

Abstract
Estrogen (E2) plays a critical role in the etiology and progression of human breast cancer. The estrogenic response is complex and not completely understood, including in terms of the involved responsive genes. Here we show that Hsp22 (synonyms: HspB8, E2IG1, H11), a member of the small heat shock protein (sHSP) superfamily, was induced by E2 in estrogen receptor–positive MCF-7 breast cancer cells, resulting in an elevated Hsp22 protein level, whereas it was not induced in estrogen receptor–negative MDA-MB-231 cells. This induction was prevented by the pure anti-estrogen ICI182780 (faslodex, fulvestrant), whereas tamoxifen, a substance with mixed estrogenic and anti-estrogenic properties, had no major inhibitory effect on this induction, nor did it induce Hsp22 on its own. Cadmium (Cd) is an environmental pollutant with estrogenic properties (metalloestrogen) that has been implicated in breast cancer. Treatment of MCF-7 cells with Cd also resulted in induction of Hsp22, and this induction was also inhibited by ICI182780. In live MCF-7 cells, Hsp22 interacted at the level of dimers with Hsp27, a related sHSP, as was shown by quantitative fluorescence resonance energy transfer measurements. In cytosolic extracts of MCF-7 cells, most of the E2- and Cd-induced Hsp22 was incorporated into high–molecular mass complexes. In part, Hsp22 and Hsp27 were components of distinct populations of these complexes. Finally, candidate elements in the Hsp22 promoter were identified by sequence analysis that could account for the induction of Hsp22 by E2 and Cd. Taken together, Hsp22 induction represents a new aspect of the estrogenic response with potential significance for the biology of estrogen receptor– positive breast cancer cells.
INTRODUCTION
Breast cancer is the most common malignancy in women and one of the leading causes of death (American Cancer Society 1993). Numerous studies have shown that endocrine factors, notably 17β-estradiol (E2), play a critical role in the etiology and progression of human breast cancer. The effect of E2 is mediated through its ability to bind to the α and β forms of the estrogen receptor (ER), a ligand-activated transcription factor. The estrogenic response is complex, both in terms of the number of responsive genes, the involved regulatory mechanisms and molecules, and even the consequences for tumor growth (Rochefort 1995; Ohlsson et al 2001; Sanchez et al 2002). Various anti-estrogens have been widely used for both research and treatment of ER-positive (ER+) breast cancer in patients. Primarily, they function through their ability to compete with available E2 for binding to the ER. The “classic” anti-estrogenic drug tamoxifen, however, acts both as partial antagonist and partial agonist (Hodges et al 2003), whereas the more recently developed drug ICI182780 (fulvestrant, faslodex) has predominantly anti-estrogenic properties (Wakeling 2000; Clarke et al 2001; Morris and Wakeling 2002). A better understanding of the estrogenic response is expected to result in new approaches to interfere with the growth of ER+ breast cancer cells.
Hsp22 (synonyms: HspB8, E2IG1, H11) belongs to the superfamily of small mammalian heat shock proteins (sHSPs) and was identified independently in 4 laboratories at approximately the same time, although in different contexts (Charpentier et al 2000; Smith et al 2000; Benndorf et al 2001; Kappé et al 2001). In one of those studies, a serial analysis of gene expression (SAGE) screen was conducted with the aim to identify genes that are regulated by E2. Hsp22 mRNA was found to be among the most highly induced transcripts in ER+ MCF-7 breast cancer cells (Charpentier et al 2000), and this up-regulation of Hsp22 mRNA was confirmed in a more recent study (Yang et al 2006). These data strongly suggest Hsp22 induction to be a hitherto unrecognized part of the estrogenic response, although to date, this induction has not been verified at the protein level.
Hsp27, a related sHSP, was observed some 2 decades ago in human and mouse breast cancer cells and is the “classic” stress-responsive sHSP. In studies with MCF-7 breast cancer cells, this protein was found to be induced by E2 and initially designated “estrogen-regulated 24K protein” (Edwards et al 1980; Ciocca et al 1984). This protein was reported to be involved in growth and differentiation of hormonally controlled tissues such as breast and endometrium (Ciocca et al 1989). In studies with Ehrlich ascitic tumor cells, a murine breast cancer cell line, this protein accumulated in the stationary phase of the tumor growth and was designated “growth-related protein p25” (Benndorf et al 1988b). The identity as Hsp27 (or Hsp25 in rodents), however, was established only later after sequencing (Benndorf et al 1988a; Fuqua et al 1989; Gaestel et al 1989). Although the role of Hsp27 in breast and other cancer has been studied extensively, general conclusions could not be drawn so far because of disparate associations of Hsp27 expression with disease prognosis and response to therapy in different tumors (reviewed in Ciocca and Vargas-Roig 2002). In line with its general ability to support cell survival in adverse conditions, Hsp27 supports survival of MCF-7 cells in the presence of the anticancer drug doxorubicin, thus contributing to drug resistance (Oesterreich et al 1993). Both Hsp27 and Hsp22 are known to interact with each other forming homo- and heterodimers, as well as high–molecular mass complexes (HMMC; Sun et al 2004), although the exact nature of these HMMCs is undefined.
Cadmium (Cd) is an industrial and environmental metal toxicant that exerts adverse effects on a number of human organs, including the reproductive organs (Darbre 2006; Takiguchi and Yoshihara 2006). At the cellular level, Cd has pleiotropic effects that include the induction of Hsp27 and the activation of the p38 mitogen-activated protein kinase (p38 MAPK) cascade, resulting in increased phosphorylation of Hsp27 (Hirano et al 2005 and references therein). Interestingly, Cd is a so-called metalloestrogen, mimicking the action of E2 both in vivo and in vitro by activating the ERα through a high-affinity interaction with the hormone-binding domain, independently of E2. For example, in female rats exposed to environmentally relevant doses, Cd induced well-characterized responses similar to E2, such as uterine weight and growth and development of the mammary glands, and it also induced expression of E2-regulated genes in MCF-7 cells (Stoica et al 2000; Choe et al 2003; Johnson et al 2003; Martin et al 2003; Brama et al 2006). Cd was suspected to add to the estrogenic burden of the human breast, and a role in breast cancer has been proposed (Garcia-Morales et al 1994; Satarug and Moore 2004; McElroy et al 2006). Consequently, it has been classified as a carcinogen in humans (Waalkes 2003; Waisberg et al 2003).
In this study, we report that Hsp22 is effectively induced at the level of the protein by E2 and Cd in the ER+ MCF-7 breast cancer cells, and this induction can be inhibited by the pure anti-estrogen ICI182780. Estrogen receptor–negative (ER−) MDA-MB-231 breast cancer cells do not show this response to E2 and Cd. We further show by quantitative fluorescence resonance energy transfer (qFRET) measurements that Hsp22 and Hsp27 interact in the cytoplasm of MCF-7 cells, and both proteins form HMMCs in cell extracts. Hsp22 induced by E2 or Cd treatment is largely incorporated into these HMMCs. Finally, by sequence analysis, we identified candidate regulatory elements in the human Hsp22 promoter that might account for the induction of Hsp22 by E2 and Cd. Taken together, our data show new aspects of the estrogenic response and of aberrant E2 signaling resulting from exposure to the environmental metal toxicant Cd in breast cancer cells.
MATERIALS AND METHODS
Vector constructs
For the qFRET experiments, Hsp22 and Hsp27 cDNAs were fused with the citrine (CIT) and cyan (CFP) fluorescent proteins with the use of the corresponding expression vectors peCITN1 and peCFPN1 (BD Biosciences, Franklin Lakes, NJ, USA) (Hoppe et al 2002). Cloning information for the resulting pCFPN1-Hsp27 and peCITN1-Hsp22 constructs was provided previously (Sun et al 2004; Fontaine et al 2006). For control purposes, the “empty” vectors peCFPN1 and peCITN1 were used.
For the hairpin Hsp22-shRNA construct, 2 complimentary oligonucleotides (5′-gatctaaaACCCTAAGGTCGGGCATGGTGttcaagagaCACCATGCCCGACCTTAGGGTtttgggc-3′; 5′-ggccgcccaaaACCCTAAGGTCGGGCATGGTGtctcttgaaCACCATGCCCGACCTTAGGGTttta-3′; sequence stretches complimentary to Hsp22 mRNA are capitalized) were designed that are specific for Hsp22 and do not match any other human sequence. These oligonucleotides were annealed, yielding a heteroduplex DNA with sticky ends (5′, BglII; 3′, NotI). This fragment was cloned into a short hairpin (shRNA) expression vector pCH1 as described previously (Velkey and O'Shea 2003). In the resulting construct (pCH1-shHsp22), the Hsp22 hairpin template is driven from the H1 promoter. This construct contained a second expression cassette with the red fluorescent protein (dsRed) driven from the simian cytomegalovirus immediate-early enhancer/promoter, thus allowing the determination of the fraction of transfected cells by fluorescence microscopy. For control, the same vector without the Hsp22-shRNA insert was used.
Cell culture and transfection
ER+ MCF-7 and ER− MDA-MB-231 breast cancer cells were grown in a 5% CO2-humidified atmosphere at 37°C in 6-well plates unless specified otherwise. In protein induction assays, for optimal response to E2 and Cd, cells were initially grown for 48 hours in Dulbecco modified Eagle medium (DMEM) in the presence of 10% fetal calf serum (complete medium) reaching ∼60% confluency. Then the medium was replaced with DMEM without fetal calf serum (FCS) and without phenol red (starvation medium), in which the cells were kept until hour 72. In this time period, cell growth slowed, yielding ∼70% confluency. This medium was replaced with DMEM without phenol red containing 10% charcoal-treated FCS (E2-deficient medium). At hour 93 or 96 (cells at ∼85% confluency), the following substances, alone or in combination as specified in Figure 1A–C, Figure 1D (lanes 1–3), and Figure 3, were added: at hour 93, 1 μM ICI182780, 10 μM 4-OH-tamoxifen (4-OH-tam) if tested as antagonist, or 0.2 μg/mL actinomycin D; and at hour 96, 0.01 μM E2, 10 μM 4-OH-tam if tested as agonist, or 20 μM CdCl2. The cells were then allowed to grow until hour 144 or hour 120 (if actinomycin D or Cd was included). For determination of the short-term (1.5-hour) effect of Cd on phosphorylation of Hsp22 and Hsp27 (Fig 1D, lanes 4–9), cells were grown for 48 hours in complete medium to reach ∼80% confluency. At hour 48, this medium was replaced with starvation medium (− serum assays; lanes 6–9) or with E2-deficient medium (+ serum assays; lanes 4, 5), and 1 μM ICI18278 was added as indicated (lanes 7, 9). At hour 51, 50 μM CdCl2 was added as indicated (lanes 5, 8, 9). Cells were harvested at hour 52.5. In preliminary experiments, the optimal timing and concentrations of all tested substances were determined (not shown).
Fig 1.

Induction of Hsp22. (A) Effects of 17β-estradiol (E2), cadmium (Cd), and ICI182780 (fulvestrant, faslodex) on induction of Hsp22 in estrogen receptor–positive (ER+) MCF-7 cells. Cells were grown consecutively in complete medium, starvation medium, and E2-deficient medium supplemented with 0.01 μM E2, 1 μM ICI182780, or 20 μM CdCl2, alone or in combination as indicated, and processed for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)/Western blotting as described in Materials and Methods. Hsp22 was induced in response to E2 and CdCl2, and induction by either agent was prevented in the presence of ICI182780. (B) Effects of E2, 4-OH-tamoxifen (4-OH-tam), and actinomycin D on induction of Hsp22 in ER+ MCF-7 cells. Cells were grown as in panel A in E2-deficient medium supplemented with 0.01 μM E2, 10 μM 4-OH-tam, or 0.2 μg/mL actinomycin D, alone or in combination as indicated, and processed for SDS-PAGE/Western blotting. Also as in panel A, Hsp22 was induced by E2, and this induction was prevented in the presence of actinomycin D. 4-OH-tam had no major effect on Hsp22 induction. (C) Effects of E2 and Cd on Hsp22 in estrogen receptor–negative (ER−) MDA-MB-231 cells. Cells were grown as in panel A in E2-deficient medium supplemented with 0.01 μM E2 or 20 μM CdCl2 as indicated and processed for SDS-PAGE/Western blotting. MDA-MB-231 cells did not contain any detectable Hsp22, nor did treatment with E2 or CdCl2 result in induction of Hsp22. (D) Effects of E2, CdCl2, ICI182780, and fetal calf serum (FCS) on phosphorylation of Hsp22 and Hsp27 in ER+ MCF-7 cells. For long-term treatment, cells were grown and treated with E2 or CdCl2 as in panel A (+ serum, lanes 1–3). For short-term treatment (see Materials and Methods), cells were either grown in complete medium supplemented with 50 μM CdCl2, as indicated, in the presence of serum (+ serum, lanes 4, 5), or they were grown consecutively in complete medium and starvation medium supplemented with 50 μM CdCl2 or 1 μM ICI182780, alone or in combination as indicated, in the absence of serum (− serum, lanes 6–9). After harvest, cells were processed for isoelectric-focusing (IEF)-PAGE/Western blotting. Hsp22 was not phosphorylated, nor could phosphorylation be induced by any of the treatments (upper panel). Hsp27 was partially phosphorylated, and short-term treatment with CdCl2 induced moderately (lane 5) or greatly (lane 8) phosphorylation (lower panel). ICI182780 did not prevent phosphorylation of Hsp27 (lane 9). sHSP isoforms: (a) nonphosphorylated; (b) monophosphorylated; (c) biphosphorylated; (d) triphosphorylated; (+) anode, (−) cathode. In panels A–D, blots of the same samples were developed for both Hsp22 and Hsp27. In panels A–C, no major changes in the expression of Hsp27 were detected. Visualization of β-actin (loading control) in A–C indicated equal loading of the samples
Fig 3.

Complex formation of Hsp22 and Hsp27 in protein extracts of estrogen receptor–positive (ER+) MCF-7 cells. Cells were grown consecutively in complete medium, starvation medium, and in 17β-estradiol (E2)–deficient medium supplemented with 0.01 μM E2 or 20 μM CdCl2 as indicated. Cells were harvested and processed for high-performance liquid chromatography (HPLC) size exclusion chromatography followed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)/Western blotting. (A) Absorbance at 280 nm over the entire separation. The positions of HPLC molecular mass marker proteins are indicated. (B) Coomassie blue-stained SDS gels loaded with equal aliquots of fractions 1–20 of the HPLC separation of control cells. The protein pattern in the various fractions indicated good separation by HPLC. The positions of SDS-PAGE molecular mass marker proteins are indicated on the left. (C, D) Western blots showing the size distribution of Hsp22 and Hsp27, respectively, in untreated control cells or in cells treated with E2 or CdCl2. Both Hsp22 and Hsp27 distributed throughout a wide molecular mass range, indicating polydispersity. Induction by E2 or CdCl2 caused a redistribution of Hsp22 toward fractions 1 and 2 (corresponding to >670 kDa)
For qFRET experiments, MCF-7 cells were grown in glass-bottom 6-well culture plates (Mattek, Ashland, MA, USA) in complete medium. Transfection was at ∼60% confluency with 0.75 μg of vector DNA for each construct and with Lipofectamine 2000 (Invitrogen, San Jose, CA, USA). Forty-eight hours later, the live cells expressing the Hsp22-CIT and Hsp27-CFP fusion proteins were washed 3 times with PBS and kept at 37°C in DMEM without phenol red for collecting the fluorescence images. With transfection efficiency at ∼10%, this procedure provided a sufficient number of cells expressing both fusion proteins that were suitable for qFRET measurements.
For drug resistance experiments, MCF-7 cells were grown in complete medium. A pellet of ∼2 × 106 cells was resuspended in 100 μL of Nucleofector reagent V (Amaxa, Gaithersburg, MD, USA) mixed with 2 μg of pCH1-shHsp22 or pCH1 vector DNA then electroporated with the program P20 of the Amaxa electroporation system (Amaxa). After electroporation, ∼15 000 cells were seeded into each well of 24-well culture plates for use in growth and drug resistance assays. The fraction of transfected cells (transfection efficiency) was estimated 48 hours after electroporation by visualizing the expressed DsRed marker dye with a fluorescence microscope. Typically, the transfection efficiency was >75%. The effect of down-regulation of Hsp22 by the pCH1-shHsp22 construct was monitored 72 hours after transfection by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blotting.
Live cell imaging and quantitative fluorescence resonance energy transfer (qFRET) measurements
MCF-7 cells were transfected to express Hsp22-CIT and Hsp27-CFP fusion proteins as described above. Live cell fluorescence imaging and qFRET measurements were as described previously with an inverted epifluorescence microscope (Eclipse TE-2000 U, Nikon, Melleville, NY, USA) (Fontaine et al 2006). For qFRET measurements, the output data were expressed as the apparent average fluorescence resonance energy transfer efficiency (AAFE). The following negative controls were used: (1) cells were cotransfected with the “empty” CIT vector (peCITN1) and the Hsp27-CFP vector (cf Fig 2B), (2) cells were cotransfected with the “empty” CFP vector (peCFPN1) and the Hsp22-CIT vector (not shown), and (3) cells were transfected with the “empty” CFP (peCFPN1) and CIT (peCITN1) vectors (not shown). The AAFE signals for all control settings were similar and defined the baseline signal. Sample AAFE values significantly different from this baseline signal indicated interaction.
Fig 2.
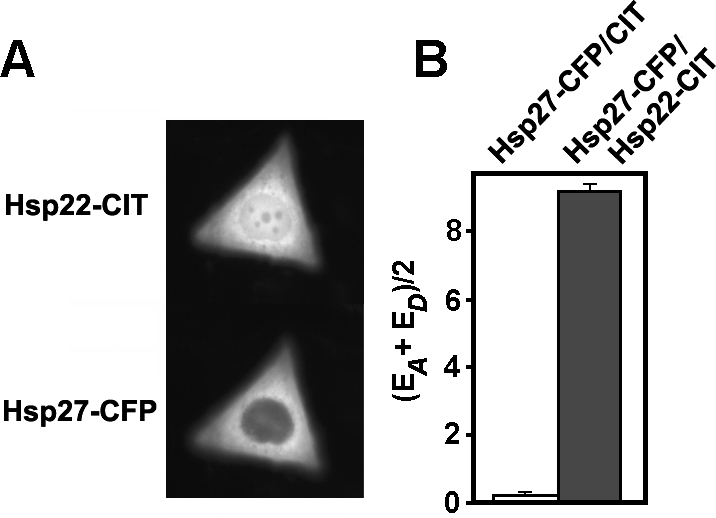
Interaction of Hsp22 and Hsp27 in estrogen receptor–positive (ER+) MCF-7 cells in vivo. Cells grown in complete medium were doubly transfected to express citrine (CIT) and cyan (CFP) fluorescent protein fusion proteins (Hsp27-CFP and Hsp22-CIT). (A) Fluorescence image of a representative cell coexpressing both sHSP fusion proteins. Expressed Hsp22-CIT was located in both the cytoplasm and nucleus, whereas expressed Hsp27-CFP was mainly located in the cytoplasm. (B) qFRET data collected from 69 doubly-transfected cells (gray bar) resulted in an average fluorescence resonance energy transfer efficiency (AAFE) value that was significantly different (P < 0.01) from the AAFE value collected from 63 control cells coexpressing Hsp27-CFP and CIT (white bar). Error bars represent standard error
MCF-7 cells expressing the fluorescent marker protein DsRed were visualized with a fluorescent microscope equipped with the exciter filter 560/55, the barrier filter 645/75, and the dichroid mirror EP 595LP. Otherwise, the configuration of the microscope was as described previously (Fontaine et al 2006).
Size exclusion chromatography
For one high-performance liquid chromatography (HPLC) run, MCF-7 cells were grown in two 180-cm2 culture dishes to ∼100% confluency. Harvested cells were pooled and lysed in 2 mL of ice-cold buffer A (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM KCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 10% glycerol, 1% CHAPS, 1% β-mercaptoethanol, 1 protease inhibitor pill) and kept on ice for 30 min. After centrifugation at 14,000 × g for 20 min at 4 °C, 500 μL of the supernatant was used for HPLC chromatography as described previously (Fontaine et al 2005). Fractions of 1.67 mL were collected as indicated in Figure 3. Proteins in all fractions were precipitated overnight at −20°C by 2 volumes ethanol containing 1% β-mercaptoethanol, and the precipitates were collected by centrifugation. The pellets were dissolved in 100 μL of buffer B (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 200 mM dithiothreitol, 0.01% bromophenol blue), briefly sonicated, and boiled for 5 min. Identical aliquots of each fraction (10% for Hsp22 detection; 1% for Hsp27 detection) were analyzed by SDS-PAGE/Western blotting for the presence of Hsp22 and Hsp27 with the use of corresponding specific antibodies (see below).
Viability assays
The resistance of MCF-7 cells to doxorubicin and CdCl2 was evaluated with the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay of cell viability. For this, cells were electroporated as described above with the construct pCH1-shHsp22 or the “empty” pCH1 vector for control. Approximately 15 000 electroporated cells were seeded into each well of 24-well plates containing 1 mL of complete medium. To determine the resistance to doxorubicin, 24 hours later this drug was added at 0.03, 0.1, 0.3, 1, 3, and 10 μM final concentrations, and incubation was continued for an additional 72 hours. Similarly, to determine resistance to CdCl2, this metal toxicant was added at 0.2, 0.5, 1, 3, 5, 10, 30, 50, and 100 μM final concentrations. No drugs were added to the controls. Seventy-two hours later, the medium was replaced by 0.5 mL of complete medium containing 500 μg/mL of MTT, and cells were incubated at 37°C for 3 hours. After aspirating the medium, the precipitated dye was dissolved in 300 μL 0.04 N HCl in isopropanol, and 200 μL of this solution was transferred to a 96-well plate. Absorbance at 570 nm was determined with a Fluoroskan Ascent plate reader (MTX Lab, Vienna, VA, USA). The determined viability values represent the net outcome of cell proliferation, cell death, and attachment to the support. For each toxicant concentration, samples were taken from 6 wells (n = 6).
Miscellaneous
The following stock solutions were used: 20 μM 17β-estradiol (E2) (Sigma, St. Louis, MO, USA) in absolute ethanol, 200 μM ICI182780 (Tocris, Ellisville, MO, USA) in dimethyl sulfoxide (DMSO), 2 mM 4-OH-tam (Sigma) in DMSO, 50 mM CdCl2 in physiological saline, and 10 mM doxorubicin (Alexis, Lausen, Switzerland) in DMSO.
SDS-PAGE, isoelectric-focusing polyacrylamide gel electrophoresis (IEF-PAGE), and Western blotting were performed as described previously (Benndorf et al 2000, 2001). For IEF-PAGE, a mixture of ampholines (Biorad, Hercules, CA, USA) was used containing ampholines 3–10 (50%), 3–5 (25%), and 5–7 (25%). For Western blotting, the polyvinylidene fluoride membranes were incubated with primary polyclonal goat anti-Hsp22 (Abcam, Cambridge, MA, USA), polyclonal rabbit anti-Hsp27 (Assay Designs, Ann Arbor, MI, USA), or monoclonal anti-β-actin antibodies (Sigma) at dilutions of 1/2000, 1/10 000, and 1/10 000, respectively, followed by incubation with suitable secondary antibodies coupled to horseradish peroxidase. Protein bands were visualized on X-ray films with the ECL reagent (Pierce, Rockford, IL, USA). For semiquantitative determination of Hsp22 induction, the original X-ray films were scanned, and the optical density was determined with NIH Image J software.
Transcription factor binding site predictions in the human Hsp22 promoter and gene (sequence NT_009775 of the Homo sapiens chromosome 12 genomic contig) were made with the web tool Transcription Element Search System site at (http://www.cbil.upenn.edu/tess; Schug and Overton 1997).
Statistics
For the qFRET method, 30 microscopic fields (with 1–3 cells each) in each group were evaluated (control group, n = 63; sample group, n = 69), and the collected data were expressed as values of mean AAFE plus or minus the standard error. In the drug resistance assays, for each drug concentration, cell viability was determined in 6 wells of 24-well plates (n = 6) by the MTT test. Three independent experiments were conducted. Data are shown as mean absorbance at 570 nm plus or minus the standard deviation.
The unpaired Student's t-test was applied to compare results between sample groups. Differences between the groups were considered statistically significant at P < 0.01.
RESULTS
Induction of Hsp22
On the basis of previous reports on E2 induction of Hsp22 mRNA in MCF-7 cells (Charpentier et al 2000; Yang et al 2006), we tested to see whether this response also translates to the level of the protein. For optimal response, MCF-7 cells were grown consecutively in complete medium, starvation medium, and in E2-deficient medium, which was supplemented with the substances as is specified in Figure 1 and in the Materials and Methods section. In the absence of E2, the amount of Hsp22 as detected by SDS-PAGE/Western blotting was relatively low (Fig 1A, lane 1). This level, however, was further decreased by treating the cells with the pure anti-estrogen ICI182780 (lane 2). E2, on the other hand, strongly increased the amount of Hsp22 (lane 3). Densitometric evaluation revealed a >3-fold increase in the amount of this protein after induction by E2. The presence of ICI182780 abolished this induction of Hsp22 by E2 (lane 4). These data suggest that Hsp22 is induced by E2 and that this induction is inhibited by the anti-estrogen ICI182780.
Hsp27 was also reported to be induced by E2, although on a lesser scale (Edwards et al 1980). We therefore included Hsp27 in this analysis. As shown in Figure 1A, Hsp27 was not subject to major regulation by E2 or ICI182780 in the conditions chosen for this experiment.
Because the metal toxicant Cd is known to mimic E2 and might also have a role in human breast cancer, we tested to see whether Hsp22 can be induced by Cd in MCF-7 cells. As shown in Figure 1A (lane 5), Cd is also a potent inducer, specifically of Hsp22, whereas it had no major effect on the abundance of Hsp27. Densitometric evaluation revealed a >2-fold increase in the amount of HSP22 after induction by Cd. Similar to the induction of Hsp22 by E2, ICI182780 effectively inhibited the induction of Hsp22 by Cd (lane 6). This suggests that the induction of Hsp22 by Cd indeed results from the estrogenic properties of the metal toxicant.
In contrast to the pure anti-estrogen ICI182780, 4-OH-tam has mixed properties and acts partially as an estrogenic agonist and partially as an antagonist to E2. When tested alone, 4-OH-tam did not cause a major induction of Hsp22 on its own (Fig 1B, lane 2) compared to untreated control cells (lane 1) and to cells treated with E2 alone (lane 3). On the other hand, 4-OH-tam only slightly attenuated the Hsp22 induction by E2 (lane 4) compared to the treatment with E2 alone (lane 3). 4-OH-tam also had no major effect on the expression of Hsp27 in the same samples, as was observed previously in MCF-7 cells (Horman et al 1997).
To determine whether the induction of Hsp22 by E2 is regulated at the level of transcription, actinomycin D, a potent inhibitor of RNA polymerase II, was included in these assays. In the presence of this drug, E2 no longer caused induction of Hsp22 (Fig 1B, lane 5), thus indicating transcriptional regulation.
In contrast to the ER+ MCF-7 cells, ER− breast cancer cells did not express significant amounts of Hsp22 mRNA (Charpentier et al 2000). In line with these data, in ER− MDA-MB-231 cells, no significant amounts of Hsp22 protein could be detected on Western blots (Fig 1C, lane 1). Treatment with neither E2 nor Cd resulted in any detectable induction of Hsp22 (lanes 2 and 3, respectively). For comparison, Hsp27 was readily detected in MDA-MB-231 cells in accordance with previous studies (Oesterreich et al. 1993; Fuqua et al. 1994), although these cells contain less Hsp27 than MCF-7 cells (not shown). Incubation with either E2 or Cd did not induce Hsp27.
For loading control, β-actin was visualized on the same blots shown in Figure 1A–C, indicating equal sample loading.
In many cells, Hsp27 is partially phosphorylated, including in MCF-7 cells (Ciocca et al 1992), and a variety of agents including Cd can increase the degree of its phosphorylation (Radloff et al 1998; Eichler et al 2005; Hirano et al 2005; Leal et al 2007). Hsp22 was also reported to be phosphorylated in vivo (Smith et al 2000). Therefore, using IEF-PAGE/Western blotting, we determined whether the same treatment of MCF-7 cells with E2 or Cd as used above can induce phosphorylation of Hsp22. Hsp22 of untreated control cells migrated as 1 major band (band a), with a pI of ∼5.0 (Fig 1D, upper panel, lane 1), as was described previously (Benndorf et al 2001). Treatment with either E2 (lane 2) or Cd (lane 3) did not result in any detectable acidic shift of this band, suggesting that phosphorylation of Hsp22 was not induced by these treatments.
For comparison, phosphorylation of Hsp27 was analyzed in the same samples. In untreated cells, the major fraction of Hsp27 migrated to the position of the unphosphorylated Hsp27 isoform (band a), and a minor fraction migrated to the position of the monophosphorylated isoforms (band b; Fig 1D, lower panel, lane 1). The samples also contained traces of biphosphorylated Hsp27 isoforms (band c). Again, treatment with either E2 (lane 2) or Cd (lane 3) did not result in any major acidic shift of the band pattern, suggesting that phosphorylation of Hsp27 was not induced by these treatments.
With respect to Hsp27, this outcome was not expected because Cd robustly induced phosphorylation of this sHSP in a number of cell types (Radloff et al 1998; Eichler et al 2005; Hirano et al 2005; Leal et al 2007). In those studies, however, cells were exposed to Cd for shorter time periods (typically 0.5–2 hours) compared with the experiments shown in Figure 1A,B. To confirm that MCF-7 cells indeed are able to respond to Cd by activating the p38 MAPK signaling pathway, resulting eventually in phosphorylation of Hsp27, cells were exposed to Cd for a shorter time period. Because FCS is known to contain Cd-binding proteins that interfere with the effective concentration of Cd ions (Trisak et al 1990), these incubations were done both in the presence and absence of FCS. Short-term exposure (1.5 hours) of MCF-7 cells to 50 μM CdCl2 in the presence of FCS caused a moderate shift toward the phosphorylated isoforms (bands b, c) compared with the control (Fig 1D, lower panel, lanes 5 and 4, respectively). In contrast, in the absence of FCS, Cd treatment shifted the band pattern greatly toward the phosphorylated Hsp27 isoforms with reduced amounts of unphosphorylated Hsp27 (band a) and the emergence of the triphosphorylated isoform (band d) compared with the control (lanes 8 and 6, respectively). This acidic shift occurred also in the presence of the anti-estrogen ICI182780, indicating that Cd-induced phosphorylation of Hsp27 does not involve ER-mediated signaling pathways (lane 9). For comparison, ICI182780 alone had no effect on the Hsp27 isoform pattern (lane 7). No phosphorylation of Hsp22 was observed (Fig 1D, upper panel, lanes 5, 8, 9) compared with the corresponding controls (lanes 4, 6, 7), even under conditions that favored phosphorylation of Hsp27.
Taken together, the collected data suggest that in ER+ MCF-7 breast cancer cells, Hsp22 could be effectively induced by E2 and Cd. This induction involved the ER because it did not occur in ER− MDA-MB-231 breast cancer cells and because it was counteracted by the anti-estrogen ICI182780. Phosphorylation of Hsp22 was not induced, even under conditions that induced phosphorylation of Hsp27.
Intracellular localization and interaction of Hsp22 and Hsp27 in MCF-7 cells
Hsp22 and Hsp27 have been shown to be interacting proteins (Irobi et al 2004; Sun et al 2004). To learn about their possible interaction in MCF-7 cells, we used the qFRET method to determine the in vivo interaction of ectopically expressed Hsp27-CFP and Hsp22-CIT fusion proteins. Twenty-four hours after transfection, expressed Hsp22-CIT localized in both the cytoplasm and the nucleus of MCF-7 cells (Fig 2A). Distribution in the cytoplasm was relatively even, with some accumulation in the perinuclear space. Most of the nucleus was also labeled, with a few unidentified nuclear domains excluding the fusion protein. The cytoplasmic distribution of Hsp27-CFP was very similar to that of Hsp22-CIT, whereas not much Hsp27-CFP was found in the nucleus compared with Hsp22-CIT (Fig 2A). Both proteins colocalized in the cytoplasm, thus providing the possibility that they might physically interact in this compartment. A similar subcellular location of both fusion proteins was found in transfected COS-7 cells (Fontaine et al 2006).
To determine directly the interaction between both proteins in live MCF-7 cells, the qFRET method was applied. Cells were doubly transfected to express both constructs, and 24 hours later, the CIT, CFP, and fluorescence resonance energy transfer images were recorded. The images were analyzed by the qFRET algorithm, and the AAFE values were determined as described previously (Hoppe et al 2002; Fontaine et al 2006). For cells expressing both sHSP fusion proteins, the obtained AAFE signal was ∼20 times greater than that of the control cells expressing the “empty” CIT vector and the Hsp27-CFP vector (Fig 2B). Statistical analysis indicated that this was a significant difference. Thus, Hsp22 and Hsp27 did interact in the cytoplasm of MCF-7 cells.
Size distribution of Hsp22 and Hsp27 in extracts of MCF-7 cells
Hsp22 and Hsp27 are known to form homo- and heterodimers, heterogeneous homo-oligomeric complexes, and possibly also hetero-oligomeric complexes, resulting usually in an elution from HPLC gel filtration columns that is characterized by a wide range of apparent molecular masses, from HMMCs down to dimers or monomers (Sun et al 2004; Fontaine et al 2005). To determine the effect of induction of Hsp22 by E2 and Cd on the sHSP complex formation in MCF-7 cells, the size distribution of both Hsp22 and Hsp27 in cell extracts was determined by gel filtration HPLC. The HPLC column used separated proteins in the molecular mass range between >670 and <23 kDa (Fig 3A), and the MCF-7 cell proteins were reasonably well separated, as revealed by the Coomassie blue– stained protein patterns of the corresponding fractions (Fig 3B). In contrast to most of the cell proteins, both Hsp22 and Hsp27 distributed throughout a wide molecular mass range with discernible peaks and “valleys” (Fig 3C,D). This applies also to the relatively small amounts of Hsp22 (cf Fig 1A, lane 1) present in cells grown in E2-deficient medium (Fig 3C, control). In these cells, most of the Hsp22 distributed in fractions 3–7 (corresponding to >670 to ∼200 kDa) and in fractions 13– 16/17 (corresponding to ∼66 to ∼30 kDa). Induction by E2 of Hsp22 (cf Fig 1A, lane 3) resulted in a pronounced shift in the distribution of Hsp22, with the new peak in the high–molecular-mass fractions 1 and 2 (corresponding to >670 kDa; Fig 3C, E2). Similarly, the induction of Hsp22 by Cd (cf Fig 1A, lane 5) caused a pronounced shift in the distribution of Hsp22 toward fractions 1 and 2 (Fig 3C, CdCl2). Thus, the increase in the cellular amounts of Hsp22, be it by E2 or Cd treatment, contributes predominantly to the formation of the HMMCs of >670 kDa.
The distribution of Hsp27 in the same fractions was similar to that of Hsp22, although not identical (Fig 3D). Interestingly, in the lower molecular mass fractions, Hsp27 peaked in fractions 12–14 (corresponding to ∼100 to ∼50 kDa), whereas Hsp22 peaked in fractions 15–17 (corresponding to ∼50 to ∼30 kDa). Treatment of the cells with either E2 or Cd did not result in a comparable shift of Hsp27 distribution into the high–molecular mass fractions 1 and 2.
Taken together, our results indicate that most Hsp22 synthesized after induction by E2 or Cd was incorporated into the HMMCs with a molecular mass >670 kDa. Although fractions of both proteins did interact in live cells (cf Fig 2), Hsp27 was not shifted in a comparable proportion into these HMMCs after induction of Hsp22. This finding, together with the slightly different size distribution and the partially different intracellular location (cf Fig 2A), suggests that both sHSPs were partially incorporated into distinct populations of sHSP complexes.
Down-regulation of Hsp22 by the RNA interference method and drug resistance
Previously it was shown that ectopic expression of Hsp22 and Hsp27 promoted cell growth and resistance to doxorubicin in breast cancer cells (Oesterreich et al 1993; Yang et al 2006). In order to explore the possibility of down-regulation of Hsp22 as a potential therapeutic strategy, we decreased the amount of Hsp22 in MCF-7 cells by the RNA interference (RNAi) method, and tested to see whether this would modulate cell growth and drug resistance. Western blot analysis revealed that MCF-7 cells grown in complete medium contained Hsp22 (insert in Fig 4, lane 1). Similar amounts of Hsp22 were found in MCF-7 cells that were electroporated with the “empty” pCH1 control vector (lane 2). In contrast, MCF-7 cells that were electroporated with the pCH1-shHsp22 vector contained reduced amounts of Hsp22 (lane 3). This down-regulation was specific for Hsp22 because the amount of Hsp27 was not affected by this procedure, as was shown by Western blotting (insert in Fig 4). Electroporation of MCF-7 cells typically yielded >75% efficiency, as estimated from the expression of the DsRed marker dye, and this transgene expression persisted during the entire duration of the experiments (not shown). β-Actin was visualized on the same blots to demonstrate equal loading.
Fig. 4.

Resistance of estrogen receptor–positive (ER+) MCF-7 cells to doxorubicin after down-regulation of endogenous Hsp22 expression. MCF-7 cells grown in complete medium were electroporated to take up control pCH1 vector (light bars) or the pCH1–short hairpin (sh)Hsp22 vector (dark bars). Approximately 15 000 electroporated cells were seeded into each well of 24-well plates containing complete Dulbecco modified Eagle medium (DMEM). Twenty-four hours later, doxorubicin was added as indicated, and after additional 72 hours, cell viability was determined by the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay. Error bars represent standard deviation. The inset shows Western blots for Hsp22, Hsp27, and β-actin of untreated control cells (lane 1) and of cells electroporated with the pCH1 control vector (lane 2) or the pCH1-shHsp22 vector (lane 3). A representative experiment is shown. Although the down-regulation of Hsp22 was effective, the resistance of MCF-7 cells to doxorubicin was not affected
Growth of MCF-7 cells with down-regulated Hsp22 in complete medium without added toxicants as determined by cell counting was not different from that of control MCF-7 cells (not shown). In the presence of various doxorubicin concentrations (between 0.03 and 10 μM), the viability of MCF-7 cells with down-regulated Hsp22 (Fig 4, dark columns) was measured by MTT assay and compared with the viability of cells transfected with the control vector (light columns). As expected, increasing concentrations of doxorubicin decreased cell viability, with virtually no viable cells left at 10 μM doxorubicin. Resistance to doxorubicin showed no major differences between cells with down-regulated Hsp22 and control cells. A similar experiment was conducted with various CdCl2 concentrations (between 0.2 and 100 μM). As reported previously (Brama et al 2006), low concentrations of Cd (between 0.5 and 3 μM) stimulated cell growth, whereas higher concentrations decreased cell viability with virtually no viable cells left at 50 μM CdCl2. No difference in Cd-induced cell growth or resistance to CdCl2 between cells with down-regulated Hsp22 and control cells could be detected (not shown). Although transient down-regulation of Hsp22 in MCF-7 cells was effective, it did not alter anchorage-dependent cell growth or resistance to doxorubicin or Cd.
Potential regulatory elements in the Hsp22 promoter
We have analyzed the DNA sequence of the proximal (up to ∼900 bp upstream) and the distal (up to ∼4000 bp upstream) promoter regions, as well as parts of the coding sequence of the human HSP22 gene for potential elements that might be involved in the regulation of the expression of Hsp22 by E2 or other factors. After binding to the ligand, the ER associates with promoter elements of target genes that are induced by E2. The classical consensus E2 response element (ERE) is composed of a palindrome of 5′-(A/G)GGTCA-3′ motifs, separated by 3 bp, and is found in some of the promoters of the E2-regulated genes. The majority of the identified elements, however, deviate from this consensus motif. The deviations include imperfect palindromes, widely spaced or direct repeats of ERE half-sites (ERE1/2), or even a single ERE1/2. Usually, such imperfect elements on their own have only weak transcriptional activity, whereas 2 or more imperfect elements throughout the regulatory sequences of E2-responsive genes can cooperate for ER binding and transactivation (Kato et al 1992). In addition, individual ERE1/2 sites can be associated with binding sites for the transcription factor Sp1 (GC boxes or GT/CACC boxes), and the significance of these associated ER/Sp1-binding sites for the response to E2 was demonstrated (Sanchez et al 2002).
Neither the proximal nor the distal promoter regions of human Hsp22 contain a perfect palindromic ERE. The proximal promoter contains 2 putative ERE1/2 sites and 4 putative Sp1-binding sites (Fig 5A). Although conventionally, the SP1 binding site consensus motif was denoted 5′-(G/T)GGGCGG(G/A)(G/A)(C/T)-3′ (Dreier et al 2001), data base–supported SP1 binding site analysis at the Transcription Element Search System web site (http://www.cbil.upenn.edu/tess) favored the motif 5′-(G/T/A)(G/A)GG(C/G)(G/A)(G/T)(G/T)(G/C)(T/A/C)-3′. The 4 putative Sp1 binding sites in the proximal promoter, as shown in Figure 5A, fit this consensus motif (2 mismatches permitted). The most downstream putative SP1 binding site is in close association with one of the ERE1/2 sites. Interestingly, the upstream ERE1/2 is embedded in a putative retinoid X receptor γ (RXRγ) binding site. Similarly extended 5′-(G/A)GGTCA-3′ sites were shown to represent common response elements for nuclear receptors with P boxes homologous to that of the ER, including RXRs (Kato et al 1995). In addition, the proximal promoter contains the common promoter elements CAAT box and TATA box, and 2 perfect palindromic putative heat shock elements (consensus sequence 5′-GAANNTTC-3′) that could account for the heat inducibility reported for Hsp22 (Chowdary et al 2004).
Fig 5.

Sequence analysis of putative regulatory elements of the human Hsp22 gene. (A) Proximal promoter with the putative promoter elements CAAT box, TATA box, SP1 binding sites, palindromic heat shock elements (HSE), 17β-estradiol response element half-sites (ERE1/2), and translation start codon atg. The proximal and distal ERE1/2 sites are associated with an SP1 site and a retinoid X receptor γ (RXRγ) site, respectively. (B) Distal Hsp22 promoter region with a cluster of 4 adjacent putative ERE1/2 sites. The second ERE1/2 site is associated with a putative SP1 binding site. (C) Section of the first intron with 2 adjacent putative ERE1/2 sites. Putative ERE1/2 sequences matching the consensus motif 5′-(G/A)GGTCA-3′ are in bold, capitalized and underlined (1 mismatch permitted). The putative combined ERE1/2/RXRγ site in panel A is in bold and doubly underlined. Putative SP1 binding sites matching the consensus motif 5′-(G/T/A)(G/A)GG(C/G)(G/A)(G/T)(G/ T)(G/C)(T/A/C/)-3′ are in bold with dotted underlining (2 mismatches permitted). Palindromic HSEs matching the consensus motif 5′-GAANNTTC-3′ are in bold with dashed underlining. Other elements are underlined
The distal promoter region contains a cluster of 4 putative ERE1/2 sites, and one of them is also in reasonably close association with a putative SP1 site (Fig 5B). These distal elements might also contribute to the E2 induction of HSP22. The functionality of similarly distant EREs was demonstrated for the chicken ovalbumin gene (Kato et al 1992). Furthermore, the gene of the human Hsp22 contains 2 adjacent putative ERE1/2 sites close to the 5′ end of the first intron, although no association with an SP1 site was found (Fig 5C).
In preliminary reporter gene assays, none of these elements was sufficient to mediate E2 induction in transfected MCF-7 cells, although most of the constructs showed expression in COS-7 cells (data not shown).
DISCUSSION
E2 has been suspected to play a role in the genesis of breast cancer, although there is not a simple link between high E2 levels and cancer risk (Coyle 2004). The current hypothesis is that E2 acts as an epigenetic factor that is involved in conversion of precancerous to cancerous cells in the course of tumor progression. Hsp22 was identified recently as a potential key player in the progression of breast cancer in humans (Yang et al 2006). Hsp22 expression is a downstream event of both erbB2 → cyclin D1 and E2 signaling pathways, and this fact was suggested to be the basis for the well known association between cyclin D1 expression and poor prognosis. Although these findings strongly support a role of Hsp22 in ER+ breast cancer, its precise role at the molecular level remains elusive.
Previous studies demonstrated induction of Hsp22 by E2 in ER+ breast cancer cells at the transcriptional level (Charpentier et al 2000; Yang et al 2006). Here, we present data concerning Hsp22 induction at the protein level. ER+ breast cancer cells showed induction of Hsp22 protein in response to E2, whereas ER− breast cancer cells did not show this response. The data indicate that E2-mediated Hsp22 induction is fairly pronounced (>3-fold) compared with other proteins. For example, Hsp27, another sHSP induced by E2 in ER+ breast cancer cells, was induced ∼1.5-fold by E2 (Edwards et al 1980). The inhibitory effects of the anti-estrogen ICI182780 and the RNA polymerase II inhibitor actinomycin D suggest involvement of ERα in the transcriptional regulation of Hsp22. The reason why in this study the expected induction of Hsp27 by E2, and also the down-regulation by ICI182780, were not seen is probably related to the nature of the MCF-7 cells used. Later passages of MCF-7 cells as used in this study were reported to contain elevated and constitutively expressed amounts of Hsp27 that were no longer regulated by E2 (Fuqua et al 1994).
It has been reported that Cd mimics mitogenic signaling by an ER-dependent mechanism. In ER+ MCF-7 cells, Cd interacted with the E2-binding domain of the ERα (Stoica et al 2000), and, as a consequence, it stimulated Erk1/2, Akt, and PDGFRα signaling and expression of the oncogenes c-jun and c-fos (Brama et al 2006). Similar to E2, Cd also regulated the expression of the progesterone receptor, cathepsin D, and of the pS2 genes in MCF-7 cells, but not in ER− MDA-MB-231 cells, unless they were transfected to express ER (Garcia-Morales et al 1994). These estrogenic cellular effects of Cd are complemented by its effects at the level of the organism: Similar to E2, Cd increased uterine weight, promoted growth and development of the mammary glands, and induced hormone-regulated genes in ovariectomized mice (Johnson et al 2003). ER antagonists such as ICI182780 blunted these responses, strongly suggesting signaling via ER. With these estrogenic properties, environmental Cd clearly has the potential to interfere with endocrine signaling and to act as an endocrine disruptor by mimicking E2. In addition to Cd, a number of other metal toxicants with estrogenic properties have been identified, including aluminum, arsenite, chromium (II), copper, lead, mercury, nickel, tin, vanadate, and others (Darbre 2006). These metalloestrogens are now considered risk factors that can contribute to aberrant E2 signaling in the human breast. In view of these facts, the pronounced induction of Hsp22 by Cd and its inhibition by ICI182780 is not entirely unexpected. These findings suggest that induction of Hsp22 by Cd indeed is related to its estrogenic properties and is mediated by the action of the ER. It can be expected that exposure to environmentally relevant concentrations of Cd could result in increased expression of Hsp22, and this situation might shed new light on the action of environmental factors in carcinogenesis.
Synthesis of Hsp27 and αB-crystallin, 2 other sHSPs, has been reported to be induced in different cell types by various metal toxicants, including Cd, arsenite, and mercury (Pittenger et al 1992; Head et al 1996; Bonham et al 2003; Croute et al 2005; Eichler et al 2005). That Cd treatment did not result in any detectable induction of Hsp27 in the experiments of this study also could be related to the nature of the MCF-7 cell line used, as mentioned above. Occasionally, cells that express readily detectable levels of Hsp27 are refractory to further induction of this sHSP by Cd, as has been shown for astroglia cells (Opanashuk and Finkelstein 1995).
In a number of studies, Cd induced a robust and swift phosphorylation of Hsp27 in various cell types, including macrophages, renal podocytes, renal mesangial cells, and adrenal chromaffin cells (Radloff et al 1998; Eichler et al 2005; Hirano et al 2005; Leal et al 2007). In these cells, Cd is likely to activate signaling pathways that do not involve the ER. Instead, Cd might interfere with calcium signaling because of the similar properties of these 2 ions. This notion is supported by the short response time of Hsp27 phosphorylation (typically within minutes and up to ∼2 hours) seen in most cells compared with the long response time (typically many hours to a day) for events involving the ER. Thus, the time course of Hsp27 phosphorylation in MCF-7 cells suggests an ER-independent signaling pathway. A further argument for at least 2 separate Cd-related signaling pathways in MCF-7 cells comes from the fact that the anti-estrogen ICI182780 did not inhibit the induction of Hsp27 phosphorylation, while it did inhibit the induction of Hsp22 synthesis. Although Hsp22 apparently can also be phosphorylated (Smith et al 2000; Benndorf et al 2001), the corresponding signaling pathways are not activated in MCF-7 cells in response to Cd.
In this study, Hsp22 was shown to interact with Hsp27 in the cytoplasm of MCF-7 cells, and induction of Hsp22 synthesis correlated with increased distribution of this sHSP in the HMMCs. The ability of Hsp22 and other sHSPs to interact with one another and to form HMMCs has been studied previously (Sun et al 2004; Fontaine et al 2005). Whatever the actual function of Hsp22 in breast cancer cells is, it might be accomplished by these HMMCs. For example, HMMC formation might be involved in the chaperone-like activity that has been reported for Hsp22 (Chowdary et al 2004; Kim et al 2004; Carra et al 2005). Apparently, Hsp22 and Hsp27 were, in part, components of distinct complex populations. This was suggested by the slightly different HPLC distribution pattern of both proteins and because the induction-caused shift of Hsp22 toward the HMMCs in fractions 1 and 2 was not paralleled by a corresponding shift of Hsp27 (Fig 3C,D).
As was demonstrated recently, ectopic stable expression of Hsp22 in NIH3T3 cells greatly enhanced colony formation in an anchorage-independent soft agar growth assay (Yang et al 2006). Also in anchorage-dependent growth assays, ectopic stable expression of Hsp22 in T47D and MCF-7 correlated with increased cell growth and reduced apoptosis, whereas no major effect of down-regulation of Hsp22 expression on MCF-7 cell growth and resistance to doxorubicin or Cd was found in the anchorage-dependent assays in this study. This could be related to the fact that the down-regulation of Hsp22, although effective, did not lower the amount of Hsp22 below a critical level needed for the maintenance of its cellular functions.
Other sHSPs have been also implicated in the biology of breast cancer. αB-crystallin was recently identified as a novel oncoprotein expressed in basal-like breast carcinomas that independently predicted shorter survival (Moyano et al 2006). Expression of Hsp27 was found to be associated with growth, resistance to doxorubicin, and invasiveness of breast cancer cells and was inversely associated with cell motility (Oesterreich et al 1993; Lemieux et al 1997). Interestingly, some cytostatic drugs can specifically induce Hsp27 in mouse breast cancer cells (Bielka et al 1994). Although Hsp27 could be a useful tumor marker in certain subsets of tumors, its suitability as a general prognostic or predictive factor in breast cancer is limited according to present knowledge, and further studies are needed (Ciocca and Vargas-Roig 2002). The data presented here would justify a more detailed study of the value of Hsp22 as a prognostic and predictive factor in breast cancer, possibly in context with expression of Hsp27 and other sHSPs.
By sequence analysis, we have identified potential ERE1/2 sites in the human Hsp22 promoter and gene that could account for the E2 regulation of Hsp22. The most likely candidate site contains an ERE1/2 site associated with a putative SP1 site in the proximal promoter region. Chromatin immunoprecipitation assays with ER-specific antibodies have indeed suggested the involvement of the proximal promoter region in E2 regulation of Hsp22, although no detailed element analysis was performed (Yang et al 2006). It should be noted that the ER functions cooperatively with a number of transcription factors such as SP1 (Sanchez et al 2002). For the Hsp27 promoter, this cooperation does not even require direct binding of the ER to DNA. Deletion of the 5′-(A/G)GGTCA-3′ motif in the composite ERE1/2/SP1 response element 5′-GGGCGG-(N)9-GGGTCA-3′ did not affect the responsiveness to E2 (Porter et al 1997). At this time, no final conclusions can be drawn on the functionality of any of the candidate elements shown in Figure 5 for Hsp22 expression and induction.
Taken together, the induction of Hsp22 by E2 and Cd in ER+ breast cancer cells represents a new aspect of the estrogenic response and of the aberrant response to environmental factors that mimic E2. Studies of new E2-regulated genes could be useful for classifying breast cancers according to their ability to respond to therapies and might lead to new therapeutic approaches for hormone-dependent breast cancers. Further studies will be needed to define the exact role of Hsp22 in breast cancer cells, both in the context of ER-mediated signaling and in the response to environmental factors.
Acknowledgments
This work was supported by a Munn Idea grant of the University of Michigan Comprehensive Cancer Center to R.B. and by the National Institutes of Health grant P01ES11188 to M.J.W. (PI) and R.B. We thank Drs J.M. Velkey and K.S. O'Shea (Ann Arbor, MI, USA) for providing us with the pCH1 vector. The help of Dr Yong Tai Hou (Shanghai, PRC) with designing the shRNA-Hsp22 construct, of Dr V. Vega-Warner (Ann Arbor) with the MTT assay, and of Dr S. Oesterreich (Houston, TX, USA) for the critical reading of the manuscript are gratefully acknowledged.
REFERENCES
- American Cancer Society. 1993 Cancer Facts and Figures—1993. American Cancer Society, Atlanta, GA. [Google Scholar]
- Benndorf R, Engel K, Gaestel M. Analysis of small Hsp phosphorylation. Methods Mol Biol. 2000;99:431–445. doi: 10.1385/1-59259-054-3:431.1064-3745(2000)099[0431:AOSHP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Benndorf R, Kraft R, Otto A, Stahl J, Böhm H, Bielka H. Purification of the growth-related protein p25 of the Ehrlich ascites tumor and analysis of its isoforms. Biochem Int. 1988a;17:225–234.0158-5231(1988)017[0225:POTGPP]2.0.CO;2 [PubMed] [Google Scholar]
- Benndorf R, Nürnberg P, Bielka H. Growth phase–dependent proteins of the Ehrlich ascites tumor analyzed by one- and two-dimensional electrophoresis. Exp Cell Res. 1988b;174:130–138. doi: 10.1016/0014-4827(88)90148-6.0014-4827(1988)174[0130:GPPOTE]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Benndorf R, Sun X, and Gilmont RR. et al. 2001 HSP22, a new member of the small heat shock protein superfamily, interacts with mimic of phosphorylated HSP27 (3DHSP27). J Biol Chem. 276:26753–26761. [DOI] [PubMed] [Google Scholar]
- Bielka H, Hoinkis G, Oesterreich S, Stahl J, Benndorf R. Induction of the small stress protein, hsp25, in Ehrlich ascites carcinoma cells by anticancer drugs. FEBS Lett. 1994;343:165–167. doi: 10.1016/0014-5793(94)80311-0.0014-5793(1994)343[0165:IOTSSP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Bonham RT, Fine MR, Pollock FM, Shelden EA. Hsp27, Hsp70, and metallothionein in MDCK and LLC-PK1 renal epithelial cells: effects of prolonged exposure to cadmium. Toxicol Appl Pharmacol. 2003;191:63–73. doi: 10.1016/s0041-008x(03)00226-6.0041-008X(2003)191[0063:HHAMIM]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Brama M, Gnessi L, and Basciani S. et al. 2006 Cadmium induces mitogenic signaling in breast cancer cell by an ERalpha-dependent mechanism. Mol Cell Endocrinol. 264:101–108. [DOI] [PubMed] [Google Scholar]
- Carra S, Sivilotti M, Chavez Zobel AT, Lambert H, Landry J. HspB8, a small heat shock protein mutated in human neuromuscular disorders, has in vivo chaperone activity in cultured cells. Hum Mol Genet. 2005;14:1659–1669. doi: 10.1093/hmg/ddi174.1460-2083(2005)014[1659:HASHSP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Charpentier AH, Bednarek AK, Daniel RL, Hawkins KA, Laflin KJ, Gaddis S, MacLeod MC, Aldaz CM. Effects of estrogen on global gene expression: identification of novel targets of estrogen action. Cancer Res. 2000;60:5977–5983.0008-5472(2000)060[5977:EOEOGG]2.0.CO;2 [PubMed] [Google Scholar]
- Choe SY, Kim SJ, Kim HG, Lee JH, Choi Y, Lee H, Kim Y. Evaluation of estrogenicity of major heavy metals. Sci Total Environ. 2003;312:15–21. doi: 10.1016/S0048-9697(03)00190-6.0048-9697(2003)312[0015:EOEOMH]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Chowdary TK, Raman B, Ramakrishna T, Rao CM. Mammalian Hsp22 is a heat-inducible small heat-shock protein with chaperone-like activity. Biochem J. 2004;381:379–387. doi: 10.1042/BJ20031958.0264-6021(2004)381[0379:MHIAHS]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciocca DR, Adams DJ, Edwards DP, Bjercke RJ, McGuire WL. Estrogen-induced 24K protein in MCF-7 breast cancer cells is localized in granules. Breast Cancer Res Treat. 1984;4:261–268. doi: 10.1007/BF01806037.0167-6806(1984)004[0261:EKPIMB]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Ciocca DR, Fuqua SA, Lock-Lim S, Toft DO, Welch WJ, McGuire WL. Response of human breast cancer cells to heat shock and chemotherapeutic drugs. Cancer Res. 1992;52:3648–3654.0008-5472(1992)052[3648:ROHBCC]2.0.CO;2 [PubMed] [Google Scholar]
- Ciocca DR, Puy LA, Fasoli LC. Study of estrogen receptor, progesterone receptor, and the estrogen-regulated Mr 24,000 protein in patients with carcinomas of the endometrium and cervix. Cancer Res. 1989;49:4298–4304.0008-5472(1989)049[4298:SOERPR]2.0.CO;2 [PubMed] [Google Scholar]
- Ciocca DR, Vargas-Roig LM. Hsp27 as a prognostic and predictive factor in cancer. Prog Mol Subcell Biol. 2002;28:205–218. doi: 10.1007/978-3-642-56348-5_11.0079-6484(2002)028[0205:HAAPAP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Clarke R, Leonessa F, Welch JN, Skaar TC. Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacol Rev. 2001;53:25–71.0031-6997(2001)053[0025:CAMPOA]2.0.CO;2 [PubMed] [Google Scholar]
- Coyle YM. The effect of environment on breast cancer risk. Breast Cancer Res Treat. 2004;84:273–288. doi: 10.1023/B:BREA.0000019964.33963.09.0167-6806(2004)084[0273:TEOEOB]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Croute F, Beau B, Murat JC, Vincent C, Komatsu H, Obata F, Soleilhavoup JP. Expression of stress-related genes in a cadmium-resistant A549 human cell line. J Toxicol Environ Health Part A. 2005;68:703–718. doi: 10.1080/15287390590925447.1528-7394(2005)068[0703:EOSGIA]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Darbre PD. Metalloestrogens: an emerging class of inorganic xenoestrogens with potential to add to the oestrogenic burden of the human breast. J Appl Toxicol. 2006;26:191–197. doi: 10.1002/jat.1135.0260-437X(2006)026[0191:MAECOI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Dreier B, Beerli RR, Segal DJ, Flippin JD, Barbas CF 3rd. Development of zinc finger domains for recognition of the 5′-ANN-3′ family of DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem. 2001;276:29466–29478. doi: 10.1074/jbc.M102604200.0021-9258(2001)276[29466:DOZFDF]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Edwards DP, Adams DJ, Savage N, McGuire WL. Estrogen induced synthesis of specific proteins in human breast cancer cells. Biochem Biophys Res Commun. 1980;93:804–812. doi: 10.1016/0006-291x(80)91148-1.0006-291X(1980)093[0804:EISOSP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Eichler TE, Ransom RF, Smoyer WE. Differential induction of podocyte heat shock proteins by prolonged single and combination toxic metal exposure. Toxicol Sci. 2005;84:120–128. doi: 10.1093/toxsci/kfi048.1096-0929(2005)084[0120:DIOPHS]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Fontaine JM, Sun X, Benndorf R, Welsh MJ. Interactions of HSP22 (HSPB8) with HSP20, αB-crystallin, and HSPB3. Biochem Biophys Res Commun. 2005;337:1006–1011. doi: 10.1016/j.bbrc.2005.09.148.0006-291X(2005)337[1006:IOHHWH]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Fontaine JM, Sun X, Hoppe AD, Simon S, Vicart P, Welsh MJ, Benndorf R. Abnormal small heat shock protein interactions involving neuropathy-associated HSP22 (HSPB8) mutants. FASEB J. 2006;20:2168–2170. doi: 10.1096/fj.06-5911fje.0892-6638(2006)020[2168:ASHSPI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Fuqua SA, Benedix MG, Krieg S, Weng CN, Chamness GC, Oesterreich S. Constitutive overexpression of the 27,000 dalton heat shock protein in late passage human breast cancer cells. Breast Cancer Res Treat. 1994;32:177–186. doi: 10.1007/BF00665768.0167-6806(1994)032[0177:COOTDH]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Fuqua SA, Blum-Salingaros M, McGuire WL. Induction of the estrogen-regulated “24K” protein by heat shock. Cancer Res. 1989;49:4126–4129.0008-5472(1989)049[4126:IOTEKP]2.0.CO;2 [PubMed] [Google Scholar]
- Gaestel M, Gross B, and Benndorf R. et al. 1989 Molecular cloning, sequencing and expression in Escherichia coli of the 25-kDa growth-related protein of Ehrlich ascites tumor and its homology to mammalian stress proteins. Eur J Biochem. 179:209–213. [DOI] [PubMed] [Google Scholar]
- Garcia-Morales P, Saceda M, and Kenney N. et al. 1994 Effect of cadmium on estrogen receptor levels and estrogen-induced responses in human breast cancer cells. J Biol Chem. 269:16896–16901. [PubMed] [Google Scholar]
- Head MW, Hurwitz L, Goldman JE. Transcription regulation of alpha B-crystallin in astrocytes: analysis of HSF and AP1 activation by different types of physiological stress. J Cell Sci. 1996;109:1029–1039. doi: 10.1242/jcs.109.5.1029.0021-9533(1996)109[1029:TROABI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Hirano S, Sun X, and DeGuzman CA. et al. 2005 p38 MAPK/HSP25 signaling mediates cadmium-induced contraction of mesangial cells and renal glomeruli. Am J Physiol Renal Physiol. 288:F1133–F1143. [DOI] [PubMed] [Google Scholar]
- Hodges LC, Cook JD, and Lobenhofer EK. et al. 2003 Tamoxifen functions as a molecular agonist inducing cell cycle–associated genes in breast cancer cells. Mol Cancer Res. 1:300–311. [PubMed] [Google Scholar]
- Hoppe A, Christensen K, Swanson JA. Fluorescence resonance energy transfer–based stoichiometry in living cells. Biophys J. 2002;83:3652–3664. doi: 10.1016/S0006-3495(02)75365-4.0006-3495(2002)083[3652:FRETSI]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horman S, Galand P, Mosselmans R, Legros N, Leclercq G, Mairesse N. Changes in the phosphorylation status of the 27 kDa heat shock protein (HSP27) associated with the modulation of growth and/or differentiation in MCF-7 cells. Cell Prolif. 1997;30:21–35. doi: 10.1046/j.1365-2184.1997.00069.x.0960-7722(1997)030[0021:CITPSO]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irobi J, Van Impe K, and Seeman P. et al. 2004 Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat Genet. 36:597–601. [DOI] [PubMed] [Google Scholar]
- Johnson MD, Kenney N, and Stoica A. et al. 2003 Cadmium mimics the in vivo effects of estrogen in the uterus and mammary gland. Nat Med. 9:1081–1084. [DOI] [PubMed] [Google Scholar]
- Kappé G, Verschuure P, Philipsen RL, Staalduinen AA, Van de Boogaart P, Boelens WC, De Jong WW. Characterization of two novel human small heat shock proteins: protein kinase– related HspB8 and testis-specific HspB9. Biochim Biophys Acta. 2001;1520:1–6. doi: 10.1016/s0167-4781(01)00237-8.0006-3002(2001)1520[0001:COTNHS]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Kato S, Sasaki H, Suzawa M, Masushige S, Tora L, Chambon P, Gronemeyer H. Widely spaced, directly repeated PuGGTCA elements act as promiscuous enhancers for different classes of nuclear receptors. Mol Cell Biol. 1995;15:5858–5867. doi: 10.1128/mcb.15.11.5858.1098-5549(1995)015[5858:WSDRPE]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Tora L, Yamauchi J, Masushige S, Bellard M, Chambon P. A far upstream estrogen response element of the ovalbumin gene contains several half-palindromic 5′-TGACC-3′ motifs acting synergistically. Cell. 1992;68:731–742. doi: 10.1016/0092-8674(92)90148-6.0092-8674(1992)068[0731:AFUERE]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Kim MV, Seit-Nebi AS, Marston SB, Gusev NB. Some properties of human small heat shock protein Hsp22 (H11 or HspB8) Biochem Biophys Res Commun. 2004;315:796–801. doi: 10.1016/j.bbrc.2004.01.130.0006-291X(2004)315[0796:SPOHSH]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Leal RB, Posser T, Rigon AP, Oliveira CS, Goncalves CA, Gelain DP, Dunkley PR. Cadmium stimulates MAPKs and Hsp27 phosphorylation in bovine adrenal chromaffin cells. Toxicology. 2007;234:34–43. doi: 10.1016/j.tox.2007.01.023.0300-483X(2007)234[0034:CSMAHP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Lemieux P, Oesterreich S, Lawrence JA, Steeg PS, Hilsenbeck SG, Harvey JM, Fuqua SA. The small heat shock protein hsp27 increases invasiveness but decreases motility of breast cancer cells. Invasion Metastasis. 1997;17:113–123.0251-1789(1997)017[0113:TSHSPH]2.0.CO;2 [PubMed] [Google Scholar]
- Martin MB, Reiter R, and Pham T. et al. 2003 Estrogen-like activity of metals in MCF-7 breast cancer cells. Endocrinology. 144:2425–2436. [DOI] [PubMed] [Google Scholar]
- McElroy JA, Shafer MM, Trentham-Dietz A, Hampton JM, Newcomb PA. Cadmium exposure and breast cancer risk. J Natl Cancer Inst. 2006;98:869–873. doi: 10.1093/jnci/djj233.0027-8874(2006)098[0869:CEABCR]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Morris C, Wakeling A. Fulvestrant (‘Faslodex’)—a new treatment option for patients progressing on prior endocrine therapy. Endocr Relat Cancer. 2002;9:267–276. doi: 10.1677/erc.0.0090267.1351-0088(2002)009[0267:FFNTOF]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Moyano JV, Evans JR, and Chen F. et al. 2006 αB-crystallin is a novel oncoprotein that predicts poor clinical outcome in breast cancer. J Clin Invest. 116:261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesterreich S, Weng CN, Qiu M, Hilsenbeck SG, Osborne CK, Fuqua SA. The small heat shock protein hsp27 is correlated with growth and drug resistance in human breast cancer cell lines. Cancer Res. 1993;53:4443–4448.0008-5472(1993)053[4443:TSHSPH]2.0.CO;2 [PubMed] [Google Scholar]
- Ohlsson H, Brunner N, Engelholm LH, Lundholt BK, Weidle U, Briand P, Lykkesfeldt AE. Identification of two estrogen regulated genes associated with growth regulation of human breast cancer. Mol Cell Endocrinol. 2001;182:1–11. doi: 10.1016/s0303-7207(01)00559-7.0303-7207(2001)182[0001:IOTERG]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Opanashuk LA, Finkelstein JN. Relationship of lead-induced proteins to stress response proteins in astroglial cells. J Neurosci Res. 1995;42:623–632. doi: 10.1002/jnr.490420504.0360-4012(1995)042[0623:ROLPTS]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Pittenger GL, Gilmont RR, Welsh MJ. The low molecular weight heat shock protein (hsp27) in rat Sertoli cells: evidence for identity of hsp27 with a germ cell–responsive phosphoprotein. Endocrinology. 1992;130:3207–3215. doi: 10.1210/endo.130.6.1597139.0013-7227(1992)130[3207:TLMWHS]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Porter W, Saville B, Hoivik D, Safe S. Functional synergy between the transcription factor Sp1 and the estrogen receptor. Mol Endocrinol. 1997;11:1569–1580. doi: 10.1210/mend.11.11.9916.0888-8809(1997)011[1569:FSBTTF]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Radloff M, Delling M, Marti T, Gercken G. Hsp27 phosphorylation is induced in alveolar macrophages exposed to CdO-coated silica particles. Biochem Biophys Res Commun. 1998;248:219–222. doi: 10.1006/bbrc.1998.8871.0006-291X(1998)248[0219:HPIIIA]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Rochefort H 1995 Oestrogen- and anti-oestrogen–regulated genes in human breast cancer. Ciba Found Symp. 191:254–265.discussion 265–268. [DOI] [PubMed] [Google Scholar]
- Sanchez R, Nguyen D, Rocha W, White JH, Mader S. Diversity in the mechanisms of gene regulation by estrogen receptors. Bioessays. 2002;24:244–254. doi: 10.1002/bies.10066.0265-9247(2002)024[0244:DITMOG]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Satarug S, Moore MR. Adverse health effects of chronic exposure to low-level cadmium in foodstuffs and cigarette smoke. Environ Health Perspect. 2004;112:1099–1103. doi: 10.1289/ehp.6751.0091-6765(2004)112[1099:AHEOCE]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug J, Overton GC 1997 TESS: Transcription Element Search Software on the WWW. Computational Biology and Informatics Laboratory Technical Report CBIL-TR-1997-1001-v0.0. School of Medicine, University of Pennsylvania, Philadelphia, PA, USA. [Google Scholar]
- Smith CC, Yu YX, Kulka M, Aurelian L. A novel human gene similar to the protein kinase (PK) coding domain of the large subunit of herpes simplex virus type 2 ribonucleotide reductase (ICP10) codes for a serine-threonine PK and is expressed in melanoma cells. J Biol Chem. 2000;275:25690–25699. doi: 10.1074/jbc.M002140200.0021-9258(2000)275[25690:ANHGST]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Stoica A, Katzenellenbogen BS, Martin MB. Activation of estrogen receptor-alpha by the heavy metal cadmium. Mol Endocrinol. 2000;14:545–553. doi: 10.1210/mend.14.4.0441.0888-8809(2000)014[0545:AOERBT]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Sun X, Fontaine J-M, Rest JS, Shelden EA, Welsh MJ, Benndorf R. Interaction of human HSP22 (HSPB8) with other small heat shock proteins. J Biol Chem. 2004;279:2394–2402. doi: 10.1074/jbc.M311324200.0021-9258(2004)279[2394:IOHHHW]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Takiguchi M, Yoshihara S. New aspects of cadmium as endocrine disruptor. Environ Sci. 2006;13:107–116.1431-6250(2006)013[0107:NAOCAE]2.0.CO;2 [PubMed] [Google Scholar]
- Trisak ST, Doumgdee P, Rode BM. Binding of zinc and cadmium to human serum albumin. Int J Biochem. 1990;22:977–981. doi: 10.1016/0020-711x(90)90203-f.0020-711X(1990)022[0977:BOZACT]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Velkey JM, O'Shea KS. RNA interference induces trophectoderm differentiation in mouse embryonic stem cells. Genesis. 2003;37:18–24. doi: 10.1002/gene.10218.1061-2289(2003)037[0018:RIITDI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Waalkes MP. Cadmium carcinogenesis. Mutat Res. 2003;533:107–120. doi: 10.1016/j.mrfmmm.2003.07.011.0027-5107(2003)533[0107:CC]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Waisberg M, Joseph P, Hale B, Beyersmann D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology. 2003;192:95–117. doi: 10.1016/s0300-483x(03)00305-6.0300-483X(2003)192[0095:MACMOC]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Wakeling AE. Similarities and distinctions in the mode of action of different classes of antioestrogens. Endocr Relat Cancer. 2000;7:17–28. doi: 10.1677/erc.0.0070017.1351-0088(2000)007[0017:SADITM]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Yang C, Trent S, Ionescu-Tiba V, Lan L, Shioda T, Sgroi D, Schmidt EV. Identification of cyclin D1– and estrogen-regulated genes contributing to breast carcinogenesis and progression. Cancer Res. 2006;66:11649–11658. doi: 10.1158/0008-5472.CAN-06-1645.0008-5472(2006)066[11649:IOCDAE]2.0.CO;2 [DOI] [PubMed] [Google Scholar]