

Abstract
The nuclear envelope consists of a doublemembraned extension of the rough endoplasmic reticulum. In this report we describe long, dynamic tubular channels, derived from the nuclear envelope, that extend deep into the nucleoplasm. These channels show cell-type specific morphologies ranging from single short stubs to multiple, complex, branched structures. Some channels transect the nucleus entirely, opening at two separate points on the nuclear surface, while others terminate at or close to nucleoli. These channels are distinct from other topological features of the nuclear envelope, such as lobes or folds.
The channel wall consists of two membranes continuous with the nuclear envelope, studded with features indistinguishable from nuclear pore complexes, and decorated on the nucleoplasmic surface with lamins. The enclosed core is continuous with the cytoplasm, and the lumenal space between the membranes contains soluble ER-resident proteins (protein disulphide isomerase and glucose-6-phosphatase).
Nuclear channels are also found in live cells labeled with the lipophilic dye DiOC6. Time-lapse imaging of DiOC6-labeled cells shows that the channels undergo changes in morphology and spatial distribution within the interphase nucleus on a timescale of minutes.
The presence of a cytoplasmic core and nuclear pore complexes in the channel walls suggests a possible role for these structures in nucleo–cytoplasmic transport. The clear association of a subset of these structures with nucleoli would also be consistent with such a transport role.
In recent years, the view that the nucleoplasm is organized into a number of morphologically distinct and functionally significant domains has gained ground, supported by increasing evidence for subnuclear localization of processes such as replication (Banfalvi et al., 1989; Mills et al., 1989; Hozak and Cook, 1994; Hutchison et al., 1994), repair (Jackson et al., 1994a , b), transcription (Jackson et al., 1993; Wansink et al., 1993), and RNA splicing and processing (Huang and Spector, 1991; Xing and Lawrence, 1993; Xing et al., 1993). In some cases these localized functions, or proteins associated with them, can be correlated with specific subnuclear structures, identified either morphologically or immunocytochemically at the light or EM level (Carmo-Fonseca et al., 1991, 1992; Spector et al., 1991; Lamond and Carmo-Fonseca, 1993; Huang and Spector, 1996; for review see Strouboulis and Wolfe, 1996). Although these intranuclear domains or structures are not bounded by membranes as cytoplasmic organelles are, they appear to offer a means of segregation of function in much the same way as organelles in the cytoplasm do; such an analogy has already been suggested (Spector, 1990; Moen et al., 1995).
Although the morphological and functional subdomains of the nucleus are not membrane bound, there is evidence that isolated nuclei contain large amounts of phospholipid and inositides and that activity of some nuclear enzymes depends on the products of intranuclear lipids metabolism for normal function (Maraldi et al., 1993). Isolated nuclei that lack the membranes of the bulk nuclear envelope still contain phospholipid as well as polyphosphoinositides. Indeed, it has been suggested that a separate inositide phosphorylation–dephosphorylation cycle might exist within the nucleus (Divecha et al., 1991; Banfic et al., 1993). Much of this nuclear lipid is presumed to exist as proteolipid complexes, most probably associated with a nucleoskeletal matrix, as it is not removed by detergent extraction with Triton X-100 (Divecha et al., 1993; Mazzotti et al., 1995).
Recent advances in immunocytochemical detection of phosphoinositols have permitted analysis of the intranuclear distribution of lipids that are not in the form of membranes (Mazzotti et al., 1995). However, there have also been suggestions that some of this nuclear lipid is in the form of recognizable bilayer membranes. Intranuclear membrane extensions have been described in plant cells (Dickinson and Bell, 1972; Li and Dickinson, 1987), insect cells (polytene cells of the Drosophila melanogaster salivary gland; Hochstrasser and Sedat, 1987; Parke and de Boni, 1992), and mammalian cells (Bourgeois et al., 1979; Stevens and Trogadis, 1986). Single and double membrane invaginations have been identified, and an association with evaginations of the nuclear envelope into the cytoplasm has also been described (Parke and de Boni, 1992). Experimental demonstration of a statistically significant association of nuclear invaginations with nucleoli has led to the suggestion that such structures might play an important role in nucleocytoplasmic transport (Bourgeois et al., 1979).
Nucleocytoplasmic transport is bidirectional and energy requiring; almost certainly the majority of this flow passes through the nuclear pore complexes (NPC)1 that stud the nuclear envelope (Dingwall and Laskey, 1992; Hinshaw et al., 1992; Rout and Wente, 1994). Despite the identification of nuclear targeting signals, little is known of the routes by which imported polypeptides move from the NPC to their specific subnuclear compartment within the nucleoplasm. Recent results have suggested that proteins capable of repeatedly shuttling between the nucleus and the cytoplasm may play an important role in these processes (Laskey and Dingwall, 1993; Schmidt-Zachmann et al., 1993). EM immunocytochemistry hints at the possibility that a nuclear skeleton may provide tracks for the shuttling movement of these proteins (Meier and Blobel, 1992).
Similarly, newly made ribosomes and mRNA probably leave the nucleus through NPCs, but their routes from the sites of intranuclear synthesis to the NPC remain obscure (Rosbash and Singer, 1993). Again, linear tracks have been observed in some experimental systems by in situ hybridization, suggesting the possible involvement of a nucleoskeleton and reinforcing a linear production line model for postsynthetic processing (Lawrence et al., 1989; Xing and Lawrence, 1993; Xing et al., 1993; Moen et al., 1995). Other experimental systems have not found evidence for a tracked route of RNA transport through the nucleoplasm, but rather, suggest a diffusion through a network of channels preferentially accessible to nascent RNA (Zachar et al., 1993). Interestingly, even in the cases where nascent transcripts formed extended intranuclear tracks, the majority of these tracks was not observed to reach the nuclear envelope (Rosbash and Singer, 1993; Xing et al., 1993, 1995). The presence of a draining network of intranuclear membrane bound channels could account for this unexpected observation.
In this study we report a combination of serial section transmission electron microscopy (TEM) and confocal laser scanning fluorescence microscopy (CLSM) experiments designed to characterize intranuclear membranes in more detail and to provide a baseline for considering the functional significance of membrane bound structures in the nucleus. Our results suggest that long, branching, intranuclear membrane channels are derived from the ER as deep, narrow invaginations of the nuclear envelope. These structures, which traverse deeply into the nucleoplasm and may pass completely through the nucleus as the nucleoplasm forms an annulus in the process, are found in all tissue culture cell types examined. Primary cells in culture also contain similar structures. The number of channels and their morphological complexity vary widely but remain characteristic for a given cell type. Examination of the channels in living cells shows that these structures are dynamic, changing position and morphology within the interphase nucleus. Some of the results described here have been presented in abstract. (Vaux, D., M. Hollinshead, and M. Fricker. 1994. The Eukaryotic Nucleus. Keystone Symposium)
Materials and Methods
Chemicals
Vectashield antioxidant mountant and fluorochrome-conjugated horse anti– mouse and horse anti–rabbit secondary antibodies were obtained from Vector Labs, Inc. (Burlingame, CA). Sterile trypsin solution, DME, and Liebowitz 15 media were obtained from GIBCO BRL (Gaithersburg, MD). Fetal calf serum was obtained from Hyclone Labs (Logan, UT), and heat inactivated for 30 min at 56°C before use. Other chemicals were analytical grade and obtained from Sigma Chemical Co. (St. Louis, MO), Merck Chemical Div. (Rahway, NJ), or BDH Chemicals Ltd. (Dagenham, Essex, England).
Fluorescent Probes
Biotinylated or directly fluorochrome conjugated Con A was obtained from Vector Labs, Inc. Fluorescein isothiocyanate-labeled immunoglobulin (FITC-IG) fraction from normal goat serum, DAPI, and propidium iodide (PI) were obtained from Sigma Chemical Co. 3,3′-dihexyloxacarbocyanine iodide (DiOC6), lysine-fixable fluorescein isothiocyanate–dextrans, fluorochrome-conjugated streptavidin, To-Pro-3 iodide, and dihydroethidium (DiHE) were obtained from Molecular Probes Inc. (Eugene, OR).
Primary Antibodies
A monoclonal antibody against a synthetic peptide corresponding to the COOH terminus of protein disulphide isomerase was obtained from culture supernatant of the 1D3 hybridoma cell line. A rabbit polyclonal antiserum against an ER membrane fraction was the generous gift of Professor Daniel Louvard (Institut Pasteur, Paris, France). A rabbit polyclonal antiserum against a peptide corresponding to the first 32 NH2-terminal amino acids of human lamin A was the generous gift of Drs. George Simos and Spyros Georgatos (European Molecular Biology Laboratory, Heidelberg, Germany). This antiserum recognizes lamins A, B, and C from a wide range of mammalian and avian species.
Cells
HeLa, Vero, NRK, 3T3, CHO, A431, J774, RAW, G8, C2, and NOR10 cell lines were maintained in culture at 37°C in 5% CO2 in DME supplemented with 10% heat inactivated fetal calf serum. Cells were removed from stock flasks with trypsin and plated onto sterile 25-mm diam coverslips at least 1 d before use. Primary cells were obtained from the peritoneal cavity of BALB/c mice by sterile lavage and placed in culture for 1–3 d before use.
Tissue Sections
Rats were perfusion fixed using a periodate/lysine/paraformaldehyde fixative, and 10 μm frozen sections from blocks of a range of tissues were collected on gelatinized glass slides, as described (the slides were provided by P. Tree and L. Darley (Sir William Dunn School of Pathology, UK), following the procedure described for mice by Hughes et al., 1995). Before use, the tissue sections were permeabilized with 0.2% (vol/vol) Triton X-100 in PBS for 15 min and quenched in fresh 50 mM ammonium chloride solution for 15 min, both at room temperature. Sections were labeled for 45 min with a mixture of 4 μM To-Pro-3 iodide and 10 μg/ml TRITC Con A in PBS at room temperature before washing four times in PBS, mounting in Vectashield, and examining by confocal microscopy.
Serial Section Electron Microscopy
For conventional microscopy, cells were washed with ice cold PBS and fixed in 0.5% glutaraldehyde in 200 mM sodium cacodylate, pH 7.4, for 30 min, and then washed in buffer and postfixed in 1% osmium tetroxide/ 1.5% potassium ferrocyanide for 60 min at room temperature. After washing in water and then staining in 0.5% mg uranyl acetate overnight at 4°C, the cells were left attached to the culture dish and flat embedded in Epon. Sections 70–90 nm thick were cut and collected either onto 50 mesh or slot grids, contrasted with lead citrate, and examined using two microscopes (EM1O at 60 kV; Philips Technologies, Cheshire, CT; EM912 at 80 kV; Zeiss, Inc., Thornwood, NY).
For the cytochemical staining of G-6-Pase, we used a minor modification (Griffiths et al., 1983) of the procedure developed by Wachstein and Meisel (1956). To optimize staining of AtT-20 cells for G-6-Pase we found it necessary to fix the cells in <1% glutaraldehyde and to incubate the fixed cells with the substrate solution for 2 h at 37°C.
Cryoimmunoelectron Microscopy
HeLa cells were fixed for 10 min at 4°C in 4% paraformaldehyde in 250 mM Hepes (pH 7.4) and then transferred into 8% paraformaldehyde in Hepes at room temperature for at least 20 min, removed from the plastic culture dish with a rubber policeman, and spun to a pellet. Fresh 8% paraformaldehyde was added to cross-link the cell pellet for another 30 min at room temperature. They were then washed in Hepes, infiltrated with sucrose, and frozen in liquid nitrogen. Thin sections of frozen cells were cut and labeled according to published procedures (Tokuyasu, 1980; Griffiths et al., 1984).
Lectin and Antibody Labeling of Cells
Cells on 25-mm coverslips were washed twice with Ca2+ and Mg2+ free PBS at room temperature and then fixed either in methanol at −20°C for 6 min or in 3% paraformaldehyde (PFA) in PBS for 10 min at room temperature. Coverslips were then washed in PBS and stored at 4°C for up to 3 d before labeling. Before labeling, PFA fixed samples were permeabilized with 0.2% NP-40 in PBS for 5 min at room temperature. For indirect lectin labeling, coverslips were first incubated with 10 μg/ml of biotinylated lectin in PBS for 30–60 min at room temperature, washed four times in PBS, and then incubated in 1 μg/ml fluorochrome conjugated streptavidin in PBS for 30–60 min at room temperature. After washing, coverslips were mounted using a glycerol based mountant containing anti-oxidants to minimize photobleaching (Vectashield; Vector Labs, Inc.). For direct labeling, fluorochrome conjugated lectins were used at 5 or 10 μg/ml in PBS for 30 min at room temperature. Indirect immunofluorescence was carried out as described previously. Most samples were also counterstained with 0.1 μg/ml DAPI for 5 min during the final wash before mounting. Samples were examined using either a microscope equipped for epifluorescence illumination and a low light level CCD camera (Axiophot; Zeiss Inc.) or a CLSM.
Scrape Loading of Cells
HeLa or NRK cells were plated in 60-mm diam tissue culture dishes and allowed to grow to 90% confluence. The monolayers were washed twice with ice cold PBS, and the cells were scraped from the dish in 0.5 ml of ice cold PBS containing 10 mg/ml of FITC conjugated normal goat immunoglobulin (150) using a soft rubber policeman. The cells were allowed to stand on ice for 5 min and then diluted in 10 ml ice cold complete medium and pelleted by centrifugation at 400 g for 5 min. The cells were resuspended in complete culture medium and plated onto 25-mm diam coverslips. The samples were either examined live or after fixation in 4% PFA.
Low Light Level Fluorescence Microscopy
The microscope (Axiophot; Zeiss Inc.) was equipped with a 50 W mercury arc lamp for epifluorescence illumination, a 100× 1.3 NA Plan Neofluar oil-immersion objective, and an Optivar set at 1.25×. Images were captured using a cooled CCD camera (CDC 242 Peltier; Hamamatsu Phototonics K.K., Hamamatsu City, Japan) under the control of software (BioVision; ImproVision Ltd., Warwick, UK) running on a Macintosh Quadra 900 computer. Optimal settings were obtained by adjustment of camera gain and offset and on-chip integration time, using the intensity histogram to ensure that the range of fluorescence intensities fell within an 8 bit recordable range. Settings were optimized individually for each fluorochrome and then held constant for all samples in a given experiment. Images were stored as 8 bit greyscale files. Serial Z axis images separated by 0.5-μm steps were collected using a motor driven focus system under computer control (BioVision, ImproVision Ltd.). The 8 bit images from each fluorochrome were median filtered with a 3 × 3 kernel and merged using the 24 bit merge capability of Adobe Photoshop. No other processing was used in the images displayed here. Vitally stained cells were also examined with the low light level CCD system attached to an inverted microscope (Axiovert; Zeiss Inc.) with a temperature controlled stage.
Confocal Microscopy
Two confocal microscope systems were used. The first was based on a scan head (MRC600; BioRad Laboratories, Cambridge, MA) attached to a Nikon Diaphot TMD inverted microscope with modification of the laser excitation path to provide co-aligned beams from a number of different lasers including 488-nm line (25 mW Argon-ion; Ion Laser Technologies Ltd., Salt Lake City, UT) and 543-nm line (1.3 mW Gre-Ne; Melles-Griot, Rochester, NY) introduced through single-mode fiber optics (Fricker and White, 1992; Fricker et al., 1994). Neutral density filters were used to roughly balance the intensities of the two beams. Fluorescence emissions were collected at 540 ± 15 nm and >600 nm using a Nikon 60× 1.4 NA Plan apochromat oil-immersion objective.
The second system comprised an MRC 1000 scan head attached to a Nikon Diaphot 300 inverted microscope with excitation at 488 nm, 568 nm, and 647 nm (15 mW Argon-Krypton; Ion Laser Technologies Ltd). Fluorescence emissions were collected at 522 ± 35 nm for fluorescein, 605 ± 32 nm for rhodamine, Texas red, and propidium iodide, and 680 ± 32 nm for To-Pro-3 using a Nikon 60× 1.4 NA Plan Apochromat oil-immersion objective.
Three dimensional (3-D) datasets were collected with an XY pixel spacing of 0.05–0.09 μm/pixel for fixed cells and 0.18 μm/pixel for live cell recordings and a focus motor (z axis) increment of 0.4 or 0.5 μm. Individual frames were averaged or Kalman filtered over four to eight frames. The gain required to visualize the intranuclear structures was often sufficient to saturate signal from the ER outside the nucleus.
Image Reconstruction and Display
3-D confocal images were median filtered using a 3 × 3 box and visualized using software (ThruView Plus; BioRad Laboratories) as a rotation series of height-coded projections (White, 1995). Typically the interior of the nucleus was viewed from the coverslip side, omitting the first two to three sections at the base of the cell that contained the thin layer of peripheral cytoplasm and nuclear envelope. An intensity threshold was manually selected to retain detail of the intranuclear channels. The first occurrence of a pixel above this threshold was recorded along the line of sight for each angle in the rotation series and displayed as a height-coded image, where intensity represents the Z position of the pixel, white nearest the viewer, black furthest away (White, 1995). Smooth animations for inspection were calculated with rotation steps of 4° and a constant tilt angle of 30°. The Z-stretch factor required to compensate for the asymmetric sampling in X, Y, and Z was calculated from the focus motor increment to account for the difference in refractive index between the oil-immersion fluid, coverslip, and mounting medium (White et al., 1996). This correction factor was calculated using a weighted average of rays across the full NA of the lens. An average factor of 0.829 was used for cells in Vectashield, and a factor of 0.7 was used for live cells. Voxel dimensions in these images are thus approximately scaled as a 1 × 1 × 1 aspect ratio.
Vital Staining and Live Cell Confocal Microscopy
For live cell analysis, small tissue culture dishes were prepared by gluing 1-cm deep Perspex cylinders (OD 25 mm, ID 19 mm) onto 25-mm diam coverslips using a silicone liquid adhesive and sterilizing them in 100% ethanol before use. This arrangement permitted the use of oil immersion objectives on an inverted microscope with a temperature controlled stage for either low light level CCD or laser scanning confocal microscopy. The cells were transferred to Liebowitz 15 medium containing 10% fetal calf serum (L10) to remove the requirement for a 5% CO2 atmosphere during examination. Similar results were obtained in short term experiments by sealing a second coverslip to the top of the dish with vacuum grease after equilibration of the contents at 5% CO2, but this system maintains pH for only 30 min. Preliminary experiments using <20 mM Hepes to buffer normal growth medium revealed substantial changes in cell morphology, as described previously (Bowers and Dahm, 1993), which were not seen when L10 was used.
Cells were labeled with dihydroethidium (diHE) at 1 μg/ml in complete culture medium for 90 min at 37°C, DiOC6 at 0.5 μg/ml in complete culture medium for 10 min at 37°C, and then examined with the CLSM at 37°C. Complete 3-D data sets were collected from a field, ∼70 × 50 μm containing a small number of cells (usually 5–10). Taking care to minimize movement of the stage and the cell monolayer, the modified tissue culture dish was then perfused with PBS, followed by methanol at −20°C, which was left on the cells for 6 min (i.e., comparable to the fixation conditions used to prepare fixed cells for labeling). 3-D datasets were collected during fixation. The DiOC6 signal was extracted completely from the cells by the methanol within 30 sec. After fixation, the dish was perfused with PBS to wash the monolayer. The cells were then labeled using fluorochromeconjugated Con A that was added through the perfusion cannula. Further 3-D datasets were collected from the original field of cells during the next 20 min as the Con A stain gradually developed.
For time course experiments, live cells were prepared and labeled as above, and then imaged using the 488 nm line of a 15 mW krypton–argon laser on a microscope (MRC 1000; BioRad Laboratories) running in low power mode and with maximum attenuation with neutral density filters (equivalent to 0.3% transmission for full power mode) it was possible to collect up to five complete 3-D images (20 sections, 4 frame Kalman filter) from a single cell before the signal intensity became unacceptably low.
Results
TEM Identifies Tubular Membrane Profiles within the Nucleoplasm
Examination of tissue culture cells sectioned parallel to the growth substrate after in situ embedding revealed frequent membrane profiles within nuclei. Fig. 1, a–j shows 10 consecutive 70-nm sections through an oriented 3T3 nucleus (a low magnification view is shown in k). The double membraned figure in the center can be followed through all sections. a grazes the nuclear envelope, giving en face views of several nuclear pore complexes; the dark structure to the left is an HRP-loaded endosome within the cytoplasm. The predominant feature running through the series is a double membraned channel surrounded by an electron dense coat and with an electron lucent core. Additional, finer strands of membrane are associated with this larger structure (for example, Fig. 1, g–j). Small electron dense particles may be found attached to the inner membrane of the lucent core (Fig. 1 e).
Figure 1.
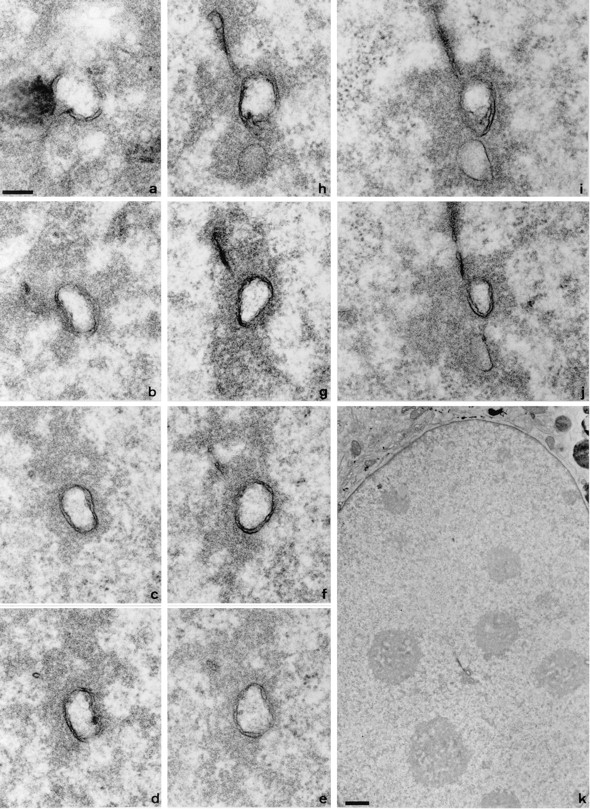
Serial section EM reveals complex membrane-bound networks within the nucleus. Consecutive 70-nm serial sections from a region of a 3T3 cell nucleus sectioned parallel to the growth substrate are shown in a to j. Section a begins at the nuclear envelope; note the en face nuclear pore complexes and the dark mass of an HRP-loaded endosome in the cytoplasm to the left of the invaginating membranes. The position of the channel within the nucleus is shown in the low magnification overview in k; clearly visible within the nucleoplasm are nucleoli that are not apparently associated with this channel. Bars: (a–j) 200 nm; (k) 1 μm.
A double membrane ring was the predominant morphology of membrane channel in 3T3, NRK, A431, and HeLa cells (Fig. 2). The annular double membranes frequently contain local fenestrations indistinguishable from nuclear pores sectioned transversely at the nuclear envelope (compare Fig. 2 a and the higher magnification inset in d with the nuclear envelope shown in e).
Figure 2.
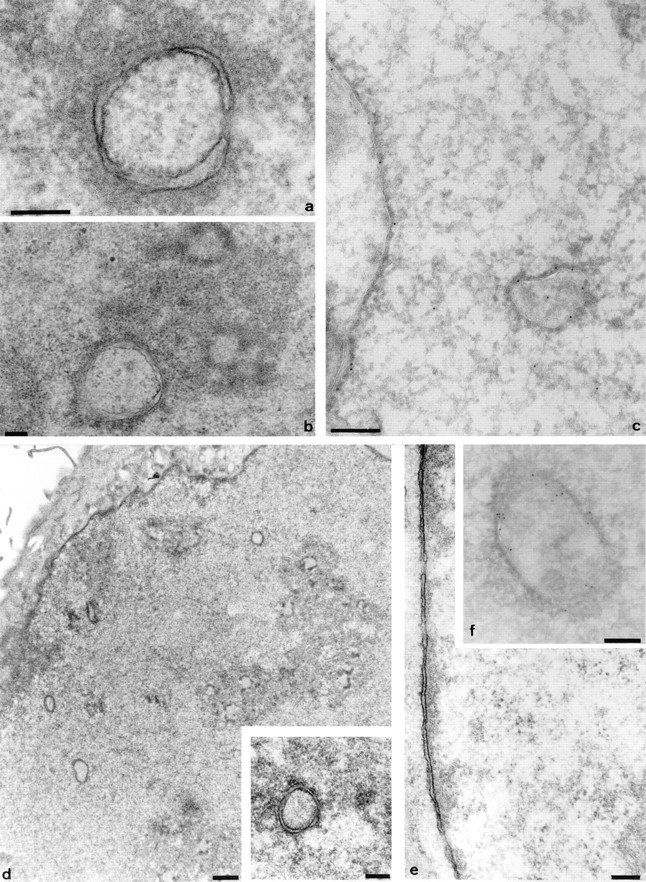
The intranuclear channels in a range of cell types contain nuclear pore complexes and are associated with an electron dense halo in the nucleoplasm. Representative profiles of channels from 3T3 cells (a), NRK cells (b), A431 cells (d and inset), and HeLa cells (f) are shown. Double membrane structures are clearly visible, together with local fenestrations, and comparable to the appearance of the nuclear pore complexes in the nuclear envelope of the 3T3 nucleus shown in e. The electron lucent core is also seen to contain small circular or crescent shaped features, best seen within the channel in b. Electron dense material in the nucleoplasm is associated with the channels (a, b and d, inset). In addition, channel association with nucleoli is shown for NRK cells (b). Thawed frozen thin section colloidal gold immunocytochemistry confirms that the channels are associated with strong immunoreactivity for lamins (c) and protein disulphide isomerase (f), as shown by the 9-nm gold particles decorating the intranuclear channels in these HeLa nuclei. Bars: 200 nm; d, 1 μm.
To test the possibility that these tubular membrane figures form extensions of ER membrane, we studied cells labeled histochemically for an ER marker, glucose-6-phosphatase (G-6-Pase). The lumen between the two membranes of the intranuclear tubules is decorated with the histochemical reaction product for G-6-Pase, confirming an ER origin (Fig. 3). Both short stubs of ER extending into the nucleoplasm and long channels that persist as double membrane extensions throughout the sections can be seen. The latter are distinct from the plate-like infoldings of the nuclear envelope that are characteristic of certain cell types, such as the polymorphonuclear leukocyte. The histochemical reaction product fills the lumen of the ER, the intermembrane space of the nuclear envelope, and the intranuclear channels, but is excluded from nuclear pores in both the nuclear envelope and the intranuclear channels. Shallow angle sections through the margins of nuclear channels (e.g., Fig. 3 a, top left) show that the number of nuclear pores per unit area in the channels is comparable to that in the bulk nuclear envelope (Fig. 3 d, top left).
Figure 3.
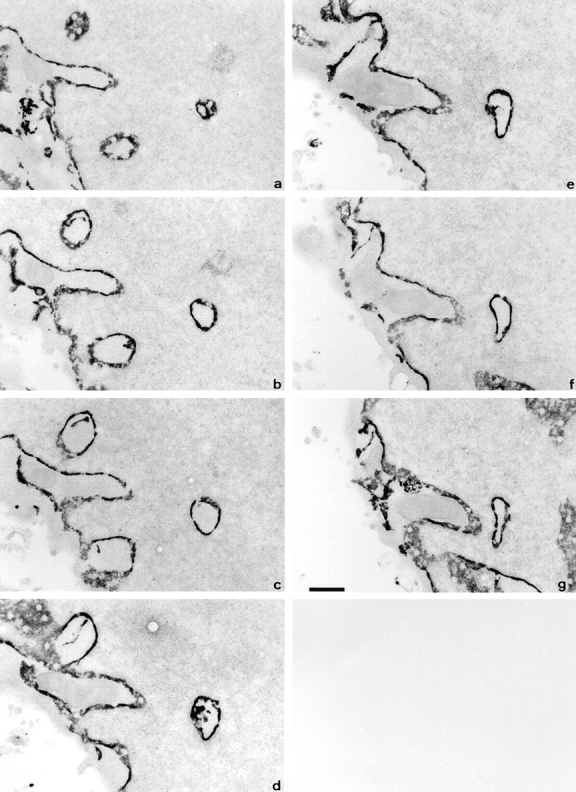
The intranuclear channels are bounded by a double membrane enclosing a G-6-Pase positive lumen. Consecutive 70 nm serial sections of part of an ATt20 nucleus sectioned parallel to the growth substrate after histochemical visualization of G-6Pase activity. Dense reaction product fills the space between the inner and outer nuclear membranes but is excluded from nuclear pore complexes. Similar reaction product is seen within the channels in the nucleoplasm. Bar, 500 nm.
The high electron density of the lumenal reaction product highlights the presence of fronds of membrane extending into the lumen of the channel as discrete clumps on the wall of a long channel (Fig. 3 d). Sometimes they appear to invade the core of a channel as a fine finger of ER from the cytoplasm, as can be well seen in the short channel at top left (Fig. 3, a–e).
Intranuclear Con A Binding Sites Form Linear Arrays
Fluorochrome conjugated Con A labels the endoplasmic reticulum and nuclear envelope. Confocal microscopy showed it also labels a limited number of nucleoplasmic sites in all cell lines examined (Fig. 4). This Con A labeling was completed via mannose-terminated oligosaccharides, because binding was abolished by competition with α-methyl mannoside (not shown).
Figure 4.
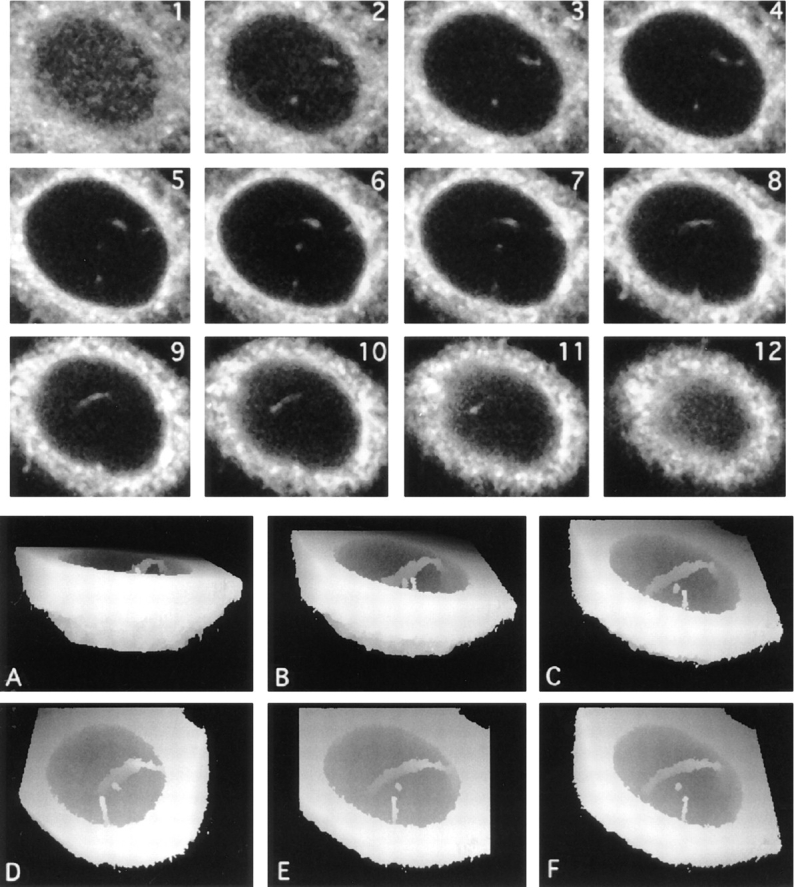
Intranuclear channels labeled with lectins recognizing ER-type oligosaccharide side chains and visualized by confocal microscopy and 3-D reconstruction. Serial optical sections of a Con-A–labeled HeLa nucleus were collected at 0.5 μm intervals by CLSM from the base of the cell (1–12), forming a 3-D XYZ image. Fluorescent structures are clearly visible within the nucleus in successive optical sections. To visualize the overall morphology of the channels, the sections nearest the base of the cell were discarded and views projected at increasing tilt angles (30° increments; A–C and F) and rotation angles (24° increments; D–F). In these height reconstructions, structures above a threshold intensity nearest the viewer are coded as white for each tilt or rotation angle and also mask underlying structures. The major nuclear channel traverses the nucleoplasm, contacting the nuclear envelope at two locations. In addition, a second, shorter tube that terminates within the nucleus is also visible.
The dataset illustrated in Fig. 4, consisting of 12 XY planes, was used to construct a height-encoded, 3-D view of nuclear volume seen from below with the basal cytoplasm removed. Different tilted views of the reconstruction confirmed that the channels were often orthogonal or near-orthogonal to the growth substrate. In addition, near horizontal Con A positive nucleoplasmic channels were also seen.
Intranuclear Membranes Are Found in Many Cell Types
To see how widespread intranuclear tubules were, we examined a range of cell types by labeling with Con A, followed by confocal microscopy and 3-D visualization. Labeled channels were found in all cell types tested, at varying frequencies. In non-synchronized populations not all cells contained detectable intranuclear Con A positive structures. However, by increasing the sensitivity (i.e., by using conditions that gave saturating levels of signal from the ER and nuclear envelope), we could detect more intranuclear structures. This suggests that some nuclear structures are either smaller than many cytoplasmic ER features or that they contain fewer accessible Con A binding sites. 3-D reconstruction of nucleoplasmic Con A positive channels from several cell types showed that some cells contained only a single tubule, and others contained a complex branching network. Fig. 5 illustrates the various degrees of complexity of the network seen in five different cell types.
Figure 5.
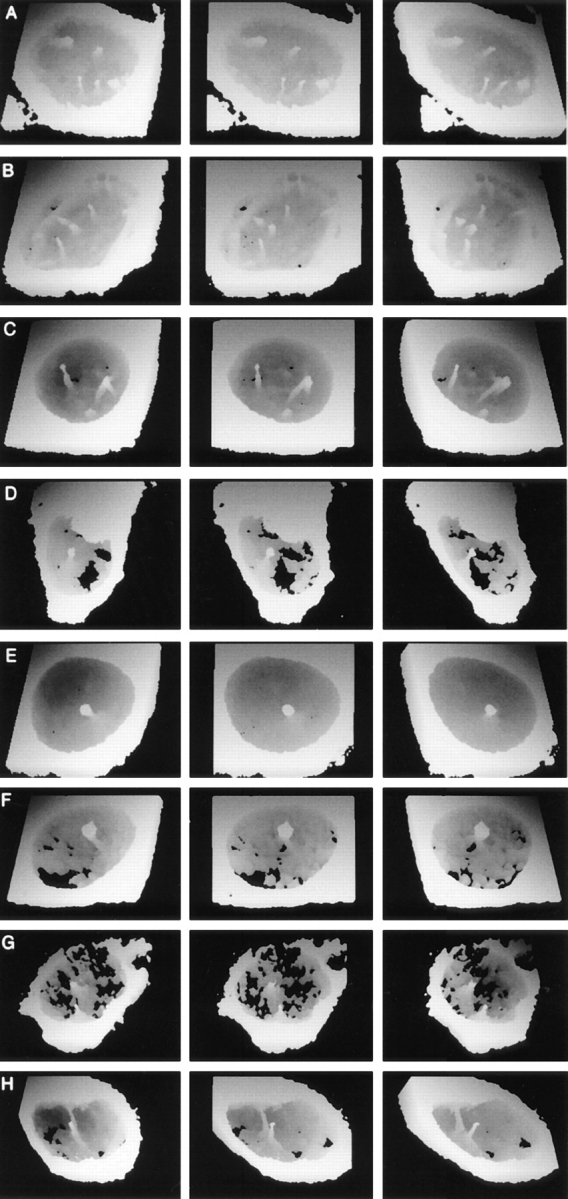
Intranuclear channels vary in number and morphology among different cell types, contain ER soluble resident proteins and ER membrane proteins, and enclose a cytosolic core. Rotation series of height coded 3-D views at 24° intervals of 3T3 (A), CHO (B), G8 (C), PEC (D), and NRK (E) cells labeled with fluorescent tagged Con A are shown. Reconstructions were made as shown in Fig. 4. The nuclear channel morphology was characteristic for each cell type and ranged from multiple branched channels (e.g., G8 cells in C) to single unbranched channels (e.g., PECs and NRK cells in D and E, respectively). In addition to being Con A positive structures, channels were also visible in an NRK nucleus labeled with a monoclonal antibody against the soluble resident ER protein, protein disulphide isomerase (F), a HeLa nucleus labeled with a polyclonal antiserum against ER membrane proteins (G), and a HeLa nucleus after scrape loading of the cell with a 150-kD fluorescent tracer (H).
NRK cells (Fig. 5 E) tend to have few intranuclear channels, almost invariably oriented vertically to the growth substrate and often offset within the nucleoplasm. HeLa cells (Fig. 4) tend to have a larger number of channels and more channels that are not vertically oriented to the growth substrate. Primary mouse peritoneal exudate cells in culture also show intranuclear membrane channels, often single and usually vertical to the growth substrate (Fig. 5 D). 3T3, CHO, and G8 cells (Fig. 5, A–C, respectively) have a more complex, frequently branched network.
We counted channels in complete 3-D datasets of >80 nuclei of each cell type (Fig. 6). We used a strict definition of a channel. It must extend at least 1.5 μm in X, Y, or Z to exclude short indentations found in some cell types (especially along the basal–nuclear margin) and residual “tails” of fluorescence along the Z axis from intense labeling of the nuclear envelope or cytoplasmic structures. More than 60% of NRK cells do not have such channels, whereas >90% of CHO cells contain at least one, with half having three or more. We found no cell type with fewer channels than NRK cells, nor any cell type with a more abundant array than CHO cells.
Figure 6.

Frequency histogram of nuclear channel distribution in a range of cell types. Cells were labeled with fluorochrome tagged Con A and 3-D datasets collected for >80 nuclei for each cell type. The number of channels defined as an intranuclear labeled structure at least 1.5 μm in x, y, or z was counted by inspection of the serial optical sections.
The differences were reproducible over time, as shown by the independent counts for NRK cells carried out 4 mo apart (Fig. 6, NRK-1 and NRK-2).
The presence of channels in >90% of the cells in an unsynchronized CHO population suggested that these structures could not be restricted to early postmitotic cells in G1. Double labeling of unsynchronized cell populations with Con A and an antibody to the cell cycle–dependent antigen, PCNA, confirmed that the abundance of channels found in S phase nuclei was not lower than the abundance measured over the entire interphase population (data not shown; a complete analysis of the cell cycle dependence of the nuclear channels as described here will be presented elsewhere (Vaux, D., M. Hollinshead, N. White, and M. Fricker, manuscript in preparation).
Channels were found in the nuclei of a predominantly differentiated, postmitotic primary cell population (3 d adherent mononuclear cells cultured from peritoneal lavage of mice; Fig. 5 D) as well as cells from established cell lines (Fig. 5, A–C and E). Similar structures were also seen in the nuclei of normal rat hepatocytes in tissue sections (Fig. 7). Varying numbers of channels per nucleus were seen in 10-μm sections of perfusion-fixed rat liver; the figure shows nuclei with a channel number ranging from 0 to 3. In this specimen the mitotic index for hepatocyte nuclei was <0.2%.
Figure 7.
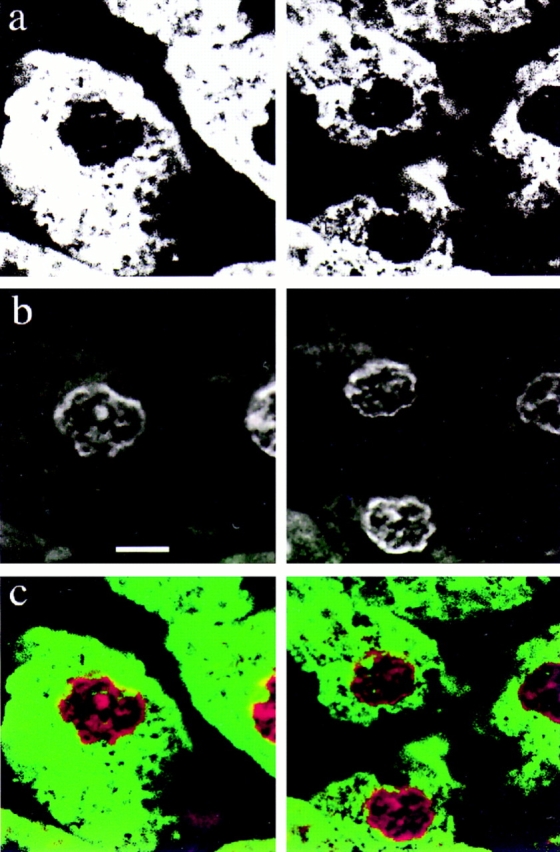
Nuclear channels are present in normal rat hepatocytes. Perfusion-fixed sections of rat liver were labeled with fluorochrome-tagged Con A in a and the nucleic acid stain To-Pro-3 in b. A two color merge of the data is shown in c. Note the association between the nucleolus and two channels in the left hand cell. Bar, 5 μm.
Composition of the Channels
The serial section TEM, G-6-Pase labeling, and presence of nuclear pores together suggest that the nuclear envelope invaginates to form these structures. If so, the invaginating membranes should contain ER membrane proteins, the annular space between the double membranes should contain soluble ER proteins, and the central lumen should correspond to the cytoplasm. The nuclear channels could be labeled with a monoclonal antibody directed against protein disulphide isomerase (PDI), a soluble resident enzyme of the ER (Fig. 5 F); similar structures were labeled by an antiserum recognizing ER membrane proteins (Fig. 5 G).
To test the possibility that the cores of the intranuclear channels are not only topologically equivalent to the cytoplasm but are in continuity with it, we preloaded the cytoplasm with a high molecular mass (150 kD) fluorescent tracer that was too large to pass through the nuclear pores. The channels were filled with the tracer (Fig. 5 H), confirming that they were extensions of the cytoplasm.
Double labeling experiments confirmed that PDI (Fig. 8 A) was contained within the structures that bound Con A (Fig. 8 B; compare arrows in A and B). PDI was also found within the intermembrane space of channels seen by EM (Fig. 2 f), with immunolabeling gold particles decorating the nucleoplasmic membrane–bound channels.
Figure 8.

Intranuclear PDI immunoreactivity co-localizes with Con-A–labeled channels. The figure shows separate eight-bit grey scale images collected from a triple labeled NRK cell using a cooled CCD camera. A shows the distribution of the PDI signal in the fluorescein channel. B shows the Con A signal in the rhodamine channel, and C shows the nucleic acid labeled with DAPI. D is a 24-bit merge of the first three channels. The arrowhead points to an intranuclear structure that can be followed through a number of focal planes in which PDI and Con A reactivity colocalize.
If the channels are invaginations of the whole nuclear envelope, then the electron dense material on the nucleoplasmic side of the double membrane seen by EM (Figs. 1–3) might be similar to peripheral heterochromatin and include a structure similar to the nuclear lamina. Confocal microscopy confirmed that the channels defined by Con A (Fig. 9 a) are associated with lamins (Fig. 9 b), often in areas poor in nucleic acids (Fig. 9 c). These results were confirmed by colloidal gold immunocytochemistry at the EM level (Fig. 2 c).
Figure 9.

Con A–labeled channels are associated with lamin immunoreactivity. Maximum projections of six consecutive optical sections at 0.3 μm Z interval from a triple labeled HeLa nucleus are shown; a shows Con A reactivity. b shows polyclonal antilamin reactivity, and c shows nucleic acid labeling with Yo-Pro 1. Associated XZ (above) and YZ (right) projections along the lines indicated by the black arrowheads are also shown. The rotation series of height-coded, tilted 3-D views at 24° intervals are presented for Con A labeling in d–f and lamin labeling in g–i. Each of the Con A sites in the nucleus is colocalized with sites of lamin reactivity, but note also that there are additional intranuclear lamin reactive sites that do not have associated Con A reactivity. Bar, 5 μm.
Additional sites of intranuclear lamin staining, unassociated with membrane or Con A labeling, were also seen (Fig. 9, a and b). These additional sites exhibited various morphologies including punctate sites and extended linear tracks, as is clearly demonstrated in the height-encoded reconstructions of the entire datasets for Con A labeling (Fig. 9, d–f) and lamin staining (Fig. 9, g–i).
Association of Some Channels with Nucleoli
This was confirmed by confocal microscopy, after staining nucleic acid with propidium iodide and channels with Con A. The channels often ran through holes in nucleic acid staining (Fig. 10, A and B) and ran up to, or past, nucleoli (Fig. 10 C). Electron microscopy suggested that some of the channels were closely associated with nucleoli (Fig. 2 b). Double labeling of tissue sections of normal liver also demonstrated an association between hepatocyte nucleoli and Con A–labeled channels (Fig. 7, merged view of the left hand cell).
Figure 10.

Some channels that terminate within the nucleus are associated with nucleoli. a shows Con A reactivity; b shows nucleic acid labeling with propidium iodide. c shows a two color merge of the data from a and b. Two XZ views across the nucleus at the level of the channels (arrowheads) are also shown.
However, not all nucleoli show a closely associated channel (Fig. 10 C, XY and XZ views), and some channels show no apparent association with nucleoli (by light microscopy in Fig. 10 C, and by EM in Fig. 1 k).
Dynamic Channels are Present in Live Cells
We next excluded the possibility that the channels were artifacts of cell preparation by visualizing them in living cells. The presence of channels in cells scrape-loaded with a 150-kD fluorescent tracer before fixation provided preliminary evidence that such structures are present in live cells (Fig. 5 H). NRK, BHK, or HeLa cells in culture were labeled with dihydroethidium and with the lipophilic dye, DiOC6, whichis selectively concentrated into cellular membranes (including the ER). The ER, the nuclear envelope (Fig. 11 a), and intranuclear structures (Fig. 11, a and b) can then be seen in confocal optical sections cut through the center of the nucleus in living cells. The labeling of nucleic acids with dihydroethidium confirms that the confocal optical sections are indeed through the center of the nuclei (Fig. 11, e and f).
Figure 11.

Nuclear channels are detectable in live cells and correspond to the Con A–labeled structures seen in fixed cells. a shows a single plane from the dataset for two DiOC6 labeled live cells; e shows the dihydroethidium label in the same plane. b and f are from data collected 20 min later. c shows the loss of the DiOC6 signal at the end of methanol fixation, while g shows that the cells remain intact and the dihydroethidium label persists, although reduced in intensity. d shows the same cells and plane after 20 min of Con A labeling. h–j show sequential rotations of reconstructions of DiOC6 labeled cells, and underneath in k–m, respectively, the same cell is shown after Con A labeling. Cutaway views of a reconstructed DiOC6 − labeled HeLa nucleus from a separate experiment are shown at three time points in n–p. Note that the morphology of the larger left hand tube alters over the 35-min period, and the smaller channel shows a progressively increasing separation from the large channel. Bars: (a–m) 25 μm; (n–p) 2 μm.
We next confirmed that the intranuclear membranes that label with DiOC6 are the same structures that were visualized after fixation using Con A, by a sequential labeling experiment carried out on the stage of the confocal microscope. Fig. 11: (a) shows a single XY plane from the dataset for two DiOC6 labeled cells; (e) shows the dihydroethidium label in the same plane. Fig. 11 (b and f) are from data collected 20 min later. The upper nucleus has changed considerably in shape. After methanol fixation, the DiOC6 signal is lost (c), but the cells remain intact; and some dihydroethidium label persists (g). After Con A labeling, the intranuclear channels originally labeled by DiOC6 are seen to be labeled with Con A. The result shown for a single plane in Fig. 11 (a–g) is confirmed by 3-D reconstruction in the lower part of Fig. 11. The DiOC6 labeled structures found in the nuclei of live cells (Fig. 11, h–j) correspond to the Con A positive structures found in fixed permeabilized cells (Fig. 11, k–m); fixation causes little apparent change to their morphology at the light microscope level.
We next observed the dynamics of the channels by incubating cells with DiOC6 and collecting repeated complete 3-D images over 35 min (Fig. 11, n–p); the channels clearly persist over this timescale. Some channels do not change position within the nucleus but do alter their morphology, while others alter their position.
Discussion
The results reported here suggest that the nucleoplasm of mammalian cells contains long, dynamic, branching membrane channels that are derived from the ER as deep, narrow invaginations of both membranes of the nuclear envelope. Nuclear invaginations with a range of morphologies have been reported in EM studies on mammalian nuclei (Bourgeois et al., 1979; Dupuy-Coin et al., 1986; Stevens and Trogadis, 1986), in insect nuclei (Hochstrasser and Sedat, 1987; Parke and de Boni, 1992), and in plant nuclei (Dickinson and Bell, 1972; Li and Dickinson, 1987).
Single image planes, whether optical sections in the confocal microscope or thin sections in the TEM, do not allow unambiguous identification of tubular channels. Consequently, all data presented here have been analyzed in three dimensions, after reconstruction from serial sections obtained using the confocal microscope, or TEM. This approach permits the unambiguous identification of tubular incursions, distinct from flat plates or folds, that would be difficult to identify without such a 3-D analysis. Furthermore, the nuclear channels are seen in live cells and so cannot be artifacts of the fixation or immunocytochemistry.
3-D analysis demonstrates that channels are entirely distinct from the folds that render the nuclei of some cell types lobulated (e.g., polymorphonuclear leukocytes). In particular, channels are unrelated to the constricting furrows in the surface of the nuclear envelope that may be associated with bands of intermediate filaments (Kamei, 1994). These latter structures are characterized by loops of bundled intermediate filaments constricting the nuclear envelope; they do not form independent intranuclear membrane profiles separated from the nuclear envelope, even in confocal reconstructions (Kamei, 1994). The absence of detectable intermediate filaments may also result in irregularities of the nuclear envelope, characterized as prominent, multiple infoldings (Sarria et al., 1994). The infoldings found in these vimentin-negative cells are clearly distinct from the deep, narrow channels described in this study.
The channels described here all consist of a cytoplasmic invagination bounded by a double membrane. This makes them quite distinct from the structures described in insect nuclei, where the invaginations are of the inner nuclear membrane only, producing a structure with a core in continuity with the ER (Hochstrasser and Sedat, 1987; Parke and de Boni, 1992). Furthermore, detailed statistical analysis demonstrates that the single-membrane invaginations found in Drosophila nuclei are always associated with an adjacent double-membraned evagination of the nuclear envelope into the cytoplasm (Parke and de Boni, 1992). These differences, together with the observation that the core of the Drosophila invaginations are frequently filled with large electron-dense granules (Hochstrasser and Sedat, 1987) not seen in mammalian cells in this study, suggest that the structures we describe are unlikely to be closely related to the invaginations described in Drosophila.
Double-membraned invaginations of the nuclear envelope in plant cells have been described (Dickinson and Bell, 1972; Li and Dickinson, 1987). These structures are developmentally restricted to a short interval in postmeiotic gymnosperm microspores and have several features that distinguish them from the channels described here. Firstly, they are very short invaginations (maximum length, 0.9 μm); secondly, they are very numerous (200–400 per nucleus) and, thirdly, they are composed of membranes devoid of nuclear pores (Li and Dickinson, 1987). These features lead us to conclude that these plant cell structures are unlikely to be related to the large double-membraned channels found in mammalian nuclei.
Invaginations of the mammalian nuclear envelope may be single membraned or double membraned (Bourgeois et al., 1979). Double-membraned nuclear invaginations have previously been described in serial section EM studies as “nucleolar channels” because of an invariant association with nucleoli (Bourgeois et al., 1979; Dupuy-Coin et al., 1986). The results presented here confirm the presence of invaginating channels terminating in, or adjacent to, nucleoli in nuclei from a variety of cell types. However, it is clear that a combined CLSM and serial TEM analysis demonstrates additional categories of intranuclear invagination, in some cases unrelated to nucleoli and even in some cases having no intranuclear termination. Some channels (tubes) terminate within the nucleoplasm, others (tunnels) pass entirely through; some nuclei contain none, others many; some are simple, others branched. These overall features may be seen by confocal microscopy. More detailed structure is revealed by serial TEM sections, where the double membrane, the embedded nuclear pore complexes, the fine membrane processes, and the closed end of the channel are all clearly visible. One of the most unexpected findings of the present study is channels that traverse the nucleoplasm and have connections to two separate points on the nuclear envelope. This novel morphology means that the nucleoplasm forms an annulus around the channel. This is topologically quite distinct from any sheet- or plate-like infolding of the nuclear envelope. In particular, these traversing channels cannot be removed from the interphase nucleus without a membrane fusion or fission event.
Composition of the Channels
EM of serial sections showed direct connections between the double-membraned intranuclear tubes and the double membrane of the nuclear envelope. The double membranes of the nuclear envelope and the channels both contain nuclear pores. This topology is consistent with a deep invagination of both the outer and inner nuclear membranes. Three lines of evidence support this view. Firstly, both the ER lumen and the lumenal space between the two membranes forming the boundary of the channels contain protein disulphide isomerase (by CLSM, Fig. 5, and by EM immunolabeling, Fig. 2), Con A binding sites (Figs. 4 and 5), and glucose 6 phosphatase (Fig. 3). Secondly, the membrane proteins of the ER were found in the peripheral ER, the bulk nuclear envelope, and the nuclear channels (Fig. 5). Thirdly, membranes of both ER and the nuclear channels accumulated the lipophilic dye DiOC6 (Fig. 11). These results identify the channels as deep invaginations of both inner and outer nuclear membranes, but cannot exclude the possibility that the composition of the membranes forming the channel differs in detail from that of the bulk nuclear envelope.
Our data strongly suggest that the majority of tubular membranes within the nucleus contain a core of material that is continuous with the bulk of the cytoplasm. The space at the center of the double-membraned channels has the electron density of cytoplasm in EM images, and such an identity would be topologically plausible. This space could be separate from bulk cytoplasm or accessible to it, depending on the organization of the junctions between the intranuclear channels and the nuclear envelope. High molecular weight tracers that are too large to enter the nucleoplasm through nuclear pores have access to the lumen of the channels after scrape-loading into the cytoplasm, confirming that the lumen is continuous with the cytoplasm. The lumen of nuclear channels is not always empty; ribosomes, small vesicles, and cytoskeletal elements have all been seen. Thus, one effect of these structures is to bring cytoplasmic space closer to the interior of the nucleus.
The nucleoplasm around the nuclear channels has an increased electron density similar to that seen in peripheral heterochromatin adjacent to the nuclear envelope (Fig. 1). Light and EM immunocytochemistry confirm that in both cases these electron-dense areas include a lamina containing nuclear lamins (Figs. 2 and 9). Several recent reports suggest that the nuclear lamin proteins are not confined to a lamina at the nuclear periphery, but may be found at focal sites throughout the nucleoplasm (Goldman et al., 1992; Bridger et al., 1993). We confirm the presence of focal lamin accumulations in the nucleoplasm and show that some of these sites correspond to channels, while others do not (Fig. 9), confirming that there are at least two classes of focal lamin sites within the nucleus, besides the peripheral nuclear lamina (Belmont et al., 1993; Bridger et al., 1993). A functional significance for intranuclear lamin B sites has been suggested by colocalization of these sites with sites of DNA replication in mid to late S phase (Moir et al., 1994). Interestingly, this association is confined to lamin B even though intranuclear lamin A–C foci are also found (Goldman et al., 1992), suggesting a still higher order of complexity for intranuclear distribution of lamins.
Distribution and Dynamics of the Channels
Nuclear channels were found in all cell types examined, including cultured primary cells (macrophages explanted from the peritoneal cavity). The number of channels and their morphological complexity vary widely but remain characteristic for a given cell type and do not change significantly over time (Fig. 6). The organization of the channels with respect to the growth substrate is characteristic for each cell type, and this also remains stable for a given cell type over time in culture. The abundance of channels orientated vertically with respect to the growth substrate could depend in part on the better visibility of such structures by light microscopy, but this cannot be the whole explanation since the same bias was seen in oriented serial EM section analyses.
Channels are not restricted to transformed cell lines in culture, as they are also found in cultured primary peritoneal exudate cells (Fig. 5 D), which are predominantly postmitotic, differentiated macrophages in these preparations, and in nuclei of normal hepatocytes in tissue sections (Fig. 7). Furthermore, the presence of channels in these postmitotic nuclei suggests that such structures are maintained in cells that are not cycling.
Within individual tissue culture cells, although the channels may persist for hours in interphase nuclei, they may also rapidly change their morphology and position within the nucleoplasm on a timescale of minutes. We cannot exclude the possibility that some of these changes are the indirect result of gross changes in nuclear shape. However, de novo production of channels in interphase nuclei was not observed.
Possible Functional Implications of Nuclear Channels
Collectively, these data leave little doubt that there are widespread incursions of double ER membranes into the depths of the nucleoplasm in many cell types. Since the nuclear envelope is disassembled into vesicles during mitosis, it is most likely that the channels are formed during nuclear envelope reassembly. Chromatin becomes coated with bound nuclear envelope vesicles while decondensation is progressing; such vesicles penetrate between chromosomes and produce a deeply convoluted layer. These convolutions may be resolved by chromatin decondensation to produce a uniform convex vesicle layer; fusion of such a layer would produce a nuclear envelope devoid of deep channels. However, if the vesicle fusion event precedes complete decondensation, a complex, channel-rich nuclear envelope may be produced. Thus, the channels may arise as a result of incomplete resolution of invaginations during nuclear envelope reassembly; differences in channel morphology between cell types could then result from differential rates of chromatin decondensation and nuclear envelope vesicle fusion. It is possible that the slow resolution of channels could continue after vesicle fusion and, indeed, perhaps continue into interphase. In this case, the channels described might simply represent nonfunctional remnants of the process of nuclear envelope reassembly; their presence might even be deleterious for the cell.
There are three pieces of evidence that suggest that, although channels may originate in this way, they are not nonfunctional remnants. First, channels persist for long periods in interphase cells. More than 90% of CHO cells in a nonsynchronized culture have one or more channel, and this proportion suggests that most interphase CHO cells still have not resolved their channels, whether they are in G1, S or G2 phases of the cell cycle. Second, channels may be found in postmitotic populations (such as the primary peritoneal exudate cells and normal tissue hepatocytes examined in this study), again implying that the structures described here are not resolved in such populations. Third, if the channels represented mitotic remnants, it is not easy to explain the association with nucleoli that has been described (Bourgeois et al., 1979; Dupuy-Coin et al., 1986). Although an answer to this question must await further functional studies, we suggest possible functions that the channels might serve.
One plausible function of such invaginations is to bring a larger proportion of the nucleoplasm close to a nuclear pore. For a spherical nucleus 5 μm diam, 60% of the nucleoplasm lies within 0.5 μm of the nuclear envelope, and the center of the nucleoplasm lies at 2.5 μm away from it. However, a single tube traversing the nucleus halves the maximum distance from any point in the nucleoplasm to the envelope and brings a further 12% of the nucleoplasm within 0.5 μm of the envelope. Three or four appropriately placed tubes would bring almost all of the nucleoplasm within a distance of 0.5 μm of the envelope.
This means that even nucleoli which are buried within the nucleus may lie close to the envelope. A function of nuclear channels in transport processes has been suggested in measles virus–infected hamster cells, where an association between tubular invaginations and nucleoli was reported (Dupuy-Coin et al., 1986). Indeed, a substantial number of channels contact the nucleolus or terminate within it, both in tissue culture cells and normal hepatocytes. However, we also find a clear category of nuclear channels of varying morphology that have no apparent relationship with nucleoli, even when the entire nuclear volume is reconstructed in three dimensions and examined in animated rotation. Such channels may be genuinely dissociated from nucleoli, with attendant functional implications. Alternatively, the channels detected at the LM level might represent only the larger “trunks” of branched structures, leaving undetected fine branches to communicate with apparently distant nucleoli. The serial section EM data confirms that there are very fine lateral branches associated with some nuclear channels, as may be clearly seen in Fig. 1.
Taken together, these results and the data presented here suggest that the traditional view of the nuclear envelope as an approximately spherical coat defining the nucleoplasm is at best an incomplete one. Deep tubular incursions into the nucleoplasm occur sufficiently often in the population to warrant inclusion in any discussion of structure–function relationships in the envelope of mammalian interphase nucleus.
Acknowledgments
We thank Peter Cook for discussion and critical reading of the manuscript and the members of our groups for helpful discussions. We thank Dr. George Simos and Dr. Spyros Georgatos (European Molecular Biology Laboratory, Heidelberg, Germany) and Professor Daniel Louvard (Institut Pasteur, Paris, France) for the generous gifts of antibody. We thank Peter Tree and Liz Darley for providing the rat tissue sections.
Abbreviations used in this paper
- CLSM
confocal laser scanning fluorescent microscopy
- G-6-Pase
glucose-6-phosphatase
- NPC
nuclear pore complex
- PDI
protein disulphide isomerase
- TEM
transmission electron microscopy, 3-D, three-dimensional
Footnotes
The work was partially funded by grants from the Medical Research Council (to D. Vaux) and a major equipment grant from the Wellcome Trust (to D. Vaux). N.S. White is a Royal Society Industry Fellow.
Please address all correspondence to David Vaux, Sir William Dunn School of Pathology, South Parks Road, Oxford, OX1 3 RE, United Kingdom. Tel.: (44) 1865 275544. Fax: (44) 1865 275501. E-mail: VAUX @MOLBIOL.OX.AC.UK
References
- Banfalvi G, Wiegant J, Sarkar N, van-Duijn P. Immunofluorescent visualization of DNA replication sites within nuclei of Chinese hamster ovary cells. Histochemistry. 1989;93:81–86. doi: 10.1007/BF00266851. [DOI] [PubMed] [Google Scholar]
- Banfic H, Zizak M, Divecha N, Irvine RF. Nuclear diacylglycerol is increased during cell proliferation in vivo. Biochem J. 1993;290:633–636. doi: 10.1042/bj2900633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont AS, Zhai Y, Thilenius A. Lamin B distribution and association with peripheral chromatin revealed by optical sectioning and electron microscopy tomography. J Cell Biol. 1993;123:1671–1685. doi: 10.1083/jcb.123.6.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois CA, Hemon D, Bouteille M. Structural relationship between the nucleolus and the nuclear envelope. J Ultrastruct Res. 1979;68:328–340. doi: 10.1016/s0022-5320(79)90165-5. [DOI] [PubMed] [Google Scholar]
- Bowers CW, Dahm LM. Buffers for cell culture. Trends Cell Biol. 1993;3:76–77. doi: 10.1016/0962-8924(93)90068-c. [DOI] [PubMed] [Google Scholar]
- Bridger JM, Kill IR, O'Farrell M, Hutchison CJ. Internal lamin structures within G1 nuclei of human dermal fibroblasts. J Cell Sci. 1993;104:297–306. doi: 10.1242/jcs.104.2.297. [DOI] [PubMed] [Google Scholar]
- Carmo-Fonseca M, Pepperkok R, Sproat BS, Ansorge W, Swanson MS, Lamond AI. In vivo detection of snRNP-rich organelles in the nuclei of mammalian cells. EMBO (Eur Mol Biol Organ) J. 1991;10:1863–1873. doi: 10.1002/j.1460-2075.1991.tb07712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo-Fonseca M, Peppercock R, Carvelho MT, Lamond AI. Transcription-dependent colocalization of the U1, U2, U4/U6, and U5 snRNPs in coiled bodies. J Cell Biol. 1992;117:1–14. doi: 10.1083/jcb.117.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson HG, Bell PR. Structures resembling nuclear pores at the orifice of nuclear invaginations in developing micropores of Pinus banksiana. . Dev Biol. 1972;27:425–429. doi: 10.1016/0012-1606(72)90181-9. [DOI] [PubMed] [Google Scholar]
- Dingwall C, Laskey R. The nuclear membrane. Science (Wash DC) 1992;258:942–947. doi: 10.1126/science.1439805. [DOI] [PubMed] [Google Scholar]
- Divecha N, Banfic H, Irvine RF. The polyphosphoinositide cycle exists in the nuclei of Swiss 3T3 cells under the control of a receptor (for IGF-I) in the plasma membrane, and stimulation of the cycle increases nuclear diacylglycerol and apparently induces translocation of protein kinase C to the nucleus. EMBO (Eur Mol Biol Organ) J. 1991;10:3207–3214. doi: 10.1002/j.1460-2075.1991.tb04883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divecha N, Banfic H, Irvine RF. Inositides and the nucleus, and inositides in the nucleus. Cell. 1993;74:405–407. doi: 10.1016/0092-8674(93)80041-c. [DOI] [PubMed] [Google Scholar]
- Dupuy-Coin AM, Moens P, Bouteille M. Three-dimensional analysis of given cell structures: nucleolus, nucleoskeleton, and nuclear inclusions. Methods Achiev Exp Pathol. 1986;12:1–25. [PubMed] [Google Scholar]
- Fricker MD, White NS. Wavelength considerations in confocal microscopy of botanical specimens. J Microsc (Oxf) 1992;166:29–42. [Google Scholar]
- Fricker MD, Tlalka M, Ermantraut J, Obermeyer G, Dewey M, Gurr S, Patrick J, White NS. Confocal fluorescence ratio imaging of ion activities in plant cells. Scanning Microsc Suppl. 1994;8:391–405. [Google Scholar]
- Goldman AE, Moir RD, Montag-Lowy M, Stewart M, Goldman RD. Pathway of incorporation of microinjected lamin A into the nuclear envelope. J Cell Biol. 1992;104:725–732. doi: 10.1083/jcb.119.4.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths G, Quinn P, Warren G. Dissection of the golgi complex I. Monensin inhibits the transport of viral membrane proteins from medial to trans golgi cisternae in baby hamster kidney cells infected with Semliki Forest virus. J Cell Biol. 1983;96:835–850. doi: 10.1083/jcb.96.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths G, McDowall AW, Back R, Dubochet J. On the preparation of cryosections for immunocytochemistry. J Ultrastruct Res. 1984;89:65–78. doi: 10.1016/s0022-5320(84)80024-6. [DOI] [PubMed] [Google Scholar]
- Hinshaw JE, Carragher BO, Milligan RA. Architecture and design of the nuclear pore complex. Cell. 1992;69:1133–1141. doi: 10.1016/0092-8674(92)90635-p. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M, Sedat JW. Three dimensional organization of Drosophila melanogasterinterphase nuclei. II. Chromosome spatial organization and gene regulation. J Cell Biol. 1987;104:1471–1483. doi: 10.1083/jcb.104.6.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hozak P, Cook PR. Replication factories. Trends Cell Biol. 1994;4:48–51. doi: 10.1016/0962-8924(94)90009-4. [DOI] [PubMed] [Google Scholar]
- Huang S, Spector DL. Nascent pre-mRNA transcripts are associated with nuclear regions enriched in splicing factors. Genes Dev. 1991;5:2288–2302. doi: 10.1101/gad.5.12a.2288. [DOI] [PubMed] [Google Scholar]
- Huang S, Spector DL. Intron-dependent recruitment of premRNA splicing factors to sites of transcription. J Cell Biol. 1996;133:719–732. doi: 10.1083/jcb.133.4.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DA, Fraser IP, Gordon S. Murine macrophage scavenger receptor: in vivo expression and function as receptor for macrophage adhesion in lymphoid and non-lymphoid organs. Eur J Immunol. 1995;25:466–473. doi: 10.1002/eji.1830250224. [DOI] [PubMed] [Google Scholar]
- Hutchison C, Bridger JM, Cox LS, Kill IR. Weaving a pattern from disparate threads: Lamin function in nuclear assembly and DNA replication. J Cell Sci. 1994;107:3259–3269. doi: 10.1242/jcs.107.12.3259. [DOI] [PubMed] [Google Scholar]
- Jackson DA, Hassan AB, Errington R, Cook P. Visualization of focal sites of transcription within human nuclei. EMBO (Eur Mol Biol Organ) J. 1993;12:1059–1065. doi: 10.1002/j.1460-2075.1993.tb05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DA, Balajee AS, Mullenders L, Cook PR. Sites in human nuclei where DNA damaged by ultraviolet light is repaired: visualization and localization relative to the nucleoskeleton. J Cell Sci. 1994a;107:1745–1752. doi: 10.1242/jcs.107.7.1745. [DOI] [PubMed] [Google Scholar]
- Jackson DA, Hassan AB, Errington RJ, Cook PR. Sites in human nuclei where DNA damaged by ultraviolet light is repaired: localization relative to transcription sites and concentrations of proliferating cell nuclear antigen and the tumour suppressor protein, p53. J Cell Sci. 1994b;107:1753–1760. doi: 10.1242/jcs.107.7.1753. [DOI] [PubMed] [Google Scholar]
- Kamei H. Relationship of nuclear invaginations to perinuclear rings composed of intermediate filaments in MIA PaCa-2 and some other cells. Cell Struct Funct. 1994;19:123–132. doi: 10.1247/csf.19.123. [DOI] [PubMed] [Google Scholar]
- Lamond A, Carmo-Fonseca C. The coiled body. Trends Cell Biol. 1993;3:198–204. doi: 10.1016/0962-8924(93)90214-l. [DOI] [PubMed] [Google Scholar]
- Laskey RA, Dingwall C. Nuclear shuttling: the default pathway for nuclear proteins? . Cell. 1993;74:585–586. doi: 10.1016/0092-8674(93)90505-k. [DOI] [PubMed] [Google Scholar]
- Lawrence JB, Singer RH, Marselle LM. Highly localized tracks of specific transcripts within interphase nuclei visualized by in situ hybridization. Cell. 1989;57:493–502. doi: 10.1016/0092-8674(89)90924-0. [DOI] [PubMed] [Google Scholar]
- Li FL, Dickinson HG. The structure and function of nuclear invaginations characteristic of microsporogenesis in Pinus banksiana. . Ann Bot (Lond) 1987;60:321–330. [Google Scholar]
- Maraldi NM, Santi S, Zini N, Ognibene A, Rizzoli R, Mazzotti G, Di Primio R, Bareggi R, Bertagnolo V, Pagliarini C, Capitani S. Decrease in nuclear phospholipids associated with DNA replication. J Cell Sci. 1993;104:853–859. doi: 10.1242/jcs.104.3.853. [DOI] [PubMed] [Google Scholar]
- Mazzotti G, Zini N, Rizzi E, Rizzoli R, Galanzi A, Ognibene A, Santi S, Matteucci A, Martelli AM, Maraldi NM. Immunocytochemical detection of phosphatidylinositol 4,5-bisphosphate localization sites within the nucleus. J Histochem Cytochem. 1995;43:181–191. doi: 10.1177/43.2.7822774. [DOI] [PubMed] [Google Scholar]
- Meier UT, Blobel G. Nopp140 shuttles on tracks between nucleolus and cytoplasm. Cell. 1992;70:127–138. doi: 10.1016/0092-8674(92)90539-o. [DOI] [PubMed] [Google Scholar]
- Mills AD, Blow JJ, White JG, Amos WB, Wilcock D, Laskey RA. Replication occurs at discrete foci spaced throughout nuclei replicating in vitro. J Cell Sci. 1989;94:471–477. doi: 10.1242/jcs.94.3.471. [DOI] [PubMed] [Google Scholar]
- Moen PTJ, Smith KP, Lawrence JB. Compartmentalization of specific pre-mRNA metabolism: an emerging view. Hum Mol Genet. 1995;4:1779–1789. doi: 10.1093/hmg/4.suppl_1.1779. [DOI] [PubMed] [Google Scholar]
- Moir RD, Montag-Lowy M, Goldman RD. Dynamic properties of nuclear lamins: lamin B is associated with sites of DNA replication. J Cell Biol. 1994;125:1201–1212. doi: 10.1083/jcb.125.6.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parke PC, de Boni U. Nuclear membrane modifications in polytene nuclei of Drosophila melanogaster: serial reconstruction and cytochemistry. Anat Rec. 1992;234:15–26. doi: 10.1002/ar.1092340103. [DOI] [PubMed] [Google Scholar]
- Rosbash M, Singer RH. RNA travel: tracks from DNA to cytoplasm. Cell. 1993;75:399–401. doi: 10.1016/0092-8674(93)90373-x. [DOI] [PubMed] [Google Scholar]
- Rout MP, Wente SR. Pores for thought: nuclear pore complex proteins. Trends Cell Biol. 1994;4:357–365. doi: 10.1016/0962-8924(94)90085-x. [DOI] [PubMed] [Google Scholar]
- Sarria AJ, Lieber JG, Nordeen SK, Evans RM. The presence or absence of a vimentin-type intermediate filament network affects the shape of the nucleus in human SW-13 cells. J Cell Sci. 1994;107:1593–1607. doi: 10.1242/jcs.107.6.1593. [DOI] [PubMed] [Google Scholar]
- Schmidt-Zachmann MS, Dargemont C, Kuehn LC, Nigg EA. Nuclear export of proteins: the role of nuclear retention. Cell. 1993;74:493–504. doi: 10.1016/0092-8674(93)80051-f. [DOI] [PubMed] [Google Scholar]
- Spector DL. Higher order nuclear organisation: three-dimensional distribution of small nuclear ribonucleoprotein particles. Proc Natl Acad Sci USA. 1990;87:147–151. doi: 10.1073/pnas.87.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector DL, Fu X-D, Maniatis T. Associations between distinct pre-mRNA splicing components and the cell nucleus. EMBO (Eur Mol Biol Organ) J. 1991;10:3467–3481. doi: 10.1002/j.1460-2075.1991.tb04911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens J, Trogadis J. Reconstructive three dimensional electron microscopy: a routine biological tool. Analyt Quant Cytol Histol. 1986;8:102–107. [PubMed] [Google Scholar]
- Strouboulis J, Wolfe AP. Functional compartmentalization of the nucleus. J Cell Sci. 1996;109:1991–2000. doi: 10.1242/jcs.109.8.1991. [DOI] [PubMed] [Google Scholar]
- Tokuyasu KT. Immunocytochemistry in ultra-thin frozen sections. Histochem J. 1980;12:381–403. doi: 10.1007/BF01011956. [DOI] [PubMed] [Google Scholar]
- Wachstein M, Meisel E. On the histochemical demonstration of glucose-6-phosphatases. J Histochem Cytochem. 1956;4:592–596. [Google Scholar]
- Wansink DG, Schul W, Kraan I, Steensel B, Driel R, Jong L. Fluorescent labeling of nascent RNA reveals transcription by RNA polymerase II in domains scattered throughout the nucleus. J Cell Biol. 1993;122:283–293. doi: 10.1083/jcb.122.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, N.S. 1995. Visualisation Systems for Multidimensional CLSM Images. In Handbook of Biological Confocal Microscopy, J.B. Pawley, editor. Plenum Publishing Corp., New York. 211–254.
- White NS, Errington RJ, Fricker MD, Wood JL. Aberration control in quantitative imaging of botanical specimens by mutlidimensional fluorescence microscopy. J Microsc (Oxf) 1996;181:99–116. [Google Scholar]
- Xing Y, Lawrence JB. Nuclear RNA tracks: structural basis for transcription and splicing? . Trends Cell Biol. 1993;3:346–353. doi: 10.1016/0962-8924(93)90105-a. [DOI] [PubMed] [Google Scholar]
- Xing Y, Johnson CV, Dobner PR, Lawrence JB. Higher level organization of individual gene transcription and RNA splicing. Science (Wash DC) 1993;259:1326–1330. doi: 10.1126/science.8446901. [DOI] [PubMed] [Google Scholar]
- Xing Y, Johnson CV, Moen PT, Jr, McNeil JA, Lawrence JB. Nonrandom gene organization: structural arrangements of specific premRNA transcription and splicing with SC-35 domains. J Cell Biol. 1995;131:1635–1647. doi: 10.1083/jcb.131.6.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachar Z, Kramer J, Mims IP, Bingham PM. Evidence for channeled diffusion of pre-mRNAs during nuclear RNA transport in metozoans. J Cell Biol. 1993;121:729–742. doi: 10.1083/jcb.121.4.729. [DOI] [PMC free article] [PubMed] [Google Scholar]