

Abstract
Vacuole inheritance in yeast involves the formation of tubular and vesicular “segregation structures” which migrate into the bud and fuse there to establish the daughter cell vacuole. Vacuole fusion has been reconstituted in vitro and may be used as a model for an NSF-dependent reaction of priming, docking, and fusion. We have developed biochemical and microscopic assays for the docking step of in vitro vacuole fusion and characterized its requirements. The vacuoles must be primed for docking by the action of Sec17p (α-SNAP) and Sec18p (NSF). Priming is necessary for both fusion partners. It produces a labile state which requires rapid docking in order to lead productively to fusion. In addition to Sec17p/Sec18p, docking requires the activity of the Ras-like GTPase Ypt7p. Unlike Sec17p/Sec18p, which must act before docking, Ypt7p is directly involved in the docking process itself.
Aconserved proteinaceous apparatus catalyzes many cellular membrane trafficking steps, such as transport from ER through the Golgi to the endosomes and plasma membrane (Beckers et al., 1989; Graham and Emr, 1991); in synaptic exocytosis (DeBello et al., 1995); secretion in chromaffin cells (Roth and Burgoyne, 1995); transcytosis (Sztul et al., 1993); and in endosome–endosome fusion (Diaz et al., 1989a ; Rodriguez et al., 1994). This apparatus has been identified through pioneering biochemical and genetic approaches in both mammalian and yeast cells (reviewed in Pfeffer, 1996; Südhof, 1995; Rothman, 1994). A core component is the NEM sensitive fusion protein (NSF)1 (Wilson et al., 1989), an ATPase which is recruited to the membranes by its interaction with soluble NSF attachment proteins (SNAP) (Clary et al., 1990; Schiavo et al., 1995). NSF and SNAPs form large 20S complexes with SNAP receptors (SNAREs; Wilson et al., 1992; Söllner et al., 1993a ). SNAREs are integral membrane proteins which may mediate the recognition between fusion partners. Although numerous fusion reactions in eukaryotic cells depend on NSF, there appear to be NSF-independent mechanisms, too: e.g., transport between trans-Golgi and the vacuole (Graham and Emr, 1991), apical transport in polarized epithelial cells (Ikonen et al., 1995), and ER–ER fusion (Latterich et al., 1995). Reassembly of the Golgi from vesicular fragments depends on both NSF and an NSF-related protein, p97 (Acharya et al., 1995; Rabouille et al., 1995). Small Raslike GTPases are also essential for most fusion reactions (reviewed in Nuoffer and Balch, 1994). Though a number of factors regulating the GTPase cycle of these proteins are known and downstream targets are being identified (Brennwald et al., 1994; Stenmark et al., 1995; Li et al., 1995; Stahl et al., 1996), their molecular roles in the trafficking process are largely unclear. One of the best hints so far comes from a yeast strain with a mutation in Ypt1p. In this strain the subunits of the v-SNARE for ER to Golgi transport (Lian et al., 1994) and the corresponding 20S complex (Lupashin, V., and G. Waters, personal communication) are disassembled. Recently, uso1p/p115 was also shown to be required for the assembly or stability of 20S complexes (Sapperstein et al., 1996). Several other factors such as PI-Kinases (reviewed in DeCamilli et al., 1996) and protein kinases/phosphatases (Tuomikosi et al., 1989; Woodman et al., 1992; Conradt et al., 1992; Stuart et al., 1993) have also been implicated in trafficking reactions.
The following steps were suggested to occur in the course of an NSF-dependent fusion reaction. Upon binding of the SNARE on a transport vesicle (v-SNARE) to its counterpart in the target membrane (t-SNARE), a small 7S complex forms which can recruit SNAPs and NSF to yield a large 20S fusion particle. Via its NSF subunit, this particle hydrolyzes ATP, resulting in its disintegration and in membrane fusion (Söllner et al., 1993b ). However, several lines of evidence are consistent with an earlier function of NSF which precedes the last step of fusion (Holz et al., 1989; Rexach and Schekman, 1991; Hay and Martin, 1992; Chamberlain et al., 1995; Hanson et al., 1995). This notion received strong support from recent studies with yeast vacuoles which could clearly separate NSF and α-SNAP action from bilayer fusion (Mayer et al., 1996).
Vacuole fusion is part of the inheritance reaction of the yeast vacuole (Conradt et al., 1992). Early in S-phase, yeast cells project tubular and vesicular structures into the growing bud. These elements fuse there to establish the new vacuole in the daughter cell (Weisman and Wickner, 1988; Gomes de Mesquita et al., 1991; Raymond et al., 1992). Isolated yeast vacuoles can perform this reaction in vitro (Conradt et al., 1992). The in vitro reaction can be monitored by a biochemical complementation assay (Conradt et al., 1992; Haas et al., 1994) which measures the sequential, coupled steps of vacuole priming, docking, and bilayer fusion (Conradt et al., 1994; Mayer et al., 1996). The reaction depends on the Ras-like GTPase Ypt7p (Haas et al., 1995) and on Sec18p and Sec17p (Haas and Wickner, 1996) which are the equivalents of α-SNAP and NSF in yeast (Wilson et al., 1989; Griff et al., 1992). Vacuole fusion is sensitive to Microcystin LR, an inhibitor of Ser/Thr-phosphatases, and to GTPγS (Conradt et al., 1992; Haas et al., 1994). Furthermore, LMA1, a heterodimer of thioredoxin and proteinase B inhibitor 2, is required (Xu and Wickner, 1996; Xu et al., 1997). LMA1 acts synergistically with Sec18p but employs neither the redox properties of thioredoxin nor the protease B inhibitor activity. Rather, it seems to use these components to perform a novel function. The Sec18p/Sec17p reaction, which leads to the release of Sec17p from the membrane (Mayer et al., 1996), activates the vacuoles for subsequent fusion. It produces a labile state which can be used for fusion only in the presence of LMA1. In its absence, the vacuoles undergo a rapid, ATP- and Sec18p-dependent inactivation. LMA1 must act together with or shortly after Sec18p to capture the labile state and prevent the inactivation of the fusion apparatus (Xu et al., 1997).
The kinetics of the Sec17p/Sec18p and Ypt7p reactions were analyzed in some detail, showing that these components act well before bilayer mixing (Mayer et al., 1996). Sec17p/Sec18p can even act before docking. However, it remained unclear whether the Sec17p/Sec18p reaction was a prerequisite for docking and whether Ypt7p was involved in docking or in a later step. We have now established a microscopic assay of docking and a biochemical assay which is based on dilution resistance. With these assays we can demonstrate that the Sec17p/Sec18p reaction must occur on both fusion partners to prime them for docking. This ATP-dependent priming reaction creates a labile state which can be guided into a productive fusion event only if docking occurs rapidly. In contrast to Sec17p/ Sec18p, which are required before docking, the Ras-like GTPase Ypt7p is required at the docking event directly. We therefore propose that Sec17p/Sec18p action creates a labile change in SNARE structure or association state on each vacuole which Ypt7p employs to mediate docking.
Materials and Methods
Materials
Sources for chemicals are described in Mayer et al. (1996). The bead beater was from Biospec Products (Bartlesville, OK). Yeast strains are described in Conradt et al. (1992, 1994), Haas et al. (1994), and Haas et al. (1995). RSY249, RSY270, and RSY272 were obtained from R. Schekman and are described in Kaiser and Schekman (1990) and Haas et al. (1996).
Biochemical Procedures
The following proteins were purified according to published procedures: Gdi1p purification from E. coli BL21 (pNB620) (Garrett et al., 1994), purification of his6-tagged Sec18p (Haas and Wickner, 1997) and affinity purification of IgG (Mayer et al., 1996). Fab-fragments of IgG were prepared as follows: Antibodies were isolated from 10 ml of rabbit serum by affinity chromatography on protein A–Sepharose (Mayer et al., 1995), dialyzed against 20 mM K-phosphate with 10 mM EDTA and concentrated to 15 mg/ml by ultrafiltration in Centriprep-30 units (Amicon, Danvers, MA). Each sample was mixed with one volume digestion buffer (22 mM cysteine/HCl in 10 mM Na-phosphate buffer, pH 7.2, freshly prepared) and one bed volume of immobilized papain (Pierce, Rockford, IL). After shaking for 12 h at 37°C, the supernatant was mixed with 2 vol of 100 mM Na-phosphate pH 8.5 and passed five times through a 10-ml protein A–Sepharose column by gravity. The flow-through was dialyzed against 150 mM KCl in PS buffer (10 mM Pipes/KOH, pH 6.8, 200 mM sorbitol). After centrifugation (5 min, 20,000 g), the supernatant was concentrated to 2 mg/ml by ultrafiltration (Centricon-30), frozen in liquid nitrogen, and stored at −20°C. Fragmentation of the IgG was monitored by SDS-PAGE. Stock solutions for inhibitors or antibodies were made in PS buffer with 150 mM KCl or in DMSO (for 2 mM microcystinLR and 10 mM FM4-64) and kept at −80°C.
Isolation of Vacuoles and Cytosol
Vacuoles and cytosol were isolated according to Mayer et al. (1996) except that cells for the cytosol preparation were lysed in a 250-ml beadbeater. Yeast cells (20,000 OD) were suspended in 150 ml of lysis buffer. Acid washed glass beads (100 g) were added and the mixture was transferred into the bead beater. Air was excluded from the lysis chamber by addition of lysis buffer. The suspension was subjected to 20 bursts (5 s each) followed by cooling periods (1 min) on ice. The lysate was clarified by centrifugation (Conradt et al., 1992). Cytosol from sec17 and sec18 strains (RSY 249, RSY 270 and RSY 272) was prepared as described (Haas and Wickner, 1996) by lysis of spheroplasts in a french press in the presence of MgATP and DTT to maintain active Sec18p.
Vacuole Fusion
Standard vacuole fusion reactions (Mayer et al.,1996) contained 3 μg of each vacuole type (isolated from strains BJ3505 and DKY6281) in 30–35 μl of “reaction mixture” (10 mM Pipes/KOH, pH 6.8, 200 mM sorbitol, 150 mM KCl, 0.5 mM MgCl2, 0.5 mM MnCl2, 0.5 mM ATP, 1.5 mg/ml cytosol, 7.5 μM pefablock SC, 7.5 ng/ml leupeptin, 3.75 μM o-phenanthroline, 37.5 ng/ml pepstatin A, 20 mM creatine phosphate, and 35 U/ml creatine kinase). The standard reaction time was reduced to 70 min at 27°C. 1 U fusion activity is defined as 1 μmol p-nitrophenol developed per min and μg BJ3505 vacuoles.
Assays for Vacuole Docking
Biochemical assay: Vacuoles were incubated under standard fusion conditions (see above). At the desired time, a 30-μl standard reaction was diluted to 600 μl with complete reaction mixture (minus vacuoles). A parallel control sample was left at its original vacuole concentration. The incubation was continued until the end of the 70-min period. Samples were then chilled on ice and adjusted to 600-μl with ice-cold reaction mixture. The vacuoles were reisolated by centrifugation (10 min 13,000 g, 2°C), resuspended in 30 μl reaction mixture, and assayed for fusion.
Microscopic assay: Reactions for microscopic analysis were performed in 8 μM Microcystin LR, 10 mM Pipes/KOH, pH 6.8, 200 mM sorbitol, 65 mM KCl, 0.25 mM MgCl2, 0.075 mM MnCl2, 0.25 mM ATP, 2 mg/ml cytosol, 7.5 μM pefablock SC, 7.5 ng/ml leupeptin, 3.75 μM o-phenanthroline, 37.5 ng/ml pepstatin A, 10 mM creatine phosphate, and 17.5 U/ml creatine kinase to minimize spontaneous, ATP-independent clustering of vacuoles. The extent of this background reaction was strain dependent, and reduction of the KCl, MgCl2, and MnCl2 concentrations contributed to minimizing the ATP-independent vacuole aggregation. The optimized conditions permit fusion with slightly reduced efficiency, but with the same kinetic properties as the standard buffer (data not shown). The samples were incubated at 27°C for 15 min and chilled on ice. Aliquots (12 μl) of each sample were mixed with the same volume of staining mixture (50 μM FM4-64 in 0.4% Seaplaque-agarose in PS buffer; kept liquid at 34°C), transferred immediately to a slide and covered with a cover glass. The slide had been prechilled on an ice-cooled aluminum block to facilitate immediate solidification of the agarose and to stop the docking reaction. After 5 min at 4°C, the samples were observed by fluorescence microscopy (Haas et al., 1995). Photos of ten random fields were taken on Kodak TMZ3200 film. Docking was quantitated by counting the number of contacts each vacuole had with other vacuoles in the same focal plane. At least 250 vacuoles were counted per condition. Docked vacuole clusters are large enough that there are generally only 1 or 2 clusters per field. They settle somewhat faster than undocked vacuoles, leading to a higher number of vacuoles in the photos (the focal plane was kept close to the slide surface). Undocked vacuoles are disperse and yield more blurred fluorescent structures, which are simply vacuoles out of the plane of focus. There is no lysis or loss of vacuolar material when vacuoles dock or when docking is prevented.
Results
Vacuole fusion in vitro requires cytosol and ATP. The vacuoles are derived from two yeast strains. One strain (DKY6281) lacks alkaline phosphatase but has all vacuolar proteases. The other strain (BJ3505) lacks vacuolar proteases A and B which are needed for processing pro- alkaline phosphatase. It therefore bears the inactive proform of this enzyme. After the sequential steps of priming, docking and bilayer fusion, the proteases from one fusion partner can process the pro-alkaline phosphatase from the other partner to the active, mature form. Fusion per se, and not the proteolytic activation of pro-alkaline phosphatase, is rate-limiting in our reactions, as there is no kinetic lag between the acquisition of reaction-resistance to fusion inhibitors such as GTPγS or microcystin-LR, which do not affect proteases, and the appearance of alkaline phosphatase activity (Mayer et al., 1996). Alkaline phosphatase activity can then be determined spectrophotometrically.
Recently, we investigated the kinetics of Sec17p and Sec18p function in vacuole fusion (Mayer et al., 1996). Both proteins were only required in an initial phase of the reaction, a phase which could even precede the docking step. However, it remained unclear if the Sec17p/Sec18p reaction is necessary to initiate the docking event. Alternatively, it could have been completed before docking, but might be required for the fusion pathway at a later stage which could occur after docking. By investigating the docking event directly, we can now address this question.
Sec18p Activity Is a Prerequisite for Vacuole Docking
We define docking as the sequence of events which occurs upon contact between the vacuoles, establishes their stable interaction, and commits them to subsequent fusion. The docking of yeast vacuoles can be assayed biochemically, employing dilution resistance as a criterion. Why is dilution resistance an assay for the association state of vacuoles? Docked vacuoles adhere stably to each other. This enables them to undergo subsequent fusion reactions despite dilution, provided that the concentrations of necessary soluble factors are not changed. On the other hand, dilution strongly reduces the probability for the formation of new interactions between free, undocked vacuoles. Hence, dilution prevents fusion between undocked vacuoles but not the fusion of docked vacuoles. The fusion signal obtained after dilution is therefore a direct measure of the association state of the vacuoles at the time of dilution. It is, however, important to verify that the suppression of fusion is indeed due to spatial separation of undocked vacuoles rather than to dilution of peripheral factors. Such factors might dissociate from the membranes but not be supplied by the reaction mixture used for dilution. Two observations confirm that fusion is suppressed only by spatial separation in diluted samples: First, the vacuoles can be reisolated from an ongoing fusion reaction and be resuspended in fresh reaction mixture (which would not contain factors that might have been released from the vacuoles in the first phase). This treatment can be performed at any phase of the reaction without interfering with subsequent fusion (Mayer et al., 1996). Second, the supernatant of large-scale, ongoing fusion reactions, which would contain dissociated peripheral factors in the necessary concentrations, can be retrieved. If used for diluting newly started reactions, it efficiently prevents fusion (data not shown). Thus, dilution resistance provides a valid criterion of the association state of the vacuoles and a tool for monitoring docking.
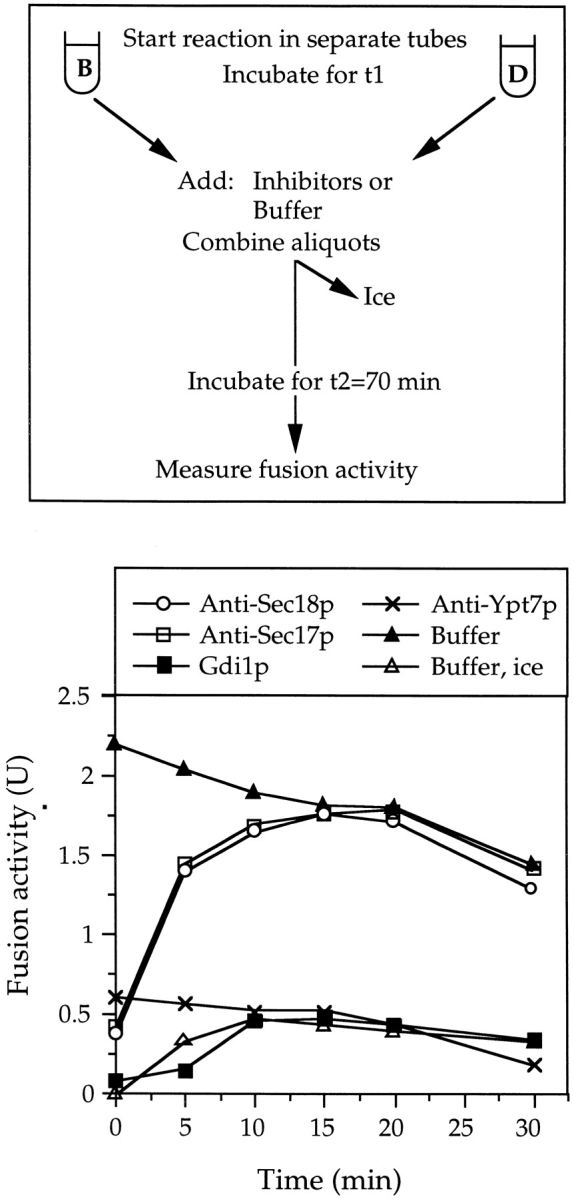
Using dilution resistance as a biochemical assay, we determined the time course of the docking reaction and its relationship to the requirements for Sec18p, Sec17p, and Ypt7p (Fig. 1). Aliquots of a fusion reaction were diluted or mixed with inhibitors at different times of a 70-min incubation. A further aliquot was placed on ice to monitor the progression of the fusion reaction. When diluted at the start, the reaction was completely prevented, but full resistance to dilution was achieved within 30 min after the onset of the reaction. In contrast, resistance to antibodies to Sec17p and Sec18p occurred after only 15 min. This suggested that the Sec17p/Sec18p reaction might be a prerequisite for achieving dilution resistance, i.e., for the docking step. The time course of dilution resistance overlapped with the time courses obtained with antibodies to Ypt7p or with Gdi1p (which binds to, and extracts, Ypt7p from the vacuoles). This close kinetic link suggested that Ypt7p might be directly involved in the docking process. To test these hypotheses, it was necessary to establish the absolute order of the requirements for Sec17p, Sec18p, and Ypt7p in relation to the docking event.
Figure 1.

Kinetics of dilution resistance and the requirements for Sec17p, Sec18p, and Ypt7p. Samples (7× the standard volume) were incubated at 27°C. At each time, one sample was divided into 30-μl standard reactions. Six aliquots were transferred to tubes containing 5 μl of the inhibitors, control buffer only, or 570 μl reaction mixture and incubated at 27°C for the rest of the 70-min reaction. Another aliquot, which received only 5 μl of control buffer, was set on ice. After 70 min, fusion was measured. The graphs present the average of four independent experiments with standard deviation. The curves for the samples which received buffer only, were set on ice or were diluted are displayed in both panels for comparison. To facilitate averaging, the data was normalized: The activity of the samples incubated at 27°C with addition of buffer only was set to 100%. The activity of the sample set on ice at 0 min is the 0% reference. In the four experiments, the maximal fusion activities in the samples with buffer only were: 1.8 ± 0.13 U, 2.1 ± 0.2 U, 1.7 ± 0.19 U and 2.2 ± 0.19 U. The background, given by samples set on ice at 0 min, varied from 0.16 to 0.29 U. Inhibitor concentrations were: Affinity-purified antibodies against Sec18p (70 μg/ml) Sec17p (70 μg/ml), or Ypt7p (120 μg/ml); Gdi1p (120 μg/ml).
We investigated, with a two step experiment, whether the docking step of the reaction must be preceded by Sec18p activity. In the first phase, the vacuoles were incubated in the presence of Fab-fragments directed against Sec18p to suppress Sec18p activity (Fig. 2 A, right panel). After 20 min, a period which permits a standard reaction to become largely dilution resistant (see Fig. 1), the vacuoles were reisolated, resuspended in fresh reaction mixture, split into aliquots, and supplemented with purified Sec18p to overcome the Fab-block. If the second incubation was then performed at standard vacuole concentration, efficient fusion occurred. However, if the vacuoles were diluted 20-fold in the second phase, fusion was suppressed. The vacuoles had not become dilution resistant, i.e., they had not been able to dock during the first incubation. In a positive control sample in which Sec18p was not inhibited in the first phase, the vacuoles had docked and the reaction had become largely dilution resistant (Fig. 2 A, left panel). Analogous approaches were used to demonstrate that the acquisition of dilution resistance depends on ATP (Fig. 2 B) and physiological temperature (Fig. 2 C). Both conditions are also required for Sec18p activity (Wilson et al., 1989; Mayer et al., 1996).
Figure 2.

Sec18p action is a prerequisite for dilution resistance. (A) Sec18p dependence. Two 3× fusion reactions were started at 27°C in the presence or absence of Fab-fragments (150 μg/ml) prepared from the total immunoglobulin fraction from antiserum directed against Sec18p. After 20 min, the samples were chilled and made chemically equivalent by supplementation with antibodies to Sec18p. The samples were then split into three standard reactions. These were either left concentrated (Undil.) or diluted 20fold (Dil.) with reaction mixture. Each received 15 μg/ml purified Sec18p and 75 μM DTT and all were incubated at 27°C. An aliquot kept on ice serves as a measure of the background reaction which had occurred during the first incubation. After 70 min, all samples were chilled on ice and adjusted to the same volume with reaction mixture. The vacuoles were reisolated by centrifugation, resuspended in 30 μl ice-cold reaction mixture and assayed for fusion. The same results were obtained if Sec18p was added to the diluted sample to 0.75 μg/ml to satisfy only the stoichiometric requirement for saturation of the antibodies (not shown). (B) ATP dependence. Two 3× fusion reactions were incubated for 20 min at 27°C in the presence or absence of the ATP regenerating system. The samples were then chilled and the regenerating system was added to the sample lacking it. The samples were split into aliquots which were diluted with reaction mixture or left concentrated. Further incubation and analysis was as in A. (C) Temperature dependence. Fusion reactions equivalent to three standard reactions were incubated for 20 min at 0°C or 27°C. The samples were split into aliquots, diluted with reaction mixture where indicated, and further processed as in A.
The requirement of the docking reaction for Sec18p could also be investigated by light microscopy of the samples (Fig. 3). Reactions were performed for 15 min in the presence of the Ser/Thr-phosphatase inhibitor Microcystin LR. This substance inhibits a late stage of vacuole fusion (Conradt et al., 1994; Mayer et al., 1996), but does not interfere with the early steps of the reaction leading to docking (data not shown). This facilitates the microscopic analysis since it prevents the consumption of clusters of docked vacuoles by ongoing fusion. The docking reaction was stopped by embedding the vacuoles in agarose which was rapidly solidified by chilling on a precooled microscopy slide. If the vacuoles were incubated at 27°C in complete reaction mixtures, almost all of them were found in clusters of up to 15 vacuoles (Fig. 3 A). This indicates a very high efficiency of the docking reaction. On average, each vacuole formed 2.5–3 contacts to other vacuoles in the focal plane (Fig. 3 C). On ice, or in the absence of ATP, only a few, small clusters appeared, with only 2–3 vacuoles per cluster and 0.25 contacts per vacuole on average. Likewise, Fab-fragments directed against Sec18p prevented the formation of clusters. The effect could be reversed by readdition of purified Sec18p. This reversal by purified Sec18p eliminates the possibility that docking was inhibited by potential unspecific side effects of the Fab, such as steric hindrance of the interaction between the vacuoles. Furthermore, temperature sensitive mutants were used to provide independent evidence that docking requires Sec18p. If cytosol and vacuoles were prepared from either sec18-1 or sec17-1 mutant strains, the docking reaction was impaired and only 0.35–0.6 contacts were formed per vacuole (Fig. 3, B and C). In contrast, the wild-type control displayed efficient clustering with 2.7 contacts per vacuole. The docking competence of sec18-1 mutant vacuoles could be restored by wild-type cytosol which provides active Sec18p. Alternatively, purified Sec18p added to the mutant cytosol was able to rescue the docking reaction (not shown). Therefore, the inability to dock was a consequence of the genetic inactivation of Sec18p and not due to indirect effects. We conclude that the Sec17p/Sec18p reaction is a prerequisite for docking.
Figure 3.


Sec18p activity is required for visible clustering of vacuoles. (A) Biochemical inhibition of Sec18p. Vacuoles were prepared from the strain RSY249 and used in 30 μl fusion reactions for microscopic analysis as described in Materials and Methods. After adding apyrase (10 U/ ml; “no ATP”), Fab fragments against Sec18p (125 μg/ml), control-Fab against Carboxypeptidase Y (125 μg/ ml), or buffer only, the samples were preincubated for 5 min on ice. One sample, blocked with Sec18p-Fab, then received purified Sec18p (35 μg/ml) to rescue the block imposed by the Fab fragments. All samples were supplemented with 75 μM DTT and incubated for 15 min at 27°C or on ice. They were then chilled on ice and mixed with low melting agarose containing the vacuole stain FM4-64. The suspension was transferred onto a prechilled microscopy slide, left at 4°C for 5 min, and analyzed by fluorescence microscopy. Pictures of 10 fields were taken for each condition. (B) Clustering in sec17-1 and sec18-1 mutants. Wildtype (RSY249), sec17-1 (RSY270), and sec18-1 (RSY272) strains were grown at 25°C. Vacuoles and cytosols were prepared from these strains as described in Materials and Methods. Fusion reactions for microscopic analysis were performed as in A using cytosol and vacuoles from the same strains. In one sample, sec18-1 vacuoles were combined with wt cytosol to rescue the defect of Sec18p. Vacuoles from the sec17-1 strain could not be reactivated by wt cytosol (not shown). (C) Quantitation of the samples shown in A and B. The average number of contacts per vacuole in the focal plane was determined from the pictures.
The Requirement for Sec18p Activity Is Symmetric
Must Sec18p activate both fusion partners for docking, or is activation needed only on one fusion partner, which can then recognize and bind its “passive” counterpart? This issue could be addressed by activating the fusion partners separately (Fig. 4). Both vacuole types were incubated in separate tubes for 15 min in the presence or absence of antibodies to Sec18p. This time is sufficient to complete the Sec18p-reaction in normal incubations (see Figs. 1, 2, and 6). The second reaction phase, performed after mixing the separate fusion partners, should then be independent of Sec18p. Efficient fusion in the second phase occurred only if both fusion partners had completed the Sec18p reaction in the first phase. Sec18p action on only one of the fusion partners was not sufficient to promote fusion in the second phase, which was performed in the presence of antibodies to prevent Sec18p action after mixing. However, if purified Sec18p was added into the second incubation to overcome the inhibition by the antibodies, the reaction could be rescued. This excludes the possibility that the vacuoles had been irreversibly damaged during the first incubation with the antibodies. Two control samples, in which the fusion partners were premixed in the first phase in the presence or absence of the antibodies, indicate that the inhibitory potential of the antibodies was sufficient to inactivate both fusion partners, and that an uninhibited reaction could indeed pass the Sec18p requirement in the first phase. Thus, Sec18p activity is needed on both fusion partners to prime them for the subsequent reactions of docking and bilayer mixing.
Figure 4.

Sec18p action is required on both fusion partners to activate them for docking and fusion. Separate samples of BJ3505 vacuoles (with pro-alkaline phosphatase) and DKY6281 vacuoles (carrying the maturation enzymes), each equivalent to three standard fusion reactions, were incubated at 27°C in the presence or absence of antibodies to Sec18p (70 μg/ml). After 15 min, the samples were chilled, supplemented with antibodies to Sec18p (to 70 μg/ml), and left on ice for 5 min. They were mixed pairwise to make combinations where only one of the fusion partners or neither of them had been preincubated with anti-Sec18p. The tubes were centrifuged briefly (2 min, 8,000 g, 4°C) and the vacuoles resuspended in fresh reaction mixture with 75 μM DTT. Each sample was split into aliquots which received either 1.5 μg/ml Sec18p to release the block of the Sec18p pathway or antibodies to Sec18p to maintain the block. The samples were incubated at 27°C or on ice (to indicate the background reaction which had occurred during the first incubation). After 70 min, fusion activity was determined. Control samples having both fusion partners already mixed during the first incubation were taken through the same procedure in parallel (right panel).
Figure 6.
The requirement for Ypt7p cannot be fulfilled while the vacuoles are in separate tubes. For each time point, two separate samples, each equivalent to five standard fusion reactions, were incubated at 27°C. One contained BJ3505 vacuoles carrying the pro-alkaline phosphatase (B) whereas the other contained DKY6281 vacuoles harboring the maturation enzyme, proteinase A (D). At the indicated times, aliquots were withdrawn from the reactions, chilled, and transferred into tubes containing Gdi1p (final concentration 100 μg/ml), antibodies to either Ypt7p (120 μg/ ml), Sec18p (80 μg/ml), or Sec17p (80 μg/ml), or control buffer only. The tubes were centrifuged briefly (1 min, 8,000 g, 4°C) and the vacuoles were resuspended gently and incubated for 70 min at 27°C or on ice. Fusion was then determined.
Priming Creates a Labile State which Is Stabilized upon Docking
The accompanying manuscript demonstrates that Sec18p action creates a labile, primed state. It is consumed productively only if LMA1, a heterodimer of thioredoxin and IB 2, is provided at or shortly after the time of priming (Xu et al., 1997). Does productive consumption of the primed state also require docking of the vacuoles? We investigated this question by starting fusion reactions with 20fold reduced vacuole concentration, but with standard concentration of all soluble components. These conditions separate the vacuoles spatially and thereby suppress docking and fusion. At different times, the vacuoles were concentrated by centrifugation and the reaction was allowed to proceed at the standard membrane concentration (Fig. 5). The Sec17p/Sec18p reaction, which is needed to activate the vacuoles for docking (Figs. 2 and 3), does proceed under these conditions, since it is independent of the membrane concentration (Mayer et al., 1996). Strikingly, the ability to fuse after reconcentration declined rapidly (Fig. 5). After only 15 min of incubation in the diluted state, the vacuoles had lost their fusion competence, whereas vacuoles in nonactivated control samples remained fusion competent. These control samples had been incubated either without ATP, or on ice, to prevent activation. We propose that docking, as well as LMA1, is required to use the labile, activated state and avoid its unproductive decay.
Figure 5.

Activation produces a labile state which requires docking for productive consumption. Fusion reactions were started in the presence or absence of ATP with 6 μg vacuoles in a total volume of 600 μl, i.e., with a 20-fold reduced vacuole concentration. After the indicated times at 27°C or on ice, the samples were centrifuged (5 min at 10,000 g, 2°C) and the vacuoles were resuspended in 30 μl complete reaction mixture, i.e., at standard vacuole concentration. After a total reaction time of 120 min at standard concentration and at 27°C, fusion activity was determined.
Ypt7p Action Is Linked to the Docking Event
The time course of acquiring resistance to antibodies to Ypt7p or to Gdi1p coincides with dilution resistance, an indicator of docking (Fig. 1). This suggested a close kinetic link of Ypt7p to the docking reaction. In addition, like for Sec17p/Sec18p (Fig. 4), Ypt7p activity is required on both fusion partners (Haas et al., 1995). We have addressed the Ypt7p requirement for the pre-docking or docking phase with two strategies. First, we investigated whether the requirement for Ypt7p could be fulfilled before the docking event. Docking between the fusion partners was prevented by incubating them in separate tubes (Fig. 6). At different times, aliquots received inhibitors, were mixed and were assayed for the effect of the inhibitor on docking (which could only start between the two partners after mixing) and fusion. Though separate, the vacuoles became resistant to antibodies to Sec17p and Sec18p with the same kinetics as in standard experiments (cf. Fig. 1), i.e., within 15 min.
This underscores the finding that Sec17p/Sec18p activity is required before, but not directly at, the docking event. However, the vacuoles remained fully sensitive to Gdi1p, or to antibodies to Ypt7p, throughout 30 min of separate preincubation (Fig. 6). This time is sufficient to completely fulfill the requirement for Ypt7p in a standard reaction (Fig. 1 and data not shown). We therefore conclude that the vacuoles cannot complete the Ypt7p step before the docking event. Rather, Ypt7p activity must be required at or after the docking event.
We could discriminate between the latter two possibilities by following the Ypt7p requirement in the microscopic docking assay (Fig. 7). If the docking reaction was performed in the presence of either Gdi1p or Fab fragments to Ypt7p, most vacuoles remained undocked and formed ∼0.5 contacts on average. Control Fab or the addition of only buffer did not interfere with the docking reaction and permitted the formation of clusters with up to 15 vacuoles. The docking reaction was highly efficient, since almost all vacuoles participated in the formation of clusters, interacting by 2.5 contacts per vacuole on average. These results demonstrate that Ypt7p is required for the docking reaction. Since the biochemical assay in Fig. 6 indicated that the requirement for Ypt7p could not be satisfied before docking, we conclude that Ypt7p must act at docking per se.
Figure 7.

Visible clustering of vacuoles in the early reaction phase depends on Ypt7p. (A) Fusion reactions for microscopic analysis were performed as in Fig. 4. Samples were incubated for 15 min at 27°C in the presence of buffer, Gdi1p (100 μg/ ml), or Fab fragments against Ypt7p (125 μg/ml) or carboxypeptidase Y (Control-Fab; 125 μg/ml). (B) Quantitation of the data in A was performed as in Fig. 3.
In summary, Sec17p and Sec18p action is a prerequisite for docking. Sec17p/Sec18p must activate both fusion partners and transform them into a labile state to enable them to participate in the following docking reaction. The docking step itself is mediated by a reaction involving Ypt7p.
Discussion
Evidence has accumulated suggesting a function of NSF and α-SNAP in an early, prefusion stage of membrane trafficking reactions (Rexach and Schekman, 1991; Hay and Martin, 1992; Chamberlain et al., 1995). A kinetic investigation of vacuole fusion in yeast revealed that Sec18p (yeast NSF) and Sec17p (yeast α-SNAP) could even act before docking (Mayer et al., 1996). However, it remained unclear if their activity was a prerequisite for docking. Sec17p/Sec18p could have initiated a reaction sequence early which fed into the fusion pathway at a later, postdocking stage. Another possibility to explain the early function of NSF and α-SNAP was that both proteins only act on a small fraction of vacuoles, causing a persistent activation of the fusion machinery. The constitutively activated fusion machinery could then catalyze multiple rounds of fusion without further need for NSF or α-SNAP. This mode of action would be more compatible with the classical model of NSF and α-SNAP as catalysts of bilayer mixing (Rothman, 1994). Therefore, it is a crucial distinction whether Sec17p/Sec18p can act, or must act, before the docking step. We addressed this question by assaying the docking event biochemically and microscopically. Docking was severely inhibited if the Sec17p/Sec18p reaction was blocked by Fab-fragments or if the reaction components were derived from temperature sensitive sec18 or sec17 mutant strains. Docking also depended on ATP and temperature, both of which are also necessary for the Sec17p/ Sec18p reaction (Wilson et al., 1989; Mayer et al., 1996). Furthermore, Sec18p must act on both partners to activate them for docking and fusion. Docking was highly efficient, bringing virtually all vacuoles into clusters. Sec17p/Sec18p must have activated all the vacuoles individually, since efficient docking occurred even in the presence of Microcystin LR which inhibits a late stage of vacuole fusion and prevents the system from going through a catalytic cycle. The early requirement of Sec17p/Sec18p can therefore not be due to a persistent activation of the fusion machinery on only a small fraction of vacuoles. Thus, the combined evidence strongly indicates that the Sec17p/Sec18p reaction is a prerequisite for docking and completes its task in this phase.
Our finding receives independent support from observations on ER to Golgi traffic. ER transport vesicles do not bind to Golgi membranes when reaction components are isolated from a sec18 or ypt1 mutant (Rexach and Schekman, 1991). Our findings are also consistent with data on the in vitro clustering of macrophage endosomes which depends on ATP, cytosol, and temperature (Diaz et al., 1989b ). On the other hand, electron microscopic investigations have received a different interpretation. It was reported that docked vesicles are still found on Golgi membranes in the absence of NSF function (Orci et al., 1989) and on synaptic membranes after competitive inhibition of α-SNAP (DeBello et al., 1995). However, this association was not shown to depend on specific proteins. Thus, it remained unclear if the apposition of the vesicles to the target membranes was indeed caused by engaging the docking apparatus which commits the membranes for fusion, or by other factors. In the case of synaptic vesicles for example, interaction with the cytoskeleton may affect the picture. These vesicles, coming from the lumen of the synaptic terminal, may move along cytoskeletal elements such as actin until they reach a position closely apposed to the plasma membrane and seemingly “dock.” The ATP-dependent priming event, which precedes the ATP independent bilayer fusion event (Holz et al., 1989; Hay and Martin, 1992), might then reflect the NSF-dependent activation and engagement of the docking apparatus.
What might happen in the Sec17p/Sec18p dependent priming reaction? The synaptic t-SNARE syntaxin can bind α-SNAP and NSF. Upon incubation with ATP, the complex is disrupted and syntaxin experiences a transient conformational change which can spontaneously revert (Hanson et al., 1995). Integrating this observation into the vacuolar system, one may speculate that the Sec17p/Sec18p reaction transiently modifies the structure or association state of SNAREs, bringing them into a binding-competent conformation. This should facilitate the interaction between cognate SNAREs and hence docking. If Sec17p/Sec18p act by inducing a transient conformational change of SNAREs, one would predict that the primed, docking-competent state is unstable. Indeed we observed that Sec18p action drives vacuoles into an unstable state (Fig. 5; see also Xu et al., 1997). This activated state can be used only if the vacuoles can dock shortly after activation (Fig. 5). The relative stability of vacuoles which were preincubated separately, though at normal concentrations (Figs. 4 and 6), may reflect their docking (BJ3505 with BJ3505 and DKY6281 with DKY6281) during preincubation, then exchange upon mixing. Alternatively, the extensive surface area of the large “rafts” of docked/fused vacuoles formed during separate preincubation may mask any loss of competence per unit area of vacuoles (Fig. 5). Besides docking, stabilization of the activated state requires LMA1 (Xu et al., 1997). LMA1 is a heterodimer of thioredoxin and proteinase B inhibitor which acts synergistically with Sec18p. LMA1 can only act together with or immediately after Sec17p/ Sec18p. We suggest that docking is mediated by a Sec17p/ Sec18p driven transient rearrangement of SNAREs into a docking competent configuration which may be stabilized by LMA1.
In contrast to Sec17p and Sec18p, which must act in a priming step before docking, Ypt7p is involved in the docking process itself. Like activation by Sec17p/Sec18p (Fig. 4), Ypt7p activity is required on both fusion partners (Haas et al., 1995). In addition, we have shown that the Ypt7p step must be preceded by the Sec17p/Sec18p reaction (Mayer et al., 1996). These results can be integrated into observations from studies of ER to Golgi transport. There, mutations in the Ras-like GTPase Ypt1p were shown to interfere with assembly or stability of the 20S SNARE complex (Waters, G., personal communication) and the subunits of the v-SNARE (Lian et al., 1994). The immediate involvement of Ypt7p in the docking reaction may thus reflect its function in assembling functional SNARE proteins from their primed subunits or in forming the complexes between the cognate, primed SNAREs. This leads us to propose an obligate order of events: Sec18p mediated release of Sec17p must prime the vacuoles by activating their SNAREs, followed by Ypt7p mediated assembly of SNARE complexes which causes docking. Subsequent fusion of the vacuoles is independent of Sec17p, Sec18p, and Ypt7p, but sensitive to various inhibitors like GTPγS and Microcystin LR (Mayer et al., 1996). To verify our hypothesis, it will be necessary to isolate the SNARE proteins, develop assays for their conformations and associations, and relate their behavior to the docking event.
Acknowledgments
We are grateful for plasmids from P. Novick, D. Gallwitz, and J. Rothman and for sec17 and sec18 mutant strains from R. Schekman. We thank P. Slusarewicz for a gift of purified Sec18p and M. Leonard for excellent technical assistance. Discussions with P. Haas, P. Slusarewicz, Z. Xu, and C. Barlowe were stimulating and helpful.
Abbreviations used in this paper
- NSF
NEN sensitive fusion protein
- SNAP
soluble NSF attachment protein
- SNARE
SNAP receptor
Footnotes
The work was supported by a grant from the National Institutes of General Medical Sciences.
Please address all correspondence to W. Wickner, Department of Biochemistry, Dartmouth Medical School, 7200 Vail Building, Hanover, NH 03755-3844. Tel.: (603) 650-1701. Fax: (603) 650-1353.
References
- Acharya U, Jacobs R, Peters J-M, Watson N, Farquhar MG, Malhotra V. The formation of golgi stacks from vesiculated golgi membranes requires two distinct fusion events. Cell. 1995;82:895–904. doi: 10.1016/0092-8674(95)90269-4. [DOI] [PubMed] [Google Scholar]
- Beckers CJM, Block MR, Glick BS, Rothman JE, Balch WE. Vesicular transport between the endoplasmic reticulum and the golgi stack requires the NEM sensitive fusion protein. Nature (Lond) 1989;339:397–398. doi: 10.1038/339397a0. [DOI] [PubMed] [Google Scholar]
- Brennwald P, Kearns B, Champion K, Keränen S, Bankaitis V, Novick P. Sec9 is a SNAP-25 like component of a yeast SNARE complex that may be the effector of Sec4 function in exocytosis. Cell. 1994;79:245–258. doi: 10.1016/0092-8674(94)90194-5. [DOI] [PubMed] [Google Scholar]
- Chamberlain LH, Roth D, Morgan A, Burgoyne RD. Distinct effects of α-SNAP, 14-3-3 proteins, and calmodulin on priming and triggering of regulated exocytosis. J Cell Biol. 1995;130:1063–1070. doi: 10.1083/jcb.130.5.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clary DO, Griff IC, Rothman JE. SNAPs, a family of NSF attachment proteins involved in intracellular membrane fusion in animals and yeast. Cell. 1990;61:709–721. doi: 10.1016/0092-8674(90)90482-t. [DOI] [PubMed] [Google Scholar]
- Conradt B, Haas A, Wickner W. Determination of four biochemically distinct, sequential stages during vacuole inheritance in vitro. J Cell Biol. 1994;126:99–110. doi: 10.1083/jcb.126.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradt B, Shaw J, Vida T, Emr S, Wickner W. In vitro reactions of vacuole inheritance in Saccharomyces cerevisiae. . J Cell Biol. 1992;119:1469–1479. doi: 10.1083/jcb.119.6.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBello WM, O'Connor V, Dresbach T, Whiteheart SW, Wang SS-H, Schweitzer FE, Betz H, Rothman JE, Augustine GJ. SNAP mediated protein-protein interactions essential for neurotransmitter release. Nature (Lond) 1995;373:626–630. doi: 10.1038/373626a0. [DOI] [PubMed] [Google Scholar]
- De Camilli P, Emr SD, McPherson PS, Novick P. Phosphoinositides as regulators in membrane traffic. Science (Wash DC) 1996;271:1533–1539. doi: 10.1126/science.271.5255.1533. [DOI] [PubMed] [Google Scholar]
- Diaz R, Mayorga LS, Weidman PJ, Rothman JE, Stahl PD. Vesicle fusion following receptor-mediated endocytosis requires a protein active in Golgi transport. Nature (Lond) 1989a;339:398–400. doi: 10.1038/339398a0. [DOI] [PubMed] [Google Scholar]
- Diaz R, Mayorga LS, Mayorga LE, Stahl PD. In vitro clustering and multiple fusion among macrophage endosomes. J Biol Chem. 1989b;264:13171–13180. [PubMed] [Google Scholar]
- Garrett MD, Zahner JE, Cheney CM, Novick PJ. GDI1 encodes a GDP dissociation inhibitor that plays an essential role in the yeast secretory pathway. EMBO (Eur Mol Biol Organ) J. 1994;13:1718–1728. doi: 10.1002/j.1460-2075.1994.tb06436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes de Mesquita DS, ten Hoopen R, Woldringh CL. Vacuolar segregation to the bud of Saccharomyces cerevisiae: an analysis of morphology and timing in the cell cycle. J Gen Microbiol. 1991;137:2447–2454. doi: 10.1099/00221287-137-10-2447. [DOI] [PubMed] [Google Scholar]
- Graham TR, Emr SD. Compartmental organization of Golgi-specific protein modification and vacuolar protein sorting events defined in a yeast sec18 (NSF) mutant. J Cell Biol. 1991;114:207–218. doi: 10.1083/jcb.114.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griff IC, Schekman R, Rothman JE, Kaiser CA. The yeast SEC17 gene product is functionally equivalent to mammalian α-SNAP protein. J Biol Chem. 1992;267:12106–12115. [PubMed] [Google Scholar]
- Haas A, Wickner W. Homotypic vacuole fusion requires Sec17p (yeast α-SNAP) and Sec18p (yeast NSF) EMBO (Eur Mol Biol Organ) J. 1996;15:3296–3305. [PMC free article] [PubMed] [Google Scholar]
- Haas A, Conradt B, Wickner W. G-protein ligands inhibit in vitro reactions of vacuole inheritance. J Cell Biol. 1994;126:87–97. doi: 10.1083/jcb.126.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas A, Schleglmann D, Lazar T, Gallwitz D, Wickner W. The GTPase Ypt7 of Saccharomyces cerevisiaeis required on both partner vacuoles for the homotypic fusion step of vacuole inheritance. EMBO (Eur Mol Biol Organ) J. 1995;14:5258–5270. doi: 10.1002/j.1460-2075.1995.tb00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson PI, Otto H, Barton N, Jahn R. The N-ethylmaleimidesensitive fusion protein and α-SNAP induce a conformational change in syntaxin. J Biol Chem. 1995;270:16955–16961. doi: 10.1074/jbc.270.28.16955. [DOI] [PubMed] [Google Scholar]
- Hay JC, Martin TF. Resolution of regulated secretion into sequential MgATP-dependent and calcium-dependent stages mediated by distinct cytosolic proteins. J Cell Biol. 1992;119:139–151. doi: 10.1083/jcb.119.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz RW, Bittner MA, Peppers SC, Senter RA, Eberhardt DA. MgATP-independent and MgATP-dependent exocytosis. J Biol Chem. 1989;264:5412–5419. [PubMed] [Google Scholar]
- Ikonen E, Tagaya M, Ullrich O, Montecucco C, Simons K. Different requirements for NSF, SNAP, and Rab proteins in apical and basolateral transport in MDCK cells. Cell. 1995;81:571–580. doi: 10.1016/0092-8674(95)90078-0. [DOI] [PubMed] [Google Scholar]
- Kaiser CA, Schekman R. Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell. 1990;61:723–733. doi: 10.1016/0092-8674(90)90483-u. [DOI] [PubMed] [Google Scholar]
- Latterich M, Fröhlich K-U, Schekman R. Membrane fusion and the cell cycle: cdc48 participates in the fusion of ER membranes. Cell. 1995;82:885–894. doi: 10.1016/0092-8674(95)90268-6. [DOI] [PubMed] [Google Scholar]
- Li C, Takei K, Geppert M, Daniell L, Stenius K, Chapman ER, Jahn R, De Camilli P, Südhof TC. Synaptic targeting of rabphilin-3A, a synaptic vesicle Ca2+/phospholipid-binding protein, depends on rab3A/3C. Neuron. 1995;13:885–898. doi: 10.1016/0896-6273(94)90254-2. [DOI] [PubMed] [Google Scholar]
- Lian JP, Stone S, Jiang Y, Lyons P, Ferro-Novick S. Ypt1p implicated in v-SNARE activation. Nature (Lond) 1994;372:698–701. doi: 10.1038/372698a0. [DOI] [PubMed] [Google Scholar]
- Mayer A, Neupert W, Lill R. Mitochondrial protein import: reversible binding of the presequence at the trans side of the outer membrane drives partial translocation and unfolding. Cell. 1995;80:127–137. doi: 10.1016/0092-8674(95)90457-3. [DOI] [PubMed] [Google Scholar]
- Mayer A, Wickner W, Haas A. Sec18p (NSF)-driven release of Sec17p (α-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Nuoffer C, Balch WE. GTPases: multifunctional molecular switches regulating vesicular traffic. Annu Rev Biochem. 1994;63:949–990. doi: 10.1146/annurev.bi.63.070194.004505. [DOI] [PubMed] [Google Scholar]
- Orci L, Malhotra V, Amherdt M, Serafini T, Rothman JE. Dissection of a single round of vesicular transport: sequential intermediates for intercisternal movement in the Golgi stack. Cell. 1989;56:357–368. doi: 10.1016/0092-8674(89)90239-0. [DOI] [PubMed] [Google Scholar]
- Pfeffer SR. Transport vesicle docking: SNARE and associates. Ann. Rev. Cell Biol. and . Dev Biol. 1996;12:441–461. doi: 10.1146/annurev.cellbio.12.1.441. [DOI] [PubMed] [Google Scholar]
- Rabouille C, Levine TP, Peters J-M, Warren G. An NSF-like ATPase, p97, and NSF mediate cisternal regrowth from mitotic golgi fragments. Cell. 1995;82:905–914. doi: 10.1016/0092-8674(95)90270-8. [DOI] [PubMed] [Google Scholar]
- Raymond CK, Roberts CJ, Moore KE, Howald I. Biogenesis of the vacuole in Saccharomyces cerevisiae. . Int Rev Cytol. 1992;139:59–120. doi: 10.1016/s0074-7696(08)61410-2. [DOI] [PubMed] [Google Scholar]
- Rexach MF, Schekman RW. Distinct biochemical requirements for the budding, targeting, and fusion of ER-derived transport vesicles. J Cell Biol. 1991;114:219–229. doi: 10.1083/jcb.114.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez L, Stirling CJ, Woodman PG. Multiple N-ethylmaleimide- sensitive components are required for endosomal vesicle fusion. Mol Biol Cell. 1994;5:773–783. doi: 10.1091/mbc.5.7.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth D, Burgoyne RD. SNAP-25 is present in a SNARE complex in adrenal chromaffin cells. FEBS Lett. 1994;351:207–210. doi: 10.1016/0014-5793(94)00833-7. [DOI] [PubMed] [Google Scholar]
- Rothman JE. Mechanisms of intracellular protein transport. Nature (Lond) 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Sapperstein SK, Lupashin VV, Schmitt HD, Waters MG. Assembly of the ER to Golgi SNARE complex requires Uso1p. J Cell Biol. 1996;132:755–767. doi: 10.1083/jcb.132.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo G, Gmachl MJS, Stenbeck G, Sollner TH, Rothman JE. A possible docking and fusion particle for synaptic transmission. Nature (Lond) 1995;378:733–736. doi: 10.1038/378733a0. [DOI] [PubMed] [Google Scholar]
- Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature (Lond) 1993a;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- Söllner T, Bennett MK, Whiteheart S, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitrothat may correspond to sequential steps of synaptic vesicle docking, activation and fusion. Cell. 1993b;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- Stahl B, Chou JH, Li C, Sudhof TC, Jahn R. Rab3 reversibly recruits rabphilin to synaptic vesicles by a mechanism analogous to raf recruitment by ras. EMBO (Eur Mol Biol Organ) J. 1996;15:1799–1809. [PMC free article] [PubMed] [Google Scholar]
- Stenmark H, Vitale G, Ullrich O, Zerial M. Rabaptin-5 is a direct effector of the small GTPase Rab5 in endocytic membrane fusion. Cell. 1995;83:423–432. doi: 10.1016/0092-8674(95)90120-5. [DOI] [PubMed] [Google Scholar]
- Stuart RA, Mackay D, Adamczewski J, Warren G. Inhibition of intra-Golgi transport in vitro by mitotic kinase. J Biol Chem. 1993;268:4050–4054. [PubMed] [Google Scholar]
- Südhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature (Lond) 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- Sztul E, Colombo M, Stahl P, Samanta R. Control of protein traffic between distinct plasma membrane domains. Requirement for a novel 108,000 protein in the fusion of transcytotic vesicles with the apical plasma membrane. J Biol Chem. 1993;268:1876–1885. [PubMed] [Google Scholar]
- Tuomikosi T, Felix M-A, Doree M, Gruenberg J. Inhibition of endocytic vesicle fusion in vitro by the cell-cycle control protein kinase cdc2. Nature (Lond) 1989;342:942–945. doi: 10.1038/342942a0. [DOI] [PubMed] [Google Scholar]
- Weisman LS, Wickner W. Intervacuole exchange in the yeast zygote: a new pathway in organelle communication. Science (Wash DC) 1988;241:589–591. doi: 10.1126/science.3041591. [DOI] [PubMed] [Google Scholar]
- Wilson DW, Wilcox CA, Flynn GC, Chen E, Kuang W-J, Henzel WJ, Block MR, Ullrich E, Rothman JE. A fusion protein required for vesicle-mediated transport in both mammalian cells and yeast. Nature (Lond) 1989;339:355–359. doi: 10.1038/339355a0. [DOI] [PubMed] [Google Scholar]
- Wilson DW, Whiteheart SW, Wiedmann M, Brunner M, Rothman JE. A multisubunit particle implicated in membrane fusion. J Cell Biol. 1992;117:531–538. doi: 10.1083/jcb.117.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodman PG, Mundy DI, Cohen P, Warren G. Cell-free fusion of endocytic vesicles is regulated by phosphorylation. J Cell Biol. 1992;116:331–338. doi: 10.1083/jcb.116.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wickner W. Thioredoxin is required for vacuole inheritance in S. cerevisiae. . J Cell Biol. 1996;132:787–794. doi: 10.1083/jcb.132.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Mayer A, Muller E, Wickner W. A heterodimer of thioredoxin and IB2 cooperates with Sec18p (NSF) to promote yeast vacuole inheritance. J Cell Biol. 1997;136:299–306. doi: 10.1083/jcb.136.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]