

Abstract
An early event in the Trypanosoma cruzi cell invasion process, the recruitment of host lysosomes, led us to investigate the involvement of signal transduction. Infective trypomastigotes were found to contain a soluble Ca2+-signaling activity for mammalian cells that is sensitive to protease inhibitors. Inhibitor and substrate utilization profiles were used to purify a candidate peptidase for involvement in this process, from which we isolated a full-length cDNA clone. The sequence revealed a novel enzyme, denominated T. cruzi oligopeptidase B, which is homologous to members of the prolyl oligopeptidase family of serine hydrolases, known to participate in the maturation of biologically active peptides. The T. cruzi oligopeptidase B was expressed as a fully active product in Escherichia coli, and antibodies to the recombinant enzyme inhibited both peptidase activity and Ca2+ signaling induced in normal rat kidney cells by trypomastigote extracts. Our data suggest that the T. cruzi oligopeptidase B participates in processing events in the cytoplasm of the parasites, generating a factor with Ca2+-signaling activity for mammalian cells.
Signal transduction events induced in host cells by intracellular pathogens are emerging as important regulators of the invasion process (Bliska et al., 1993). As opposed to intracellularly targeted toxins, which have been extensively studied, very little is known about pathogen products that trigger signaling pathways through mammalian surface receptors. Experimental evidence suggests that factors capable of modulating the behavior of mammalian cells are produced by certain intracellular bacteria (Galan, 1994; Menard et al., 1996; Yamamoto et al., 1996), but their molecular nature and mechanism of action are largely unknown. In the case of Shigella, an isolated complex containing the gene products IpaB and IpaC was shown to induce membrane ruffling and macropinocytosis in epithelial cells, similar to what is observed during bacterial internalization (Menard et al., 1996). Although the mode of action and cellular target of the Ipa complex have not yet been determined, it was proposed that these secreted proteins may bind to a surface receptor (Menard et al., 1996).
Recent findings from several laboratories indicate that the cell invasion mechanism of the protozoan parasite Trypanosoma cruzi involves receptor-mediated signal transduction (Ming et al., 1995; Rodriguez et al., 1995; Barr et al., 1996). Entry of T. cruzi into nonphagocytic mammalian cells occurs by recruitment and fusion of host lysosomes at the parasite attachment site, an unusual process that results in formation of a parasitophorous vacuole with lysosomal properties (Tardieux et al., 1992; Rodriguez et al., 1996). The parasite resides in this lysosome-derived vacuole for a short period after invasion, after which it escapes into the cytoplasm, where replication occurs (Meirelles and de Souza, 1983; Ley et al., 1990). T. cruzi invasion of nonphagocytic mammalian cells is restricted to two life cycle stages: metacyclic forms that are transmitted by the insect vector, and trypomastigotes that are released from infected host cells. Epimastigotes are noninfective forms that replicate in the insect vector, and amastigotes are the intracellular stages that replicate inside host cells. Although T. cruzi exhibits tropism for specific cell types in the vertebrate host, it is able to infect many different cell types in culture (Brener, 1973).
Consistent with their infective capabilities, metacyclics (Dorta et al., 1995) and trypomastigotes (Tardieux et al., 1994; Burleigh and Andrews, 1995; Barr et al., 1996) contain a soluble factor that induces transient increases in the cytosolic free calcium concentration ([Ca2+]i)1 of a variety of mammalian cell types (Burleigh and Andrews, 1995). In response to live trypomastigotes or trypomastigote soluble extracts, Ca2+ is mobilized from intracellular stores in an IP3-mediated (Rodriguez et al., 1995), pertussis toxin-sensitive pathway (Tardieux et al., 1994). Prevention of these transients by buffering host cell intracellular free Ca2+ (Tardieux et al., 1994) or depleting intracellular Ca2+ stores (Rodriguez et al., 1995) results in inhibition of parasite invasion. Furthermore, rapid rearrangements in the host cell actin cytoskeleton are observed as a consequence of trypomastigote-induced Ca2+ transients (Rodriguez et al., 1995). Since experimental depolymerization of host cell actin microfilaments results in enhancement of invasion by T. cruzi (Schenkman et al., 1991; Tardieux et al., 1992), the available evidence supports the postulated role for Ca2+ signaling in facilitating cell invasion by these parasites.
Further characterization of the soluble trypomastigote Ca2+-signaling activity revealed that the induction of Ca2+ transients in mammalian cells is coupled to the activity of a parasite peptidase (Burleigh and Andrews, 1995). On the basis of protease inhibitor profile and substrate specificity, an 120-kD peptidase was identified in T. cruzi trypomastigotes as a candidate for involvement in mammalian cell signaling (Burleigh and Andrews, 1995). However, the purified peptidase had no Ca2+-signaling activity on its own, and it was found to be present at similar levels in epimastigotes, a noninvasive life cycle stage of T. cruzi, which does not signal mammalian cells (Burleigh and Andrews, 1995). Removal of a heparin-binding fraction from the trypomastigote soluble extract resulted in a depletion of Ca2+ signaling but not of peptidase activity, suggesting that, in addition to the peptidase, a factor present only in invasive forms of the parasite is required for signaling (Burleigh and Andrews, 1995). This second component could be a precursor that is processed by the peptidase, resulting in a mature Ca2+ agonist. Alternatively, it could function as a recognition molecule, perhaps involved in targeting the peptidase to the host cell membrane where its subsequent activity would result in the induction of Ca2+ transients.
The goal of the present study was to clarify these issues, by characterizing the peptidase at the molecular level and by determining its intracellular location and requirement for Ca2+ signaling. Cloning and nucleotide sequencing of a full-length cDNA clone revealed that the T. cruzi peptidase is a cytosolic enzyme closely related to members of the prolyl oligopeptidase family of serine endopeptidases, some of which were previously shown to function in eukaryote prohormone processing (Fuller et al., 1988; Kreil, 1990). Antibodies to the recombinant peptidase inhibit both peptidase activity and Ca2+ signaling in mammalian cells by trypomastigote extracts, providing direct evidence for participation of the T. cruzi oligopeptidase B in this signaling pathway.
Materials and Methods
Cells and Parasites
Normal rat kidney (NRK) fibroblasts were maintained in 10 mM Hepesbuffered DME containing 10% FBS at 37°C in a humidified atmosphere containing 5% CO2. Trypomastigotes from the T. cruzi Y strain were obtained from supernatants of infected monolayers of LLC-MK2 cells as previously described (Andrews et al., 1987). Amastigotes were generated in culture by incubating freshly harvested trypomastigotes in liver infusion tryptose medium containing 10% FBS at 37°C, 5% CO2 for 24 h. Epimastigotes (Y strain and Dm28c) were cultured in liver infusion tryptose medium at 28°C (Nogueira and Cohn, 1976). Metacyclic parasites were purified from stationary phase cultures of epimastigotes (Dm28c) by passing parasites through a DEAE-cellulose column (Yoshida, 1986).
Preparation of Parasite Soluble Extract
Freshly harvested T. cruzi were washed in Hepes-buffered Ringer's solution containing 0.5% BSA (Heuser, 1989), resuspended at 2 × 108 parasites per ml in Dulbecco's PBS containing 2 mM CaCl2 and 1 mM MgCl2 (PBS2+), and then killed by heating at 56°C for 5 min and frozen at −80°C. Freeze-thawed parasites were disrupted by sonication (model 250; Branson Ultrasonics, Danbury, CT) on ice with a microtip for two 15-s bursts at a setting of 2.5. Unbroken cells and nuclei were removed by centrifugation at 750 g for 10 min at 4°C. The postnuclear supernatant was centrifuged at 100,000 g at 4°C for 45 min using a Ti50.1 rotor (Beckman Instruments, Fullerton, CA), and the resulting supernatant fraction was passed over a Con A–Sepharose column (Pharmacia, Uppsala, Sweden). The unbound material was then collected, aliquoted, and stored at −80°C.
Peptidase Purification and Amino Acid Sequencing
Purification of the 120-kD T. cruzi peptidase was carried out as previously described (Burleigh and Andrews, 1995). Briefly, epimastigotes, grown and harvested as described above, were resuspended in PBS2+ at a concentration of 5 × 108/ml, heated to 56°C for 5 min, and then immediately frozen at −80°C. Thawed parasite suspensions were adjusted to 20 μM N-[N- (l-3-trans-carboxirane-2-carbonyl)-l-leucyl]-agmatine (E-64) to inhibit the T. cruzi major lysosomal protease, cruzain (Cazzulo et al., 1989), before preparation of the parasite soluble fraction as described above. The flow-through fraction from Con A–Sepharose (50 ml) was applied to a 10ml column of DEAE-cellulose equilibrated in PBS2+. Unbound material containing the 120-kD enzyme was subjected to ammonium sulfate precipitation (30–52% saturation) for 2 h on ice. Precipitated protein was recovered by centrifugation at 15,000 g for 30 min, and pellets were resuspended in PBS2+ containing 20 μM E-64, filtered through a 0.22-μm filter unit (Millipore Products Division, Bedford, MA) and desalted on an Econo-Pac® 10DG column (BioRad Laboratories, Hercules, CA) equilibrated in PBS2+. 4 ml of desalted material was applied to a 1.6 cm × 60 cm Sephacryl-300 column (Pharmacia) equilibrated in PBS2+ and proteins were eluted in the same buffer, collected as 2-ml fractions. Peptidase activity was assayed using the synthetic substrate Z-phenylalanyl-arginyl-7amino-4-methyl coumarin (Z-Phe-Arg-AMC) (Enzyme Systems Products, Livermore, CA) as described previously (Burleigh and Andrews, 1995). Peptidase-containing fractions were pooled and concentrated by ammonium sulfate precipitation (70% saturation). The pellet was resuspended in 25 mM Bis-Tris, pH 6.4, desalted, and applied to a Mono-Q HR 5/5 column (Pharmacia). Bound material was eluted with a gradient of 0–250 mM NaCl, 25 mM Bis-Tris, pH 6.4. The 120-kD enzyme eluted at a salt concentration of ∼75–90 mM NaCl. These fractions were pooled and applied to a Mono-P HR 5/5 chromatofocusing column (Pharmacia) and bound material eluted with a pH gradient of 6.4–4.0 formed with Polybuffer 74 (Pharmacia). At this point in the purification scheme, it was possible to detect a significant 120-kD band in Coomassie-stained SDSPAGE gels that hydrolyzed the synthetic substrate Z-Phe-Arg-AMC in a gel overlay assay (Burleigh and Andrews, 1995). Thus, the 120-kD band containing purified peptidase (∼10 μg) was excised from an SDS-PAGE gel and submitted to the W.M. Keck Biotechnology Resource Laboratory at Yale University for in-gel tryptic digestion and amino acid sequence analysis of HPLC-purified peptides.
Isolation of a Peptidase cDNA Clone
Total cellular RNA was isolated from T. cruzi using a guanidinium-thiocyanate extraction method (Chomczynski and Sacchi, 1987). Degenerate oligonucleotide primers were designed in forward and reverse orientations based on the amino acid sequence of two tryptic fragments of the peptidase: DYMNSYSPVD and DEPVFWA. These primers were used to amplify DNA from first strand cDNA that was synthesized from 10 μg total trypomastigote RNA using AMV reverse transcriptase under conditions suggested by the manufacturer (Boehringer Mannheim Corp., Indianapolis, IN). For PCR amplification of peptidase DNA, 5-μl aliquots of trypomastigote cDNA were used as template, and PCR reactions were carried out using the GeneAmp kit (Perkin-Elmer Corp., Norwalk, CT) with 35 cycles of: 1-min annealing at 40°C, 1-min extension at 72°C, 1-min denaturation at 94°C, and a final 5-min extension step at 72°C, using a PTC-100 automated thermocycler (MJ Research, Inc., Watertown, MA). One combination of forward and reverse primers generated a 750-bp PCR product, which was directly cloned into the TA-cloning vector pCRII® (Invitrogen, San Diego, CA) and sequenced from the T7 and SP6 primer sites in the plasmid by the W.M. Keck Biotechnology Resource Laboratory to confirm its authenticity. The 750-bp fragment was labeled by incorporation of [32P]dCTP (Amersham Corp., Arlington Heights, IL) during PCR amplification and used as a probe to screen a T. cruzi amastigote cDNA library constructed in λ ZAPII (Stratagene, La Jolla, CA; generously provided by S. Teixeira and J. Donelson, University of Iowa, Iowa City). Plaques of positive clones were isolated, and pBluescript plasmids containing cDNAs were excised from the phage according to manufacturer's specifications (Stratagene). A clone with the largest insert (2.6-kb EcoRI-XhoI fragment), which contained the full-length peptidase cDNA, was completely sequenced in both directions by the W.M. Keck Biotechnology Resource Laboratory at Yale University.
Expression of Recombinant Peptidase in Escherichia coli
The peptidase gene was amplified from the full-length cDNA clone in pBluescript using the LA-PCRTM kit (Takara Biomedicals, Otsu, Shiga, Japan) using forward (5′-GCT ACC GTG CTC GAG ATG AAG TGT3′) and reverse (5′-GTG ACA CCC TAA CTC GAG CAA TAA-3′) primers designed with internal Xho I sites (bold); the initiation codon of the forward primer is underlined. The PCR product was cloned directly into pCRII (Invitrogen), and then the 2.2-kb XhoI-XhoI fragment was excised and subcloned into the pET19b expression vector (Novagen, Inc., Madison, WI). Restriction analysis was carried out to ensure correct orientation of the gene, and sequencing from the T7 promoter in the plasmid demonstrated that the ATG was in frame. The peptidase was expressed as an NH2-terminal polyhistidine-tagged fusion protein in E. coli BL21 by induction of a log phase culture with 1 mM isopropylthio-β-d-galactoside for 16 h at 27°C. Bacteria were harvested and the recombinant T. cruzi oligopeptidase (rPEP) was purified on nickel-agarose resin according to the manufacturer's specifications (Novagen, Inc.).
Generation and Affinity Purification of Anti-Peptidase Antibodies
All antibodies to the T. cruzi oligopeptidase B were raised by the Pocono Rabbit Farm & Laboratory, Inc. (Canadensis, PA). Antibodies to the denatured 120-kD peptidase were obtained by immunizing a guinea pig with polyacrylamide gel slices containing 5 μg of protein for the initial immunization and 2 μg for subsequent boosts. Serum was collected biweekly and analyzed for peptidase-specific antibodies on Western blots of trypomastigote soluble fractions. Neutralizing antibodies were raised by immunizing a rabbit with 200 μg purified rPEP in complete Freund's adjuvant, followed by monthly boosts of 50 μg in incomplete Freund's adjuvant. Silver stain of an overloaded SDS-PAGE gel confirmed the absence of contaminants in the rPEP preparation obtained by nickel affinity chromatography (not shown). IgG from antipeptidase sera was purified using protein A–Sepharose (Pharmacia) according to a standard procedure (Harlow and Lane, 1988). For affinity purification of antibodies, purified rPEP (750 μg) was coupled to 400 μl of ECH-Sepharose resin (Pharmacia) by addition of 0.1 M of the cross-linking reagent 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDAC; Sigma Chemical Co., St. Louis, MO). The beads were washed and unreacted groups were blocked with 100 mM ethanolamine for 1 h at 37°C. The rPEP-Sepharose was washed extensively with dH2O and equilibrated in 10 mM Tris-HCl, pH 7.5, before incubating overnight at 4°C with antipeptidase IgG (5 ml of IgG at 1 μg/ml). Unbound IgG was removed by washing the rPEP-Sepharose column with 10 ml of 10 mM Tris-Cl, pH 7.5, followed by 10 ml of 500 mM NaCl, 10 mM Tris-Cl, pH 7.5. Antipeptidase IgG was eluted with 5 ml 100 mM glycine, pH 2.5, and neutralized with 500 μl 1 M Tris-HCl, pH 8.0.
SDS-PAGE and Immunoblotting
Parasite soluble fractions or rPEP were treated with Laemmli sample buffer (Laemmli, 1970) containing 5% β-2-mercaptoethanol at 25° or 100°C for 5 min before separation on 7.5% polyacrylamide gels. Proteins were transferred to ImmobilonTM membranes (Millipore Corp.) according to the method of Towbin et al. (1979). After an overnight incubation at 4°C in blocking buffer (5% nonfat milk, 20 mM Tris, pH 7.4, 15 mM NaCl, 0.05% sodium azide), blots were incubated with 25 μg/ml guinea pig anti– peptidase IgG or 5 μg/ml rabbit anti–rPEP IgG in blocking buffer for 2 h at room temperature. Blots were washed five times with 20 mM Tris, pH 7.4, 150 mM NaCl, and 0.1% Tween 20 (TBST) for 5 min each wash, and then incubated with a 1:4,000 dilution of alkaline phosphatase–labeled goat anti–guinea pig IgG or goat anti–rabbit IgG (Kirkegaard & Perry Laboratories, Gaithersburg, MD) for 1 h in blocking buffer. After a 30min wash with TBST, immunoreactive bands were detected by incubating blots with the alkaline phosphatase substrate 5-bromo-4-chloro-3-indolyl1-phosphate/nitro blue tetrazolium (BCIP/NBT; Promega Corp., Madison, WI).
Peptidase Activity Assays
Peptidase activity was carried out either as a soluble assay using 7-amino4-methyl coumarin (AMC) or 7-amino-4-trifluoromethyl coumarin (AFC)– conjugated peptide substrates (Enzyme Systems Products), or by direct detection of peptide-AMC hydrolysis in SDS-PAGE gels as described previously (Burleigh and Andrews, 1995). For kinetic analysis, rPEP was diluted with 50 mM Tris-Cl, pH 8.0, 150 mM NaCl to a final concentration of 6 × 10−9 M, and substrates were added in the range of 2–15 μM. Initial velocity (vo) of substrate hydrolysis was recorded as the release of AMC over 60 s (during which the rate of AMC hydrolysis was linear) in a model F-2000 spectrofluorimeter (Hitachi Instruments Inc., Danbury, CT) at excitation and emission wavelengths of 380 and 440 nm, respectively. Km values were calculated from Lineweaver-Burke plots (Segel, 1976). Inhibitor sensitivity was carried out by incubation of rPEP (5 ng) in 500 μl with varying concentrations of inhibitors at room temperature for 10 min before adding to 1.5 ml 10 μM Z-Phe-Arg-AMC in TBS and measuring hydrolysis of substrate for 5 min in the spectrofluorimeter, as described above.
Detection of Expressed Products of the Peptidase Gene in T. cruzi
First strand cDNA was synthesized from total RNA isolated from different T. cruzi life cycle stages as described above for trypomastigotes. 1 μl of first strand cDNA synthesis reactions from the three life cycle stages (trypomastigotes, amastigotes, and epimastigotes) was used as template for PCR with a primer pair (forward primer: 5′-ATGAAGTGTGGTCCCATTGCCACG-3′; reverse primer: 5′-CATTTCCACCTCCGAAGAAGTGTC-3′) designed to produce a full-length amplification product. Amplification conditions were as follows: 40 cycles of 1 min at 94°C; 1 min at 50°C; 3 min at 72°C; and a final step of 7 min at 72°C. Southern blot analysis of PCR products separated by electrophoresis on a 1% agarose gel was carried out to determine the specificity of the resulting 2.1-kb bands. Blots were hybridized with the 2.5-kb EcoRI-XhoI fragment excised from the full-length cDNA clone. The EcoRI-XhoI fragment was purified from a 0.7% agarose gel using the CompassTM DNA purification kit (American Bioanalytical, Natick, MA), and 50 ng was used in a random priming reaction (Boehringer Mannheim Corp.) with 50 μCi[32P]dCTP (Amersham Corp.). Hybridization was carried out overnight at 42°C in 50% formamide, 6× SSC, 0.5% SDS, and 5× Denhardt's solution. The blots were washed to a final stringency of 0.6× SSC at 55°C before exposure to X-OMat film (Eastman Kodak Co., Rochester, NY).
Genomic DNA Isolation and Southern Blots
Genomic DNA was isolated from epimastigotes (Y strain) following published methods (Medina-Acosta and Cross, 1993). To determine the number of genomic copies of the T. cruzi peptidase gene, 5 μg aliquots of total genomic DNA were digested for 16 h with restriction enzymes, and digested DNA was separated on 0.9% agarose gels in 0.5× Tris-borateEDTA buffer and transferred to Hybond-N membranes by capillary blotting in 20× SSC for 16 h. Blots were hybridized with the full-length peptidase cDNA probe.
Inhibition of Peptidase and Ca2+-Signaling Activities with Anti-recombinant Peptidase Antibodies
Inhibition of oligopeptidase B activity in trypomastigote soluble extract (TSE) with IgG from rabbits immunized with the purified recombinant enzyme was carried out in solution by adding 0.75 μg/ml to 250 μg/ml purified IgG to 50 μl TSE in a final volume of 100 μl. After a 60-min incubation at 4°C, oligopeptidase B activity was measured by adding 200 μl TBS containing 20 μM Z-Phe-Arg-AMC and 10 μM E-64, and incubated for 10 min at 37°C. The reactions were stopped by adding 1 ml ethanol and AMC release was measured as described above. To test for the effect of antirPEP antibodies on the soluble Ca2+-signaling activity in these samples, TSE was preincubated with 200 μg/ml preimmune IgG or anti-rPEP IgG for 60 min at 4°C, and then added to NRK fibroblasts preloaded with 5 μM fura-2/AM, 0.01% pluronic F-127 (Molecular Probes Inc., Eugene OR), and 2.5 mM probenecid (Sigma Chemical Co.). Changes in intracellular calcium concentration of NRK cells were continuously measured in a spectrofluorimeter equipped with intracellular cation determination software as described previously (Burleigh and Andrews, 1995). Ca2+-signaling activity of trypomastigote soluble extracts was also assayed at the single cell level, by time lapse fluorescence video microscopy. NRK cells were plated 48 h before the assay at 1.3 × 105 per dish, on 3.5-cm glass bottom microwells (MatTek Corp., Ashland, MA), and loaded with 5 μM fluo3/AM (Molecular Probes Inc.) for 45 min at 37°C. After loading, the dishes were washed in Hepes-buffered Ringer's solution (Heuser, 1989) and placed on the heated stage of an Axiovert 135 microscope (Carl Zeiss, Inc., Thornwood, NY). Fluorescence images were collected with a video camera (CCD-C72; Dage/MTI, Michigan City, IN) through a ×40 objective and recorded on an optical memory disk (TQ-3031F; Panasonic, Secaucus, NJ), at time-lapse intervals of 1 s using a computer-controlled shutter system (Metamorph software; Universal Imaging Corp., Westchester, PA). Parasite soluble extracts (50 μl) preincubated with preimmune or immune anti-rPEP IgG (200 μg/ml) were added to the cells at 50 s after initiation of the time-lapse recording. Pseudocolored images were transferred to Adobe Photoshop (Adobe Systems, Inc., Mountain View, CA), composed and printed in a high resolution color printer (XLS 8600PS; Eastman Kodak Co.).
EM Immunolocalization
Trypomastigotes freshly harvested from LLC-MK2 tissue-culture supernatants (1 × 107 per ml) were fixed with an equal volume of 2× fixative (0.2% glutaraldehyde, 8% formaldehyde, 200 mM Hepes, pH 7.4) and centrifuged for 10 min at 3,000 g. The supernatant was removed, and fresh 1× fixative was added to the pellet for an additional 2 h. Pellets were infiltrated with 2.1 M sucrose/PBS and frozen in liquid nitrogen. Cryosectioning and labeling of parasites was carried out as described previously (Webster and Grab, 1988) using affinity-purified antipeptidase IgG at 20 μg/ml and 10 nm protein A–gold. Micrographs were taken using a 410 TEM (Phillips Instrument Co., Marwar, NJ) operating at 80 kV.
Results
Isolation of a Full-Length cDNA Clone Encoding the T. cruzi Peptidase Implicated in Ca2+ Signaling
The T. cruzi peptidase was purified several hundredfold from epimastigote soluble extracts in several FPLC steps as described previously (Burleigh and Andrews, 1995). The 120-kD band, shown to have peptidase activity in a gel overlay assay (Burleigh and Andrews, 1995), was excised from an SDS-PAGE gel and subjected to trypsin digestion, peptide purification by HPLC, and amino acid sequencing of selected peptides. Degenerate oligonucleotides designed based on amino acid sequences of internal tryptic peptides of the 120-kD peptidase were used in reverse transcriptase (RT)–PCR reactions with total cellular RNA from trypomastigotes as template. The resulting 750-bp PCR product was cloned, sequenced from both ends, and found to contain homologous sequences to known peptidases by searching the available sequence databases. This 750-bp PCR product was used as a probe to screen a T. cruzi cDNA library, from which several positive clones were obtained. The clone with the largest insert (2.6 kb) was completely sequenced in both directions (GenBank accession number U69897) and a 2215-bp open reading frame was identified that contained an A-T rich region with a stop codon (TAA) 42 bp upstream of the ATG designated as the initiation codon (Fig. 1). Several other clones containing smaller inserts (2.1 and 1.4 kb) were partially sequenced and found to be peptidase cDNA clones lacking complete 5′ ends (not shown).
Figure 1.

Nucleotide sequence of cloned cDNA and deduced amino acid sequence of the T. cruzi oligopeptidase B. The deduced amino acid sequence is shown below the nucleotide sequence. Both are numbered on the right. Amino acid sequences obtained for tryptic peptides of the purified peptidase (underlined). The stop codons that limit the open reading frame (bold); polyadenylation signals are highlighted (double underline). These sequence data are available from GenBank under accession number U69897.
Recombinant T. cruzi Oligopeptidase B Expressed in E. coli Has Identical Biochemical Properties to the Native Enzyme
The peptidase cDNA sequence (Fig. 1), which predicts a polypeptide of 714 amino acids with a calculated molecular mass of 81,011 daltons, was cloned into pET-19b and expressed in E. coli. Preliminary characterization of the purified polyhistidine-tagged rPEP revealed that the specific activity of the enzyme, using Z-Phe-Arg-AMC as a substrate, was similar to that described for the native peptidase purified from epimastigotes (Burleigh and Andrews, 1995). However, because the T. cruzi peptidase was shown to migrate in an SDS-PAGE gel as a band of 120 kD, it was of interest to determine the relationship between the 81-kD polypeptide predicted by the cDNA sequence and the native molecule. To accomplish this, polyclonal antibodies raised to gel-extracted T. cruzi 120-kD peptidase were used to probe Western blots of trypomastigote soluble proteins and rPEP separated by SDS-PAGE under nonreducing and reducing conditions. Immunoreactive bands of ∼120 and 80 kD were detected in both the soluble trypomastigote extract and purified rPEP under nonreducing conditions (Fig. 2 A, lanes 1). However, when samples were fully reduced and boiled, the 120-kD band was no longer detectable and the 80-kD one was the major band (Fig. 2 A, lanes 2). The two lower molecular weight bands observed in reduced boiled samples are likely to be degradation products, since they occur in both the native enzyme and rPEP samples (Fig. 2 A, lanes 2). These results demonstrate that the T. cruzi oligopeptidase B, both parasite derived and recombinant, migrates with an apparently higher molecular weight on SDS-PAGE gels when electrophoresis is carried out under conditions known to preserve peptidase activity (Burleigh and Andrews, 1995), but migrates at the expected size of 80 kD when fully denatured.
Figure 2.

Molecular size determination of native and recombinant T. cruzi oligopeptidase B. (A) Immunoblots of oligopeptidase B partially purified from epimastigotes (T. cruzi, lanes 1 and 2) or 0.6 μg purified recombinant peptidase (rPEP, lanes 1 and 2) with guinea pig polyclonal anti–oligopeptidase B IgG (25 μg/ml) followed by acid phosphatase–conjugated anti–guinea pig IgG. Samples were reduced with 5% β-mercaptoethanol and kept at either 25°C (lanes 1) or heated for 5 min at 100°C (lanes 2) before SDS-PAGE. (B) FPLC fractionation of a trypomastigote soluble extract on Sephacryl-300. Proteins were eluted in PBS2+ while A280 nm was continuously monitored (open squares). The peak absorbance value (fraction 41) was taken as 100% and the baseline as 0% protein. Peptidase activity (black circles) was assayed in 20 μl aliquots of each fraction using ZFR-AMC as a substrate, as described in Materials and Methods. Fractions containing peptidase activity (64-76) were pooled, concentrated, reduced, boiled for 5 min, separated by SDS-PAGE, and immunoblotted with anti–oligopeptidase B guinea pig IgG as described above. The elution of molecular mass standards is indicated by letters (a) catalase = 232 kD; (b) aldolase = 158 kD; (c) albumin = 67 kD; (d) ribonuclease A = 13.7 kD.
The T. cruzi oligopeptidase B eluted from S-300 gel filtration columns in fractions where proteins of ∼180 kD would be expected (Fig. 2 B). The only detectable immunoreactivity on a Western blot probed with anti-peptidase antibodies was associated with the pooled peak of peptidase activity in fractions 64–76 (Fig. 2 B). No immunoreactivity was detected in the pool containing fractions 77–89, which would contain monomeric proteins in the 60–100-kD range, suggesting that the peptidase is not present in monomeric form in parasite extracts. A similar profile was observed for elution of the purified recombinant oligopeptidase B from an S-300 column (not shown).
Previous studies of the native T. cruzi oligopeptidase B revealed that this enzyme is specific for peptide substrates with basic amino acids residues in the P1 position (Ashall, 1990; Santana et al., 1992; Burleigh and Andrews, 1995). Kinetic analyses using purified rPEP were carried out with three Arg-containing synthetic substrates. The calculated Km values for these substrates were similar: Z-Phe-ArgAMC and Z-Gly-Gly-Arg-AMC had Km values of 6 ± 2 μM, and Z-Arg-Arg-AMC of 5 ± 1 μM. No hydrolysis of Z-GlyPro-AMC, Z-Gly-Phe-AFC, or Z-Ala-AFC by the recombinant peptidase was detected, indicating specificity for cleavage at the carboxyl side of arginine residues, as previously reported (Ashall, 1990; Santana et al., 1992; Burleigh and Andrews, 1995). Sensitivity of the recombinant oligopeptidase B to several different protease inhibitors was then examined. The concentration of inhibitor that resulted in a 50% reduction of the initial rate of hydrolysis of Z-Phe-Arg-AMC (IC50) was determined: 0.02 μM leupeptin, 0.05 μM antipain, 0.33 μM p-chloromercuribenzoic acid, 0.6 μM N-α-p-tosyl-l-lysine-chloromethyl ketone, 10 μM chymostatin, 225 μM diisopropyl fluorophosphate, 0.25 μM Z-Phe-Arg-fluoromethylketone, and 0.2 μM acetylArg-Arg-chloromethylketone. Millimolar concentrations of PMSF, aprotinin, α2-macroglobulin, EDTA, and E-64 did not inhibit recombinant oligopeptidase B activity.
The results presented above demonstrate that the biochemical properties of the recombinant oligopeptidase B are identical to those of the peptidase purified from T. cruzi (Burleigh and Andrews, 1995), thereby confirming the authenticity of the cDNA clone and indicating that it contains the entire open reading frame (Fig. 1).
T. cruzi Oligopeptidase B Is Homologous to Members of the Prolyl Oligopeptidase Family of Serine Hydrolases
Comparison of the peptidase cDNA sequence with available databases revealed that the T. cruzi oligopeptidase B shares significant homology with a subgroup of serine hydrolases referred to as the prolyl oligopeptidase family. The GxSxGGzz consensus sequence (where x = any amino acid and z = a hydrophobic residue; Barrett and Rawlings, 1992) that harbors the active site serine residue in all members of this enzyme family was also found in the T. cruzi sequence (Fig. 3). Since the greatest degree of homology between members of the prolyl oligopeptidase family has been shown to occur within the COOH-terminal third of the molecule (Barrett and Rawlings, 1992), this region of the T. cruzi peptidase was aligned with the protease II from E. coli (Kanatani et al., 1991) and Moraxella lacunata (Yoshimoto et al., 1995), and prolyl endopeptidases from Flavobacterium meningosepticum (Yoshimoto et al., 1991), Aeromonas hydrophila (Kanatani et al., 1993), and porcine brain (Rennex et al., 1991). From the partial sequence alignment (Fig. 3), it is apparent that these molecules share significant homology, particularly in the vicinity of the active site serine residues (Fig. 3, asterisk). The T. cruzi peptidase was more similar overall to the Moraxella and E. coli peptidases, having 34% and 30% identical residues, respectively. Less identity to the T. cruzi enzyme was observed with peptidases from Flavobacterium (22%), Aeromonas (21%), and pig brain (18%). The T. cruzi sequence contains conserved Asp and His residues at positions 647 and 682, which have been proposed to form the catalytic triad with Ser in this family of enzymes (Stone et al., 1991; Yoshimoto et al., 1995). It was on the basis of sequence similarity with the Moraxella and E. coli protease II enzymes (since renamed oligopeptidase B; Tsuru and Yoshimoto, 1994) that the T. cruzi enzyme was named oligopeptidase B.
Figure 3.

The T. cruzi oligopeptidase B is homologous to members of the prolyl oligopeptidase family of serine hydrolases. Residues 412– 683 of the T. cruzi oligopeptidase B (top) are aligned with Moraxella lacunata (residues 385–655; Yoshimoto et al., 1995) and Escherichia coli protease II (residues 384– 653; Kanatani et al., 1991), and prolyl oligopeptidases from Flavobacterium meningosepticum (residues 409– 676; Yoshimoto et al., 1991), Aeromonas hydrophila (residues 390–657; Kanatani et al., 1993), and porcine brain (residues 403–681; Rennex et al., 1991). Identical residues are boxed, and the active site serine residue included in the consensus sequence GxSxGGzz is denoted by an asterisk (*).
Oligopeptidase B Gene is Present as a Single Copy and Expressed in All Life Cycle Stages of T. cruzi
Southern blot analysis of T. cruzi genomic DNA probed with the full-length oligopeptidase B cDNA revealed a simple pattern after single enzyme digestions. A single band was produced with several different enzymes, and two bands were obtained after digestion with SacI, ScaI, or PvuI (Fig. 4 A). These results are consistent with the predicted restriction map of the peptidase cDNA (not shown), suggesting that the peptidase gene is represented as a single copy in the T. cruzi Y strain genome.
Figure 4.

The T. cruzi oligopeptidase B gene is present as a single copy in the genome and expressed in all life cycle stages of the parasite. (A) Southern blot of T. cruzi genomic DNA digests probed with the full-length oligopeptidase B cDNA labeled with [32P]dCTP. The blot was washed to a final stringency of 0.6× SSC at 60°C and exposed to film for 5 d. (B) Expression of the oligopeptidase B gene in life cycle stages of T. cruzi. (I) Amplification of homologous sequences from total parasite RNA by RT-PCR. PCR products were separated on agarose, blotted to nylon, and probed with the fulllength peptidase cDNA, revealing a single band of 2.1 kb in three life cycle stages of T. cruzi. (II) Detection of peptidase activity in parasite soluble extracts separated by SDS-PAGE. Total soluble extract was prepared from each parasite life cycle stage, and 3 × 107 parasite equivalents were loaded in each lane. Oligopeptidase B activity was detected directly in a gel overlay assay as described in the Materials and Methods. Active purified recombinant peptidase (0.6 μg; rPEP) migrated with an apparent molecular mass of 120 kD, similar to oligopeptidase B activity expressed in each developmental stage of T. cruzi: epimastigotes (Epi), metacyclics (Meta), amastigotes (Amast), and trypomastigotes (Trypo).
From previous work it was known that oligopeptidase B is expressed in trypomastigotes and epimastigotes (Burleigh and Andrews, 1995). To determine whether the peptidase gene was expressed in other life cycle stages of T. cruzi, RT-PCR was carried out using total RNA prepared from each stage, and specific oligonucleotide primers predicted to result in the amplification of a full-length PCR product. PCR products were separated on agarose gels, blotted, and probed with a full-length cDNA probe (Fig. 4, B-I) that revealed a single 2.1-kb band present in the three main life cycle stages of T. cruzi. For direct detection of oligopeptidase B activity in the life cycle stages of the parasite, soluble extracts were prepared and separated on SDS-PAGE gels, and peptidase activity was detected in a gel overlay assay using Z-Phe-Arg-AMC as the substrate. As shown in Fig. 4, B-II, peptidase activity migrating at an apparent molecular mass of 120 kD was found in all parasite life cycle stages. The 120-kD band was previously shown to be the only peptidase detectable with this substrate in Con A–unbound soluble fractions of T. cruzi (Burleigh and Andrews, 1995). Purified recombinant oligopeptidase B was also included in the gel (Fig. 4, B-II, rPEP) to demonstrate that the active product of the cloned peptidase gene expressed in E. coli also migrates at 120 kD, identical to the enzyme isolated from T. cruzi (Fig. 2 A; Burleigh and Andrews, 1995).
Antibodies to the Recombinant T. cruzi Oligopeptidase B Inhibit Peptide Hydrolysis and Ca2+-Signaling Activity of Trypomastigote Soluble Extracts
Polyclonal antibodies raised against gel-purified 120-kD peptidase were unable to inhibit the activity of the enzyme in solution or to precipitate native enzyme in the absence of detergent (data not shown). Therefore, highly purified active rPEP was used to immunize rabbits, and IgG prepared from immune sera was tested for peptidase-blocking activity. As shown in Fig. 5 A, anti-rPEP IgG proved capable of inhibiting activity of oligopeptidase B in TSE, whereas preimmune rabbit IgG added at the same final concentrations had no effect. The maximum inhibitory effect (90%) achieved with 100 μg/ml anti-rPEP IgG (Fig. 5 A) was not increased with higher concentrations of IgG (up to 500 μg/ml) or by extending the preincubation time (not shown). On a Western blot of total soluble trypomastigote proteins separated by SDS-PAGE under reducing conditions, anti-rPEP IgG specifically recognized the 80-kD peptidase (Fig. 5 B, αrPEP). Additional bands of lower intensity appearing on this blot are also recognized by the same concentration of IgG prepared from preimmune rabbit sera (Fig. 5 B, Pre), with the exception of a band of ∼40 kD that is most likely an oligopeptidase B degradation product.
Figure 5.

Antibodies to the recombinant peptidase inhibit peptidase activity and Ca2+ signaling in mammalian cells by trypomastigote extracts. (A) Oligopeptidase B activity in TSE pretreated with increasing concentrations of preimmune rabbit IgG (open circles) or immune rabbit IgG against rPEP (black circles). Purified IgG ranging from 0.75–500 μg/ml was added to TSE followed by a 1-h incubation at 4°C. Hydrolysis of the Z-Phe-Arg-AMC oligopeptidase B substrate was measured at 37°C for 10 min, as described in Materials and Methods. The values for nmol AMC released are plotted as a function of IgG concentration ([IgG] μg/ml). (B) Total trypomastigote soluble extract (6 × 106 parasite equivalents) was heated to 100°C in Laemmli sample buffer in the presence of 5% β2-mercaptoethanol before separation on a 10% SDS-PAGE gel and immunoblotting with 5 μg/ml αrPEP (αrPEP) or 5 μg/ml IgG prepared from the preimmune rabbit serum (Pre) as described in the Materials and Methods. The arrow indicates the 80-kD oligopeptidase B. (C) Effect of anti-rPEP on the Ca2+-signaling activity of TSE on NRK fibroblasts. TSE was preincubated with 200 μg/ml preimmune IgG (black line) or anti-rPEP IgG (grey line) for 1 h at 4°C. The ability of TSE to induce [Ca2+]i transients in NRK cells loaded with the Ca2+-sensitive dye fura-2/AM was measured in a spectrofluorometric assay, as described in Materials and Methods. Results are plotted as the ratio of the fluorescence emission of fura-2/AM at 510 nm after alternating excitation at 340 nm/380 nm against time (s). TSE was added to the cells at the points indicated by the arrow. The results of six independent experiments are shown.
To test whether the inhibition of oligopeptidase B activity by anti-rPEP would affect the Ca2+-signaling activity of trypomastigote extracts (Tardieux et al., 1994; Burleigh and Andrews, 1995), TSE was pretreated with either preimmune or immune IgG at a final concentration of 200 μg/ml, and then added to fura-2/AM-loaded NRK fibroblasts. Preincubation of TSE with anti-rPEP antibodies resulted in a significant delay in the induction of cytosolic free Ca2+ transients, when compared with treatment with preimmune IgG (Fig. 5 C). In addition, the level of the response was significantly reduced in four out of six experiments (Fig. 5 C).
To directly visualize the kinetics and intensity of the Ca2+ transients triggered in NRK cells by trypomastigote soluble extracts, fluorescence video microscopy at the single cell level was performed after pretreatment with peptidase-blocking antibodies or preimmune IgG (Fig. 6). Similar to the spectrofluorimetric data, pretreatment of TSE with anti-rPEP antibodies (Fig. 6, panels I) resulted in a delayed response, and fewer cells responded in comparison with cells preincubated with preimmune IgG (Fig. 6, panels PI). The time required for the onset of a Ca2+ transient after addition of control or immune IgG-treated TSE was determined in eight separate experiments (Table I). NRK cells receiving anti-rPEP IgG-treated TSE consistently showed a delayed response when compared with cells receiving preimmune treated TSE. Therefore, this data demonstrate a direct link between the activity of T. cruzi oligopeptidase B and the generation of Ca2+ transients in mammalian cells. As observed with the native purified T. cruzi oligopeptidase B (Tardieux et al., 1994; Burleigh and Andrews, 1995), the recombinant peptidase had no Ca2+-signaling activity by itself when added to NRK cells at a specific activity equivalent to trypomastigote extracts (not shown).
Figure 6.

Antibodies to the recombinant T. cruzi oligopeptidase B delay the onset of [Ca2+]i transients in NRK cells. Time-lapse fluorescence microscopy images of NRK cells loaded with the Ca2+-sensitive dye fluo-3/AM. Images shown were acquired 40 s before, and 20, 30, 50, and 100 s after exposure to TSE. In the two experiments shown on the top of the figure, TSE was preincubated for 1 h at 4°C with 200 μg/ml preimmune rabbit IgG (PI); in the two experiments shown at the bottom, preincubation was carried out with 200 μg/ml anti-rPEP immune IgG (I). Fluorescence intensity is pseudocolored, with the highest [Ca2+]i shown in red and the lowest in blue. Bar, 10 μm.
Table I.
Effect of Anti-rPEP IgG on the Time of Onset of [Ca2+]i Elevation Triggered in NRK Cells by Trypomastigote Soluble Extracts
| Experiments | Onset of response‡ | |||
|---|---|---|---|---|
| Preimmune IgG* | Anti-rPEP IgG | |||
| s | ||||
| 1 | 7 | 30 | ||
| 2 | 18 | NR | ||
| 3 | 16 | NR | ||
| 4 | 18 | 34 | ||
| 5 | 18 | 47 | ||
| 6 | 19 | 85 | ||
| 7 | 19 | 51 | ||
| 8 | 30 | 51 | ||
Preincubation of trypomastigote soluble extract (50 μl) with 200 μg/ml (experiments 1–6) or 500 μg/ml (experiments 7 and 8) of purified preimmune or immune IgG was carried out at 4°C for 1 h before addition of NRK cells.
Time point in which the first {Ca2+]i elevation was detected at the single cell level in fluo-3–loaded NRK fibroblasts, during a time-lapse video recording of fluorescence intensity after addition of pretreated trypomastigote extracts. NR, no response was observed in a period of 150 s.
T. cruzi Oligopeptidase B Is Localized in the Cytoplasm of Trypomastigotes
The cDNA sequence of the T. cruzi oligopeptidase B predicts a soluble, hydrophilic protein with no secretion signal sequence (Fig. 1). Consistent with this finding, >90% of oligopeptidase B is released in soluble form and found in the supernatant of 100,000 g centrifugations when trypomastigotes are disrupted by sonication (not shown). For determining the precise localization of the enzyme in the parasites, ultrathin cryosections of aldehyde-fixed trypomastigotes and amastigotes were labeled with affinity- purified guinea pig anti–oligopeptidase B IgG, followed by 10 nm protein A–gold. Labeling was observed throughout the cells, and appeared to be restricted to the cytosol in both trypomastigotes (Fig. 7 A) and amastigotes (Fig. 7 B). No labeling was detected on the surface of the parasites or in the flagellar pocket. In addition, no label was associated with organelles such as the ER, Golgi complex, nucleus, or kinetoplast. This pattern of labeling is consistent with a cytoplasmic localization.
Figure 7.
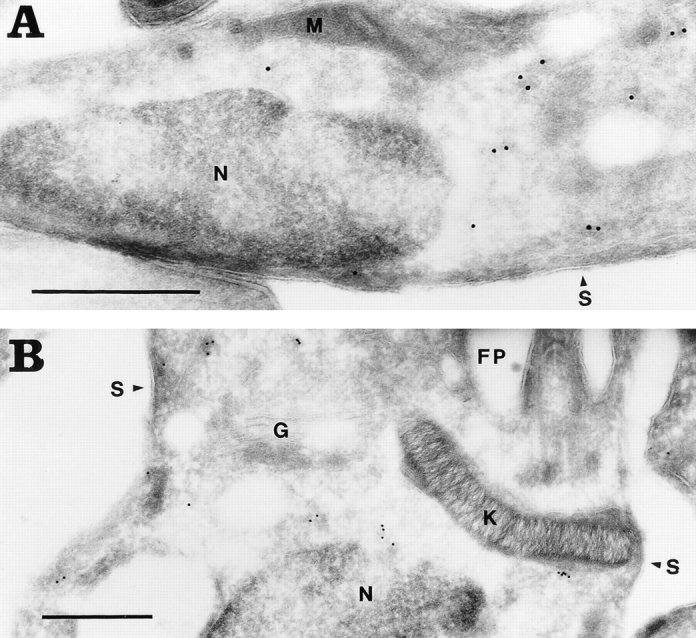
The T. cruzi oligopeptidase B is localized in the cytoplasm of the parasites. Ultrathin cryosections of aldehyde-fixed T. cruzi were labeled with affinitypurified guinea pig anti–oligopeptidase B IgG followed by protein A–10 nm gold. The subcellular localization of oligopeptidase B in trypomastigotes (A) is similar to that of amastigotes (B). No labeling is detected in the nucleus (N), kinetoplast (K), mitochondrion (M), Golgi complex (G), on the parasite surface (S), or in the flagellar pocket (FP). Bar, 0.5 μm.
Discussion
The unusual invasion mechanism recently elucidated for T. cruzi, which involves recruitment and fusion of host cell lysosomes at the parasite entry site (Tardieux et al., 1992; Rodriguez et al., 1996), led us to search for signal transduction events triggered specifically by the forms capable of invading mammalian cells. A soluble factor that generates IP3 and releases Ca2+ from the intracellular stores of mammalian cells was detected and found to be present only in the infective trypomastigote and metacyclic life cycle stages (Tardieux et al., 1994; Burleigh and Andrews, 1995; Dorta et al., 1995; Barr et al., 1996). Buffering of host cell free intracellular Ca2+ (Tardieux et al., 1994) or depletion of intracellular Ca2+ stores (Rodriguez et al., 1995) blocked trypomastigote entry, suggesting that the Ca2+-signaling activity, detected also with intact parasites, is essential for invasion.
Results from a previous study using protease inhibitors (Burleigh and Andrews, 1995) suggested that the activity of a T. cruzi 120-kD peptidase was coupled to the Ca2+-signaling activity of trypomastigotes. Here, we describe the cloning of the gene encoding this peptidase, whose sequence revealed its relationship to a family of serine oligopeptidases that includes hormone-processing enzymes (Fuller et al., 1988; Kreil, 1990). Most importantly, using specific antibodies generated against the recombinant T. cruzi oligopeptidase B, we provide direct evidence for involvement of this enzyme in the generation of Ca2+ transients in mammalian cells by trypomastigote extracts. Our present results are consistent with the hypothesis that the T. cruzi oligopeptidase B, although not capable of signaling by itself, is required for generating a Ca2+-signaling factor for mammalian cells (Burleigh and Andrews, 1995).
The T. cruzi oligopeptidase B shares significant sequence homology with members of the recently defined prolyl oligopeptidase family (Barrett and Rawlings, 1992). This family of serine hydrolases contains both endo- and exopeptidases, soluble and membrane-associated enzymes that exhibit variable but quite restricted substrate specificities. Prolyl oligopeptidases contain a consensus sequence GxSxGGzz that includes the active site serine residue, and they have a proposed mechanism of catalysis involving a Ser-Asp-His charge relay system (Barrett and Rawlings, 1992). The highest degree of sequence conservation among these enzymes, including the T. cruzi oligopeptidase B, is observed within the COOH-terminal third of the molecule, where the catalytic residues are situated. The T. cruzi oligopeptidase B is most similar to the Moraxella and E. coli peptidases in terms of primary sequence and substrate specificity, cleaving substrates on the COOH-terminal side of basic amino acids (Pacaud and Richaud, 1975; Yoshimoto et al., 1995). Less identity was found between the T. cruzi enzyme and the Flavobacterium, Aeromonas, and pig brain peptidases, all of which are post-prolyl cleaving enzymes (Rennex et al., 1991; Yoshimoto et al., 1991; Kanatani et al., 1993). As additional peptidases containing the GxSxGGzz consensus sequence are identified, a division in the prolyl oligopeptidase family may emerge, based on sequence identity as well as substrate specificity. Since the T. cruzi oligopeptidase B is the first protozoan member of the prolyl oligopeptidase family to be cloned and sequenced, it would be interesting to determine if similar peptidases are present in other ancient eukaryotes. The extensive sequence similarity observed between the T. cruzi oligopeptidase B and the bacterial enzymes was somewhat surprising, although an additional example of a closer relationship of a trypanosome gene with bacterial but not eukaryotic homologues has recently appeared in the literature (Stebeck et al., 1996).
Physiological substrates have been identified for only a few members of the prolyl oligopeptidase family of serine peptidases. Some of them, such as the human dipeptidyl aminopeptidase IV and aminopeptidase P, were shown to cleave biologically active peptides in vitro, which led to a proposed role for these enzymes in the final degradation of mature peptide agonists (Mentlein, 1988). However, there is significant evidence that members of this family also participate in the final steps of maturation of regulatory peptides (Fuller et al., 1988; Mentlein, 1988; Kreil, 1990). One of the best characterized examples is the Saccharomyces cerevisiae dipeptidyl aminopeptidase A (DPAP A), which is involved in the maturation of the yeast mating hormone α-factor (Fuller et al., 1988). DPAP A is a transGolgi enzyme that removes dipeptides from the NH2 terminus of pro–α-factor to release the 17–amino acid active hormone as the last step in the α-factor processing cascade (Fuller et al., 1988). Studies of substrate preferences for prolyl oligopeptidases have shown that these enzymes preferentially hydrolyze small peptides with an upper limit of 30 amino acids, and therefore have been described as “true oligopeptidases” (Barrett and Rawlings, 1992; Polgar, 1992). A notable exception is the E. coli oligopeptidase B, which was shown to hydrolyze aspartokinase I–homoserine dehydrogenase at a hinge region separating the two functional domains of the polypeptide (Veron et al., 1972; Pacaud and Richaud, 1975). However, cleavage of this protein by the E. coli peptidase does not occur in the presence of threonine, an allosteric effector for the dehydrogenase, suggesting that in larger substrates conformational changes may be required for the exposure of bonds susceptible to cleavage.
Purification and characterization of a high molecular weight alkaline peptidase from T. cruzi that hydrolyzes substrates on the carboxyl side of basic amino acid residues was reported previously, although no biological role was proposed for it (Ashall, 1990; Santana et al., 1992). Based on the reported characteristics, it is likely that the enzyme described in those studies is the same as the one we have now cloned and expressed. Expression of the fulllength T. cruzi oligopeptidase B cDNA in E. coli resulted in the overproduction of active peptidase with identical properties to the enzyme purified from soluble parasite extracts (Burleigh and Andrews, 1995). Both enzymes migrated in SDS-PAGE gels at an apparent molecular mass of 120 kD when active, but were reduced to the predicted size of 80 kD when fully denatured. On gel filtration columns, the enzymes eluted with a molecular mass fraction of ∼180 kD, suggesting dimerization.
Polyclonal antibodies directed to the purified active recombinant peptidase were effective in blocking the peptidase activity in trypomastigote soluble extracts and also resulted in inhibition of the Ca2+ transients elicited in NRK fibroblasts by this fraction. Under conditions where 90% of the oligopeptidase B activity was inhibited, the generation of Ca2+ transients was significantly delayed and often reduced in intensity, an observation that was made while measuring the transient increase of cytosolic free Ca2+ in a population spectrofluorimetric assay, and also at the single cell level. Our previous results, which strongly implicated a role for oligopeptidase B in the signaling mechanism (Burleigh and Andrews, 1995), have now been confirmed. The results presented here provide direct evidence for the involvement of the T. cruzi oligopeptidase B in generating Ca2+ transients in mammalian cells.
Purified oligopeptidase B was unable to elicit Ca2+ transients in mammalian cells (Burleigh and Andrews, 1995), and, similarly, the recombinant oligopeptidase B had no effect. It was previously demonstrated that removal of a heparin-binding fraction from the trypomastigote soluble extract resulted in depletion of Ca2+ signaling activity, whereas oligopeptidase B activity persisted (Burleigh and Andrews, 1995). It was therefore postulated that a factor(s) expressed only in invasive forms of T. cruzi would be processed by oligopeptidase B, as an obligatory step to generate an active Ca2+ agonist for mammalian cells. Assuming that the antibodies against the recombinant oligopeptidase B directly interfere with activity of the native peptidase toward its natural substrate, as indicated by the strong inhibition observed on hydrolysis of synthetic peptide substrates, the delayed Ca2+ response observed in the presence of anti-rPEP IgG may reflect a slower rate of agonist generation by the residual peptidase activity (10%). We reported previously that serial dilutions of trypomastigote soluble extracts result in proportionately decreased Ca2+-signaling responses in mammalian cells (Burleigh and Andrews, 1995). Here we show that after a 90% reduction of oligopeptidase B activity in parasite extracts, Ca2+ transients still occur, albeit at a slower rate and reaching lower levels. This is consistent with a scenario in which the substrate is the limiting factor, with the peptidase being required only in small amounts as a result of continuous turnover during the catalytic process.
The pattern of Ca2+ signaling induced in mammalian cells by intact trypomastigotes is asynchronous and repetitive (Tardieux et al., 1994), in contrast with parasite-soluble extracts that trigger a major simultaneous Ca2+ transient in all cells of the population (Burleigh and Andrews, 1995). This suggests that the processed trypomastigote signaling factor may be released from live parasites in small amounts and delivered to host cells in a localized fashion. This scenario is consistent with the recent demonstration that recruitment of host cell lysosomes, a process proposed to be regulated by [Ca2+]i transients, is highly localized, involving only lysosomes located at ∼10 μm from the parasite invasion site (Rodriguez et al., 1996).
The postulated role of the T. cruzi oligopeptidase B in the processing of an immature agonist of trypomastigotes is consistent with its structural relationship to enzymes with a very restricted substrate specificity, including the yeast DPAP A that has a known role in prohormone processing. The lack of a secretion signal sequence and the cytosolic localization of the T. cruzi oligopeptidase B imply that the postulated processing steps must occur in the cytoplasm of the parasites, with the putative active factor being released by an unconventional mechanism. An alternative pathway for the extracellular release of polypeptides lacking classical secretory signal sequences is emerging (Cooper and Barondes, 1990; Muesch et al., 1990; Rubartelli et al., 1992; Florkiewicz et al., 1995; Siders and Mizel, 1995). Some of the proteins in this group have clearly defined signaling roles outside cells, for example, interleukin (IL)-1β (Siders and Mizel, 1995) and basic FGF (Florkiewicz et al., 1995). In the case of IL-1β, conversion from pro-IL-1β to the active IL-1β is known to be mediated by a cytosolic cysteine protease, ICE (Kostura et al., 1989; Cerretti et al., 1992). Little is known about the export mechanism of cytoplasmically processed polypeptides, except that agents that perturb Golgi function do not block it, and that vesiculation of the plasma membrane has been implicated in some cases (Cooper and Barondes, 1990; Florkiewicz et al., 1995). A recent report identified genes involved in nonclassical protein export in yeast (Cleves et al., 1996), and at least one of these genes resembles small bacterial membrane proteins that function as transporters (Lewis, 1994). Now that we have determined that the T. cruzi oligopeptidase B, involved in triggering Ca2+ signaling in mammalian cells, is cytosolic, a search for the processed factor and its release mechanism may provide useful new information about this intriguing process.
Acknowledgments
We acknowledge the contribution to this work by members of the W.M. Keck Foundation Biotechnology Resource Laboratory at Yale University School of Medicine, for carrying out protein microsequencing, oligonucleotide synthesis, and DNA sequencing. We are grateful to S. Teixeira and J. Donelson for providing the T. cruzi cDNA library, A.K. Ma for excellent technical assistance, H. Tan for photography, L. Iadorola and L. Hartnell for EM, A.M. Quinn for assistance with sequence alignments, and C. Tschudi and E. Ullu for helpful discussions.
Abbreviations used in this paper
- AFC
7-amino-4-trifluoromethyl coumarin
- AMC
7-amino-4-methyl coumarin
- Z-Phe-Arg-AMC
Z-phenylalanyl-arginyl-7-amino-4-methyl coumarin
- [Ca2+]i
intracellular free calcium concentration
- E-64
N-[N-(l-3-trans-carboxirane-2-carbonyl)-l-leucyl]- agmatine
- IL
interleukin
- NRK
normal rat kidney
- rPEP
recombinant T. cruzi oligopeptidase B
- RT
reverse transcriptase
- TSE
trypomastigote soluble extract
Footnotes
This work was funded by National Institutes of Health grant R01AI32056 to N.W. Andrews. B. Burleigh was supported by a postdoctoral fellowship from the Medical Research Council of Canada.
Address all correspondence to Norma W. Andrews, Department of Cell Biology, Yale University School of Medicine, 333 Cedar Street, New Haven, CT 06520-8002. Tel.: (203) 785-4314. Fax: (203) 785-7226. E-mail: norma_andrews@qm.yale.edu
References
- Andrews NW, Hong KS, Robbins ES, Nussenzweig V. Stagespecific surface antigens expressed during the morphogenesis of vertebrate forms of Trypanosoma cruzi. . Exp Parasitol. 1987;64:474–484. doi: 10.1016/0014-4894(87)90062-2. [DOI] [PubMed] [Google Scholar]
- Ashall F. Characterization of an alkaline peptidase of Trypanosoma cruziand other trypanosomatids. Mol Biochem Parasitol. 1990;38:77–87. doi: 10.1016/0166-6851(90)90207-3. [DOI] [PubMed] [Google Scholar]
- Barr SC, Han WP, Andrews NW, Lopez JW, Ball BA, Pannabecker TL, Gilmour RF., Jr A factor from Trypanosoma cruzi induces repetitive cytosolic free Ca2+transients in isolated primary canine cardiac myocytes. Infect Immun. 1996;64:1770–1777. doi: 10.1128/iai.64.5.1770-1777.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett AJ, Rawlings ND. Oligopeptidases, and the emergence of the prolyl oligopeptidase family. Biol Chem Hoppe-Seyler. 1992;373:353–360. doi: 10.1515/bchm3.1992.373.2.353. [DOI] [PubMed] [Google Scholar]
- Bliska JB, Galan JE, Falkow S. Signal transduction in the mammalian cell during bacterial attachment and entry. Cell. 1993;73:903–920. doi: 10.1016/0092-8674(93)90270-z. [DOI] [PubMed] [Google Scholar]
- Brener Z. Biology of Trypanosoma cruzi. . Annu Rev Microbiol. 1973;27:347–383. doi: 10.1146/annurev.mi.27.100173.002023. [DOI] [PubMed] [Google Scholar]
- Burleigh BA, Andrews NW. A 120 kDa alkaline peptidase from Trypanosoma cruzi is involved in the generation of a novel Ca2+-signaling factor for mammalian cells. J Biol Chem. 1995;270:5172–5180. doi: 10.1074/jbc.270.10.5172. [DOI] [PubMed] [Google Scholar]
- Cazzulo JJ, Couso R, Raimondi A, Wernstedt C, Hellman U. Further characterization and partial amino acid sequence of a cysteine proteinase from Trypanosoma cruzi. . Mol Biochem Parasitol. 1989;33:33–42. doi: 10.1016/0166-6851(89)90039-x. [DOI] [PubMed] [Google Scholar]
- Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, March C, Kronheim SR, Druck T, Cannizzaro LA. Molecular cloning of the interleukin-1 beta converting enzyme. Science (Wash DC) 1992;256:97–100. doi: 10.1126/science.1373520. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Cleves AE, Cooper DNW, Barondes SH, Kelly RB. A new pathway for protein export in Saccharomyces cerevisiae. . J Cell Biol. 1996;133:1017–1026. doi: 10.1083/jcb.133.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DNW, Barondes SH. Evidence for export of a muscle lectin from cytosol to extracellular matrix and for a novel secretory mechanism. J Cell Biol. 1990;110:1681–1691. doi: 10.1083/jcb.110.5.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorta ML, Ferreira AT, Oshiro MEM, Yoshida N. Ca2+ signal induced by Trypanosoma cruzimetacyclic trypomastigote surface molecules implicated in mammalian cell invasion. Mol Biochem Parasitol. 1995;73:285–289. doi: 10.1016/0166-6851(94)00123-5. [DOI] [PubMed] [Google Scholar]
- Florkiewicz RZ, Majack RA, Buechler RD, Florkiewicz E. Quantitative export of FGF-2 occurs through an alternative, energy-dependent non-ER/Golgi pathway. J Cell Physiol. 1995;162:388–399. doi: 10.1002/jcp.1041620311. [DOI] [PubMed] [Google Scholar]
- Fuller RS, Sterne RE, Thorner J. Enzymes required for yeast prohormone processing. Annu Rev Physiol. 1988;50:345–362. doi: 10.1146/annurev.ph.50.030188.002021. [DOI] [PubMed] [Google Scholar]
- Galan JE. Interactions of bacteria with non-phagocytic cells. Curr Opin Immunol. 1994;6:590–595. doi: 10.1016/0952-7915(94)90146-5. [DOI] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1988. Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 726 pp.
- Heuser JE. Changes in lysosome shape and distribution correlated with changes in cytoplasmic pH. J Cell Biol. 1989;108:855–864. doi: 10.1083/jcb.108.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatani A, Masuda T, Shimoda T, Misoka F, Sheng X, Lin, Yoshimoto T, Tsuru D. Protease II from Escherichia coli: sequencing and expression of the enzyme gene and characterization of the expressed enzyme. J Biochem (Tokyo) 1991;110:315–320. doi: 10.1093/oxfordjournals.jbchem.a123577. [DOI] [PubMed] [Google Scholar]
- Kanatani A, Yoshimoto T, Kitazono A, Kokubo T, Tsuru D. Prolyl endopeptidase from Aeromonas hydrophila: cloning, sequencing, and expression of enzyme gene, and characterization of the expressed enzyme. J Biochem (Tokyo) 1993;113:790–796. doi: 10.1093/oxfordjournals.jbchem.a124120. [DOI] [PubMed] [Google Scholar]
- Kostura MJ, Tocci MJ, Limjuco G, Chin J, Cameron P, Hillman AG, Chartrain NA, Schmidt JA. Identification of a monocyte specific pre-interleukin 1 beta convertase activity. Proc Natl Acad Sci USA. 1989;86:5227–5231. doi: 10.1073/pnas.86.14.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreil G. Processing of precursors by dipeptidylaminopeptidases: a case of molecular ticketing. Trends Biochem Sci. 1990;15:23–26. doi: 10.1016/0968-0004(90)90126-v. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (Lond) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lewis K. Multidrug resistance pumps in bacteria: variations on a theme. Trends Biochem Sci. 1994;19:119–123. doi: 10.1016/0968-0004(94)90204-6. [DOI] [PubMed] [Google Scholar]
- Ley V, Robbins ES, Nussenzweig V, Andrews NW. The exit of Trypanosoma cruzifrom the phagosome is inhibited by raising the pH of acidic compartments. J Exp Med. 1990;171:401–413. doi: 10.1084/jem.171.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Acosta E, Cross GAM. Rapid isolation of DNA from trypanosomatid protozoa using a simple ‘mini-prep' procedure. Mol Biochem Parasitol. 1993;59:327–329. doi: 10.1016/0166-6851(93)90231-l. [DOI] [PubMed] [Google Scholar]
- Meirelles MNL, de Souza W. Interaction of lysosomes with endocytic vacuoles in macrophages simultaneously infected with Trypanosoma cruzi and Toxoplasma gondii. . J Submicrosc Cytol Pathol. 1983;17:327–334. [PubMed] [Google Scholar]
- Menard R, Dehio C, Sansonetti PJ. Bacterial entry into epithelial cells: the paradigm of Shigella. . Trends Microbiol. 1996;4:220–226. doi: 10.1016/0966-842X(96)10039-1. [DOI] [PubMed] [Google Scholar]
- Mentlein R. Proline residues in the maturation and degradation of peptide hormones and neuropeptides. FEBS (Fed Eur Biochem Soc) Lett. 1988;234:251–256. doi: 10.1016/0014-5793(88)80092-9. [DOI] [PubMed] [Google Scholar]
- Ming M, Ewen ME, Pereira MEA. Trypanosome invasion of mammalian cells requires activation of the TGFβ signaling pathway. Cell. 1995;82:287–296. doi: 10.1016/0092-8674(95)90316-x. [DOI] [PubMed] [Google Scholar]
- Muesch A, Hartmann E, Rohde K, Rubartelli A, Sitia R, Rappoport TA. A novel pathway for secretory proteins? . Trends Biochem Sci. 1990;15:86–88. doi: 10.1016/0968-0004(90)90186-f. [DOI] [PubMed] [Google Scholar]
- Nogueira N, Cohn Z. Trypanosoma cruzi:mechanism of entry and intracellular fate in mammalian cells. J Exp Med. 1976;143:1402–1420. doi: 10.1084/jem.143.6.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacaud M, Richaud C. Protease II from Escherichia coli. . J Biol Chem. 1975;250:7771–7779. [PubMed] [Google Scholar]
- Polgar L. Unusual secondary specificity of prolyl oligopeptidase and the different reactivities of its two forms toward charged substrates. Biochemistry. 1992;31:7729–7735. doi: 10.1021/bi00148a038. [DOI] [PubMed] [Google Scholar]
- Rennex D, Hemmings BA, Hofsteenge J, Stone SR. cDNA cloning of porcine brain prolyl endopeptidase and identification of the active-site seryl residue. Biochemistry. 1991;30:2195–2203. doi: 10.1021/bi00222a025. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Rioult MG, Ora A, Andrews NW. A trypanosomesoluble factor induces IP3 formation, intracellular Ca2+mobilization and microfilament rearrangement in host cells. J Cell Biol. 1995;129:1263–1273. doi: 10.1083/jcb.129.5.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Samoff E, Rioult MG, Chung A, Andrews NW. Host cell invasion by trypanosomes requires lysosomes and microtubule/kinesin-mediated transport. J Cell Biol. 1996;134:349–362. doi: 10.1083/jcb.134.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubartelli A, Bajetto A, Allavena G, Wollman E, Sitia R. Secretion of thioredoxin by normal and neoplastic cells through a leaderless secretory pathway. J Biol Chem. 1992;267:24161–24164. [PubMed] [Google Scholar]
- Santana JM, Grellier P, Rodier M-H, Schrevel J, Teixeira A. Purification and characterization of a new 120-kD alkaline proteinase of Trypanosoma cruzi. . Biochem Biophys Res Commun. 1992;187:1466–1473. doi: 10.1016/0006-291x(92)90467-y. [DOI] [PubMed] [Google Scholar]
- Schenkman S, Robbins ES, Nussenzweig V. Attachment of Trypanosoma cruzito mammalian cells requires parasite energy, and invasion can be independent of the target cell cytoskeleton. Infect Immun. 1991;59:645–654. doi: 10.1128/iai.59.2.645-654.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segel, I.H. 1976. Biochemical Calculations. Second edition. John Wiley & Sons, Inc., New York. 214–236.
- Siders WM, Mizel SB. Interleukin-1β secretion. J Biol Chem. 1995;270:16258–16264. doi: 10.1074/jbc.270.27.16258. [DOI] [PubMed] [Google Scholar]
- Stebeck CE, Frevert U, Mommsen TP, Vassella E, Roditi I, Pearson TW. Molecular characterization of glycosomal NAD+-dependent glycerol 3-phosphate dehydrogenase from Trypanosoma brucei rhodesiense. . Mol Biochem Parasitol. 1996;76:145–158. doi: 10.1016/0166-6851(95)02555-3. [DOI] [PubMed] [Google Scholar]
- Stone SR, Rennex D, Wikstrom P, Shaw E, Hofsteenge J. Inactivation of prolyl endopeptidase by a peptidylchloromethane. Biochem J. 1991;276:837–840. doi: 10.1042/bj2760837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardieux I, Webster P, Ravesloot J, Boron W, Lunn JA, Heuser JE, Andrews NW. Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell. 1992;71:1117–1130. doi: 10.1016/s0092-8674(05)80061-3. [DOI] [PubMed] [Google Scholar]
- Tardieux I, Nathanson MH, Andrews NW. Role in host cell invasion of Trypanosoma cruzi–induced cytosolic free Ca2+transients. J Exp Med. 1994;179:1017–1022. doi: 10.1084/jem.179.3.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuru D, Yoshimoto T. Oligopeptidase B: protease II from Escherichia coli. . Methods Enzymol. 1994;244:201–215. doi: 10.1016/0076-6879(94)44017-4. [DOI] [PubMed] [Google Scholar]
- Veron M, Falcoz-Kelly F, Cohen GN. The threonine-sensitive homoserine dehydrogenase and aspartokinase activities of Escherichia coliK12. Eur J Biochem. 1972;28:520–527. doi: 10.1111/j.1432-1033.1972.tb01939.x. [DOI] [PubMed] [Google Scholar]
- Webster P, Grab DJ. Intracellular colocalization of variant surface glycoprotein and transferrin-gold in Trypanosoma brucei. . J Cell Biol. 1988;106:279–288. doi: 10.1083/jcb.106.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Klein TW, Friedman H. Induction of cytokine granulocyte-macrophage colony-stimulating factor and chemokine macrophage inflammatory protein 2 mRNAs in macrophages by Legionella pneumophila or Salmonella typhimuriumattachment requires different ligand-receptor systems. Infect Immun. 1996;64:3062–3068. doi: 10.1128/iai.64.8.3062-3068.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida N. Trypanosoma cruzi: recognition of trypomastigote surface antigens by lytic antisera from mice resistant to acute infection. Exp Parasitol. 1986;61:184–191. doi: 10.1016/0014-4894(86)90151-7. [DOI] [PubMed] [Google Scholar]
- Yoshimoto T, Kanatani A, Shimoda T, Inaoka T, Kokubo T, Tsuru D. Prolyl endopeptidase from Flavobacterium meningosepticum: cloning and sequencing of the enzyme gene. J Biochem (Tokyo) 1991;110:873–878. doi: 10.1093/oxfordjournals.jbchem.a123682. [DOI] [PubMed] [Google Scholar]
- Yoshimoto T, Tabira J, Kabashima T, Inoue S, Ito K. Protease II from Moraxella lacunata: cloning, sequencing, and expression of the enzyme gene, and crystallization of the expressed enzyme. J Biochem (Tokyo) 1995;117:654–660. doi: 10.1093/oxfordjournals.jbchem.a124759. [DOI] [PubMed] [Google Scholar]