s; 20 × [95°C × 30 s, 55°C × 60 s, 72°C x 90 s]; 72°C × 10 min) were performed in 50 μl using 5 μg/ml pcDL-CAepi, 1.5 mM Mg, 1 μM each primer, 100 mM dNTPs (Pharmacia Fine Chemicals, Piscataway, NJ), 10% DMSO, and 1.25 U of an 8:1 unit ratio mixture of Taq DNA polymerase (Promega, Madison, WI) and Pfu DNA polymerase (Stratagene, La Jolla, CA) made fresh for each experiment. PCR products were purified via QX Matrix (Qiagen, Chatsworth, CA), combined, and used as template for a second round of PCR using only the standard primers, performed as above except with 10 amplification cycles. Full-length recombinant PCR products were cloned into plasmid pCRII (Invitrogen, San Diego, CA). The sequence of the entire coding region was verified (Sequenase 2.0; United States Biochemical Corp., Cleveland, OH), and inserts from correct clones were isolated as NarI–SacI fragments and cloned into NarI- and SacI-digested pcDL-CAepi. Plasmids for transfections were prepared with Qiagen DNA purification columns.
W86 Mutants.
Codon 86 was randomized by the same bimolecular recombinant PCR strategy described above, using primers possessing the sequence NN(C/T) on the coding strand. Approximately 50 pCRII transformants were picked and colony sequenced with the CircumVent thermal cycle sequencing kit (New England Biolabs Inc., Beverly, MA) using a 32P–end labeled primer that spanned nucleotides 275–293 in the CENP-A cDNA to determine the sequence of codon 86. 13 different mutants were recovered (Y, F, I, L, V, C, N, D, H, R, S , P, and G) and cloned into pcDL-CAepi as above. Constructs that failed to localize were sequenced completely to verify that loss of function was due to mutation at codon 86 (Sequenase 2.0; United States Biochemical Corp.).
Transfer of CENP-A Helix II to Histone H3.
A trimolecular recombinant PCR strategy was used to replace helix II codons 85–112 of histone H3 with the corresponding codons 87–114 of CENP-A. Three fragments were generated in the first round of PCR. Fragment 1, the 5′ fragment, containing codons 1–84 of histone H3, was amplified from plasmid pcDLH3HA1 using the standard 5′ primer as above and a 30-mer primer at the 3′ end corresponding to codons 80–84 of histone H3 plus codons 87–91 of CENP-A (the insertion of two codons in CENP-A relative to histone H3 accounts for the difference in residue numbering). Fragment 2, the central fragment corresponding to helix II of CENP-A, was amplified from pcDLCAHA1 using a 5′ primer complementary to the 3′ primer of fragment 1 and a primer at the 3′ end corresponding to the last five codons of CENP-A helix II and codons 113–117 of histone H3. Fragment 3, the 3′ fragment encoding the COOH-terminal portion of histone H3 and the HA-1 epitope, was amplified from pcDL-H3HA1 using a 5′ primer corresponding to the last five codons of CENP-A helix II and codons 113–117 of histone H3 and the standard 3′ primer an oligo in the 3′ untranslated region of CENP-A. The three fragments were purified, and then combined in equimolar amounts to provide the template for a second round of PCR using the standard 5′ and 3′ primers; the product was isolated and subcloned for expression as described above. The complete coding region sequence of the resulting plasmid was verified by sequencing.
Plasmid pMH3-CAHA1 was constructed by replacing the histone H3 coding region of pMH3.2-614 with an epitope-tagged CENP-A fragment. PCR primers for CENP-A were constructed incorporating an NcoI site at the ATG initiator codon at the 5′ end and a single copy of the HA-1 epitope followed by an AflIII site at the 3′ end. The amplified product was cloned into NcoI–AflIII-digested pMH3.2-614 and verified by DNA sequencing.
Immunofluorescence
For analysis of protein localization in transfected cells, immunofluorescence microscopy was performed 18–72 h after transfection, as described previously (Sullivan et al., 1994). Endogenous centromere antigens were visualized with a human anticentromere antiserum, hACA-M detected with a rhodamine–coupled secondary antibody, while HA epitope–tagged proteins were visualized with mAb 12CA5 (a kind gift from Dr. Ian Wilson, The Scripps Research Institute, La Jolla, CA) and fluorescein-coupled secondary antibody.
Immunochemistry
Immunoblots were performed as described previously (Sullivan et al., 1994) using human anticentromere serum hACA-M at a dilution of 1:2,000 and mAb 12CA5 at a concentration of 5 μg/ml. Blots were developed using HRP-coupled secondary antibodies (Amersham Corp., Arlington Heights, IL) and a chemiluminescence detection reagent (Pierce Chemical Co., Rockford, IL).
For immunoprecipitation analysis, protein expression in a stable pUHD10.3-CAepi transformant was induced for 3 d. Nuclei from 3–5 × 107 cells were isolated according to Masumoto et al. (1989), washed in buffer A ( 5 mM Hepes, pH 7.5, 10 μM leupeptin, 1.5 μM aprotinin, 1 mM DTT), and centrifuged at 3,000 g. The nuclear pellet was resuspended in 500 μl digestion buffer at a concentration of 0.5–1 × 108/ml (buffer A containing 200 U/ml micrococcal nuclease, 1 mM CaCl2) and incubated at 37°C for 5 min. Digestion was stopped by addition of EDTA to a final concentration of 10 mM. After centrifugation at 8,000 g, the supernatant was collected, and the pellet was resuspended in buffer A and subjected to two additional rounds of extraction by sonication for 10 s followed by centrifugation and collection of the supernatants. Supernatants were pooled in a siliconized Eppendorf tube, supplemented with 0.1% NP-40 and 25 μg of mAb 12CA5, and mixed end over end for 2 h at 4°C. A 100-μl aliquot of protein A–Sepharose (Pharmacia Fine Chemicals) previously equilibrated with buffer A was added and incubated for an additional 2 h at 4°C. The beads were collected by centrifugation and the supernatant was saved. Immunoprecipitates were washed five times with buffer A, and then resuspended in SDS-PAGE sample buffer. Equivalent amounts of all soluble fractions and one half of the immunoprecipitated proteins were analyzed by Western blotting.
Isolation of a Human CENP-A Genomic Sequence
A human Caucasian male placental genomic DNA library prepared in Lambda Fix II (Stratagene; a kind gift from Edward Chan, The Scripps Research Institute) was screened by PCR (Israel, 1993) using CENP-A primers that span a small intron. Two phage with overlapping inserts spanning 20 kb of genomic DNA were isolated and characterized by restriction mapping using a series of probes derived from the CENP-A cDNA (to be described in detail elsewhere). A 2,878-bp EcoRI fragment containing a 5′ flanking genomic sequence was isolated and sequenced by a combination of manual and automated methods (GenBank accession number U82609). The 2.9-kb fragment was found to contain 1,101 bp upstream of the start of our CENP-A cDNA clone, the first 250 bp of the cDNA and 1,527 bp of the first intron in CENP-A.
Cell Cycle Analysis
HeLa cells were grown in 10-cm dishes to ∼60% confluence. The first block was initiated by replacing medium with complete DME containing 2 mM thymidine. After 15 h, cells were released by washing twice with dPBS and adding normal complete DME, and were allowed to grow for 9 h. Cells were blocked a second time for 15 h as above. After release as above, samples were collected at 2 h intervals for 16 h by trypsinization and washed twice with PBS, and pellets were kept at −70°C until preparation of RNA. For time points exhibiting an increased mitotic index (8–12 h after release), cells were also recovered from the media and the washes before trypsinization.
RNA was isolated by acidic guanidinium thiocyanate/phenol-chloroform extraction (Xie and Rothblum, 1991). CENP-A mRNA was assayed by RNase protection using a probe constructed by cloning a 155-bp EcoR1– ApaI fragment containing the 5′ end of the CENP-A cDNA into pBSSK(+) (Stratagene). Plasmid was linearized with XbaI for transcription by T7 RNA polymerase (Maxiscript kit; Ambion, Austin, TX) and α-[32P]UTP (Amersham Corp.) according to the manufacturer's instructions. The probe length was 203 bp with a protected fragment length of 153 bp. RNase protection asays (HybSpeed RPA kit; Ambion) were performed using 10 μg of total HeLa RNA isolated from synchronized cells. Hybridization of probe (350K cpm/rxn) and RNA was carried out for 1 h at 68°C in siliconized tubes followed by digestion with an RNase A/T1 mixture used at a dilution of 1:100 from the supplied concentration. End-labeled size markers were prepared from an HaeIII digest of pBS-SK(+). Reactions were electrophoresed on 6% sequencing gels and exposed for 2 h on a phosphor imaging screen from Molecular Dynamics (Sunnyvale, CA). ImageQuant software (Molecular Dynamics) was used to quantitate signal intensities. Histone H3 mRNA abundance was determined in the same samples by Northern blot analysis, using the coding region of plasmid pMH3.2-614 as a probe, and similarly quantitated.
Results
Structural Determinants of Centromeric Targeting
The histone fold domain consists of a set of three α helices (H I, H II, H III) separated by two turn/β sheet structures (strand A, strand B); histone H3 and, by homology, CENP-A contain an additional α helix at the NH2 terminus of the fold domain (N helix; Fig. 1 A) (Arents et al., 1991). To evaluate CENP-A targeting within the context of this structure, we prepared a set of substitution derivatives by replacing CENP-A sequences within the fold with the homologous histone H3 sequences (Fig. 1 A). Mutations were constructed using an epitope-tagged version of CENP-A carrying three copies of the influenza hemagglutinin HA-1 epitope (Wilson et al., 1984) at the COOH terminus, allowing us to monitor the expression (Fig. 1 B) and localization of CENP-A derivatives in transfected cells (Fig. 2; WT).
Figure 1.

Mutations constructed for analysis of the CENP-A histone fold domain. (A) A diagram of the structural organization of the histone fold domain is shown at top over a sequence comparison between CENP-A and histone H3. Below is a table detailing the specific amino acid substitutions for each of the CENP-A mutants analyzed. Only the residues changed in CENP-A are shown. (B) Efficient expression of CENP-A mutants in HeLa cells. Plasmids containing mutant CENP-A sequences were transiently transfected into HeLa cells for analysis of protein expression by Western blotting with mAb 12CA5: (1) wildtype CA-HA1; (2) HN1; (3) HN2; (4) HH1; (5) HSA; (6) HSA5; (7) HSAΔ; (8) HSA3; (9) HH2; (10) HH2.1; (11) HH2.2; (12) HH2.3; (13) HSB; (14) HC. Molecular mass markers are shown (left) with sizes in kD noted.
Figure 2.

Sequences required for centromere assembly are located in the central portion of the histone fold domain. Each panel shows a representative nucleus from a transfected cell visualized by confocal microscopy. CENP-A–HA1 derivatives were localized with mAb 12CA5 (green, left) and endogenous centromere proteins were detected with a human autoantiserum (red, right). The center of each panel shows a merge of the two immunofluorescence signals to evaluate antigen codistribution, where yellow indicates colocalization. WT, wild-type CENP-A; NΔ, amino-terminal deletion mutant CA-APA; hI, helix I mutant HH1; sA, strand A mutant HSA; hII, helix II mutant HH2; sB, strand B mutant HSB; C, carboxyl-terminal mutant HC. The lower right panel illustrates the relative positions of these structural elements in CENP-A. Note that CENP-A derivatives that fail to target often required increased exposure to collect an image of CENP-A–HA1 distribution, accomplished by increasing the slit width on the confocal microscope, resulting in an apparent increase in the total nuclear fluorescence intensity as compared with targeting derivatives. Nevertheless, for these nontargeting derivatives, no qualitative differences in distribution were observed between cells that expressed the transfected gene products at the limits of detection vs high level expressors.
We first asked whether the histone fold domain is sufficient to direct centromeric targeting. In previous experiments, the NH2-terminal tail of CENP-A was replaced with that of histone H3, which, although lacking sequence homology, shares its highly basic character with CENP-A (Sullivan et al., 1994). To determine if a basic NH2-terminal tail is dispensable for targeting, codons 4–31 of CENP-A were excised. The resulting protein showed no impairment of targeting to centromeres, demonstrating that a basic NH2-terminal tail is dispensable for this function (Fig. 2; NΔ). Thus, the COOH-terminal portion of CENP-A, corresponding to the histone fold homology domain, is both necessary and sufficient for assembly of CENP-A at centromeres.
Within the histone fold domain, we initially examined four regions corresponding to secondary structure segments of the domain based on the data of Arents et al. (1991) and Richmond et al. (1993): helices I and II, strand A, and strand B (Fig. 1 A). We also tested the COOH terminus, which is longer in CENP-A and divergent from histone H3. Helix III was not tested since only a single conservative (Ile-Val) substitution is found in this segment of CENP-A. The two most conserved regions, helix I (Fig. 2; hI) and strand B (Fig. 2; sB), could be substituted without affecting targeting, as could the COOH terminus (Fig. 2; C). The strand A substitution, residues 75–86, was profoundly deficient in targeting ability (Fig. 2; sA). It retained a small degree of targeting specificity that was observed as a slight increase in centromere staining over an essentially uniform nuclear incorporation in a minority of cells (see also Fig. 3 D below). Substitution of helix II of the histone fold domain resulted in a complete loss of targeting to centromeres (Fig. 2; hII). These data demonstrate that sequences in the central portion of the histone fold domain are primarily responsible for targeting CENP-A to centromeres.
Figure 3.
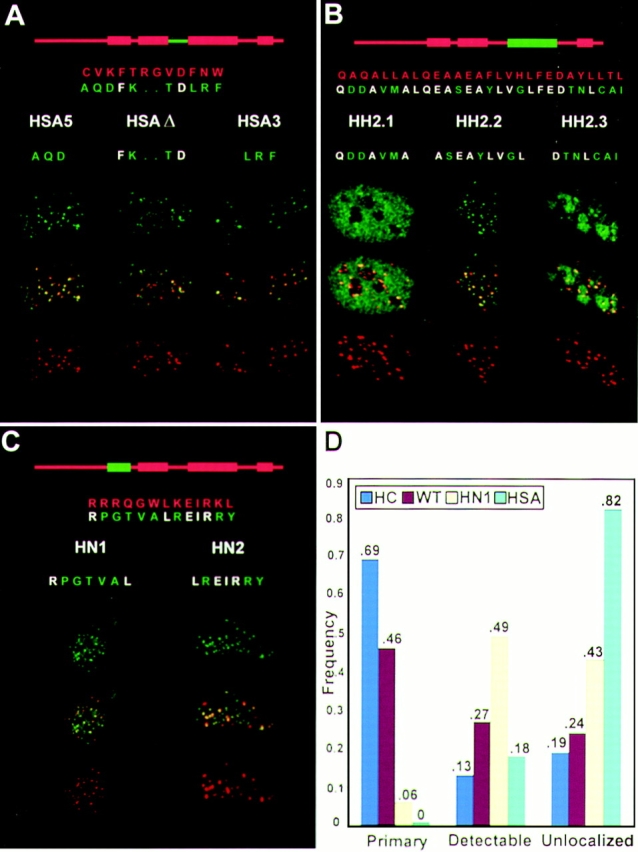
Analysis of sequence components within the histone fold structures required for targeting. The strand A and helix II elements, as well as the N-helix of CENP-A, were further dissected to identify discrete sequences required for targeting. A–C show immunofluorescence results. Each panel shows a diagram of CENP-A with the test region highlighted in green over an alignment of the sequence of CENP-A (red) with the corresponding sequence of histone H3, with divergent residues highlighted in green. Typical immunofluorescence results are shown for individual mutants below the alignment. Images are as in Fig. 2, except they are oriented vertically with CENP-A mutants on top (green), endogenous centromeres at bottom (red), and a merge of both signals shown in the center of each group. Yellow indicates colocalization. (A) Strand A subregions. (B) Helix II subregions. (C) N-helix subregions. (D) A chart of the frequency of different localization patterns obtained for four CENP-A derivatives. Wide field images (161 μm × 215 μm) were collected using a ×40 objective. At least 80 transfected cells were examined for each construct and CENP-A–HA1 distribution was scored as described in the text. The frequency of cells in which the test protein was observed to be primarily localized (left group), detectably localized (center group), or unlocalized (right group) to the centromeres is plotted. A legend at the top left specifies each construct as it appears in Fig. 1. For control constructs WT and HC, there was a close correlation between total fluorescence intensity and the degree of nucleoplasmic staining, indicating that mislocalization of these proteins was due to overexpression.
The two segments identified in this experiment comprise a large contiguous region at the center of the domain and contain most of the divergent amino acids that distinguish CENP-A from histone H3. To further refine identification of targeting sequences, strand A and helix II were each divided into NH2-terminal, central, and COOH-terminal portions, each containing three to five CENP-A specific residues that were substituted with histone H3 sequences as above (Fig. 1 A). This analysis showed the NH2 and COOH termini of the long central helix II were necessary for targeting CENP-A, but replacement of residues in the central portion of the helix had no effect (Fig. 3 B). In contrast, none of the strand A subregion mutations, including the deletion of two amino acids in the center of this region, showed any significant impairment of targeting (Fig. 3 A). Thus, the CENP-A–specific sequences at the two ends of helix II, residues 88–92 and 109–114, are necessary for assembly at centromeres, while strand A, residues 75–86, can accommodate significant change in amino acid sequence and length without disruption of targeting activity.
One residue in this region, Trp86, was selected for specific mutagenesis. This residue is notable because Trp is absent in the core histones, but is present at this same position in CSE4 (Stoler et al., 1995). This codon was randomized with a PCR procedure, and 13 mutants encoding different amino acids were recovered and tested for targeting (data not shown). Replacement of Trp86 with the aromatic residues Tyr or Phe (the amino acid normally found in this position in histone H3) had no effect on targeting. Aliphatic residues showed intermediate levels of targeting roughly correlated with hydrophobicity, while hydrophilic and charged residues failed to target at all. This experiment rules out a specific role for this Trp residue in centromeric targeting, but demonstrates a requirement for an aromatic amino acid.
Histone H3 contains an additional α helix at the amino terminus of the histone fold domain, the N-helix. Secondary structure prediction reveals a putative α helix in this segment of CENP-A, spanning residues 43–55. When this region was replaced along with the entire NH2-terminal tail of histone H3, the resulting protein, H3-CA, retained targeting activity but was less efficient at localizing to centromeres (Sullivan et al., 1994). Two additional replacement mutants were constructed, HN1 and HN2, to ask whether a secondary targeting element could be identified in this region (Fig. 1 A). Mutant HN1, replacing the NH2terminal portion of this helix, had a distribution similar to H3-CA, with localization at centromeres detected over variable levels of nonspecific nuclear staining (Fig. 3, C and D). Mutant HN2, spanning the COOH-terminal portion of the N-helix, targeted normally.
Quantitative assessment of the relative roles of the different targeting elements of CENP-A is complicated by the fact that levels of gene expression vary considerably within the transiently transfected cell population. Even for wild-type CENP-A–HA1, a substantial fraction of cells was observed in which overexpression results in uniform nuclear staining. To compare the targeting defects of the strand A and HN1 mutations, we assayed populations of transfected cells, judging the distribution of epitopetagged CENP-A as being primarily localized at centromeres (e.g., Fig. 2; WT and C), detectably localized (ranging from Fig. 2; sA, to Fig. 3 C), or unlocalized (e.g., Fig. 2, hII). Data are presented in histogram form in Fig. 3 D. For two control constructs assayed simultaneously, the majority of cells had primarily localized epitope with the remaining cells approximately evenly distributed in the other two classes (Fig. 3 D; WT and HC). For the N-helix mutant, only a small proportion of cells (6%) exhibited primarily localized mutant protein, while most cells (49%) showed detectably localized centromeric CENP-A over varying levels of general nuclear staining (Fig 3 D; HN1). For the strand A mutant, no cells were observed with staining primarily at centromeres, and only 18% showed any detectable targeting above the general nuclear staining (Fig. 3 D; HSA). These results suggest that the predicted N-helix of CENP-A contains sequences that are required for efficient targeting to centromeres but to a lesser extent than sequences of strand A or helix II.
Since the long central helix II was the only region that was absolutely required for targeting to centromeres, we sought to determine whether it could act by itself to direct histone H3 preferentially to centromeres. A derivative of histone H3 was constructed by replacing residues 85–112 of histone H3 with the corresponding residues of CENP-A (87–114). The resulting protein showed no ability to localize to centromeres, not even at the level of the strand A mutant of CENP-A (data not shown). Thus, while helix II sequences are required for targeting CENP-A to centromeres, they function only in conjunction with other components of CENP-A.
Self-association of CENP-A Predicts Formation of Homotypic Nucleosomes
The COOH-terminal segment of histone H3 helix II provides a unique function within the nucleosome, mediating protein–protein association at the dyad axis that links the two symmetric halves of the nucleosome (Camerini-Otero and Felsenfeld, 1977; Xie et al., 1996). A requirement of this sequence for targeting CENP-A was revealed by mutant HH2.3 (Fig 3 B), suggesting that protein–protein interactions within the nucleosome are important for CENP-A function. To ask whether CENP-A exhibits selfassociation properties, we constructed a stable HeLa cell line that inducibly expresses the epitope-tagged CENP-A derivative, CENP-A–HA1. Upon induction, cells accumulate CENP-A–HA1 (Fig. 4 A) at their centromeres (data not shown), allowing us to assay protein interactions under conditions in which CENP-A was primarily localized at centromeres. Chromatin was solubilized from isolated nuclei by micrococcal nuclease digestion followed by brief sonication to release centromeric chromatin. After immunoprecipitation from this soluble chromatin extract using mAb 12CA5, fractions were analyzed by SDS-PAGE and immunoblot analysis using human anti–centromere antibodies, allowing detection of both epitope-tagged and endogenous CENP-A (Fig. 4, B and C). Under conditions in which CENP-A–HA1 was present at a lower abundance than endogenous CENP-A, the immunoprecipitated fraction always contained equimolar quantities of endogenous CENP-A recovered with CENP-A–HA1, as judged by Western blot signal intensity (Fig. 4 B). When CENP-A–HA1 was overexpressed relative to endogenous CENP-A, the endogenous protein was still recovered in immunoprecipitates, but in diminished quantities (Fig. 4 C). These data provide strong evidence for CENP-A self-association in vivo. A nucleosome containing CENP-A could in principle be either heterotypic, containing one copy each of CENP-A and histone H3, or homotypic with two copies of CENP-A. Recovery of endogenous CENP-A in the presence of a vast amount of the potential competitor histone H3 demonstrates a preference for self-association. The presence of equimolar amounts of endogenous and epitope-tagged proteins under the conditions of Fig. 4 B, where the quantitatively minor CENP-A–HA1 is essentially doping the CENP-A pool, indicates that this association is highly efficient—essentially all CENP-A–HA1 is present in an equimolar complex. Competition by CENP-A–HA1 when it is quantitatively overexpressed, as in Fig. 4 C, is further evidence for efficient CENP-A/CENP-A self-association. We conclude that CENP-A nucleosomes are homotypic for CENP-A.
Figure 4.
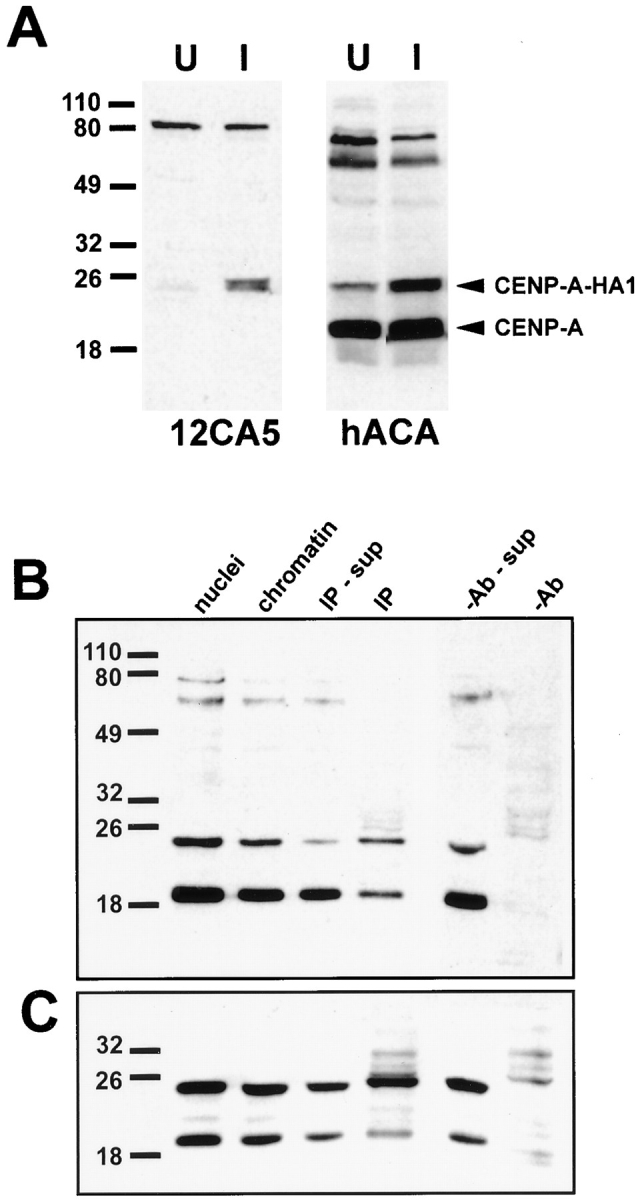
Self-association in vivo indicates that CENP-A nucleosomes are homotypic. (A) Inducible expression of CENP-A– HA1 in stably transformed HeLa cells. HeLa tTA-CAHA cells were grown in the presence (U) or absence (I) of tetracycline for a period of 2 d. Total cell protein was then analyzed by Western blotting with mAb 12CA5 (left) and with a human anticentromere serum (right). The positions of CENP-A and the plasmidderived CENP-A–HA1 are noted (right). Induction results in accumulation of CENP-A–HA1. (B) HeLa tTA-CAHA cells were induced by removal of tetracycline as in A. A soluble chromatin fraction was prepared from isolated nuclei for immunoprecipitation as described in Materials and Methods. After immunoprecipitation with mAb 12CA5, fractions were analyzed by Western blotting with human anticentromere serum to reveal both endogenous and epitope-tagged CENP-A. Fractions are: nuclei, whole nuclei; chromatin, micrococcal nuclease solubilized nuclear extract; IP-sup, supernatant after immunoprecipitation; IP, proteins recovered by immunoprecipitation; -Ab-sup, supernatant from mock immunoprecipitation without mAb 12CA5; -Ab, proteins recovered by mock immunoprecipitation without mAb 12CA5. (C) Western blot analysis of an immunoprecipitation experiment after induction of CENP-A–HA1 to levels higher than endogenous CENP-A. Fractions are as in B.
Regulatory Determinants of Centromeric Targeting
The assembly of normal histone H3 into chromatin takes place concurrently with DNA replication, as histone H3/H4 tetramers are deposited on newly synthesized DNA within minutes (Worcel et al., 1978). For our initial experiments, we reasoned that expression of CENP-A during S phase would be appropriate, and we prepared a construct in which the coding region of CENP-A was placed under the regulatory signals of a mouse S-phase–dependent histone H3 gene (Taylor et al., 1986; Harris et al., 1991) (Fig. 5). Surprisingly, even at low levels of expression, CENP-A synthesized from this plasmid failed to accumulate at centromeres but was distributed throughout the nucleus (Fig. 5). Since we observe targeting in cells that express CENP-A–HA1 constitutively, we interpret these results to show that uncoupling CENP-A expression from normal histone expression in S phase is an important component of the CENP-A targeting mechanism.
Figure 5.
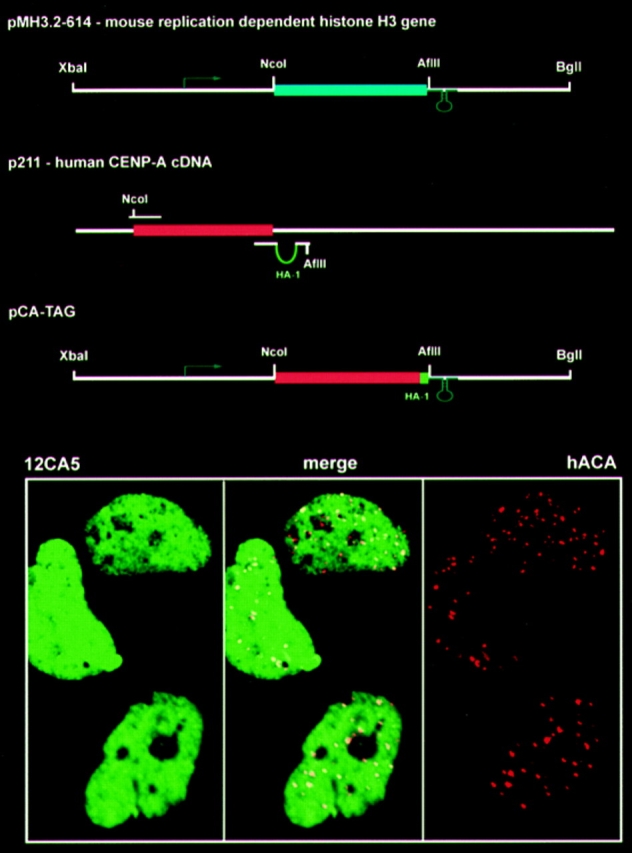
Restriction of CENP-A expression to S phase abolishes centromeric targeting. (Top) Construction of plasmid pCA-TAG, which expresses CENP-A under the regulatory elements of a replication-dependent histone H3 gene, MH3.2-614. The construct was prepared by replacing the intronless histone H3 coding region with the CENP-A cDNA coding region, adding an HA-1 epitope (green) and maintaining both the promoter (arrow) and 3′ untranslated (loop) regulatory components of MH3.2-614. (Bottom) Results of immunofluorescence analysis of cells transiently transfected with pCA-TAG, with CENP-A–HA1 on the left (green), endogenous centromeres on the right (red), and a merge of the two signals in the center. Localization of CENP-A– HA1 at centromeres could not be detected in cells expressing the protein even at the lower limits of detection.
The cell cycle–dependent expression of CENP-A was examined directly in cells synchronized at the G1/S boundary using a double thymidine block procedure. CENP-A mRNA was detected using an RNase protection assay (Fig. 6 A) while histone H3 transcripts were detected by Northern blot analysis (Fig. 6 B). HeLa cells released from a block at the G1/S boundary take 7 h to complete S phase, and spend 3.5 h in G2 and ∼1 h in mitosis (Rao and Johnson, 1970). A plot of the relative abundance of each transcript as a function of time after release (Fig. 6 C) showed that accumulation of histone H3 mRNA paralleled previously published analyses, peaking in mid S phase 4–5 h after release from the thymidine block, followed by a rapid decline to baseline levels by 8–10 h (Harris et al., 1991). In contrast, CENP-A mRNA accumulation did not begin until mid S phase and reached maximal levels 8–10 h after release. CENP-A mRNA levels also rapidly declined between 10 and 12 h after release. CENP-A protein was assayed in parallel by Western blot analysis using a human autoantiserum, showing a gradual increase in abundance starting 4–6 h after release, consistent with an approximate doubling of the CENP-A pool (data not shown).
Figure 6.

CENP-A expression occurs late in the cell cycle, after histone H3, and may be driven by a cell cycle–dependent promoter element. HeLa cells were synchronized at the G1/S boundary using a double thymidine block, and then released into S phase. Samples were collected at 2-h intervals for a period of 16 h, and total RNA was isolated. (A) CENP-A mRNA was detected using an RNase protection assay with a probe spanning the 5′ end of the cDNA, resulting in a protected band of the predicted size, 158 bp. A second, shorter protected band that paralleled the main band in abundance was observed, probably corresponding to an alternative transcription start site or promiscuous digestion of the 5′ end of the probe/transcript hybrid. Lanes are: M, markers; A, asynchronous culture; 0–16, time points in hours after release; P, undigested probe. (B) RNA was also analyzed by Northern blotting and probed with a histone H3 coding region probe. Samples correspond to the time course in A and are aligned under the appropriate lanes. (C) Signals from A and B were quantitated using a phosphorimager. The relative abundance of histone H3 (grey) and CENP-A (black) transcripts, using the lowest value for each transcript as the baseline, is plotted as a function of time. (D) Sequence of a segment of genomic DNA flanking the first exon of CENP-A, aligned with cell cycle–regulatory elements identified for three late S/G2-regulated genes. Regions of homology are boxed, and the distance of the last displayed nucleotide from the transcription start site, or the 5′ end of the cDNA for CENP-A, is shown at right.
The pattern of mRNA accumulation observed for CENP-A is similar to that of several cell cycle related gene products, including cdc2, cdc25C, and cyclin A (Dalton, 1992; Zwicker et al., 1995). A common repressor-mediated transcriptional control mechanism has recently been identified among these three cell cycle–regulated genes, conferred by a conserved DNA sequence motif that spans 15 bp located within 20 nucleotides 5′ of the transcription start site (Lucibello et al., 1995; Zwicker et al., 1995). This element contains two conserved segments, seven and five nucleotides in length, separated by a 3-bp linker of unconserved sequence (Fig. 6 D). A genomic clone for human CENP-A was isolated, and a 2.9-kb fragment containing the first exon and 1.1 kb of 5′ flanking genomic DNA was subjected to DNA sequence analysis. A sequence nearly identical to the cell cycle repressor motif was found 11 bp upstream of the 5′ end of the CENP-A cDNA (Fig. 6 D). In CENP-A, the two conserved elements of the motif shared 100% identity with the cell cycle repressor motif. Curiously, these were separated by 8 bp rather than 3 bp, precisely an additional half helical turn of the DNA, as compared with cdc2, cdc25C, and cyclin A. Nevertheless, coupled with the observation that CENP-A mRNA accumulates with a similar kinetic pattern during the cell cycle, it is reasonable to propose that this motif is involved in linking CENP-A gene activity to the cell cycle. Taken together, these results strongly suggest that expression late in the cell cycle is necessary for proper assembly of CENP-A at centromeres.
Discussion
Three pieces of evidence suggest that CENP-A acts as a core histone, replacing histone H3 within the histone octamer. The first is the biochemical demonstration that CENP-A copurifies with nucleosomes and with the histone H3/H4 tetramer during fractionation of chromatin (Palmer and Margolis, 1985; Palmer et al., 1987). The second is the high degree of amino acid sequence homology shared by CENP-A and histone H3, specifically within the histone fold domain (Palmer et al., 1991; Sullivan et al., 1994). Finally, association with chromatin and a genetic interaction with normal histone H4 suggest that CSE4, the putative S. cerevisiae homologue of CENP-A, is a nucleosomal protein (Stoler et al., 1995; Smith et al., 1996). From these considerations, it is logical to consider the overall organization of CENP-A as similar to that of histone H3 within the histone octamer (Arents et al., 1991; Arents and Moudrianakis, 1993; Richmond et al., 1993).
Structural Basis for CENP-A Assembly at Centromeres
At a structural level, the ability of CENP-A to assemble into centromeric chromatin is specified solely by the histone fold domain. As with the other core histones, histone H3 makes several contacts with nucleosomal DNA as it winds over the surface of the histone octamer (Mirzabekov et al., 1978; Shick et al., 1980; Richmond et al., 1984, 1993; Hill and Thomas, 1990; Arents and Moudrianakis, 1993). Two of the three targeting elements of CENP-A correspond to histone H3–DNA contact sites. The first of these is near the site where DNA enters and exits the octamer, corresponding to the position of the N-helix (Fig. 7 A, peach), which acts as a weak targeting element in our experiments (Richmond et al., 1984, 1993; Hill and Thomas, 1990). A second major H3–DNA contact takes place at the position of strand A and the NH2 terminus of helix II, one of the most concentrated sites of divergence between CENP-A and histone H3 (Mirzabekov et al., 1978; Shick et al., 1980; Richmond et al., 1984, 1993). These sequences form a fairly broad strip on the surface of the nucleosome lying directly across the DNA path (Fig. 7 A, yellow and tan). Strand A is a part of a parallel β sheet structure that has been proposed to act as a specific DNA binding element of the histone octamer (Arents and Moudrianakis, 1993), while the NH2 terminus of helix II is directly adjacent to this region and exposed on the surface. Since small substitutions in strand A had no significant effect on centromeric targeting, it is unlikely that specific side-chain interactions with DNA are required for this region's contribution to the targeting function. Rather, it may act by imparting some general structural features to this portion of the core particle, perhaps influencing the structure of the NH2 terminus of helix II.
Figure 7.

Model of CENP-A targeting elements. (A) A model of a (CENP-A/H4)2 heterotetramer. The sequences of CENP-A and histone H4 were modeled onto atomic coordinates of dTAFII and projected as an alpha carbon backbone trace. Histone H4 (white) and CENP-A (green) are shown with targeting features highlighted: N-helix (peach), strand A (yellow), NH2 terminus of helix II (tan), and COOH terminus of helix II (orange). The image in the top left is a “front” view perpendicular to the superhelical axis, while the top right shows a view down the superhelical axis. Below is a “bottom” view showing the dyad axis. DNA winds spool-like around the core starting from the left of the N-helix in the top right view, over the top to contact the strand A–N helix II segment of the “back” copy of CENP-A, across the dyad axis and up across the “front” copy strand A–N helix II segment, and over the top again to exit along the back N-helix. Compare with figures in Arents and Moudrianakis (1993). Images were generated using rasmol (Sayle and Milner-White, 1995). (B) Diagram of proposed DNA–CENP-A contacts in the nucleosome core particle. The 146-bp core particle associated DNA is shown. Histone H3 contact sites mapped by cross-linking (Mirzabekov et al., 1978) are shown as black bars, illustrating symmetric contacts made with each DNA strand by the two copies of histone H3 in the core particle. Approximate H3–DNA contact sites observed by x-ray crystallography (Richmond et al., 1993) are shown as grey bars, and the dyad axis is indicated by an orange line at the center. The structural segments of CENP-A involved in these contacts are denoted by colored bars as in A. (Arrowheads) Location of structural distortions in nucleosomal DNA noted by Richmond et al. (1984) and reviewed by Wolfe (1995), and thought to play a role in establishing the translational position of DNA on the histone octamer. Note the congruence of CENP-A targeting elements with DNA contact sites and their symmetry around the dyad axis.
A third region of CENP-A that is necessary for targeting is the COOH-terminal portion of the long central helix II (Fig. 7, orange). This region, largely buried in the interior of the H3/H4 tetramer, forms an important protein– protein interaction between the two copies of histone H3, directly on the dyad axis of the nucleosome (Camerini- Otero and Felsenfeld, 1977). This is the only homotypic interchain interaction that can be detected by contact site cross-linking experiments with nucleosome core particles, indicating that the H3/H4 tetramer is held together primarily by this H3–H3 interaction (for review see van Holde, 1989). The role of this region in mediating protein–protein interaction is quite apparent in the structure of the dTAFII62/dTAFII42 heterotetramer, a component of TFIID whose structure is strikingly similar to the heterotetrameric histone H3/H4 core of the nucleosome (Xie et al., 1996). In this structure, two molecules of dTAFII42, the histone H3 homologue, make an extensive contact at the COOH terminus of helix II, which links the two symmetric halves of the heterotetramer. In CENP-A, this region, 109-AYLLTL114, presents more hydrophobic and bulky side chains than the corresponding region of histone H3, 107-TNLCAI112. Thus, this element is situated to affect the protein–protein interactions across the dyad axis of a CENP-A nucleosome, differentiating it from histone H3.
DNA Recognition by Specialized Nucleosomes: A Model
Taken together, these structural considerations suggest a model for the selective recognition of centromeric DNA by CENP-A driven by specialized DNA contact surfaces and self-association. We propose that the specific function of the COOH terminus of helix II is to promote CENP-A– CENP-A self-association, presumably in the context of a (CENP-A/H4)2 heterotetramer, to form a homotypic CENP-A nucleosome. A homotypic CENP-A nucleosome will possess a duplicated set of differentiated DNA contact sites arrayed across the nucleosome surface. The repetition and geometry of these sites provides the possibility for cooperative interaction of the specialized DNA binding surfaces of CENP-A nucleosome, allowing what individually may be only weakly selective binding sites to sum to a significant affinity for centromeric DNA sequence or structure (Fig. 7 B).
Two predictions of this model are that (a) CENP-A should form homotypic nucleosomes, and (b) target DNA should have a repeating substructure that matches the specialized surfaces of CENP-A. Experimental support for the self-association of CENP-A has been obtained by coimmunoprecipitation of endogenous with transfected CENP-A (Fig. 4). While we have not yet determined experimentally the DNA sequences or structures to which CENP-A is bound, it is very likely that they include the satellite DNA component of mammalian chromosomes. Satellite DNA is unconserved at the level of primary sequence (Beridze, 1982). Theoretical analysis of satellite DNAs, however, reveals a substructure comprised of two 50–60-bp bending elements that are separated by 20–30 bp of low bending potential that is conserved among satellites from numerous species (Fitzgerald et al., 1994). Recognition of such conserved structural features of DNA by CENP-A might explain how centromere structure and function are conserved without apparent DNA sequence conservation. The strong strand A–helix II targeting site corresponds to a region where nucleosomal DNA is deformed, bending more sharply across the protein surface than flanking regions (Fig. 7 B, arrows) (Richmond et al., 1984; Wolffe, 1995). DNA bending or curvature is known to be an important determinant of histone octamer positioning on DNA (Shrader and Crothers, 1989; Sivolob and Khrapunov, 1995). Furthermore, analysis of the nucleosomal positioning signal in the ribosomal 5S RNA gene suggests that the regions 2–3 helical turns on either side of the dyad axis, very close to the predicted site of interaction with strand A, play a dominant role in specifying octamer position on the DNA (FitzGerald and Simpson, 1985). DNA recognition by CENP-A may therefore be a specialized implementation of general nucleosomal positioning features. These considerations support the notion that some of the molecular recognition events that specify centromere formation in higher eukaryotes take place at the level of DNA structure rather than DNA sequence, per se, and that these occur in the context of a specialized nucleosome.
Chromatin Assembly and the Specification of Centromeres
Structural recognition alone is not sufficient to explain the specific localization of CENP-A to centromeres, since overexpression of CENP-A results in a distribution throughout the nucleus. Thus, there does not appear to be an efficient mechanism to degrade ectopically localized CENP-A as is observed for CENP-C (Lanini and McKeon, 1995). Rather, our evidence points to regulation of the timing of CENP-A synthesis as an important feature of the targeting mechanism. Restricting expression of CENP-A to S phase abolished targeting, and analysis of steady state CENP-A mRNA abundance revealed that, indeed, it is uncoupled from normal histone expression, beginning late in S phase and extending through G2. Replication of centromeric chromatin therefore occurs through a process that is at least partially independent of normal chromatin replication. One reason for this may be simply to couple CENP-A synthesis with centromere DNA replication, which occurs in mid to late S phase (O'Keefe et al., 1992). A second possibility is that the temporal offset is required to promote the assembly of homotypic CENP-A nucleosomes, by expression at a time when concentrations of potentially competitive histone H3 are diminished. A third explanation for the role of temporal segregated from bulk histone synthesis is that a unique replication pathway for centromeric chromatin is part of the process by which cells recognize and propagate centromeres as distinct functional compartments of the chromosomes. Epigenetic features of centromere structure and function have been identified through analysis of position effect variegation in Drosophila (Spradling and Karpen, 1990; Henikoff, 1992) and of activation of deficient centromere sequences in S. pombe (Steiner and Clarke, 1994). Understanding how CENP-A chromatin replication is linked to the maintenance and function of centromeres on human chromosomes will provide new insight into the question of what constitutes an animal cell centromere.
Why Histone H3?
The heart of the nucleosome is the histone (H3-H4)2 heterotetramer. As discussed above, the heterotetramer possesses most of the DNA binding properties of the nucleosome as well as the information required for positioning (FitzGerald and Simpson, 1985; Dong and van Holde, 1991; Wolffe, 1995) and is deposited first onto DNA after replication, followed by the slower addition of histone H2AH2B dimers (Worcel et al., 1978). Histones H3 and H4 are thus uniquely situated to play a primary role in nucleosomal DNA recognition. Of all the four core histones, only histone H3 has the opportunity to direct its own selfassembly through homotypic interactions (Camerini-Otero and Felsenfeld, 1977; Arents et al., 1991; Xie et al., 1996). Homotypic H3–H3 interactions are therefore a key to harnessing the cooperative binding potential inherent in the dyad symmetry of the nucleosome. Additional histone H3 variants have been identified at the sequence level in Caenorhabditis elegans (Gown et al., 1996) and as a mouse cDNA (GenBank accession number AA008158). The mouse sequence contains a histone fold domain that is only 80% identical to that of mammalian CENP-A and may correspond to mouse CENP-A or represent yet another histone H3 homologue. Taken together, these observations reveal that histone H3 occupies a unique niche in the structure of the nucleosome, one that may provide an important element of adaptability for the structural differentiation of the chromatin fiber.
In summary, analysis of the histone fold domain structures of CENP-A that are required for its localization into centromeres reveals that this process depends upon the specialization of key elements of the histone H3 molecule: DNA binding surfaces and the unique H3–H3 homotypic dimer interface. Examining these features in the context of a histone octamer reveals how these elements can combine to provide modified DNA binding sites distributed in a cooperative array spanning ∼120 bp of nucleosomal DNA. In addition to providing a framework for understanding how centromeric chromatin may be built upon a nucleosomal DNA recognition mechanism, these experiments focus on the unique aspects of histone H3 within the nucleosome. Thus, understanding the relationships between structure and function for the specialized centromeric CENP-A nucleosome may provide new insight into the functions that histone H3 provides for chromatin in general.
Acknowledgments
We thank H. Damke, S. Schmid, and H. Bujard for their gifts of tTA vectors and HeLa cell line, and E. Chan for the human genomic library.
This work was supported by National Institutes of Health grant GM39068 to K.F. Sullivan, and in part by a grant from the Markey Charitable Trust to the Department of Cell Biology.
Footnotes
1. Abbreviation used in this paper: HA, hemagglutinin.
Address all correspondence to Kevin F. Sullivan, Department of Cell Biology, The Scripps Research Institute, 10550 N. Torrey Pines Rd., La Jolla, CA 92037. Tel.: (619) 784-2350. Fax: (619) 784-2345. e-mail: ksulli van@scripps.edu
References
- Arents G, Moudrianakis EN. Topography of the histone octamer surface: repeating structural motifs utilized in the docking of nucleosomal DNA. Proc Natl Acad Sci USA. 1993;90:10489–10493. doi: 10.1073/pnas.90.22.10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arents G, Burlingame RW, Wang BC, Love WE, Moudrianakis EN. The nucleosomal core histone octamer at 3.1 A resolution: a tripartite protein assembly and a left-handed superhelix. Proc Natl Acad Sci USA. 1991;88:10148–10152. doi: 10.1073/pnas.88.22.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel, F.M., R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl. 1995. Current Protocols in Molecular Biology. John Wiley and Sons, New York.
- Beridze, T. 1982. Satellite DNA. Springer-Verlag, Berlin. 149 pp.
- Bloom K. The centromere frontier: kinetochore components, microtubule-based motility, and the CEN-value paradox. [Review] Cell. 1993;73:621–624. doi: 10.1016/0092-8674(93)90242-i. [DOI] [PubMed] [Google Scholar]
- Bloom KS, Carbon J. Yeast centromere DNA is in a unique and highly ordered structure in chromosomes and small circular minichromosomes. Cell. 1982;29:305–317. doi: 10.1016/0092-8674(82)90147-7. [DOI] [PubMed] [Google Scholar]
- Bloom K, Hill A, Jones E. Conditional dicentric chromosomes in yeast. Prog Clin Biol Res. 1989;318:149–158. [PubMed] [Google Scholar]
- Cai M, Davis RW. Yeast centromere binding protein CBF1, of the helix-loop-helix protein family, is required for chromosome stability and methionine prototrophy. Cell. 1990;61:437–446. doi: 10.1016/0092-8674(90)90525-j. [DOI] [PubMed] [Google Scholar]
- Camerini-Otero RD, Felsenfeld G. Histone H3 disulfide dimers and nucleosome structure. Proc Natl Acad Sci USA. 1977;74:5519–5523. doi: 10.1073/pnas.74.12.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke L, Carbon J. Isolation of a yeast centromere and construction of functional small circular chromosomes. Nature (Lond) 1980;287:504–509. doi: 10.1038/287504a0. [DOI] [PubMed] [Google Scholar]
- Dalton S. Cell cycle regulation of the human cdc2 gene. EMBO (Eur Mol Biol Organ) J. 1992;11:1797–1804. doi: 10.1002/j.1460-2075.1992.tb05231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Luna S, Soria I, Pulido D, Ortin J, Jimenez A. Efficient transformation of mammalian cells with constructs containing a puromycinresistance marker. Gene (Amst) 1988;62:121–126. doi: 10.1016/0378-1119(88)90585-9. [DOI] [PubMed] [Google Scholar]
- Dong F, van Holde KE. Nucleosome positioning is determined by the (H3-H4)2 tetramer. Proc Natl Acad Sci USA. 1991;88:10596–10600. doi: 10.1073/pnas.88.23.10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw WC, Rothfield N. Identification of a family of human centromere proteins using autoimmune sera from patients with scleroderma. Chromosoma (Berl) 1985;91:313–321. doi: 10.1007/BF00328227. [DOI] [PubMed] [Google Scholar]
- Earnshaw WC, Sullivan KF, Machlin PS, Cooke CA, Kaiser DA, Pollard TD, Rothfield NF, Cleveland DW. Molecular cloning of cDNA for CENP-B, the major human centromere autoantigen. J Cell Biol. 1987;104:817–829. doi: 10.1083/jcb.104.4.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw WC, Ratrie H, Stetten G. Visualization of centromere proteins CENP-B and CENP-C on a stable dicentric chromosome in cytological spreads. Chromosoma (Berl) 1989;98:1–12. doi: 10.1007/BF00293329. [DOI] [PubMed] [Google Scholar]
- Fitzgerald DJ, Dryden GL, Bronson EC, Williams JS, Anderson JN. Conserved patterns of bending in satellite and nucleosome positioning DNA. J Biol Chem. 1994;269:21303–21314. [PubMed] [Google Scholar]
- FitzGerald PC, Simpson RT. Effects of sequence alterations in a DNA segment containing the 5 S RNA gene from Lytechinus variegatuson positioning of a nucleosome core particle in vitro. J Biol Chem. 1985;260:15318–15324. [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gown AM, Jiang JJ, Matles H, Skelly M, Goodpaster T, Cass L, Reshatof M, Spaulding D, Coltrera MD. Validation of the S-phase specificity of histone (H3) in situ hybridization in normal and malignant cells. J Histochem Cytochem. 1996;44:221–226. doi: 10.1177/44.3.8648081. [DOI] [PubMed] [Google Scholar]
- Hahnenberger KM, Baum MP, Polizzi CM, Carbon J, Clarke L. Construction of functional artificial minichromosomes in the fission yeast Schizosaccharomyces pombe. . Proc Natl Acad Sci USA. 1989;86:577–581. doi: 10.1073/pnas.86.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris ME, Bohni R, Schneiderman MH, Ramamurthy L, Schumperli D, Marzluff WF. Regulation of histone mRNA in the unperturbed cell cycle: evidence suggesting control at two posttranscriptional steps. Mol Cell Biol. 1991;11:2416–2424. doi: 10.1128/mcb.11.5.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SC. A structural taxonomy of DNA-binding domains. [Review] Nature (Lond) 1991;353:715–719. doi: 10.1038/353715a0. [DOI] [PubMed] [Google Scholar]
- Hegemann JH, Shero JH, Cottarel G, Philippsen P, Hieter P. Mutational analysis of centromere DNA from chromosome VI of Saccharomyces cerevisiae. . Mol Cell Biol. 1988;8:2523–2535. doi: 10.1128/mcb.8.6.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S. Position effect and related phenomena. [Review] Curr Opin Genet Dev. 1992;2:907–912. doi: 10.1016/s0959-437x(05)80114-5. [DOI] [PubMed] [Google Scholar]
- Hieter P, Pridmore D, Hegemann JH, Thomas M, Davis RW, Philippsen P. Functional selection and analysis of yeast centromeric DNA. Cell. 1985;42:913–921. doi: 10.1016/0092-8674(85)90287-9. [DOI] [PubMed] [Google Scholar]
- Hill CS, Thomas JO. Core histone-DNA interactions in sea urchin sperm chromatin. The NH2-terminal tail of H2B interacts with linker DNA. Eur J Biochem. 1990;187:145–153. doi: 10.1111/j.1432-1033.1990.tb15288.x. [DOI] [PubMed] [Google Scholar]
- Hyman AA, Middleton K, Centola M, Mitchison TJ, Carbon J. Microtubule-motor activity of a yeast centromere-binding protein complex. Nature (Lond) 1992;359:533–536. doi: 10.1038/359533a0. [DOI] [PubMed] [Google Scholar]
- Israel DI. A PCR-based method for high stringency screening of DNA libraries. Nucleic Acids Res. 1993;21:2627–2631. doi: 10.1093/nar/21.11.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanini L, McKeon F. Domains required for CENP-C assembly at the kinetochore. Mol Biol Cell. 1995;6:1049–1059. doi: 10.1091/mbc.6.8.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le MH, Duricka D, Karpen GH. Islands of complex DNA are widespread in Drosophilacentric heterochromatin. Genetics. 1995;141:283–303. doi: 10.1093/genetics/141.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner J. A zinc finger protein, essential for chromosome segregation, constitutes a putative DNA binding subunit of the Saccharomyces cerevisiae kinetochore complex, Cbf3. EMBO (Eur Mol Biol Organ) J. 1994;13:5203–5211. doi: 10.1002/j.1460-2075.1994.tb06851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner J, Carbon J. A 240 kD multisubunit protein complex, CBF3, is a major component of the budding yeast centromere. Cell. 1991;64:717–725. doi: 10.1016/0092-8674(91)90501-o. [DOI] [PubMed] [Google Scholar]
- Li X, Nicklas RB. Mitotic forces control a cell-cycle checkpoint. Nature (Lond) 1995;373:630–632. doi: 10.1038/373630a0. [DOI] [PubMed] [Google Scholar]
- Lucibello FC, Truss M, Zwicker J, Ehlert F, Beato M, Muller R. Periodic cdc25C transcription is mediated by a novel cell cycle-regulated repressor element (CDE) EMBO (Eur Mol Biol Organ) J. 1995;14:132–142. doi: 10.1002/j.1460-2075.1995.tb06983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann RK, Grunstein M. Histone H3 N-terminal mutations allow hyperactivation of the yeast GAL1 gene in vivo. EMBO (Eur Mol Biol Organ) J. 1992;11:3297–3306. doi: 10.1002/j.1460-2075.1992.tb05408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marschall LG, Clarke L. A novel cis-acting centromeric DNA element affects S. pombecentromeric chromatin structure at a distance. J Cell Biol. 1995;128:445–454. doi: 10.1083/jcb.128.4.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masumoto H, Masukata H, Muro Y, Nozaki N, Okazaki T. A human centromere antigen (CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J Cell Biol. 1989;109:1963–1973. doi: 10.1083/jcb.109.5.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrew J, Diehl B, Fitzgerald-Hayes M. Single base-pair mutations in centromere element III cause aberrant chromosome segregation in Saccharomyces cerevisiae. . Mol Cell Biol. 1986;6:530–538. doi: 10.1128/mcb.6.2.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JR. Structural and mechanical control of mitotic progression. [Review] Cold Spring Harbor Symp Quant Biol. 1991;56:613–619. doi: 10.1101/sqb.1991.056.01.070. [DOI] [PubMed] [Google Scholar]
- Middleton K, Carbon J. KAR3-encoded kinesin is a minus-end- directed motor that functions with centromere binding proteins (CBF3) on an in vitro yeast kinetochore. Proc Natl Acad Sci USA. 1994;91:7212–7216. doi: 10.1073/pnas.91.15.7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzabekov AD, Shick VV, Belyavsky AV, Bavykin SG. Primary organization of nucleosome core particle of chromatin: sequence of histone arrangement along DNA. Proc Natl Acad Sci USA. 1978;75:4184–4188. doi: 10.1073/pnas.75.9.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell PJ, Tjian R. Transcriptional regulation in mammalian cells by sequence-specific DNA binding proteins. [Review] Science (Wash DC) 1989;245:371–378. doi: 10.1126/science.2667136. [DOI] [PubMed] [Google Scholar]
- Miyazaki WY, Orr-Weaver TL. Sister-chromatid cohesion in mitosis and meiosis. [Review] Annu Rev Genet. 1994;28:167–187. doi: 10.1146/annurev.ge.28.120194.001123. [DOI] [PubMed] [Google Scholar]
- Murphy TD, Karpen GH. Localization of centromere function in a Drosophilaminichromosome. Cell. 1995;82:599–609. doi: 10.1016/0092-8674(95)90032-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklas RB, Ward SC, Gorbsky GJ. Kinetochore chemistry is sensitive to tension and may link mitotic forces to a cell cycle checkpoint. J Cell Biol. 1995;130:929–939. doi: 10.1083/jcb.130.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa O, Matsumoto T, Chikashige Y, Yanagida M. Characterization of Schizosaccharomyces pombeminichromosome deletion derivatives and a functional allocation of their centromere. EMBO (Eur Mol Biol Organ) J. 1989;8:3045–3052. doi: 10.1002/j.1460-2075.1989.tb08455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe RT, Henderson SC, Spector DL. Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J Cell Biol. 1992;116:1095–1110. doi: 10.1083/jcb.116.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer DK, Margolis RL. Kinetochore components recognized by human autoantibodies are present on mononucleosomes. Mol Cell Biol. 1985;5:173–186. doi: 10.1128/mcb.5.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer DK, O'Day K, Wener MH, Andrews BS, Margolis RL. A 17-kD centromere protein (CENP-A) copurifies with nucleosome core particles and with histones. J Cell Biol. 1987;104:805–815. doi: 10.1083/jcb.104.4.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer DK, O'Day K, Trong HL, Charbonneau H, Margolis RL. Purification of the centromere-specific protein CENP-A and demonstration that it is a distinctive histone. Proc Natl Acad Sci USA. 1991;88:3734–3738. doi: 10.1073/pnas.88.9.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluta AF, Mackay AM, Ainsztein AM, Goldberg IG, Earnshaw WC. The centromere: hub of chromosomal activities. Science (Wash DC) 1995;270:1591–1594. doi: 10.1126/science.270.5242.1591. [DOI] [PubMed] [Google Scholar]
- Polizzi C, Clarke L. The chromatin structure of centromeres from fission yeast: differentiation of the central core that correlates with function. J Cell Biol. 1991;112:191–201. doi: 10.1083/jcb.112.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao PN, Johnson RT. Mammalian cell fusion: studies on the regulation of DNA synthesis and mitosis. Nature (Lond) 1970;225:159–164. doi: 10.1038/225159a0. [DOI] [PubMed] [Google Scholar]
- Richmond TJ, Finch JT, Rushton B, Rhodes D, Klug A. Structure of the nucleosome core particle at 7 A resolution. Nature (Lond) 1984;311:532–537. doi: 10.1038/311532a0. [DOI] [PubMed] [Google Scholar]
- Richmond TJ, Rechsteiner T, Luger K. Studies of nucleosome structure. Cold Spring Harbor Symp Quant Biol. 1993;58:265–272. doi: 10.1101/sqb.1993.058.01.031. [DOI] [PubMed] [Google Scholar]
- Rieder CL, Cole RW, Khodjakov A, Sluder G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol. 1995;130:941–948. doi: 10.1083/jcb.130.4.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders M, Fitzgerald-Hayes M, Bloom K. Chromatin structure of altered yeast centromeres. Proc Natl Acad Sci USA. 1988;85:175–179. doi: 10.1073/pnas.85.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayle RA, Milner-White EJ. RASMOL: biomolecular graphics for all. Trends Biochem Sci. 1995;20:374–375. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]
- Sears DD, Hegemann JH, Shero JH, Hieter P. Cis-acting determinants affecting centromere function, sister-chromatid cohesion and reciprocal recombination during meiosis in Saccharomyces cerevisiae. . Genetics. 1995;139:1159–1173. doi: 10.1093/genetics/139.3.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shick VV, Belyavsky AV, Bavykin SG, Mirzabekov AD. Primary organization of the nucleosome core particles. Sequential arrangement of histones along DNA. J Mol Biol. 1980;139:491–517. doi: 10.1016/0022-2836(80)90143-6. [DOI] [PubMed] [Google Scholar]
- Shrader TE, Crothers DM. Artificial nucleosome positioning sequences. Proc Natl Acad Sci USA. 1989;86:7418–7422. doi: 10.1073/pnas.86.19.7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivolob AV, Khrapunov SN. Translational positioning of nucleosomes on DNA: the role of sequence-dependent isotropic DNA bending stiffness. J Mol Biol. 1995;247:918–931. doi: 10.1006/jmbi.1994.0190. [DOI] [PubMed] [Google Scholar]
- Smith MM, Yang P, Soledad M, Santisteban, Boone PW, Goldstein AT, Megee PC. A novel histone H4 mutant defective in nuclear division and mitotic chromosome transmission. Mol Cell Biol. 1996;16:1017–1026. doi: 10.1128/mcb.16.3.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorger PK, Severin FF, Hyman AA. Factors required for the binding of reassembled yeast kinetochores to microtubules in vitro. J Cell Biol. 1994;127:995–1008. doi: 10.1083/jcb.127.4.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling AC, Karpen GH. Sixty years of mystery. Genetics. 1990;126:779–784. doi: 10.1093/genetics/126.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner NC, Clarke L. A novel epigenetic effect can alter centromere function in fission yeast. Cell. 1994;79:865–874. doi: 10.1016/0092-8674(94)90075-2. [DOI] [PubMed] [Google Scholar]
- Stoler S, Keith KC, Curnick KE, Fitzgerald-Hayes M. A mutation in CSE4, an essential gene encoding a novel chromatin-associated protein in yeast, causes chromosome nondisjunction and cell cycle arrest at mitosis. Genes & Dev. 1995;9:573–586. doi: 10.1101/gad.9.5.573. [DOI] [PubMed] [Google Scholar]
- Sullivan KF, Glass CA. CENP-B is a highly conserved mammalian centromere protein with homology to the helix-loop-helix family of proteins. Chromosoma (Berl) 1991;100:360–370. doi: 10.1007/BF00337514. [DOI] [PubMed] [Google Scholar]
- Sullivan KF, Hechenberger M, Masri K. Human CENP-A contains a histone H3 related histone fold domain that is required for targeting to the centromere. J Cell Biol. 1994;127:581–592. doi: 10.1083/jcb.127.3.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal M, Shimron F, Yagil G. Unwound regions in yeast centromere IV DNA. J Mol Biol. 1994;243:179–189. doi: 10.1006/jmbi.1994.1645. [DOI] [PubMed] [Google Scholar]
- Taylor JD, Wellman SE, Marzluff WF. Sequences of four mouse histone H3 genes: implications for evolution of mouse histone genes. J Mol Evol. 1986;23:242–249. doi: 10.1007/BF02115580. [DOI] [PubMed] [Google Scholar]
- Tyler-Smith C, Willard HF. Mammalian chromosome structure. [Review] Curr Opin Genet Dev. 1993;3:390–397. doi: 10.1016/0959-437x(93)90110-b. [DOI] [PubMed] [Google Scholar]
- van Holde, K.E. 1989. Chromatin. Springer-Verlag, New York. 246 pp.
- Willard HF. Evolution of alpha satellite. [Review] Curr Opin Genet Dev. 1991;1:509–514. doi: 10.1016/s0959-437x(05)80200-x. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Niman HL, Houghten RA, Cherenson AR, Connolly ML, Lerner RA. The structure of an antigenic determinant in a protein. Cell. 1984;37:767–778. doi: 10.1016/0092-8674(84)90412-4. [DOI] [PubMed] [Google Scholar]
- Wolffe, A.P. 1995. Chromatin Structure and Function. Academic Press, San Diego. 213 pp.
- Worcel A, Han S, Wong ML. Assembly of newly replicated chromatin. Cell. 1978;15:969–977. doi: 10.1016/0092-8674(78)90280-5. [DOI] [PubMed] [Google Scholar]
- Xie WQ, Rothblum LI. Rapid, small-scale RNA isolation from tissue culture cells. Biotechniques. 1991;11:324–327. [PubMed] [Google Scholar]
- Xie X, Kokubo T, Cohen SL, Mirza UA, Hoffmann A, Chait BT, Roeder RG, Nakatani Y, Burley SK. Structural similarity between TAFs and the heterotetrameric core of the histone octamer. Nature (Lond) 1996;380:316–322. doi: 10.1038/380316a0. [DOI] [PubMed] [Google Scholar]
- Zwicker J, Lucibello FC, Wolfraim LA, Gross C, Truss M, Engeland K, Muller R. Cell cycle regulation of the cyclin A, cdc25C and cdc2 genes is based on a common mechanism of transcriptional repression. EMBO (Eur Mol Biol Organ) J. 1995;14:4514–4522. doi: 10.1002/j.1460-2075.1995.tb00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]