

Abstract
Decorin is a member of the expanding group of widely distributed small leucine-rich proteoglycans that are expected to play important functions in tissue assembly. We report that mice harboring a targeted disruption of the decorin gene are viable but have fragile skin with markedly reduced tensile strength. Ultrastructural analysis revealed abnormal collagen morphology in skin and tendon, with coarser and irregular fiber outlines. Quantitative scanning transmission EM of individual collagen fibrils showed abrupt increases and decreases in mass along their axes, thereby accounting for the irregular outlines and size variability observed in cross-sections. The data indicate uncontrolled lateral fusion of collagen fibrils in the decorindeficient mice and provide an explanation for the reduced tensile strength of the skin. These findings demonstrate a fundamental role for decorin in regulating collagen fiber formation in vivo.
Small leucine-rich proteoglycans (SLRPs)1 belong to an expanding family of secreted proteoglycans that comprise structurally related but genetically distinct products, including decorin, biglycan, fibromodulin, lumican, epiphycan, and keratocan (28). The SLRPs share a common structural architecture that can be divided into three domains. The amino-terminal domain contains the negatively charged glycosaminoglycan chains, dermatan or chondroitin sulfate, or tyrosine sulfate. This region of the molecule, which contains four conserved cysteine residues, might be involved in binding to cationic domains of cell surfaces and extracellular matrix proteins. The cysteinefree central domain comprises ∼80% of the protein and is composed of eight to ten tandem repeats of leucine-rich regions. This region is perhaps the best studied insofar as it has been recently shown that specific amino acid sequences located between repeats 4 and 6 are responsible for binding to type I collagen (51, 59). If the SLRPs fold in a manner similar to the leucine-rich ribonuclease inhibitor, the concave face of the molecule could interact with other proteins as the ribonuclease inhibitor does with its substrate (33). The carboxyl end domain contains two cysteine residues, and its function still remains to be elucidated.
The evidence favoring protein–protein interaction as the major function of SLRPs is persuasive. For example, decorin binds noncovalently to the surface of fibrillar collagen, primarily type I (53), and retards the rate and degree of collagen fibrillogenesis in vitro (66). This specific interaction is mediated by the protein core (42), whereas the glycosaminoglycan chain of decorin extends laterally from adjacent collagen fibrils, thereby maintaining interfibrillar spacing (52). This lateral orientation has also been demonstrated in collagen fibrils reconstituted in vitro in the presence of decorin (55). Thus, coordinated expression of decorin and associated collagens may regulate an orderly matrix assembly.
Decorin purportedly binds to collagen types II (66), III (60), and VI (6), fibronectin (48), C1q (34), and transforming growth factor-β (20). Moreover, decorin has been implicated in the control of cell proliferation by inducing arrest of tumor cells in the G1 phase of the cell cycle (14, 47).
To gain further insights into the functional role of decorin and to explore tissue specificity and functional redundancy during development, we generated mice disrupted at the decorin gene locus. The nullizygous animals were viable but showed skin fragility with marked reduction in tensile strength. Compared with normal skin, the collagen fiber network was more loosely packed with abnormal collagen fibers varying in diameter along their shafts. These observations provide the first genetic evidence that decorin is essential for proper collagen fibrillogenesis and demonstrate an important role for this proteoglycan in a process fundamental to animal development. Our results also provide insights into how disruption of collagen fibrillogenesis might result in pathology and predict the potential existence of a human genetic disease caused by mutations in the decorin gene locus.
Materials and Methods
Targeting Vector and Identification of Targeting Events
A 5.5 XbaI decorin genomic fragment isolated from a λFIX II genomic mouse 129Sv library (49), isogenic to the embryonic stem (ES) cells, was used to construct the decorin targeting vector. This genomic fragment, encompassing exons 1 and 2, was ligated into pBluescript KS with a deleted EcoRV site, and the resulting fragment was designated pMD. A plasmid containing Pgk-neo (58) was digested with XhoI and EcoRV, and the XhoI site was made blunt-ended with Klenow polymerase. The resulting Pgk-neo fragment of 1.6 kb was ligated into a unique EcoRV site of exon 2, thereby dividing the genomic fragment into two arms of homology of ∼3.8 and ∼1.7 kb to the targeted locus, respectively. To enrich for targeting events, the diphtheria toxin-A cassette driven by the thymidine kinase promoter (70) was added downstream of the targeting vector into the XhoI site of the multiple cloning site of pMD. Linearization of this targeting vector was done with NotI before electroporation into the ES cells. The R1-ES cells (39) were cultured in standard ES cell culture conditions (2), in DME supplemented with 15% FCS, 0.1 mM β-mercaptoethanol, and 1,000 U/ml of human lymphocyte inhibitory factor (h-LIF) on neomycin-resistant primary cultures of embryonic fibroblast feeders. LIF was produced from the CHO cell line stably transfected with the h-LIF cDNA. Cells were electroporated with 40 μg/ml of decorin targeting vector. The resulting G418-resistant clones were isolated by ring-cloning and expanded (3). Approximately 100 clones were analyzed by Southern blot analysis for the detection of the correct targeting events (see below).
Germ-Line Transmission, Husbandry, and Genotype Determination
Targeted ES cells were introduced into mouse blastocysts according to standard procedures. The resulting chimeras were bred to B1/Swiss females. Tail DNA from B1/Swiss female agouti progeny was tested for the presence of the targeted allele by Southern blot analysis. Heterozygous mice from 129Sv X B1/Swiss mixed background were mated to homozygosity. The genotype of each mouse was determined by Southern blotting and/or PCR analysis of mouse tail DNA. Genomic DNA from mouse tail fragments was digested with EcoRI and separated on 0.8% agarose gels. A 310-bp probe encompassing a sequence just 5′ to the targeting vector was prepared by PCR using the T7 primer from a subcloned 5.5-kb genomic fragment of mouse decorin phage clone F 11-1 (49) and an antisense primer specific to exon 1. This probe detects a 7- or 6.5-kb sequence of DNA digested with EcoRI derived from normal wild-type and targeted animals, respectively. Alternatively, PCR was used to detect the presence of homologous recombination of the decorin gene. Sense and antisense primers corresponding to exon 2 of murine decorin (5′-CCTTCTGGCACAAGTCTCTTGG-3′ and 5′-TCGAAGATGACACTGGCATCGG-3′) were used to detect a 161-bp fragment, indicating lack of homologous recombination. An additional primer corresponding to the Pgk promoter of the Pgk-neo cassette (5′-TGGATGTGGAATGTGTGCGAG-3′) was used to detect a 250-bp fragment, indicating the presence of homologous recombination. The reaction mix of 28 μl consisted of 100–500 ng genomic DNA, 100 ng each of the three primers, 1.8 mM MgCl2, 1× buffer, 360 μm dinucleotide triphosphates, and 2.5 U of Taq polymerase. Reaction conditions were: 1 min at 95°C, 20 s at 57°C, and 30 s at 72°C for 35 cycles.
Hematological Analysis, RNA Extraction, Proteoglycan Purification, and Immunoblotting
Approximately 0.5 ml of blood was retrieved by cardiac puncture of homozygous animals and their respective control litter mates (n = 6). A full body profile, including hematological and clinical chemistry, was performed as described before (4). Differential white blood cell counts were determined manually on blood smears after Wright staining. RNA was isolated from mouse tissues using a “Tri” reagent (Molecular Research Center, Cincinnati, OH), and separated on agarose formaldehyde gels. Proteoglycans were extracted from ∼50 mg of adult mouse tail tissue in 4 M guanidine HCl, 100 mM sodium acetate buffer, pH 6.0, containing the following protease inhibitors: 1 mM PMSF, 5 mM benzamidine HCl, 5 mM iodoacetamide, 5 μg/ml leupeptin, 5 μg/ml pepstatin, and 10 mM EDTA. After a 64-h extraction at 4°C with gentle rotation, the unextracted residue was removed by centrifugation, while the supernatant was dialyzed against 0.1 M NaCl, 0.1 M Tris HCl, pH 7.3, for 72 h at 4°C. An aliquot of 100 μl of the dialysate was incubated with protease-free chondroitinase ABC (165 mU/ml) overnight at 37°C in the presence of chondroitin 4-S and 6-S (0.1 μg/ml) as carrier. Total protein was precipitated with 10 vol of cold 100% ethanol and collected by centrifugation. The resulting pellet was air dried and resuspended in 40 μl SDS-PAGE buffer. Proteins were separated on 8–10% polyacrylamide gels and transferred to nitrocellulose (Hybond ECL; Amersham Corp., Arlington Heights, IL) for 1 h at 700 mA. Immunoblots were incubated overnight at 4°C in a blocking solution containing TBS-T (20 mM Tris-HCl, pH 7.6, 137 mM NaCl, 0.1% Tween 20) and 5% nonfat dry milk. After washing the membrane with TBS-T, it was incubated for 1 h in a 1:5,000 dilution of a rabbit anti–mouse decorin antiserum (LF-113), which was generated against a synthetic decorin peptide from the amino-terminal region spanning residues 36–49 of the mouse protein (16). The membrane was extensively washed with TBS-T, and then incubated for 1 h with goat anti–rabbit IgG conjugated to HRP (Sigma Chemical Co., St. Louis, MO) diluted to 1:10,000 in TBS-T. After further washing, the antigen/antibody complexes were detected with ECL detection reagents (Amersham Corp.) according to the manufacturer's instructions. Exposure of the immunoblot to XAR5 film (Eastman Kodak Co., Rochester, NY) was for 2–10 s.
Morphological and Immunohistochemical Studies
Tissues were fixed in 10% buffered formaldehyde and processed for light microscopy (27). For immunohistochemistry, paraffin sections were warmed to 50°C for 30 min, dewaxed in xylene, and rehydrated through decreasing concentrations of ethanol. Endogenous peroxidase activity was blocked by a 15-min preincubation with methanolic H2O2 (0.5% vol/vol). After washing in TBS, pH 7.5, the sections were incubated with normal goat serum (1:200) for 30 min, washed extensively with TBS, and incubated with the rabbit polyclonal LF-113 anti-decorin or the LF-106 antibiglycan antisera (16) at 1:1,000 dilution for 18 h at 4°C. The LF-106 is directed against a murine biglycan synthetic peptide, amino acids 50–64, conjugated to horseshoe crab hemocyanin (16). An HRP-conjugated goat anti–rabbit IgG (Sigma Chemical Co.) was applied at 1:200 dilution for 45 min. A 3-amino-ethyl carbazole substrate kit (AEC; Vector Laboratories Inc., Burlingame, CA) was used to visualize the specific peroxidase activity. The sections were washed in water, counterstained with 0.2% methylene blue, and mounted with Gel/Mount aqueous medium (Biomeda Co., Foster City, CA) before photography. For EM, small portions of skin, tail tendon, or cornea were fixed in 3% glutaraldehyde, 25 mM sodium acetate buffer, pH 5.7, containing 0.3 M MgCl2 and 0.05% cuprolinic blue (53). The unosmicated tissues were rinsed three times in buffer containing MgCl2, en bloc stained with 1% sodium tungstate, dehydrated in graded alcohols, and embedded in Spurr's epoxy resin. Thin-sections were observed with a transmission electron microscope 100B (JEOL USA, Peabody, MA), with or without further staining with uranyl acetate (27) or sodium tungstate (53). For quantitative studies, collagen fibril diameters were measured on photographic prints with a calibrated final magnification of 90,000. Several hundred micrographs were taken from the skin, tendon, and cornea of five homozygous, two heterozygous and three wildtype animals. A total of 2,803 collagen profiles from wild-type, heterozygous, and homozygous skin was measured, and histograms were generated. For scanning transmission EM (STEM) analysis, 4-mm2 pieces of skin were finely minced, suspended in 1 ml Tris-buffered (pH 7.4) saline supplemented with 50 mM EDTA and 100 mM sucrose, and subjected to mild disruption in a hand-held Dounce homogenizer. The supernatants were then sampled for EM. Fibrils were adsorbed to 400-mesh carbonfilmed grids, washed with ultrapure water, and air dried. The unstained fibrils were examined by STEM and mass mapping procedures (21, 22, 23).
Biomechanical Test of Skin Tensile Strength
Dorsal skin was carefully excised and pinned out on a styrofoam support. Using a plastic template, dumbbell-shaped skin specimens (4 cm × 2 cm) oriented parallel to the spine were dissected with the aid of a scalpel and placed between two microscope slides. The specimens were kept moist with cold saline and transported to the bioengineering laboratory on ice. The overall thickness of the specimens was measured with a micrometer while the skin was between glass slides and found not be significantly different among animals. A tensile machine displacement controller manufactured by Comten Industries Inc. (St. Petersburg, FL) was used to measure tensile strength of the skin specimens, which were gripped between specially designed clamps equipped with Velcro-covered faces to prevent slippage of the tissue. The skin specimens were kept continuously moist with room temperature saline and stretched at a low speed rate of 1 mm/s (10%/s) until failure. Tensile strength was computed from maximal load or force in newton (N) at failure divided by the initial cross-sectional area of the specimen in the reduced test area (32, 46).
Results
Generation of the Decorin Null Mice
The genomic locus encoding the murine decorin gene was targeted with a replacement construct containing ∼5.5 kb of DNA sequence interrupted in exon 2 by the neomycin resistance cassette driven by the Pgk promoter (Fig. 1 A). We made use of a positive/negative selection scheme by using neomycin resistance for positive selection and diphtheria toxin-A driven by the thymidine kinase promoter for negative selection of nonhomologous recombinants (70). 110 G418-resistant ES clones were isolated and expanded. Homologous recombination was detected by Southern blotting with a single genomic probe flanking the construct. This probe detected a 6.5-kb EcoRI fragment specific for the targeting vector (the neo cassette has an internal EcoRI site), and a 7-kb EcoRI fragment specific for the wild-type allele. Among the first 51 clones that were screened by Southern blotting, 13 (∼25% targeting frequency) had the predicted 6.5-kb EcoRI band corresponding to the targeted allele (not shown). Two independent ES clones with a normal karyotype were used to produce male chimeras that were able to affect germ-line transmission of the disrupted allele when bred to C57BL/6 females. Crosses of heterozygous parents resulted in the birth of mice that were homozygous for the mutant Dcn gene based on either Southern blotting (Fig. 1 B) or PCR (Fig. 1 C) of tail biopsy DNA.
Figure 1.


Disruption of the decorin gene locus in mouse ES cells and generation of decorin-deficient mice. (A) Targeting strategy. A 5.5-kb XbaI genomic fragment encoding exons 1 and 2 (filled boxes, not in scale) was used to construct the targeting vector. The predicted structure of the disrupted allele (bottom panel) shows the 5′ probe used to detect the diagnostic allele of 6.5 kb in contrast with the wild-type allele of 7.0 kb. This was due to the presence of a new EcoRI site in the Pgk-neo cassette. Abbreviations for restriction endonucleases: E, EcoRI; X, XbaI; N, NotI; Xh, XhoI. (B) Southern blot analysis of tail DNA isolated from two separate litters of mice including wild-type (+/ +), heterozygous (+/−), and homozygous (−/−) animals. Lanes 1–3 are from 3-mo-old animals derived from the breeding of two heterozygous mice, while lanes 4–9 are from newborn animals derived from the breeding of a heterozygous male and a homozygous female. The targeted allele of 6.5 kb is labeled by an asterisk. The DNA was separated on a 0.75% agarose gel, transferred to a nitrocellulose filter, and hybridized under high stringency to a PCRgenerated probe 5′ to the targeting vector. The size of molecular weight markers is indicated in the left margin in kb. (C) PCR detection of the targeted allele using primers specific for exon 2 (a and c, top scheme) or Pgk-neo (primer b). By using primer a and c, a fragment of 161 bp was identified in the wild-type (lane 1) and heterozygous animal (lane 2). However, the combination of primer b and c gave rise to a larger fragment of 250 bp, encompassing the 3′ end of the neo cassette and the 3′ end of exon 2, which was detected only in the heterozygous (lane 2) and homozygous (lane 3) animals. The products were separated on a 6% nonreducing polyacrylamide gel and stained with ethidium bromide. The size in bp is indicated in the left margin.
Homozygous Mutant Mice Are Viable
Mice heterozygous for the decorin knockout mutation (129Sv X Bl/Swiss mixed background) were mated to homozygosity. The resulting progeny (n = 69) showed a standard Mendelian transmission: 16 homozygotes (23%), 37 heterozygotes (54%), and 16 wild type (23%). This Mendelian distribution was subsequently observed in several progenies derived from mating heterozygous animals. To confirm the disruption of both decorin alleles, the absence of decorin-specific transcript was checked by Northern blotting analysis. Total RNA from several major tissues, including manubrium sterni (which contains cartilage, bone, and bone marrow), heart, liver, kidney, and skin, an organ that is known to express high levels of decorin message (49), showed no detectable decorin-specific transcript (Fig. 2 A). Thus, both copies of the decorin gene were successfully disrupted by homologous recombination initiated by our targeting vector. As expected, the levels of decorin were slightly reduced in the Dcn+/− animals when normalized on the housekeeping gene GAPDH. Moreover, when the Northern blots were hybridized with either a full-length cDNA probe encoding murine biglycan or a reverse transcriptase–PCR-generated probe encoding a portion of murine lumican, no significant change in either transcript was observed (not shown). These findings indicate that neither biglycan nor lumican (murine fibromodulin probe was not available) is upregulated to compensate for the lack of decorin in any of the tissues examined so far. The results obtained at the mRNA levels were confirmed by the absence of the immunoreactive decorin proteoglycan from whole tail extracts (Fig. 2 B). When partially purified proteoglycans were separated by 7.5% SDS gel electrophoresis and immunoblotted, no decorin immunoreactivity was observed by using a specific antipeptide antibody (16) either before (Fig. 2 B, lane 3) or after (Fig. 2 B, lane 6) chondroitinase ABC treatment, in spite of the fact that approximately equal amounts of protein were loaded on the gel. Collectively, these results indicate that disruption of both alleles of the mouse decorin gene results in abrogation of decorin message and protein.
Figure 2.

Absence of decorin mRNA and protein in animals harboring a homozygous disruption of the decorin gene. (A) Northern blot analysis using total RNA prepared from selected tissues as indicated of wild-type (+/+), heterozygous (+/−), and homozygous (−/−) adult animals. High stringency hybridization was performed using cDNAs encoding the full-length decorin or the housekeeping gene GAPDH as 32P-labeled probes (49). Notice the lack of decorin transcripts in the Dcn−/− animals (lanes 9–12, 15, and 16). (B) Immunoblot analysis of guanidine HCl extracts of tail tissues before or after chondroitinase ABC (+ABCase) digestion using an anti-decorin antibody. Notice the presence of a proteoglycan centering around 94 kD and the presence of a 45-kD unprocessed protein core in the wild-type (lane 1) and heterozygous animal (lane 2); no immunoreactive material was detected in the homozygous animal (lane 3). After chondroitinase ABC digestion, the high molecular mass proteoglycan was converted into two broad bands of 45–50 kD (lanes 4 and 5). Decorin-specific epitopes were detected with a polyclonal antibody (LF-113) directed against a synthetic decorin peptide from the amino-terminal region spanning residues 36–49 of the mouse protein (16).
Lack of Decorin Leads to Thinning and Fragility of the Skin
Initial analysis of the Dcn−/− mice did not reveal any gross anatomical abnormalities. The animals grew to normal size, were fertile, and did not exhibit obvious behavioral deficiencies. A complete serum chemistry and hematological profile of 2-mo-old homozygous and control mice did not show any significant difference in the levels of albumin, globulin, electrolytes, sodium, and chloride, in the hepatic enzymes, ALT and AST, or in the number of white and red blood cells, hematocrit and hemoglobin. Radiographical studies of whole animals did not reveal any overt bone abnormalities in the decorin-deficient animals (not shown). However, closer analysis of the Dcn−/− mice revealed skin laxity and significant fragility. When 3-moold mice were sacrificed by cervical dislocation, the simple application of pressure to the cervical region caused an abrupt rupture of the skin (Fig. 3 A, arrows). In addition the skin of the tail was detached in its entirety from the underlying soft tissues (Fig. 3 A, arrowheads). Light microscopic observation of the skin from the tail showed a sharp detachment of the skin between the deeper dermis and the fascia, with clean, sharp edges along the dissection (Fig. 3 B, arrowheads). Random sampling of abdominal skin from several Dcn−/− mice revealed dermal thinning and loose connective tissue in the hypodermal layer (Fig. 3 D). Thus, skin laxity observed in the decorin-deficient animals could be due at least in part to accumulation of loose connective tissue in the dermal and hypodermal layers. In contrast, age-matched Dcn+/+ mice littermates (Fig. 3 C) or Dcn+/− animals (not shown) showed no such structural changes. To confirm the absence of decorin, immunohistochemical analysis using the anti-decorin antibody showed no detectable epitopes in the skin of Dcn−/− animals (Fig. 3 D). In addition, all the tissues tested, including skeletal muscle, adipose tissue, and uterus (Fig. 3, D and F), as well as Fallopian tubes and ovaries (not shown), revealed no decorin immunoreactivity. In contrast, the wild-type animals showed the expected immunoreactivity in the dermis (Fig. 3 C, asterisks), in the myoepithelial layer and connective tissues surrounding mammary ducts (Fig. 3 E, arrows), and in the adventitia of small blood vessels (Fig. 3 E, arrowheads). In the Dcn−/− animals, staining with an antibody against biglycan showed the presence of this epitope in the endometrium and intramural blood vessels (Fig. 3 G), as well as in the epidermal layer and follicular epithelium (Fig. 3 H).
Figure 3.
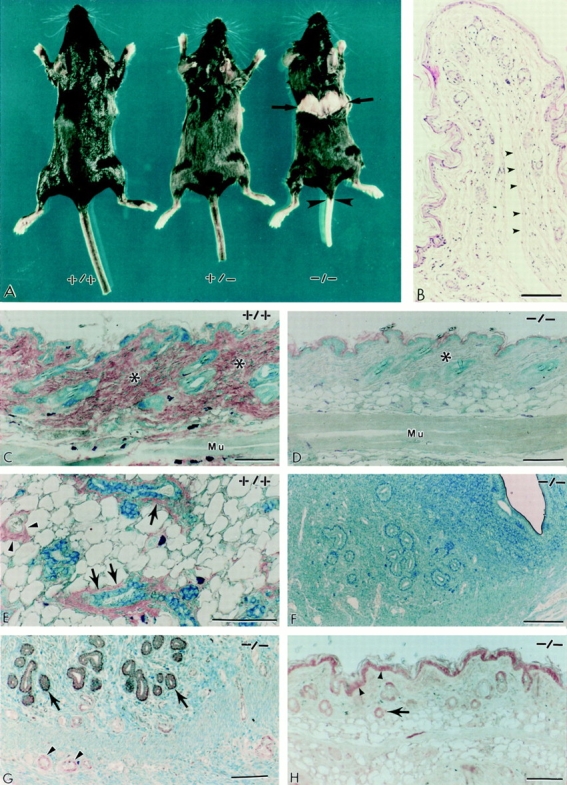
The phenotype of decorin-deficient mice reveals thinning and fragility of the skin. (A) Gross photograph of wild-type (+/+), heterozygous (+/−), and homozygous (−/−) littermates. Notice the sharp rupture of the back skin in the −/− animal (arrows) and the detachment of the tail skin (arrowheads) that occurred during cervical dislocation. The skin fragility was never observed in either +/+ or +/− animals (n > 300). B is a cross-section of the detached tail skin. Notice the sharp and bloodless line of rupture (arrowheads) along the deeper dermis. Immunohistochemical analysis of skin and skeletal muscle (C and D), mammary gland (E), and uterus (F) from wild-type (+/+) and decorin-deficient (−/−) animals using the LF-113 anti-decorin antibody. Notice the intense immunoreactivity in the dermis of a wild-type animal (C, asterisks) and the fine immunoreactivity on either side of the skeletal muscle (Mu). In contrast, the −/− dermis (D, asterisk), the subcutaneous and perimysial connective tissues, are totally unreactive as are the uterine wall and mucosa (F). As expected, the anti-decorin antibody labeled specifically the adventitia of small blood vessels (E, arrowheads) and the myoepithelial cells and fine connective tissue surrounding mammary ducts (E, arrows). Immunodetection of biglycan using an anti-peptide (LF-106) antibody in uterus (G) and skin (H) from Dcn−/− mice. Notice the “normal” expression of biglycan in the endometrium (G, arrows) and in the intramural small blood vessels (G, arrowheads). As expected, in skin, both the epidermis (H, arrowheads) and the follicular epithelium (H, arrow) were labeled by the anti-biglycan antibodies. Sections were reacted with LF-113 or LF-106 antisera at 1: 1,000 dilution, and then visualized with peroxidase-conjugated IgG (1:200) followed by counterstaining with 0.2% methylene blue. Bar, 100 μm.
The rupture of the skin similar to that shown in Fig. 3 A had a considerable penetrance, insofar as it was observable in >50% of all the Dcn−/− mice. In contrast, skin fragility was never observed in the heterozygous, wild-type animals; nor was it encountered in other animals (n > 300) with similar genetic background but harboring a wild-type allele. These data indicated that, to obtain the skin fragility phenotype, both decorin alleles had to be inactivated.
Skin of Decorin-deficient Animals Exhibits Reduced Tensile Strength
It is generally accepted that skin strength correlates directly with the overall organization, content, and physical properties of the collagen fibril network (15). Therefore, to achieve a more objective measurement of skin fragility, we determined the tensile strength in the physiological range of loading (low rate of loading) in portions of dorsal skin from six animals, three wild-type and three Dcn−/− mice. Two fresh specimens per animal of identical dumbbell-shape were generated on a plastic template, and load/ displacement curves were determined by subjecting them to a stress rate of 1 mm/s (10%/s) with constant recording by a Comten tensile machine displacement controller. Tensile strength was expressed as the maximal load or force in newton (N) before skin failure divided by the initial crosssectional area (1 cm2) of the specimen in the central test area (46). A typical experiment derived from stretching fresh dorsal skin from two age-matched animals is shown in Fig. 4. The skin from the decorin-deficient animals exhibited a premature failure at ∼7 N, in contrast with the skin from the wild type that ruptured at ∼27 N. The overall values of tensile strength measurements were significantly reduced in Dcn−/− animals (7 N ± 2) as compared with the wild-type animals (21 N ± 5). The data further suggest that the decorin-deficient mice exhibited a reduction in ductility, i.e., the percentage of deformation before failure (calculated from the displacement values in Fig. 4). The normal values centered around 170% vs 110% for the decorin-deficient animals. These independent biomechanical parameters thus confirm the observation of skin fragility reported above and indicate that the decorin-deficient phenotype is due to a decline in the intrinsic tensile strength of the skin.
Figure 4.

The skin of decorin-deficient mice has reduced tensile strength. Freshly excised, dumbbell-shaped samples (4 × 2 cm) of dorsal skin oriented parallel to spine from +/+ (•) or −/− (○) animals were generated on a plastic template, thereby producing skin specimens with a narrow central region (1 cm2) where failure would occur by avoiding stress concentration at the jaws of the grip. The samples were gripped into a tensile testing machine (model 4Z387A; Comten Industries) and stretched to tensile failure at a constant strain rate of 1 mm/s, equivalent to 10%/s. A data collecting system linked to a Macintosh Quadra computer (Apple Computer Inc., Cupertino, CA) was used to generate load/displacement curves. Peak of curve represents point of failure. Tensile strength was computed from maximal load at failure divided by the initial cross-sectional area of the specimen in the reduced test area. Six age-matched animals were analyzed with two specimens per animal. Data points represent a typical experiment from one animal each.
A Structural Basis for Skin Fragility in the Decorin-deficient Mice: Abnormal Morphology of Collagen Fibrils and Reduction of Collagen-bound Proteoglycans
Ultrastructural analysis of dermal collagen from the skin of 3-mo-old Dcn−/− animals revealed aberrant organization of collagen fibrils with several distinct structural abnormalities. First, dermal collagen was less orderly packed (Fig. 5 A) than age-matched wild type (Fig. 5 C). Second, there was great variability in shape and size (Fig. 5, A and B); individual fibrils exhibited irregular outlines in crosssection compared with the circular outline of controls. In some instances, the cross-sectional profiles of collagen fibrils in affected dermis reached very large diameters up to 240–260 nm (Fig. 5 A, asterisks) and showed scalloped edges (Fig. 5 A, arrowhead) or focal lateral fusion with thinner fibrils (Fig. 5 B, arrowheads). In contrast, heterozygous (not shown) or wild-type skin (Fig. 5 C) showed collagen fibrils with fairly uniform, circular cross-sectional profiles. Third, there was the coexistence of thin collagen fibrils (Fig. 5 A, circles) intermingled with large ones, whereas in the wild type the average size was quite uniform. As a result of the overall round shape of the collagen fibrils, it was possible to measure the average diameter of several hundred cross-sectional profiles. The results demonstrated that the average collagen diameter did not significantly vary between wild-type (Fig. 5 D) and homozygous animals (Fig. 5 E), 116 ± 18 (n = 788) vs 119 ± 35 (n = 795), respectively. However, the Dcn−/− animals exhibited a wider range with profiles varying between 40 and 260 nm, confirming the qualitative data shown above. In contrast, the wild-type animals contained profiles ranging between 40 and 180 nm. The heterozygous animals had a distribution and an average collagen diameter (118 ± 14; n = 1,220) nearly identical to the wild type (not shown).
Figure 5.
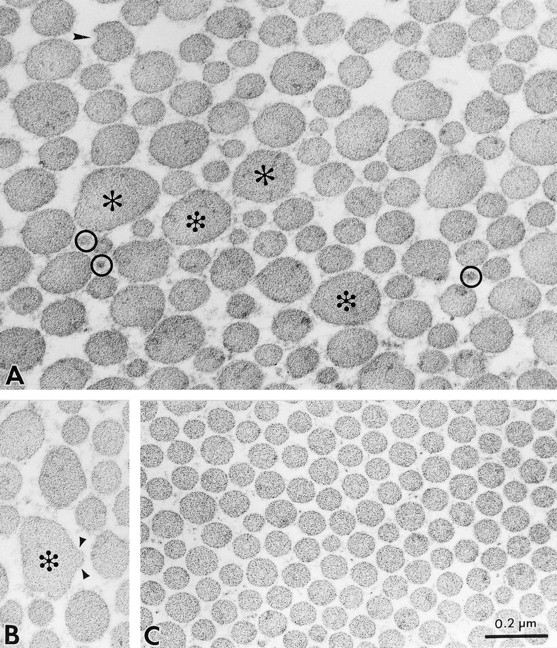
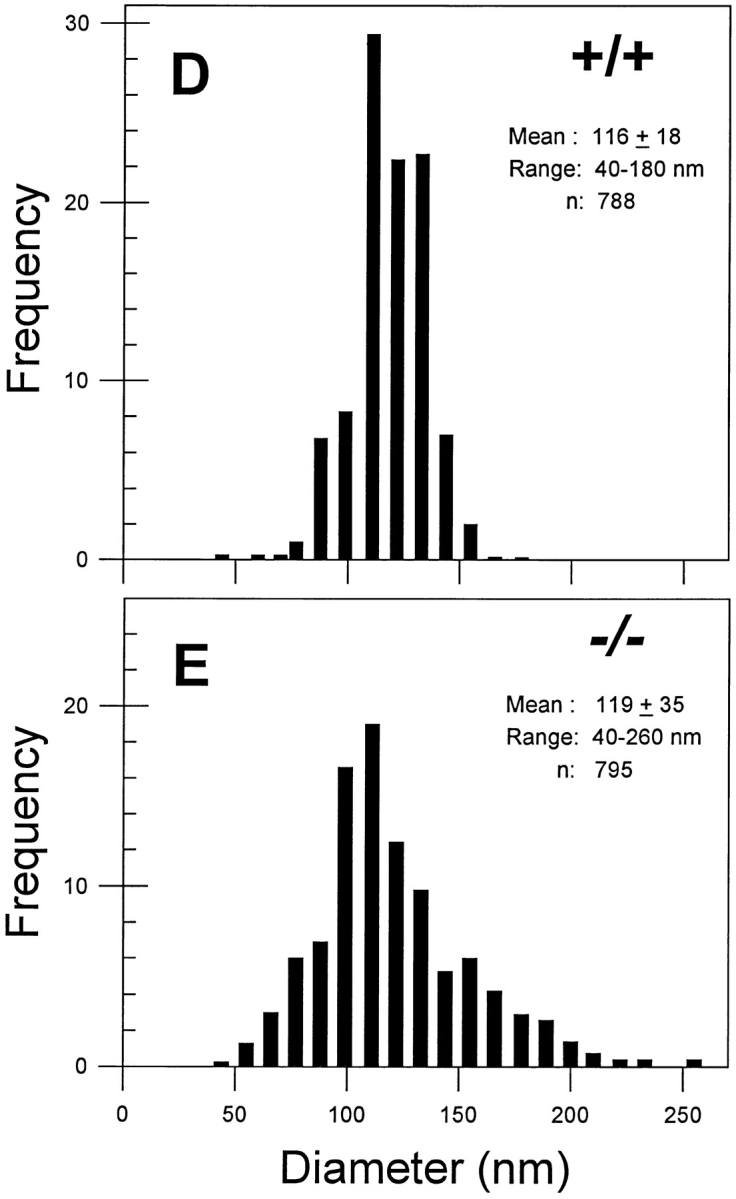
Ultrastructural analysis of skin from the decorin-deficient mice reveals abnormal collagen fibrillogenesis. (A–C) Transmission electron micrographs of dermal collagen from Dcn−/− (A and B) and Dcn+/+ (C) animals. Notice the presence of larger (>200 nm) and irregular fibrils (A and B, asterisks) and the coexistence of smaller (30–40 nm) fibrils (A, circles) in the Dcn−/−. Note also the presence of coarser fibrils exhibiting lateral fusion to an adjacent tapered segment (B, arrowheads). In contrast, collagen from the wild-type mouse (C) showed a more compact and uniform pattern of fibril diameter and distribution. The Dcn+/− collagen pattern was identical to the Dcn+/+ animals (not shown). (D and E) Distribution of collagen fibril diameter in dermal collagen from Dcn+/+ (D) and Dcn−/− (E) animals. Notice that, although the mean diameter is not significantly different between the groups, the range and distribution is quite different in the Dcn−/− animals. Heterozygous animals had a distribution and an average collagen diameter (118 ± 14; n = 1,220) nearly identical to the wild type (not shown). Bar, 0.2 μm.
Ultrastructural analysis of tail tendon collagen revealed even more bizarre profiles and shapes. Individual fibrils exhibited very irregular, ragged outlines in cross-section compared with normal skin. The collagen fibrils reached 660 nm in diameter (Fig. 6, A and B) in contrast with the wild-type animals where the average was about 200 nm (Fig. 6 C). The aberrant fibrils appeared to be due to multiple and concurrent lateral fusions of their central thick segments to thinner fibrils (Fig. 6 B). Moreover, intermingled with these gigantic fibrils were numerous thin fibrils with an average diameter of 40–60 nm. These thin fibrils were less frequently detected in tendons of age-matched controls. Since decorin is also expressed in the cornea, we analyzed the ultrastructure of the corneal collagen in wildtype, heterozygous, and homozygous animals. We found no significant changes in either packing or overage size of corneal collagen in the Dcn−/− animals (not shown).
Figure 6.
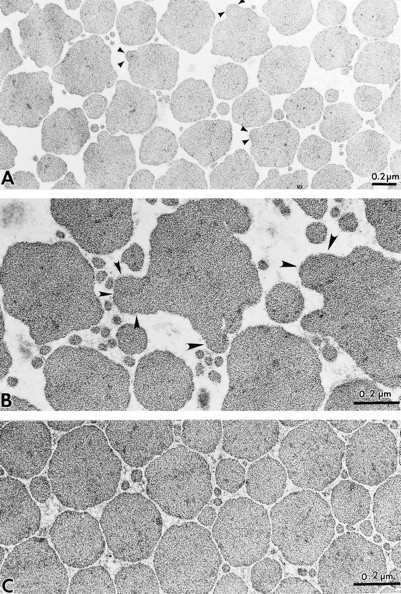

Decorin-deficient mice reveal abnormal collagen in tail tendon and a decrease in collagen-associated proteoglycans. (A) Lower power view of cross-sectional area of adult tail tendon collagen from Dcn−/− reveals abnormal shape and multiple lateral fusions. Higher power view of Dcn−/− tendons shows bizarre, gigantic fibrils (>660 nm in largest diameter) with multiple lateral fusions (B, arrowheads), in contrast with the more uniform collagen fibrils of the wild-type animals (C) with an average diameter of ∼200 nm. Transmission electron micrographs of longitudinally sectioned collagen fibrils from Dcn+/+ (D) and Dcn−/− (E) tendon, after fixation in the presence of 0.05% cuprolinic blue and 0.3 M MgCl2, and staining with uranyl acetate to visualize the collagen binding pattern. Notice that the typical, 67-nm periodicity of the type I collagen is maintained in the Dcn−/− mice (E). However, numerous d bands are not occupied by proteoglycan granules (E, spaces between arrowheads and unfilled arrowheads), in contrast with the wild type where nearly every d band is occupied (D). Bar 0.2 μm.
To investigate further the role of proteoglycans in collagen fibrillogenesis, we processed skin and tendon in the presence of cuprolinic blue, a cationic dye that preserves proteoglycans in an extended configuration in the presence of 0.3 M MgCl2 (53). Under such high salt concentration, binding of the dye is specific for proteoglycans with sulfate esters. Using this method, decorin has been localized to the d and e bands in the D-gap zone of collagen quarter-stagger in tendon, cornea, and pericardium (52, 53, 56), a localization confirmed by immunoelectron microscopy (43, 56) and in situ staining of isolated molecules (35). When collagen fibrils from tail tendons were fixed in the presence of cuprolinic blue and stained with uranyl acetate to visualize the banding pattern, it was evident that the collagen of affected animals (Fig. 6 E) exhibited the 67-nm periodicity characteristic of type I collagen. As expected, in the Dcn+/+ animals, all the d bands were occupied (Fig. 6 D, rows of arrowheads). In contrast, in the Dcn−/− animals, several d bands contained no orthogonally arrayed proteoglycan granules (Fig. 6 E, spaces between arrowheads and also unfilled arrows). Moreover, we found a significant reduction of proteoglycan granules and filaments around dermal collagen fibrils of homozygous animals when compared with wild type (not shown). The proteoglycans appeared smaller in average length in the Dcn−/−, likely reflecting the presence of keratan-sulfate– containing proteoglycans, such as fibromodulin and lumican, that are known to be expressed in both tendon and skin (28). Collectively, these results provide a structural basis for the phenotype described above and further indicate that the abnormal collagen morphology is associated with a decrease in collagen-bound proteoglycans. The loss of such important protein/protein interaction sites could significantly contribute to the abnormal collagen fibrillogenesis described above.
Mass Mapping of Dermal Fibrils from Decorin-deficient Animals Reveals Pronounced Nonuniformity in the Axial Mass Distribution
Next, we wished to determine whether the collagen fibrils from the skin of Dcn−/− animals were uniform along their axes. This was investigated by determining the axial mass distributions of unstained, isolated fibrils using STEM. Dispersed fibrils of up to ∼55 μm (∼800 D-periods; 1 D = 67 nm) in length were prepared, and montages of STEM images were recorded along the entire lengths of each fibril. Consecutive mass per unit length measurements (averages over two D-periods) were made along each fibril. Typical results are shown in Fig. 7. Fibrils from Dcn−/− animals showed pronounced nonuniformity along their axes compared with the Dcn+/+ control fibrils. A typical unstained fibril from the homozygous animals showed repeated bulges along the axis (Fig. 7 A). Axial mass distributions of these fibrils revealed a series of mass peaks, often with a factor of two or three times the mass per unit length of the adjacent parts of the fibril, and each typically extending 50–80 D-periods along the fibril (Fig. 7, D–F). In contrast, fibrils from the wild-type animals appeared uniform in STEM images (Fig. 7 C), and axial mass measurements confirmed that these fibrils lacked the mass peaks seen regularly in the homozygous animals (Fig. 7 G). A degree of nonuniformity existed in control fibrils (Fig. 7 G), although this was much less pronounced than that seen in Dcn−/− mice (Fig. 7, D–F). To quantify the changes in mass per unit length along individual fibrils, a simple measure of fibril uniformity was defined as the nonuniformity index, which was equal to the range of mass per unit length for each fibril divided by the mean mass per unit length (Fig. 7 H). Analysis of fibrils from Dcn+/+ mice showed that the nonuniformity index decreased with increasing fibril mass per unit length (i.e., increasing fibril diameter). Therefore, the fibrils with the most relatively uniform shafts were the larger diameter fibrils. In contrast, analysis of fibrils from Dcn−/− mice showed that the index increased with increasing fibril mass per unit length. The lumpy nature of axial mass distribution profiles of the fibrils from the Dcn−/− animals is consistent with a lateral fusion of short fibril segments with a longer fibril. The occurrence of lateral associations of long fibrils is also indicated by the observation of twisted pairs of fibrils (Fig. 7 B). These results strongly indicate that the larger and irregular skin fibrils seen in transverse sections are likely to be a consequence of lateral fusion, producing a fibril whose morphology changes continuously along its axial length.
Figure 7.
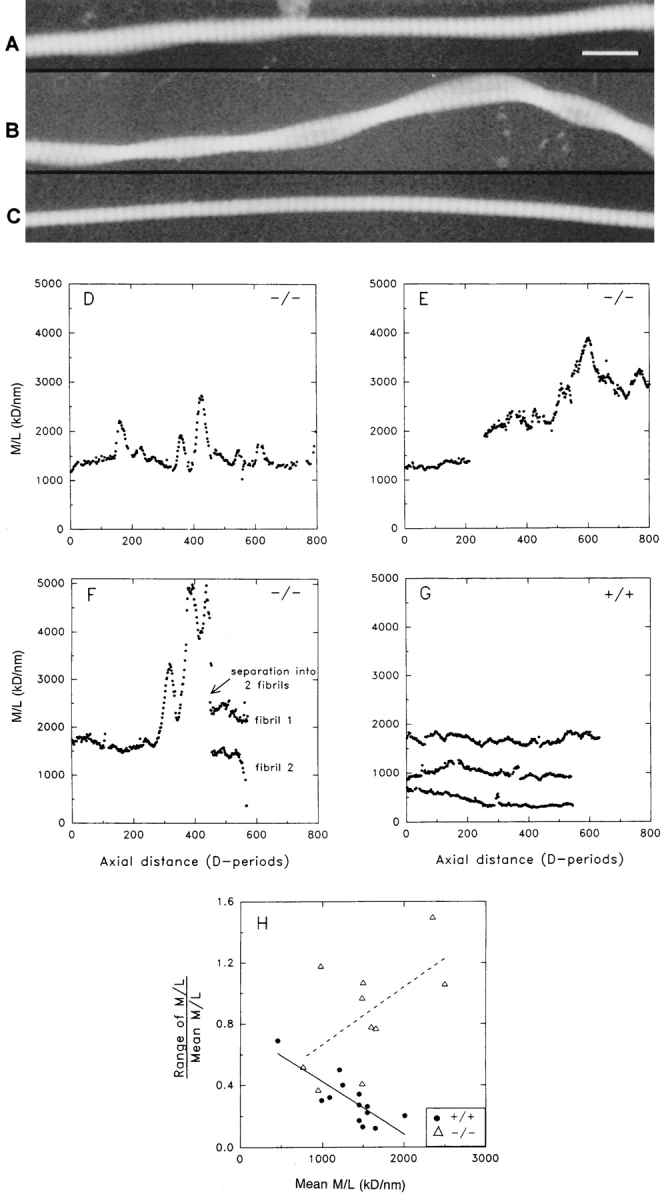
Mass mapping of collagen fibrils isolated from the skin of decorin-deficient animals reveals pronounced nonuniformity in the axial mass distributions with repeated bulges. (A–C) Typical dark-field STEM images of isolate unstained fibrils from mouse skin. A and B show typical images for fibrils from a decorin-deficient mouse. A shows a single fibril with distinct bulging at two locations over 60 D-periods (1 D = 67 nm) length. B is consistent with two fibrils twisted together in close contact but not fused. C shows a typical fibril from control skin with uniform diameter along its length. (D–G) Axial mass distributions of single fibrils obtained by measurements from STEM images. A measurement of mass per unit length was made every two D-periods along the entire available length of each fibril. Missing points occur where parts of a fibril were clearly contaminated. (D–F) Typical axial mass distribution profiles from the decorin-deficient sample or (G) from wild-type animals. The decorin-deficient sample shows pronounced mass peaks in the fibrial axial mass distribution, which are not found in the control. (H) Scatter plot of the relative variation in M/L along single fibrils against the corresponding mean M/L. First order regression lines are shown superimposed on each set of data. The data from the decorin-deficient samples show an overall increase in the relative intrafibrillar variation in M/L with increasing lateral size (Mean M/L); this contrasts with the control sample that shows the opposite trend. Bar, 200 μm.
Discussion
Collagen provides the principal source of mechanical strength in connective tissues and is often implicated in diseases where such mechanical support is defective (44). The molecular interactions that take place among various types of fibril-forming collagens and members of the SLRP gene family constitute a fundamental regulatory mechanism for the assembly of heterotypic fibrils. The results of the present investigation clearly show the developmental significance of decorin as a key regulator of collagen fibrillogenesis and validate previous observations that the core protein of decorin plays a central role in regulating fibrillogenesis (10, 66, 67). Given the existence of several members of the SLRP gene family that could conceivably compensate for the lack of decorin function, the observed phenotype was not entirely predictable. We observed skin fragility and abnormal fibrillogenesis in dermal and tendon collagen, but not in corneal collagen, indicating tissue specificity and a requirement for decorin for proper skin and tendon development and maintenance of normal tensile strength.
Decorin Gene Knockout Causes Abnormal Collagen Structure and Skin Fragility
The cross-sections of fibrils in skin and tendon showed regular, uniform fibrils in control animals. In contrast, in the decorin-deficient animals, the collagen fibrils were coarser and irregular in size and shape. Furthermore, they were often haphazardly arranged with increased interfibrillar spaces. These structural changes were associated with a reduction in collagen-bound proteoglycans in both dermal and tendon collagen. Of note, the STEM data showed that the fibrils in null mice were not uniform in diameter but had bulges along their shafts. These data thus explain the variability in size and shape seen in cross-sections of the skin and tendon collagen. There are two possible mechanisms to explain the formation of bulges along the axis of individual collagen fibrils. First, the bulges (abrupt increases in mass per unit length) could occur when early fibrils fuse laterally onto an existing larger fibril. Our quantitative data (Fig. 7 H) show that, as the fibril diameter increases in control animals, the bulge factor decreases. Thus, with increasing fibril diameter there is less possibility of fibril fusion. This is the reverse in the decorin-deficient mice; with increasing fibril diameter, the bulge factor increases. Interestingly, at a fixed decorin/collagen ratio, with increasing fibril diameter, the surface area/volume ratio decreases and the concentration of decorin at the fibril surface increases. Therefore, in control mice, as the fibril diameter increases, the concentration of decorin at the fibril surface increases and fibril fusion is inhibited. This mechanism of restricting fibril fusion to small diameter fibrils is lost in the decorin-deficient mice. The second possible explanation is that collagen fibrils might have a natural tendency to be irregular. Decorin, in some unknown way, would keep the fibrils regular in outline. There is in vitro evidence against this interpretation because fibrils formed from purified collagen solutions tend to be fairly uniform in cross-section (31). Furthermore, collagen fibrils assembled in vitro from purified acid-soluble calf skin collagen participate in lateral association and fusion in the absence of decorin (unpublished observations). The most severely affected tissue in decorin-deficient mice is skin, which cannot withstand sudden tensile strain as its normal counterpart does. The abnormal packing of collagen together with the nonuniformity of fiber diameter may have a considerable influence on the biomechanics of this tensile tissue. Skin fragility in these mutant animals could be ascribed to this anomalous collagen network, which would allow full body development and viability, but would lead to a reduced tensile strength with potential complications such as increased incidence of injury, infection, and scarring. Biomechanical measurements using physiological stress rates (i.e., slow stretching rates of 1 mm/s) revealed at least a threefold reduction in tensile strength of dorsal skin samples with a concurrent 35–40% reduction in ductility. Of note, using similar low speed stress rates, it has been recently shown that the tensile strength of rat skin correlates with insoluble collagen and is decreased when cross-linking of collagen is blocked by lathyrogen treatment (15). This is similar to the decrease in mechanical strength detected in lathyritic rat tail tendon (19).
The presence of small proteoglycans occupying the d band of collagen fibrils in the decorin knockout animals suggests that other members of the SLRP gene family may bind the same sites. However, neither biglycan nor lumican were upregulated at the mRNA level, thus suggesting that there is no obligatory compensation for the lack of decorin by at least two members of the SLRP gene family.
The absence of overt abnormalities in the cornea of Dcn−/− animals may be explained by the larger requirement for keratan sulfate in this transparent structure and by the presence of at least five members of the SLRP gene family, which could control more tightly the fibril diameter necessary for transparency. Of note, β-d-xyloside, a xylose analogue that interferes with the synthesis of chondroitin/ dermatan sulfate but not keratan sulfate proteoglycans, causes no alterations in fibril diameter but induces disruption of lamellar organization and fibril packing in developing avian cornea (17). These findings therefore suggest that decorin may play a less prominent role in the regulation of collagen fibril diameter in cornea, but is important in maintaining the interfibrillar spacing and lamellar cohesiveness.
Decorin: A Key Regulator of Collagen Fibrillogenesis
Connective tissue interactions, including those between proteoglycans and collagen, are evolutionarily conserved and physiologically relevant. High affinity interaction of dermatan sulfate proteoglycan with collagen was inferred three decades ago when extraction with 6 M urea or digestion with collagenase was required for efficient isolation of this proteoglycan from the skin (61). Moreover, it is firmly established that (a) fibrillogenesis is a multistep assembly process (64); (b) collagen molecules incubated in vitro at neutral pH and physiologic temperatures aggregate and develop into insoluble fibrils of various sizes and lengths (31); (c) unipolar and bipolar fibrils occur in vivo (23); (d) collagen fibrils grow from paraboloidal tips and this mechanism is highly regulated (23, 30); (e) the size and shape of fibrils are enzymatically controlled by the procollagen C-proteinase (26); and (f) this process can be modulated by skin proteoglycans (62). When the kinetics of in vitro fibrillogenesis were monitored by turbidimetry, it was discovered that decorin from tendon (66) or cornea (45) significantly delayed collagen fibrillogenesis. The lateral assembly of triple-helical collagen molecules was also delayed by decorin resulting in thinner fibrils (8, 9, 65). During development, the ratio of dermatan sulfate to hydroxyproline decreases as the fibril cross-sectional areas increase (53), an observation corroborated by the finding that fibril growth is associated with a significant decrease in fibril- associated decorin (8). The corollary, that thicker fibrils are associated with less dermatan sulfate/unit weight of collagen, can be inferred from the fact that the upper dermis of calf skin containing finer fibrils has twice the dermatan sulfate/collagen ratio of deeper dermis containing coarser fibrils (53). Of note, in human skin, the papillary (upper) dermis expresses higher levels of decorin than the reticular (deeper) layer (50). Collectively, these findings favor a role of decorin in permitting lateral association of fibril segments, a concept also supported by the three-dimensional molecular model of decorin (69). Binding of decorin to collagen would interfere with further lateral accretion of collagen molecules and retard/stop the growth of fibrils. Therefore, decorin would play a pivotal role in regulating the orderly assembly and growth of collagen fibrils, which in turn would affect the tensile strength of connective tissues. The abnormal collagen morphology and the STEM data fully support this interpretation.
Relation of the Decorin-deficient Phenotype to Dermatosparaxis and Ehlers-Danlos Syndrome
Dermatosparaxis, “torn skin” syndrome, is a recessively inherited connective tissue disorder that results from the lack of activity of type I procollagen N-proteinase, the enzyme that removes the amino-terminal propeptide from type I procollagen (36). Initially identified in cattle more than 25 years ago, this disorder has now been reported in sheep, cats, and dogs (5, 13, 18). Affected animals have skin fragility, joint laxity, and often succumb prematurely to sepsis after avulsion of portions of the skin (18). The hallmark of the disease is the presence of abnormal collagen fibrils that are thinner and haphazardly arrayed, and often in cross-section resemble hieroglyphs. A similar syndrome has been now reported in three patients with skin fragility and twisted, ribbon-like collagen fibrils (40, 41, 57). The human syndrome, designated Ehlers-Danlos syndrome (EDS) VIIC, differs from the EDS VIIA and VIIB insofar as the latter two are caused by mutations of the NH2-terminal propeptide of collagen α1(I) and α2(I) genes, respectively (12). The mutations in EDS types VIIA and B result in the skipping of exon 6, which contains the cleavage sites for the N-proteinase, thereby leading to accumulation of unprocessed collagen. Intuitively, it might have been expected that a deficiency in N-proteinase activity and the lack of the cleavage site for the enzyme would have given rise to the same disease. However, EDS types VIIA and B are distinct from dermatosparaxis in that (a) joint hypermobility, rather than skin fragility, is the principal clinical feature, and (b) ultrastructural analysis of the dermal collagen shows rough-bordered collagen fibrils rather than hieroglyphs (24, 68). A contribution factor to the skin fragility in dermatosparaxis may be the steric blocking of a factor to the gap zone of the collagen fibrils insofar as the amino-terminal propeptide would interfere with this region by forming a hairpin (29). In dermatosparactic skin, several proteoglycan filaments are not closely bound to collagen fibrils but are floating rather freely in the interfibrillar spaces (54). Thus, dermatosparactic animals (by blocking access to decorin) and decorin null animals (by lacking decorin) could share a common pathogenetic mechanism, i.e., the obstruction/deficiency of a key regulatory molecule in collagen fibrillogenesis. Further support for this concept is provided by the fact that the dermatan sulfate/hydroxyproline ratio in dermatosparactic skin is reduced from 4.7 to 3.6 (38). Since the surface of the abnormal hieroglyphic collagen is two to four times higher than that of cylindrical fibrils of normal skin, dermatosparactic animals should also have a significant decrease in collagen-associated decorin.
Transgenic mice homozygous for an in-frame deletion of exon 6 of the collagen α2(V) gene exhibit phenotypic and ultrastructural features resembling EDS and some aspects of the decorin-deficient phenotype (1). The mutant animals develop skin fragility with disorganized dermal fibrils of variable diameter; however, the affected animals show various degrees of kypho-lordosis and corneal abnormalities, not observed in the decorin null mice. Several independent studies have now confirmed the linkage of α1(V) collagen gene to EDS I and II (11, 37, 63). It is known that type V collagen localizes at the periphery of the coarser collagen fibrils, composed predominantly of type I, and it has been proposed that the interaction between collagen types I and V regulated fibril diameter (7). Thus, genetic defects in type V collagen could explain the observation that collagen fibrils in skin of EDS I and II patients are larger than normal and irregular in outline. These observations indicate that “modifiers” of collagen fibrillogenesis, such as type V collagen and decorin, might have profound biological consequences in vivo.
Conclusions
This study has been fruitful in establishing a functional role for a secreted proteoglycan in maintaining the structural integrity of the cutis and perhaps tendon sheaths. Tensile strength is certainly a major property of connective tissues, and our data demonstrate that decorin stabilizes the fibrillar matrix in vivo. Decorin governs collagen fibril growth and could influence higher order matrix assembly. Understanding how changes in decorin alter the biological properties of collagen and disrupt the finely balanced network of molecular interactions that govern fibril assembly and architecture may lead to the discovery of novel genetic disorders caused by members of the SLRP gene family, and could potentially lead to the development of medical strategies for their treatment. In addition to helping to elucidate the biological role of decorin, the decorin-deficient mouse could represent a useful animal model to investigate matrix assembly, wound healing, and tumorigenesis. On the basis of our animal phenotype, we predict the existence of a human skin fragility syndrome, to be included in the EDS category, which could be caused by either a recessive deletion or a mutation in the collagen binding domain of the decorin gene at the 12q23 locus.
Acknowledgments
We thank T. Scholzen for help in the initial stages of this work; C. Clark and R. Trelstad for their critical reading of the manuscript; M. Mathiak, I. Eichstetter, and B. Tuma for valuable assistance; S. Kalidindi for help with biomechanical measurements; M. Pisano for valuable discussion and advice; L. Fisher for the generous gift of antisera; K. Soderberg for ES cell work; J. Avis for embryo manipulation; M. Verloop and R. Jenkins for animal procedures; M. Wilson-Heiner for genotyping; T. Doetschman for the gift of the neomycin-resistant mouse colony; A. Nagy for the R1 cells; and the Genetics Institute for the gift of CHO-LIF cell line.
This work was supported by National Institutes of Health grants RO1 CA39481 and RO1 CA47282 (R.V. Iozzo), AR41816 and CA30199 (H. Baribault), and by Wellcome Trust 019512 and the Medical Research Council ICS-95-14 (K.E. Kadler).
Abbreviations used in this paper
- EDS
Ehlers-Danlos syndrome
- ES
embryonic stem
- SLRP
small leucine-rich proteoglycan
- STEM
scanning transmission EM
Footnotes
K.G. Danielson and H. Baribault contributed equally to this work.
References
- 1.Andrikopoulos K, Liu X, Keene DR, Jaenisch R, Ramirez F. Targeted mutation in the col5a2gene reveals a regulatory role for type V collagen during matrix assembly. Nat Genet. 1995;9:31–36. doi: 10.1038/ng0195-31. [DOI] [PubMed] [Google Scholar]
- 2.Baribault H, Oshima RG. Polarized and functional epithelia can form after the targeted inactivation of both mouse keratin 8 alleles. J Cell Biol. 1991;115:1675–1684. doi: 10.1083/jcb.115.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baribault H, Price J, Miyai K, Oshima RG. Mid-gestational lethality in mice lacking keratin 8. Genes & Dev. 1993;7:1191–1202. doi: 10.1101/gad.7.7a.1191. [DOI] [PubMed] [Google Scholar]
- 4.Baribault H, Penner J, Iozzo RV, Wilson-Heiner M. Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes & Dev. 1994;8:2964–2973. doi: 10.1101/gad.8.24.2964. [DOI] [PubMed] [Google Scholar]
- 5.Bavinton JH, Peters DE, Ramshaw AM. A morphologic study of a mild form of ovine dermatosparaxis. J Invest Dermatol. 1985;84:391–395. doi: 10.1111/1523-1747.ep12265475. [DOI] [PubMed] [Google Scholar]
- 6.Bidanset DJ, Guidry C, Rosenberg LC, Choi HU, Timpl R, Höök M. Binding of the proteoglycan decorin to collagen type VI. J Biol Chem. 1992;267:5250–5256. [PubMed] [Google Scholar]
- 7.Birk DE, Fitch JM, Babiarz JP, Doane KJ, Linsenmayer TF. Collagen fibrillogenesis in vitro: interaction of types I and V collagen regulates fibril diameter. J Cell Sci. 1990;95:649–657. doi: 10.1242/jcs.95.4.649. [DOI] [PubMed] [Google Scholar]
- 8.Birk DE, Nurminskaya MV, Zycband EI. Collagen fibrillogenesis in situ: fibril segments undergo post-depositional modifications resulting in linear and lateral growth during matrix development. Dev Dyn. 1995;202:229–243. doi: 10.1002/aja.1002020303. [DOI] [PubMed] [Google Scholar]
- 9.Birk DE, Hahn RA, Linsenmayer C, Zycband EI. Characterization of collagen fibril segments from chicken embryo cornea, dermis and tendon. Matrix Biol. 1996;15:111–118. doi: 10.1016/s0945-053x(96)90152-3. [DOI] [PubMed] [Google Scholar]
- 10.Brown DC, Vogel KG. Characteristics of the in vitro interaction of a small proteoglycan (PG II) of bovine tendon with type I collagen. Matrix. 1989;9:468–478. doi: 10.1016/s0934-8832(11)80016-8. [DOI] [PubMed] [Google Scholar]
- 11.Burrows NP, Nicholls AC, Yates JRW, Gatward G, Sarathachandra P, Richards A, Pope FM. The gene encoding collagen α1(v) (COL5A1)is linked to mixed Ehlers-Danlos syndrome type I/II. J Invest Dermatol. 1996;106:1273–1276. doi: 10.1111/1523-1747.ep12348978. [DOI] [PubMed] [Google Scholar]
- 12.Byers PH. Ehlers-Danlos syndrome: recent advances and current understanding of the clinical and genetic heterogeneity. J Invest Dermatol. 1994;103:47S–52S. doi: 10.1111/1523-1747.ep12399038. [DOI] [PubMed] [Google Scholar]
- 13.Counts DF, Byers PH, Holbrook KA, Hegreberg GA. Dermatosparaxis in a Himalayan cat: I. Biochemical studies of dermal collagen. J Invest Dermatol. 1980;74:96–99. doi: 10.1111/1523-1747.ep12519991. [DOI] [PubMed] [Google Scholar]
- 14.De Luca A, Santra M, Baldi A, Giordano A, Iozzo RV. Decorin-induced growth suppression is associated with upregulation of p21, an inhibitor of cyclin-dependent kinases. J Biol Chem. 1996;271:18961–18965. doi: 10.1074/jbc.271.31.18961. [DOI] [PubMed] [Google Scholar]
- 15.Dombi GW, Haut RC, Sullivan WG. Correlation of highspeed tensile strength with collagen content in control and lathyritic rat skin. J Surg Res. 1993;54:21–28. doi: 10.1006/jsre.1993.1004. [DOI] [PubMed] [Google Scholar]
- 16.Fisher LW, Stubbs JT, III, Young MF. Antisera and cDNA probes to human and certain animal model bone matrix noncollagenous proteins. Acta Orthop Scand. 1995;66:61–65. [PubMed] [Google Scholar]
- 17.Hahn RA, Birk DE. β-D xyloside alters dermatan sulfate proteoglycan synthesis and the organization of the developing avian corneal stroma. Development (Camb) 1992;115:383–393. doi: 10.1242/dev.115.2.383. [DOI] [PubMed] [Google Scholar]
- 18.Hanset R. Dermatosparaxis of the calf, a genetic defect of the connective tissue. Hoppe-Seyler's Z Physiol Chem. 1971;352:13–15. [Google Scholar]
- 19.Haut RC. The effect of a lathyritic diet on the sensitivity of tendon to strain rate. J Biomech Eng. 1985;107:155–174. doi: 10.1115/1.3138537. [DOI] [PubMed] [Google Scholar]
- 20.Hildebrand A, Romaris M, Rasmussen LM, Heinegård D, Twardzik DR, Border WA, Ruoslahti E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor β. Biochem J. 1994;302:527–534. doi: 10.1042/bj3020527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holmes DF. Mass mapping of extracellular matrix assemblies. Biochem Soc Trans. 1995;23:720–725. doi: 10.1042/bst0230720. [DOI] [PubMed] [Google Scholar]
- 22.Holmes DF, Mould AP, Chapman JA. Morphology of sheetlike assemblies of pN-collagen, pC-collagen and procollagen studied by scanning transmission electron microscopy mass measurements. J Mol Biol. 1991;220:111–123. doi: 10.1016/0022-2836(91)90385-j. [DOI] [PubMed] [Google Scholar]
- 23.Holmes DF, Chapman JA, Prockop DJ, Kadler KE. Growing tips of type I collagen fibrils formed in vitroare near-paraboloidal in shape, implying a reciprocal relationship between accretion and diameter. Proc Natl Acad Sci USA. 1992;89:9855–9859. doi: 10.1073/pnas.89.20.9855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holmes DF, Watson RB, Steinmann B, Kadler KE. EhlersDanlos syndrome type VIBB. Morphology of type I collagen fibrils formed in vivo and in vitrois determined by the conformation of the N-propeptide. J Biol Chem. 1993;268:15758–15765. [PubMed] [Google Scholar]
- 25.Holmes DF, Lowe MP, Chapman JA. Vertebrate (chick) collagen fibrils formed in vivocan exhibit a reversal in molecular polarity. J Mol Biol. 1994;235:80–83. doi: 10.1016/s0022-2836(05)80016-6. [DOI] [PubMed] [Google Scholar]
- 26.Holmes DF, Watson RB, Chapman JA, Kadler KE. Enzymic control of collagen fibril shape. J Mol Biol. 1996;261:93–97. doi: 10.1006/jmbi.1996.0443. [DOI] [PubMed] [Google Scholar]
- 27.Iozzo RV. Biosynthesis of heparan sulfate proteoglycan by human colon carcinoma cells and its localization at the cell surface. J Cell Biol. 1984;99:403–417. doi: 10.1083/jcb.99.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iozzo RV, Murdoch AD. Proteoglycans of the extracellular environment: clues from the gene and protein side offer novel perspectives in molecular diversity and function. FASEB (Fed Am Soc Exp Biol) J. 1996;10:598–614. [PubMed] [Google Scholar]
- 29.Kadler KE. Learning how mutations in type I collagen genes cause connective tissue disease. Int J Exp Path. 1993;74:319–323. [PMC free article] [PubMed] [Google Scholar]
- 30.Kadler KE, Hojima Y, Prockop DJ. Collagen fibrils in vitro grow from pointed tips in the C- to N-terminal direction. Biochem J. 1990;268:339–343. doi: 10.1042/bj2680339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kadler KE, Holmes DF, Trotter JA, Chapman JA. Collagen fibril formation. Biochem J. 1996;316:1–11. doi: 10.1042/bj3160001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kato YP, Christiansen DL, Hahn RA, Shieh S, Goldstein JD, Silver FH. Mechanical properties of collagen fibres: a comparison of reconstituted and rat tendon fibres. Biomaterials. 1989;10:38–40. doi: 10.1016/0142-9612(89)90007-0. [DOI] [PubMed] [Google Scholar]
- 33.Kobe B, Deisenhofer J. A structural basis of the interactions between leucine-rich repeats and protein ligands. Nature (Lond) 1995;374:183–186. doi: 10.1038/374183a0. [DOI] [PubMed] [Google Scholar]
- 34.Krumdieck R, Höök M, Rosenberg LC, Volanakis JE. The proteoglycan decorin binds C1q and inhibits the activity of the C1 complex. J Immunol. 1992;149:3695–3701. [PubMed] [Google Scholar]
- 35.Lan, Y., C. Cummings, J.K. Sheehan, K.E. Kadler, D.F. Holmes, and J.A. Chapman. 1993. Visualisation of individual proteoglycan-collagen interactions. In Dermatan Sulphate Proteoglycans. J.E. Scott, editor. Portland Press, London. 183–192.
- 36.Lichtenstein JR, Martin GR, Kohn LD, Byers PH, McKusick VA. Defect in conversion of procollagen to collagen in a form of Ehlers-Danlos syndrome. Science (Wash DC) 1973;182:298–300. doi: 10.1126/science.182.4109.298. [DOI] [PubMed] [Google Scholar]
- 37.Loughlin J, Irven C, Hardwick LJ, Butcher S, Walsh S, Wordsworth P, Sykes B. Linkage of the gene that encodes the α1 chain of type V collagen (COL5A1) to type II Ehlers-Danlos syndrome (EDS II) . Hum Mol Genet. 1995;4:1649–1651. doi: 10.1093/hmg/4.9.1649. [DOI] [PubMed] [Google Scholar]
- 38.Matsunaga E, Shinkai H, Nusgens B, Lapiere CM. Acidic glycosaminoglycans, isolation and structural analysis of a proteodermatan sulfate from dermatosparactic calf skin. Collagen Relat Res. 1986;6:467–479. doi: 10.1016/s0174-173x(87)80046-8. [DOI] [PubMed] [Google Scholar]
- 39.Nagy A, Rossant J, Nage R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nusgens BV, Verellen-Dumoulin C, Hermanns-Lê T, De Paepe A, Nuytinck L, Piérard GE, Lapière CM. Evidence for a relationship between Ehlers-Danlos type VII C in humans and bovine dermatosparaxis. Nat Genet. 1992;1:214–217. doi: 10.1038/ng0692-214. [DOI] [PubMed] [Google Scholar]
- 41.Petty EM, Seashore MR, Braverman IM, Spiesel SZ, Smith LT, Milstone LM. Dermatosparaxis in children. A case report and review of the newly recognized phenotype. Arch Dermatol. 1993;129:1310–1315. doi: 10.1001/archderm.129.10.1310. [DOI] [PubMed] [Google Scholar]
- 42.Pogány G, Vogel KG. The interaction of decorin core protein fragments with type I collagen. Biochem Biophys Res Commun. 1992;189:165–172. doi: 10.1016/0006-291x(92)91539-3. [DOI] [PubMed] [Google Scholar]
- 43.Pringle GA, Dodd CM. Immunoelectron microscopic localization of the core protein of decorin near the d and e bands of tendon collagen fibrils by use of monoclonal antibodies. J Histochem Cytochem. 1990;38:1405–1411. doi: 10.1177/38.10.1698203. [DOI] [PubMed] [Google Scholar]
- 44.Prockop DJ, Kivirikko KI. Collagens: molecular biology, diseases, and potentials for therapy. Annu Rev Biochem. 1995;64:403–434. doi: 10.1146/annurev.bi.64.070195.002155. [DOI] [PubMed] [Google Scholar]
- 45.Rada JA, Cornuet PK, Hassell JR. Regulation of corneal collagen fibrillogenesis in vitro by corneal keratan sulfate proteoglycan (lumican) and decorin core proteins. Exp Eye Res. 1993;56:635–648. doi: 10.1006/exer.1993.1081. [DOI] [PubMed] [Google Scholar]
- 46.Rigby BJ. Effect of cyclic extension on the physical properties of tendon collagen and its possible relation to biological ageing of collagen. Nature (Lond) 1964;202:1072–1074. doi: 10.1038/2021072a0. [DOI] [PubMed] [Google Scholar]
- 47.Santra M, Skorski T, Calabretta B, Lattime EC, Iozzo RV. De novodecorin gene expression suppresses the malignant phenotype in human colon cancer cells. Proc Natl Acad Sci USA. 1995;92:7016–7020. doi: 10.1073/pnas.92.15.7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmidt G, Robenek H, Harrach B, Glössl J, Nolte V, Hörmann H, Richter H, Kresse H. Interaction of small dermatan sulfate proteoglycan from fibroblasts with fibronectin. J Cell Biol. 1987;104:1683–1691. doi: 10.1083/jcb.104.6.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scholzen T, Solursh M, Suzuki S, Reiter R, Morgan JL, Buchberg AM, Siracusa LD, Iozzo RV. The murine decorin. Complete cDNA cloning, genomic organization, chromosomal assignment and expression during organogenesis and tissue differentiation. J Biol Chem. 1994;269:28270–28281. [PubMed] [Google Scholar]
- 50.Schönherr E, Beavan LA, Hausser H, Kresse H, Culp LA. Differences in decorin expression by papillary and reticular fibroblasts in vivo and in vitro. . Biochem J. 1993;290:893–899. doi: 10.1042/bj2900893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schönherr E, Hausser H, Beavan L, Kresse H. Decorin-type I collagen interaction. Presence of separate core protein-binding domains. J Biol Chem. 1995;279:8877–8883. doi: 10.1074/jbc.270.15.8877. [DOI] [PubMed] [Google Scholar]
- 52.Scott JE. Proteoglycan-fibrillar collagen interactions. Biochem J. 1988;252:313–323. doi: 10.1042/bj2520313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scott JE, Orford CR. Dermatan sulphate-rich proteoglycan associates with rat tail-tendon collagen at the d band in the gap region. Biochem J. 1981;197:213–216. doi: 10.1042/bj1970213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scott JE, Haigh M, Nusgens B, Lapière CM. Proteoglycan: collagen interactions in dermatosparactic skin and tendon. An electron histochemical study using cupromeronic blue in a critical electrolyte concentration method. Matrix. 1989;9:437–442. doi: 10.1016/s0934-8832(11)80012-0. [DOI] [PubMed] [Google Scholar]
- 55.Scott PG, Winterbottom N, Dodd CM, Edwards E, Person CH. A role for disulphide bridges in the protein core in the interaction of proteodermatan sulphate and collagen. Biochem Biophys Res Commun. 1986;138:1348–1354. doi: 10.1016/s0006-291x(86)80431-4. [DOI] [PubMed] [Google Scholar]
- 56.Simionescu D, Iozzo RV, Kefalides NA. Bovine pericardial proteoglycan: biochemical, immunochemical and ultrastructural studies. Matrix. 1989;9:301–310. doi: 10.1016/s0934-8832(89)80006-x. [DOI] [PubMed] [Google Scholar]
- 57.Smith LT, Wertelecki W, Milstone LM, Petty EM, Seashore MR, Braverman IM, Jenkins TG, Byers PH. Human dermatosparaxis: a form of Ehlers-Danlos syndrome that results from failure to remove the amino-terminal propeptide of type I procollagen. Am J Hum Genet. 1992;51:235–244. [PMC free article] [PubMed] [Google Scholar]
- 58.Soriano P, Montgomery C, Geske R, Bradley A. Targeting disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 59.Svensson L, Heinegård D, Oldberg Å. Decorin binding sites for collagen type I are mainly located in leucine rich repeats 4-5. J Biol Chem. 1995;270:20712–20716. doi: 10.1074/jbc.270.35.20712. [DOI] [PubMed] [Google Scholar]
- 60.Thieszen SL, Rosenquist TH. Expression of collagens and decorin during aortic arch artery development: implications for matrix pattern formation. Matrix Biol. 1994;14:573–582. doi: 10.1016/s0945-053x(05)80006-x. [DOI] [PubMed] [Google Scholar]
- 61.Toole BP, Lowther DA. The organisation of hexamine-containing compounds in bovine skin. Biochim Biophys Acta. 1966;121:315–325. doi: 10.1016/0304-4165(66)90120-6. [DOI] [PubMed] [Google Scholar]
- 62.Toole BP, Lowther DA. The effect of chondroitin sulphateprotein in the formation of collagen fibrils in vitro. . Biochem J. 1968;109:857–866. doi: 10.1042/bj1090857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Toriello HV, Glover TW, Takahara K, Byers PH, Miller DE, Higgins JV, Greenspan DS. A translocation interrupts the COL5A1gene in a patient with Ehlers-Danlos syndrome and hypomelanosis of Ito. Nat Genet. 1996;13:361–365. doi: 10.1038/ng0796-361. [DOI] [PubMed] [Google Scholar]
- 64.Trelstad RL. Multistep assembly of type I collagen fibrils. Cell. 1982;28:197–198. doi: 10.1016/0092-8674(82)90334-8. [DOI] [PubMed] [Google Scholar]
- 65.Vogel KG, Trotter JA. The effects of proteoglycans on the morphology of collagen fibrils formed in vitro. . Collagen Relat Res. 1987;7:105–114. doi: 10.1016/s0174-173x(87)80002-x. [DOI] [PubMed] [Google Scholar]
- 66.Vogel KG, Paulsson M, Heinegård D. Specific inhibition of type I and type II collagen fibrillogenesis by the small proteoglycan of tendon. Biochem J. 1984;223:587–597. doi: 10.1042/bj2230587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vogel KG, Koob TJ, Fisher LW. Characterization and interactions of a fragment of the core protein of the small proteoglycan (PGII) from bovine tendon. Biochem Biophys Res Commun. 1987;148:658–663. doi: 10.1016/0006-291x(87)90927-2. [DOI] [PubMed] [Google Scholar]
- 68.Watson RB, Wallis GA, Holmes DF, Viljoen D, Byers PH, Kadler KE. Ehlers-Danlos syndrome type VIIB. Incomplete cleavage of abnormal type I procollagen by N-proteinase in vitroresults in the formation of copolymers of collagen and partially cleaved pNcollagen that are near circular in cross-section. J Biol Chem. 1992;267:9093–9100. [PubMed] [Google Scholar]
- 69.Weber IT, Harrison RW, Iozzo RV. Model structure of decorin and implications for collagen fibrillogenesis. J Biol Chem. 1996;271:31767–31770. doi: 10.1074/jbc.271.50.31767. [DOI] [PubMed] [Google Scholar]
- 70.Yagi T, Ikawa Y, Yoshida K, Shigetani Y, Takeda N, Mabuchi I, Yamamoto T, Aizawa S. Homologous recombination at c-fynlocus of mouse embryonic stem cells with the use of diphtheria toxin A-fragment gene in negative selection. Proc Natl Acad Sci USA. 1990;87:9918–9922. doi: 10.1073/pnas.87.24.9918. [DOI] [PMC free article] [PubMed] [Google Scholar]