

Abstract
The Saccharomyces cerevisiae mating pheromone a-factor is a prenylated and carboxyl methylated extracellular peptide signaling molecule. Biogenesis of the a-factor precursor proceeds via a distinctive multistep pathway that involves COOH-terminal modification, NH2-terminal proteolysis, and a nonclassical export mechanism. In this study, we examine the formation and fate of a-factor biosynthetic intermediates to more precisely define the events that occur during a-factor biogenesis. We have identified four distinct a-factor biosynthetic intermediates (P0, P1, P2, and M) by metabolic labeling, immunoprecipitation, and SDSPAGE. We determined the biochemical composition of each by defining their NH2-terminal amino acid and COOH-terminal modification status. Unexpectedly, we discovered that not one, but two NH2-terminal cleavage steps occur during the biogenesis of a-factor. In addition, we have shown that COOH-terminal prenylation is required for the NH2-terminal processing of a-factor and that all the prenylated a-factor intermediates (P1, P2, and M) are membrane bound, suggesting that many steps of a-factor biogenesis occur in association with membranes. We also observed that although the biogenesis of a-factor is a rapid process, it is inherently inefficient, perhaps reflecting the potential for regulation. Previous studies have identified gene products that participate in the COOH-terminal modification (Ram1p, Ram2p, Ste14p), NH2-terminal processing (Ste24p, Axl1p), and export (Ste6p) of a-factor. The intermediates defined in the present study are discussed in the context of these biogenesis components to formulate an overall model for the pathway of a-factor biogenesis.
In Saccharomyces cerevisiae, the peptide mating pheromones a-factor and α-factor function to promote conjugation between cells of the opposite mating type, MATa and MATα (Marsh et al., 1991; Sprague and Thorner, 1992). Like the peptide hormones secreted by higher eukaryotes, the yeast mating pheromones are initially synthesized as larger precursors that undergo posttranslational modification and proteolytic processing before their export from the cell. Despite their functional equivalence as signaling molecules, the a-factor and α-factor pheromones are structurally quite dissimilar and exemplify distinct paradigms for biogenesis. The maturation of α-factor is well characterized and involves the “classical” secretory pathway (ER→ Golgi→ secretory vesicles; Julius et al., 1984). Subsequent to its translocation across the ER membrane, the α-factor precursor undergoes signal sequence cleavage, glycosylation, a series of proteolytic processing steps in the lumenal compartments of the secretory pathway, and then exits the cell via exocytosis (Fuller et al., 1986; Sprague and Thorner, 1992). In contrast to our extensive understanding of α-factor maturation, our view of the events involved in a-factor biogenesis is still incomplete. An important difference between the two pheromones is that secretion of a-factor is mediated by a “nonclassical” export mechanism (Kuchler et al., 1989; McGrath and Varshavsky, 1989; Michaelis, 1993). The purpose of the present study is to delineate the steps of a-factor biogenesis that occur before its export, by the identification and characterization of a-factor biosynthetic intermediates.
Mature bioactive a-factor is a prenylated and methylated dodecapeptide, derived by the posttranslational maturation of a precursor encoded by the similar and functionally redundant genes MFA1 and MFA2 (Brake et al., 1985; Michaelis and Herskowitz, 1988). The structures of the precursor and mature forms of a-factor derived from MFA1 are shown in Fig. 1. The a-factor precursor can be subdivided into three functional segments: (a) the mature portion (shaded in Fig. 1), which is ultimately secreted; (b) the NH2-terminal extension; and (c) the COOH-terminal CAAX motif (C is cysteine, A is aliphatic, and X is one of many residues). As shown here, and also suggested by our previous studies, the biogenesis of a-factor occurs by an ordered series of events involving first COOH-terminal CAAX modification, then NH2-terminal processing, and finally export from the cell (He et al., 1991; Michaelis, 1993; Sapperstein et al., 1994).
Figure 1.

Structure of precursor and mature forms of a-factor encoded by MFA1. The a-factor precursor encoded by MFA1 is shown with the NH2-terminal extension, COOH-terminal CAAX motif, and mature portion (shaded gray) indicated. Every fifth residue is numbered. Mature a-factor derived from this precursor is modified on its COOH-terminal cysteine residue by a farnesyl moiety and a carboxyl methyl group, as indicated.
The COOH-terminal maturation of the a-factor precursor is directed by its CAAX sequence. The CAAX motif is present at the COOH terminus of numerous eukaryotic proteins, most notably the Ras proteins, and is known to signal a triplet of posttranslational modifications. These include prenylation of the cysteine residue, proteolysis of the COOH terminal AAX residues (VIA for a-factor), and methylation of the newly exposed cysteine carboxyl group (Clarke, 1992; Zhang and Casey, 1996). The yeast enzymes that mediate the modification of CAAX-terminating proteins are known from genetic and biochemical studies. RAM1 and RAM2 encode the subunits of the cytosolic farnesyltransferase enzyme (Fujiyama et al., 1987; He et al., 1991; Powers et al., 1986; Schafer et al., 1990). An “AAX” endoprotease has been detected as a membrane-associated activity in yeast extracts, although the corresponding gene(s) remains elusive (Ashby et al., 1992; Hrycyna and Clarke, 1992). STE14 encodes the prenylcysteine-dependent carboxyl methyltransferase that mediates methylation, the final step in modification of CAAX proteins; Ste14p is also membrane associated (Hrycyna and Clarke, 1990; Hrycyna et al., 1991; Marr et al., 1990; Sapperstein et al., 1994). In mutants (ram1, ram2, and ste14) defective in CAAX modification, biologically active a-factor is not produced.
The events involved in the NH2-terminal proteolytic processing of the a-factor precursor are less well-defined than those of COOH-terminal maturation. It was recently shown that a protease encoded by the AXL1 gene is required for one step of the NH2-terminal processing of a-factor (Adames et al., 1995). Axl1p belongs to the insulin-degrading enzyme (IDE)1 subfamily of proteases; an AXL1 homologue, Ste23p, was also found to perform a role at least partially redundant to that of Axl1p in a-factor processing (Adames et al., 1995). Recently, we have identified another gene, STE24, whose product participates in the NH2-terminal processing of the a-factor precursor in a manner distinct from Axl1p and Ste23p (Fujimura-Kamada and Michaelis, 1997). Based on a priori inspection of the precursor and mature forms of a-factor (Fig. 1), a single NH2-terminal proteolytic cleavage event (between residues N21 and Y22) might have been predicted; however, we provide evidence in the present study that the proteolytic processing of the NH2terminal extension of the a-factor precursor occurs in two distinct steps.
The final event in a-factor biogenesis is the export of the fully matured pheromone from the cell. The absence of a canonical NH2-terminal signal sequence in the MFA1 and MFA2 sequences, as well as the lack of effect upon a-factor secretion of sec mutants blocked at various steps in the classical secretory pathway, led to the suggestion of a nonclassical export mechanism for a-factor export (McGrath and Varshavsky, 1989; Sterne, 1989). Indeed, a-factor export is now known to be mediated by Ste6p, a member of the ATP-binding cassette (ABC) superfamily of proteins (Kuchler et al., 1989; McGrath and Varshavsky, 1989). ABC proteins carry out the ATP-dependent membrane translocation of a variety of compounds, including small peptides, hydrophobic drugs, and even prenylcysteine derivatives, by an uncharacterized mechanism (Gottesman and Pastan, 1993; Zhang et al., 1994). It is notable that a-factor undergoes COOH-terminal modification and NH2-terminal proteolytic maturation before Ste6p-mediated membrane translocation. This order of events contrasts with those of the biogenesis of the α-factor precursor and other classical secretory substrates, which undergo ER membrane translocation first and are matured only subsequently.
In the present study, we aimed to elucidate the events that occur during a-factor biogenesis, before its export from the cell. Our approach was to identify a-factor biosynthetic intermediates, determine their chemical composition and localization properties, and examine the efficiency of their formation and the effects of an a-factor CAAX mutation on their formation. In addition to identifying the biosynthetic intermediates we expected, which include the unmodified a-factor precursor (P0), the COOHterminally modified a-factor precursor (P1), and mature a-factor (M), we unexpectedly uncovered a novel and unanticipated intermediate. This species, designated P2, is fully COOH-terminally modified and has had only a segment of its NH2-terminal extension proteolytically removed. The existence of the P2 intermediate provides evidence that an additional unpredicted step occurs during the NH2-terminal processing of the a-factor precursor. The biosynthetic intermediates we identify here, considered together with known a-factor biogenesis components, are presented in terms of a comprehensive model for the a-factor biogenesis pathway.
Materials and Methods
Yeast Strains and Media
The yeast strains used in this study are listed in Table I. The MATa a-factor null strains SM1229 (mfa1-Δ1::LEU2 mfa2-Δ1::URA3; Michaelis and Herskowitz, 1988), SM1458 (mfa1-Δ1::LEU2 mfa2-Δ2::lacZ), and SM2331 (mfa1-Δ1 mfa2-Δ1) were used to harbor plasmids carrying either wildtype or mutant MFA1.
Table I.
S. cerevisiae Strains Used in This Study
| Strains | Relevant Genotype | Source | ||
|---|---|---|---|---|
| S288C | MATa gal2 | Mortimer and Johnston, 1986 | ||
| Σ1278B | MATa | Brandriss and Magasanik, 1979 | ||
| SK-1 | MATa ho::LYS2 lys2 ura3 | Alani et al., 1987 | ||
| SM1058* | MATa MFA1 MFA2 trp1 leu2 ura3 his4 can1 | Michaelis and Herskowitz, 1988 | ||
| SM1059‡ | MATα MFA1 MFA2 trp1 leu2 ura3 his4 can1 | Michaelis and Herskowitz, 1988 | ||
| SM1179‡ | MATa MFA1 mfa2-Δ2::lacZ trp1 leu2 ura3 his4 can1 | This study | ||
| SM1196‡ | MATα MFA1 mfa2-Δ2::lacZ trp1 leu2 ura3 his4 can1 | This study | ||
| SM1225‡ | MATa mfa1-Δ1::LEU2 MFA2 trp1 leu2 ura3 his4 can1 | Michaelis and Herskowitz, 1988 | ||
| SM1227‡ | MATa MFA1 mfa2-Δ1::URA3 trp1 leu2 ura3 his4 can1 | Michaelis and Herskowitz, 1988 | ||
| SM1229‡ | MATa mfa1-Δ1::LEU2 mfa2-Δ1::URA3 trp1 leu2 ura3 his4 can1 | Michaelis and Herskowitz, 1988 | ||
| SM1458‡ | MATa mfa1-Δ1::LEU2 mfa2-Δ2::lacZ trp1 leu2 ura3 his4 can1 | This study | ||
| SM1585‡ | MATa mfa1-Δ1::LEU2 mfa2-Δ2::lacZ trp1 leu2 ura3 his4 can1 p[2μ MFA1 URA3] | Transformant of SM1458 with pSM219 | ||
| SM1680‡ | MATa mfa1-Δ1::LEU2 mfa2-Δ2::lacZ trp1 leu2 ura3 his4 can1 p[2μ mfa1-ΔVIA URA3] | Transformant of SM1458 with pSM258 | ||
| SM1682‡ | MATa mfa1-Δ1::LEU2 mfa2-Δ2::lacZ trp1 leu2 ura3 his4 can1 p[2μ mfa1-C33S URA3] | Transformant of SM1458 with pSM257 | ||
| SM1710‡ | MATa mfa1-Δ1::LEU2 mfa2-Δ2::lacZ trp1 leu2 ura3 his4 can1 p[CEN MFA1 URA3] | Transformant of SM1458 with pSM233 | ||
| SM1762‡ | MATa mfa1-Δ1::LEU2 mfa2-Δ2::URA3 trp1 leu2 ura3 his4 can1 p[2μ MFA1 TRP1] | Transformant of SM1229 with pSM463 | ||
| SM1829‡ | MATa mfa1-Δ1::LEU2 mfa2-Δ2::lacZ trp1 leu2 ura3 his4 can1 p[CEN MFA1 TRP1] | Transformant of SM1229 with pSM464 | ||
| SM1865‡ | MATa ram1::URA3 trp1 leu2 ura3 his4 can1 | He et al., 1991 | ||
| SM1932‡ | MATa mfa1-Δ1::LEU2 mfa2-Δ2::lacZ trp1 leu2 ura3 his4 can1 p[2μ mfa1-I23M URA3] | Transformant of SM1458 with pSM490 | ||
| SM2165‡ | MATa MFA1 mfa2-Δ2-Δ1 trp1 leu2 ura3 his4 can1 | This study | ||
| SM2331‡ | MATa mfa1-Δ1 mfa2-Δ1 trp1 leu2 ura3 his4 can1 | This study | ||
| SM2891‡ | MATa mfa1-Δ1 mfa2-Δ1 trp1 leu2 ura3 his4 can1 p[CEN MFA1 URA1] | Transformant of SM2331 with pSM233 | ||
| SM2892‡ | MATa MFA1 mfa2:-Δ2:lacZ trp1 leu2 ura3 his4 can1 p[CEN URA3] | Transformant of SM1179 with pSM316 | ||
| SM2893‡ | MATa mfa1-Δ1 mfa2-Δ1 trp1 leu2 ura3 his4 can1 p[2μ MFA1 URA3] | Transformant of SM2331 with pSM219 | ||
| SM3019‡ | MATa mfa1-Δ1::LEU2 MFA2 trp1 leu2 ura3 his4 can1 p[CEN URA3] | Transformant of SM1225 with pSM316 | ||
| SM3020‡ | MATa mfa1-Δ1 mfa2-Δ1 trp1 leu2 ura3 his4 can1 p[CEN URA3] | Transformant of SM2331 with pSM630 | ||
| SM3021‡ | MATa mfa1-Δ1 mfa2-Δ1 trp1 leu2 ura3 his4 can1 p[2μ MFA2 URA3] | Transformant of SM2331 with pSM628 |
SM1058 was formerly designated EG123 (Siliciano and Tatchell, 1984). SM1059 was formerly designated 246.1.1 (Siliciano and Tatchell, 1984).
These strains are isogenic with SM1058.
The a-factor null strain SM1458 was produced in several steps: (a) To generate a disruption allele of MFA2 that is marked by the Escherichia coli lacZ gene, a 3.3-kb BamHI fragment from pMC1871 (Casadaban et al., 1983) was ligated into pSM29 (Michaelis and Herskowitz, 1988) that had been digested with BamHI to drop out a 0.5-kb BamHI fragment containing the COOH-terminal MFA2 coding region and its downstream sequences. The resulting plasmid, pSM38, contains an mfa2 null allele (mfa2-Δ2::lacZ) in which the COOH-terminal seven codons (32–38) of MFA2 are deleted, and the MFA2 amino terminal region is fused in frame to lacZ; for simplicity, this allele is designated mfa2::lacZ. (b) To integrate the mfa2 null mutation by one-step gene replacement, a HindIII restriction digestion of pSM38 was cotransformed, together with 5 μg of the 2μ LEU2 plasmid YEp13, into SM1058. Leu+ colonies were tested for blue color on an X-Gal filter to determine which cotransformants also contained the mfa2::lacZ gene replacement; blue colonies represented 2% of the total transformants. (c) One blue cotransformant that had been allowed to lose YEp13 was designated SM1179; the presence of the expected chromosomal mfa2::lacZ mutation in this strain was confirmed by Southern analysis. (d) Finally, two sequential crosses were carried out to produce the a-factor null mutant SM1458. SM1179 (MATa mfa2::lacZ) was crossed to the otherwise isogenic strain SM1059 (MATα MFA2), and a MATα segregant, SM1196, was obtained. Next, SM1196 (MATα MFA1 mfa2::lacZ) was crossed to SM1225 (MATa mfa1::LEU2) and an a-factorless MAT a segregant, designated SM1458 (MATa mfa1::LEU2 mfa2::lacZ), was chosen. SM1458 is a convenient recipient for the MFA1-containing TRP1 and URA3 plasmids used in this study.
The a-factorless strain SM2331 (mfa1-Δ1 mfa2-Δ1) contains the same deletions as those present in SM1229 (mfa1-Δ1::LEU2 mfa2-Δ1::URA3), except that in SM2331, these deletions are unmarked. SM2331 was constructed using URA3 integrating plasmids pDH6 (mfa1-Δ1) and pDH9 (mfa2-Δ1), kindly provided by D. Hagen and G. Sprague (University of Oregon, Eugene, OR). pDH6 contains the 1.5-kb EcoR1-XbaI fragment from pSM82 (Michaelis and Herskowitz, 1988), in which codons 2–36 of MFA1 are deleted and marked by a BamH1 recognition site. pDH9 contains a genomic DNA fragment that was originally 3.5 kb, from which the 0.8-kb BamH1-SphI fragment including and flanking MFA2 was deleted. To construct the strain SM2331, two sequential two-step gene disruptions were carried out: First, pDH9 (URA3 mfa2-Δ1) was linearized with HindIII and transformed into SM1058. From one Ura+ transformant, Ura− derivatives were selected on 5-FOA and examined by Southern analysis, yielding strain SM2165, which contains the mfa2-Δ1 mutation. Second, pDH6 (URA3 mfa1-Δ1) linearized with SphI was transformed into SM2165, a Ura+ transformant was obtained, and Ura− derivatives were selected on 5-FOA. Mating of the Ura− derivatives was tested, and one mating-defective strain, SM2331, was shown by Southern analysis to contain both the mfa1-Δ1 and mfa2-Δ1 deletions.
Complete media (YEPD), synthetic dropout media (SC-URA and SC-TRP), and SD minimal media were prepared as described previously (Michaelis and Herskowitz, 1988), except that the drop-out media lack l-methionine and l-cysteine. Where necessary, SD media was supplemented with l-histidine (20 μg/ml), l-tryptophan (20 μg/ml) or l-leucine (30 μg/ml), and uracil (20 μg/ml). Strains were grown at 30°C, unless otherwise specified.
Plasmids and DNA Manipulation
The plasmids used in this study are listed in Table II. Plasmid pSM118, used for in vitro transcription and translation of a-factor, was constructed by cloning a 0.45-kb Sau3A MFA1-containing fragment from pSM39 (Michaelis and Herskowitz, 1988) into the BamHI site of pGEM1 (Promega, Madison, WI). In pSM118, a-factor expression is driven by the SP6 promoter, which lies 85 bases upstream of the MFA1 ATG. Plasmid pSM217, a 2μ URA3 vector, was constructed by digesting pRS306 (Sikorski and Hieter, 1989) with AatII, made blunt-ended by Klenow polymerase, and ligating to it a 1.9-kb filled-in HindIII-PstI fragment from YEp24, which contains 2μ DNA. To construct various a-factor plasmids, the 1.6-kb EcoRI-XbaI fragment bearing MFA1 was subcloned from pSM18 (Michaelis and Herskowitz, 1988) into the EcoR1 and XbaI sites in the polylinker of pRS316 (Sikorski and Hieter, 1989), pSM217, pRS314 (Sikorski and Hieter, 1989), and pSM363, yielding the MFA1 plasmids pSM233, pSM219, pSM464, and pSM463, respectively.
Table II.
Plasmids Used in This Study
| Plasmid | Vector | MFA1 genotype | Yeast marker | Source | ||||
|---|---|---|---|---|---|---|---|---|
| pDH6 | pRS306 | mfa1-Δ1 | URA3 | D. Hagen and G. Sprague | ||||
| pDH9 | pRS306 | mfa2-Δ1 | URA3 | D. Hagen and G. Sprague | ||||
| pMC1871 | pBR322-LacZ | − | − | Casadaban et al. 1983 | ||||
| pRS316 | CEN | − | URA3 | Sikorski and Hieter, 1989 | ||||
| pRS314 | CEN | − | TRP1 | Sikorski and Hieter, 1989 | ||||
| pRS424 | 2μ | − | TRP1 | Christianson et al., 1992 | ||||
| pSM18 | pUC18 | MFA1 | − | Michaelis and Herskowitz, 1988 | ||||
| pSM29 | pUC18 | MFA2 | − | Michaelis and Herskowitz, 1988 | ||||
| pSM38 | pUC18 | mfa2-Δ2::lacZ | − | This study | ||||
| pSM32 | pUC18 | mfa1-Δ1 | − | Michaelis and Herskowitz, 1988 | ||||
| pSM118 | pGEM1 | SP6-MFA1 | − | This study | ||||
| pSM217 | 2μ | − | URA3 | This study | ||||
| pSM219 | 2μ | MFA1 | URA3 | This study | ||||
| pSM233 | CEN | MFA1 | URA3 | This study | ||||
| pSM257 | 2μ | mfa1-C33S | URA3 | This study | ||||
| pSM258 | 2μ | mfa1-ΔVIA | URA3 | This study | ||||
| pSM463 | 2μ | MFA1 | TRP1 | This study | ||||
| pSM464 | CEN | MFA1 | TRP1 | This study | ||||
| pSM490 | 2μ | mfa1-I23M | URA3 | This study | ||||
| pSM628 | 2μ | MFA2 | URA3 | This study | ||||
| pSM630 | CEN | MFA2 | URA3 | This study |
E. coli and yeast transformations were performed essentially as described previously (Elble, 1992; Rose et al., 1990). All cloning was carried out directly in low melting temperature agarose, as described (Sambrook et al., 1989). The E. coli strains MH1 (Hall et al., 1984) and DH5α (Hanahan, 1983) were used to propagate plasmids.
Site-directed Mutagenesis
MFA1 mutations were generated by site-directed oligonucleotide mutagenesis (Kunkel et al., 1987). The oligonucleotide sequences used to generate a-factor mutants are shown here with the mutagenic residues underlined. C33S: 5′-AAGCAATAACACTTGCTGGGTCCC-3′ (oSM24); I23M: 5′-GACACCTTTGATCATATAGTTGTCC-3′ (oSM70); ΔVIA (36-38): 5′-ACGCAGAAACTA/ACATGCTGGGTC-3′ (oSM25). The plasmid pSM233 (CEN MFA1 URA3) was the template for generating mutants. Preparation of single-stranded phage, extraction of DNA, and the in vitro elongation reaction were carried out using the Muta-gene Phagemid in vitro mutagenesis kit from Bio Rad Laboratories (Hercules, CA). DNA sequencing was carried out on double-stranded plasmid, propagated in DH5α, and prepared by the boiling mini prep procedure (Sambrook et al., 1989) using the Sequenase kit from Amersham (Arlington Heights, IL). The primer used to sequence a-factor mutations was oSM17: 5′-CTGTAACTGTTTCTCGG-3′, which anneals to the 5′ upstream region of MFA1. Desired mutations were obtained at a frequency between 10 and 50%. Mutants were subcloned from the pSM233 parental plasmid into pSM217 to produce 2μ plasmid versions, which were designated pSM257 (mfa1-C33S), pSM490 (mfa1-I25M), and pSM258 (mfa1-ΔVIA). The sequence of all MFA1 mutant plasmids was reverified after reisolation from yeast strains.
Generation of Rabbit Polyclonal a-Factor Antiserum
The dodecapeptide corresponding to the unmodified mature a-factor sequence encoded by MFA1, YIIKGVFWDPAC, was chemically synthesized by Dr. P. Shenbagamurthi at The Johns Hopkins School of Medicine Protein-Peptide-DNA Facility (Baltimore, MD). To generate a-factor antiserum, the unconjugated dodecapeptide was resuspended in Freund's complete adjuvant and directly injected into rabbits by Hazelton Research Products, Inc. (Denver, PA). Subsequent boosts were carried out in Freund's incomplete adjuvant. The immune serum was initially screened by ELISA tests versus the a-factor dodecapeptide and was further analyzed by immunoprecipitation. Antiserum from rabbit antisera designated Ab-9-137 and Ab-499 were shown to immunoprecipitate all a-factor species with equivalent efficiency (Chen, 1993).
Metabolic Labeling and Preparation of Intracellular and Extracellular a-Factor
Yeast cells were grown to log phase in synthetic media to OD600 0.5–1.0. 5 OD600 U of cells were used for each sample to be analyzed, unless otherwise indicated. Before labeling, cells were harvested by centrifugation at 5,000 g for 5 min, washed once with SD media, and resuspended in 0.5 ml fresh media in a polypropylene tube at room temperature. All labelings were performed at 30°C, unless otherwise specified.
Steady-state labeling was carried out in SC dropout media for 30 min with 150 μCi [35S]cysteine (1,160 Ci/mmol, 11 mCi/ml; ICN Biomedicals, Inc., Irvine, CA) per sample (5 OD600 U of cells). Approximately 25% of the added radioactivity is incorporated into cells during this period; [35S]cysteine uptake remains linear for up to ∼90 min. Pulse labeling was carried out in SD media plus necessary supplements for a short time (1–5 min) with 150 μCi [35S]cysteine added for each time point to be analyzed. In SD media, the half-time of [35S]cysteine incorporation is ∼7 min. The chase was initiated by addition of 10 μl of 50× chase mix (1 M cysteine, 1 M methionine) per each time point. Immediately after labeling, cells were split into separate tubes representing each time point of the chase. It was necessary to treat each sample separately because a-factor sticks to the walls of the polypropylene tubes and total extracellular a-factor must be recovered by combining the culture fluid and tube wash for each time point of the chase (see below). The chase was terminated by addition of 0.5 ml ice-cold azide stop mix (40 mM cysteine, 40 mM methionine, 20 mM NaN3, 500 μg/ml BSA [Miles Laboratories, Elkhart, IN]). Samples were kept at 4°C after termination of the chase. Labeled cells and culture fluid were moved from the “chase tube” to a new tube and separated by centrifugation in a microfuge at top speed (13,600 g) for 1 min; the chase tube was retained for the tube wash (see below). The pellet was designated the intracellular (I) fraction, and the culture supernatant together with the material obtained from the tube wash (below) is designated the extracellular (E) fraction.
Proteins from the I and E fractions were concentrated and denatured before immunoprecipitation by the following procedures. The pellet (I fraction) was washed once with 1 ml cold distilled H2O and resuspended in 1 ml cold distilled H2O. Cells were lysed by the addition of 150 μl cold NaOH– β-mercaptoethanol (1.0 ml 2 M NaOH; 80 μl β-mercaptoethanol), followed by incubation on ice for 15 min. Proteins in the I fraction were precipitated by adding 150 μl cold 50% TCA and incubating on ice for 15 min. After centrifugation at 13,600 g for 15 min at 4°C, the supernatant was removed, the precipitated proteins were resuspended in 30 μl 2× Laemmli sample buffer (20% glycerol, 10% β-mercaptoethanol, 4.3% SDS, 0.125 M Tris-HCl, pH 6.8, and 0.2% bromophenol blue), and the sample was neutralized by the addition of 2 M Tris base when necessary, followed by heating at 100°C for 3 min. Proteins from the culture fluid portion of the E fraction (above) were precipitated by addition of an equal volume of 20% cold TCA, incubation at 4°C for 15 min, and centrifugation at 13,600 g for 15 min at 4°C. The pellet was resuspended in 20 μl 2× Laemmli sample buffer and neutralized with 2 M Tris base. Since a significant portion of extracellular a-factor remains bound to the polypropylene chase tube retained above, this tube was rinsed four times with distilled H2O, and bound proteins were eluted from its sides with 0.5 ml n-propanol (Sterne, 1989). The n-propanol eluate was dried in a speed vacuum concentrator with heat. This tube wash sample was resuspended in 10 μl 1× Laemmli sample buffer and combined with 20 μl of the E fraction already in sample buffer (above) and heated at 100°C for 3 min. By combining the a-factor from culture fluid and a-factor eluted from the chase tube, quantitative recovery of extracellular a-factor is achieved.
Immunoprecipitation of a-Factor
For immunoprecipitation, I and E samples were diluted with 1.3 ml immunoprecipitation buffer (50 mM Tris-HCl, pH 7.5, 1.0% Triton X-100, 150 mM NaCl, 1 mM PMSF, 0.5% Trasylol) and cleared of insoluble material by centrifuging for 1 min at 13,500 g. Samples were transferred into new tubes, avoiding the insoluble pellet, a-factor rabbit antiserum (10 μl) of Ab-9-137 or Ab-499 (see Figs. 8 and 9) was added, and antigen–antibody complex formation was allowed to proceed overnight at 4°C. This amount of antiserum was shown to represent an excess of antibody in these immunoprecipitation reactions (Chen, 1993). It should be noted that a-factor does not bind to polypropylene in the presence of 1% Triton X-100. To collect immunoprecipitates, a 45-μl aliquot of protein A–Sepharose CL-4B beads (Pharmacia Fine Chemicals, Piscataway, NJ) suspended in immunoprecipitation buffer (1:3 beads/total volume ratio) was added and the tubes were gently agitated at 4°C for 60 min. The beads were collected by a 15-s spin at 13,600 g, washed four times with immunoprecipitation wash buffer A (0.1% Triton X-100, 0.02% SDS, 150 mM NaCl, 50 mM TrisHCl, pH 7.5, 5 mM EDTA, 1 mM PMSF, 0.5% Trasylol), and once with immunoprecipitation wash buffer B (150 mM NaCl, 50 mM Tris-HCl, pH 7.5, 5 mM EDTA, 1 mM PMSF, 0.5% Trasylol). After the final wash, bound immune complexes were released from the beads by the addition of 30 μl 2× Laemmli sample buffer. Samples were heated at 100°C for 3 min before SDS-PAGE.
Figure 8.

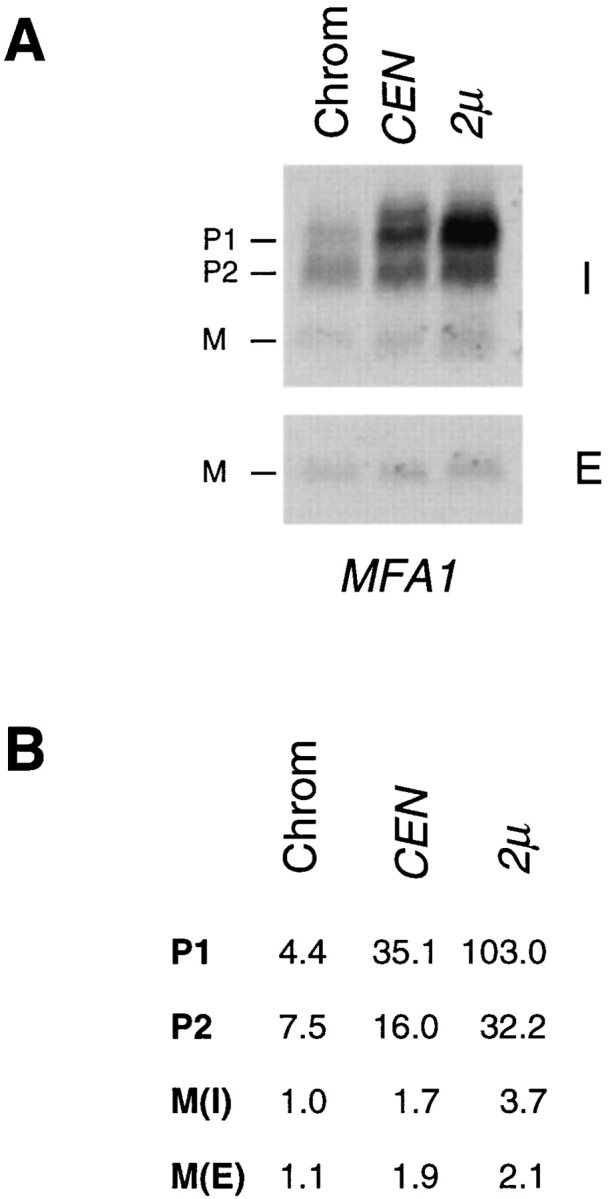
Kinetic analysis of a-factor biogenesis in strains expressing MFA1 or MFA2 at low (chromosomal), intermediate (CEN), or high (2μ) levels. Cells were pulse labeled with [35S]cysteine for 5 min, and the label was chased for the times indicated. Intracellular (I) and extracellular (E) fractions were prepared, and proteins were subjected to immunoprecipitation, SDS- PAGE, and exposed to a Phosphorimager screen for 24 h. The P1, P2, and M species are indicated. A constant gray scale value is used for all the scans shown, so that the relative level of a-factor species in each strain can be qualitatively compared by direct visual inspection. The strains examined here express MFA1 or MFA2 only, from the chromosome (SM2892 and SM3019), a CEN plasmid (SM2891 and SM3020), and a 2μ plasmid (SM2893 or SM3021).
Figure 9.

Comparison of the overall a-factor biogenesis pattern in yeast strains derived from distinct genetic lineages. Strains SM1058, S288C, SK-1, and Σ1278B were pulse labeled with [35S]cysteine for 5 min. Intracellular (I) and extracellular (E) a-factor species were immunoprecipitated immediately or after a 15-min chase and analyzed by SDS-PAGE and autoradiography.
SDS-PAGE Analysis of a-Factor
It was necessary to modify the classical SDS-PAGE method (Laemmli, 1970) to resolve the I species of a-factor, which range in molecular mass from ∼1.6 to 4.5 kD. In addition to the small size of a-factor, the lipid modification and extreme hydrophobicity of a-factor contribute to its unusual migration properties. Four aspects of the SDS-PAGE system were optimized. First, different acrylamide percentages (12–20%) were tested, keeping the acrylamide/Bis ratio constant at 30:08. Separation of the P1, P2, and M species of a-factor was found to be optimal at 15–16% acrylamide, which represented a compromise between separation of precursor (P1, P2) from mature (M) a-factor versus compression of P1 and P2 precursor bands. The second critical parameter is the pH of the 4× Laemmli lower stock buffer used in the separation gel. Whereas resolution of the P1 and P2 species of a-factor is optimal at pH 8.8, these species cannot be resolved at pH 9.0. A third important factor is that a-factor gels must be run at a relatively high current (constant current = 50–65 mA) to achieve optimal separation. Electrophoresis at a lower current results in “fuzzy” a-factor bands. Finally, the separation of a-factor intermediates is very sensitive to sample overloading. In general, immunoprecipitates derived from <1 OD600 U of cells were loaded in each gel lane. The addition of significantly more protein resulted in broad fuzzy bands. Electrophoresis of a-factor was carried out according to the modifications of the standard Laemmli procedure described above, using prestained low range molecular weight markers (Gibco BRL, Gaithersburg, MD) to standardize gel runs.
After electrophoresis, gels were fixed in 10% acetic acid for 15 min, rinsed in distilled H2O for 5 min, and soaked in 0.7 M Na salicylate, pH 7.0, for 15 min. Gels were dried under a vacuum at 80°C and subjected to autoradiography at −80°C. Alternatively, the salicylate step was omitted and dried gels were subjected to Phosphorimager analysis (Molecular Dynamics, Inc., Sunnyvale, CA).
In Vitro Transcription and Translation
To generate the unmodified a-factor precursor, an SP6-MFA1 construct was transcribed and translated in vitro. Briefly, 10 μg of pSM118 (SP6- MFA1) was linearized by EcoRI-HindIII digestion, extracted twice with phenol, once with phenol/CHCl3 (1:1), precipitated with ethanol, and resuspended in TE, pH 8.0. The SP6-MFA1 DNA was transcribed (Riboprobe System Kit; Promega) with 1 μl SP6 RNA polymerase (20 U/μl) at 37°C for 60 min. After transcription, the RNA was extracted twice with phenol, once with phenol/CHCl3 (1:1), precipitated with ethanol, and resuspended in DEPC-treated H2O. Before translation, the RNA template was heated at 65°C for 10 min and then translated using 25 μl wheat germ extract (Wheat Germ Extract System; Promega) in the presence of 100 mM potassium acetate, 0.1 mM cold amino acid–cysteine mix, and 25 μCi [35S]cysteine (1,160 Ci/mmol, 11 mCi/ml; ICN Biomedicals). After incubation at 25°C for 2 h, an equal volume of cold 50% TCA was added, precipitated proteins were pelleted, the pellet was resuspended in 30 μl 2× Laemmli sample buffer, and neutralized by the addition of 2 M Tris base, if necessary. Precursor forms of a-factor were immunoprecipitated using a-factor antiserum, as described above.
Vapor Phase Equilibrium Assay to Detect a-Factor Methyl Esters
To determine whether a particular species of a-factor contained a carboxyl methyl ester modification, [3H]S-adenosyl methionine (SAM) labeling and methylation assays were carried out, based on a previously described procedure (Clarke et al., 1988; Hrycyna et al., 1991). 10 OD600 U of yeast cells were double labeled with 300 μCi [35S]cysteine and 100 μCi [3H]methyl AdoMet (84 Ci/mmol, 0.55 mCi/ml) from Dupont/NEN (Boston, MA) for 5 min in SD media with appropriate supplements. For the mfa1-C33S mutant (strain SM1682 that contains pSM257), 300 μCi Trans-35S label from Amersham (Arlington Heights, IL) was substituted for [35S]cysteine. After centrifugation at 13,600 g for 1 min, the cell pellet, representing the I fraction, was washed with cold distilled H2O, resuspended in cell lysis buffer (50 mM potassium phosphate buffer, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 1 mM β-mercaptoethanol, 1 mM PMSF, 0.5% Trasylol), and lysed in the presence of 0.5 g baked zirconium beads (0.5 mm in diameter) obtained from Biospec Products (Bartlesville, OK) by vortexing six times in 1-min bursts at 4°C, followed by chilling on ice. The cell lysate was removed from the beads. The remaining lysate was obtained by washing twice with 0.5 ml cell lysis buffer, and all three supernatant fractions were pooled. 10 μl of a-factor antiserum was added to this lysate (1.4 ml total) directly. Immunoprecipitation, SDS-PAGE, fixation, and autoradiography were carried out as described above, except that the salicylate treatment step was omitted.
After visualization by autoradiography, intracellular a-factor intermediates in the dried gel were cut out as 0.4-cm slices, placed in a polypropylene microcentrifuge tube, and mixed with 500 μl 1 M NaOH to hydrolyze methyl esters to methanol. Immediately after the addition of NaOH, the microfuge tube, with its cap open, was placed in a 20-ml scintillation vial containing 5 ml of ASC2 scintillation fluid from Amersham, and the vial was tightly capped. Care was taken not to allow the scintillation fluid to directly contact the NaOH gel slices. After 24–36 h at 37°C, the microcentrifuge tube was removed, and the radioactivity that had been partitioned by diffusion of [3H]methanol into the scintillation fluid was assayed in a liquid scintillation counter. Total [35S]cysteine incorporation was assayed by adding 1 ml of Protosol tissue solubilizer from New England Biolabs (Beverly, MA) to the gel slices after completion of the methylation assay and allowing them to incubate for 6 h at 55°C. The entire tube with its contents was directly placed into 10 ml of OCS organic counting scintillation fluid from Amersham, and was counted in a liquid scintillation counter.
Radiolabeled Peptide Sequence Analysis
10 OD600 U of yeast cells were labeled with 500 μCi l-[2,3,4,5-3H]proline (91 Ci/mmol, 1 mCi/ml; Amersham), l-[4,5-3H]lysine (102 Ci/mmol, 5 mCi/ ml; Amersham), or Trans-35S label (1,144 Ci/mmol, 12 mCi/ml; ICN Pharmaceuticals, Inc., Irvine, CA) for 5 min in SD media with appropriate supplements. Labeled cells and culture fluid were collected, and I and E fractions were prepared as described above for [35S]cysteine-labeled samples. After electrophoresis, the gel was placed in transfer buffer (25 mM TrisHCl, pH 8.2, 192 mM glycine, 10% methanol [vol/vol]) to equilibrate for 15 min. Transfer of proteins from the gel to Immobilon polyvinyldifluoride transfer membrane from Millipore (Bedford, MA) was carried out at 70 V for 30 min. After removal of excess glycine by rinsing the blot in distilled H2O for 10 min, the membrane was air dried and subjected to autoradiography. 3H-labeled bands were visualized on film, and the film was used as a template to excise slices of polyvinyldifluoride membranes containing the desired species. Polyvinyldifluoride slices were loaded directly to a Gas-Phase Sequencer (model 470; Applied Biosystems, Inc., Foster City, CA). A 150-μl sample from each sequencing cycle containing phenylthiohydantoin (PTH)-derivatized amino acids was added to 5 ml scintillation fluid, and the radioactivity of the [35S]PTH-methionine, [3H]PTHproline, or [3H]PTH-lysine was counted in ASC2 scintillation fluid in a liquid scintillation counter. A background value, established by counting a blank sample, was subtracted from the total counts that were determined for each sequencing sample.
Cell Fractionation
10 OD600 U of yeast cells were labeled with 300 μCi [35S]cysteine for 5 min in SD media with appropriate supplements. Labeled cells were washed once in cold distilled H2O and resuspended in 100 μl cell fractionation buffer (50 mM potassium phosphate, pH 7.4, 150 mM NaCl, 1 mM β-mercaptoethanol, 1 mM PMSF, and 0.5% Trasylol). Cells were disrupted with zirconium beads by vortexing six times in 1-min bursts at 4°C, the crude extract was transferred into a new tube, and cellular debris was removed by centrifugation at 1,000 g for 10 min. This supernatant is designated the total cellular extract (T). To determine the nature of the association of a-factor with the membrane, the T extract was incubated at 4°C for 15 min alone or in the presence of one of the following compounds: 0.5% Triton X-100, 0.1 M Na2CO3, pH 12.5, 1.6 M urea, or 2 M NaCl. The treated extract was centrifuged at 100,000 g for 1 h; the supernatant was designated the soluble fraction (S). The protein in the S fraction was concentrated by TCA precipitation, resuspended in 30 μl 2× Laemmli sample buffer, and neutralized with 2.0 M Tris base. The 100,000 g pellet, designated the particulate fraction (P), was washed once with cold cell fractionation buffer and resuspended in 30 μl 2× Laemmli sample buffer. The T, S, and P fractions were subjected to immunoprecipitation and SDS-PAGE, as described above. An equivalent amount of each fraction, normalized according to the OD600 U, was loaded in each lane on SDS-PAGE.
Quantitative Analysis of a-Factor Biosynthesis
Dried gels from pulse-chase experiments were exposed to a Molecular Dynamics Phosphor Screen for several days, and the resulting data were quantified using Molecular Dynamics Image Quant software.
The identity of the intracellular precursor (P1 and P2) species and of mature (M) a-factor are shown in Fig. 5. M(I) and M(E) refer to intracellular and extracellular mature a-factor, respectively. The “processing efficiency” shown in Table III refers to the percentage of a-factor precursor initially synthesized [P1+P2]0′ that is ultimately proteolytically cleaved to the M form [M(I)+M(E)]15′. The time point of 15-min chase was chosen because the level of the total M form does not increase after this time. The processing efficiency is calculated by the following formula:
Figure 5.

The deduced structure of a-factor intermediates. The SDS-PAGE analysis of immunoprecipitated intracellular (I) and extracellular (E) species of a-factor present after a brief (5min) pulse labeling is shown (right). The deduced structure of each a-factor biosynthetic intermediate is indicated (left), based on the results from the carboxyl methylation assay (Fig. 3) and the radiolabeled peptide sequence analysis (Fig. 4). The strain labeled is SM1710, which contains pSM233 (CEN MFA1).
Table III.
Processing Efficiency and Overall Export Efficiency of a-Factor
Values were calculated from cells expressing chromosomal MFA1 or MFA2 only (SM2891 and SM2892, respectively) and are averaged from Phosphorimager analysis of two gels, one of which is shown in Fig. 8 A. The calculations are based on the assumption that each a-factor molecule contains only a single radiolabeled residue ([35S]cysteine). Even if some metabolic conversion of [35S]cysteine to [35S]methionine could occur, the overall values would not be altered significantly; only P1, which contains an NH2-terminal Met, would be affected.
The “processing efficiency” is the percentage of a-factor precursor that is initially synthesized within a 5-min pulse [P1 + P2]0′ that is ultimately converted after 15 min to mature a-factor [M(I) + M(E)]15′.
The “export efficiency” is the percentage of a-factor initially synthesized [P1 + P2 + M(I) + M(E)]0′ that is ultimately exported to culture fluid as mature a-factor [M(E)]30′.
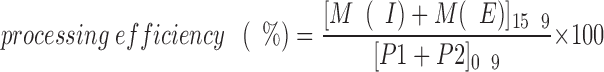
The “export efficiency” shown in Table III indicates the fraction of the total intracellular a-factor initially synthesized at T0′ [P1+P2+M(I)]0′ that is ultimately exported to the culture fluid after secretion is complete [M(E)]30′, expressed as a percentage. The export efficiency is calculated by the following formula:
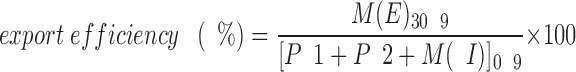
The precise half-time (t 1/2) of a-factor export is complex to measure, since the data indicates that there are two separate rates for this process, an initial rapid rate followed by a slow rate. We observe here that 80% of export is completed by 15 min of chase.
Examining a-Factor Biosynthesis After α-Factor Induction
10 OD600 U of log-phase MAT a cells (SM1227) were resuspended in SD media plus the appropriate supplements. 2 OD600 U of cells were immediately set aside as an untreated control sample. Synthetic α-factor (Sigma Immunochemicals, St. Louis, MO) was added to the remaining 8 OD600 U of cells to a final concentration of 25 μM, and α-factor–treated cells were incubated at 30°C. At various time points after the α-factor addition, aliquots (2 OD600 U) were metabolically labeled with 150 mCi [35S]cysteine for 5 min followed by a 45-min chase. Control cells were labeled the same way, except in the absence of α-factor. The I fraction was prepared from half of the pulse-labeled cells, whereas the E fraction was prepared from the remaining half of the culture that had completed the chase. I and E fractions were immunoprecipitated with a-factor antiserum and subjected to SDS-PAGE analysis. The data were quantitated by Phosphorimager analysis.
Results
Identification of a-Factor Biosynthetic Intermediates
As a first step in characterizing the a-factor biogenesis pathway, we used metabolic labeling to identify a-factor biosynthetic intermediates. Cells were radiolabeled with [35S]cysteine under steady-state conditions. Intracellular and extracellular species of a-factor were immunoprecipitated with a-factor antiserum. The immunoprecipitates were analyzed by a modified SDS-PAGE procedure (see Materials and Methods) that was optimized for the separation of a-factor intermediates. Because of their unusually small size, a-factor and its biosynthetic intermediates are difficult to resolve by standard SDS-PAGE techniques. Fig. 2 shows the pattern of a-factor biosynthetic species generated in a strain carrying a single a-factor gene MFA1. In the intracellular fraction (Fig. 2, lane 4), three species are apparent, two that are precursor sized, P1 and P2, and one that is mature sized, M. In the extracellular fraction (Fig. 2, lane 5), only mature a-factor is observed. Whereas the three intracellular species (P1, P2, and M) are present in cells carrying an a-factor gene (MFA1; Fig. 2, lanes 4 and 5), they are absent in an a-factor null mutant strain (Δmfa1 Δmfa2; lanes 2 and 3), and can be competed away by inclusion of an excess of cold a-factor synthetic peptide (lanes 6 and 7) in the immunoprecipitation. Thus, P1, P2, and M appear to be authentic MFA1-derived a-factor species. Analogous gel bands are also present in strains expressing solely MFA2 (see Fig. 8). The P1 and P2 a-factor species migrate between the 3- and 6-kD molecular mass standards, compatible with the predicted molecular mass (∼4.5 kD) for the a-factor precursor. M migrates at a position near to that of the 3-kD marker. This is somewhat slower than predicted for mature a-factor (1.6 kD), perhaps caused in part by the hydrophobicity of a-factor.
Figure 2.
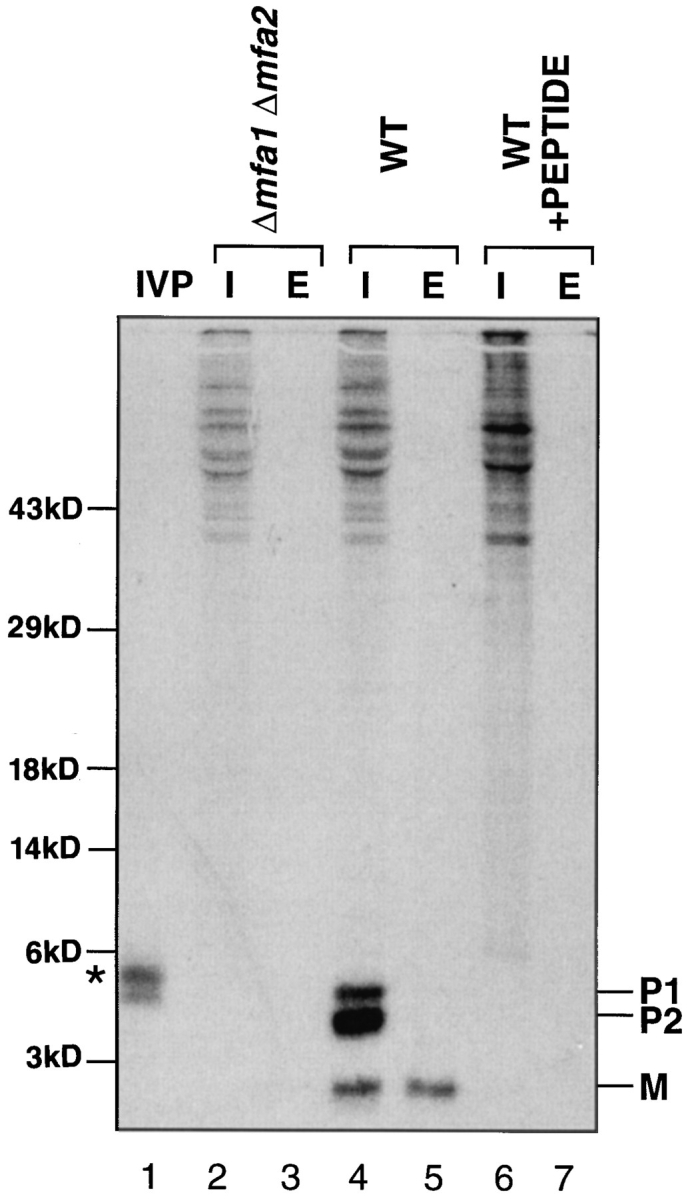
Identification of a-factor biosynthetic intermediates. Cells were labeled with [35S]cysteine under steady-state conditions and intracellular (I) and extracellular (E) extracts were prepared. Proteins were immunoprecipitated with a-factor antiserum (Ab-9-137), separated by 16% SDS-PAGE, and visualized by autoradiography, as described in Materials and Methods. The in vitro precursor (IVP; lane 1) was generated by transcription and translation of MFA1 from pSM118. Strains are transformants of an a-factor deletion strain (Δmfa1 Δmfa2) and contain either no plasmid (lanes 2 and 3; SM1458) or an a-factor plasmid, pSM464 (CEN MFA1; lanes 4–7; SM1829). In the peptide competition experiment (lanes 6 and 7), the labeled extracts are the same as in lanes 4 and 5, respectively, except that a 50-fold excess of cold synthetic dodecapeptide (YIIKGVFWDPAC) corresponding to mature a-factor was added before immunoprecipitation. The a-factor precursor species are designated P1 and P2, and the band corresponding to mature a-factor is designated M. Molecular mass markers are indicated. The band in lane 1 just above P1, marked by an asterisk, most likely represents the unmodified a-factor precursor P0.
For comparison, the in vitro transcribed and translated product derived from the MFA1 gene was immunoprecipitated and analyzed by SDS-PAGE. Two slowly migrating species are apparent (Fig. 2, lane 1). The species with the lowest mobility (marked by an asterisk) most likely corresponds to the primary a-factor translation product, P0. It should be noted that the P0 form of the a-factor precursor cannot generally be detected in vivo under steady-state labeling conditions, probably because of its rapid conversion to P1 (thus, it is absent in Fig. 2, lane 4); however, P0 can be detected in vivo after a short pulse label (see Fig. 5) or when a-factor farnesylation is blocked by a CAAX mutation (Fig. 6) or by farnesyltransferase mutations (ram1 or ram2; He et al., 1991). The higher mobility a-factor species present in Fig. 2, lane 1 likely corresponds to the partially or fully COOH-terminally modified precursor, presumably resulting from prenyltransferase activity and possibly protease activity present in the wheat germ translation extract.
Figure 6.

Fractionation and solubilization properties of intracellular a-factor. In A, cells were labeled with [35S]cysteine for 5 min and lysates were prepared. The total cellular lysate (T) was separated into particulate (P) and soluble (S) fractions by centrifugation at 100,000 g for 1 h at 4°C. The a-factor species were immunoprecipitated and subjected to SDS-PAGE analysis. Strains examined in lanes 1–9 (SM1585, SM1682, and SM1680) carry either a wild-type or mutant MFA1 plasmid, as indicated. Strain SM1865, examined in lanes 10–12, is a Δram1 mutant. In B, a strain containing a wild-type MFA1 plasmid (SM1762) was labeled with [35S]cysteine for 5 min. The total lysate (T) was subjected to the indicated treatments or to no treatment (control, lanes 1–3) and subsequently separated into particulate (P) and soluble (S) fractions by centrifugation at 100,000 g for 1 h at 4°C. The a-factor intermediates were immunoprecipitated and analyzed by SDSPAGE and autoradiography.
P1, P2, and M are COOH-terminally Modified Intermediates
We wished to directly determine the COOH-terminal modification status of the intermediates observed in Fig. 2. Because methylation is obligatorily the last step in the COOH-terminal modification of CAAX proteins, it can be used as a marker for the completion of a-factor COOHterminal processing (Hrycyna and Clarke, 1992). To determine whether the a-factor biosynthetic intermediates P1, P2, and M are methylated, a MATa strain carrying a plasmid-borne wild-type MFA1 gene was double labeled with the methyl group donor [3H]methyl AdoMet and [35S]cysteine. We also examined in parallel a strain carrying a plasmid-borne CAAX mutant, mfa1-C33S. Since the mfa1C33S CAAX mutation blocks the farnesylation of a-factor (Chen, 1993), it will also prevent AAX cleavage and methylation. Intracellular a-factor was immunoprecipitated, and biosynthetic intermediates were separated by SDSPAGE. After visualization by autoradiography, 35S-labeled bands corresponding to P0, P1, P2, and M were excised from the gel and the extent of methylation of each species was determined by the vapor phase equilibrium assay (Clarke et al., 1988; Hrycyna et al., 1991). This assay specifically detects ester-linked methyl groups, which are liberated as [3H]methanol after base hydrolysis (see Materials and Methods). The ratio between 3H and 35S for each species reflects its relative level of methylation.
The P1, P2, and M species derived from wild-type MFA1 all contain COOH-terminal methyl esters, as evidenced by the presence of [3H]methanol counts (Fig. 3 A). Conversely, the P0 species synthesized by the mfa1-C33S CAAX mutant lacks methylation, as expected (Fig. 3 B). This experiment demonstrates that all of the wild-type a-factor biosynthetic intermediates (P1, P2, and M) are methylated and thus have undergone complete COOHterminal modification. It is likely that the differences in the apparent extent of methylation of the three species, as defined by their 3H/35S ratios, is caused in part by the overexpression of MFA1, and resultant incomplete methylation of P1 in the experiment shown in Fig. 3. In a similar experiment in which MFA1 was expressed from a low copy number CEN plasmid, the 3H/35S ratios of P1, P2, and M were closer to one another (P1 and P2 differed by only 1.3-fold; P2 and M were equivalent; data not shown). These results suggest that at chromosomal levels of expression, the relative extent of methylation for the P1, P2, and M species are likely to be identical. Since P1, P2, and M have all completed COOH-terminal modification, their differing SDS-PAGE mobilities must reflect differences in their NH2-terminal processing status.
Figure 3.
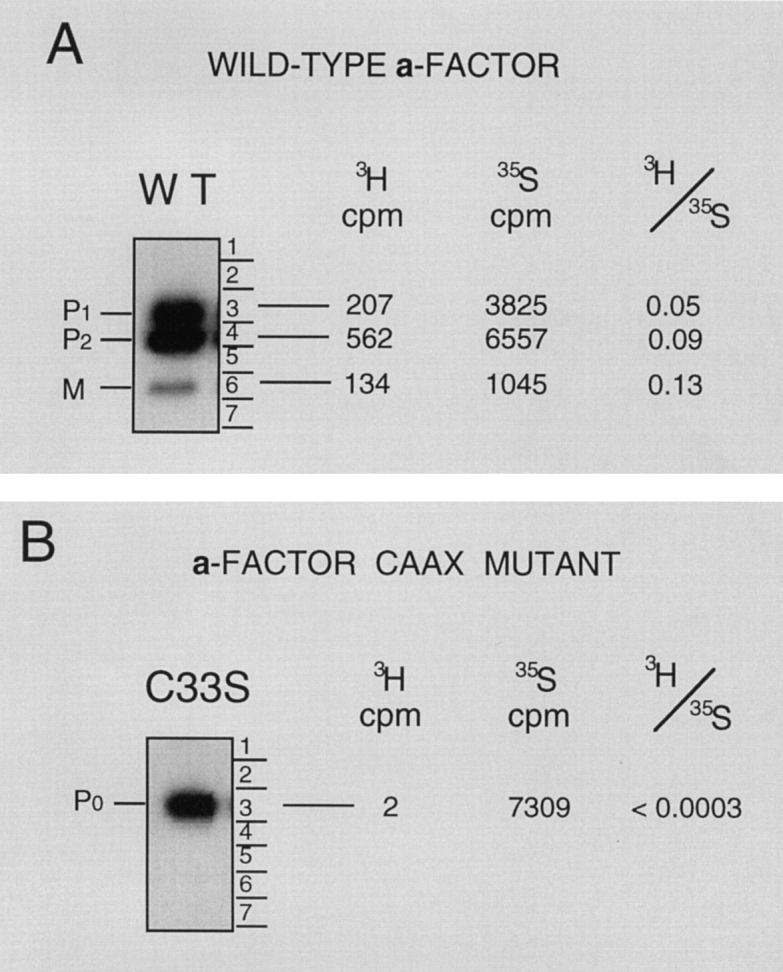
Carboxyl methylation assay to determine the COOHterminal modification status of a-factor biosynthetic intermediates. The methylation status of a-factor biosynthetic intermediates was determined in a strain containing a wild-type MFA1 plasmid (A, SM1585) or a plasmid with the a-factor CAAX mutation mfa1-C33S (B, SM1682), in which prenylation and subsequent modification of a-factor is prevented. Cells were double labeled with [35S]cysteine and [3H-methyl]AdoMet (A) or with Trans35-label and [3H-methyl]AdoMet (B), respectively. Cell extracts were prepared, proteins were immunoprecipitated with a-factor antibodies, separated by 16% SDS-PAGE, and the dried gel was subjected to autoradiography to detect the indicated intracellular a-factor biosynthetic intermediates. Each a-factor intermediate was excised from the gel, and the relative degree of carboxyl methylation was determined. Incorporation of 3H into methyl esters was determined by subjecting the excised gel slice to the vapor phase equilibrium assay in which 3H-labeled methyl ester groups are converted to [3H]methanol counts. Total 35S incorporation was determined by solubilization of the gel slice. The 3H/35S ratio determined by these two tests is shown.
P1 Has an Intact NH2-terminal Extension, P2 Has a Partially Processed NH2-terminal Extension, and M Is Fully Processed
To further define the structure of the a-factor biosynthetic intermediates, we determined the NH2-terminal amino acid of each a-factor species by radiolabeled peptide sequence analysis (Fig. 4). An a-factor deletion strain carrying wild-type MFA1 on a high copy number plasmid was labeled with [3H]lysine or [3H]proline (Fig. 4, A and B, respectively). Radiolabeled a-factor intermediates were separated by SDS-PAGE, followed by blotting onto a polyvinyldifluoride membrane. Excised segments of the polyvinyldifluoride membrane containing a single a-factor species were subjected to automated gas phase Edman degradation analysis. In addition to strains carrying wild-type MFA1, a strain carrying a mutant plasmid, mfa1-I23M, which contains an engineered methionine codon within the mature portion of a-factor, was labeled with [35S]methionine and analyzed (Fig. 4 C). This mutant was included for analysis because the higher energy of 35S as compared to 3H permitted optimal detection of radiolabeled residues. The amount of extracellular a-factor produced by mfa1-I23M is comparable to that of wild-type, indicating that a-factor maturation and secretion are relatively normal for the mutant (Nouvet, F., and S. Michaelis, unpublished observation).
Figure 4.

Radiolabeled peptide sequence analysis to establish the NH2-terminal processing status of intracellular species of a-factor. The a-factor species in immunoprecipitates from cells labeled with [3H]lysine (A), [3H]proline (B), or Trans35S label (methionine:cysteine = 20:1) (C) were separated by 16% SDS-PAGE and transferred onto polyvinyldifluoride membrane. The intracellular a-factor intermediates P1, P2, and M were visualized by autoradiography, excised, and subjected to Edman degradation analysis. The radioactive material from each cycle was quantitated by scintillation counting as shown in the bar graph. Within the a-factor sequence, the radiolabeled amino acid used for labeling is shaded darkly, and the deduced P1→ P2 and P2→ M cleavage sites are indicated. The strain labeled in A and B is SM1762, which carries the wild-type a-factor plasmid pSM463 (2μ MFA1). The strain labeled in C is SM1932, which harbors an a-factor substitution mutant plasmid, pSM490 (2μ mfa1-I23M).
When the [3H]lysine-labeled P1 species was subjected to Edman degradation, peaks of radioactivity were detected in cycles 11 and 13 (Fig. 4 A). This is the result expected if P1 begins with the first methionine of the a-factor precursor. It should be noted that the apparent “trailing” of a peak into a succeeding cycle (as seen here in cycle 11 trailing into 12, and 13 into 14) is not uncommon with this technique, and occurs when the degradation within a particular cycle is incomplete. For [3H]proline-labeled P1, distinct peaks of radioactivity are detected in cycles 3 and 10 (Fig. 4 B), and for [35S]methionine-labeled P1, produced from the I23M mutant a-factor, peaks are detected in cycles 1 and 23 (Fig. 4 C). Taken together, these results indicate that the P1 species of a-factor contains an intact NH2terminal extension.
We next determined the NH2-terminal sequence of P2. For [3H]lysine-labeled P2, peaks are present in cycles 4 and 6 (Fig. 4 A). This result is consistent with the NH2terminal residue of P2 coinciding with residue A8 of the a-factor precursor, as indicated by the designation “P1→ P2 cleavage site” in Fig. 4, A and B. The identity of the NH2terminal residue of P2 as A8 was further confirmed by analysis of [3H]proline labeled P2, in which a peak is present in cycle 3 (Fig. 4 B). Thus, rather unexpectedly, the P2 species appears to be the product of an unpredicted proteolytic cleavage step within the NH2-terminal extension of the a-factor precursor; such an intermediate was not anticipated a priori from our previous knowledge of the sequence of precursor and mature forms of a-factor.
A somewhat different result was obtained upon sequence analysis of the [35S]methionine-labeled P2 species derived from the I23M mutant. Peaks were detected both in cycles 16 and 18 (Fig. 4 C), suggesting that in this mutant, P2 actually consists of two species, one beginning at residue A6 and the other at A8. The I23M version of a-factor may undergo an aberrant processing reaction, in which an anomalous cleavage occurs between residues T5 and A6 in addition to the normal cleavage between T7 and A8. Alternatively, it is possible that there is a very minor cleavage site between T5 and A6, even for the wild-type precursor, and that this site is used more efficiently by the mutant. It is not clear why the I23M mutation, which is quite distant from the P2 cleavage site, has an effect on the P1→ P2 conversion. Interestingly, we have found that another mfa1 mutation near this region, I24N, also has an effect on this processing step, resulting in a slower than normal rate of P1→ P2 conversion (Nouvet, F., A. Kistler, and S. Michaelis, manuscript in preparation).
We also investigated the NH2-terminal sequence of the intracellular mature species, M. For [3H]lysine-labeled M, radioactivity was recovered in cycle 4 (Fig. 4 A). Correspondingly, for [35S]methionine-labeled M derived from the I23M mutant, radioactivity was recovered in cycle 2 (Fig. 4 C). (The [3H]proline signal for M was too low to count.) These results indicate that intracellular M has the same NH2 terminus (Y22) as that previously established for extracellular M by mass spectrometry (Anderegg et al., 1988); thus, intracellular M has undergone complete removal of the NH2-terminal extension.
In summary, NH2-terminal radiolabeled peptide sequence analysis (Fig. 4), in combination with the assessment of the carboxyl methylation status (Fig. 3) of the a-factor intermediates, indicates that their structure is as follows (Fig. 5): P0 is the unmodified, full-length a-factor precursor; P1 is the COOH-terminally modified precursor with an intact NH2-terminal extension; P2 is generated by a proteolytic cleavage step within the NH2-terminal extension, which removes seven amino acids; and M represents COOH-terminally modified, mature a-factor with its NH2-terminal extension completely removed. It is notable that this analysis revealed a novel precursor species, P2, which was not predicted from the primary structure of the a-factor precursor.
P1, P2, and M All Behave as Peripheral Membrane Proteins
Prenylation is necessary but not sufficient to mediate the membrane localization of at least several CAAX proteins (Hancock et al., 1990). To determine whether a-factor intermediates are soluble or membrane-bound when prenylation is prevented by an a-factor CAAX mutation, cells were labeled with [35S]cysteine, and the total cellular extract (T) was separated into particulate (P) and soluble (S) fractions by centrifugation at 100,000 g. We examined a wild-type strain and strains that could not carry out prenylation, including two different a-factor CAAX mutants (mfa1-ΔVIA and mfa1-C33S) and a ram1 mutant that we had analyzed previously (He et al., 1991). As shown in Fig. 6 A, the mutant strains synthesize exclusively P0, which fails to reach the membrane (Fig. 6 A, lanes 4–6, 7–9, and 10–12). In contrast, in cells bearing wild-type MFA1, all the a-factor intermediates, P1, P2, and M, are mainly detected in the particulate (P) fraction (Fig. 6 A, lanes 1–3), suggesting that all of these species are membrane associated.
To further examine the nature of a-factor membrane association, the total cellular lysate was subjected to a variety of extraction conditions before centrifugation (Fig. 6 B). The nonionic detergent Triton X-100 efficiently solubilizes a-factor (Fig. 6 B, lanes 4–5), presumably because of the disruption of membrane structure. a-Factor was also solubilized by treatment with 0.1 M Na2CO3, pH 12.5 (Fig. 6 B, lanes 6 and 7). Since 0.1 M Na2CO3 normally only releases peripheral membrane proteins (Fujiki et al., 1982), this result suggests that a-factor does not behave as an integral membrane protein. a-Factor was not solubilized by 1.6 M urea (Fig. 6 B, lanes 8 and 9) nor by 1 M NaCl (Fig. 6 B, lanes 10 and 11), suggesting that, in contrast to typical peripheral membrane proteins, ionic interactions are not critical for a-factor membrane association. For the most part, all the a-factor intermediates exhibit similar fractionation and extraction properties, suggesting a common mechanism for their membrane association. More detailed subcellular fractionation analysis is needed to determine whether the different species of a-factor are localized to the same or to distinct membrane compartment(s).
COOH-terminal Prenylation Precedes and Is Obligatory for the NH2-terminal Processing of a-Factor
Having established the identity and fractionation properties of the a-factor biosynthetic intermediates, we next addressed the order of a-factor maturation events. Does COOH-terminal modification occur first, followed by NH2terminal processing, or vice versa? The answer is evident by inspection of Fig. 6 A, which provides a comparison of a-factor biogenesis profiles under conditions when prenylation of the a-factor precursor does or does not occur. When prenylation is blocked, either because of an a-factor CAAX mutation, mfa1-ΔVIA or C33S (Fig. 6 A, lanes 4 and 7), or because of a defective prenylation enzyme (ram1; Fig. 6 A, lane 10), only a single species of a-factor (P0) is generated. In contrast, three species (P1, P2, and M) are evident for wild-type MFA1 (Fig. 6 A, lane 1). The absence of the NH2-terminally cleaved intermediates, P2 and M, in the prenylation mutants indicates that prenylation is a prerequisite step in a-factor biogenesis, without which further NH2-terminal processing does not occur.
We also examined the order of appearance of P1 and P2 in a wild-type strain (Fig. 7). If NH2-terminal processing is contingent upon COOH-terminal prenylation, then we would expect that in a pulse-chase experiment, P2 would appear only after the formation of P1. Indeed, in a very brief (1-min) pulse labeling experiment, P2 becomes apparent only after 2–4 min of chase, whereas P1 is present at the very earliest chase time. By 8 min, about half of the P1 synthesized has been converted to P2, and further P1→ P2 conversion stops. These data are consistent with the results from the prenylation mutants, which indicate that COOH-terminal prenylation is required for the NH2terminal processing of a-factor.
Figure 7.

Kinetic analysis demonstrating P1→ P2 conversion of the a-factor precursor. Strain SM1762, which carries pSM463 (2μ MFA1), was pulse labeled with [35S]cysteine for 30 s, chased for the indicated time points (in minutes), and extracts were prepared. The intracellular fraction was subjected to immunoprecipitation, SDS-PAGE, and autoradiography.
Kinetic Analysis of a-Factor Biogenesis in Strains Expressing Chromosomal MFA1 or MFA2: P2→ M Conversion Occurs at Low Efficiency
To obtain an overall view of a-factor biogenesis, we conducted pulse-chase experiments using strains expressing either MFA1 or MFA2 from the chromosome. Cells were pulse labeled with [35S]cysteine for 5 min, subjected to a chase for up to 60 min, and samples from several time points were immunoprecipitated and analyzed by SDSPAGE (Fig. 8 A). The data was quantified by phosphorimager analysis and was used to quantitate two parameters of a-factor biogenesis (Table III): One parameter is designated processing efficiency and measures the percentage of mature a-factor, both intracellular M(I) and extracellular M(E), that is formed relative to the total amount of a-factor synthesized in a 5-min pulse. The second parameter is designated overall export efficiency and it measures the fraction of mature a-factor that is ultimately secreted to the extracellular culture fluid, relative to the total amount of a-factor synthesized. As discussed below, our measurements (Table III) suggest an inherent inefficiency in particular steps of the a-factor biogenesis pathway.
In general, the a-factor precursors encoded by the chromosomal MFA1 and MFA2 genes exhibit properties of maturation and export that are similar to one another (Fig. 8 A), although the total amount of a-factor produced from the chromosomal MFA2 gene is significantly less (by about threefold) than for MFA1. Another difference between MFA1- and MFA2-encoded a-factor is that only the P2 precursor, and not P1, can be detected for chromosomal MFA2 (Fig. 8 A). Most likely, the absence of P1 for chromosomally expressed MFA2 simply reflects the rapid and quantitative P1→ P2 conversion that occurs when the total amount of a-factor synthesized is low. When the MFA2 level is increased by expression from CEN and 2μ plasmids, the MFA2 P1 species becomes apparent (Fig. 8, B and C).
A striking feature apparent from the kinetic analysis of chromosomally expressed a-factor (Fig. 8 A) is that the overall export efficiency is surprisingly low. Less than 15% of the total amount of a-factor synthesized in a 5-min pulse (8% for MFA1 and 14% for MFA2, Table III) ever appears as mature a-factor in the culture fluid, even after export is essentially complete. What step(s) of a-factor biogenesis can account for this low level of efficiency? Visual inspection of the a-factor biogenesis gel profile (Fig. 8 A, also B and C) reveals that P2 is an abundant species, whereas only a modest amount of mature a-factor is generated. Quantitation of the processing efficiency confirms that only a small portion of the precursor initially synthesized is ultimately converted to mature a-factor (11% for MFA1, 30% for MFA2; Table III). Thus, the P2→ M NH2terminal processing step appears to be the least efficient, and as a result, the rate-limiting step in a-factor biogenesis; therefore, this is the step at which regulation, if it exists, would most likely be imposed. An additional factor that contributes to the low overall export efficiency of a-factor is that only about half of the intracellular M(I) that is generated is actually secreted to become extracellular M(E) (Fig. 8 A). The remaining intracellular M(I) simply remains inside the cell. It is not clear why the entire pool of intracellular mature a-factor is not capable of being exported.
Processing and Biogenesis of a-Factor Is Similar among Unrelated Strains of S. cerevisiae
To ensure that the low processing and biogenesis efficiency we observe is not caused by a peculiarity of our strain background, we examined a-factor biosynthesis in yeast strains derived from distinct genetic lineages. We compared the a-factor biogenesis profiles of three widely used S. cerevisiae strains: S288C (Mortimer and Johnston, 1986), SK1 (Alani et al., 1987), and Σ1278B (Brandriss and Magasanik, 1979) to that of SM1058 (also known as EG123 [Siliciano and Tatchell, 1984]), which is our main laboratory strain and is itself derived from S288C. Cells were pulse labeled with [35S]cysteine and were processed with or without a chase to examine the amount of a-factor initially synthesized, as well as the percentage of a-factor that is processed and exported to the culture fluid. The status of intracellular and extracellular a-factor was analyzed by immunoprecipitation, SDS-PAGE, and Phosphorimager analysis (Fig. 9). The overall pattern observed in each strain is the preponderance of precursor species (P1 and P2) versus mature a-factor (M), just as in our strain, SM1058. Thus, the conversion of the a-factor precursor into mature a-factor appears to be inefficient in all of the strains tested.
Expression of Increasing Amounts of a-Factor Results in a Back-Up at the P1→ P2 Processing Step
To determine whether the overall pattern of maturation or export is altered when a-factor is overexpressed, we examined a-factor biogenesis in strains expressing chromosomal, CEN, and 2μ MFA1 or MFA2. Overall, as can be seen in Fig. 8, A–C, very few differences are apparent between MFA1- and MFA2-encoded a-factor (with the exception that P1 is difficult to discern for chromosomal MFA2). Because of their overall similarity, further quantitation is reported for MFA1 only. For quantitation, samples from strains expressing increasing levels of MFA1 were compared on a single gel (Fig. 10 A). The total amount of a-factor synthesized in each case was determined. For CEN and 2μ MFA1 plasmids, the total amount of a-factor synthesized is 4- and 10-fold greater, respectively, than for chromosomal MFA1. The fourfold increase for CEN MFA1 versus chromosomal MFA1 is in good agreement with the heightened level of expression we have observed for other genes (e.g., STE6, STE14) cloned into CEN vectors, and is consistent with the notion that the copy number of these CEN plasmids is greater than one per cell.
Figure 10.
Comparison of the a-factor biosynthesis profile in strains expressing MFA1 at low (chromosomal), intermediate (CEN), or high (2μ) levels. To quantitate the total amount of precursor plus mature a-factor synthesized in each of the MFA1bearing strains shown in Fig. 8, and to determine the relative amounts of each a-factor intermediate, the intracellular immunoprecipitates from the 0-min chase time point and the extracellular immunoprecipitates from the 15-min chase point shown in Fig. 8 were electrophoresed side by side and quantitated by Phosphorimager analysis. A shows the Phosphorimager scan. B shows the quantitation of a-factor intermediates from A. Values are expressed relative to chromosomal intracellular mature a-factor, M(I), which is set at 1.0. M(E) refers to extracellular mature a-factor. These data illustrate two points: the low level of mature (M(I) + M(E)) relative to precursor species, indicative of the low efficiency of P2→ M processing, and the buildup of P1 when a-factor is expressed at very high levels.
Strikingly, as a-factor production increases, the level of P1 increases disproportionately to that of other species (Fig. 10 A); whereas the species P2, M(I), and M(E) each undergo a modest increase in level (of about two- to fourfold as copy number increases from chromosomal to 2μ MFA1), P1 accumulates to extremely high levels (23-fold; Fig. 10 B). Thus, when expression of a-factor is relatively low, it is the inherent inefficiency of the P2→ M processing step that limits the amount of mature a-factor that can be generated. When a-factor is overexpressed, however, a more severe bottleneck develops at the preceding NH2terminal processing step (P1→ P2), resulting in the accumulation of a disproportionately high level of P1. Hence, both of the a-factor NH2-terminal processing steps, P1→ P2 and P2→ M, have the capacity to serve as potential points of regulation to limit (or increase) the amount of mature a-factor available for export.
α-Factor Induction Does Not Affect the Processing or Export Efficiency of a-Factor
The transcription of many genes involved in mating, including MFA1 and MFA2, is stimulated upon pheromone treatment (Sprague and Thorner, 1992). To examine whether any steps in the a-factor biosynthetic pathway are specifically affected by α-factor induction, we incubated cells with α-factor for varying lengths of time, and carried out metabolic labeling and immunoprecipitation of a-factor, as described in Materials and Methods. As can be seen in Fig. 11, within 5 min after treatment with α-factor, we detected a significant increase in the overall level a-factor production (Fig. 11, lane 1 vs. lane 2). The induction reached its peak 30 min after α-factor addition (Fig. 11, lane 3) and a-factor production returned to its lower constitutive level by 90 min (Fig. 11, lane 4). Quantitative analysis (data not shown) revealed that both the precursor (P1 and P2) and mature (M) species of a-factor were elevated to a comparable level at each time point (about fivefold at 30 min). However, neither “processing efficiency” nor the “export efficiency” appear to vary upon α-factor treatment. Thus, the increase in extracellular a-factor after induction (Fig. 11, lane 1 vs. lane 3) is caused primarily by the heightened expression of the a-factor genes, rather than by any alteration of the a-factor processing or biogenesis efficiency. Therefore, it does not appear to be the case that the excess intracellular pool of a-factor precursor becomes “recruited” after pheromone induction.
Figure 11.

Examination of a-factor biogenesis after α-factor induction. Strain SM1227 (MFA1 mfa2::URA3) was preincubated with 25 μM α-factor (except for the 0-min sample). At the indicated times, which represent minutes after the addition of pheromone, cells were pulse labeled with [35S]cysteine for 5 min, and the label was chased for 45 min. (For the 0-min control sample, the pulse chase was carried out in the absence of any α-factor.) The intracellular (I) fraction was prepared from a portion of the 5-min pulse-labeled cells to examine the total amount of a-factor synthesized, and the extracellular (E) fraction was prepared from an equivalent portion of the culture that had completed the 45-min chase to examine the portion of the a-factor synthesized in the pulse that ultimately underwent maturation and export. Fractions were immunoprecipitated with a-factor antiserum and subjected to SDS-PAGE analysis.
Discussion
The maturation and secretion of the yeast mating pheromone a-factor represents a novel paradigm for the biogenesis of a secreted signaling molecule. In the present study, we wished to elucidate the major events of the a-factor biogenesis pathway by characterizing a-factor biosynthetic intermediates. Using metabolic labeling, immunoprecipitation, and SDS-PAGE analysis, we identified four distinct a-factor intermediates (Fig. 2), determined the structure of these species (Fig. 5), including the COOH-terminal modification status and NH2-terminal residue of each (Figs. 3 and 4, respectively), examined their membrane association (Fig. 6 B), and determined their order of formation with respect to COOH- and NH2-terminal processing events (Fig. 6 A). A notable insight that emerged from this analysis was the identification of an unanticipated a-factor biosynthetic intermediate, P2, in which a portion of the NH2terminal extension of the a-factor precursor (the first 7 of 21 residues) is removed, presumably by a specific endoprotease. The existence of this novel partially NH2-terminally processed a-factor intermediate predicts that not just one, but two NH2-terminal cleavage steps occur during a-factor biogenesis.
A Model for a-Factor Biogenesis
The identification of a-factor biosynthetic intermediates carried out here, together with other studies identifying components of the a-factor maturation and export machinery, allow us to formulate a working model for the a-factor biogenesis pathway (Fig. 12). The precursor and mature species of a-factor that we characterized in this study are presented together with the cellular components that are postulated to mediate their formation. According to our model, the unmodified a-factor precursor (P0) first undergoes COOH-terminal modification (prenylation, proteolytic trimming, and carboxyl methylation) mediated by Ram1p/Ram2p (He et al., 1991; Schafer et al., 1990), the AAX protease (Ashby et al., 1992; Hrycyna and Clarke, 1992), and Ste14p (Hrycyna et al., 1991; Sapperstein et al., 1994), respectively, to yield the fully COOH-terminally modified and membrane associated precursor (P1). (It should be noted that our analysis has not identified distinct COOH-terminal intermediates that are only partly modified, either because their formation and disappearance is very rapid or because they do not exhibit distinctive mobilities in our gel system.) Subsequent to the completion of COOH-terminal modification, NH2-terminal proteolytic processing occurs and appears to involve two sequential steps. The first NH2-terminal cleavage event is the unanticipated processing step uncovered by this study, which generates the NH2-terminally “clipped” intermediate (P2); we provide evidence elsewhere that formation of P2 is dependent on the Ste24p protease (Fujimura-Kamada et al., 1997). The second NH2-terminal processing event is Axl1p dependent (Adames et al., 1995) and generates mature a-factor, which is then exported from the cell by the ABC transporter Ste6p (Berkower and Michaelis, 1991; Kuchler et al., 1989). It is this extracellular a-factor species that interacts with MATα cells to initiate the mating process. Several aspects of the a-factor biogenesis model depicted in Fig. 12 are discussed below in the context of the findings presented in this study.
Figure 12.

Model for the pathway of a-factor biogenesis. The precursor species (P0, P1, and P2) and mature (M) species of a-factor are indicated, and the components of the a-factor biogenesis machinery are shown. During a-factor biogenesis, unmodified a-factor precursor (P0) undergoes COOH-terminal modification (prenylation, proteolytic trimming of AAX, and carboxyl methylation) to yield the fully modified membrane-associated species (P1). Next, NH2-terminal proteolytic processing occurs in two distinct steps, the first removing seven residues from the NH2 terminus to yield the intermediate precursor (P2), and the second cleavage generating mature (M) a-factor, which undergoes export from the cell. The biogenesis components are indicated. The CAAX processing machinery includes the Ram1p/Ram2p farnesyltransferase, the genetically unidentified AAX protease, and the Ste14p methyltransferase. The gene products required for the NH2-terminal processing of the a-factor precursor are the Ste24p and Axl1p proteases. The export of a-factor is mediated by the ABC transporter Ste6p. Whereas Ram1p/Ram2p resides in the cytosol, all the other a-factor biogenesis components appear to be membrane associated, although the precise cellular membrane(s) in which they reside are not known. For the sake of simplicity, a single cellular membrane is shown here. It is unlikely, however, that the plasma membrane is the site of a-factor processing. For more details, see the Discussion.
The NH2-terminal Processing of a-Factor Involves Two Sequential Steps
The unexpected a-factor biosynthetic intermediate (P2) identified in this work reveals that the NH2-terminal cleavage of the a-factor precursor is more complex than we originally envisioned and takes place in two sequential steps. The first cleavage (P1→ P2) occurs between residues T7 and A8 of the MFA1-encoded a-factor precursor. This event is followed by a second cleavage (P2→ M) between residues N21 and Y22 to yield mature a-factor.
But is P2 truly an obligatory intermediate in the biogenesis of mature a-factor, or is it a dead-end product? In principle, demonstration of a precursor–product relationship between P2 and M by pulse-chase kinetic studies would resolve this issue, yet the fact that the P2 does not quantitatively chase to M (Fig. 8) precludes a clear-cut answer to this question. Other studies in our laboratory do, however, provide convincing evidence that P2 is indeed an essential intermediate leading to the formation of mature (M) a-factor. First, we have isolated a-factor mutants (i.e., mfa1-A8G) that are blocked in the P1→ P2 processing step and are unable to generate mature (M) a-factor (Nouvet, F., A. Kistler, and S. Michaelis, manuscript in preparation). Second, we have isolated a new sterile mutant (ste24) that is defective in the cellular machinery that carries out P1→ P2 processing. This mutant also fails to generate mature (M) a-factor (Fujimura-Kamada et al., 1997). Together, these “substrate” and “machinery” mutants provide compelling evidence that mature a-factor is derived from P2, not P1, and that the P1→ P2 cleavage step (and thus the P2 intermediate) cannot be circumvented.
Recently, the P2→ M step of a-factor processing was shown to be dependent on the AXL1 gene product, which also plays a role in axial bud site selection in haploid yeast (Adames et al., 1995; Fujita et al., 1994). Axl1p belongs to the human insulin-degrading enzyme family of zinc metalloproteases, known to cleave small peptide substrates in vitro and in vivo, most notably insulin (Becker and Roth, 1995). In an axl1 mutant, P2→ M processing fails to occur, whereas Ste24p-dependent P1→ P2 processing is unaffected, indicating that the two steps of a-factor NH2-terminal processing involve distinct proteases (Adames et al., 1995; Fujimura-Kamada et al., 1997). Like Axl1p, Ste24p is a zinc metalloprotease. It will be of considerable interest to determine the cellular location of the Axl1p and Ste24p proteases and to ask whether they directly interact with one another and have other common substrates, aside from a-factor.
We demonstrate here that the P1→ P2 cleavage of the a-factor precursor encoded by MFA1 occurs between residues T7 and A8, presumably by an endoproteolytic cleavage mechanism. Interestingly, we found that a mutation in the mature portion of MFA1, I23M, which alters a residue quite distant from the P1→ P2 cut site, results in a slightly altered cleavage site; cleavage occurs between residues T5 and A6, instead of T7 and A8 (Fig. 4). It is possible that both TA sites are legitimate cleavage sites, with the former preferred by wild-type MFA1, and the preference reversed in the mutant context. The MFA2-encoded a-factor precursor, like that of MFA1, also gives rise to a P2 intermediate (Fig. 8), although its precise cleavage site has not yet been determined. Ultimately, a systematic analysis of a-factor missense mutations located at, near, and distant from the P1→ P2 and P2→ M cleavage sites will allow us to define the minimal rules for recognition and cleavage of a-factor by its NH2-terminal processing proteases.
Dependence of NH2-terminal Processing upon COOH-terminal Prenylation
COOH-terminal prenylation is known to play a critical role for the membrane association of CAAX proteins (Zhang and Casey, 1996). We provide evidence in this study and elsewhere that in the biogenesis of the a-factor precursor, COOH-terminal prenylation is essential for subsequent NH2-terminal processing (He et al., 1991). This conclusion is based on the analysis both of a-factor mutants and of prenyltransferase mutants. In this study, we generated two a-factor CAAX mutants, mfa1-C33S and mfa1-ΔVIA, in which prenylation of a-factor does not occur. We show that in these mutants, the nonprenylated precursor P0 fails to associate with the membrane and fails to undergo NH2-terminal proteolytic processing (Fig. 6). Likewise, in mutants defective in the cellular prenylation machinery (Ram1p-Ram2p), the P0 a-factor precursor is not NH2-terminally processed (He et al., 1991). Together, these data provide convincing evidence that COOH-terminal prenylation is a prerequisite for subsequent NH2terminal events of a-factor biogenesis. For other CAAX proteins, prenylation also serves as a key modification that is required for downstream events. For instance, for Ras proteins that are modified by both a prenyl and a palmitate moiety, the addition of palmitate requires previous prenylation (Hancock et al., 1990). Likewise, a proteolytic processing event required for the functional maturation of the mammalian and chicken nuclear lamin A protein depends on its previous prenylation (Beck et al., 1990; Hennekes and Nigg, 1994; Sinensky et al., 1994).
Why might NH2-terminal proteolytic processing of the a-factor precursor depend on COOH-terminal prenylation? One possibility is that the machinery that carries out NH2-terminal processing is membrane associated. According to this view, NH2-terminal processing can only occur when a-factor reaches the membrane, which in turn requires that it be prenylated. Indeed, evidence supporting a membrane location for most of the a-factor processing machinery is accumulating (as discussed in the next section). A second possibility is that the COOH-terminal prenyl moiety serves as a specific recognition determinant for the a-factor NH2-terminal protease(s), such that cleavage cannot occur if the prenyl group is absent from the precursor, regardless of its localization. Investigation of this possibility will require in vitro characterization of the substrate recognition properties of the a-factor NH2-terminal proteases. A role for prenylation in modulating protein–protein interactions is not unprecedented, and has been demonstrated for Ras and adenylate cyclase in yeast (Horiuchi et al., 1992), as well as for substrate recognition by the Ste14p methyltransferase (Hrycyna et al., 1991; Tan et al., 1991).
It should be noted that in contrast to prenylation, carboxyl methylation is not required for the NH2-terminal processing of a-factor, nor for its membrane association, because in a ste14 mutant, which lacks methyltransferase activity, the NH2-terminal processing and membrane association of a-factor occur normally (Sapperstein et al., 1994). Only the export of a-factor is blocked in ste14 mutants, which has led to the suggestion that the methyl group may provide a specific recognition determinant for the a-factor transporter Ste6p (Sapperstein et al., 1994). Likewise, studies on the membrane localization of Ras indicate that carboxyl methylation contributes to, but is not absolutely required for, its membrane association and palmitoylation (Hancock et al., 1991).
The Biogenesis of a-Factor Is Intimately Associated with Membrane(s)
Where do the events of a-factor biogenesis take place? Our finding that all prenylated a-factor species, P1, P2, and M, are membrane associated suggests that the cellular machinery that mediates a-factor maturation is likely to be localized to a membrane, as depicted in Fig. 12. This view is further supported by the molecular and biochemical characterization of a-factor biogenesis components known to function after the Ram1p/Ram2p prenyltransferase, which appears to be cytosolic. The COOH-terminal biogenesis components (the AAX protease and Ste14p), as well as the NH2-terminal proteases (Ste24p and Axl1), all fractionate with membranes (Ashby et al., 1992; Hrycyna and Clarke, 1990, 1992; Schmidt, W., J. Romano, K. FujimuraKamada, and S. Michaelis, unpublished observations). Furthermore, hydropathy analysis of Ste14p and Ste24p predict multiple membrane spans, suggesting that these are both integral membrane proteins (Fujimura-Kamada et al., 1997; Sapperstein et al., 1994). Together, these results implicate a membrane location as the major site of a-factor maturation. Analysis of a-factor biogenesis in a ste6 mutant indicates that the maturation of a-factor is completed before its membrane translocation; thus, a-factor processing must be carried out on the cytosolic face of a membrane(s).
What is the precise cellular site where a-factor biogenesis takes place? While Fig. 12 schematically depicts biogenesis of a-factor occurring at the plasma membrane, other possibilities can certainly be considered. Either the plasma membrane or the membranes of intracellular organelles could provide a site for the processing of the a-factor precursor. Presumably, the COOH-terminal events shared by Ras, a-factor, and other prenylated proteins occur in a common location, although very little is known about the intracellular trafficking of any CAAX-terminating proteins. Detailed fractionation analysis is underway to assign the precise cellular location(s) of the several a-factor intermediates, as well as to determine whether COOH-terminal trimming, methylation, and NH2-terminal proteolytic processing occur on the same or distinct cellular membranes. Ste6p is localized to the plasma membrane in endocytosis mutants (end3, end4, and sac6) that are blocked for Ste6p internalization to the vacuole (Berkower et al., 1994; Kolling and Hollenberg, 1994; Paddon et al., 1996). In these mutants, a-factor export occurs normally, suggesting that the plasma membrane is the likely site of a-factor export.
Is There a Fundamental Connection between the COOH-terminal Modification of a-Factor and its Nonclassical Mode of Export?
Two features of a-factor distinguish it from most other extracellular proteins. One is that a-factor is prenylated and methylated; these modifications appear to be critical for receptor binding because a-factor lacking either modification is severely reduced in activity (Marcus et al., 1991). The second is that the export of a-factor is mediated by a nonclassical exporter, the ABC transporter Ste6p, instead of by the machinery of the classical secretory pathway (Michaelis, 1993). Close mammalian relatives of Ste6p, human MDR1 and mouse MDR3, mediate the transport across membranes of diverse hydrophobic compounds, including hydrophobic drugs and prenylcysteine derivatives (Gottesman and Pastan, 1993; Zhang et al., 1994). Indeed, mouse MDR3 can complement the a-factor export defect of a ste6 mutant (Raymond et al., 1992). A related protein, human MDR2, functions as a phosphatidylcholine flippase (Reutz and Gros, 1994). It is probable that the a-factor transporter Ste6p and the mammalian MDR proteins share common mechanistic features that make them particularly suitable for the translocation of highly hydrophobic substrates. Notably, at this time, a-factor is the only secreted protein or peptide known to be prenylated. An interesting possibility is that a-factor might actually be precluded from undergoing membrane translocation via the classical secretory machinery (Rapoport, 1992); for instance, a-factor may be sufficiently hydrophobic because of its prenyl group, so that it would actually “clog” the ER translocation pore. Instead, a-factor and certain other hydrophobic compounds may thus have an absolute requirement for a novel, nonclassical translocation mechanism, such as that provided by the ABC transporter Ste6p.
The a-Factor Biogenesis Process Exhibits Surprisingly Low Efficiency and Distinctive Kinetic Properties
A striking aspect of the a-factor biogenesis pathway revealed by kinetic analysis (Fig. 8) is its low efficiency. Only a small proportion (<15%) of the a-factor precursor initially synthesized during a pulse label actually appears in the culture fluid as mature a-factor. The low biogenesis efficiency can be explained primarily by the inefficiency of the P2→ M processing step, in that most of the P2 remains uncleaved and is simply slowly degraded over time. Similarly, the a-factor export step is also somewhat inefficient in that we observe that a substantial fraction of intracellular mature a-factor is never exported from the cell (Fig. 8). Why is a-factor biogenesis so inefficient at multiple steps in the processing pathway? One view is that a-factor biogenesis is processive, requiring an intimate “hand-off” of the substrate from one component to the next. Accordingly, any newly formed a-factor intermediate that is not successfully “handed off” from one biogenesis component to the next falls “out of the loop” and cannot reenter the pathway. We have demonstrated that the low efficiency of a-factor processing and export is not simply a peculiarity of our strain background, since it is also the case in strains from a variety of genetic lineages; in addition both MFA1- and MFA2-encoded a-factor exhibit this property.
The P2→ M NH2-terminal processing step and, under certain circumstances (i.e., overexpression), the P1→ P2 step, can be considered to be rate limiting for production of mature a-factor. As such, these would be ideal steps at which to exert regulation. One obvious time at which optimizing a-factor production might be critical is during mating. We examined a-factor biogenesis after α-factor treatment. Although we did detect a modest increase in the overall level of a-factor that was synthesized, processed, and secreted, the ratio of a-factor intermediates (and thus the processing efficiency) was not dramatically different from untreated cells. However, the finding that α-factor treatment alone does not affect these parameters may not necessarily reflect a lack of modulation of the a-factor pathway during actual mating. It will be important to look at the processing efficiency during the mating process per se, particularly in light of recent evidence that a high level of a-factor is necessary for efficient completion of cell fusion during late steps in mating (Brizzio et al., 1996).
Detection of a-Factor Intermediates
The success of this study relied upon the development of an electrophoresis system suitable to achieve the high resolution separation of metabolically labeled a-factor intermediates, which are difficult to separate by conventional gel electrophoresis because of their very small size (ranging between 12 and 36 amino acids) and their hydrophobicity. We expect that the gel system developed here for the separation of a-factor intermediates will prove to be useful for examining the biogenesis of other small prenylated peptides, including fungal mating pheromones, and perhaps as yet undiscovered prenylated mammalian hormones.
Implications from a-Factor for the Biogenesis of Other Fungal Mating Pheromones
Despite its seeming novelty, the a-factor biogenesis pattern may actually be commonplace in eukaryotic organisms. In recent years, more than half a dozen fungal mating pheromones have been shown either by DNA sequence analysis or by mass spectrometry to share certain features in common with a-factor, notably a COOH-terminal CAAX motif or a prenylated cysteine (with or without a carboxyl methyl group; reviewed in Caldwell et al., 1995). Examples of such pheromones include: Schizosaccharomyces pombe M-factor (Davey, 1992), Ustilago maydis a1 and a2 factors (Bolker et al., 1992; Spellig et al., 1994), Cryptococcus neoformans α-factor (Moore and Edman, 1993), Tremella mesenterica tremerogens A-10 and a-13 (Sakagami et al., 1981), Tremella brasilliensis tremerogens A-9291-I, 2, and 3 (Ishibashi et al., 1984), and Rhodotorucine toruloides rhodotorucine A (Akada et al., 1989; Kamiya et al., 1979). We have recently discovered another fungal pheromone, L-factor, produced by the yeast Saccharomycodes ludwigii. Interestingly, L-factor is capable of inducing mating type-specific responses in MATα cells of S. cerevisiae despite the fact S. ludwigii and S. cerevisiae do not appear to be closely related organisms (Kistler, A., and S. Michaelis, manuscript in preparation).
It is of historical note that the first evidence that isoprenoids could modify polypeptides came from studies of the fungal pheromones tremerogen and rhodotorucine (Kamiya et al., 1979; Sakagami et al., 1981). Although originally thought to represent a fungal specialization, these findings heralded studies that ultimately led to the discovery that numerous eukaryotic proteins are prenylated and that prenylation imparts functionally significant properties to these proteins (Zhang and Casey, 1996). In recent years, DNA sequence analysis has highlighted yet another feature that is common to at least several fungal pheromones (i.e., S. pombe M-factor, U. maydis a1 and a2 factors, and C. neoformans α-factor) in addition to their CAAX motif. Like the S. cerevisiae a-factor precursor, these pheromones are predicted to contain an NH2-terminal extension that lacks the features of a canonical signal sequence. Thus, it is tempting to propose, by analogy to S. cerevisiae a-factor, that these pheromones will all prove to be COOHterminally modified, NH2-terminally processed, and secreted by a nonclassical mechanism. Were this the case, then these pheromones might make use of a similar array of biogenesis components as does S. cerevisiae a-factor. To date, evidence supporting this notion is most compelling for S. pombe M-factor. Mass spectrometry has shown that M-factor is a prenylated and carboxyl methylated nonamer, and the M-factor gene sequences provide evidence for an NH2-terminal extension (30 and 32 amino acids long for mfm1 and mfm2, respectively) in addition to a COOHterminal CAAX motif (Davey, 1992). Perhaps not surprisingly, two S. pombe genes required for M-factor production, mam4 and mam1, have been shown to encode homologues of S. cerevisiae a-factor biogenesis components STE14 and STE6, respectively (Davey, J., and M. Yamamoto, personal communication).
It is not understood why S. cerevisiae a-factor and α-factor use distinct pathways for biogenesis despite their functional equivalence. Interestingly, while S. pombe cells of opposite mating type (M and P), like S. cerevisiae MAT a and MATα cells, produce a prenylated and a nonprenylated pheromone, respectively, this situation is not universal among fungi. In U. maydis, for instance, cells of the opposite mating type both encode mating factors that terminate with a CAAX motif (Spellig et al., 1994). Interestingly, in the case of the pathogenic fungus C. neoformans, pathogenicity is cell type specific, with the α cell type exhibiting the higher degree of virulence (Moore and Edman, 1993). If it is indeed the case that the C. neoformans α-factor pheromone is itself directly responsible for the organism's pathogenicity, then understanding the mechanism of pheromone biogenesis might provide a rational basis for therapeutic intervention.
Acknowledgments
We are grateful to Walter Schmidt, Konomi Fujimura-Kamada, Julia Romano, and Nancy Craig for comments on this manuscript, and to B.J. Earles, who carried out the Edman degradation reactions.
This work was supported by a grant (GM41223) to S. Michaelis from the National Institutes of Health.
Abbreviations used in this paper
- ABC
ATP-binding cassette
- E
extracellular
- I
intracellular
- M
mature species of a-factor
- M(E)
extracellular mature a-factor
- M(I)
intracellular mature a-factor
- P0
unmodified a-factor precursor
- P1
fully COOH-terminally modified precursor species of a factor
- P2
fully COOH-terminally modified, partially NH2-terminally cleaved precursor species of a-factor
- PTH
phenylthiohydrantoin
- SAM
S-adenosyl-l-methionine
- 5-FOA
5-fluoroorotic acid
Footnotes
Please address all correspondence to S. Michaelis, Department of Cell Biology and Anatomy, The Johns Hopkins University School of Medicine, 725 North Wolfe Street, Baltimore, MD 21205. Tel.: (410) 955-8286. Fax: (410) 955-4129. E-mail: susan_michaelis@quail.bs.jhu.edu
References
- Adames N, Blundell K, Ashby MN, Boone C. Role of yeast insulin-degrading enzyme homologs in propheromone processing and bud site selection. Science (Wash DC) 1995;270:464–467. doi: 10.1126/science.270.5235.464. [DOI] [PubMed] [Google Scholar]
- Akada R, Minomi K, Kai J, Yamashita I, Miyakawa T, Fukui S. Multiple genes coding for precursors of rhodotorucine A, a farnesyl peptide mating pheromone of the basidiomycetous yeast Rhodosporidium toruloides. . Mol Cell Biol. 1989;9:3491–3498. doi: 10.1128/mcb.9.8.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alani E, Cao L, Kleckner N. A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics. 1987;116:541–545. doi: 10.1534/genetics.112.541.test. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderegg RJ, Betz R, Carr SA, Crabb JW, Duntze W. Structure of Saccharomyces cerevisiae mating hormone a-factor. Identification of S-farnesyl cysteine as a structural component. J Biol Chem. 1988;263:18236–18240. [PubMed] [Google Scholar]
- Ashby MN, King DS, Rine J. Endoproteolytic processing of a farnesylated peptide in vitro. Proc Natl Acad Sci USA. 1992;89:4613–4617. doi: 10.1073/pnas.89.10.4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck LA, Hosick TJ, Sinensky M. Isoprenylation is required for the processing of the lamin A precursor. J Cell Biol. 1990;110:1489–1499. doi: 10.1083/jcb.110.5.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker AB, Roth RA. Insulysin and pitrilysin: insulin-degrading enzymes of mammals and bacteria. Methods Enzymol. 1995;248:693–703. doi: 10.1016/0076-6879(95)48046-3. [DOI] [PubMed] [Google Scholar]
- Berkower C, Loayza D, Michaelis S. Metabolic instability and constitutive endocytosis of STE6, the a-factor transporter of Saccharomyces cerevisiae. . Mol Biol Cell. 1994;5:1185–1198. doi: 10.1091/mbc.5.11.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkower C, Michaelis S. Mutational analysis of the yeast a-factor transporter STE6, a member of the ATP binding cassette (ABC) protein superfamily. EMBO (Eur Mol Biol Organ) J. 1991;10:3777–3785. doi: 10.1002/j.1460-2075.1991.tb04947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolker M, Urban M, Kahmann R. The a mating type locus of U. maydisspecifies cell signaling components. Cell. 1992;68:441–450. doi: 10.1016/0092-8674(92)90182-c. [DOI] [PubMed] [Google Scholar]
- Brake, A.J., C. Brenner, R. Najarian, P. Laybourn, and J. Merryweather. 1985. Structure of genes encoding precursors of the yeast peptide mating pheromone a-factor. In Protein Transport and Secretion. M. Gething, editor. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. 103–108.
- Brandriss MC, Magasanik B. Genetics and physiology of proline utilization in Saccharomyces cerevisiae: enzyme induction by proline. J Bacteriol. 1979;140:498–503. doi: 10.1128/jb.140.2.498-503.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizzio V, Gammie AE, Nijbroek G, Michaelis S, Rose MD. Cell fusion during yeast mating requires high levels of a-factor and α-factor mating pheromones. J Cell Biol. 1996;135:1727–1739. doi: 10.1083/jcb.135.6.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell GA, Naider F, Becker JM. Fungal lipopeptide mating pheromones: a model system for the study of protein prenylation. Microbiol Rev. 1995;59:406–422. doi: 10.1128/mr.59.3.406-422.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadaban MJ, Martinez AA, Shapira SK, Chou J. Beta-galactosidase gene fusions for analyzing gene expression in Escherichia coliand yeast. Methods Enzymol. 1983;100:293–308. doi: 10.1016/0076-6879(83)00063-4. [DOI] [PubMed] [Google Scholar]
- Chen, P. 1993. Biogenesis of yeast mating pheromone a-factor. PhD thesis. Johns Hopkins University School of Medicine, Baltimore, MD, 251 pp.
- Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene (Amst) 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- Clarke S. Protein isoprenylation and methylation at carboxyl-terminal cysteine residues. Annu Rev Biochem. 1992;61:355–386. doi: 10.1146/annurev.bi.61.070192.002035. [DOI] [PubMed] [Google Scholar]
- Clarke, S., J.P. Vogel, R.J. Deschenes, and J. Stock. 1988. Posttranslational modification of the Ha-ras oncogene protein: evidence for a third class of protein carboxyl methyltransferases [Published erratum appears in Proc. Natl. Acad. Sci. USA. 85:7556 (1988).] [DOI] [PMC free article] [PubMed]
- Davey J. Mating pheromones of the fission yeast Schizosaccharomyces pombe: purification and structural characterization of M-factor and isolation and analysis of two genes encoding the pheromone. EMBO (Eur Mol Biol Organ) J. 1992;11:951–960. doi: 10.1002/j.1460-2075.1992.tb05134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elble R. A simple and efficient procedure for transformation of yeasts. BioTechniques. 1992;13:18–20. [PubMed] [Google Scholar]
- Fujiki Y, Hubbard AL, Fowler S, Lazarow PB. Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J Cell Biol. 1982;93:97–102. doi: 10.1083/jcb.93.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura-Kamada K, Nouvet FJ, Michaelis S. A novel membraneassociated metalloprotease, Ste24p, is required for the NH2-terminal processing of the yeast mating pheromone a-factor. J Cell Biol. 1997;136:271–285. doi: 10.1083/jcb.136.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita A, Oka C, Arikawa Y, Katagai T, Tonouchi A, Kuhara S, Misumi Y. A yeast gene necessary for bud-site selection encodes a protein similar to insulin-degrading enzymes. Nature (Lond) 1994;372:567–570. doi: 10.1038/372567a0. [DOI] [PubMed] [Google Scholar]
- Fujiyama A, Matsumoto K, Tamanoi F. A novel yeast mutant defective in the processing of ras proteins: assessment of the effect of the mutation on processing steps. EMBO (Eur Mol Biol Organ) J. 1987;6:223–228. doi: 10.1002/j.1460-2075.1987.tb04742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller, R., A. Brake, R. Sterne, R. Kunisawa, D. Barnes, M. Flessel, and J. Thorner. 1986. Post-translational processing events in the maturation of yeast pheromone precursors. In Yeast Cell Biology. J. Hicks, editor. Alan R. Liss, New York. 461–476.
- Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- Hall MN, Hereford L, Herskowitz I. Targeting of E. colibetagalactosidase to the nucleus in yeast. Cell. 1984;36:1057–1065. doi: 10.1016/0092-8674(84)90055-2. [DOI] [PubMed] [Google Scholar]
- Hanahan D. Studies on transformation of Escherichia coliwith plasmids. J Mol Biol. 1983;166:557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- Hancock JF, Cadwallader K, Marshall CJ. Methylation and proteolysis are essential for efficient membrane binding of prenylated p21Kras(B) EMBO (Eur Mol Biol Organ) J. 1991;10:641–646. doi: 10.1002/j.1460-2075.1991.tb07992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock JF, Paterson H, Marshall CJ. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell. 1990;63:133–139. doi: 10.1016/0092-8674(90)90294-o. [DOI] [PubMed] [Google Scholar]
- He B, Chen P, Chen SY, Vancura KL, Michaelis S, Powers S. RAM2, an essential gene of yeast, and RAM1 encode the two polypeptide components of the farnesyltransferase that prenylates a-factor and Ras proteins. Proc Natl Acad Sci USA. 1991;88:11373–11377. doi: 10.1073/pnas.88.24.11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennekes H, Nigg EA. The role of isoprenylation in membrane attachment of nuclear lamins. A single point mutation prevents proteolytic cleavage of the lamin A precursor and confers membrane binding properties. J Cell Sci. 1994;107:1019–1029. doi: 10.1242/jcs.107.4.1019. [DOI] [PubMed] [Google Scholar]
- Horiuchi H, Kaibuchi K, Kawamura M, Matsuura Y, Suzuki N, Kuroda Y, Kataoka T, Takai Y. The posttranslational processing of ras p21 is critical for its stimulation of yeast adenylate cyclase. Mol Cell Biol. 1992;12:4515–4520. doi: 10.1128/mcb.12.10.4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrycyna CA, Clarke S. Farnesyl cysteine C-terminal methyltransferase activity is dependent upon the STE14 gene product in Saccharomyces cerevisiae. . Mol Cell Biol. 1990;10:5071–5076. doi: 10.1128/mcb.10.10.5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrycyna CA, Sapperstein SK, Clarke S, Michaelis S. The Saccharomyces cerevisiae STE14 gene encodes a methyltransferase that mediates C-terminal methylation of a-factor and RAS proteins. EMBO (Eur Mol Biol Organ) J. 1991;10:1699–1709. doi: 10.1002/j.1460-2075.1991.tb07694.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrycyna CA, Clarke S. Maturation of isoprenylated proteins in Saccharomyces cerevisiae.Multiple activities catalyze the cleavage of the three carboxyl-terminal amino acids from farnesylated substrates in vitro. J Biol Chem. 1992;267:10457–10464. [PubMed] [Google Scholar]
- Ishibashi Y, Sakagami Y, Isogai A, Suzuki A. Structures of Tremerogens A-9291-I and A-9291-VIII: peptidyl sex hormones of Tremella brasiliensis. . Biochemistry. 1984;23:1399–1404. doi: 10.1139/o83-101. [DOI] [PubMed] [Google Scholar]
- Julius D, Schekman R, Thorner J. Glycosylation and processing of prepro-α-factor through the yeast secretory pathway. Cell. 1984;36:309–318. doi: 10.1016/0092-8674(84)90224-1. [DOI] [PubMed] [Google Scholar]
- Kamiya Y, Sakurai A, Tamura S, Takahashi N, Tsuchiya E, Abe K, Fukui S. Structure of Rhodotorucine A, a peptidyl factor, inducing mating tube formation in Rhodosporidium toruloides. . Agricult Biol Chem. 1979;43:363–369. [Google Scholar]
- Kolling R, Hollenberg CP. The ABC-transporter Ste6 accumulates in the plasma membrane in a ubiquitinated form in endocytosis mutants. EMBO (Eur Mol Biol Organ) J. 1994;13:3261–3271. doi: 10.1002/j.1460-2075.1994.tb06627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchler K, Sterne RE, Thorner J. Saccharomyces cerevisiaeSTE6 gene product: a novel pathway for protein export in eukaryotic cells. EMBO (Eur Mol Biol Organ) J. 1989;8:3973–3984. doi: 10.1002/j.1460-2075.1989.tb08580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient sitespecific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (Lond) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Marcus S, Caldwell GA, Miller D, Xue CB, Naider F, Becker JM. Significance of C-terminal cysteine modifications to the biological activity of the Saccharomyces cerevisiae a-factor mating pheromone. Mol Cell Biol. 1991;11:3603–3612. doi: 10.1128/mcb.11.7.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr RS, Blair LC, Thorner J. Saccharomyces cerevisiae STE14 gene is required for COOH-terminal methylation of a-factor mating pheromone. J Biol Chem. 1990;265:20057–20060. [PubMed] [Google Scholar]
- Marsh L, Neiman AM, Herskowitz I. Signal transduction during pheromone response in yeast. Annu Rev Cell Biol. 1991;7:699–728. doi: 10.1146/annurev.cb.07.110191.003411. [DOI] [PubMed] [Google Scholar]
- McGrath JP, Varshavsky A. The yeast STE6gene encodes a homologue of the mammalian multidrug resistance P-glycoprotein. Nature (Lond) 1989;340:400–404. doi: 10.1038/340400a0. [DOI] [PubMed] [Google Scholar]
- Michaelis S. STE6, the yeast a-factor transporter. Semin Cell Biol. 1993;4:17–27. doi: 10.1006/scel.1993.1003. [DOI] [PubMed] [Google Scholar]
- Michaelis S, Herskowitz I. The a-factor pheromone of Saccharomyces cerevisiaeis essential for mating. Mol Cell Biol. 1988;8:1309–1318. doi: 10.1128/mcb.8.3.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore TDE, Edman JC. The α-mating type locus of Cryptococcus neoformanscontains a peptide pheromone gene. Mol Cell Biol. 1993;13:1962–1970. doi: 10.1128/mcb.13.3.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer RK, Johnston JR. Genealogy of principal strains of the yeast genetic stock center. Genetics. 1986;113:35–43. doi: 10.1093/genetics/113.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddon C, Loayza D, Vangelista L, Solari R, Michaelis S. Analysis of the localization of STE6/CFTR chimeras in Saccharomyces cerevisiaemodel for the cystic fibrosis defect CFTR ΔF508. Mol Microbiol. 1996;19:1007–1017. doi: 10.1046/j.1365-2958.1996.444973.x. [DOI] [PubMed] [Google Scholar]
- Powers S, Michaelis S, Broek D, Santa AS, Field J, Herskowitz I, Wigler M. RAM, a gene of yeast required for a functional modification of RAS proteins and for production of mating pheromone a-factor. Cell. 1986;47:413–422. doi: 10.1016/0092-8674(86)90598-2. [DOI] [PubMed] [Google Scholar]
- Rapoport TA. Transport of proteins across the endoplasmic reticulum membrane. Science (Wash DC) 1992;258:931–936. doi: 10.1126/science.1332192. [DOI] [PubMed] [Google Scholar]
- Raymond M, Gros P, Whiteway M, Thomas DY. Functional complementation of yeast ste6 by a mammalian multidrug resistance mdrgene. Science (Wash DC) 1992;256:232–234. doi: 10.1126/science.1348873. [DOI] [PubMed] [Google Scholar]
- Reutz S, Gros P. Phosphatidylcholine translocase: a physiological role for the mdr2gene. Cell. 1994;77:1071–1081. doi: 10.1016/0092-8674(94)90446-4. [DOI] [PubMed] [Google Scholar]
- Rose, M.D., F. Winston, and P. Hieter. 1990. Methods in Yeast Genetics. A Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 198 pp.
- Sakagami Y, Yoshida M, Isogai A, Suzuki A. Peptidal sex hormones inducing conjugation tube formation in compatible mating-type cells of Tremella mesenterica. . Science (Wash DC) 1981;212:1525–1527. doi: 10.1126/science.212.4502.1525. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning. A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sapperstein S, Berkower C, Michaelis S. Nucleotide sequence of the yeast STE14 gene, which encodes farnesylcysteine carboxyl methyltransferase, and demonstration of its essential role in a-factor export. Mol Cell Biol. 1994;14:1438–1449. doi: 10.1128/mcb.14.2.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer WR, Trueblood CE, Yang CC, Mayer MP, Rosenberg S, Poulter CD, Kim SH, Rine J. Enzymatic coupling of cholesterol intermediates to a mating pheromone precursor and to the ras protein. Science (Wash DC) 1990;249:1133–1139. doi: 10.1126/science.2204115. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano P, Tatchell K. Transcription and regulatory signals at the mating type locus in yeast. Cell. 1984;37:969–978. doi: 10.1016/0092-8674(84)90431-8. [DOI] [PubMed] [Google Scholar]
- Sinensky M, Fantle K, Trujillo M, McLain T, Kupfer A, Dalton M. The processing pathway of prelamin A. J Cell Sci. 1994;107:61–67. doi: 10.1242/jcs.107.1.61. [DOI] [PubMed] [Google Scholar]
- Spellig T, Bolker M, Lottspeich F, Frank RW, Kahmann R. Pheromones trigger filamentous growth in Ustilago maydis. . EMBO (Eur Mol Biol Organ) J. 1994;13:1620–1627. doi: 10.1002/j.1460-2075.1994.tb06425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague, G.F., Jr., and J. Thorner. 1992. Pheromone response and signal transduction during mating process of Saccharomyces cerevisiae. In The Molecular Biology of the Yeast Saccharomyces. 2nd ed. J.R. Broach, J.R. Pringle, and E.W. Jones, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 810 pp.
- Sterne, R.E. 1989. A novel pathway for peptide hormone biogenesis: processing and secretion of the mating pheromone a-factor by Saccharomyces cerevisiae. Ph.D. thesis. University of California, Berkeley, CA. 215 pp.
- Tan EW, Perez-Sala D, Canada FJ, Rando RR. Identifying the recognition unit for G protein methylation. J Biol Chem. 1991;266:10719–10722. [PubMed] [Google Scholar]
- Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- Zhang L, Sachs CW, Fine RL, Casey PJ. Interaction of prenylcysteine methyl esters with the multidrug resistance transporter. J Biol Chem. 1994;269:15973–15976. [PubMed] [Google Scholar]