

Abstract
The Arp2/3 complex, first isolated from Acanthamoeba castellani by affinity chromatography on profilin, consists of seven polypeptides; two actinrelated proteins, Arp2 and Arp3; and five apparently novel proteins, p40, p35, p19, p18, and p14 (Machesky et al., 1994). The complex is homogeneous by hydrodynamic criteria with a Stokes' radius of 5.3 nm by gel filtration, sedimentation coefficient of 8.7 S, and molecular mass of 197 kD by analytical ultracentrifugation. The stoichiometry of the subunits is 1:1:1:1:1:1:1, indicating the purified complex contains one copy each of seven polypeptides. In electron micrographs, the complex has a bilobed or horseshoe shape with outer dimensions of ∼13 × 10 nm, and mathematical models of such a shape and size are consistent with the measured hydrodynamic properties. Chemical cross-linking with a battery of cross-linkers of different spacer arm lengths and chemical reactivities identify the following nearest neighbors within the complex: Arp2 and p40; Arp2 and p35; Arp3 and p35; Arp3 and either p18 or p19; and p19 and p14. By fluorescent antibody staining with anti-p40 and -p35, the complex is concentrated in the cortex of the ameba, especially in linear structures, possibly actin filament bundles, that lie perpendicular to the leading edge. Purified Arp2/3 complex binds actin filaments with a K d of 2.3 μM and a stoichiometry of approximately one complex molecule per actin monomer. In electron micrographs of negatively stained samples, Arp2/3 complex decorates the sides of actin filaments. EDC/NHS cross-links actin to Arp3, p35, and a low molecular weight subunit, p19, p18, or p14. We propose structural and topological models for the Arp2/3 complex and suggest that affinity for actin filaments accounts for the localization of complex subunits to actinrich regions of Acanthamoeba.
Actin is one of the most abundant, highly conserved and well characterized of all eukaryotic proteins. The actin-based cytoskeleton determines the shape, passive mechanical properties, and motility of most eukaryotic cells. The recently discovered families of actin- related proteins (Arps)1 appear to be highly conserved as well, but their functions are as yet poorly understood (Mullins et al., 1996; Schroer et al., 1996). The three major classes of actin-related proteins—Arp1, Arp2, and Arp3— are grouped according to their degree of similarity to actin. Arp1 is most closely related to actin (50–60% sequence identity), and this class is by far the most well characterized. Arp1 is associated with the dynactin complex, a multimeric protein complex required for dynein-based vesicle motility. Ultrastructural and topological analysis of the dynactin complex has revealed that Arp1 forms short (37 nm) filaments, similar to actin filaments, that may function as attachment sites for cargo vesicles (Schroer and Sheetz, 1991; Schafer et al., 1994; Schroer et al., 1996).
Arp2 and Arp3 are less closely related to actin (40–50% and 30–40% sequence identity, respectively) and appear to be incapable of forming actin-like filaments (LeesMiller et al., 1992; Schwob and Martin, 1992). The genes for Arp2 and Arp3 are ubiquitous in eukaryotes, apparently as ancient as actin and essential in yeast (for recent reviews see Frankel and Mooseker, 1996; Mullins et al. 1996). Both Arp2 and Arp3 are major cellular proteins in Acanthamoeba castellani, present at micromolar levels and concentrated in the actin-rich cortex (Machesky et al., 1994; Kelleher et al., 1995). The primary structures of Arp2 and Arp3 suggest that both proteins fold like actin with a similar three-dimensional core structure, but with different residues exposed on the surfaces (Kabsch et al., 1985; Kelleher et al., 1995). We expect, therefore, that each protein interacts with a different set of accessory proteins, and with very few conventional actin-binding proteins (Kelleher et al., 1995; Mullins et al., 1996). Arp2 and Arp3 are both components of a multiprotein particle, the Arp2/3 complex, initially purified from Acanthamoeba castellani by affinity chromatography on the actin-binding protein, profilin (Machesky et al., 1994). In addition to Arp2 and Arp3, the purified complex contains five polypeptides with apparent molecular masses of 40, 35, 19, 18, and 14 kD. Partial amino acid sequences of p40, p35, p19, and p18 show no significant homology to previously described sequences (Machesky et al., 1994; Mullins, R.D., J. Vandekerckhove, and T.D. Pollard, unpublished results).
Neither Arp2 nor Arp3 has been purified for functional studies, so their biochemical activities and cellular functions are poorly understood. Arp2 null mutants in Saccharomyces cerevisiae arrest late in the cell cycle and heterozygote diploid cells have delayed cytokinesis (Schwob and Martin, 1992). Arp2 colocalizes with actin in both Acanthamoeba castellani (Machesky et al., 1994; Kelleher et al., 1995) and S. cerevisiae (Moreau et al., 1996). The complex binds profilin affinity columns, and an apparent homologue of the Acanthamoeba p40 has been cloned in Schizosaccharomyces pombe based on suppression of a profilin mutation (Balasubramanian et al., 1996). The Arp2/3 complex, therefore, appears to be a ubiquitous component of eukaryotic cells and may be involved in actin cytoskeletal dynamics.
To better understand the function of the Arp2/3 complex in living cells, we have studied its ultrastructure, topology, and interaction with actin filaments. We used analytical ultracentrifugation and electron microscopy of rotary-shadowed specimens to determine the structure and the native molecular weight of purified Arp2/3 complex. We also used a variety of chemical cross-linkers to identify Arp2- and Arp3-specific binding proteins and to establish nearest neighbor relationships for the subunits. Based on chemical cross-linking and electron microscopy, we propose a model for the spatial organization of these subunits. Finally, we show that purified Arp2/3 complex binds to the sides of actin filaments with micromolar affinity at a 1:1 stoichiometry with actin monomers. This actinbinding activity is sufficient to explain the localization of the complex to actin-rich structures in Acanthamoeba and is further biochemical evidence for the involvement of the complex in cytoskeletal function.
Materials and Methods
Purification
We purified Arp2/3 complex in two steps, according to the method described in Fig. 1 of Machesky et al. (1994). Ameba extracts were dialyzed into 25 mM Tris-HCl, pH 8.0, 7.5 mM Na4P2O7, 0.5 mM DTT, 0.5 mM phenyl methyl sulfonyl fluoride and chromatographed on a column of DEAE-cellulose equilibrated with 25 mM Tris-HCl, pH 8.0, 10 mM KCl, and 1 mM DTT. Flow-through from the DEAE-cellulose column was collected and fractionated by poly-l-proline Sepharose affinity chromatography (Kaiser et al., 1989). Elution with 0.4 M MgCl2 released Arp2/3 complex from the column. Profilin was subsequently eluted with 8 M urea. Purified Arp2/3 complex was dialyzed into buffer containing 150 mM NaCl, 0.2 mM MgCl2, 0.2 mM ATP, 1.0 mM DTT, 10 mM imidazole, pH 7.5 (buffer C) and concentrated by dialysis against solid sucrose. 800 g of wet cells yielded ∼2 mg purified complex.
Figure 1.

SDS-PAGE of fractions taken at each step in the purification of Arp2/3 complex from Acanthamoeba. Bands are stained with Coomassie blue. (A) An extract of soluble proteins (lane 1) was first passed over a DE-52 anion-exchange column in a low ionic strength buffer. Fractions that flowed through DEAE (lane 2) were pooled and loaded onto a poly-l-proline–Sepharose affinity column. Arp2/3 complex (lane 3) was eluted from the column with 0.4 M MgCl2. (B) Purified Arp2/3 complex, Arp3, Arp2, and five associated proteins.
Preparation of Actin
Ameba actin was purified from DEAE column fractions (Pollard, 1984). Rabbit skeletal muscle actin was purified from acetone powder by the method of Spudich and Watt (1971) as modified by MacLean-Fletcher and Pollard (1980).
Actin Copelleting Assay
Arp2/3 complex was prespun at 70,000 rpm in a (model TLA 100.2; Beckman Instrs., Fullerton, CA) rotor (200,000 g) for 1 h at 4°C. The supernatant was then incubated with various concentrations of filamentous actin, either already polymerized or polymerized in the presence of Arp2/3 complex. The mixture was then spun at 200,000 g for 30 min at 24°C to pellet actin filaments. Supernatants and pellets were collected and proteins were separated by SDS-PAGE. Actin and various complex members were quantitated in supernatant and pellet fractions by two-dimensional scanning densitometry of wet, Coomassie-stained gels. Data from densitometry scans were analyzed by the program Collage (Fotodyne, New Berlin, WI), and dissociation constants were calculated by least squares fitting of a rectangular hyperbola.
Other Interactions with Actin
Concentrations of Arp2/3 complex in the 0.1–0.5 μM range were tested for interaction with actin in the following assays. Sequestration: we measured critical concentration at 24°C in 50 mM KCl, 2 mM MgCl2, 1 mM EGTA, 0.2 mM ATP, 10 mM imidazole, pH 7.0. Nucleation: we measured the time course of spontaneous polymerization of 0.5–1.0 μM actin by measuring fluorescence of pyrene-labeled actin (Pollard and Cooper, 1986) at 24°C in 50 mM KCl, 2 mM MgCl2, 1 mM EGTA, 0.2 mM ATP, 10 mM imidazole, pH 7.0. Severing: we measured the high shear viscosity of 2 μM actin in an Ostwald-type viscometer (MacLean-Fletcher and Pollard, 1980) at 24°C in 50 mM KCl, 2 mM MgCl2, 1 mM EGTA, 0.2 mM ATP, 10 mM imidazole, pH 7.0.
Antibody Preparation
Two New Zealand white rabbits (Bunnyville Farms, Littletown, PA), JH48 and JH49, were immunized subcutaneously in 10–20 locations with either p35 (JH48) or p40 (JH49). Proteins were purified for injection by gel electrophoresis. Arp2/3 complex was purified by poly-l-proline affinity chromatography (see Fig. 1) and protein subunits were separated by SDSPAGE. The gel was stained with 0.2% Coomassie blue, 0.1% SDS, 192 mM glycine, 25 mM Tris base, pH 8.3, and destained in distilled, deionized water. Gel slices were excised, frozen in liquid nitrogen, and pulverized with a mortar and pestle. The powder containing ∼50 μg protein was added to 0.5 ml Freund's complete adjuvant and emulsified with a probe sonicator (Branson, VWR, Bridgeport, NJ). This mixture was injected subcutaneously at multiple sites after a preimmune bleed. 6 wk later, the rabbit was boosted with a similar emulsion made with Freund's incomplete adjuvant. Immune serum from each rabbit reacted with multiple bands on immunoblots of whole cell lysates but with only a single band on immunoblots of purified Arp2/3 complex.
Monospecific rabbit antibodies against Acanthamoeba Arp2 and Arp3 were prepared by Kelleher et al. (1995). Mouse mAbs that react with ameba actin were provided by Dr. James Lessard of the University of Cincinnati (Cincinnati, OH) (Lessard, 1988).
Immunofluorescence Microscopy
Antibodies used for immunofluorescence studies were affinity purified by incubation with p35 and p40 on immunoblot strips (Pollard, 1984). Cy-2 labeled goat anti–mouse and Cy-3 labeled goat anti–rabbit secondary antibodies were obtained from Biological Detection Incorporated (Pittsburgh, PA). BODIPY-labeled phalloidin was obtained from Molecular Probes (Eugene, OR). Micrographs were made using a microscope (model Orthoplan; Leitz, Inc., Rockleigh, NJ) with a 1.4 N.A. Planapo 63 × oil immersion objective (Carl Zeiss, Inc., Thornwood, NY). Confocal images were made by an argon ion laser confocal system (model MRC-600; BioRad Labs, Hercules, CA) attached to a microscope (model Optiphot; Nikon Inc., Melville, NY).
Chemical Cross-linking
Chemical cross-linkers, disuccinimidyl glutarate (DSG), disuccinimidyl tartarate (DST), dithiobis(succinimidyl propionate) (DSP), bismaleimidohexane (BMH), and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC), along with N-hydroxysuccinimide (NHS) were obtained from Pierce (Rockford, IL). Stock solutions of cross-linkers (10×) were made fresh in dry DMSO immediately before use. Final concentration of DMSO in all reactions was ⩽10%. All cross-linking reactions were carried out for 1 h at room temperature.
Analytical Ultracentrifugation
Experiments were carried out at 20–23°C in an analytical ultracentrifuge model E or Optima series XLA (Beckman Instrs., Fullerton, CA) equipped with a real-time, video-based data acquisition system and Rayleigh optics (Liu and Stafford, 1992; Stafford et al., 1995). Data collection and analysis, including determination of diffusion coefficient and molecular weight from sedimentation velocity data, were carried out as described previously (Stafford, 1992, 1994). Analytical ultracentrifugation was carried out in one of two buffers: 100 mM NaCl, 100 mM glycine, 10 mM Tris, pH 7.0, 0.2 mM MgCl2, 0.2 mM ATP, 0.1 mM EGTA (buffer B) or in 20 mM NaCl, 10 mM imidazole, pH 7.0, 0.2 mM MgCl2, 0.2 mM ATP, 0.1 mM EGTA. Amino acid analysis was performed by Boston Analytical Biotechnology (Boston, MA). The partial specific volume of 0.74 cm3/gm and hydration of 0.32 g/g used in analysis of ultracentrifugation data were calculated from the mass averaged contributions of individual amino acids (Kuntz and Kauzmann, 1974; Perkins, 1986).
Rotary Shadowing
Eight rotary-shadowing runs were performed on purified Arp2/3 complex obtained from three different protein preparations. Purified Arp2/3 complex in buffer C was mixed with an equal volume of glycerol and then immediately sprayed by a nebulizer (Fowler and Erickson, 1979; Tyler and Branton, 1980; Fowler and Aebi, 1983) onto the clean surface of a freshly cleaved mica chip. Samples were then dried under vacuum at 1–10 × 10−7 torr in a (model BAF 400 D; Balzers, Fürstentum, Liectenstein) vacuum evaporation unit. Time between dilution and spraying was typically <3 min. Dried specimens were rotary-shadowed with platinum at an angle of 5 or 6° and stabilized with carbon. The carbon-platinum replicas were then floated off the mica surface in distilled water containing approximately 1 part per 10,000 photoflo (Eastman Kodak Company, Rochester, NY) to reduce surface tension and picked up from below on 200- or 300-mesh electron microscope grids (Ernest Fullam Inc., Latham, NY). Electron micrographs were made using a JEM 100CX (JEOL U.S.A., Inc., Peabody, MA) at a nominal magnification of 83,000× or an EM10A (Carl Zeiss, Inc.) at a nominal magnification of 80,000×. Actual magnification on both microscopes was determined from electron micrographs of negatively stained tropomyosin paracrystals taken at the same settings.
Negative Staining
Negative staining of Acanthamoeba actin filaments in the presence and absence of Arp2/3 complex was carried out according to the method of Aebi et al. (1981). Briefly, carbon-coated electron microscope grids made hydrophilic by glow discharge for 20 s in a reduced atmosphere of air (Aebi and Pollard, 1987) were incubated for 30 s on drops containing ∼0.1 mg/ml protein. Grids were then stained for a total of 30 s by incubation on five successive drops of 1% uranyl formate. Uranyl formate was made fresh before each use and protected from light. Electron micrographs were made on a microscope (model 100 CX; JEOL U.S.A., Inc.) at a nominal magnification of 46,000×.
Results
Purification of the Arp2/3 Complex
We purified Arp2/3 complex in two steps (Fig. 1 A). Most of the Arp2 and Arp3 in ameba extracts is soluble (Kelleher et al., 1995) and voids along with profilin when passed through a DEAE-cellulose anion-exchange column (Machesky et al., 1994). This step removes the majority of cellular protein from the extract, including all of the actin. When the DEAE flow-through fraction passes through a poly-l-proline–Sepharose column, all of the profilin (Kaiser et al., 1989) and about 10% of the Arp2/3 complex bind. We presume that the bound fraction of the complex is retained by profilin and is not bound directly to the column since profilin coupled directly to Sepharose binds the complex (Machesky et al., 1994) and purified complex does not bind poly-l-proline Sepharose (data not shown). The remainder of the complex that is not retained on the poly-l-proline column can be purified by conventional chromatography (Kelleher, J., R.D. Mullins, V.K. Vinson, and T.D. Pollard, in manuscript in preparation). Elution of the poly-l-proline column with 400 mM MgCl2 releases purified Arp2/3 complex (Fig. 1 B). Subsequent elution with 8 M urea releases pure profilin. In a previous study (Machesky et al., 1994), four polypeptides from the complex bound to immobilized profilin and six polypeptides copurified by poly-l-proline affinity chromatography. In the present study, we report that all seven members of the Arp2/3 complex previously purified by ion-exchange chromatography also copurify by poly-l-proline affinity chromatography (Fig. 1 B). Arp2/3 complex purified in this manner is entirely free of contaminating actin and profilin as judged by immunoblotting with monoclonal antibodies.
Composition and Stoichiometry.
To determine if the seven polypeptides purified by poly-l-proline affinity chromatography formed a single homogeneous complex, we chromatographed purified Arp2/3 complex on size-exclusion columns. Sephacryl S300 and Superdex S200 columns equilibrated with either B buffer or C buffer (see Materials and Methods) were calibrated with size standards, including catalase (Rs = 5.2 nm) and mouse IgG (Rs = 5.8 nm), by the method of Ackers (1967). On both columns, all seven polypeptides of the purified Arp2/3 complex eluted together as a single symmetric peak with a Stokes' radius of 5.3 ± 0.1 nm (Fig. 2; n = 4, all values are reported ± standard deviation). In addition, monospecific antibodies against Arp2, p40, and p35 coimmunoprecipitated all seven members of the complex from whole ameba cell extracts (data not shown).
Figure 2.

Analytical gel filtration of Arp2/3 complex. All seven polypeptides of the purified Arp2/3 complex migrate as a single peak with a Stokes' radius of 5.3 nm. Conditions: 200 μl of purified Arp2/3 complex at 0.5 mg/ml was run on a 1 × 55-cm column of Sephacryl S300 equilibrated with 100 mM NaCl, 100 mM glycine, 0.2 mM MgCl2, 0.1 mM EGTA, 0.1 mM ATP, 1 mM DTT, 10 mM Tris-HCl, pH 7.0. Fractions of 0.4 ml were collected and protein concentration measured by the Bradford (1976) assay. Two column runs were averaged to produce the trace. Fractions from one run were precipitated with methanol/chloroform, and proteins were separated by SDS-PAGE and stained with Coomassie brilliant blue (inset).
To determine subunit stoichiometry, we separated the components of purified Arp2/3 complex by gel electrophoresis in SDS and quantitated proteins by two-dimensional densitometry of Coomassie-stained gels. Assuming equal dye binding, the four highest molecular weight components—Arp3, Arp2, p40, and p35—are present in equimolar proportions (Table I) as reported in preliminary studies (Machesky et al., 1994). In addition, p14 also appears to be 1:1 with these components. Since p18 and p19 are not resolved completely on our gels, we integrated the two together. The average stoichiometry of p18 plus p19 is 1.6 (Table I), so the stoichiometry of each subunit is ∼0.8. This slightly low value may be due to differences in dye binding since Coomassie staining of p18 is consistently darker than p19 (Fig. 1 B).
Table I.
Stoichiometry of Arp2/3 Complex Subunits
| Subunit | 1 | 2 | 3 | 4 | 5 | Mean ± SD | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Arp3 | 0.96 | 0.99 | 0.91 | 1.08 | 1.03 | 0.99 ± 0.06 | ||||||
| Arp2 | 1.03 | 1.10 | 0.90 | 1.07 | 0.93 | 1.00 ± 0.09 | ||||||
| p40 | 0.92 | 0.99 | 0.96 | 0.93 | 1.00 | 0.96 ± 0.03 | ||||||
| p35 | 1.09 | 0.96 | 1.23 | 0.92 | 1.04 | 1.05 ± 0.12 | ||||||
| p19 + p18 | 0.72 | 1.19 | 2.23 | 1.89 | 2.15 | 1.63 ± 0.66 | ||||||
| p14 | ND | ND | 1.07 | 0.74 | 1.11 | 0.97 ± 0.20 |
Five different preparations of Arp2/3 complex (samples 1–5) were analyzed by SDSPAGE. Gels were stained with Coomassie brilliant blue, densitometrically scanned in two dimensions, and digitized. Intensities were integrated over each band using the image analysis program Collage (Fotodyne, New Berlin, WI). Integrated values were divided by the apparent molecular weight of each subunit. For each of the five scans, the values obtained for Arp3, Arp2, p40, and p35 were averaged; this number was used to normalize the values for all six bands.
Physical Properties of the Arp2/3 Complex
Analytical Ultracentrifugation.
At concentrations above 70 nM, the Arp2/3 complex sediments in B buffer as a single homogeneous peak at 8.7 ± 0.2 S (n = 6). Fig. 3 shows the time-independent distributions of sedimentation coefficients in purified Arp2/3 samples at concentrations between 0.07 and 1.9 μM. When normalized by dilution factor, these distributions are identical at the highest concentrations. Dilution to 70 nM or below causes the distribution of sedimentation coefficients to broaden and shift to lower values, consistent with dissociation of one or more subunits (Fig. 3, inset). By fitting sedimentation velocity profiles collected at concentrations above 100 nM to single Gaussian peaks (Stafford, 1992, 1994), we determined the diffusion coefficient of the complex to be 3.92 cm2s−1. From the sedimentation and diffusion coefficients obtained by sedimentation velocity experiments, we calculated a molecular mass of 197 ± 10 kD for the Arp2/3 complex. The hydrodynamic parameters measured by ultracentrifugation predict a Stokes' radius of 5.4 ± 0.3 nm, which is in good agreement with the 5.3 nm value determined by gel filtration.
Figure 3.

Plots of the distribution of sedimentation coefficients of the Arp2/3 complex determined at various protein concentrations by sedimentation velocity ultracentrifugation. Conditions: The Arp2/3 complex was centrifuged in a Beckman model E centrifuge, AN-F Ti rotor at 40,000 rpm, 20°C, in buffer containing 100 mM NaCl, 100 mM glycine, 0.2 mM MgCl2, 0.1 mM EGTA, 0.1 mM ATP, 1 mM DTT, 10 mM Tris-HCl, pH 7.0. The samples were diluted by successive factors of three, giving a range of concentrations from 1.9 to 0.07 μM. (Inset) Plot of peak S value as a function of protein concentration. The sedimentation coefficient is stable over a wide range of concentrations but shifts to lower values below 70 nM.
Electron Microscopy.
We studied the ultrastructure of the Arp2/3 complex by rotary replicating it with platinum and visualizing it by electron microscopy (Fig. 4). In typical fields, the Arp2/3 complex appears globular and slightly asymmetric with an average aspect ratio of 1.2 ± 0.1 (n = 24). The maximum dimension of the shadowed complex is 19.3 ± 1.5 nm and the minimum dimension is 16.0 ± 1.0 nm (n = 24). When actin filaments are included with the complex, their measured diameter is 16 nm, so we estimate that the shadowing procedure adds ∼3 nm of platinum to each side of a shadowed particle. Correcting for this coating, the average outer dimensions of the Arp2/3 complex are 13.3 × 10.0 nm. Most of the replicated complex molecules are bilobed in appearance with a cleft that divides the molecule into two approximately equal halves. In fields containing multiple molecules of Arp2/3 complex, these clefts are randomly oriented, suggesting they are not systematic shadowing artifacts (Fig. 4 B). Fig. 5 shows representative morphologies of rotary-shadowed molecules. Half of the molecules (51%, n = 251 molecules from eight fields and two separate protein preparations) appear compact and globular, and only slightly bilobed (Fig. 5 A). The other half (49%) exhibit a more clearly defined cleft. Of the latter half, 90% have a narrow central cleft extending ∼75% along the length of the molecule, leaving the two halves of the molecule closely opposed (Fig. 5 B). The remaining 10% have a distinct horseshoe shape with a prominent central cleft separating the halves of the molecule (Fig. 5 C).
Figure 4.

Electron microscopy of purified Arp2/3 complex prepared by rotary replication. Conditions: Arp2/3 complex at 0.5 mg/ml in 150 mM NaCl, 0.2 mM MgCl2, 0.1 mM EGTA, 0.1 mM ATP, 1 mM DTT, 10 mM imidazole, pH 7.4, was mixed 1:1 with glycerol and sprayed onto freshly cleaved mica chips. Samples were vacuum-dried and rotary-replicated with platinum at 6° and carbon-coated at 60°. (A) Field of particles. (B) Higher magnification view of three molecules showing distinct bilobed structure.
Figure 5.
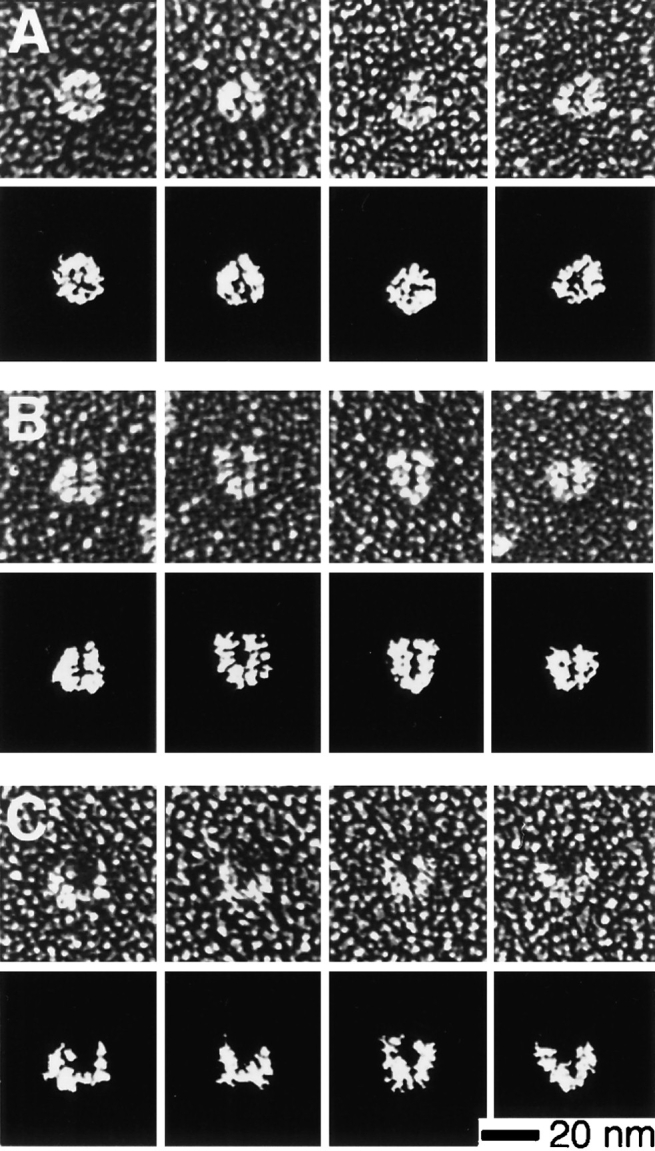
Gallery of electron micrographs and camera lucida interpretations of purified Arp2/3 complex particles prepared by rotary replication. Conditions: See Fig. 4. (A) Approximately half (51%, n = 251) of the molecules surveyed displayed a compact, globular morphology with only a slightly bilobed appearance. (B) Nearly half of the molecules (44%) displayed a distinct bilobed morphology with a clearly defined central cleft. (C) A smaller number of molecules (5%) had a splayed, horseshoe shape with a large central cleft.
Determination of Nearest-Neighbor Subunits in the Arp2/3 Complex by Chemical Cross-linking
We used a battery of chemical cross-linkers with various spacer arm lengths and chemical reactivities to probe subunit topology of the Arp2/3 complex. We used low protein concentrations (1–2 μM) to minimize intermolecular crosslinking. We analyzed reaction products by SDS-PAGE and immunoblotting with subunit-specific antibodies. Five different cross-linkers produced heterodimers of subunits from the complex.
The sulfhydryl-reactive cross-linker BMH cross-links Arp2 to p40. Incubation of Arp2/3 complex with BMH generates a new 83-kD band and significantly reduces the amount of Arp2 and p40 visible on Coomassie-stained polyacrylamide gels (Fig. 6). Of the four monospecific antibodies against complex members, only those raised against Arp2 and p40 react with this cross-linked band. The spacer arm length of BMH is relatively long (1.9 nm), so, to control for intercomplex cross-linking, we ran control and cross-linked samples of Arp2/3 complex over a Superdex S200 size-exclusion column. Both samples eluted with the same partition coefficient, well within the fractionation range of the column, indicating no significant dimerization of complex in the cross-linked sample. By SDS-PAGE, the peak column fractions contained the 83-kD cross-linked product but no detectable, uncross-linked Arp2 or p40 (data not shown).
Figure 6.

Chemical cross-linking of subunits in the Arp2/3 complex with the 1.61 nm, sulfhydryl-reactive cross-linker BMH. SDS-PAGE of reaction products. Conditions: Cross-linking was performed for 1 h at 24°C with 0.3 mg/ml purified Arp2/3 complex, 1.5 mM BMH, 10% DMSO, 150 mM NaCl, 0.2 mM MgCl2, 10 mM imidazole, pH 7.5. Reaction was quenched with an equal volume of 1 M Tris-HCl, pH 8.0. (Left) Control samples. (Right) BMH–cross-linked samples. C, Coomassie-stained polyacrylamide gel; p40, immunoblot probed with anti-p40; Arp2, immunoblot probed with anti-Arp2; *, cross-linked product. Arp2 and p40 are specifically cross-linked in high yield, forming a band of ∼85 kD recognized by both antibodies.
The amine-reactive, oxidizable cross-linker DST crosslinks Arp2 to p35 and Arp3 to p18 or p19. These reaction products appear as two bands at 95 and 67 kD (Fig. 7 A). The larger product reacts with antibodies to Arp2 and p35 (Fig. 7 B). To determine if one or more of the low molecular weight components of the complex was also involved, we cut the cross-linked band out of a wet gel, extracted the protein, and incubated it with sodium periodate to oxidize DST cross-links. When analyzed by SDS-PAGE, the oxidized sample contained bands corresponding to Arp2, p35, as well as two bands that appear to be breakdown products of p35. None of the bands, however, correspond to low molecular weight components of the complex (Fig. 7 C). We conclude, therefore, that this product is indeed a heterodimer of Arp2 and p35.
Figure 7.

Chemical cross-linking of the Arp2/3 complex with the 0.64 nm, amine-reactive cross-linker DST. SDS-PAGE of reaction products. Conditions: Cross-linking was performed for 1 h at 24°C with 0.3 μg/ml purified Arp2/3 complex, 3 mM DST, 10% DMSO, 150 mM NaCl, 0.2 mM MgCl2, 10 mM imidazole, pH 7.5. Reaction was quenched with an equal volume of 1 M Tris-HCl, pH 8.0. (A) Coomassie-stained polyacrylamide gel. Lane 1, control; lane 2, DST–cross-linked samples. (B) Immunoblots of crosslinked bands probed with antibodies to Arp3, Arp2, p40, and p35. (C) Cross-linked bands were excised from wet gels, flash frozen, pulverized, and incubated overnight in 100 mM sodium periodate to oxidize DST cross-links. Uncross-linked polypeptides were separated by SDS-PAGE and stained with silver. Positions of the complex subunits from adjacent lanes on the same gel are marked. Arp2 is cross-linked to p35 and Arp3 to either p18 or p19.
Only antibodies against Arp3 react with the smaller DST–cross-linked product (Fig. 7 B), suggesting that DST cross-links Arp3 to a subunit for which we have no specific antibodies. To determine if this is the case, we extracted the cross-linked band from a wet gel, oxidized the crosslink with sodium periodate, and analyzed the products. We recovered two polypeptides, the larger corresponding in size to Arp3, and the smaller to either p18 or p19 (Fig. 7 C). After this harsh treatment, the smaller polypeptide did not run as a sharp band, and it was not possible, based on comparison to native complex in the same gel, to determine precisely the identity of the lower band. We conclude that the smaller DST–cross-linked product is a heterodimer of Arp3 and either p18 or p19.
The amine-reactive cross-linkers DSP and DSG crosslink p19 and p14. Cross-linking of Arp2/3 complex by either of these cross-linkers produces a 29-kD band (Fig. 8). DSP-induced cross-links can be reversed by reduction with β-mercaptoethanol. We excised the 29-kD band from a wet gel, extracted and boiled it in sample buffer containing β-mercaptoethanol, and ran it on a second gel. Bands were detected at 19 and 14 kD, suggesting that the 29-kD product is a heterodimer of p14 and p19.
Figure 8.

Chemical cross-linking of the Arp2/3 complex (lane C) with the amine-reactive cross-linkers DSP and DSG. Conditions: Cross-linking was carried out for 1 h at 24°C with 0.14 mg/ml purified Arp2/3 complex, 1 mM DSP or DSG, 10% DMSO, 150 mM NaCl, 0.2 mM MgCl2, 0.1 mM EGTA, 10 mM imidazole, pH 7.5. Reaction was quenched with an equal volume of 1 M Tris-HCl, pH 8.0, and reaction products separated by SDS-PAGE and stained with Coomassie brilliant blue. The DSP–cross-linked band (*) was excised from a wet gel, flash-frozen, pulverized, and boiled in sample buffer containing β-mercaptoethanol to reduce the DSP cross-link. Uncross-linked polypeptides were separated by SDS-PAGE and stained with silver (Reduced). Positions of the complex subunits run in adjacent lanes on the same gel are marked. The 29-kD cross-linked product produced by DSP is a heterodimer of p19 and p14.
The zero-length heterobifunctional cross-linker EDC, used with NHS, produces several cross-linked products recognized by monospecific antibodies to complex subunits (Fig. 9). Four sets of these bands appear to be heterodimers. Antibodies against Arp2 and p40 both recognize an 85-kD band (Fig. 9 B, lanes 1 and 2). Combined with the BMH cross-linking result from above, this is strong evidence for a direct association of Arp2 with p40.
Figure 9.

Chemical cross-linking of the Arp2/3 complex with the zero-length, heterobifunctional cross-linker EDC. Conditions: Cross-linking was carried out for 1 h at 24°C with 0.3 mg/ml purified Arp2/3 complex, 5 mM EDC, 5 mM NHS, 10% DMSO, 150 mM NaCl, 0.2 mM MgCl2, 0.1 mM EGTA, 10 mM imidazole, pH 7.5. Reaction was quenched with an equal volume of 1 M Tris-HCl, pH 8.0. Reaction products were analyzed by SDSPAGE and immunoblotting. Immunoblots were probed with monospecific antibodies against Arp3, Arp2, p40, and p35 at 1: 12,000 dilution. (A) Control complex. (B) Cross-linked complex. Cross-linking produces four new sets of bands: (i) a band of 85 kD recognized by antibodies against Arp2 and p40, (ii) a band of 75 kD recognized by antibodies against p40 and p35, (iii) a band of 88 kD recognized by antibodies against p35 and Arp3, and (iv) a doublet of bands at ∼66 and 70 kD recognized only by antibodies against Arp3.
EDC/NHS also produces a 75-kD band recognized by monospecific antibodies against p35 and p40 (Fig. 9 B, lanes 2 and 3) and a prominent band at 88 kD recognized by antibodies against Arp3 and p35 (Fig. 9 B, lanes 3 and 4). These results indicate that p35 is in close proximity to p40 and to Arp3. Two additional EDC/NHS-generated bands, at 66 and 70 kD, are recognized by antibodies against Arp3. The relatively small apparent molecular weights of these bands and the fact that they are not recognized by any other monospecific antibodies indicates they are heterodimers of Arp3 cross-linked to small molecular weight members of the complex. The two bands run close together on SDS-PAGE at approximately the sizes expected for heterodimers of Arp3 with p18 and p19. Also, DST cross-links one or both of these subunits to Arp3, suggesting that the two new bands are composed of Arp3 crosslinked to p18 and to p19.
EDC/NHS also produce additional bands with higher apparent molecular weights. These appear to be higher order structures—heterotrimers, heterotetramers, or larger.
Immunofluorescence Localization of p35 and p40 in Acanthamoeba
Dual localization of actin filaments with BODIPY-phalloidin and of p35 or p40 with fluorescent antibodies showed both p35 and p40 throughout the cytoplasm, concentrated around unidentified intracellular vacuoles and enriched along with actin in the cell cortex (Fig. 10). Most of the staining in the cell cortex is associated with actin filament fibrils. Some fibrils were in microspikes projecting from the cell surface (Fig. 10 C″), while others were deeper in the cell cortex (e.g., compare Fig. 10, A and A″). Confocal images of 0.8-μm sections (Fig. 10, E′ and F′) confirmed the dramatic enrichment of p35 and p40 in the cell cortex and their association with radially projecting fibrils.
Figure 10.


Immunofluorescence localization of p35 and p40 in Acanthamoeba castellani. Cells fixed with 1% formaldehyde in 97.5% ice-cold methanol were stained with affinity-purified rabbit antibodies against either p35 (A and B) or p40 (C and D). (A–D) Phase contrast microscopy. (A″–D″) Localization of p35 and p40 detected with cy3-conjugated goat anti–rabbit secondary antibodies. (A′–D′) Actin filament localization with BODIPY-conjugated phalloidin. Both p35 and p40 were present throughout the cytoplasm and enriched in the cell cortex especially in fibrous structures within microspikes (C″) or entirely within the cell (A″). (E′and F′) Confocal images of 0.8 μm sections of fixed amebas stained with antibodies against p40 (E′ and F′). (E and F) Corresponding phase images. The confocal images emphasize the enrichment of p40 in regions of the cell cortex and the fibrous character of the staining pattern.
Interaction of the Arp2/3 Complex with Actin
Purified Arp2/3 complex binds to rabbit skeletal muscle actin filaments in a pelleting assay but has no detectable activity in assays for actin monomer sequestration, nucleation of actin filament assembly, or severing actin filaments (see Materials and Methods). All seven bands of the complex pellet with actin filaments, regardless of whether the actin is polymerized in the presence of complex or polymerized before it is added to complex (Fig. 11), indicating that copelleting is not due to copolymerization of Arp2 and/or Arp3 with actin. The dependence of the pelleting of p14, p18, p19, and p35 on actin concentration fit a hyperbolic curve yielding an apparent K d of 2.3 ± 0.3 μM (R2 = 0.975). The other bands behaved similarly but were not resolved well enough from actin for accurate quantitation.
Figure 11.

Pelleting of Arp2/3 complex with actin filaments. Conditions: 50 mM KCl, 1 mM MgCl2, 1 mM EGTA, 10 mM imidazole, pH 7.0. A stock solution of 25 μM polymerized actin was diluted to various concentrations with 0.25 μM purified Arp2/3 complex. After 15 min at room temperature, we centrifuged the samples for 30 min at 24°C in a TLA 100 rotor at 200,000 g. Supernatants were analyzed by SDS-PAGE for depletion of complex subunits. Coomassie brilliant blue–stained gels were digitized with a Microtek ScanMaker III flat-bed scanner, and densities of gel bands were integrated by computer. Data shown are for depletion of p18+p19 and were fit to a rectangular hyperbola yielding an apparent K d of 2.3 ± 0.3 μM (R2 = 0.975).
We did not rigorously determine the number of Arp2/3 complex–binding sites on an actin filament, but determined that 1 μM filamentous actin bound 0.45 ± 0.04 μM of a 2.6 μM solution of Arp2/3 complex (n = 6 bands analyzed from two copelleting experiments, data not shown). This is in good agreement with the value of 0.48 μM predicted from a model in which each actin monomer in the filament binds one molecule of Arp2/3 complex with a K d of 2.3 μM. We conclude that the Arp2/3 complex binds to the sides of actin filaments at a stoichiometry of one actin monomer per Arp2/3-binding site.
By electron microscopy of negatively stained samples, Arp2/3 complex binds to the sides of actin filaments. At low concentrations of actin (100 nM), Arp2/3 complex decorates Acanthamoeba actin filaments along their entire lengths (Fig. 12). The diameter of decorated filaments is 24 ± 5 nm (n = 11). The pattern of decoration was irregular for most filaments observed (Fig. 12 B) but occasionally had a periodicity of ∼36 nm or one-half the pitch of the two-start helix in the actin filament structure.
Figure 12.

Electron micrographs of single, negatively stained actin filaments in the absence (A) and presence (B) of Arp2/3 complex. Conditions: 20 μM actin was polymerized at 24°C for 30 min in 50 mM KCl, 1 mM MgCl2, 0.2 mM ATP, 1 mM EGTA, 10 mM imidazole, pH 7.0, and then diluted to 0.3 μM in the same buffer or buffer containing 2.3 μM Arp2/3 complex. Lightly glow-discharged, carboncoated grids were incubated on a drop of this solution and then stained with 1% uranyl formate according to the method of Aebi and Pollard (1987). The diameter of actin filaments decorated with Arp2/3 complex is 24 ± 3 nm (n = 15), compared to 7.5 ± 0.8 nm (n = 15) for actin filaments alone.
To determine which subunits of the Arp2/3 complex mediate binding to actin filaments, we cross-linked Arp2/3 complex to Acanthamoeba actin filaments with the zerolength chemical cross-linker EDC. We cross-linked Arp2/3 complex alone, actin filaments alone, and a mixture of Arp2/3 complex and actin filaments. In the Arp2/3 and actin mixture, monoclonal antiactin antibodies recognized several new bands not present when actin was cross-linked alone. Three prominent new bands at 93, 79, and 59 kD (Fig. 13 A) appeared to be actin monomers cross-linked to single subunits of the Arp2/3 complex. Antibodies against Arp3 also recognized a 93-kD band not present when Arp2/3 complex was cross-linked in the absence of actin (Fig. 13 B, lanes 3–5), and antibodies against p35 recognized a 79-kD band not present when Arp2/3 complex alone was cross-linked (Fig. 13 B, lanes 1–3). These results indicate a direct association between Arp3 and actin and between p35 and actin. The actin-containing, cross-linked band at 59 kD is not recognized by monospecific antibodies against Arp3, Arp2, p40, or p35, indicating that it is an actin monomer cross-linked to one of the small molecular weight subunits of the complex, p19, p18, or p14.
Figure 13.
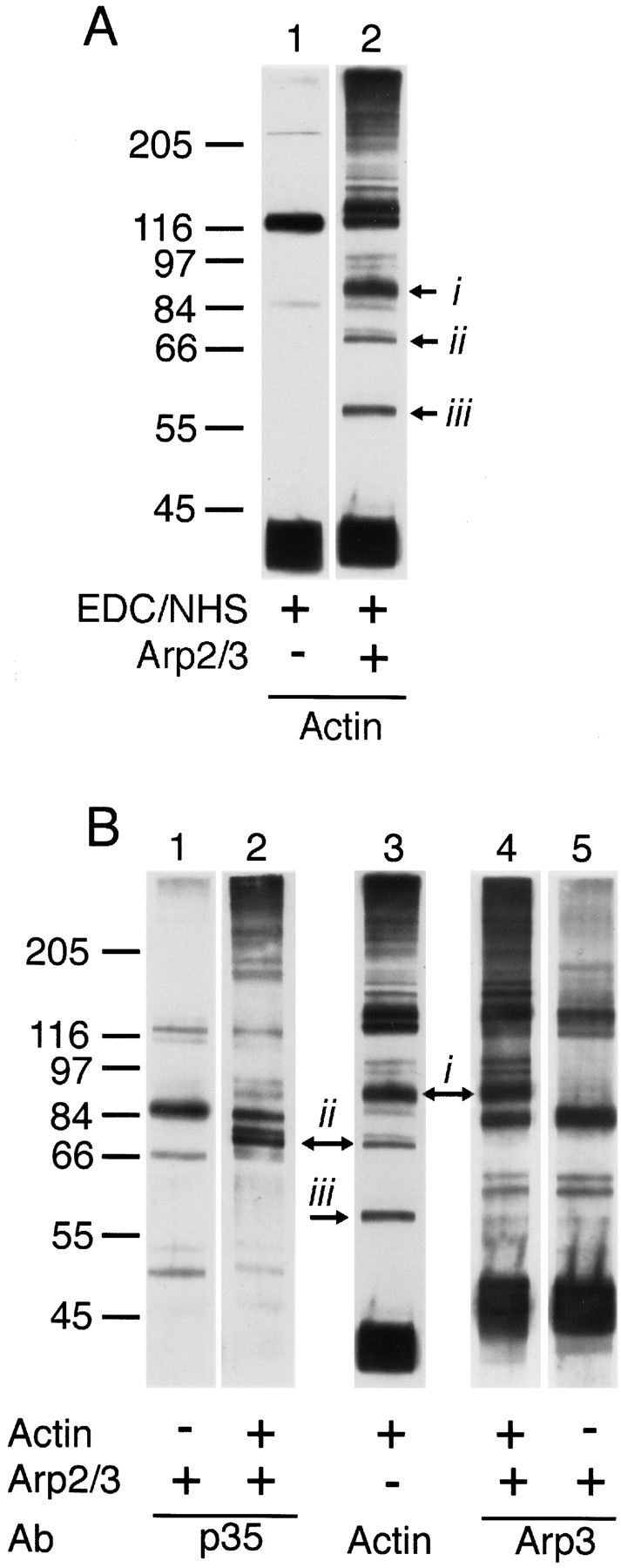
EDC cross-links actin to Arp3, to p35, and to one of the low molecular weight subunits of the Arp2/3 complex. Conditions: Cross-linking was carried out for 1 h at 24°C with 2 μM purified Arp2/3 complex, 4 μM filamentous actin, 5 mM EDC, 5 mM NHS, 10% DMSO, 50 mM KCl, 0.2 mM MgCl2, 0.1 mM EGTA, 10 mM imidazole, pH 7.5. Reaction was quenched with an equal volume of 1 M Tris-HCl, pH 8.0, and the products analyzed by SDS-PAGE and immunoblotting. Immunoblots were probed with monospecific antibodies against Arp3, p35, and actin at dilutions of 1:12,000, 1:6,700, and 1:10,000, respectively. Purified Arp2/3 complex and Acanthamoeba actin were incubated with EDC and NHS, and the reactions were quenched. (A) Immunoblots of EDC/NHS-cross-linked samples probed with monoclonal antiactin antibodies. Lane 1, EDC-treated actin filaments; lane 2, EDC-treated mixture of actin filaments and Arp2/3 complex showing three prominent new bands at (i) 97, (ii) 79, and (iii) 59 kD. (B) Immunoblots of EDC/NHS-cross-linked samples probed with antibodies against p35 (lanes 1 and 2), actin (lane 3), or Arp3 (lanes 4 and 5). The cross-linking reactions contained Arp2/3 complex, actin filaments, or both as indicated with +. Arrows denote new bands recognized by the anticomplex antibodies that correspond to new bands recognized by antiactin antibodies.
Interaction of Arp2/3 Complex with Profilin
The Arp2/3 complex has previously been shown to bind immobilized profilin (Machesky et al., 1994). The binding, however, appears to be rather weak. Profilin does not coimmunoprecipitate out of cell extracts with any of the four monospecific complex antibodies (data not shown). The amine-reactive cross-linkers DST and DSG, under conditions in which they cross-linked actin to profilin, did not cross-link profilin to any complex subunit (data not shown). In addition, profilin added to mixtures of actin filaments, and Arp2/3 complex did not measurably copellet under a 30-min spin at 200,000 g nor did it inhibit copelleting of Arp2/3 complex (data not shown). Fluorescence anisotropy experiments with rhodamine-labeled Acanthamoeba profilin-II suggest that the affinity of Arp2/3 complex for profilin is in the low micromolar range (Kelleher, J.F., R.D. Mullins, V.K. Vinson, and T.D. Pollard, manuscript in preparation).
Discussion
Subunit Composition of the Arp2/3 Complex
Machesky et al. (1994) originally purified four proteins— Arp3, Arp2, p40, and p35—by profilin affinity chromatography. Purification by conventional ion exchange chromatography, using anti-Arp3 antibodies as an assay, yielded the same set of proteins along with three additional polypeptides, p19, p18, and p14. Affinity chromatography on poly-l-proline yielded the original four proteins and variable amounts of p19 and p14. The present study shows that affinity chromatography on poly-l-proline can be used to purify all seven polypeptides. The two Arps, p40, p35, and p14 are present in equimolar ratios judging from Coomassie blue–stained gels. The stoichiometries of p19 and p18 are less certain. The staining of p19 is consistently lighter than p18 so either (a) p19 does not run at its true molecular weight on SDS-PAGE, (b) p19 does not bind dye at the ratio of other complex members, or (c) p19 is not present at 1:1 stoichiometry. The fact that p19 is never absent from a preparation of Arp2/3 complex and that the complex runs as a sharp, symmetrical peak on gel filtration columns argues against c and indicates that p19 is indeed present at 1:1 stoichiometry.
Hydrodynamic Properties of the Arp2/3 Complex
The Arp2/3 complex, purified by affinity chromatography on poly-l-proline, sediments as a homogeneous particle at 8.7 S and runs as a single peak at 5.3-nm Stokes' radius on calibrated gel filtration columns. The molecular mass determined by analytical ultracentrifugation is 197 kD, consistent with the presence of no more than one copy each of the seven polypeptides in the complex. The sedimentation coefficient is constant over a large concentration range, but at low concentration (⩽70 nM), the distribution of the sedimentation coefficients broadens and its peak shifts to lower values. We interpret this shift as a partial dissociation of the complex and estimate that the K d of the most weakly bound subunit(s) of the complex is on the order of 70 nM. The cytoplasmic concentrations of Arp2 and Arp3 are ⩾2 μM (Machesky et al., 1994; Kelleher et al., 1995), so the particle should be stable in the cytoplasm.
Hydrodynamic data indicate that the Arp2/3 complex is not spherical. Two models are typically used to approximate nonspherical particles in hydrodynamic studies: a cigar-shaped, prolate ellipsoid of revolution and a discshaped, oblate ellipsoid of revolution. Our physical data are consistent with either a prolate ellipsoid of revolution with an axial ratio of 4:1, maximum length of 19.5 nm, and diameter 4.9 nm, or an oblate ellipsoid of revolution with an axial ratio of 1:4.3, thickness of 2.9 nm, and diameter of 12.6 nm. Electron microscopy rules out an elongated morphology for the complex and suggests a more compact, horseshoe shape with outer dimensions approximating the diameter of the oblate ellipsoid model.
To determine if such a shape is consistent with our physical characterization of the Arp2/3 complex, we constructed hydrodynamic models of various shapes and calculated their sedimentation coefficients (Fig. 14). The models all consisted of five identical, rigidly coupled spheres 4.52 nm in diameter. A partial specific volume of 0.74 cm3/gm gives each model a molecular mass of 197 kD. Using a hydration value of 0.323 g/g, calculated from amino acid analysis (see Materials and Methods), we calculated frictional and sedimentation coefficients according to the method of Riseman and Kirkwood (1956). We considered four general models (Fig. 14): (a) a globular, double-tetrahedral arrangement; (b) an extended linear model; (c) a planar, open horseshoe structure; and (d) a tightly closed, horseshoe arrangement.
Figure 14.

Hydrodynamic bead models of the Arp2/3 complex. Models assume five rigidly linked, identical 4.52-nm-diam spheres, each with a partial specific volume of 0.74 cm3/gm. Each model, thus, has a molecular mass of 197 kD. The partial specific volume and hydration (0.323 g/g) used in all calculations were based on amino acid analysis. Hydrodynamic parameters for each configuration were calculated by the method of Riseman and Kirkwood (1956). (A) Globular, double-tetrahedral arrangement, 10.3 S. (B) Linear model, 8.1 S. (C) Open horseshoe-shaped model, 8.7 S. (D) Compact horseshoe-shaped model, 9.5 S. The best agreement with experimental data was given by model C.
The calculated sedimentation coefficients of the globular and extended models were significantly higher (10.3 S) and lower (8.1 S), respectively, than the values measured for the Arp2/3 complex. Of the four models, we obtained best agreement with our experimental data from the open horseshoe structure (Fig. 14 C). This arrangement is planar and symmetric and can be defined by two angles: α, the angle that a line from the center of the middle sphere to the center of an adjacent sphere makes with the horizontal, and β, the angle that a line from the center of an outermost sphere to the center of the adjacent sphere makes with the horizontal (see angles illustrated in Fig. 14, C and D). To obtain a general horseshoe shape, we require that α ⩽ β ⩽ 90°. We determined all combinations of α and β, within these constraints, that give our model exact agreement with measured hydrodynamic data. The limiting cases of these models are α = 30°, β = 90° and α = 54°, β = 54°. In the example shown in Fig. 14 C, α = 45°, β = 76°, and the model's outer dimensions are 12 × 13 nm. This structure is slightly more splayed than most of the Arp2/3 complex molecules we observed but agrees with the approximate shape and dimensions determined from electron microscopy. The models used in our calculations were based on the hydrodynamic properties of ideal spheres, but irregularities in the surfaces of proteins (rugosity) contribute to hydrodynamic drag and reduce the sedimentation coefficient. Including a contribution from rugosity in our hydrodynamic calculations would make a more tightly closed horseshoe model, with morphology somewhere between the models of Fig. 14, C and D, consistent with the measured sedimentation coefficient.
Subunit Topology
Chemical cross-linking produced seven heterodimers of Arp2/3 complex subunits. Based on these cross-links, we propose a nearest-neighbor subunit topology (Fig. 15). The four largest molecular weight subunits, Arp3, Arp2, p40, and p35, are in close proximity. Zero-length crosslinks can be made between Arp3 and p35, p35 and p40, and Arp2 and p40. In addition, a 0.64-nm cross-link can be made between p35 and Arp2, suggesting a relatively compact arrangement of these subunits (Fig. 15). Based on electron microscopy and analytical ultracentrifugation, this arrangement appears to be roughly planar.
Figure 15.

Chemical crosslinking model of the Arp2/3 complex. Lines represent chemical cross-links made between subunits with various cross-linkers. Line lengths are proportional to cross-linker spacer arms and subunit sizes are proportional to apparent molecular weights. Both subunits denoted with a * and one of the three subunits denoted with a + can be cross-linked to actin by EDC/NHS.
Cross-linking identified p40 as an Arp2-binding protein and p35 as an Arp3-binding protein. The fact that p35 can be cross-linked to both Arps and to p40 suggests that it may function to stabilize or spatially organize the complex. Previous studies have focused on the possibility that Arp2 and Arp3 form a heterodimer analogous to two actin monomers within a filament. We did not find a direct interaction between the two actin related proteins, but the fact that p35 can be cross-linked to each indicates that they may be in close proximity.
Arp2 is efficiently cross-linked to p40 by the sulfhydrylreactive cross-linker BMH as well as by EDC/NHS. The BMH cross-link is quite specific since Arp2 and Arp3 each have eight cysteine residues in similar positions, but BMHcross-links p40 only to Arp2. Structural models of Arp2 (Kelleher et al., 1995) based on the crystal structure of actin (Kabsch et al., 1985) suggest that only three cysteines are available for cross-linking at the surface of the molecule, cys103, cys340, and cys387. Model structures place cys103 and cys387 in a region corresponding to subdomain 1 and cys340 in a region corresponding to subdomain 3 of an actin monomer. All three residues are on the front surface in the standard view of the actin monomer (Kabsch et al., 1985). Therefore, p40 faces the surface of Arp2 that corresponds to the surface of an actin monomer exposed on the outside of an actin filament.
The relative positions of the three smallest molecular weight subunits are more ambiguous. We can cross-link p19 to p14 and we can cross-link two of the three low molecular weight subunits to Arp3. Based on apparent molecular weights, we believe these subunits are p18 and p19. Fig. 15 illustrates our favored arrangement of p18, p19, and p14 within the complex.
Actin Interaction
Here we present the first evidence of direct interaction between the Arp2/3 complex and actin filaments. Biochemical and electron microscopic experiments reveal that the Arp2/3 complex binds to the sides of actin filaments with micromolar affinity. In Acanthamoeba cells, the cytoplasmic concentration of filamentous actin is 100 μM (Pollard and Cooper, 1986) and that of Arp2/3 complex is 2 μM (Kelleher et al., 1995). From these values and the measured K d of 2.3 μM, we estimate that 98% of the complex in Acanthamoeba cells is bound to actin filaments. By immunofluorescence, the complex localizes preferentially to regions of the cell with high actin filament concentration (Machesky et al., 1994; Kelleher et al., 1995 and this study). We also find that both p35 and p40 localize to actin filament fibrils in the cell periphery and to microspikes projecting from the cell surface.
The morphology of the leading edge of motile Acanthamoeba cells is similar to locomoting tissue culture cells that extend a lamellapod, an actin-rich extension of the cell cortex, devoid of cytoplasmic organelles, in which the majority of actin filaments are oriented with their barbed ends facing the cell membrane (Oliver et al., 1978; Small et al., 1978). At the trailing edges of locomoting cells, organelles and vesicles approach more closely to the plasma membrane. Based on these morphological criteria, the cells in Fig. 10, C and F, appear to be moving toward the upper left hand corners of the micrographs. The staining pattern of p40 in both cells is consistent with a localization to actin bundles in the advancing edge of the cell. Further work is required to determine if Arp2/3 complex is directly involved in the process of cell locomotion or lamellapod extension.
Three subunits of the complex—Arp3, p35, and either p19, p18, or p14—were found, by chemical cross-linking, to be closely associated with actin. Sequence analysis of Arp3 suggests that it cannot copolymerize with actin, but that both Arp3 and Arp2 conserve homology to regions of actin that are exposed at the barbed ends of actin filaments. Based on this observation, Kelleher et al. (1995) suggested that Arp2 and Arp3 might form a heterodimer, containing a structural motif similar to the barbed end of an actin filament. Such a structure might regulate actin polymerization in vivo by forming a cryptic nucleus for filament formation. At present, our biochemical data do not support this type of activity for the purified complex, but it is possible that this activity, if it exists, requires posttranslational modifications, a cofactor, or a second messenger. Further experiments are required to address this hypothesis.
Arp2/3 complex binds to the sides of actin filaments. The fact that three subunits of the complex interact with actin suggests that the complex makes multiple contacts with a single filament and/or it cross-links filaments into networks or bundles. Work in progress indicates that the complex organizes actin filaments into bundles in vitro (Kelleher, J.F., R.D. Mullins, V.K. Vinson, and T.D. Pollard, manuscript in preparation), much like the linear structures that we observe in the cortex of fixed cells.
Arp2/3 complex–decorated actin filaments have a diameter of 24 nm, so Arp2/3 complex molecules project a maximum distance of 7 nm from the filament surface. The largest dimension of the complex is ∼13 nm. Therefore, the long axis of the complex must be inclined to within 33° of the filament axis. Also, decoration of actin filaments does not appear as a regular, repeating pattern. This irregularity may be caused by incomplete occupation of Arp2/3binding sites at the concentrations used for electron microscopy or by the presence of multiple actin-binding sites within the complex, each binding with a unique orientation.
The function of the Arp2/3 complex in living cells is still a mystery, but its interaction with profilin, direct binding to actin filaments, and localization to actin filament fibrils in the cell cortex suggest that it plays a role in regulating the actin cytoskeleton.
Acknowledgments
We are grateful to members of the Pollard lab for technical advice and helpful discussions, especially to J. Kelleher for affinity-purifying antibodies, D. Kaiser for assistance with poly-l-proline affinity chromatography, E. DeLaCruz for purifying rabbit skeletal muscle actin, and P. Maupin for assistance with rotary shadowing and electron microscopy.
This work was supported by National Institutes of Health research grant GM-26338 to T.D. Pollard and by a research fellowship from the Jane Coffin Childs Memorial Fund for Medical Research to R.D. Mullins.
Abbreviations used in this paper
- Arps
actin-related proteins
- BMH
bismaleimidohexane
- DSG
disuccinimidyl glutarate
- DSP
dithiobis(succinimidyl propionate)
- DST
disuccinimidyl tartarate
- EDC
1-ethyl-3-(3dimethylaminopropyl)-carbodiimide hydrochloride
- NHS
N-hydroxysuccinimide
Footnotes
Address all correspondence to R. Dyche Mullins, Department of Cell Biology and Anatomy, Johns Hopkins University School of Medicine, 725 N. Wolfe St., Baltimore, Maryland 21205. Tel.: (410) 955-5672. Fax: (410) 955-4129. E-mail: dmullins@welchlink.welch.jhu.edu
References
- Ackers GK. A new calibration procedure for gel filtration columns. J Biol Chem. 1967;242:3237–3238. [Google Scholar]
- Aebi U, Pollard TD. A glow discharge unit to render electron microscope grids and other surfaces hydrophilic. J Electron Micro Tech. 1987;7:29–33. doi: 10.1002/jemt.1060070104. [DOI] [PubMed] [Google Scholar]
- Aebi U, Fowler WE, Isenberg G, Pollard TD, Smith PR. Crystalline actin sheets: their structure and polymorphism. J Cell Biol. 1981;91:340–351. doi: 10.1083/jcb.91.2.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian MK, Feoktistova A, McCollum D, Gould K. Fission yeast Sop2p: a novel and evolutionarily conserved protein that interacts with Arp3p and modulates profilin function. EMBO (Eur Mol Biol Organ) J. 1996;15:6426–6437. [PMC free article] [PubMed] [Google Scholar]
- Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Fowler WE, Aebi U. Preparation of single molecules and supramolecular complexes for high resolution metal shadowing. J Ultrastruct Res. 1983;83:319–334. doi: 10.1016/s0022-5320(83)90139-9. [DOI] [PubMed] [Google Scholar]
- Fowler WE, Erickson HP. The trinodular structure of fibrinogen: confirmation by both shadowing and negative stain electron microscopy. J Mol Biol. 1979;134:241–249. doi: 10.1016/0022-2836(79)90034-2. [DOI] [PubMed] [Google Scholar]
- Frankel S, Mooseker M. The actin related proteins. Curr Opin Cell Biol. 1996;8:30–37. doi: 10.1016/s0955-0674(96)80045-7. [DOI] [PubMed] [Google Scholar]
- Kabsch W, Mannherz HG, Suck D. Three-dimensional structure of the complex of actin and DNase I at 4.5Å resolution. EMBO (Eur Mol Biol Organ) J. 1985;4:2113–2118. doi: 10.1002/j.1460-2075.1985.tb03900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser DA, Goldschmidt-Clermont PJ, Levine BA, Pollard TD. Characterization of renatured profilin purified by urea elution from poly-lproline agarose columns. Cell Motil. 1989;14:251–262. doi: 10.1002/cm.970140211. [DOI] [PubMed] [Google Scholar]
- Kelleher JF, Atkinson SJ, Pollard TD. Sequences, structural models, and cellular localization of the actin-related proteins Arp2 and Arp3 from Acanthamoeba. . J Cell Biol. 1995;131:385–397. doi: 10.1083/jcb.131.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz ID, Kauzmann W. Hydration of proteins and polypeptides. Adv Protein Chem. 1974;28:239–345. doi: 10.1016/s0065-3233(08)60232-6. [DOI] [PubMed] [Google Scholar]
- Lees-Miller JP, Henry G, Helfman DM. Identification of act2, an essential gene in the fission yeast Schizosaccharomyces pombethat encodes a protein related to actin. Proc Natl Acad Sci USA. 1992;89:80–83. doi: 10.1073/pnas.89.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard JL. 2 monoclonal antibodies to actin - one muscle selective and one generally reactive. Cell Motil. 1988;10:349–362. doi: 10.1002/cm.970100302. [DOI] [PubMed] [Google Scholar]
- Liu S, Stafford WF. A real-time video-based Rayleigh optical system for an analytical ultracentrifuge allowing imaging of the entire centrifugal cell. Biophys J. 1992;61:A476. [Google Scholar]
- Machesky LM, Ampe C, Vandekerckhove J, Pollard TD. Purification of a cortical complex containing two unconventional actins from Acanthamoebaby affinity chromatography on profilin-agarose. J Cell Biol. 1994;127:107–115. doi: 10.1083/jcb.127.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean-Fletcher S, Pollard TD. Viscometric analysis of the gelation of Acanthamoebaextracts and purification of two gelation factors. J Cell Biol. 1980;85:414–428. doi: 10.1083/jcb.85.2.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau V, Madania A, Martin RP, Winsor B. The Saccharomyces cerevisiaeactin-related protein Arp2 is involved in the actin cytoskeleton. J Cell Biol. 1996;134:117–132. doi: 10.1083/jcb.134.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RD, Kelleher JF, Pollard TD. Actin' like actin? . Trends Cell Biol. 1996;6:208–212. doi: 10.1016/0962-8924(96)20017-0. [DOI] [PubMed] [Google Scholar]
- Oliver JM, Krawiec JA, Becker EL. The distribution of actin during chemotaxis in rabbit neutrophils. J Reticuloendothel Soc. 1978;24:697–704. [PubMed] [Google Scholar]
- Perkins SJ. Protein volumes and hydration effects. Eur J Biochem. 1986;157:169–180. doi: 10.1111/j.1432-1033.1986.tb09653.x. [DOI] [PubMed] [Google Scholar]
- Pollard TD. Polymerization of ADP-actin. J Cell Biol. 1984;99:769–777. doi: 10.1083/jcb.99.3.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, Cooper JA. Actin and actin-binding proteins. A critical evaluation of mechanisms and functions. Annu Rev Biochem. 1986;55:987–1035. doi: 10.1146/annurev.bi.55.070186.005011. [DOI] [PubMed] [Google Scholar]
- Riseman, J., and J.G. Kirkwood. 1956. The statistical mechanical theory of irreversible processes in solutions of macromolecules. In Rheology: Theory and Applications. J. Firisch, editor. New York, Academic Press. 495–523.
- Schafer DA, Gill SR, Cooper JA, Heuser JE, Schroer TA. Ultrastructural analysis of the dynactin complex: an actin-related protein is a component of a filament that resembles actin. J Cell Biol. 1994;126:403–412. doi: 10.1083/jcb.126.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer TA, Sheetz MP. Two activators of microtubule-based vesicle transport. J Cell Biol. 1991;115:1309–1318. doi: 10.1083/jcb.115.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer TA, Bingham JB, Gill SR. Actin-related protein 1 and cytoplasmic dynein-based motility—what's the connection? . Trends Cell Biol. 1996;6:212–215. doi: 10.1016/0962-8924(96)20014-5. [DOI] [PubMed] [Google Scholar]
- Schwob, E., and R.P. Martin. 1992. New yeast actin-like gene required late in the cell cycle. 355:179–182. [DOI] [PubMed]
- Small JV, Isenberg G, Celis JE. Polarity of actin at the leading edge of cultured cells. Nature (Lond) 1978;272:638–639. doi: 10.1038/272638a0. [DOI] [PubMed] [Google Scholar]
- Spudich JA, Watt S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- Stafford WF. Boundary analysis in sedimentation transport experiments: a procedure for obtaining sedimentation coefficient distributions using the time derivative of the concentration profile. Anal Biochem. 1992;203:295–301. doi: 10.1016/0003-2697(92)90316-y. [DOI] [PubMed] [Google Scholar]
- Stafford WF. Boundary analysis in sedimentation velocity experiments. Methods Enzymol. 1994;240B:478–506. doi: 10.1016/s0076-6879(94)40061-x. [DOI] [PubMed] [Google Scholar]
- Stafford WF, Mabuchi K, Takahashi K, Tao T. Physical characterization of calponin: a circular dichroism, analytical untracentrifuge, and electron microscopy study. J Biol Chem. 1995;270:10576–10579. doi: 10.1074/jbc.270.18.10576. [DOI] [PubMed] [Google Scholar]
- Tyler J, Branton D. Rotary shadowing of extended molecules dried from glycerol. J Ultrastruct Res. 1980;71:95–102. doi: 10.1016/s0022-5320(80)90098-2. [DOI] [PubMed] [Google Scholar]
- Wang Y-L. Exchange of actin subunits at the leading edge of living fibroblasts: possible role of treadmilling. J Cell Biol. 1985;101:597–602. doi: 10.1083/jcb.101.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]