

Abstract
Yeast protein, Bee1, exhibits sequence homology to Wiskott-Aldrich syndrome protein (WASP), a human protein that may link signaling pathways to the actin cytoskeleton. Mutations in WASP are the primary cause of Wiskott-Aldrich syndrome, characterized by immuno-deficiencies and defects in blood cell morphogenesis. This report describes the characterization of Bee1 protein function in budding yeast. Disruption of BEE1 causes a striking change in the organization of actin filaments, resulting in defects in budding and cytokinesis. Rather than assemble into cortically associated patches, actin filaments in the buds of Δbee1 cells form aberrant bundles that do not contain most of the cortical cytoskeletal components. It is significant that Δbee1 is the only mutation reported so far that abolishes cortical actin patches in the bud. Bee1 protein is localized to actin patches and interacts with Sla1p, a Src homology 3 domain–containing protein previously implicated in actin assembly and function. Thus, Bee1 protein may be a crucial component of a cytoskeletal complex that controls the assembly and organization of actin filaments at the cell cortex.
Proteins that are conserved between yeast and mammals are likely to carry out fundamental cellular functions. The complete yeast genome database provides a facile route for the identification of these proteins, which can then be studied directly in yeast through combined genetic and biochemical approaches. In recent years, yeast has been increasingly attractive for studying actin cytoskeleton function in cell morphogenesis. During mitotic growth, yeast cells undergo highly reproducible changes in cell shape that are accompanied by actin cytoskeleton rearrangements (Adams and Pringle, 1984; Kilmartin and Adams, 1984). Actin assembly and organization in yeast are modulated by a set of actin-binding proteins similar to those that operate in mammalian cells (for review see Welch et al., 1994). Examples of these actin-binding proteins include Sac6p, a fimbrin homologue that bundles actin filaments (Adams et al., 1989); Cap1p and Cap2p, subunits of a protein complex that caps the barbed (high-affinity) ends of actin filaments (Amatruda et al., 1990); and Cof1p, a cofilin homologue that severs actin filaments (Moon et al., 1993). These actin-binding proteins localize to cortical actin patches, membrane-associated structures that are concentrated at sites of cell surface growth (Adams and Pringle, 1984; Kilmartin and Adams, 1984). It has been speculated that the cortical patches may be involved in membrane activities, such as exocytosis and endocytosis (for review see Bretscher et al., 1994).
In addition to the conserved actin-binding proteins, building blocks of protein interactions common to mammalian cytoskeletal and signaling molecules are also found in yeast. Sla1p, a protein implicated in actin assembly in yeast (Holtzman et al., 1993; Li et al., 1995), contains three Src homology 3 (SH3)1 domains and a COOH-terminal repeat structure similar to a region in bindin, a sea urchin sperm adhesion protein. Disruption of SLA1 results in aberrant morphology of actin patches and temperature-sensitive cell growth. Despite the fact that a significant number of yeast actin cytoskeletal components have been identified, little is known about how they interact with each other in vivo to control actin filament assembly and organization.
A search of the yeast genome database revealed an open reading frame that encodes a protein with sequence homology to Wiskott-Aldrich syndrome protein (WASP) (Symons et al., 1996). Mutations in WASP result in WiskottAldrich syndrome (WAS) (Derry et al., 1994, 1995), a severe immuno-deficiency and platelet deficiency disease (for review see Kirchhausen and Rosen, 1996; RemoldO'Donnell et al., 1996). Lymphocytes from WAS patients are morphologically abnormal with decreased size and density of microvilli (Kenney et al., 1986; Molina et al., 1992), suggesting that WASP may be required for blood cell morphogenesis. Recently, a more ubiquitously expressed homologue of WASP, N-WASP, was identified (Miki et al., 1996). Overexpression of either WASP in tissue culture cells induces the formation of actin-rich structures to which the overexpressed WASP localizes, suggesting that WASP may interact directly with actin cytoskeletal components (Miki et al., 1996; Symons et al., 1996).
WASP homologues share several functional domains. WASP homology domain 1 (WH1), an NH2-terminal domain that exhibits sequence similarity to pleckstrin homology domains, can interact with phospholipids in vitro (Miki et al., 1996). Both WASP and N-WASP contain a GTPase-binding domain (GBD) that interacts with the activated form of Cdc42 (Kolluri et al., 1996; Symons et al., 1996), a small GTP-binding protein involved in regulating actin cytoskeleton rearrangements (for review see Nobes and Hall, 1995). Downstream from the GBD, there is a proline-rich region that interacts with the SH3 domains of several signaling proteins (Rivero-Lezcano et al., 1995; Banin et al., 1996; Bunnell et al., 1996; Miki et al., 1996). The presence of these functional regions in WASP suggests that WASP may act as a molecular scaffold that links various signaling pathways to the actin cytoskeleton.
The yeast WASP-like protein contains all of the functional regions listed above except the GBD that interacts with Cdc42p (Symons et al., 1996). I have named this protein Bee1 because of its similarity to WASP. Reported here are the results of studies on the cellular function of Bee1 protein (Bee1p) in yeast. Bee1p is a component of the cortical actin cytoskeleton and plays an essential role in the organization of actin filaments at the cell cortex. BEE1-disrupted cells are defective in budding and cytokinesis, most likely as a result of the inability to assemble cortical actin patches. Thus, Bee1 protein may share a structural function with WASP.
Materials and Methods
Strains and Media and Genetic Manipulations
Yeast strains used in this work are listed in Table I. Yeast cell culture and genetic techniques were carried out by methods described by Sherman et al., 1974.
Table I.
Yeast Strains
| Name | Genotype | Source | ||
|---|---|---|---|---|
| RLY1 | MATa ura3-52 his3-Δ200 leu2-3,112 lys2-801 | Drubin lab | ||
| RLY156 | MATa/α ura3-52/ura3-52 his3-Δ200/his3-Δ200 | this work | ||
| leu2-3, 112/leu2-3,112 lys2-801/lys2-801 | ||||
| BEE1/Δbee1::LEU2 | ||||
| RLY157 | MATa ura3-52 his3-Δ200 leu2-3,112 lys2-801 | this work | ||
| Δbee1::LEU2 | ||||
| RLY160 | MATa ura3-52 his3-Δ200 leu2-3,112 lys2-801 | this work | ||
| Δbee1::LEU2 pBEE1-myc |
All of the above strains are congenic in S288C background.
Cloning, Plasmid, and Strain Construction
Yeast genomic DNA was prepared from strain RLY1 as described (Hoffman and Winston, 1987). A 2.4-kb DNA fragment containing the BEE1 coding region (NCBI accession No. 1101757) and 282-bp 5′ and 180-bp 3′ flanking sequences was amplified from yeast genomic DNA by PCR using a 5′ primer, 5′-BamHI-TACTTGAAATTGTGTCTCTG-3′ (Primer 1), and a 3′ primer, 5′-XbaI-ATCATTGTAGCCCGACTATT-3′ (Primer 2). This fragment was cut with BamHI and XbaI and cloned into pRS316 (Sikorski and Hieter, 1989) and Bluescript SK to yield pRL101 and pRL88, respectively. To construct the BEE1 knock-out plasmid, pRL88 was cut with BsmI, removing 84% of the BEE1 coding region (amino acids 102–633 and 11 bp of 3′ UTR), and blunted and ligated with a DNA fragment containing the LEU2 gene (Berben et al., 1991), yielding pRL90. pRL102, a yeast vector for COOH-terminal 6-myc tagging was constructed by ligating the BamHI-SnaBI fragment that contains six copies of myc epitope from CS+MT (Roth et al., 1991) into vector pRS306 (Sikorski and Hieter, 1989) between BamHI and XbaI (blunt). To construct a plasmid that expresses COOH-terminal myc-tagged Bee1p, a BEE1 fragment was PCRamplified from yeast genomic DNA using Primer 1 and a 3′ primer of sequence BamHI-CCAATCATCACCATTGTCC (Primer 3). The latter corresponds to the COOH terminus of Bee1p. This fragment was cut with BamHI and ligated into the BamHI site of pRL102. The orientation of the insert was determined, and the resulting plasmid in which the myc tag was at the 3′ end of BEE1 was named pRL108. pRL108 was cut with PvuII and the fragment that contains BEE1-myc was ligated into pRS423 (Sikorski and Hieter, 1989) between the PvuII sites, yielding pRL111. pRL111 complements the Δbee1 mutation.
To construct Δbee1 strain, PRL90 was cut with XbaI and BamHI and transformed into a Leu2− diploid yeast strain. The diploid was sporulated and the tetrads were dissected and analyzed. The tetrads showed 2:2 segregation for the Leu+ phenotype. To confirm BEE1 deletion in the Leu+ colonies, genomic DNA was prepared from a tetrad and analyzed by PCR using Primers 1 and 3. Genomic DNA from the Leu− colonies gave a 2.2kb fragment, corresponding to the segment flanked by the two primers. Genomic DNA from the Leu+ colonies did not yield any PCR product, as expected for BEE1 deletion since the sequence corresponding to Primer 3 was within the deleted part of BEE1 gene in PRL90. In addition, the phenotypes of the Leu+ colonies can be fully complemented by PRL101, a centromere-containing plasmid carrying full-length BEE1.
Immunofluorescence
Cells were fixed directly in growth media by the addition of 37% formaldehyde to 5% final concentration. Immunofluorescence staining was carried out essentially as described (Drubin et al., 1988). Rhodamine-conjugated donkey anti–goat and FITC-conjugated donkey anti–rabbit and donkey anti–mouse secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA).
Phalloidin Staining of Yeast Cells
Cells were fixed directly in growth media by the addition of 37% formaldehyde to 5% final concentration. Staining with rhodamine-labeled phalloidin (Molecular Probes, Eugene, OR) was carried out as described (Lillie and Brown, 1994).
Permeabilization of Yeast Cells and In Vitro Actin Assembly
Cell growth and permeabilization, rhodamine-actin preparation, and in vitro rhodamine-actin assembly assay were carried out as described (Li et al., 1995).
Fluorescence Microscopy
Fluorescence imaging was performed on a microscope (model Axiophot; Carl Zeiss, Inc., Thornwood, NY) with a HB100 W/Z high-pressure mercury lamp and a 100× Plan Neofluar oil immersion objective (Carl Zeiss, Inc.). Image acquisition was performed using Northern Exposure (Phase 3 Imaging Systems, Milford, MA).
Video Microscopy
5 μl of an exponentially growing yeast culture was mixed with 5 μl of 1.6% low melting agarose in YPD (kept at 37°C) in a well of a multitest slide (ICN Biomedicals, Inc., Costa Mesa, CA). The slide was covered with a glass coverslip and sealed with nail polish. The cells were observed with a 63×/1.4NA plan-Apochromat objective on a microscope (model Axiovert 135; Carl Zeiss, Inc.). Images were collected with a cooled CCD (Photometics Ltd., Tucson, AZ). Image acquisition and data analysis were carried out using Metamorph 2.0 (Universal Imaging Corp., West Chester, PA).
Electron Microscopy
10 ml of exponentially growing cells were harvested and the cell pellet was resuspended directly in 1 ml of 1% glutaraldehyde in 200 mM cacodylate and 100 mM KPO4 buffer, pH 6.5. Cells were fixed for 30 min at room temperature and were washed three times with 100 mM KPO4, pH 7.5. Cells were treated with 0.2 mg/ml zymolyase 20T (Seikagaku Corp., Tokyo, Japan) for 30 min at 37°C. Cells were washed twice in 200 mM cacodylate buffer, pH 7.4, and incubated at room temperature for 1 h in 1.5% potassium ferrocyanide/1% osmium tetroxide (in water). The cells were washed with water (3 × 15 min), incubated for 1 h in 2% uranyl acetate (in water, at room temperature), washed again in water (3 × 15 min), and dehydrated in ethanol 70%/90%/100% (15 min each). The samples were then infiltrated in Epon/Araldite mixed 1:1 with propyleneoxide for 2 h, transferred to pure Epon/Araldite in an embedding mould, and polymerized overnight at 60°C. Ultra-thin sections were cut on a Reichert Ultracut S microtome (Leica Inc., Deer Lake, IL), stained with uranyl acetate and lead citrate, and examined in a transmission electron microscope (model 1200 EX; JEOL U.S.A., Inc., Peabody, MA) at 80 kV.
Yeast Extract Preparation and Immunoprecipitation
Yeast cells were lysed by the liquid nitrogen–grinding method (Sorger and Pelham, 1987) in UBT (50 mM KHepes, pH 7.5, 100 mM KCl, 3 mM MgCl2, 1 mM EGTA, 0.5% Triton X-100) supplemented with protease inhibitors as described (Li et al., 1995). The cell lysate was centrifuged at 300,000 g for 60 min. The resulting high-speed supernatant was usually at a concentration of ∼10 mg/ml. 40 μl high-speed extract was incubated, for 1 h at 4°C, with 20 μl protein A–Sepharose beads (Pharmacia LKB Biotechnology, Piscataway, NJ) bound with mouse anti-myc monoclonal antibody (ascites; Evan et al., 1985) or, as a control, mouse anti-hemagglutinin monoclonal antibody (ascites; BABCO, Berkeley, CA), or affinitypurified rabbit anti-Sla1 antibody (a gift from D. Drubin, University of California, Berkeley, CA) or, as a control, affinity-purified rabbit anti– glutathione S-transferase antibody. The beads were washed four times with UBT and once with UBT + 0.5 M KCl and then incubated with 50 μl UBT + 1 M KCl for 10 min. The eluted proteins were precipitated with 10% trichloroacetic acid and resuspended in 20 μl protein gel sample buffer. 10 μl of each sample and 5 μl of the extract were loaded onto a 12.5% polyacrylamide gel. Immunoblot analysis was carried out using the enhanced chemiluminescence detection kit (Amersham Corp., Arlington Heights, IL).
Results
BEE1 Is Required for Budding and Cytokinesis
A 2.4-kb yeast genomic DNA fragment was cloned that contains the BEE1 coding region and its flanking sequences. A knock-out construct was made in which 84% of the BEE1 coding region was replaced by the LEU2 gene. The knock-out construct was transformed into a Leu2− diploid yeast strain. The diploid was sporulated and the tetrads were dissected and analyzed. When the spores were grown at room temperature, all of the tetrads had two colonies that grew at a wild-type rate and two colonies that grew much more slowly (Fig. 1 A). The fast growing colonies all had a Leu− phenotype, whereas the slow growing ones were all Leu+ (data not shown), indicating that the slow growth results from the integration of the knockout construct. Deletion of BEE1 gene in the Leu+ strains was further confirmed by PCR analysis and complementation of the slow growth by a centromere plasmid carrying BEE1 gene (see Materials and Methods). At elevated temperatures (34°C and above), Δbee1 cells are unable to grow (Fig. 1 B). The temperature-sensitive growth phenotype is common to many mutations affecting actin cytoskeleton function (e.g., Adams et al., 1991; Holtzman et al., 1993).
Figure 1.

Effects of BEE1 deletion on colony growth. (A) Δbee1 mutation results in poor colony growth. RLY156 (see Table I) was sporulated and tetrads were dissected and grown on YPD for 4 d at room temperature. (B) Temperature sensitivity of Δbee1 cells. An equal density of RLY1 (wild-type) and RLY157 (Δbee1) liquid cultures were spotted on YPD plates and grown at 23, 30, 34, and 37°C for 3 d.
Δbee1 cells grown at 23°C were examined by light microscopy. The cells were heterogeneous in size and morphology, and many were larger than wild-type cells (Fig. 2 A). About 37% of Δbee1 cells had more than one nucleus. A significant fraction of the cells (∼24%, compared to only 3% in wild-type population) were large budded with divided nuclei. There were also clumps of cells that could not be separated with a microdissection needle. These morphological characteristics suggest that the Δbee1 mutation may affect budding and cytokinesis. In addition to the above morphologies, a small fraction (∼5%) of the cells appeared amorphous and lysed. Cell death does not appear to be the main cause of the slow colony growth at 23°C, however, because >85% of Δbee1 cells can form colonies (data not shown).
Figure 2.

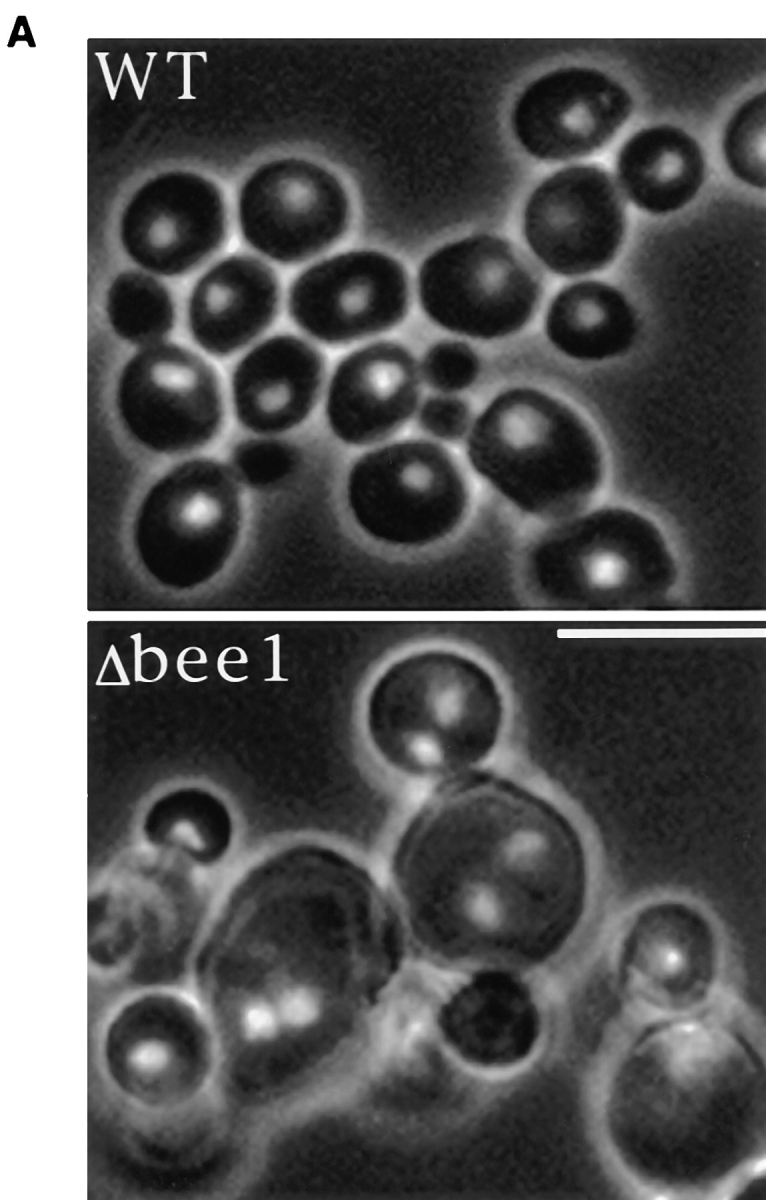
Δbee1 mutant cells exhibit defects in bud growth and cytokinesis. (A) Superimposed phase contrast and 4′,6-diamidino-2-phenylindole staining images of wild type (RLY1) and Δbee1 (RLY157) cells, photographed at the same magnification. Bar, 10 μm. (B) Comparison of bud emergence rates of RLY1 and RLY157 cells. 40 unbudded RLY1 or RLY157 cells were aligned on YPD agar with a microdissection needle at time 0. At each time point, the fractions of cells that have budded were scored and plotted over time. (C) Comparison of the rates of bud growth of RLY1 and RLY157 cells. Exponentially growing cultures of RLY1 or RLY157 cells were embedded in YPD-agarose and bud growth was recorded by time-lapse video as described in Materials and Methods. Bud lengths were measured by Metamorph 2.0 (Universal Imaging Corp.) and plotted over time. The plots for two RLY1 and two RLY157 cells are shown. (D) Comparison of cytokinesis rates of RLY1 and RLY157 cells. 40 large-budded RLY1 or RLY157 cells were aligned on YPD agar with a microdissection needle at time 0. At each time point, the completion of cytokinesis for each cell was determined by attempting to separate the bud from the mother with the dissection needle. The fractions of cells that completed cytokinesis were plotted over time.
To better understand the Δbee1 deficiency, the rates of bud emergence, bud growth, and cytokinesis were compared between Δbee1 and wild-type cells. Because many Δbee1 cells are in clumps, it was easier to observe the above events on solid media. 40 unbudded Δbee1 or wildtype cells of average sizes were aligned on agar with a dissection needle, and bud emergence was scored every 30 min. Surprisingly, Δbee1 cells did not show any apparent defect in bud emergence (Fig. 2 B). The slightly faster bud emergence rate of Δbee1 cells can be explained by the larger sizes of Δbee1 G1 cells, which presumably result from a cytokinesis delay in the previous cell cycle (see below).
To estimate the rate of bud growth, individual budding cells (starting from a bud size of ∼1 μm) were followed by video microscopy. Bud length was measured and plotted as a function of time. Fig. 2 C shows bud growth of two Δbee1 and wild-type cells. Bud length increased linearly before reaching about 2.5 μm. The estimated rates of bud length increase within the linear period were 19 ± 6 nm/ min for Δbee1 cells (sample size: 17 cells) and 53 ± 16 nm/ min for wild-type cells (sample size: 16 cells). Since the buds in both strains are roughly spherical, the rate at which the surface area increases in Δbee1 cells was about 1/7 of that in wild-type cells.
The increase in the fraction of large-budded cells with divided nuclei in Δbee1 liquid culture suggests that there might be a delay in cytokinesis. But it was also possible that the large-budded cells were actually two contacting G1 cells. To directly test whether Δbee1 causes a delay in cell division, 40 large-budded (bud size >1/2 of mother size) Δbee1 or wild-type cells were aligned on agar, and cell division was determined by attempting to separate the bud from the mother with a microdissection needle. The rate at which cell separation completes in Δbee1 cells, estimated from the data shown in Fig. 2 D, was only 40% of that in wild-type cells. This delay in cell division was not likely to result from an S phase or a mitotic block, since in Δbee1 liquid cell culture, there was not an increase in the fraction of large-budded cells with undivided nuclei, characteristics of S or M phase–arrested cells.
Bee1p Is Essential for the Assembly of Cortical Actin Patches
Because actin is involved in both budding and cytokinesis, the organization of actin filaments in Δbee1 cells was examined by rhodamine-phalloidin staining. In wild-type cells, actin patches are concentrated in the bud during polarized growth (Fig. 3 A, a) and at the septum during cytokinesis (Fig. 3 B, a). In Δbee1 cells, actin filaments appear to be assembled at the right regions, i.e., the bud or the septum. However, actin is organized into structures strikingly different from those in wild-type cells. Rather than form cortical patches, actin filaments (F-actin) in Δbee1 cells assemble into thick cables that do not seem to be restricted to the cell cortex (Fig. 3 A, b and B, b). A few scattered actin patches are seen in some cells, but these patches are in the mother rather than the bud. Bars of actin are also found in the mother of some Δbee1 cells (not shown). The amount of F-actin in Δbee1 buds, judged by the intensity of rhodamine fluorescence, appears to be higher than that in wild-type buds. This result suggests that Bee1p plays an important role in the organization of actin filaments but is not required for the polarized distribution of F-actin.
Figure 3.

Actin cytoskeleton defects caused by Δbee1 mutation. The images show rhodamine-phalloidin staining of budding cells (A) and dividing cells (B) of strain RLY1 (wild type) (a) and RLY157 (Δbee1) (b). The panels on the right in B are superimposed 4′,6-diamidino-2-phenylindole and phase images of the cells shown on the left. Bar, 10 μm.
Actin assembly in the cell is regulated by many actinbinding proteins. The altered F-actin organization and level in Δbee1 cells may correlate with changes in the localization of cellular actin-binding proteins. In wild-type cells, fimbrin (Sac6p), cofilin (Cof1p), and capping protein (Cap2p) are all localized in cortical actin patches (Drubin et al., 1988; Amatruda and Cooper, 1992; Moon et al., 1993). In Δbee1 cells, Sac6p is associated with the actin bundles in the buds, whereas Cof1p and Cap2p do not appear to be enriched in the buds (Fig. 4). This suggests that the actin bundles in the buds of Δbee1 cells have a different biochemical composition from that of cortical actin patches.
Figure 4.

Immunofluorescence localization of actin-binding proteins in Δbee1 cells. Double immunofluorescence staining of RLY1 (WT) and RLY157 (Δbee1) was carried out using goat anti-actin (a gift from D. Drubin) and rabbit anti-Sac6p (Drubin et al., 1988), rabbit anti-Cof1p (Moon et al., 1993), and rabbit anti-Cap2p (Amatruda and Cooper, 1992) primary antibodies and FITC-conjugated anti– rabbit and rhodamine-conjugated anti–goat secondary antibodies. I and III show actin staining. II and IV show Sac6p (top), Cof1p (middle), and Cap2 (bottom) staining of the same cells as those shown on the left in I and III, respectively. Bar, 10 μm.
Δbee1 Cells Accumulate Post-Golgi Vesicles in the Bud
To further understand the Δbee1 phenotype, the cells were examined by electron microscopy. As shown in Fig. 5 B, Δbee1 mutant cells accumulate a large number of vesicles mostly in the bud. Wild-type cells, by contrast, do not accumulate any vesicles (Fig. 5 A). In some Δbee1 buds, the vesicles appear to form linear arrays (Fig. 5 C), perhaps along the aberrant actin bundles. The vesicles accumulated in Δbee1 cells are similar in size to post-Golgi vesicles (80–100 nm) (Novick et al., 1980). The accumulation of these vesicles may reflect an exocytosis defect, which would explain the slow rates of bud growth and cytokinesis in Δbee1 cells.
Figure 5.
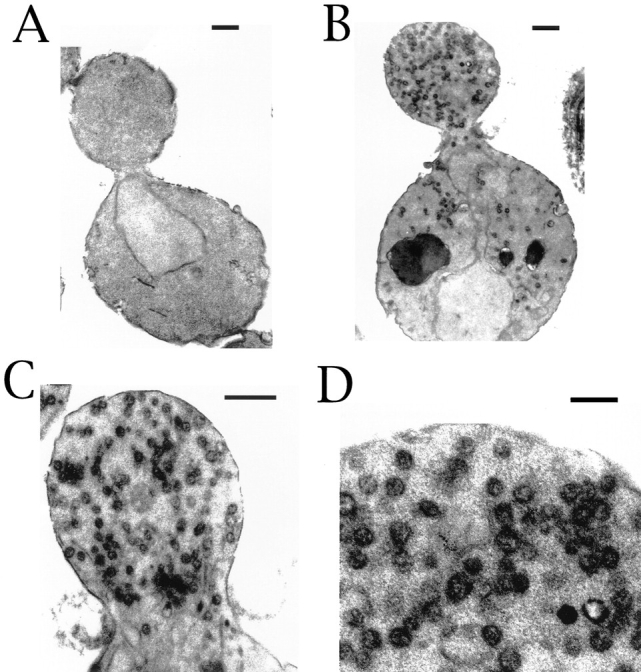
Δbee1 cells accumulate vesicles in the bud. Thin-sectioning electron microscopy was carried out on RLY1 (wild type) (A) and RLY157 (Δbee1) (B–D) as described in Materials and Methods. D shows a portion of the bud in B at a higher magnification. Bars: (A– C) 500 nm; (D) 200 nm.
Bee1p Localizes to Cortical Actin Patches
To ask whether Bee1p interacts directly with the cortical actin cytoskeleton, Bee1p cellular localization was determined by using a multicopy plasmid that expresses myctagged Bee1p. This plasmid complements the slow growth and temperature-sensitive phenotypes of the null mutant (not shown), suggesting that myc-tagged Bee1p is functional. Immunofluorescence staining with an anti-myc antibody showed that Bee1p exhibits patchlike staining. This staining pattern is not seen in cells that do not express myc-tagged Bee1p (Fig. 6 A) or in cells that express an unrelated myc-tagged protein (data not shown), suggesting that the staining is specific for myc-Bee1p. Double staining using anti-actin and anti-myc antibodies showed that the majority of Bee1p patches colocalize with actin patches (Fig. 6 B), indicating that Bee1p is in fact a component of the cortical actin patches. Bee1p does not appear to be enriched on actin cables in the mother.
Figure 6.

Immunofluorescence localization of myctagged Bee1p. Double immunofluorescence staining of RLY1 (expressing untagged Bee1p, A) and RLY160 (expressing myc-tagged Bee1p, B) was carried out using mouse anti-myc (right) and goat anti-actin primary antibodies (left), and FITC-conjugated anti–mouse and rhodamine-conjugated anti– goat secondary antibodies. Bar, 10 μm.
Bee1p Interacts with Sla1p, an SH3 Domain–containing Cortical Cytoskeletal Component
The domain structure of WASP suggests that the WASP family of proteins may provide a molecular scaffold that brings together various structural and signaling proteins. I tested whether Bee1p interacts with any known components of cortical actin patches. Myc-tagged Bee1p was immunoprecipitated from detergent-solubilized high-speed supernatant of myc-Bee1p–expressing cells using protein A beads bound with anti-myc antibody. Proteins that coprecipitated with myc-Bee1p and were eluted from the beads by 1 M KCl were analyzed by immunoblotting using antibodies against yeast proteins required for actin cytoskeletal function, including Actin, Sac6p, Cap2p, Cof1p, Sla1p, and Bem1 (Pringle et al., 1995). Only Sla1p and actin were detected in the high-salt eluate of anti-myc beads but not detected in the eluate of the control anti–hemagglutinin epitope beads (Fig. 7 a).
Figure 7.

Interaction of Bee1p with Sla1 and actin as detected by immunoprecipitation. RLY160 extract preparation and immunoprecipitation using either mouse anti-myc antibody (Evan et al., 1985) (a) or rabbit anti-Sla1p antibody (a gift from D. Drubin) (b) were carried out as described in Materials and Methods. Immunoblot analysis of the immunoprecipitated proteins was carried out using antibodies against Sla1p, myc, Bem1p (Pringle et al., 1995), actin (Boehringer Mannheim Corp., Indianapolis, IN), and Cof1p (Moon et al., 1993). Lane 1, extract before immunoprecipitation; lane 2, eluate of the control beads (see Materials and Methods); lane 3, eluate of anti-myc beads (a) or eluate of antiSla1 beads (b).
Anti-Sla1p antibody-bound protein A beads also precipitated myc-Bee1p (Fig. 7 b). Actin, however, was not detected in the Sla1p immunoprecipitate (not shown). Sla1p is an SH3 domain–containing protein involved in cortical actin assembly and organization (Holtzman et al., 1993; Li et al., 1995). The interaction between Bee1p and Sla1p may be mediated through the SH3 domains of Sla1p and the proline-rich domain of Bee1p. Bem1p, a SH3 domain– containing protein involved in budding (Bender and Pringle, 1991), did not coimmunoprecipitate with Bee1p (Fig. 7 a), indicating that not all SH3-containing proteins interact with Bee1p.
Bee1p Is Required for In Vitro Actin Assembly in Permeabilized Yeast Cells
We previously established a permeabilized cell assay for cortical actin assembly in vitro (Li et al., 1995). Permeabilized Δsla1 cells fail to assemble actin into the bud, suggesting that Sla1 may be involved in stimulating actin polymerization at cortical patches. Δbee1 cells should also exhibit an in vitro actin assembly defect since Sla1 is not localized to the bud in these cells (Li, R., unpublished result). Permeabilized Δbee1 and wild-type cells were prepared and the rhodamine-actin assembly assay was carried out as described (Li et al., 1995). While >60% of permeabilized wild-type cells were able to incorporate rhodamine-actin into patches in the bud, permeabilized Δbee1 were completely deficient in actin assembly in vitro (Fig. 8).
Figure 8.

Bee1p is required for actin assembly in permeabilized yeast cells. RLY1 and RLY157 cells that had been grown at 23°C were permeabilized and assayed for rhodamine actin assembly as described in Materials and Methods. Rhodamine fluorescence images are shown. Bar, 10 μm.
Discussion
The Function of Bee1 Protein in Cortical Actin Patch Assembly
The results presented in this report showed that Bee1p is a component of cortical actin patches and is essential for the organization of actin filaments. Although null mutations in many cytoskeletal proteins result in a less polarized cortical actin distribution, accumulation of aberrant actin structures, and disappearance of actin cables, Δbee1 is the only mutation reported so far that abolishes cortical actin patches in the bud. Electron microscopy studies on wild-type cells showed that at least some cortical actin patches are membrane invaginations associated with highly organized actin filaments (Mulholland et al., 1994). The actin structures that replace cortical patches in the buds of Δbee1 cells are bundles of actin filaments that, under light microscope, do not appear to be restricted to the cell periphery. These aberrant actin structures lack many cortical patch components including cofilin, capping protein, and Sla1 and therefore are fundamentally different from actin patches.
What might be the role of Bee1p in cortical actin patch assembly? WH1, shared between WASP and Bee1p, can potentially interact with phospholipids in the cell (Miki et al., 1996). One simple model is that Bee1p localizes to the cell cortex via WH1 and brings together a set of cytoskeletal proteins that locally organize actin filaments into defined membrane-associated structures. Cofilin is an actin-depolymerizing factor usually associated with dynamic actin in the cell (Bamburg and Bray, 1987; Yonezawa et al., 1987), whereas capping protein can prevent elongation from the barbed ends of actin filaments (Caldwell et al., 1989; Amatruda and Cooper, 1992). The lack of these actin-binding proteins may contribute to the alteration of actin structures in the buds of Δbee1 cells. There is no evidence that Bee1p can directly recruit cofilin or capping protein. The interactions of cofilin and capping protein with actin are inhibited, at least in vitro, by phosphatidylinositol 4,5-bisphosphate (PIP2) (Yonezawa et al., 1990; Amatruda and Cooper, 1992). Cofilin activity is also regulated by phosphorylation (for review see Moon and Drubin, 1995). It is possible that Bee1p can locally modulate the level of PIP2 or the phosphorylation state of cofilin by interacting with the cofilin kinase or phosphatase.
Understanding how Bee1p functions will depend on the identification of its interacting proteins. So far, there is good evidence that one cytoskeletal component, Sla1, physically interacts with Bee1p. It is not known whether this interaction is direct, but an interaction between the three SH3 domains of Sla1 with the proline-rich domain of Bee1p is an appealing possibility. The proline-rich domain of Bee1p may also interact with profilin, a possibility that is currently under examination. The action of Bee1p, however, can not be mediated entirely through its interaction with Sla1, since the phenotype of an SLA1 null mutant is less severe than that of Δbee1. Δsla1 cells grow at a near wild-type rate at 23°C and contain cortical actin patches, although often in large clusters.
Bee1p, like N-WASP (Miki et al., 1996), also coimmunoprecipitates with actin. I have not been able to detect any interaction between Bee1p and F-actin. It is likely that Bee1p and N-WASP are actin monomer–binding proteins, which may explain the in vitro F-actin–depolymerizing activity of N-WASP (Miki et al., 1996). The result that actin does not coimmunoprecipitate with Sla1p suggests that actin and Sla1p may be in different Bee1p-containing complexes.
In vitro analysis in permeabilized yeast cells revealed an activity that promotes actin assembly in the bud. Although the exact biochemical nature of this activity is not completely understood, the activity is distinct from the ends of actin filaments and is dependent on Sla1p (Li et al., 1995). The result that permeabilized Δbee1 cells are also defective in actin assembly in vitro suggests that this activity may depend on the interaction between Sla1p and Bee1p. If Sla1p and Bee1p are important for the polarized actin assembly, why is the F-actin content in Δbee1 buds higher than that in the mothers? One explanation might be that actin cables in Δbee1 cells remain oriented toward the bud. The cables may provide a transport system (for review see Bretscher et al., 1994) that delivers cytoskeletal proteins and/or actin monomers to the bud, maintaining the polarity of actin distribution. In the absence of Bee1p, actin assembly in the bud may be stimulated by an aberrant mechanism that was not detected in the permeabilized cells.
The Role of Cortical Actin in Budding and Cytokinesis
The accumulation of cortical actin patches at growth sites and at the plane of cytokinesis led to the view that these actin structures are somehow required for cell surface generation (Adams and Pringle, 1984; Kilmartin and Adams, 1984). The lack of cortical patches but not actin cables in Δbee1 cells provides an opportunity to better define the role of cortical patches in polarized cell growth and division. The results reported here suggest that disruption of cortical patches does not prevent cell polarity establishment but slows down polarized cell surface growth and cytokinesis.
The phenotypes of act1 mutants first implicated actin in exocytosis (Novick and Botstein, 1985), but the mechanism of this involvement is unclear. Vesicle accumulation was also observed in other cytoskeletal mutants, including Δtpm1 (tropomyosin gene disruption) (Liu and Bretscher, 1989) and myo2-66 (a mutation in MYO2 which encodes a class V myosin) (Johnston et al., 1991). Δtpm1 and myo266 cells accumulate vesicles predominantly in the mother, probably reflecting a defect in vesicle transport into the bud due to the lack of actin cables in Δtpm1 cells or the loss of Myo2 motor function in myo2-66 cells. Δbee1 cells, by contrast, accumulate post-Golgi vesicles in the bud like late sec mutants (Novick et al., 1980). This suggests that the lack of cortical actin patches in Δbee1 cells does not affect the transport of vesicles to the bud but may affect the membrane fusion step of exocytosis. It is also possible, however, that the vesicles are sequestered by the aberrant actin bundles in Δbee1 buds and are unable to fuse with the plasma membrane. Experiments are currently being carried out to test the above possibilities and to identify the cargo of Δbee1 vesicles.
The Relation between Bee1p and WASP
Is Bee1p a true homologue of WASP? The phenotype of bee1 null mutants resembles, at a superficial level, some of the cellular defects of WAS. For example, T cells from WAS patients are characterized by size heterogeneity, reduced number of the actin-rich microvilii, and the appearance of abnormal surface structures. Overexpression of N-WASP in Cos7 cells reduces the number of actin stress fibers but results in F-actin accumulation in cortical areas (Miki et al., 1996), consistent with a role for N-WASP in cortical actin assembly. Expression of WASP in yeast, however, does not rescue the Δbee1 defect (Li, R., and T. Kirchhausen, unpublished result), suggesting that the functions of Bee1p and WASP, like their sequences, are not completely homologous. The most significant difference between WASP and Bee1p is the lack of a recognizable Cdc42binding motif in the sequence of the latter. This is not surprising considering that yeast cells lacking Cdc42 function are unable to polarize (Adams et al., 1990), whereas Δbee1 cells can, suggesting that Cdc42p and Bee1p may not be involved in the same pathway. Recent studies in tissue culture cells showed that the interaction between Cdc42p and WASP is probably not required for the effects of Cdc42p on actin organization (Lamarche et al., 1996). Therefore, the significance of Cdc42–WASP interaction awaits further investigation.
Acknowledgments
This work was supported by Harcourt General Charitable Foundation.
Abbreviations used in this paper
- Bee1p
Bee1 protein
- GBD
GTPasebinding domain
- SH3
Src homology 3
- WAS
Wiskott-Aldrich syndrome
- WASP
Wiskott-Aldrich syndrome protein
- WH1
WASP homology domain 1
Footnotes
I am very grateful to Tom Kirchhausen for communicating the sequence similarity between Bee1p and WASP before its publication and for his encouragement throughout these studies. I am indebted to David Drubin, John Cooper, and John Pringle for generously providing antibodies. I am grateful to Susanna Rankin and Le Ma for their help in light microscopy, and Maria Ericsson for her expert assistance in electron microscopy. I am grateful to the members of my laboratory, and Dan Sun and Tika Li for support and encouragement. I thank Christine Field, Dan Finley, Ryn Miake-Lye, Tim Mitchison, David Pellman, Tom Rapaport, and Dirk Winter for their comments on the manuscript.
Address all correspondence to Rong Li, Department of Cell Biology, Harvard Medical School, 240 Longwood Ave., Boston, MA 02115. Tel: (617) 432-0640. Fax: (617) 432-1144. E-mail: RLi@warren.med.harvard.edu
References
- Adams AE, Botstein D, Drubin DG. A yeast actin-binding protein is encoded by SAC6, a gene found by suppression of an actin mutation. Science (Wash DC) 1989;243:231–233. doi: 10.1126/science.2643162. [DOI] [PubMed] [Google Scholar]
- Adams AE, Botstein D, Drubin DG. Requirement of yeast fimbrin for actin organization and morphogenesis in vivo. . Nature (Lond) 1991;354:404–408. doi: 10.1038/354404a0. [DOI] [PubMed] [Google Scholar]
- Adams AE, Johnson DI, Longnecker RM, Sloat BF, Pringle JR. CDC42 and CDC43, two additional genes involved in budding and the establishment of cell polarity in the yeast Saccharomyces cerevisiae. . J Cell Biol. 1990;111:131–142. doi: 10.1083/jcb.111.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams AEM, Pringle JR. Relationship of actin and tubulin distribution to bud growth in wild-type and morphogenetic mutant Saccharomyces cerevisiae. . J Cell Biol. 1984;98:934–945. doi: 10.1083/jcb.98.3.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatruda JF, Cooper JA. Purification, characterization, and immunofluorescence localization of Saccharomyces cerevisiaecapping protein. J Cell Biol. 1992;117:1067–1076. doi: 10.1083/jcb.117.5.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatruda JF, Cannon JF, Tatchell K, Hug C, Cooper JA. Disruption of the actin cytoskeleton in yeast capping protein mutants. Nature (Lond) 1990;344:352–354. doi: 10.1038/344352a0. [DOI] [PubMed] [Google Scholar]
- Bamburg JR, Bray D. Distribution and cellular localization of actin depolymerizing factor. J Cell Biol. 1987;105:2817–2825. doi: 10.1083/jcb.105.6.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin S, Truong O, Katz DR, Waterfield MD, Brickell PM, Gout I. Wiskott-Aldrich syndrome protein (WASP) is a binding partner for C-src family protein-tyrosine kinases. Curr Biol. 1996;6:981–988. doi: 10.1016/s0960-9822(02)00642-5. [DOI] [PubMed] [Google Scholar]
- Bender A, Pringle JR. Use of a screen for synthetic lethal and multicopy suppressee mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. . Mol Cell Biol. 1991;11:1295–1305. doi: 10.1128/mcb.11.3.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berben G, Dumont J, Gilliquet V, Bolle P, Hilger F. The YDp plasmid: a uniform set of vectors bearing versatile gene disruption cassettes for Saccharomyces cerevisiae. . Yeast. 1991;7:475–477. doi: 10.1002/yea.320070506. [DOI] [PubMed] [Google Scholar]
- Bretscher A, Drees B, Harsay E, Schott D, Wang T. What are the basic functions of microfilaments? Insights from studies in budding yeast. J Cell Biol. 1994;126:821–825. doi: 10.1083/jcb.126.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnell, S.C., P.A. Henry, R. Kolluri, and T. Kirchhausen. 1996. Identification of Itk/Tsk SH3 domain ligands. J. Biol. Chem. In press. [DOI] [PubMed]
- Caldwell JE, Heiss SG, Mermall V, Cooper JA. Effects of CapZ, an actin capping protein of muscle, on the polymerization of actin. Biochemistry. 1989;28:8506–8514. doi: 10.1021/bi00447a036. [DOI] [PubMed] [Google Scholar]
- Chenevert J, Corrado K, Bender A, Pringle J, Herskowitz I. A yeast gene (BEM1)necessary for cell polarization whose product contains two SH3 domains. Nature (Lond) 1992;356:77–79. doi: 10.1038/356077a0. [DOI] [PubMed] [Google Scholar]
- Derry JMJ, Ochs HJ, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. 1994;78:635–644. [PubMed] [Google Scholar]
- Derry JM, Kerns JA, Weinberg KI, Ochs HD, Volpini V, Estivill X, Walker AP, Francke U. WASP gene mutations in Wiskott-Aldrich syndrome and X-linked thrombocytopenia. Hum Mol Genet. 1995;4:1127–1135. doi: 10.1093/hmg/4.7.1127. [DOI] [PubMed] [Google Scholar]
- Drubin DG, Miller KG, Botstein D. Yeast actin-binding proteins: evidence for a role in morphogenesis. J Cell Biol. 1988;107:2551–2561. doi: 10.1083/jcb.107.6.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-mycproto-oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman CS, Winston F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. . Gene. 1987;57:267–272. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- Holtzman DA, Yang S, Drubin DG. Synthetic-lethal interactions identify two novel genes, SLA1 and SLA2, that control membrane cytoskeleton assembly in Saccharomyces cerevisiae. . J Cell Biol. 1993;122:635–644. doi: 10.1083/jcb.122.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston GC, Prendergast JA, Singer RA. The Saccharomyces cerevisiae MYO2gene encodes an essential myosin for vectorial transport of vesicles. J Cell Biol. 1991;113:539–551. doi: 10.1083/jcb.113.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney D, Cairns L, Remold-O'Donnell E, Peterson J, Rosen FS, Parkman R. Morphological abnormalities in the lymphocytes of patients with Wiskott-Aldrich syndrome. Blood. 1986;68:1329–1332. [PubMed] [Google Scholar]
- Kilmartin J, Adams AEM. Structural rearrangements of tubulin and actin during the cell cycle of the yeast Saccharomyces. . J Cell Biol. 1984;98:922–933. doi: 10.1083/jcb.98.3.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen T, Rosen FS. Disease mechanism: unravelling Wiskott-Aldrich syndrome. Curr Biol. 1996;6:676–678. doi: 10.1016/s0960-9822(09)00447-3. [DOI] [PubMed] [Google Scholar]
- Kolluri R, Tolias KF, Carpenter CL, Rosen FS, Kirchhausen T. Direct interaction of the Wiskott-Aldrich syndrome protein with the GTPase Cdc42. Proc Natl Acad Sci USA. 1996;93:5615–5618. doi: 10.1073/pnas.93.11.5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarche N, Tapon N, Stowers L, Burbelo PD, Aspenstrom P, Bridges T, Chant J, Hall A. Rac and Cdc42 induce actin polymerization and G1 cell cycle progression independently of p65PAKand the JNK/SAPK MAP kinase cascade. Cell. 1996;87:519–529. doi: 10.1016/s0092-8674(00)81371-9. [DOI] [PubMed] [Google Scholar]
- Li R, Zheng Y, Drubin D. Regulation of cortical actin cytoskeleton assembly during polarized cell growth in budding yeast. J Cell Biol. 1995;128:599–615. doi: 10.1083/jcb.128.4.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillie SH, Brown SS. Immunofluorescence localization of the unconventional myosin, Myo2p, and the putative kinesin-related protein, Smy1p, to the same regions of polarized growth in Saccharomyces cerevisiae. . J Cell Biol. 1994;125:825–842. doi: 10.1083/jcb.125.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HP, Bretscher A. Disruption of the single tropomyosin gene in yeast results in the disappearance of actin cables from the cytoskeleton. Cell. 1989;57:233–242. doi: 10.1016/0092-8674(89)90961-6. [DOI] [PubMed] [Google Scholar]
- Miki H, Miura K, Takenawa T. N-WASP, a novel actin depolymerizing protein, regulates the cortical cytoskeletal rearrangement in a PIP2- dependent manner downstream of tyrosine kinases. EMBO (Eur Mol Biol Organ) J. 1996;15:5326–5335. [PMC free article] [PubMed] [Google Scholar]
- Molina IJ, Kenney DM, Rosen FS, Remold-O'Donnell E. T cell lines characterize events in the pathogenesis of the Wiskott-Aldrich syndrome. J Exp Med. 1992;176:867–874. doi: 10.1084/jem.176.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon A, Drubin DG. The ADF/cofilin proteins: stimulus-responsive modulators of actin dynamics. Mol Biol Cell. 1995;6:1423–1431. doi: 10.1091/mbc.6.11.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon AL, Janmey PA, Louie KA, Drubin DG. Cofilin is an essential component of the yeast cortical cytoskeleton. J Cell Biol. 1993;120:421–435. doi: 10.1083/jcb.120.2.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland J, Preuss D, Moon A, Wong A, Drubin D, Botstein D. Ultrastructure of the yeast actin cytoskeleton and its association with the plasma membrane. J Cell Biol. 1994;125:381–391. doi: 10.1083/jcb.125.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, Rac and Cdc42 GTPases: regulators of actin structures, cell adhesion and motility. Biochem Soc Trans. 1995;23:456–459. doi: 10.1042/bst0230456. [DOI] [PubMed] [Google Scholar]
- Novick P, Botstein D. Phenotypic analysis of temperature-sensitive yeast actin mutants. Cell. 1985;40:405–416. doi: 10.1016/0092-8674(85)90154-0. [DOI] [PubMed] [Google Scholar]
- Novick P, Field C, Schekman R. Identification of 23 complementation groups required for posttranslational events in the yeast secretory pathway. Cell. 1980;21:205–215. doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- Pringle JR, Bi E, Harkins HA, Zahner JE, De Virgilio C, Chant J, Corrado K, Fares H. Establishment of cell polarity in yeast. CHS Sym Quan Biol. 1995;60:729–744. doi: 10.1101/sqb.1995.060.01.079. [DOI] [PubMed] [Google Scholar]
- Remold-O'Donnell E, Rosen FS, Kenney DM. Defects in Wiskott-Aldrich syndrome blood cells. Blood. 1996;87:2621–2631. [PubMed] [Google Scholar]
- Rivero-Lezcano OM, Marcilla A, Sameshima JH, Robbins KC. Wiskott-Aldrich syndrome protein physically associates with Nck through Src homology 3 domains. Mol Cell Biol. 1995;15:5725–5731. doi: 10.1128/mcb.15.10.5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth MB, Zahler AM, Stolk JA. A conserved family of nuclear phosphoproteins localized to sites of polymerase II transcription. J Cell Biol. 1991;115:587–596. doi: 10.1083/jcb.115.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, F., G. Fink, and C. Lawrence. 1974. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorger PK, Pelham HR. Purification and characterization of a heat-shock element binding protein from yeast. EMBO (Eur Mol Biol Organ) J. 1987;6:3035–3041. doi: 10.1002/j.1460-2075.1987.tb02609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons M, Derry JMJ, Karlak B, Jiang S, Lemahieu V, McCormick F, Francke U, Abo A. Wiskott-Aldrich syndrome protein, a novel effector for the GTPase Cdc42Hs, is implicated in actin polymerization. Cell. 1996;84:723–734. doi: 10.1016/s0092-8674(00)81050-8. [DOI] [PubMed] [Google Scholar]
- Welch MD, Holtzman DA, Drubin DG. The yeast actin cytoskeleton. Curr Opin Cell Biol. 1994;6:110–119. doi: 10.1016/0955-0674(94)90124-4. [DOI] [PubMed] [Google Scholar]
- Yonezawa N, Nishida E, Koyasu S, Maekawa S, Ohta Y, Yahara I, Sakai H. Distribution among tissues and intracellular localization of cofilin, a 21kDa actin-binding protein. Cell Struct Funct. 1987;12:443–452. doi: 10.1247/csf.12.443. [DOI] [PubMed] [Google Scholar]
- Yonezawa N, Nishida E, Iida K, Yahara I, Sakai H. Inhibition of the interactions of cofilin, destrin, and deoxyribonuclease I with actin by phosphoinositides. J Biol Chem. 1990;265:8382–8386. [PubMed] [Google Scholar]