

Abstract
β-Catenin is essential for the function of cadherins, a family of Ca2+-dependent cell–cell adhesion molecules, by linking them to α-catenin and the actin cytoskeleton. β-Catenin also binds to adenomatous polyposis coli (APC) protein, a cytosolic protein that is the product of a tumor suppressor gene mutated in colorectal adenomas. We have expressed mutant β-catenins in MDCK epithelial cells to gain insights into the regulation of β-catenin distribution between cadherin and APC protein complexes and the functions of these complexes. Full-length β-catenin, β-catenin mutant proteins with NH2-terminal deletions before (ΔN90) or after (ΔN131, ΔN151) the α-catenin binding site, or a mutant β-catenin with a COOH-terminal deletion (ΔC) were expressed in MDCK cells under the control of the tetracycline-repressible transactivator. All β-catenin mutant proteins form complexes and colocalize with E-cadherin at cell–cell contacts; ΔN90, but neither ΔN131 nor ΔN151, bind α-catenin. However, β-catenin mutant proteins containing NH2-terminal deletions also colocalize prominently with APC protein in clusters at the tips of plasma membrane protrusions; in contrast, full-length and COOH-terminal– deleted β-catenin poorly colocalize with APC protein. NH2-terminal deletions result in increased stability of β-catenin bound to APC protein and E-cadherin, compared with full-length β-catenin. At low density, MDCK cells expressing NH2-terminal–deleted β-catenin mutants are dispersed, more fibroblastic in morphology, and less efficient in forming colonies than parental MDCK cells. These results show that the NH2 terminus, but not the COOH terminus of β-catenin, regulates the dynamics of β-catenin binding to APC protein and E-cadherin. Changes in β-catenin binding to cadherin or APC protein, and the ensuing effects on cell morphology and adhesion, are independent of β-catenin binding to α-catenin. These results demonstrate that regulation of β-catenin binding to E-cadherin and APC protein is important in controlling epithelial cell adhesion.
β-Catenin is a ubiquitous protein in multicellular organisms and was originally identified through its association with the cytoplasmic domain of cadherins, a family of Ca2+-dependent cell adhesion proteins (McCrea and Gumbiner, 1991; McCrea et al., 1991; Nagafuchi and Takeichi, 1989; Ozawa et al., 1989). Cadherin-mediated intercellular adhesion initiates structural and functional changes in cells and is important for maintaining tissue integrity during embryonic development and in adult organisms (Nelson et al., 1992; Takeichi, 1990, 1991). These functions of cadherin require intracellular attachment of cadherin to the actin cytoskeleton that is dependent on binding of cadherin to catenins (Hirano et al., 1987; Nagafuchi and Takeichi, 1988; Ozawa et al., 1990); β-catenin mediates the linkage of cadherins to α-catenin, which in turn interacts with the actin cytoskeleton (Aberle et al., 1994; Hülsken et al., 1994; Jou et al., 1995; Rimm et al., 1995).
Coordination of intercellular adhesion and cell migration is important during embryonic development. For example, during gastrulation, neurulation, and organogenesis, sheets of cells move past neighboring cells without losing cell–cell contact (Gumbiner, 1992). Significantly, embryonic development of mice lacking β-catenin is disrupted at gastrulation (Haegel et al., 1995). These embryos form a trophectoderm and develop through preimplantation stages presumably because of the contribution of maternal β-catenin or replacement of β-catenin in adherens junctions by its homologue plakoglobin. However, β-catenin–deficient embryos have severe defects in cell adhesion at gastrulation (Haegel et al., 1995). It is also noteworthy that human cancer cell lines expressing a mutated form of β-catenin, which binds E-cadherin but not α-catenin, are defective in E-cadherin–mediated adhesion (Oyama et al., 1994). The importance of β-catenin in embryogenesis is also revealed in studies in Drosophila. A homologue of β-catenin in Drosophila, armadillo, was originally identified through its role in transduction of the wingless/Wnt cell–cell signal that mediates cell fate determination. Drosophila embryos that are zygotically null for armadillo develop with severe segment polarity defects (Noordermeer et al., 1994; Peifer et al., 1991; Peifer and Wieschaus, 1990; Siegfried et al., 1994). Similar to the requirement of β-catenin in vertebrate embryogenesis, armadillo is required for adherens junction assembly in Drosophila embryogenesis. A combination of maternal and zygotic nulls of armadillo disrupts formation of an organized epithelium early in Drosophila embryogenesis (Cox et al., 1996; Peifer et al., 1993).
In addition to binding to cadherin, β-catenin has also been shown recently to bind to the product of the adenomatous polyposis coli (APC)1 tumor suppressor gene. The APC/β-catenin complex contains α-catenin but not cadherin (Rubinfeld et al., 1993; Su et al., 1993). APC protein and E-cadherin compete for binding to β-catenin in transient expression assays (Hülsken et al., 1994). APC protein is a 310-kD cytoplasmic protein that is mutated in patients with the inherited colon cancer syndrome familial adenomatous polyposis. Mutations in APC protein also represent an early event in a high percentage of sporadic colon cancers (Groden et al., 1991; Kinzler et al., 1991; Polakis, 1995). The cellular function(s) of APC protein are not known. In addition to binding catenins, APC protein also binds to microtubules in vitro and in vivo (Munemitsu et al., 1994; Polakis, 1995; Smith et al., 1994). In epithelial cells, APC protein localizes to the ends of microtubule bundles in actively migrating plasma membrane protrusions and to the tips of membranes involved in early cell– cell contact formation (Näthke et al., 1996). The subcellular localization of APC protein indicates that it is involved in cell migration.
To examine domains in β-catenin that are important for regulating its subcellular distribution and function(s) in cadherin and APC protein complexes, we expressed NH2- and COOH-terminal–deleted β-catenin in MDCK cells. Our results demonstrate that deletion of the COOH-terminal domain does not affect the subcellular distribution of β-catenin. Deletion of the NH2-terminal domain stabilizes β-catenin in APC protein and E-cadherin complexes. NH2-terminal–deleted β-catenin prominently colocalizes with APC protein in clusters at the tips of plasma membrane protrusions. MDCK cells expressing NH2-terminal– deleted β-catenin are inhibited in early cell–cell contact formation and compaction when plated at low cell densities. Both colocalization with APC protein and inhibition of adhesion by NH2-terminal–deleted β-catenin mutant proteins are independent of β-catenin binding to α-catenin.
Materials and Methods
Antibodies
A rabbit polyclonal antiserum β-cat.N was generated in rabbits against the NH2-terminal 14 amino acids of β-catenin. Rabbit polyclonal antibodies raised against the COOH-terminal 14 amino acids of α-catenin and β-catenin (β-cat.C) have been described previously (Hinck et al., 1994b ). A mAb against the COOH-terminal 212 amino acids of β-catenin (β-cat.C) was obtained from Transduction Laboratories (Lexington, KY). A mouse mAb KT3 against a SV-40 large T antigen epitope was kindly provided by Gernot Walter (University of California, San Diego), and has been described previously (MacArthur and Walter, 1984). A polyclonal antibody against the cytoplasmic domain of E-cadherin has been described previously (Marrs et al., 1994). Polyclonal antibodies against APC protein were kindly provided by Paul Polakis (ONYX Pharmaceuticals, Richmond, VA) and Inke Näthke (Stanford University), and have been described previously (Näthke et al., 1996; Rubinfeld et al., 1993). Antisera were affinity purified with antigen for immunofluorescence as indicated.
Plasmid Construction
Mouse β-catenin cDNA (Butz et al., 1992) in pBluescript SKII+ (Stratagene, La Jolla, CA) was used to generate deletion mutants (see Fig. 1). The deduced amino acid sequence is identical to a partial dog β-catenin sequence (amino acids 51–311) and identical to the human β-catenin sequence except for one amino acid difference at position 706 (Hülsken et al., 1994). To detect β-catenin and β-catenin mutants immunologically, an oligonucleotide encoding the amino acid sequence KPPTPPPEPET from SV-40 large T antigen (KT3 epitope tag) was added to the 3′ termini of all cDNAs (MacArthur and Walter, 1984). Nucleotide sequences of PCR- derived sequences in constructs shown below were confirmed by direct sequencing (PAN Facility, Beckman Center, Stanford University School of Medicine). The cDNA construct for ΔN131 (see below) was found to contain a PCR-introduced point mutation at nucleotide 553 that led to a conservative amino acid change of Val175 to Leu175.
Figure 1.

Schematic representation of KT3-tagged full-size and mutant β-catenin proteins. NH2- and COOH-terminal domains and the 13 internal armadillo-like repeats of β-catenin are indicated. A stretch of unrepeated amino acids between repeat 10 and 11 (empty box) is shown. The binding sites for α-catenin, E-cadherin, APC, and the epitopes for rabbit β-catenin antisera β-cat.N, β-cat.C, and mouse mAb KT3 are indicated. The epitope for antiserum β-cat.N is deleted in ΔN90, ΔN131, and ΔN151, and these mutant proteins are not detected by β-cat.N. A mouse mAb raised against the 212 COOH-terminal amino acids of β-catenin was used instead of antiserum β-cat.C, and both β-cat.C antibodies do not detect ΔC. The following amino acids are deleted in the mutants: ΔC, 696–781; ΔN90, 1–90; ΔN131, 1–131; ΔN151, 1–151.
ΔN131 was constructed by PCR using oligonucleotides containing suitable restriction sites (5′-SacII/3′-XbaI) for cloning into the pUDH10-3 vector (Gossen and Bujard, 1992). The oligonucleotide defining the start site of the cDNA (5′-CCATCGATTCTAGACCGCGGCCACCATGGCTTTGAAACATGCAGTTGTCAATTTG) contained a Kozak consensus sequence for translation initiation (Kozak, 1989). The oligonucleotide defining the stop site of the cDNA (5′-CCATCGATTCTAGATTAGGTCTCGGGCTCAGGAGGAGGAGTAGGAGGCTTGATATC C A G - GTCAGTATCAAACCAGGC) codes for 13 additional amino acids including the KT3 epitope after the COOH terminus of the mutant protein (indicated in italics) followed by a stop codon; the underlined sequence is derived from β-catenin. In two cloning steps, the BglII and, subsequently, Eco47III/SpeI fragments of the original cDNA were exchanged for the respective PCR-derived fragments of ΔN131 in pUDH10-3. ΔN131 was used to construct a pBluescript KSII+ vector for addition of the KT3 epitope sequence to the 3′ termini of other cDNAs. The Bsp106I/StuI fragment of ΔN131 was cloned into pBluescript KSII+-HA (Elferink et al., 1993). The EcoRV fragment containing the HA tag sequence and the complete ΔN131 sequence except for the KT3 epitope was subsequently removed. This resulted in the vector pBluescript KSII+-KT3 in which the sequence for the KT3 epitope and a stop codon are preceded 5′-upstream by EcoRV/SmaI/BamHI restriction sites to allow the addition of cDNAs in all three reading frames.
ΔNΔC was constructed by cloning the blunted BglII fragment of the original β-catenin cDNA into pBluescript KSII+-KT3/SmaI. The resulting EcoRI/XbaI fragment, in which the sequence for the KT3 epitope tag is fused to the sequence of the last repeat in the core region of β-catenin, was exchanged with the respective EcoRI/XbaI fragment in pUdH10-3/ ΔN131 to obtain pUDH10-3/ΔNΔC.
ΔC was constructed by cloning the 5′ EcoRI fragment with the start codon of the original β-catenin cDNA into pUDH10-3. The resulting vector contained the sequence encoding the NH2-terminal half of β-catenin. The 3′-terminal BglII/XbaI fragment of this β-catenin sequence was replaced with the 3′ BglII/XbaI fragment of ΔNΔC to obtain the expression vector pUDH10-3/ΔC.
A vector expressing full-length KT3-tagged β-catenin* was constructed by exchanging the SpeI/BamHI fragment encoding the COOH-terminal part of ΔC in pUDH10-3/ΔC against the respective fragment encoding the COOH-terminal part of ΔN131.
ΔN90 was constructed by PCR using the oligonucleotide 5′-CCATCGATTCTAGACCGCGGCCACCATGGCTCAGAGGGTCCGAGCT for the 5′ terminus of the cDNA and the same oligonucleotide as that used for the 3′ terminus of the cDNA of ΔN131. The SacII/XbaI PCR fragment was cloned into pUDH10-3. To replace PCR-derived sequences with original cDNA, the SphI/XbaI fragment of ΔN90 was exchanged with the SphI/XbaI fragment of full-length KT3-tagged β-catenin.
ΔN151 was constructed by PCR using the oligonucleotide 5′-CCATCGATTCTAGACCGCGGCCACCATGGCAATTCCTGAGCTGACA for the 5′ terminus of the cDNA and the same oligonucleotide as that used for the 3′ terminus of the cDNA of ΔN131. The SacII/XbaI PCR fragment was cloned into pUDH10-3. To replace PCR-derived sequences with original cDNA, the Eco47III/XbaI fragment of ΔN151 was exchanged with the Eco47III/XbaI fragment of full-length KT3-tagged β-catenin. PCRderived sequences were confirmed by sequencing.
Cell Transfection
MDCK cells (type II) were transfected with pIgR cloned into the pCB6, which uses the cytomegalovirus promoter to drive pIgR expression and a G418 drug resistance marker. A well-polarized clone expressing a uniformly high level of pIgR was selected and designated B3. The B3 cells were then cotransfected with plasmid pUHD15-1 (Gossen and Bujard, 1992) coding for the tTa transactivator and pSV2-puro in a ratio of 10:1, respectively. Transfection was done by the calcium phosphate precipitation method, and cells were plated onto 150-mm dishes. Drug-resistant clones were selected in the presence of 5 μg/ml puromycin (Sigma Chemical Co., St. Louis, MO). 180 clones were picked using cloning rings and then expanded. Each clone was transiently transfected with plasmid pUHC13-3 consisting of the luciferase gene under control of the tetracycline-regulated promoter in the presence and absence of 1 μg/ml tetracycline, using the lipofectamine reagent (GIBCO BRL, Gaithersburg, MD). After 48 h, cells were homogenized and tested for luciferase activity. Clone T23 showed 180-fold more luciferase activity in the absence of tetracycline as compared with in its presence. This clone was chosen for further studies. T23 cells were cotransfected with vectors for expression of the tetracycline-repressible transactivator tTA (Gossen and Bujard, 1992) and a plasmid containing a gene for resistance to puromycin. Parental MDCK clone T23 was cotransfected with the respective expression vectors for β-catenin mutant proteins (see above; ΔN90, ΔN131, ΔN151, ΔNΔC, ΔC, β-catenin*) and plasmid pHMR272 carrying a gene for resistance to hygromycin (Gossen and Bujard, 1992) using the lipofectamine protocol from GIBCO BRL. Clones were selected in 300 μg/ml hygromycin (Boehringer Mannheim Biochemicals, Indianapolis, IN). Low passage aliquots of drug-resistant MDCK clones were frozen and stored in liquid nitrogen; clones were passaged in DME medium containing 10% FCS (Gemini, Calabasas, CA) and 300 μg/ml hygromycin without doxycycline (Dox) for a maximum of 4–6 wk. In some experiments, expression of β-catenin mutant proteins was repressed by addition of 1 μg/ml tetracycline (Tet) or 20 ng/ml Dox (Sigma Chemical Co.) to the culture medium. Cells were cultured 1–3 d without hygromycin before an experiment.
Immunoprecipitation and Immunoblotting
MDCK cells were maintained in DME medium supplemented with 10% FCS and passaged by mild trypsinization. For preparation of SDS lysates, MDCK cells were cultured for 4 d with or without 20 ng/ml Dox; cells were extracted in hot SDS buffer containing 1% SDS, 10 mM Tris-HCl, pH 7.5, and 2 mM EDTA, and then scraped from the petri dish with a rubber policeman. Samples were boiled for 15 min, and insoluble material was removed by centrifugation at 12,000 g for 15 min. Protein concentrations were determined using the bicinchoninic acid protein assay reagent kit (BCA; Pierce Chemical Co., Rockford, Il). Lysates were boiled for 5 min after adding an equal volume of SDS reducing sample buffer (Laemmli, 1970).
For preparation of Triton X-100 lysates, MDCK cells were cultured with or without 20 ng/ml Dox for 4 d or as indicated, and then extracted for 15 min at 4°C with 1% Triton X-100, 20 mM Tris-HCl, pH 8, 140 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol (vol/vol), 10 μg/ml DNase and RNase, 1 mM sodium vanadate, 50 mM NaF, and a protease inhibitor mix (1 mM Pefabloc, 1 mM benzamidine, and 10 μg/ml each of aprotinin, pepstatin, and leupeptin); DNase, RNase, Pefabloc, pepstatin, and leupeptin were purchased from Boehringer Mannheim Biochemicals, and all other reagents were from Sigma Chemical Co. After extraction, cells were scraped from the petri dish with a rubber policeman. Insoluble material was removed by centrifugation at 12,000 g for 30 min. Protein concentrations were determined using the BCA protein assay reagent kit (Pierce Chemical Co.). Portions of lysates were boiled for 5 min after adding an equal volume of SDS reducing sample buffer. Other portions of lysates were precleared by incubation with 5 μl of nonimmune rabbit serum and 100 μl Pansorbin (Calbiochem-Novabiochem Corp., La Jolla, CA) for 1 h at 4°C. After removing the Pansorbin by centrifugation, lysates were incubated for 2–3 h at 4°C with either 30 μl protein A–Sepharose (Pharmacia Fine Chemicals, Piscataway, NJ) coupled to E-cadherin or β-catenin antibody, or 60 μl protein A–Sepharose coupled to α-catenin or APC protein antibody. Immunoprecipitates were washed once with wash buffer (0.5% NP-40, 20 mM Tris-HCl, pH 8, 150 mM NaCl), once with wash buffer containing 1 M LiCl, and once with wash buffer. Immunoprecipitates were boiled in 30 or 60 μl SDS reducing sample buffer.
Lysates and immunoprecipitates were separated by SDS-PAGE in 7.5% polyacrylamide gels (Laemmli, 1970). Gels were transferred to nitrocellulose membranes with a pore size of 0.45 μm (Schleicher & Schuell Inc., Keene, NH) in a buffer containing 25 mM Tris-HCl, pH 7.4, 50 mM glycine, and 20% methanol. Membranes were stained with Ponceau S to check for protein transfer, and then destained in 20 mM Tris-HCl, pH 7.4, 0.5% Tween-20, 140 mM NaCl (TTS). Membranes were blocked overnight at 4°C in TTS with 5% powdered milk. Primary antibodies were incubated with membranes for 2 h at room temperature (RT) in TTS with 5% powdered milk. Membranes were washed four times for 10 min each with TTS, and then incubated with the appropriate secondary antibodies coupled to HRP (Amersham Corp., Arlington Heights, IL) in TTS, 5% powdered milk, and 5% serum from the animal species that was used to generate the secondary antibody. Membranes were washed for at least 2 h with six changes of TTS, and antibody reactivity was visualized with enhanced chemiluminescence reagent (Amersham Corp.). Bands on ECLexposed X-OMAT AR film (Eastman Kodak Co., Rochester, NY) were analyzed with a GS 300 transmittance/reflectance scanning densitometer (Hoefer Scientific Instruments, San Francisco, CA), or scanned with a ScanJet IIc (Hewlett-Packard Co., Palo Alto, CA) and analyzed with NIH Image. All reagents were from Sigma Chemical Co. unless indicated otherwise.
Immunofluorescence and Phase-Contrast Microscopy
For immunofluorescence, MDCK cells were cultured and plated on 35mm tissue-culture dishes with collagen-coated coverslips in DME medium supplemented with 10% FCS with or without Tet as indicated. For immunofluorescence of low density cultures, 1 × 105 cells were plated and fixed after 24 h. Part of a culture of clone ΔN131-D was preincubated for 24 h in 1 μg/ml Tet before plating onto coverslips and was kept in medium containing Tet until fixation. Cells were washed once in Dulbecco's PBS (0.9 mM CaCl2, 2.7 mM KCl, 1.5 mM KH2PO4, 0.5 mM MgCl2, 137 mM NaCl, and 8.1 mM NaHPO4) and fixed for 20 at RT in 2% formaldehyde in Dulbecco's PBS. Cells were washed four times for 5 min each in Dulbecco's PBS with 50 mM NH4Cl, blocked, and permeabilized for 20 min in Dulbecco's PBS with 50 mM NH4Cl, 1% BSA, 2% goat serum, and 0.05% Saponin (blocking buffer). Cells were incubated 1 h at RT with primary antibody in blocking buffer and washed four times for 5 min each in Dulbecco's PBS with 50 mM NH4Cl. Cells were incubated 1 h at RT with fluorescein- or rhodamine-conjugated goat secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) in blocking buffer, and then washed four times for 5 min each in Dulbecco's PBS with 50 mM NH4Cl. Antibodies were used in the following dilutions: affinity-purified E-cadherin antiserum 1:50; affinity-purified α-catenin antiserum 1:50; mouse mAb KT3 1:50; β-cat.N antiserum 1:200; and secondary antibodies 1:200. For KT3/APC protein double immunofluorescence, 5 × 105 cells were plated onto collagen-coated coverslips in 35-mm tissue-culture dishes and fixed after 48 h. Cells were washed once in Dulbecco's PBS, fixed for 5 min at −20°C in precooled methanol, washed four times in Dulbecco's PBS, and blocked in Dulbecco's PBS with 1% BSA and 2% goat serum (blocking buffer). Cells were incubated for 1 h at RT with primary antibody in blocking buffer, washed four times for 5 min each in Dulbecco's PBS, incubated 1 h at RT with fluorescein- or rhodamine-conjugated goat secondary antibodies (Jackson ImmunoResearch Laboratories) in blocking buffer, and washed four times for 5 min each in Dulbecco's PBS. Antibodies were used in the following dilutions: mouse mAb KT3 1:50; and APC protein antiserum 1:200. Cells were mounted in a mixture of 16.7% Mowiol (Calbiochem-Novabiochem Corp.), 33% glycerol, and 0.1% phenylenediamine in Dulbecco's PBS without CaCl2 and MgCl2 and viewed with an Axiophot inverted fluorescence microscope (Carl Zeiss Inc., Thornwood, NY) using a ×63 oil immersion objective. For phase-contrast microscopy of low density cultures, each clone was split and cultured for 3 d with or without 20 ng/ml Dox. 5 × 105 cells were plated onto 100-mm tissue-culture dishes in medium with or without 20 ng/ml Dox, and photomicrographs of the cultures were taken 24 and 48 h after plating with an Axiovert 10 phase-contrast microscope (Carl Zeiss Inc.).
Results
Expression of β-Catenin Mutant Proteins in MDCK Cells
Mutant β-catenin proteins lacking NH2-terminal 90 (ΔN90), 131 (ΔN131), 151 (ΔN151) amino acids, or COOH-terminal 86 amino acids (ΔC), and full-length β-catenin (β-catenin*) were expressed in MDCK cells (Fig. 1). The sequence for the SV-40 large T antigen epitope recognized by the mAb KT3 (MacArthur and Walter, 1984) was added to the 3′ termini of all cDNA constructs to distinguish protein products from endogenous β-catenin. All constructs were expressed under the control of the Tet-repressible transactivator; in the presence of either Tet or Dox, expression of exogenous protein is completely repressed (Gossen and Bujard, 1992).
MDCK clones cultured for 4 d without or with doxycycline (−/+ Dox) were analyzed for expression of mutant β-catenins (Fig. 2). Mutant β-catenins (Fig. 2, a–c; marked with stars in β-cat.C, β-cat.N, and KT3 blots) were detected in protein extracts from clones cultured without Dox, but not in extracts from clones cultured with Dox. 15–20 clones were analyzed for every construct. Mutant β-catenin levels in clones expressing ΔN90, ΔN131, or ΔN151 β-catenin were on average higher than that in clones expressing β-catenin* and ΔC β-catenin. However, the level of ΔN151 in clone ΔN151-D was similar to β-catenin* in clone β-catenin*–10 (Fig. 2 c; KT3 blot). In the clones shown in Fig. 2, the ratios of ΔN90, ΔN131, ΔN151, or ΔC β-catenin to MDCK endogenous β-catenin were 5:1, 1:1, 0.5:1, and 1:1, respectively (Fig. 2 a, lanes 7, 9, and 11; Fig. 2 b, lane 5).
Figure 2.

Dox-repressible expression of β-catenin mutant proteins in MDCK cells. MDCK clones were cultured for 4 d without or with Dox (−/+ Dox) and extracted with 1% SDS. 15-μg protein lysates were subjected to SDS-PAGE and immunoblotted with different antibodies (see Fig. 1). β-catenin mutant proteins (stars in a–c) were expressed only in cultures without Dox; endogenous β-catenin was expressed in all cultures (a and b). Expression levels of exogenous full-length β-catenin*, ΔN90, ΔN131, and ΔN151 were compared with that of endogenous β-catenin by immunoblotting with mAb β-cat.C (a). Exogenous β-catenin* and ΔC were compared with endogenous β-catenin by immunoblotting with β-cat.N (b). The expression levels of mutant β-catenin proteins in different clones were compared by immunoblotting with the tag antibody KT3 (c). The β-cat.C blot was reblotted with E-cadherin antiserum (d). The KT3 blot was reblotted with α-catenin antiserum (e). Molecular mass standards are indicated in kD.
Expression of E-cadherin leads to an increase in the cellular levels of catenins (Herrenknecht et al., 1991; Weißig, 1993). Therefore, we analyzed whether expression of β-catenin mutant proteins affected levels of endogenous β-catenin, E-cadherin, or α-catenin by comparing their amounts in lysates from clones cultured without or with Dox. Expression of ΔN90, ΔN131, or ΔN151 did not significantly affect the level of endogenous β-catenin (Fig. 2, a and b, compare lanes 7 to 8, 9 to 10, and 11 to 12, respectively). Expression of ΔC β-catenin resulted in a small but reproducible decrease in amount of endogenous β-catenin (Fig. 2, a and b, lanes 5 and 6). Although full-length exogenous β-catenin* and endogenous β-catenin were not always well separated by SDS-PAGE because of similarity in their electrophoretic mobilities, a decrease of endogenous β-catenin in response to the expression of β-catenin* was detectable in some of these clones (Fig. 2 a, lanes 3 and 4).
The amount of E-cadherin was slightly higher in clones expressing ΔN90 β-catenin (ΔN90-A) and ΔN131 β-catenin (ΔN131-D) compared with levels in β-catenin–10* and ΔC clones (Fig. 2 d). Differences in E-cadherin levels may be the result of clonal variation since the amount of E-cadherin in a particular clone was not significantly affected after repression of mutant β-catenin expression by addition of Dox (Fig. 2 d; compare lanes 7 to 8, 9 to 10, and 11 to 12, respectively). Expression of mutant β-catenins did not have a significant effect on amounts of α-catenin in all clones except ΔN131-D in which the α-catenin level was slightly decreased when cells were cultured without Dox (Fig. 2 e, lanes 9 and 10).
Mutant β-Catenin Proteins Compete with Endogenous β-Catenin for Binding to E-Cadherin and α-Catenin
Binding of β-catenin mutants to E-cadherin and α-catenin was analyzed by coimmunoprecipitation of protein complexes (Fig. 3). The binding site for E-cadherin in β-catenin is located within the armadillo repeat domain of β-catenin (Aberle et al., 1994; Hülsken et al., 1994) and was retained in all mutant proteins. Accordingly, all β-catenin mutant proteins were coimmunoprecipitated by E-cadherin antibody (Fig. 3 a; KT3 blot). Binding of ΔN90 and ΔN131 β-catenin to E-cadherin resulted in a reduction in amount of endogenous β-catenin complexed with E-cadherin compared with the amount of endogenous β-catenin in similar complexes isolated from the same clones cultured with Dox (Fig. 3 b; β-cat.C blot; compare lanes 5 and 6, and 7 and 8, respectively). The binding site for α-catenin is located between amino acids 120 and 151 in β-catenin (Aberle et al., 1994, 1996) and was incomplete or missing in ΔN131 and ΔN151 β-catenin. Accordingly, ΔN131 and ΔN151 β-catenin were not coimmunoprecipitated by α-catenin antibody (Fig. 3 c; KT3 blot). Binding of ΔN90 and ΔC β-catenin to α-catenin resulted in a reduction in amount of endogenous β-catenin bound to α-catenin compared with endogenous β-catenin in complexes with α-catenin that were isolated from the same clones cultured in the presence of Dox (Fig. 3 d; β-cat.C blot; compare lanes 5 and 6, and 11 and 12, respectively). These results confirm that ΔN90 and ΔC β-catenin retained both E-cadherin and α-catenin binding sites, and that ΔN131 and ΔN151 β-catenin retained the E-cadherin, but not α-catenin, binding site; all mutant β-catenin proteins competed with endogenous β-catenin for these binding partners.
Figure 3.

β-catenin mutant proteins compete with endogenous β-catenin for binding to E-cadherin and α-catenin. MDCK clones were cultured 4 d without or with Dox (−/+) and extracted with 1% Triton X-100 lysis buffer. Protein lysates were split: 400-μg lysates were immunoprecipitated with E-cadherin antiserum to analyze binding of mutant β-catenin proteins to E-cadherin (a and b). 400 μg protein lysates were immunoprecipitated with α-catenin antiserum to analyze binding of mutant β-catenin proteins to α-catenin (c and d). Equivalent fractions of the immunoprecipitates were subjected to SDS-PAGE and blotted with mAbs KT3 (a and c) or β-cat.C (b and d). β-catenin mutant proteins are indicated (stars).
NH2-terminal–deleted Mutant Proteins of β-Catenin Are Enriched in APC Protein Complexes
Binding of mutant β-catenins to APC protein was also analyzed by coimmunoprecipitation. All mutant β-catenins were coimmunoprecipitated by APC protein antibody (Fig. 4). However, ΔN90, ΔN131, and ΔN151 β-catenin were significantly enriched in APC protein immunoprecipitates compared with endogenous β-catenin, ΔC, or exogenous β-catenin*. Three times more ΔN90, ΔN131, or ΔN151 was coimmunoprecipitated with APC protein than endogenous β-catenin (Fig. 4 c). Note that total amounts of mutant β-catenin in lysates used for immunoprecipitation were either similar to, or less than that of, endogenous β-catenin (Fig. 4 a); the ratios of ΔN90, ΔN131, or ΔN151 β-catenin to endogenous β-catenin in Triton X-100 lysates were 1.6:1, 1.3:1, and 0.1:1, respectively. Very little ΔC β-catenin was detected in APC protein complexes, although as much ΔC as ΔN151 β-catenin was present in the Triton X-100 lysates of each clone (compare Fig. 4 b and 4 d; KT3 blots).
Figure 4.
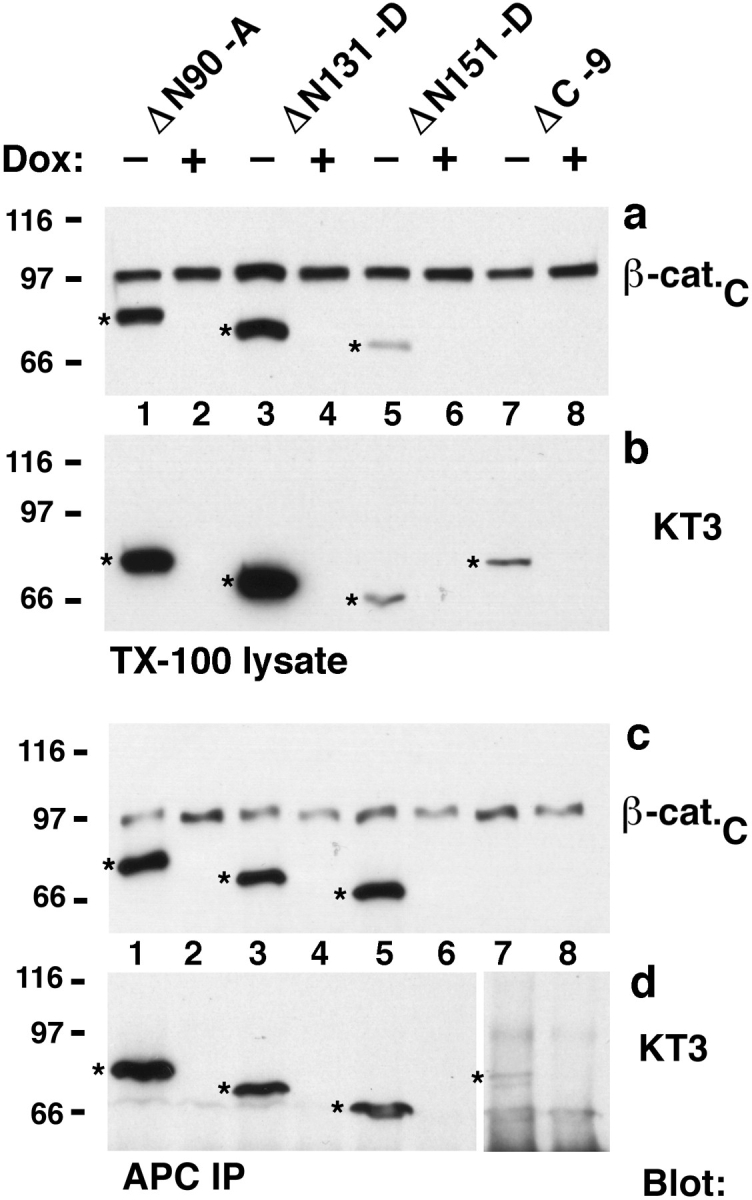

ΔN90, ΔN131, and ΔN151 are enriched in APC protein complexes. Aliquots of the same Triton X-100 lysates as described in Fig. 3 were either subjected to SDS-PAGE or used for immunoprecipitation with APC antiserum (a–d). 9-μg protein lysates were subjected to SDS-PAGE and immunoblotted with the indicated mAbs to compare the ratio of β-catenin mutant proteins to endogenous β-catenin (a), and to each other (b). 1,500-μg protein lysates were immunoprecipitated with APC antiserum. Equivalent fractions of the immunoprecipitates were subjected to SDS-PAGE and blotted with the indicated mAbs to analyze binding of mutant β-catenin proteins to APC (c–d). β-catenin mutant proteins are indicated (stars). A longer exposure of the blot in (d) is shown for lanes 7 and 8. Triton X-100 lysates were prepared from clones β-catenin*–7 and ΔN131-7, and aliquots of the lysates were immunoprecipitated with antiserum β-cat.C or APC antiserum (e). Immunoprecipitates were subjected to SDSPAGE and blotted with antiserum β-cat.C to analyze the ratio of exogenous β-catenin* and ΔN131 to endogenous β-catenin. β-catenin mutant proteins are indicated (stars).
These data indicate that, compared with endogenous β-catenin, NH2-terminal–deleted β-catenins bind preferentially to APC protein. In addition, the amount of ΔN151 β-catenin that is complexed with APC protein is very similar to that of ΔN90 and ΔN131 β-catenin, even though there is much less ΔN151 β-catenin in clone ΔN151-D. Therefore, we suspect that binding of NH2-terminal–deleted β-catenin to APC protein approaches a maximum.
To further explore the degree of enrichment of mutant β-catenin in APC protein complexes, amounts of β-catenin* and ΔN131 β-catenin bound to APC protein were compared in two different clones (β-cat.*-7 and ΔN131-7; Fig. 4 e). Portions of Triton X-100 lysates of these clones were used to immunoprecipitate both full-length and deleted β-catenin with β-cat.C antiserum, or to coimmunoprecipitate APC protein complexes. In β-cat.C immunoprecipitates, the ratios of endogenous β-catenin to β-catenin* in the β-catenin*-7 clone, and those of endogenous β-catenin to ΔN131 in the ΔN131-7 clone, were ∼1:1. In APC protein immunoprecipitates, the ratio of endogenous β-catenin to β-catenin* was also ∼1:1, but, in contrast, the ratio of endogenous β-catenin to ΔN131 β-catenin was ∼1:3. ΔN131 β-catenin was considerably more abundant in APC protein immunoprecipitates than either endogenous β-catenin or exogenous full-length β-catenin* (Fig. 4 e).
NH2-terminal Deletions Result in Increased β-Catenin Stability in APC Protein and E-Cadherin Complexes
Enrichment of ΔN90, ΔN131,and ΔN151 β-catenins bound to APC protein could be caused by increased stability of these β-catenins in the complex. To test this hypothesis, expression of β-catenin mutant proteins was repressed by addition of Dox to cultures for 0, 6, 12,or 18 h. Amounts of mutant β-catenin remaining in Triton X-100 lysates of these cells, and in E-cadherin and APC protein complexes isolated from those lysates, were analyzed by immunoprecipitation and/or Western blotting (Fig. 5).
Figure 5.

Increased stability of ΔN90, ΔN131, and ΔN151 in the E-cadherin– and APC protein–bound pools. MDCK clones were cultured 0, 6, 12, or 18 h with Dox and extracted with 1% Triton X-100 lysis buffer. Protein lysates were split: 1,500-μg protein lysates were immunoprecipitated with APC antiserum (first column), and 500-μg lysates were immunoprecipitated with E-cadherin antiserum (second column). Equivalent fractions of the immunoprecipitates and 50-μg (β-cat.*-10,-7, ΔC-9) or 25-μg (ΔN90-A, ΔN131-D, ΔN151-D) protein lysates (third column) were subjected to SDS-PAGE and immunoblotted with mAb KT3. For β-catenin*–10, –7, and ΔC-9 blots, three times more of the APC immunoprecipitations were used than for the ΔN90-A, ΔN131-D, and ΔN151-D blots, and the blots were exposed longer.
A significant increase in the relative stabilities of NH2terminal–deleted β-catenins compared with full-length β-catenin* or ΔC β-catenin was detected in E-cadherin and APC protein complexes from lysates. This difference in stability was most pronounced in the APC protein– bound pools. Very little or no full-length β-catenin* or ΔC β-catenin were detected in the APC protein complex after only 6 h of treatment with Dox. In contrast, there was little or no decrease in amounts of ΔN90, ΔN131, or ΔN151 β-catenin in the APC protein complex after 18 h of treatment with Dox. In the E-cadherin–bound pools of lysates, 10– 12% of the original amount of full-length β-catenin* and ΔC β-catenin was detected after 12 h of treatment with Dox. In contrast, amounts of ΔN90, ΔN131, or ΔN151 β-catenin bound to E-cadherin were not or little reduced after 18 h of treatment with Dox.
In the total Triton X-100 lysates, the amounts of all mutant β-catenin proteins were reduced after 12 to 18 h of treatment with Dox. The percentage of remaining mutant protein after 12 h of treatment with Dox is 17% and 18% of β-catenin* in clones β-catenin*–7 and β-catenin*–10, respectively; 18% of ΔC β-catenin; 44% of ΔN90 β-catenin; 32% of ΔN131 β-catenin; and 26% of ΔN151 β-catenin. The close similarity in the decrease in amounts of β-catenin* in two independent clones indicates that the rate and efficiency of Dox repression of gene expression were similar in different MDCK clones. Assuming that this is also the case in other clones, the half-life of ΔC is similar to that of full-length β-catenin*, whereas ΔN90, ΔN131, and ΔN151 β-catenin are slightly more stable than full-length β-catenin*. The mutant β-catenin proteins in the total TX100 lysates represent the sum of the E-cadherin– and APC-bound, and unbound pools. Although ΔN90, ΔN131, or ΔN151 β-catenin was very stable in E-cadherin and APC protein complexes, their overall stability in the lysates was not very different from that of full-length β-catenin* and ΔC β-catenin. This indicates that pools of ΔN90, ΔN131, and ΔN151 β-catenin that are not bound to either E-cadherin or APC protein turn over at a rate similar to that of full-length β-catenin* and ΔC β-catenin.
The high stability of the NH2-terminal–deleted β-catenins in the APC protein complexes correlated with the enrichment of these mutant proteins in the APC protein complexes compared with endogenous β-catenin in the same cells (Fig. 4). Surprisingly, we could not detect an enrichment of NH2-terminal–deleted β-catenins in the E-cadherin pool when compared with endogenous β-catenin in the same cells (see Fig. 3 c), even though NH2-terminal–deleted β-catenins were more stable than full-length exogenous β-catenin* in the E-cadherin complexes of different clones (Fig. 5). We note that most of the E-cadherin in MDCK cells is normally bound to endogenous β-catenin in a 1:1 molar ratio (Hinck et al., 1994a ). In contrast, most of the APC protein appears to be free of endogenous β-catenin (Näthke et al., 1996). Therefore, NH2-terminal– deleted β-catenins must compete with endogenous β-catenin to bind to E-cadherin, but they may be enriched in the APC protein pool by binding unoccupied sites and remaining stabilized in these complexes. This may explain the difference in the ratios of the mutant proteins to endogenous β-catenin when comparing the E-cadherin– bound or APC-bound pools in the same cultures (Figs. 3 and 4).
NH2-terminal–deleted β-Catenin Prominently Localizes in Clusters with APC Protein at Tips of Plasma Membrane Protrusions
Our immunoprecipitation studies show that NH2-terminal–deleted β-catenin proteins form stable complexes with E-cadherin and APC protein. We examined the subcellular distribution of mutant β-catenins by immunofluorescence microscopy. Mutant β-catenins were epitope tagged and can be distinguished from endogenous β-catenin with the monoclonal anti-tag antibody KT3. We examined two types of cell cultures: low density cultures in which small groups of cells had initiated cell–cell contacts (Fig. 6), and high density cultures in which mature cell–cell contacts had been established (Fig. 7).
Figure 6.
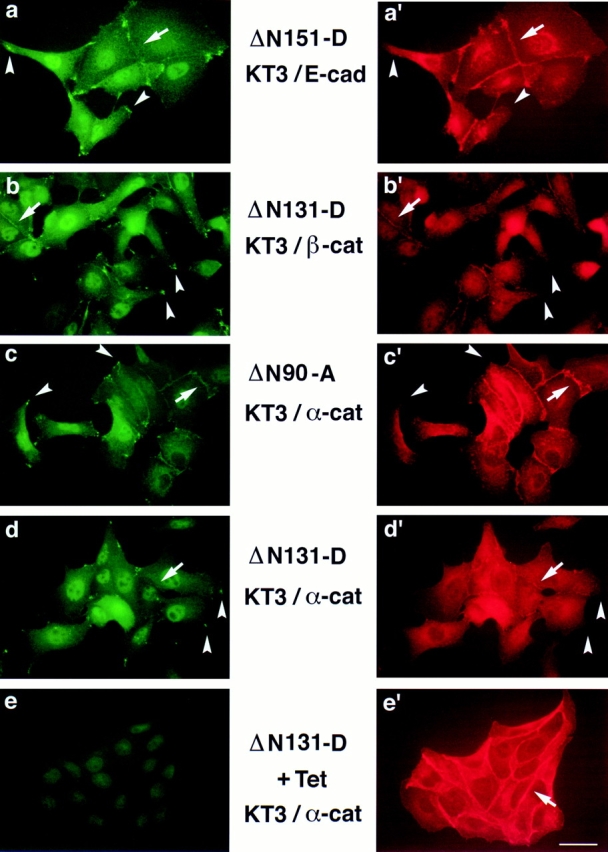
ΔN151, ΔN131, and ΔN90 localize to clusters near the plasma membrane in extending membranes of MDCK cells. MDCK clones were double stained with mAb KT3 against the epitope tag in β-catenin mutant proteins (a–e) and antiserum against E-cadherin (a′), antiserum β-cat.N against endogenous β-catenin (b′), and antiserum against α-catenin (c′–e′). Areas with ΔN151, ΔN131, and ΔN90 clusters are indicated by arrowheads (a–d). These clusters are not detected with E-cadherin, endogenous β-catenin, or α-catenin antisera (a′–d′, arrowheads). ΔN131 expression was repressed in ΔN131-D by 48 h incubation with Tet before fixation (e). (Arrows) Areas of intercellular contact that are stained by antisera to E-cadherin (a′), endogenous β-catenin (b′), α-catenin (c′–e′), and sometimes weakly by mAb KT3 (a–d). Bar, 10 μm.
Figure 7.
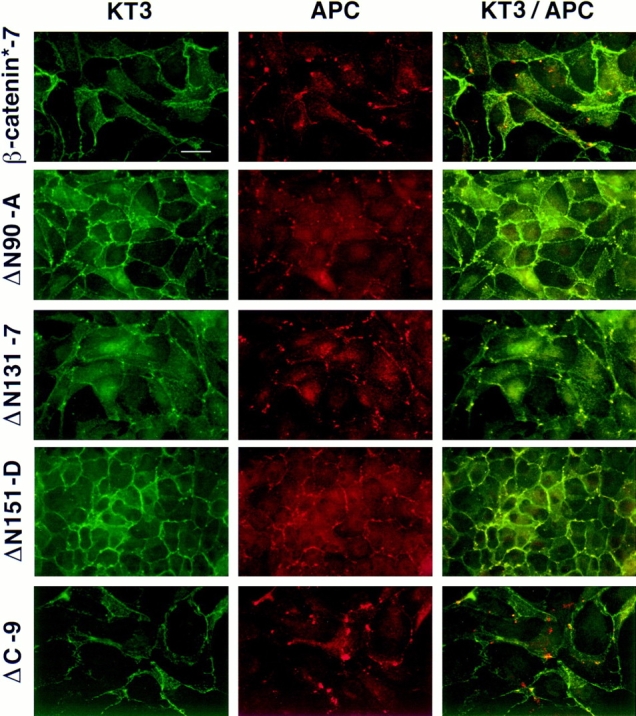
ΔN90, ΔN131, and ΔN151 colocalize with APC protein in MDCK cells. MDCK clones were double stained with mAb KT3 against the epitope tag in β-catenin mutant proteins and antiserum against APC protein. Full-length β-catenin* and ΔC localize to the lateral membranes of cells and show little overlap with APC protein clusters (top and bottom panel). ΔN90, ΔN131, and ΔN151 localize to lateral membranes of cells and colocalize with APC protein in clusters (middle panels). Bar, 10 μm.
ΔN90, ΔN131, and ΔN151 β-catenins prominently localized to clusters at the outer boundary of cell–cell contacts and at the tips of plasma membrane protrusions (Fig. 6, a–d, arrowheads). Neither E-cadherin, endogenous β-catenin, nor α-catenin colocalize with the NH2-terminal– deleted β-catenins in theses clusters (Fig. 6, a′–d′, arrowheads). This distinctive subcellular distribution is very similar to that of APC protein in MDCK cells (Näthke et al., 1996). Therefore, we examined colocalization of the mutant β-catenin proteins with APC protein in these clusters (Fig. 7). ΔN90, ΔN131, and ΔN151 β-catenins colocalized precisely with APC protein in almost all clusters at the tips of plasma membrane protrusions; incidences of APC protein clusters without ΔN90, ΔN131, or ΔN151 β-catenins were very rare (Fig. 7, middle panels). However, fulllength β-catenin* and ΔC β-catenin localized to intercellular contacts, and there was little overlap in distributions between these proteins and APC protein (Fig. 7, top and bottom panel).
In low density cultures, ΔN90, ΔN131, and ΔN151 β-catenins were poorly localized at intercellular contacts, compared with strong staining for E-cadherin and α-catenin at the same sites (Fig. 6). In these cell–cell contact areas, NH2-terminal–deleted β-catenin mutant proteins colocalized with E-cadherin, endogenous β-catenin, and α-catenin (Fig. 6, a–d, and a′–d′, arrows). Localization of ΔN90, ΔN151, and ΔN131 β-catenins to intercellular contacts was much more prominent in high density cultures (Fig. 7; KT3 immunofluorescence). The distribution of β-catenin* and ΔC was identical to that of endogenous β-catenin (data not shown).
Note that some nuclear staining was detected with the KT3 antibody (see Fig. 6, a–d), but that the staining persisted in the absence of mutant protein expression (see Fig. 6 e), indicating that nuclear staining was not specific to mutant β-catenin in these cells.
Distinct Differences in Morphologies of Cells Expressing NH2-terminal–deleted β-Catenin Compared With Cells Expressing β-Catenin* and ΔC β-Catenin
We examined the morphology and colony formation of cells expressing different mutant β-catenins. In low density cultures, the morphology of cells expressing ΔN90, ΔN131, or ΔN151 β-catenins was distinctly different from that of the same cells treated with Dox (i.e., without mutant β-catenin expression; Fig. 8), parental MDCK cells, and cells expressing β-catenin* or ΔC β-catenin (data not shown). Cells treated with Dox, parental MDCK cells, and cells expressing β-catenin* or ΔC β-catenin produced compact cell colonies; note that cells at the edges of colonies rarely extended membrane protrusions onto the surrounding cell-free surface. In addition, we found very few individual cells that were not associated with colonies in these cultures. Neither addition of Dox to the cultures nor addition of the KT3 tag to full-length β-catenin affected the morphology of either parental MDCK cells (Fig. 8, top panel) or β-catenin*–expressing cells (data not shown), respectively. In contrast, the surface area of cells expressing ΔN90, ΔN131, or ΔN151 β-catenins (i.e., in the absence of Dox) was greater than that of control cells, indicating that the former were poorly compacted. Cells loosely associated in colonies also had many membrane extensions projecting onto the surrounding cell-free surface. In addition, many individual cells were not associated directly with colonies and were dispersed throughout the culture; in general, these cells have a more fibroblastic morphology than control cells (Fig. 8). Repression of ΔN90, ΔN131, or ΔN151 β-catenin expression by treatment with Dox reverted the morphology of cells to compacted colonies typical of parental MDCK cells (Fig. 8, top panels; see also Fig. 6, e and e′).
Figure 8.

Effect of ΔN90, ΔN131, and ΔN151 expression on colony formation in low density MDCK cultures. Cultures of MDCK clones were untreated or pretreated 3 d with Dox and plated at low density without (−) or with (+) Dox. Cell morphology was analyzed 24 and 48 h after plating. Expression of ΔN90, ΔN131, and ΔN151 delayed formation of tight round colonies in low density cultures (first and third columns). This effect could be reversed by repression of expression of ΔN90, ΔN131, and ΔN151 with Dox (second and fourth columns). Dox itself had no effect on the morphology of the parental cells (top panel). Bar, 40 μm.
At higher cell densities in which there was less free surface available for cell migration, differences in morphologies of cells expressing ΔN90, ΔN131, or ΔN151 β-catenins and control cells were less evident. Cells expressing ΔN90, ΔN131, or ΔN151 β-catenins established compact colonies and, eventually, formed complete monolayers similar to those of control cells (see Fig. 7).
Discussion
β-Catenin binds independently to E-cadherin and APC protein, and one function of these interactions is to link E-cadherin to the actin cytoskeleton via α-catenin (Aberle et al., 1994; Hülsken et al., 1994; Jou et al., 1995; Rimm et al., 1995). In the case of E-cadherin, this hierarchy of protein interactions is important for regulating E-cadherin function in establishing cell–cell adhesion (Ozawa and Kemler, 1992). The role of catenin binding in APC protein function(s) is not known, but it has been suggested that APC protein may regulate cellular β-catenin levels (Munemitsu et al., 1995; Papkoff et al., 1996). In this study, we examined interactions of β-catenin mutant proteins with E-cadherin, α-catenin, and APC protein, and investigated their subcellular distributions and effects on the morphology of MDCK epithelial cells. Our results provide novel insights into the roles of β-catenin in regulating E-cadherin and/or APC protein functions: (a) NH2-terminal deletion of β-catenin results in stabilization of β-catenin in APC protein complexes; (b) full-length β-catenin poorly colocalizes with APC protein, but NH2-terminal–deleted β-catenin strongly colocalizes with APC protein in clusters at the tips of membrane protrusions; (c) expression of NH2-terminal–deleted β-catenin affects cell morphology and intercellular adhesion; (d) the effects on cell morphology are independent of β-catenin binding to α-catenin; and (e) alterations in β-catenin binding to APC protein, the subcellular location of mutant β-catenin/APC protein complexes, and the resulting changes in cell morphology imply that APC protein is involved in cell migration and that β-catenin binding to APC protein regulates this function.
Regulation of β-Catenin Distribution between Different Cellular Pools
Epitope-tagged ΔN90, ΔN131, and ΔN151 and ΔC β-catenin were expressed in MDCK cells using a Dox/Tet-repressible expression system. As an additional control, epitopetagged full-length β-catenin (β-catenin*) was expressed in MDCK cells. Expression of β-catenin mutant proteins had no significant effect on cellular levels of E-cadherin and α-catenin. However, a small but reproducible reduction in the amount of endogenous β-catenin was observed in cells expressing full-length β-catenin* or ΔC β-catenin (Fig. 2). All β-catenin mutant proteins competed with MDCK endogenous β-catenin for binding to E-cadherin (Figs. 3 and 4). Exogenous full-length β-catenin*, ΔN90, and ΔC β-catenin competed with MDCK endogenous β-catenin for binding to α-catenin, whereas ΔN131 and ΔN151 β-catenin were not associated with α-catenin (Fig. 3).
Comparison of the amounts of NH2-terminal–deleted and endogenous β-catenin coimmunoprecipitated in a complex with APC protein revealed that mutant β-catenins were specifically enriched. By comparison, endogenous β-catenin was not enriched in the same coimmunoprecipitates (Fig. 4). The enrichment of mutant β-catenin in the APC protein complex was independent of binding of mutant β-catenin to α-catenin. Our data suggest that the NH2-terminal domain of β-catenin is important for regulating its stability in the APC protein complex (Fig. 5). Similar observations have been reported recently by Munemitsu et al. (1996). APC protein stimulates β-catenin degradation in some cell lines (Munemitsu et al., 1995). Although the mechanism regulating the dynamics of β-catenin interactions with APC protein remains to be elucidated, phosphorylation of APC protein (Rubinfeld et al., 1996) and/or β-catenin by glycogen synthase kinase 3β (GSK3β) may be important. GSK3β associates with the APC protein/βcatenin complex in some cell lines (Rubinfeld et al., 1996). The NH2 terminus of β-catenin contains a GSK3β phosphorylation site, and we are currently investigating the role of this site for β-catenin stability in the APC complex. Interestingly, β-catenin mutants lacking this site are more stable than wild-type β-catenin in Xenopus embryos (Yost et al., 1996), indicating that the phosphorylation state of β-catenin regulates β-catenin turnover. Our data also show that NH2-terminal–deleted β-catenin is more stable in E-cadherin complexes than full-length and ΔC β-catenin (Fig. 5). Regulation of β-catenin stability in E-cadherin complexes may be determined by mechanism(s) similar to those in the APC protein complexes.
NH2-terminal–deleted β-Catenin Mutant Proteins Colocalize with APC Protein in Clusters at Tips of Plasma Membrane Protrusions
Recent studies by Näthke et al (1996) demonstrated that APC protein localizes to clusters of puncta at the tip of actively migrating cellular protrusions and at the outer boundaries of newly formed cell–cell contacts. This distribution indicates that one function of APC protein is to regulate specific types of cell migration. Whereas endogenous β-catenin is seldom detected in the APC protein clusters, ΔN90, ΔN131, and ΔN151 β-catenin prominently colocalized with APC protein in these clusters (Fig. 7). This colocalization was independent of mutant β-catenin binding to α-catenin, since neither ΔN131 nor ΔN151 β-catenin bound α-catenin. Surprisingly, localization of ΔN90 β-catenin to APC protein clusters was not accompanied by strong colocalization of α-catenin, even though this mutant β-catenin retained an α-catenin binding site and formed complexes with α-catenin (Fig. 6, c and c′).
Preliminary studies show that localization of ΔN90, ΔN131, and ΔN151 β-catenin in APC protein clusters is disrupted by nocodazole but not by cytochalasin D (data not shown), suggesting that linkage of the APC protein complex to microtubules, rather than to actin filaments, may be more important for APC protein localization and, perhaps, function (see also Näthke et al., 1996).
Expression of NH2-terminal–deleted β-Catenin Inhibits Colony Formation in Low Density Cultures
We examined the effects of mutant β-catenin expression on MDCK cell morphology. Cultures of cells were initiated from single cells that normally migrate, form contacts with other cells, and, by 24 h, establish small colonies. Three general differences in morphology were detected in cells expressing ΔN90, ΔN131, or ΔN151 β-catenin compared with control cells. At low density, cells were dispersed, more fibroblastic in morphology, and less efficient at forming colonies. At high density, cells formed monolayers similar in morphology to those of controls (data not shown). Differences in morphology of cells expressing mutant β-catenin compared with control cells may be due to a combination of decreased cell–cell adhesion and either decreased movement resulting in a lower probability of cells contacting each other, or increased migration resulting in dispersion of cells.
Poor colony formation and decreased compaction of cells within colonies may be caused by interference of E-cadherin function by β-catenin mutants. E-cadherin plays a key role in the initial formation of cell–cell contacts in MDCK cells (Gumbiner et al., 1988; McNeill et al., 1993), and linkage of E-cadherin to the cortical actin cytoskeleton via β-catenin and α-catenin is essential for cell–cell adhesion (Hirano et al., 1987; Nagafuchi and Takeichi, 1988; Ozawa et al., 1990). ΔN131 and ΔN151 β-catenin may disrupt E-cadherin function because these mutant proteins cannot bind α-catenin and, hence, the actin cytoskeleton to E-cadherin. This inhibitory effect may be overcome at higher cell densities because cells are forced into contacts and there is sufficient E-cadherin bound to endogenous β-catenin to function in cell–cell contact. However, cells expressing ΔN90 β-catenin, which does bind α-catenin, exhibited the same morphology as that of ΔN131 and ΔN151 β-catenin–expressing cells. This result indicates that expression of NH2-terminal–deleted β-catenin affects colony formation in low density cultures independently of the binding of β-catenin to α-catenin. Furthermore, this phenotype correlates with the dominant colocalization of all three NH2-terminal–deleted β-catenin mutant proteins with APC protein in clusters at the ends of membrane protrusions. Previous studies have shown that these APC protein clusters localize to the ends of microtubules, and that this localization is independent of the actin cytoskeleton (Näthke et al., 1996). A role of clusters of APC protein in cell migration and/or early contact formation has been suggested (Näthke et al., 1996). Based upon the results of the present study, we suggest that stable binding of β-catenin to APC protein may affect a function of APC protein in these clusters in microtubule organization and/or cell migration. Thus, in low density cultures, altered migratory behavior of cells may reduce the time and/or number of intercellular contacts that enable cells to initiate stable cell– cell adhesion, thereby giving rise to the observed defect in colony formation. Furthermore, APC protein may be directly involved in the initiation of stable adhesion between extending membranes of these cells (Näthke et al., 1996). A detailed analysis of the role of the APC/β-catenin complex underlying this phenotype is currently under investigation.
Role of β-Catenin in Signaling Pathways and Embryonic Development
NH2-terminal–deleted β-catenin mutant proteins have been used to study the function of β-catenin in cell signaling during embryonic development (Fagotto et al., 1996; Funayama et al., 1995). In Drosophila, the β-catenin homologue armadillo was originally identified through its role in transduction of the wingless/Wnt cell–cell signal that mediates cell fate determination (Noordermeer et al., 1994; Peifer et al., 1991; Peifer and Wieschaus, 1990; Siegfried et al., 1994). In vertebrates, the wingless/Wnt signaling pathway may be involved in the formation of the dorsal-ventral axis during Xenopus embryogenesis, and β-catenin seems to be a downstream component of this pathway (Cui et al., 1995; Funayama et al., 1995; Heasman et al., 1994; McCrea et al., 1993; Sokol et al., 1991). The cellular mechanisms by which β-catenin receives or transmits the wingless/Wnt signal are not well understood. There is evidence that β-catenin has distinct roles in Wingless/Wnt signaling and cell adhesion, and that β-catenin binding to cadherin antagonizes its signaling function (Cox et al., 1996; Fagotto et al., 1996; Orsulic and Peifer, 1996). In contrast to these results, ectopic Wnt expression in mammalian cell lines leads to stabilization of β-catenin or its homologue plakoglobin in the cadherin/catenin complex and to increased adhesion (Bradley et al., 1993; Hinck et al., 1994b ). Ectopic expression of NH2-terminal–deleted β-catenin mutant proteins similar to the ones used in this study or of β-catenin with a mutated GSK3β phosphorylation site induced formation of a secondary dorsal body axis in Xenopus embryos (Fagotto et al., 1996; Funayama et al., 1995; Guger and Gumbiner, 1995; Yost et al., 1996). These investigators noted that mutant β-catenin protein was found in the nucleus. Yost et al. (1996) showed also that inhibition of GSK3β by overexpression of a dominant negative mutant causes nuclear localization of endogenous β-catenin in Xenopus embryos. This localization may be important for regulating cell fate determination at the gene transcription level.
In this study, we have shown that mutant β-catenin proteins form stable complexes with cadherin and APC protein. Although we have not directly analyzed the function of endogenous β-catenin in APC protein complexes, our results indicate strongly that a change in the dynamics of β-catenin binding to APC protein resulting in stabilization of the β-catenin/APC protein complex coincides with alterations in cell adhesion, migration, and morphology. Given the possibility that one function of APC protein is in regulating cell migration (Näthke et al., 1996), we suggest that the dynamics of β-catenin binding to APC protein plays a key role in regulating APC protein function in these events during complex cell movements. In support of this idea, we have shown recently (Pollack, A., A. Barth, W.J. Nelson, and K.E. Mostov, manuscript submitted for publication) that expression of these mutant β-catenins inhibits hepatocyte growth factor/scatter factor–induced tubulogenesis of MDCK cysts. The initial stage of tubulogenesis involves migration of long cell extensions from the cyst into the surrounding matrix; subsequently, these extensions reorganize into tubules. Expression of NH2-terminal–deleted β-catenin inhibits formation of these cell extensions and subsequent tubule formation. Significantly, mutant β-catenin and APC protein colocalize prominently at the tips of short membrane extensions that do not migrate further. Precise control of cell–cell adhesion and migration is important during complex morphogenetic processes such as gastrulation and neurulation. Thus, in addition to affecting signaling pathways, β-catenin mutant proteins may alter morphogenetic movements of cells by impairing the functions of E-cadherin and/or APC protein in cell adhesion and migration.
Acknowledgments
This work was supported by grants to W.J. Nelson from the National Institutes of Health (NIH) and American Cancer Society, and by grants to K.E. Mostov from the NIH. A. Barth was supported by a NATO fellowship from the Deutscher Akademischer Austauschdienst and an American Heart Association fellowship.
Abbreviations used in this paper
- APC
adenomatous polyposis coli
- Dox
doxycycline
- GSK3β
glycogen synthase kinase 3β
- RT
room temperature
- Tet
tetracycline
Footnotes
We are very thankful to Drs. Inke Näthke and Paul Polakis for providing APC protein antisera and for helpful discussions. We also thank Dr. Rolf Kemler for providing the β-catenin cDNA; Dr. Hermann Bujard for providing the plasmids for the tetracycline-repressible expression system; Dr. Gernot Walter for providing the mAb KT3; and Drs. Helen McNeill and Eugenio de Hostos for their advice during preparation of the manuscript.
A.I.M. Barth and A.L. Pollack contributed equally to this work.
References
- Aberle, H., S. Butz, J. Stappert, H. Weissig, R. Kemler, and H. Hoschuetzky. 1994. Assembly of the cadherin-catenin complex in vitro with recombinant proteins. J. Cell Sci. 3655–3663. [DOI] [PubMed]
- Aberle H, Schwartz H, Hoschuetzky H, Kemler R. Single amino acid substitutions in proteins of the armadillo gene family abolish their binding to α-catenin. J Biol Chem. 1996;271:1520–1526. doi: 10.1074/jbc.271.3.1520. [DOI] [PubMed] [Google Scholar]
- Bradley RS, Cowin P, Brown AMC. Expression of Wnt-1 in PC12 cells results in modulation of plakoglobin and E-cadherin and increased cellular adhesion. J Cell Biol. 1993;123:1857–1866. doi: 10.1083/jcb.123.6.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butz S, Stappert J, Weissig H, Kemler R. Plakoglobin and betacatenin: distinct but closely related [letter] Science (Wash DC) 1992;257:1142–1144. doi: 10.1126/science.257.5073.1142-a. [DOI] [PubMed] [Google Scholar]
- Cox RT, Kirkpatrick C, Peifer M. Armadillo is required for adherens junction assembly, cell polarity, and morphogenesis during Drosophilaembryogenesis. J Cell Biol. 1996;134:133–148. doi: 10.1083/jcb.134.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Brown JD, Moon RT, Christian JL. Xwnt-8b: a maternally expressed XenopusWnt gene with a potential role in establishing the dorsoventral axis. Development (Camb) 1995;121:2177–2186. doi: 10.1242/dev.121.7.2177. [DOI] [PubMed] [Google Scholar]
- Elferink LA, Peterson MR, Scheller RH. A role for synaptotag- min (p65) in regulated exocytosis. Cell. 1993;72:153–159. doi: 10.1016/0092-8674(93)90059-y. [DOI] [PubMed] [Google Scholar]
- Fagotto F, Funayama N, Gluck U, Gumbiner BM. Binding to cadherins antagonizes the signaling activity of β-catenin during axis formation in Xenopus. . J Cell Biol. 1996;132:1105–1114. doi: 10.1083/jcb.132.6.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funayama N, Fagotto F, McCrea P, Gumbiner BM. Embryonic axis induction by the armadillo repeat domain of β-catenin: evidence for intracellular signaling. J Cell Biol. 1995;128:959–968. doi: 10.1083/jcb.128.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- Guger KA, Gumbiner BM. β-catenin has Wnt-like activity and mimics the Nieuwkoop signaling center in Xenopusdorsal-ventral patterning. Dev Biol. 1995;172:115–125. doi: 10.1006/dbio.1995.0009. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM. Epithelial morphogenesis. Cell. 1992;69:385–387. doi: 10.1016/0092-8674(92)90440-n. [DOI] [PubMed] [Google Scholar]
- Gumbiner B, Stevenson B, Grimaldi A. The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J Cell Biol. 1988;107:1575–1587. doi: 10.1083/jcb.107.4.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R. Lack of beta-catenin affects mouse development at gastrulation. Development (Camb) 1995;121:3529–3537. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- Heasman J, Crawford A, Goldstone K, Garner HP, Gumbiner B, McCrea P, Kintner C, Noro CY, Wylie C. Overexpression of cadherins and underexpression of beta-catenin inhibit dorsal mesoderm induction in early Xenopusembryos. Cell. 1994;79:791–803. doi: 10.1016/0092-8674(94)90069-8. [DOI] [PubMed] [Google Scholar]
- Herrenknecht K, Ozawa M, Eckerskorn C, Lottspeich F, Lenter M, Kemler R. The uvomorulin-anchorage protein alpha-catenin is a vinculin homologue. Proc Natl Acad Sci USA. 1991;88:9156–9160. doi: 10.1073/pnas.88.20.9156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinck L, Näthke IS, Papkoff J, Nelson WJ. Dynamics of cadherin/catenin complex formation: novel protein interactions and pathways of complex assembly. J Cell Biol. 1994a;125:1327–1340. doi: 10.1083/jcb.125.6.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinck L, Nelson WJ, Papkoff J. Wnt-1 modulates cell–cell adhesion in mammalian cells by stabilizing β-catenin binding to the cell adhesion protein cadherin. J Cell Biol. 1994b;124:729–741. doi: 10.1083/jcb.124.5.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano S, Nose A, Hatta K, Kawakami A, Takeichi M. Calciumdependent cell–cell adhesion molecules (cadherins): subclass specificities and possible involvement of actin bundles. J Cell Biol. 1987;105:2501–2510. doi: 10.1083/jcb.105.6.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hülsken J, Birchmeier W, Behrens J. E-cadherin and APC compete for the interaction with β-catenin and the cytoskeleton. J Cell Biol. 1994;127:2061–2069. doi: 10.1083/jcb.127.6.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jou TS, Stewart DB, Stappert J, Nelson WJ, Marrs JA. Genetic and biochemical dissection of protein linkages in the cadherin-catenin complex. Proc Natl Acad Sci USA. 1995;92:5067–5071. doi: 10.1073/pnas.92.11.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D. Identification of FAP locus genes from chromosome 5q21. Science (Wash DC) 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- Kozak M. The scanning model for translation: an update. J Cell Biol. 1989;108:229–241. doi: 10.1083/jcb.108.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (Lond) 1970;227:680–687. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- MacArthur H, Walter G. Monoclonal antibodies specific for the carboxy terminus of simian virus 40 large T antigen. J Virol. 1984;52:483–491. doi: 10.1128/jvi.52.2.483-491.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs JA, Andersson-Fisone C, Jeong MC, Nabi IR, Zurzolo C, Rodriguez-Boulan E, Nelson WJ. Plasticity in epithelial cell phenotype: modulation by differential expression of cadherin cell adhesion molecules. J Cell Biol. 1994;129:509–517. doi: 10.1083/jcb.129.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrea PD, Gumbiner B. Purification of a 92-kD cytoplasmic protein tightly associated with the cell–cell adhesion molecule E-cadherin (uvomorulin) J Biol Chem. 1991;266:4514–4520. [PubMed] [Google Scholar]
- McCrea PD, Turck CW, Gumbiner B. A homolog of the armadillo protein in Drosophila(plakoglobin) associated with E-cadherin. Science (Wash DC) 1991;254:1359–1361. doi: 10.1126/science.1962194. [DOI] [PubMed] [Google Scholar]
- McCrea PD, Brieher WM, Gumbiner BM. Induction of a secondary body axis in Xenopusby antibodies to β-catenin. J Cell Biol. 1993;123:477–484. doi: 10.1083/jcb.123.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill H, Ryan TA, Smith SJ, Nelson WJ. Spatial and temporal dissection of immediate and early events following cadherin-mediated epithelial cell adhesion. J Cell Biol. 1993;120:1217–1226. doi: 10.1083/jcb.120.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munemitsu S, Souza B, Muller O, Albert I, Rubinfeld B, Polakis P. The APC gene product associates with microtubules in vivo and promotes their assembly in vitro. Cancer Res. 1994;54:3676–3681. [PubMed] [Google Scholar]
- Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA. 1995;92:3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munemitsu S, Albert I, Rubinfeld B, Polakis P. Deletion of aminoterminal structure stabilizes β-catenin in vivoand promotes the hyperphosphorylation of the APC tumor suppressor protein. Mol Cell Biol. 1996;16:4088–4094. doi: 10.1128/mcb.16.8.4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagafuchi A, Takeichi M. Cell binding function of E-cadherin is regulated by the cytoplasmic domain. EMBO (Eur Mol Biol Organ) J. 1988;7:3679–3684. doi: 10.1002/j.1460-2075.1988.tb03249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagafuchi A, Takeichi M. Transmembrane control of cadherin- mediated cell adhesion: a 94-Kd protein functionally associates with a specific region of the cytoplasmic domain of E-cadherin. Cell Regul. 1989;1:37–44. doi: 10.1091/mbc.1.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näthke IS, Adams CL, Polakis P, Sellin JH, Nelson WJ. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. J Cell Biol. 1996;134:165–179. doi: 10.1083/jcb.134.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson WJ, Wilson R, Wollner D, Mays R, McNeill H, Siemers K. Regulation of epithelial cell polarity: a view from the cell surface. Cold Spring Harbor Symp Quant Biol. 1992;57:621–630. doi: 10.1101/sqb.1992.057.01.068. [DOI] [PubMed] [Google Scholar]
- Noordermeer J, Klingensmith J, Perrimon N, Nusse R. Dishevelled and armadillo act in the wingless signalling pathway in Drosophila. . Nature (Lond) 1994;367:80–83. doi: 10.1038/367080a0. [DOI] [PubMed] [Google Scholar]
- Orsulic S, Peifer M. An in vivo structure-function study of armadillo, the β-catenin homologue, reveals both separate and overlapping regions of the protein required for cell adhesion and for wingless signaling. J Cell Biol. 1996;134:1283–1300. doi: 10.1083/jcb.134.5.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyama T, Kanai Y, Ochiai A, Akimoto S, Oda T, Yanagihara K, Nagafuchi A, Tsukita S, Shibamoto S, Ito F, et al. A truncated beta-catenin disrupts the interaction between E-cadherin and alpha-catenin: a cause of loss of intercellular adhesiveness in human cancer cell lines. Cancer Res. 1994;54:6282–6287. [PubMed] [Google Scholar]
- Ozawa M, Kemler R. Molecular organization of the uvomorulin– catenin complex. J Cell Biol. 1992;116:989–996. doi: 10.1083/jcb.116.4.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa M, Baribault H, Kemler R. The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. EMBO (Eur Mol Biol Organ) J. 1989;8:1711–1717. doi: 10.1002/j.1460-2075.1989.tb03563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa M, Ringwald M, Kemler R. Uvomorulin-catenin complex formation is regulated by a specific domain in the cytoplasmic region of the cell adhesion molecule. Proc Natl Acad Sci USA. 1990;87:4246–4250. doi: 10.1073/pnas.87.11.4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papkoff J, Rubinfeld B, Schryver B, Polakis P. Wnt-1 regulates free pools of catenins and stabilizes APC-catenin complexes. Mol Cell Biol. 1996;16:2128–2134. doi: 10.1128/mcb.16.5.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peifer M, Wieschaus E. The segment polarity gene armadillo encodes a functionally modular protein that is the Drosophilahomolog of human plakoglobin. Cell. 1990;63:1167–1178. doi: 10.1016/0092-8674(90)90413-9. [DOI] [PubMed] [Google Scholar]
- Peifer M, Rauskolb C, Williams M, Riggleman B, Wieschaus E. The segment polarity gene armadillo interacts with the wingless signaling pathway in both embryonic and adult pattern formation. Development (Camb) 1991;111:1029–1043. doi: 10.1242/dev.111.4.1029. [DOI] [PubMed] [Google Scholar]
- Peifer M, Orsulic S, Sweeton D, Wieschaus E. A role for the Drosophilasegment polarity gene armadillo in cell adhesion and cytoskeletal integrity during oogenesis. Development (Camb) 1993;118:1191–1207. doi: 10.1242/dev.118.4.1191. [DOI] [PubMed] [Google Scholar]
- Polakis P. Mutations in the APC gene and their implications for protein structure and function. Curr Opin Genet Dev. 1995;5:66–71. doi: 10.1016/s0959-437x(95)90055-1. [DOI] [PubMed] [Google Scholar]
- Rimm DL, Koslov E, Kebriaei P, Cianci D, Morrow SJ. Alpha1(E)-catenin is an actin binding and bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci USA. 1995;92:8813–8817. doi: 10.1073/pnas.92.19.8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P. Association of the APC gene product with beta-catenin. Science (Wash DC) 1993;262:1731–1734. doi: 10.1126/science.8259518. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science (Wash DC) 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- Siegfried E, Wilder EL, Perrimon N. Components of wingless signaling in Drosophila. . Nature (Lond) 1994;367:76–80. doi: 10.1038/367076a0. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Levy DB, Maupin P, Pollard TD, Vogelstein B, Kinzler KW. Wild-type but not mutant APC associates with the microtubule cytoskeleton. Cancer Res. 1994;54:3672–3675. [PubMed] [Google Scholar]
- Sokol S, Christian JL, Moon RT, Melton DA. Injected Wnt RNA induces a complete body axis in Xenopusembryos. Cell. 1991;67:741–752. doi: 10.1016/0092-8674(91)90069-b. [DOI] [PubMed] [Google Scholar]
- Su LK, Vogelstein B, Kinzler KW. Association of the APC tumor suppressor protein with catenins. Science (Wash DC) 1993;262:1734–1737. doi: 10.1126/science.8259519. [DOI] [PubMed] [Google Scholar]
- Takeichi M. Cadherins: a molecular family important in selective cell– cell adhesion. Annu Rev Biochem. 1990;59:237–252. doi: 10.1146/annurev.bi.59.070190.001321. [DOI] [PubMed] [Google Scholar]
- Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science (Wash DC) 1991;251:1451–1455. doi: 10.1126/science.2006419. [DOI] [PubMed] [Google Scholar]
- Weißig, H. 1993. Expressions- und Strukturanalysen an β-catenin, einem zytoplasmatischen Ankerprotein des Zelladhäsionsmoleküls Uvomorulin. Diplomarbeit, Fakultät für Biologie der Alberts-Ludwigs-Universität Freiburg.
- Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of β-catenin is regulated in Xenopusembryos by glycogen synthase kinase 3. Genes & Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]