

Abstract
In this report we examine the biological and molecular basis of the control of sympathetic neuron differentiation and survival by NGF and neurotrophin-3 (NT-3). NT-3 is as efficient as NGF in mediating neuritogenesis and expression of growth-associated genes in NGF-dependent sympathetic neurons, but it is 20–40fold less efficient in supporting their survival. Both NT-3 and NGF induce similar sustained, long-term activation of TrkA, while NGF is 10-fold more efficient than NT-3 in mediating acute, short-term TrkA activity. At similar acute levels of TrkA activation, NT-3 still mediates neuronal survival two- to threefold less well than NGF. However, a mutant NT-3 that activates TrkC, but not TrkA, is unable to support sympathetic neuron survival or neuritogenesis, indicating that NT3–mediated TrkA activation is necessary for both of these responses. On the basis of these data, we suggest that NGF and NT-3 differentially regulate the TrkA receptor both with regard to activation time course and downstream targets, leading to selective regulation of neuritogenesis and survival. Such differential responsiveness to two ligands acting through the same Trk receptor has important implications for neurotrophin function throughout the nervous system.
The neurotrophic factor hypothesis postulates that interactions between a developing peripheral neuron and its target organ play an essential role in neuronal competition and cell death. This hypothesis is perhaps best exemplified by developing sympathetic neurons, which are absolutely dependent upon NGF, one member of the neurotrophin family of growth factors (7, 28, 42, 47), during the period of target competition (9, 43–45, 62). During this developmental window that occurs neonatally, NGF is believed to bind to its cognate receptors on the terminals of sympathetic neurons and to regulate their afferent input density via two primary mechanisms. First, NGF stimulates arborization and synaptogenesis via appropriate input neurons. Secondly, NGF serves as a discriminating mechanism that allows the elimination of neurons that have failed to sequester adequate target territory. This latter function is accomplished by an NGF-induced signal transduction cascade that prevents neuronal apoptosis (19, 20).
Target-derived NGF initiates these responses by binding to two different cell surface receptors: the tyrosine kinase receptor TrkA (33, 34, 36), a member of the Trk family of receptors (2), and the p75 neurotrophin receptor (12). TrkA binds preferentially to NGF, but it can also bind the structurally related neurotrophin-3 (NT-3)1 (47) in 3T3 fibroblasts (15), while the p75 receptor binds all of the neurotrophins (56, 57). It is clear from studies on cultured neurons that NGF binding to TrkA alone is sufficient to mediate many of the prototypic biological responses (31). Moreover, all sympathetic neurons are lost in TrkA −/− mice (61), as they are in NGF −/− mice (16). However, the p75 receptor likely also plays a role since recent evidence indicates that it modulates TrkA tyrosine kinase activity (3, 71), that it signals on its own to modulate ceramide (22, 23) and NFκB (10), and, finally, that there are deficits in sympathetic innervation in the p75 −/− mice (41).
Although the vast majority of sympathetic neurons have an absolute requirement for NGF during the period of target competition, three different lines of evidence indicate that these neurons may well “see” other cellular sources of neurotrophins, and that these other neurotrophins may play important biological roles. First, neurotrophins are made by Schwann cells (1, 30, 49), and sympathetic neurons themselves express both brain-derived neurotrophic factor (BDNF) and NT-3 mRNAs (60), raising the possibility of autocrine/paracrine interactions. Secondly, although neonatal sympathetic neurons do not respond to NT-3 with survival, as do their embryonic counterparts (8, 21), they express high affinity NT-3 binding sites (18); TrkC mRNA, which encodes the preferred Trk receptor for NT-3 (40), is expressed at low levels in the neonatal superior cervical ganglion (SCG). Finally, NT-3 −/− mice have 50% fewer sympathetic neurons (25, 26, 66) and display deficits in sympathetic target innervation that can be rescued by exogenous NT-3 (24).
In this paper we demonstrate that NT-3 only very poorly supports the survival of NGF-dependent neonatal sympathetic neurons, but that when survival is maintained by limiting quantities of NGF, NT-3 selectively mediates neuritogenesis and expression of genes associated with morphological growth. We have examined the biochemical basis of this differential biological responsiveness. Sympathetic neurons express relatively high levels of the TrkA receptor and low levels of the TrkC receptor. NGF activates TrkA in a graded fashion, while NT-3 activates TrkA and, to a much lesser extent, TrkC. Both of these neurotrophins induce similar sustained activation of TrkA, while NGF is 10-fold more efficient than NT-3 in mediating short-term TrkA activity. This TrkA activation is necessary for NT-3 to mediate sympathetic neuron survival and neuritogenesis, as demonstrated using a mutant NT-3 that activates only TrkC. However, even at similar acute levels of TrkA activation, NT-3 mediates neuronal survival at levels two- to threefold less well than NGF. These data suggest that NGF and NT-3 differentially regulate the TrkA receptor both with regard to activation time course and downstream targets, leading to selective regulation of neuritogenesis and survival. Such differential responsiveness to two ligands acting through the same Trk receptor has important implications for neurotrophin function throughout the nervous system.
Materials and Methods
Primary Neuronal Cultures
Mass cultures of pure sympathetic neurons from the SCG of postnatal day 1 rats were prepared as previously described (46). Neurons were plated on rat tail collagen-coated tissue-culture dishes: 4-, 12-, or 24-well plates (Falcon Labware, Becton Dickinson & Co., Lincoln Park, NJ) for neurite extension assays, 6-well plates for biochemistry, and 48-well plates for MTT assays. Low density SCG cultures for neurite extension assays were coated with poly-d-lysine and laminin (both from Collaborative Biomedical Products, Bedford, MA). For gene induction and protein analysis, ∼300,000 cells were plated per well of a 6-well dish.
In a few experiments, the neurons were cultured directly in 30 ng/ml NT-3 or BDNF, but, in the vast majority, NGF-dependent neurons were selected by culturing for 5 d in the presence of 10 ng/ml NGF. For survival assays, at the end of this 5-d selection, neurons were washed once for two h in neurotrophin-free media, and then fed with 400 μl of neurotrophincontaining media. Analysis of survival was performed 48 h later by using nonradioactive cell proliferation (MTT) assays (Celltitre 96; Promega, Madison, WI). 20 μl of the MTT reagent was added to each well and left for 90 min, followed by the addition of 200 μl of solubilization solution to lyse the cells. Each condition was repeated in quadruplicate. In each assay, baseline (0% survival) was considered 0 ng/ml NGF, and 10 ng/ml was considered 100% survival. All other conditions were related to these values.
For neurite extension assays, after 5 d, the neurons were switched into media containing 10 ng/ml NGF plus or minus the appropriate other neurotrophin. For the short-term biochemistry experiments, the medium was changed to L15 medium plus 0.1% BSA on the day of the experiment, left on for 3 h, and changed again 45 min before the induction began. Neurotrophin was then added to the cultures for 10 min, the medium was decanted off, and the cells were stored on ice. Each well was rinsed once with cold TBS, and 0.5–1.0 ml of lysis buffer was added. For the long-term biochemistry experiments, the medium was switched on day 5 to medium containing 10 ng/ml NGF plus the appropriate additional neurotrophin. Lysis was performed as described for the short-term experiments.
Analysis and Quantification of Process Outgrowth
Analysis of the effects of neurotrophins on neuronal growth was examined by measuring three parameters: process network density, total neurite length, and cell body area. The quantitative analysis of cell process density on high and low density cultures was performed using common statistics applied to random sets of lines, in particular the number of intersection points per unit area. In the microscope, the network of neural processes appears as a random set of lines in a plane. The number of visible cross-links and bifurcations of cell processes per unit area can therefore be considered as a quantitative measure of the cell process density. However, since the number of neurites is a direct function of the number of neurons, only those regions in the culture having a similar cell density are comparable. Thus, in each experiment, 10–15 sampling windows (10 mm2) were analyzed, each containing seven neuronal cell bodies. All interceptions and bifurcations of neurites within these windows were counted, resulting in an estimated value of neuritic process density. Statistical comparison of the mean values of density was performed using ANOVA (F-test).
Total neurite length and cell body area were measured in low density cultures within defined areas controlled for cell body number. Results were analyzed both within groups of sister cultures with different treatments and by pooling results of similar treatments from different groups of sister cultures. Similar results were obtained, and the pooled data are therefore presented. The t test and ANOVA were used to determine statistical significance.
RNA Preparation and Northern Blot Analysis
Total cytoplasmic RNA was collected from cultured neurons as described previously (46). Equal amounts of RNA (1–5 μg) were electrophoresed on a 1.2% agarose-formaldehyde gel, transferred to nitrocellulose, and probed with a 32P-labeled antisense RNA probe for Tα1 α-tubulin, p75 neurotrophin receptor, or tyrosine hydroxylase, as previously described (46). To confirm that equal amounts of RNA were loaded in each lane, ethidium bromide was added to the sample buffer before electrophoresis. Results were quantitated using scanning laser densitometry.
PCR Analysis and Sequencing
Total RNA was isolated from adult rat brain, liver, neonatal SCG, and cultured rat sympathetic neurons as described above. cDNAs were synthesized by reverse transcription using the SuperScriptII reverse transcriptase kit (GIBCO BRL, Gaithersburg, MD). PCR amplification was carried out on cDNAs using the following oligonucleotide primers specific for the intracellular domain of TrkC: 5′-GAGGATTTTGTAGATC-3′ and 5′-CCACGCCAGGCCAAGG-3′ as well as a set of pan-trk primers: 5′-TACGGTCGACGGTCCAAA-3′ and 5′-TGGCTTGTGGCAATTGTG-3′. Taq DNA polymerase and nucleotides were obtained from Promega. The PCR products were subcloned into the PCR-II vector using the TA Cloning Kit (Invitrogen, San Diego, CA). Random insert-containing clones were sequenced with the Sanger dideoxy protocol, using the Sequenase DNA Sequencing Kit (Amersham Corp., Arlington Heights, IL).
Antibodies
Anti-TrkCin2 was generated in rabbits using a peptide derived from the intracellular portion of rat TrkC, amino acids 88–108. This region of TrkC is distinct from the other Trk family members, but conserved amongst species. Anti-TrkCout is directed against the extracellular domain of TrkC and is described in Kaplan et al. (34). Although both of these antibodies are effective for Western blots, neither immunoprecipitates. For immunoprecipitations, we used anti-TrkA antibody RTA (14), anti-pan Trk antibody 203 (29), two different immunoprecipitating anti-TrkC antibodies that are described in Tsoulfas et al. (68), and anti-p75 antibody MC192 (11). For Western blots, we also used anti-TrkCin2 (as described above), anti-phosphotyrosine (UBI, Lake Placid, NY), and a rabbit anti–p75 antibody directed against the intracellular domain of the rat p75 neurotrophin receptor (the kind gift of P. Barker, McGill University, Montreal, Canada).
Immunoprecipitation and Western Blot Analysis
Sympathetic neurons were lysed in TBS lysis buffer (38) containing 137 mM NaCl, 20 mM Tris (pH 8.0), 1% (vol/vol) NP-40, 10% (vol/vol) glycerol, 1 mM PMSF, 10 μg/ml aprotinin, 0.2 μg/ml leupeptin, 5 mM Phenanthroline, and 1.5 mM sodium vanadate. For anti-pan Trk and anti-p75 immunoprecipitations, one well of a 6-well dish was lysed in 1 ml of buffer, while, for anti-TrkA or anti-TrkC immunoprecipitations, each of two wells was lysed in 500 μl and combined. The cells were rocked for 20 min at 4°C, scraped, and collected in microfuge tubes. Each sample was vortexed for 10 s, and then cleared by centrifugation. The lysates were normalized for protein concentration using a BCA protein assay reagent (Pierce Chemical Co., Rockford, IL). Total Trk protein was immunoprecipitated using 3 μl of anti-pan Trk 203, TrkA using 2 μl of anti-TrkA RTA, TrkC using 3 μl each of two TrkC-specific antibodies, and p75 neurotrophin receptor using 3 μl of antibody MC192. The immunoprecipitates were collected with protein A–Sepharose (Pharmacia, Uppsala, Sweden) for 1.5 h at 4°C followed by centrifugation. In some experiments, glycosylated proteins were precipitated from cell lysates with wheat germ lectin-agarose as described (35, 38).
For Western blot analysis, precipitates were washed three times with cold lysis buffer, boiled in sample buffer (2% SDS, 100 mM DTT, 10% glycerol, and 0.05% bromophenol blue) for 5 min, and electrophoresed on 8.0% SDS polyacrylamide minigels. For experiments characterizing the biochemical specificity of Trk antibodies, lysates of Sf9 cells expressing equivalent quantities of mammalian TrkA, TrkB, or TrkC were probed in Western blotting experiments with Trk antibodies. After electrophoresis, proteins were transferred to 0.2-μm nitrocellulose for 1 h at 0.5 A, and the membranes were washed twice (10 min each) in TBS. For all antibodies except anti-phosphotyrosine, for which membranes were blocked in 2% BSA (Sigma Chemical Co., St. Louis, MO), membranes were blocked in 5% nonfat dry milk in TBS for 2.5 h. Membranes were then washed twice (10 min each) in TBS, and the primary antibodies were used overnight at 4°C at dilutions of 1:5,000 for anti-phosphotyrosine, 1:1,000 or 1:2,000 for anti-pan Trk 203, 1:2,000 for anti-p75, and 1:2,000 for TrkCin2. Membranes were incubated in secondary antibody for 1.5 h at room temperature, and antibodies were used at dilutions of 1:10,000 or 1:20,000 for a goat anti–mouse HRP antibody (Boehringer Mannheim Biochemicals, Indianapolis, IN; used for anti-phosphotyrosine), 1:10,000 for a goat anti– rabbit HRP antibody (Boehringer Mannheim Biochemicals; used for antiTrkA, anti-TrkC, and anti-p75), and 1:2,000 for protein A–HRP (Sigma Chemical Co; used for anti-pan Trk). Detection was carried out using enhanced chemiluminescence (Amersham Corp.) and XAR x-ray film (Eastman Kodak Co., Rochester, NY). Results were quantitated by scanning laser densitometry.
Cultured Cell Lines
PC12 cell lines (27) were propagated in DME (GIBCO BRL) supplemented with 6% bovine calf serum and 6% horse serum in 100-mm tissueculture plates (Falcon Labware) at 7% CO2 in a 37°C chamber. Sf9 cells were infected with recombinant baculovirus vectors expressing human TrkA (65), rat TrkB (72), and rat TrkC (68) as described (65).
Results
NT-3 Selectively Promotes Neurite Extension in NGF-dependent Sympathetic Neurons
To determine whether sympathetic neurons responded to either NT-3 or BDNF after they become dependent upon target-derived NGF, we selected the NGF-dependent population of neonatal sympathetic neurons by culturing in 10 ng/ml NGF for 5 d (Fig. 1 A) and examined neurotrophinmediated survival and neurite extension. To assay for survival responses, after selection in NGF, neurons were switched to 30 ng/ml NT-3 or BDNF. BDNF was not sufficient to support the survival of NGF-dependent neurons; by 2 d after the switch, all of the neurons in the cultures were dead, as monitored by counting phase-bright cell bodies (Fig. 1 D). In contrast, 25–30 ng/ml NT-3 was sufficient to support the survival of a small population of NGFdependent neurons (Fig. 2 A).
Figure 1.

NGF-dependent sympathetic neurons respond to NT-3 but not to BDNF. Phase-contrast micrographs of cultures of pure sympathetic neurons from the postnatal day 1 rat SCG maintained in 10 ng/ml NGF for 5 d (A) and then supplemented with 30 ng/ml NT-3 (B) or 30 ng/ml BDNF (C). NT-3 enhanced the number of neurites compared with BDNF when examined 2 d after addition. In similar cultures where the NGF was replaced with 30 ng/ml BDNF (D), obvious cell body and process deterioration was evident.
Figure 2.

(A) NT-3 is 20–40-fold less efficient than NGF at supporting survival of NGF-dependent sympathetic neurons. Results of a colorimetric MTT assay to measure mitochondrial function and cell survival. Neonatal sympathetic neurons were cultured in 10 ng/ml NGF for 5 d in 48-well dishes, washed free of neurotrophin-containing medium, and then incubated for 2 d in various concentrations of NGF and/or NT-3, as indicated on the x axis. Each point represents the values pooled from three independent sets of survival assays, each of which was performed in quadruplicate. In these assays, absolute values are normalized so that the value obtained with 0 neurotrophin is 0% survival, while that obtained with 10 ng/ml NGF (in which the neurons were originally selected) is considered 100% survival. Error bars represent SEM; * = P < 0.05, ** = P < 0.01, and *** = P < 0.001. For NGF and NT-3 alone, comparisons were made using a t test with the 0 neurotrophin control. For the NGF/NT-3 combinations, comparisons were made against the survival supported by the same concentrations of NGF (no brackets) and NT-3 (brackets) alone. (B–D) NT-3 is equivalent to NGF in mediating neuritogenesis of NGF-dependent sympathetic neurons. (B) Three separate experiments were performed to determine the effect of NT-3 or BDNF on process outgrowth in sympathetic neurons. Sympathetic neurons were plated at moderate density on a collagen substrate, selected in 10 ng/ml NGF for 5 d, and then supplemented with either 30 ng/ml NT-3 or BDNF as indicated. The number of fields examined for each point ranged from 9–14. In all three experiments, significantly more neurite intersections were observed after exposure to 30 ng/ml NT-3 plus 10 ng/ml NGF (*** = P < 0.001) than 10 ng/ml NGF alone. (C and D) To confirm the results presented in B, and to directly compare NT-3 to NGF, neurons were plated at low density on a poly-d-lysine/laminin substrate, selected in 10 ng/ml NGF for 5 d, and then either maintained in 10 ng/ml NGF or switched to 30 ng/ml NGF or to 10 ng/ml NGF plus 30 ng/ml NT-3. Two independent experiments were performed, and 8–12 fields were quantitated per treatment in each experiment. The results of the two experiments were pooled. (C) 2–2.5-fold more neurite intersections were observed after exposure to either 30 ng/ml NGF or to 10 ng/ml NGF plus 30 ng/ ml NT-3 than after exposure to 10 ng/ml NGF alone (*** = P < 0.001). These NGF- and NT-3–induced increases were statistically similar to each other (P = 0.45). (D) Total neurite length was also significantly increased after exposure to 30 ng/ml NGF or to 10 ng/ml NGF plus 30 ng/ml NT-3, relative to 10 ng/ml NGF (*** = P < 0.001). Again, the increase was statistically similar for 30 ng/ml NGF vs 10 ng/ ml NGF plus 30 ng/ml NT-3 (P = 0.28). (E) NGF is more effective than NT-3 at mediating cell body hypertrophy. Cell body size of sympathetic neurons was quantitated in the experiments described in C and D. In this case, NT-3 elicited a small but statistically significant increase of ∼10%, whereas 30 ng/ml NGF led to an ∼25% hypertrophy of the sympathetic neuron cell bodies (** = P < 0.01, *** = P < 0.001). The hypertrophy observed in 10 ng/ml NGF plus 30 ng/ml NT-3 was significantly different from that seen in 30 ng/ml NGF (P = 0.0006). (F and G) Recombinant mutant NT-3 that activates TrkC but not TrkA is insufficient to support sympathetic neuron survival (F) or neuritogenesis (G). (F) Results of a colorimetric MTT assay to measure mitochondrial function and cell survival in response to NGF, NT-3, or a mutant NT-3 that binds only TrkC (mut. NT-3) (58, 59). Sympathetic neurons were cultured in 10 ng/ml NGF for 5 d, washed free of neurotrophin-containing media, and then incubated for 2 d in various concentrations of neurotrophins, as indicated on the x axis. Each point represents the values pooled from three independent sets of survival assays, each of which was performed in quadruplicate. Growth of sympathetic neurons in 50 or 100 ng/ml mutant NT-3 resulted in significantly less survival than 100 ng/ml NT-3 (*** = P < 0.001). In these assays, absolute values are normalized with regard to NGF-mediated survival, as described for A. (G) To compare sympathetic neuron process outgrowth in response to mutant NT-3 that activates only TrkC (mut. NT-3), neurons were cultured in 10 ng/ml NGF for 5 d, and then maintained for an additional 2 d in 10 ng/ml NGF plus or minus 30 ng/ml NT-3 or mutant NT-3. Two independent experiments were performed, and eight fields were quantitated per treatment. As shown in D, total neurite length was significantly increased after exposure to 10 ng/ml NGF plus 30 ng/ml NT-3, relative to 10 ng/ml NGF alone (*** = P < 0.0005). In contrast, no increase in neurite length was observed with mutant NT-3 addition relative to 10 ng/ml NGF alone (P = 0.14).
To quantitate the level of neuronal survival in NT-3 relative to NGF, we used MTT assays that measure mitochondrial function (Fig. 2 A). NGF-dependent sympathetic neurons were selected in 10 ng/ml NGF for 5 d, switched to varying concentrations of NGF or NT-3, and assayed 2 d after the switch. NGF mediated the survival of sympathetic neurons over a concentration range of 1.25– 10 ng/ml; 1.25 ng/ml NGF maintained ∼20% survival, 2.5 ng/ml 40–45% survival, 5.0 ng/ml 60–65% survival, and 10 ng/ml 100% survival. NT-3 maintained survival ∼20–40fold less efficiently than NGF; 10 ng/ml NT-3 maintained 10–15% survival, 25 ng/ml ∼20%, 50 ng/ml ∼25%, and 100 ng/ml ∼40%. A comparison of these survival curves indicated that 25 ng/ml NT-3 was roughly equivalent to 1.25 ng/ml NGF, and 100 ng/ml NT-3 to 2.5 ng/ml NGF.
To determine whether NT-3 or BDNF addition could mediate neurite extension independent of survival, sympathetic neurons were plated on collagen and selected in 10 ng/ml NGF for 5 d, and then 30 ng/ml NT-3 or BDNF was added in the presence of 10 ng/ml NGF for an additional 2 d. The addition of NT-3 led to a robust increase in the density of neuritic processes (Fig. 1 B), with a 2- to 2.5-fold increase in neuritic density in each of the three separate experiments (Fig. 2 B). In contrast, addition of 30 ng/ml BDNF had no measurable effect (Figs. 1 C and 2 B).
To more precisely define the effect of NT-3 on neuritogenesis, sympathetic neurons were plated at low density on poly-d-lysine/laminin, selected for 5 d in 10 ng/ml NGF, and then switched to 10 ng/ml NGF plus 30 ng/ml NT-3, or to 30 ng/ml NGF. 2 d later, the process network density, total neurite length, and cell body size were all measured. As seen in the higher density cultures (Fig. 2 B), the process network density was increased 2- to 2.5-fold in the presence of 10 ng/ml NGF plus 30 ng/ml NT-3 (Fig. 2 C). A statistically similar increase was noted with 30 ng/ml NGF. Similar results were obtained from measurements of total neurite length; both NT-3 and NGF mediated an ∼1.5-fold increase (Fig. 2 D). In contrast, NGF and NT-3 differentially regulated cell body size (Fig. 2 E). Neurons cultured in 10 ng/ml NGF plus 30 ng/ml NT-3 displayed a small but significant (P = 0.002) increase of ∼10%, whereas neurons cultured in 30 ng/ml NGF hypertrophied ∼25–30%, an increase that was significantly greater than that obtained with NGF plus NT-3 (P < 0.001). Thus, although NT-3 was approximately equivalent to NGF in its ability to promote neurite extension, it was significantly less effective in promoting cell body hypertrophy, and it was 20–40-fold less efficient at promoting neuronal survival.
NT-3 Enhances Survival of NGF-dependent Sympathetic Neurons
The regulation of neurite extension by both NGF and NT-3 raised the possibility that, while NT-3 on its own only poorly supported survival, it might enhance survival in the presence of limiting quantities of NGF. To test this possibility, neurons were first cultured in 10 ng/ml NGF for 5 d, and then were switched into a variety of concentrations of NGF and/or NT-3 (Fig. 2 A). 1.25 ng/ml NGF plus 25 ng/ml NT-3, each of which support ∼20% survival, together supported ∼35% neuronal survival. Similarly, 2.5 ng/ml NGF plus 50 ng/ml NT-3, which support ∼40% and 25% neuronal survival, respectively, together supported ∼65% survival. Finally, 2.5 ng/ml NGF plus 100 ng/ml NT-3, each of which support ∼40% survival, together supported 75% neuronal survival. Thus, NT-3 can enhance, in an approximately additive fashion, the survival of NGF-dependent sympathetic neurons when quantities of NGF are limiting.
NT-3 Selectively Induces Growth-associated Gene Expression
We have previously demonstrated that, in neonatal sympathetic neurons, NGF regulates the expression of the mRNAs encoding tyrosine hydroxylase, p75 neurotrophin receptor, and Tα1 α-tubulin in a graded, concentration- dependent fashion (46). To determine whether NT-3 regulated gene expression as it does neurite extension, sympathetic neurons were selected in 10 ng/ml NGF for 5 d, 10 or 30 ng/ml NT-3 was added in addition to 10 ng/ml NGF, and RNA was isolated at time points ranging from 6–48 h after addition. Northern blot analysis revealed that the addition of 30 ng/ml NT-3 for 6 h led to a 5- to 10-fold increase in Tα1 α-tubulin mRNA (Fig. 3 A), one member of the α-tubulin multigene family whose expression is regulated as a function of neuronal growth (48, 50, 51). This increase was maintained at 24 and 48 h, consistent with the robust increase in neuritic process density induced by NT-3 addition, and was concentration dependent: 10 ng/ml NT-3 elicited no significant increase in Tα1 α-tubulin mRNA (Fig. 3 D). The magnitude of the increase observed with 30 ng/ml NT-3 was similar to that observed upon addition of 200 ng/ml NGF (Fig. 3 D). We have previously demonstrated that levels of Tα1 mRNA increase in a concentration-dependent fashion with increasing levels of NGF to plateau at 100–200 ng/ml (46). Thus, 30 ng/ml NT-3 was capable of eliciting as large an increase in Tα1 mRNA as saturating quantities of NGF. In contrast with NT-3, the addition of 30 ng/ml BDNF had no effect on expression of Tα1 α-tubulin mRNA (data not shown).
Figure 3.
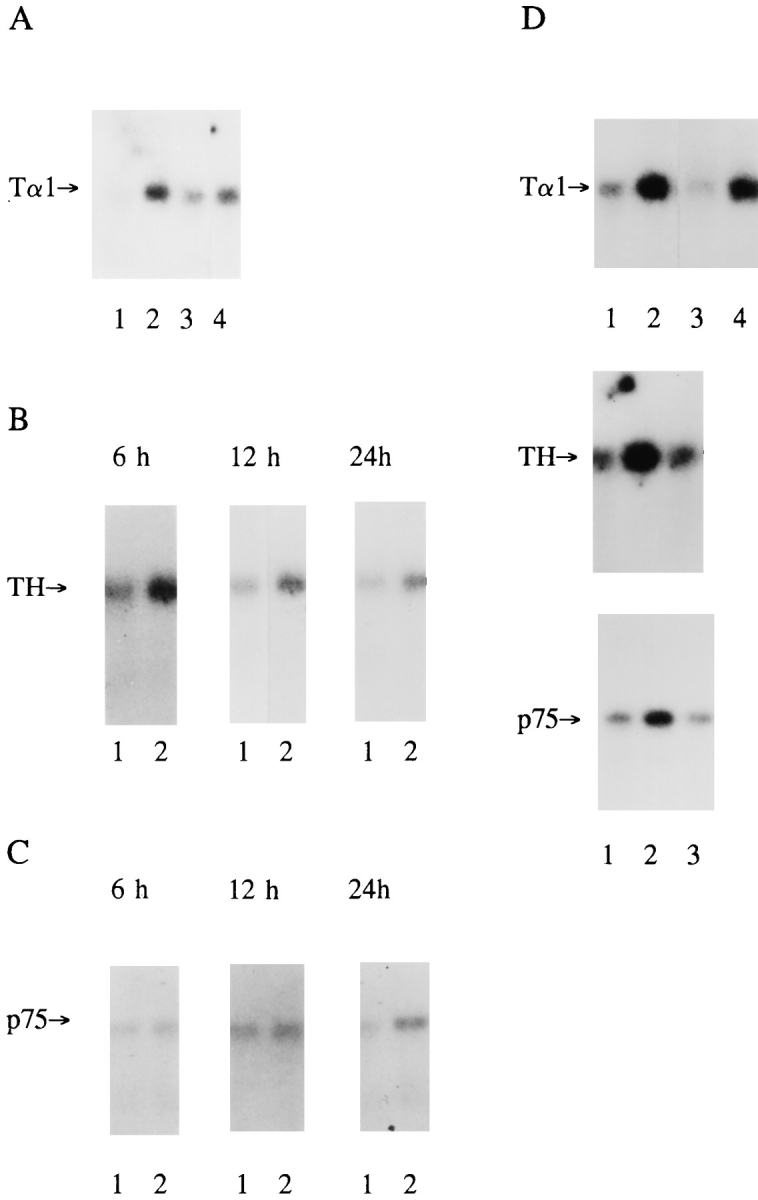
NT-3 selectively regulates expression of genes associated with morphological growth in a concentration-dependent fashion. (A) Expression of Tα1 α-tubulin mRNA (Tα1) in equal amounts of total RNA isolated from neonatal sympathetic neurons cultured for 5 d in the presence of 10 ng/ml NGF (lane 1) followed by addition of 30 ng/ml NT-3 for 6 h (lane 2), 24 h (lane 3), or 48 h (lane 4). (B and C) Expression of tyrosine hydroxylase (TH) (B) and p75 neurotrophin receptor (p75) (C) mRNAs in response to the addition of 30 ng/ml NT-3 for 6 , 12, or 24 h. In all panels, lane 1 includes total RNA from neurons maintained for the entire time in 10 ng/ml NGF, while lane 2 includes an equal amount of total RNA isolated from sister cultures treated with 10 ng/ml NGF plus 30 ng/ml NT-3. (D) Expression of Tα1 α-tubulin, tyrosine hydroxylase, and p75 neurotrophin receptor mRNAs after 24 h of treatment with 10 ng/ml NGF (lane 1), 10 ng/ml NGF plus 30 ng/ml NT-3 (lane 2), or 10 ng/ml NGF plus 10 ng/ml NT-3 (lane 3). Lane 4 for Tα1 mRNA contains an equal amount of total RNA isolated from neurons treated with 200 ng/ml NGF for 24 h.
The addition of 30 ng/ml NT-3 also led to a smaller, approximately two- to threefold increase in the expression of tyrosine hydroxylase mRNA (Fig. 3 B). This increase, which was not elicited by 10 ng/ml NT-3 (Fig. 3 D), was first observed at 6 h, and was subsequently maintained for 48 h. Addition of BDNF had no effect on expression of tyrosine hydroxylase mRNA (data not shown).
In contrast with Tα1 α-tubulin and tyrosine hydroxylase mRNAs, NT-3 had a lesser effect on the expression of p75 neurotrophin receptor mRNA (Fig. 3 C) with an ∼1.5- to 2-fold increase after 24 h in the presence of 30 but not 10 ng/ml NT-3. This reduced response is particularly striking given that increased NGF induces a robust increase in expression of p75 receptor mRNA in sympathetic neurons in culture and in vivo (46, 50, 53). A similar difference was noted at the protein level (see Fig. 6 F). p75 receptor levels were increased approximately two- to threefold in neurons cultured in 10 ng/ml NGF plus 30 ng/ml NT-3, whereas 40 and 100 ng/ml NGF led to increases of ∼5-fold and 15fold, respectively. Thus, NT-3 was equivalent to NGF in inducing expression of Tα1 α-tubulin mRNA, but was less effective at inducing expression of the p75 neurotrophin receptor.
Figure 6.

(A–D) NT-3 induces a prolonged autophosphorylation of TrkA equivalent to that induced by NGF. Neonatal sympathetic neurons were cultured for 5 d in 10 ng/ml NGF, and then switched to neurotrophincontaining media for an additional 2 d. Cellular lysates were immunoprecipitated (IP) either with anti-pan Trk or anti-TrkA. Immunoprecipitates were then analyzed by Western blot analysis with anti-phosphotyrosine or antipan Trk (1°Ab). All samples were normalized for equal amounts of protein. (A) Trk autophosphorylation in response to 2 d of constant exposure to NGF and/or NT-3. Note that all three of these lanes are from the same autoradiograph of the same Western blot. (B) NGF and NT-3 do not alter Trk receptor levels in sympathetic neurons. In two separate experiments, total Trk levels in NGF-dependent sympathetic neurons were similar regardless of neurotrophintreatment, as evidenced when neurons are cultured in various concentrations of NGF, NT-3, and/or BDNF for 2 d. (C) TrkA autophosphorylation in response to 2 d of constant exposure to NGF and/or NT-3. (D) The same immunoprecipitates as in C were probed with antipan Trk on a separate Western blot to ensure that approximately equal amounts of TrkA were present in each sample. (E) NGF and NT-3 do not alter TrkA or TrkC levels. To determine whether TrkA or TrkC levels changed with increased levels of neurotrophins, NGF-dependent sympathetic neurons were cultured for 48 h in various concentrations of NGF and/or NT-3. Lysates were immunoprecipitated with the anti-pan Trk antibody, and then analyzed by Western blots with antibodies specific to TrkA or TrkC. The appropriately sized Trk bands are indicated by arrows. (F) NGF and NT-3 alter the ratio of Trk/p75 in NGF-dependent sympathetic neurons. After immunoprecipitation with anti-pan Trk, the lysates shown in B were immunoprecipitated for the p75 neurotrophin receptor using the mAb MC192. Immunoprecipitates were then probed on a Western blot with a polyclonal anti-p75 antibody.
The NGF-dependent Population of Neonatal Sympathetic Neurons Express Low Levels of Full-Length, Kinase Domain–intact TrkC
Neonatal, NGF-dependent sympathetic neurons express both TrkA and the p75 neurotrophin receptor. To determine whether they also express the TrkC receptor, we performed Western blot analysis. To perform these studies, an anti-peptide antibody was generated against sequences from the intracellular domain of the full-length TrkC receptor. The specificity of this antibody was assessed by Western blot analysis of lysates of Sf9 insect cells expressing human TrkA, rat TrkB, or rat TrkC (Fig. 4 A). This anti-peptide antibody, anti-TrkCin2, recognized only rat TrkC and not human TrkA or rat TrkB expressed in Sf9 cells (Fig. 4 A, left), while anti-pan Trk antibody 203 (29) recognized all three Trk receptors (Fig. 4 A, right). To ensure that anti-TrkCin2 also did not recognize rat TrkA, PC12 cell lysates were immunoprecipitated with anti-pan Trk 203, and the precipitates were analyzed on Western blots. Anti-TrkCin2 did not recognize rat TrkA from PC12 cells, while anti-pan Trk 203 did (data not shown).
Figure 4.

Full-length TrkC is expressed in NGF-dependent sympathetic neurons. (A, left) Specificity of the anti-TrkCin2 antibody, which recognizes the intracellular domain of TrkC, was tested by probing lysates of insect cells expressing human TrkA, rat TrkB, or rat TrkC on Western blots. (A, right) Anti-pan Trk antibody 203 recognizes all three Trk family members on a similar Western blot. (B) Western blot analysis of full-length TrkC in NGF-dependent sympathetic neurons as detected by probing WGA precipitates with anti-TrkCin2. Lane 1 includes protein from sympathetic neurons cultured for 5 d in 10 ng/ml NGF, and lane 2 includes protein from sister cultures induced with 30 ng/ml NT-3 for 10 min. The immunoreactive TrkC band is detected at 145 kD, the size of full-length TrkC.
To determine whether NGF-dependent sympathetic neurons express full-length TrkC protein, they were cultured for 5 d in 10 ng/ml NGF, and then glycosylated proteins in the cellular lysates were precipitated using WGA. Western blot analysis of these WGA precipitates with anti-TrkCin2 revealed the presence of an immunoreactive protein of 145 kD, the size of full-length TrkC (Fig. 4 B). Confirmation that this band represented bona fide TrkC was obtained using a second antibody that is specific to the extracellular domain of TrkC (anti-TrkCout; data not shown). This second antibody, which recognizes both truncated and full-length TrkC, detected little or no truncated TrkC in the cellular lysates.
TrkC is an alternatively spliced protein, with some of the splice variants encoding proteins with kinase domain inserts (40, 67, 69). To determine whether the full-length TrkC expressed by NGF-dependent sympathetic neurons contained a kinase insert, we sequenced across the kinase domain in seven independent TrkC clones isolated from these cells by reverse transcriptase–PCR. In all cases, the clones represented TrkC mRNAs with intact kinase domains. Similarly, PCR analysis across the kinase domain demonstrated that only non–insert-containing TrkC mRNA could be amplified in RNA from cultured sympathetic neurons, while insert-containing TrkC mRNA could be easily amplified in RNA isolated from rat brain under the same conditions (data not shown). Thus, NGF-dependent sympathetic neurons express full-length, non–insert-containing TrkC.
Levels of the TrkC receptor were, however, very low relative to the TrkA receptor, as previously reported (8, 21). TrkA was easily detectable by Northern blots (Toma, J., D. Rogers, D. Senger, R. Campenot, and F. Miller, manuscript submitted for publication), immunocytochemistry (39), and Western blots of anti-pan Trk immunoprecipitates (see Fig. 6 E), while TrkC was difficult to detect using these same techniques (data not shown; see Fig. 6 E).
NT-3 Preferentially Induces TrkA Autophosphorylation in Sympathetic Neurons
These studies indicated that NGF-dependent sympathetic neurons express two Trk receptors that might be responsible for mediating the functional effects of NT-3: relatively high levels of TrkA, and low levels of full-length, kinaseintact TrkC. To determine whether either of these two receptors was activated by NT-3, we analyzed Trk receptor autophosphorylation in response to neurotrophin addition. Specifically, sympathetic neurons were selected for 5 d in 10 ng/ml NGF, washed for 3 h in neurotrophin-free media, and then treated with varying concentrations of NGF and/or NT-3 for 10 min. Cell lysates were quantitated with regards to protein concentration, and levels of Trk receptor autophosphorylation were determined by immunoprecipitation of lysates with anti-pan Trk, followed by Western blot analysis with anti-phosphotyrosine. Blots were subsequently reprobed with anti-pan Trk to ensure that equal amounts of Trk protein were present in all samples, and both phosphotyrosine and Trk protein levels were quantitated from these blots densitometrically. These experiments demonstrated that NGF addition from 10–100 ng/ml resulted in graded increases in TrkA autophosphorylation over the entire concentration range (Fig. 5 A; data not shown), consistent with the biological response curve we have previously documented (46).
Figure 5.

(A–D) NT-3 exposure leads to the autophosphorylation of both TrkA and TrkC on NGF-dependent sympathetic neurons. Neonatal sympathetic neurons were cultured for 5 d in 10 ng/ml NGF, washed for 3 h in neurotrophin-free media, and then induced with neurotrophins for 10 min. Cell lysates were immunoprecipitated (IP) with either anti-pan Trk 203 (pantrk), anti-TrkA, or anti-TrkC. Immunoprecipitates were analyzed by Western blot analysis with anti-phosphotyrosine or antipan Trk 203 as the primary antibodies (1°Ab). All samples were normalized for equal amounts of protein. (A) Trk autophosphorylation increased in a graded fashion in response to 10 ng/ml NGF, and in response to 30 ng/ml NGF, relative to base line levels of autophosphorylation with 0 NGF. (Numbers, left) Molecular weight markers; (Trk) size of the Trk band. (B) Trk autophosphorylation in sympathetic neurons in response to varying concentrations of neurotrophins. Note the presence of a single autophosphorylated Trk band in the samples treated with NT-3. (C) TrkA and TrkC autophosphorylation in response to NGF vs NT-3. Approximately 300,000 neurons were lysed and analyzed for TrkA-specific autophosphorylation in response to no neurotrophin addition, 10 ng/ml NGF, or 30 ng/ml NT-3. The same lysates were then immunoprecipitated for TrkC-specific autophosphorylation in response to 10 ng/ml NGF or 30 ng/ml NT-3. Note that the right two lanes containing the TrkC immunoprecipitates are both from the same autoradiograph of the same gel. (D) The same immunoprecipitates as in C were probed with anti-pan Trk on a separate Western blot to ensure that approximately equal amounts of TrkA were present in each sample. (E) TrkA in PC12 cells is not autophosphorylated in response to NT-3. PC12 cells were induced with NGF and/or NT-3 for 10 min, and cellular lysates were immunoprecipitated with anti-pan Trk 203, followed by a Western blot with anti-phosphotyrosine.
Similar experiments revealed that 30 ng/ml NT-3 also led to increased Trk receptor autophosphorylation on NGF-dependent sympathetic neurons. When 30 ng/ml NT-3 and 10 ng/ml NGF were added together, a small but significant increase in Trk autophosphorylation was observed relative to 10 ng/ml NGF alone (Fig. 5 B). In contrast, 30 ng/ml NT-3 plus 30 ng/ml NGF resulted in a level of Trk autophosphorylation similar to that induced by 30 ng/ml NGF alone. Interestingly, 100 ng/ml NT-3 was capable of eliciting short-term Trk autophosphorylation at levels comparable to 10 ng/ml NGF (Fig. 5 B), even though it was approximately two- to threefold less efficient at inducing neuronal survival (Fig. 2 A).
To determine whether the NT-3-induced increase in Trk autophosphorylation was due to TrkA and/or TrkC activation, we used antibodies specific to TrkA or TrkC for immunoprecipitation, followed by Western blot analysis with anti-phosphotyrosine. These studies revealed that a 10min treatment of NGF-dependent sympathetic neurons with 30 ng/ml NT-3 alone was sufficient to induce autophosphorylation of TrkA at levels ∼2.5-fold lower than 10 ng/ml NGF (Fig. 5 C). Western blotting of the same samples with a pan Trk antibody confirmed that the level of Trk protein was similar, within 25%, in each lane (Fig. 5 D). A similar, more detailed analysis revealed that 30 ng/ml NT-3 elicited approximately the same level of short-term TrkA autophosphorylation as did 2.5 ng/ml NGF (data not shown), a 10-fold difference that is similar to that observed for NT-3 vs NGF on total Trk autophosphorylation (Fig. 5 B).
Similar experiments using TrkC-specific antibodies for immunoprecipitation revealed that 30 ng/ml NT-3 also led to tyrosine phosphorylation of the 145-kD TrkC band (Fig. 5 C, TrkC IP lanes). In contrast to NT-3, treatment with 10 ng/ml NGF had no effect on TrkC activity (Fig. 5 C). Thus, NT-3 led to autophosphorylation of both TrkC and TrkA.
In contrast with sympathetic neurons, when PC12 cells were treated with 30 ng/ml NT-3 for 10 min, no autophosphorylation of TrkA was observed (Fig. 5 E), in agreement with previous findings (32). Moreover, when 30 ng/ml NT-3 was added in the presence of 10 ng/ml NGF, TrkA autophosphorylation was not increased above the level obtained with 10 ng/ml NGF alone (Fig. 5 E).
NT-3–mediated Sympathetic Neuron Survival and Neuritogenesis Requires TrkA Activation
These data indicated that NT-3 activated TrkA and, to a lesser degree, TrkC on NGF-dependent sympathetic neurons. To determine which of these two receptors mediated the observed biological responses to NT-3, we used a mutant NT-3 that binds to TrkC but not to TrkA (58, 59). As reported, when fibroblasts expressing either TrkA or TrkC were exposed to 30 ng/ml of this mutant NT-3, TrkC but not TrkA was autophosphorylated. When, instead, the same fibroblasts were exposed to 30 ng/ml NT-3, both TrkA and TrkC were autophosphorylated (data not shown).
To determine whether activation of TrkC alone was sufficient to mediate survival, sympathetic neurons were cultured for 5 d in 10 ng/ml NGF, and then were switched to varying concentrations of NGF, NT-3, or mutant NT-3 for 2 d. Quantitation of neuronal survival using MTT assays revealed that the mutant NT-3 was incapable of eliciting significant levels of neuronal survival at concentrations of up to 100 ng/ml (Fig. 2 F), while 100 ng/ml NT-3 mediated ∼45% survival. These data therefore indicate that TrkA activation is necessary for NT-3–mediated survival of sympathetic neurons.
To determine whether activation of TrkA was also necessary for NT-3–mediated neuritogenesis, sympathetic neurons were cultured at low density for 5 d in 10 ng/ml NGF, and then were maintained for an additional 2 d in 10 ng/ml NGF plus or minus 30 ng/ml NT-3 or mutant NT-3. Quantitation of total neurite length per cell body (Fig. 2 G) revealed that, as seen in previous experiments (Fig. 2 D), NT-3 caused an increase in total neurite outgrowth that was significantly greater than NGF alone (P < 0.0005). In contrast, the amount of neurite outgrowth observed in NGF plus mutant NT-3 was similar to that observed with NGF alone. Thus, TrkA activation was also necessary for NT-3–mediated neuritogenesis.
Differences in Acute and Sustained TrkA Activation in Response to NGF vs NT-3
Together, these results indicated that NGF, acting through TrkA, mediated sympathetic neuron survival and neuritogenesis, whereas NT-3, also acting at TrkA, was 20–40-fold less efficient at mediating survival, but equally effective at inducing neuritogenesis. Moreover, even when NT-3 was used at concentrations that elicited levels of short-term TrkA autophosphorylation that were similar to NGF, it was two- to threefold less effective in maintaining neuronal survival. One potential explanation for these results is that NGF-mediated Trk activation is more sustained than NT-3–mediated activation. To address this possibility, neurons were selected in 10 ng/ml NGF for 5 d, and then were switched for an additional 2 d to either 40 ng/ml NGF or to 30 ng/ml NT-3 plus 10 ng/ml NGF. Anti-pan Trk immunoprecipitations of cellular lysates followed by probing with anti-phosphotyrosine revealed that 30 ng/ml NT-3 plus 10 ng/ml NGF resulted in levels of long-term total Trk autophosphorylation that were similar to 40 ng/ml NGF (Fig. 6 A). Moreover, in both conditions, the Trk autophosphorylation was greatly increased above that obtained with 10 ng/ml NGF alone (Fig. 6 A), which was itself sustained at higher levels than when neurons were washed free of neurotrophins for several hours (data not shown). These results were very different from those observed after 10 min of induction, when the level of total Trk autophosphorylation obtained with 30 ng/ml NT-3 plus 10 ng/ml NGF was much lower than that with 30 ng/ml NGF, and was only slightly increased relative to 10 ng/ml NGF (Fig. 5 B).
To determine whether the differences in the time course of Trk autophosphorylation in response to NGF vs NT-3 were due to activation of TrkA vs TrkC, similar experiments were performed using the TrkA-specific antibody. Western blot analysis of anti-TrkA immunoprecipitates with anti-phosphotyrosine revealed that, as observed for total Trk autophosphorylation, 30 ng/ml NT-3 plus 10 ng/ml NGF was as efficient at sustaining TrkA autophosphorylation as was 40 ng/ml NGF (Fig. 6 C). Moreover, both of these conditions led to much higher TrkA autophosphorylation than did 10 ng/ml NGF, a pattern that was again very distinct from that observed at short time points (Fig. 5 C). Western blot analysis of the same immunoprecipitations with the pan Trk antibody confirmed that similar amounts of TrkA protein were present in each sample (Fig. 6 D). Thus, although 30 ng/ml NT-3 led to only a low level of TrkA autophosphorylation in the short term and would not mediate survival of these neurons, it was as efficient at sustaining long-term TrkA autophosphorylation as 30 ng/ml NGF.
To ensure that the differences between long-term and short-term Trk autophosphorylation profiles were not due to alterations in Trk receptor levels in the long-term experiments, 5-d-old NGF-dependent neurons were switched to a variety of concentrations of NGF and/or NT-3 for 48 h, and Trk levels were assessed by Western blot analysis of anti-pan Trk immunoprecipitates with the pan Trk antibody. These studies revealed that levels of Trk protein were constant over this time course, irrespective of the concentration of NGF added and/or the presence of NT-3 or BDNF (Fig. 6 B). To confirm this result, cell lysates were immunoprecipitated with a pan Trk antibody, followed by Western blot analysis with TrkA- or TrkC-specific antibodies. As observed for total Trk protein levels, the addition of 100 ng/ml NGF or NT-3 for 2 d had no effect on TrkA or TrkC levels (Fig. 6 E).
In contrast, levels of the p75 neurotrophin receptor varied considerably as a function of the level of NGF and/or NT-3. As previously reported for the mRNA (46, 52), levels of p75 protein were dramatically increased by concentrations of NGF ranging from 10–110 ng/ml (Fig. 6 E). Significant but smaller increases occurred with the addition of 30 or 100 ng/ml NT-3 to 10 ng/ml NGF (Fig. 6 E), consistent with the small changes in mRNA levels (Fig. 3 C). Thus, although Trk levels remained constant, variations in neurotrophin exposure resulted in alterations in the p75/ Trk receptor ratio, as previous reported in vivo (53).
Discussion
The data presented in this paper support a number of conclusions. First, we demonstrate that NT-3 is as effective as NGF in mediating neuritogenesis and growth-associated gene expression in NGF-dependent sympathetic neurons, but it is 20–40-fold less efficient at supporting their survival. NGF is also considerably more effective than NT-3 at mediating cell body hypertrophy and induction of the p75 neurotrophin receptor. Second, we demonstrate that sympathetic neurons respond to NGF from 10–100 ng/ml with graded increases in TrkA autophosphorylation. This concentration curve correlates precisely with our previously published biological response curve (46). Third, these studies surprisingly indicate that NT-3 exposure leads to autophosphorylation of TrkA at concentrations that do not activate TrkA in a PC12 cell environment. At short exposure time points, NGF is ∼10-fold more efficient than NT-3 at inducing TrkA autophosphorylation, but, when exposure is maintained for 48 h, the activation obtained in response to NT-3 is equivalent to that elicited by NGF. Moreover, NT-3, at concentrations that elicit short-term Trk autophosphorylation similar to that induced by NGF, mediates survival approximately two- to threefold less efficiently than NGF. Fourth, we demonstrate that NGF- dependent sympathetic neurons express full-length, kinase domain–intact TrkC that is also autophosphorylated in response to NT-3, albeit at low levels relative to TrkA. Fifth, although NT-3 leads to both TrkA and TrkC activation, it is apparently the TrkA activation that is essential for neuronal survival and neuritogenesis. Finally, long-term exposure to NGF and NT-3 does not alter either TrkA or TrkC levels in sympathetic neurons, although it does lead to differences in the p75/Trk receptor ratio. Together, these data indicate that NGF and NT-3 differentially regulate the survival and differentiation of NGF-dependent sympathetic neurons while activating the same Trk receptor.
Evidence presented here indicates that the NGF-induced graded biological response curve previously documented for NGF-dependent sympathetic neurons (46) can be completely explained on the basis of graded increases in TrkA activation. These graded increases in TrkA autophosphorylation occur at concentrations of NGF varying from 1 (Belliveau, D., and F.D. Miller, unpublished data) to 100 ng/ml. At the low end of this curve (1–10 ng/ml), the first response to be elicited is neuronal survival. On a per neuron basis, the survival response is clearly a step-function and likely occurs at a given threshold of TrkA activation. Once the survival threshold is reached, neurite outgrowth and gene expression are then regulated in a graded fashion as a function of receptor activation over the concentration range from 10–100 ng/ml. Although the breadth of this dose–response curve may seem somewhat surprising, it is very similar to that observed in PC12 cells that are overexpressing the TrkA receptor (29) (Kaplan, D., unpublished observations). Biologically, such an extended concentration curve makes sense; during the period of target innervation, neurons need to sequester a small amount of NGF to ensure their survival, but, thereafter, they are confronted with innervating an increasingly large target area as the animal grows (see 54), necessitating graded regulation of the neuronal growth response.
The more surprising result is the selectivity of the NT-3– mediated neuronal response, as demonstrated here. NGF elicits survival, neuritogenesis, cell body hypertrophy, and induction of Tα1 α-tubulin and p75 receptor mRNAs. In contrast, NT-3 mediates neuritogenesis and induction of Tα1 mRNA as effectively as NGF, but only poorly elicits survival, cell body hypertrophy, and p75 receptor induction. A number of pieces of evidence presented here indicate that NT-3 mediates these responses by activating TrkA. First, total TrkA levels are much higher than TrkC levels on sympathetic neurons, and the pattern of total Trk activation stimulated by NT-3 is essentially identical to that seen for TrkA. Second, and most importantly, selective activation of TrkC, using an NT-3 mutant that does not bind to TrkA (58), does not mediate survival or neuritogenesis, indicating that TrkA activation is necessary for these two NT-3–mediated responses. Further support for this conclusion derives from recent data indicating that TrkA activation is sufficient to maintain the survival of sympathetic neurons derived from TrkC −/− mice (17). Thus, NGF and NT-3, both of which predominantly activate TrkA in NGF-dependent sympathetic neurons, as shown here, mediate different biological responses.
Why does NT-3 activate TrkA in a sympathetic neuron, when it will not do so in PC12 cells (data shown here) (32), a transformed cell line that is the “nearest neighbor” to sympathetic neurons? Sympathetic neurons express only the insert-containing form of the TrkA receptor (Barker, P., and F. Miller, unpublished observations) (4, 13), which is preferentially activated by NT-3 (13). However, since PC12 cells also express the same TrkA receptor variant (4, 13), the difference in responsiveness to NT-3 must be a function of the cellular environment. In fact, a number of lines of evidence indicate that the p75 receptor may be responsible. In fibroblasts that do not express p75, the TrkA receptor is activated by NT-3 at relatively low concentrations (37, 64). In PC12 cells that have lost most of their p75 receptor (6) and in PC12 cells that overexpress TrkA (29), NT-3 also activates TrkA. Thus, it appears that, when the p75/TrkA receptor ratio is low, NT-3 acts at TrkA, but, when this ratio is high, NT-3 is excluded from TrkA. The fact that the ratio of p75/TrkA is ∼10-fold higher in PC12 cells than it is in a sympathetic neurons (73) is consistent with this explanation.
Since NGF and NT-3 both function through TrkA to mediate sympathetic neuron survival and neuritogenesis, then what is the reason for the selectivity of the NT-3 response relative to NGF? We suggest that these two neurotrophins differentially regulate the TrkA receptor both with regard to activation time course and downstream targets. In support of this hypothesis, we provide evidence that the profile of TrkA activation differs for these two factors. In the short term, NGF is ∼10-fold better than NT-3 at stimulating TrkA autophosphorylation, while, in the long term, NT-3 is as effective as NGF. However, in spite of the equivalent sustained TrkA signaling, NGF and NT-3 mediate different responses. This is in contrast with the model of differential signaling by mitogenic and differentiation factors for PC12 cells. This hypothesis proposes that sustained signaling elicits neurite outgrowth, while transient signaling elicits cell proliferation (55). In studies reported here, NGF and NT-3 mediate different biological responses while both stimulating sustained TrkA activation. The differences in biological responses seen here may instead be due to NGF inducing a strong acute TrkA activation that does not occur with NT-3.
We also suggest that differential activation of TrkA by NGF and NT-3 may lead to differing profiles of downstream substrate activation, and that NT-3 may simply not activate pathways essential to survival as efficiently as NGF does. Support for this hypothesis derives from the observation that, when NT-3 and NGF are used at concentrations that elicit equivalent short-term TrkA activation, NGF is still approximately two- to threefold better at maintaining neuronal survival. Thus, differences in absolute levels of short-term activation are not sufficient by themselves to fully explain the differential biological responsiveness.
This hypothesis, that NGF and NT-3 differentially activate TrkA to mediate differing neuronal responses, does not exclude the potential involvement of the TrkC and p75 receptors. Although our data indicate that TrkA activation is necessary for NT-3–mediated survival and neuritogenesis, it is still formally possible that simultaneous activation of low levels of TrkC with TrkA may somehow “dampen” certain aspects of TrkA-mediated signaling. We consider this possibility unlikely, and suggest that the p75 neurotrophin receptor is much more likely to play such a modulatory role (53). If the liganded p75 receptor signals, as recent data suggests (10, 22, 23), to mediate apoptosis (5) (Miller, F., and P. Barker, unpublished observations), then the ultimate survival of a neuron may depend upon the relative activation of TrkA vs p75 receptors. Since NT-3 binds less efficiently to TrkA than NGF, and more efficiently to the p75 receptor than NGF (57), it may well be that the TrkA signal induced by NT-3 is insufficient, in this cellular context, to override the p75-driven signal, thereby resulting in neuronal death. We have, in fact, obtained recent data demonstrating that, in low amounts of NGF, selective activation of the p75NTR leads to sympathetic neuron apoptosis (Belliveau, D., S. Bamji, M. Majdan, R. Aloyz, C. Pozniak, J. Kohn, C. Causing, and F.D. Miller, manuscript submitted for publication).
Together with the data derived from the NT-3 −/− mice (25), these experiments indicate that NT-3 is essential for the normal development of sympathetic neurons and provide insights into the cellular mechanisms whereby it mediates its effects. During neurogenesis and before target contact, sympathetic neurons express relatively high levels of TrkC and relatively low levels of TrkA, and respond to NT-3 with survival (8, 21, 70, 73). Since NT-3 is expressed within the sympathetic ganglia during this developmental window, potentially in sympathetic neurons themselves (60), there may be a TrkC-mediated autocrine/paracrine loop that participates in regulating neuronal number and survival before target contact. As sympathetic neurons become postmitotic and extend their axons toward their targets, TrkA levels greatly increase and TrkC levels greatly decrease (8, 21), coincident with the onset of NGF dependency. Development of an absolute requirement for target-derived NGF provides a cellular mechanism for establishing the appropriate target innervation density: if sympathetic neurons still responded to NT-3 with survival, this would subvert the competition. Instead, our data suggest that only those neurons that successfully compete for target-derived NGF would be competent to respond to NT-3, which would then make them more “fit” to compete for additional target territory. We suggest that the developmental difference in responsiveness to NT-3 is a consequence of the developmental switch in receptor repertoire: NT-3 signaling through TrkC in sympathetic neuroblasts mediates survival, while NT-3 signaling through TrkA in postmitotic neurons selectively mediates neuritogenesis. Is there any evidence that such a hypothesis is true in vivo, given that NT-3 only mediates neuritogenesis at concentrations of 30 ng/ml in culture? In fact, a recent report by El-Shamy et al. (24) demonstrates that NT-3 is necessary for appropriate sympathetic target innervation in vivo. In transgenic animals carrying one functional copy of the NT-3 gene (NT-3 +/− mice), certain sympathetic targets are insufficiently innervated, a deficit that can be rescued by administration of exogenous NT-3.
Thus, even the prototypic NGF-dependent sympathetic neurons, which display an absolute dependence upon NGF during the period of target competition, differentially respond to two neurotrophins that activate the same Trk receptor. Such biochemical convergence and functional cross talk of several neurotrophins at one Trk receptor could provide a partial explanation for the differences noted between Trk receptor vs neurotrophin gene knockouts (63). Thus, neuronal interdependence on multiple neurotrophins, acting through one Trk receptor, may be the rule rather than the exception.
Acknowledgments
We gratefully acknowledge Patrik Ernfors for sharing his unpublished data with us, Phil Barker and members of the Miller laboratory for many stimulating conversations during the course of this work, and Audrey Speelman, Rahul Varma, and Marta Majdan for their excellent technical assistance. We would also like to thank Drs. Mikael Rydén and Carlos Ibáñez for the gift of the purified recombinant mutant NT-3, and Drs. Pantelis Tsoulfas, Luis Parada, and Lou Reichardt for their gifts of antibody reagents.
This work was supported by grants from the Medical Research Council (MRC), and the Canadian NeuroSciences Network to F.D. Miller. F.D. Miller is a Killam Scholar, D.J. Belliveau is supported by an MRC:Genentech fellowship, C. Lachance by a Fondes de Recherche et Santé de Québec (FRSQ) fellowship, and J. Kohn by Jeanne Timmins Costello and FRSQ studentships.
Abbreviations used in this paper
- BNDF
brain-derived neurotrophic factor
- NT-3
neurotrophin-3
- SCG
superior cervical ganglion
Footnotes
D.J. Belliveau and I. Krivko contributed equally to this work.
References
- 1.Acheson A, Barker PA, Alderson RF, Miller FD, Murphy RA. Detection of brain-derived neurotrophic factor-like activity in fibroblasts and Schwann cells: inhibition by antibodies to NGF. Neuron. 1991;7:265–275. doi: 10.1016/0896-6273(91)90265-2. [DOI] [PubMed] [Google Scholar]
- 2.Barbacid M. The trk family of neurotrophin receptors. J Neurobiol. 1994;25:1386–1403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- 3.Barker PA, Shooter EM. Disruption of NGF binding to the low affinity neurotrophin receptor p75LNTR reduces NGF binding to TrkA on PC12 cells. Neuron. 1994;13:203–215. doi: 10.1016/0896-6273(94)90470-7. [DOI] [PubMed] [Google Scholar]
- 4.Barker PA, Lomen-Hoerth C, Gensch EM, Meakin SO, Glass DJ, Shooter EM. Tissue-specific alternative splicing generates two isoforms of the trkA receptor. J Biol Chem. 1993;268:15150–15157. [PubMed] [Google Scholar]
- 5.Barrett GL, Bartlett PF. The p75 nerve growth factor receptor mediates survival or death depending on the stage of sensory neuron development. Proc Natl Acad Sci USA. 1994;91:6501–6505. doi: 10.1073/pnas.91.14.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benedetti M, Levi A, Chao MV. Differential expression of nerve growth factor receptors leads to altered binding affinity and neurotrophin responsiveness. Proc Natl Acad Sci USA. 1993;90:7859–7863. doi: 10.1073/pnas.90.16.7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berkemeier LR, Winslow JW, Kaplan DR, Nikolics K, Goeddel DV, Rosenthal A. Neurotrophin-5: a novel neurotrophic factor that activates trk and trkB. Neuron. 1991;7:857–866. doi: 10.1016/0896-6273(91)90287-a. [DOI] [PubMed] [Google Scholar]
- 8.Birren SJ, Lo L, Anderson DJ. Sympathetic neuroblasts undergo a developmental switch in trophic dependence. Development (Camb) 1993;119:597–610. doi: 10.1242/dev.119.3.597. [DOI] [PubMed] [Google Scholar]
- 9.Campenot RB. Development of sympathetic neurons in compartmentalized cultures. 1. Local control of neurite growth by nerve growth factor. Dev Biol. 1982;93:1–12. doi: 10.1016/0012-1606(82)90232-9. [DOI] [PubMed] [Google Scholar]
- 10.Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhauser N, BohmMatthaei R, Baeuerle PA, Barde Y-A. Selective activation of NF-κB by nerve growth factor through the neurotrophin receptor p75. Science (Wash DC) 1996;272:542–545. doi: 10.1126/science.272.5261.542. [DOI] [PubMed] [Google Scholar]
- 11.Chandler CE, Parsons LM, Hosang M, Shooter EM. A monoclonal antibody modulates the interaction of nerve growth factor with PC12 cells. J Biol Chem. 1984;259:6882–6889. [PubMed] [Google Scholar]
- 12.Chao MV. The p75 neurotrophin receptor. J Neurobiol. 1994;25:1373–1385. doi: 10.1002/neu.480251106. [DOI] [PubMed] [Google Scholar]
- 13.Clary DO, Reichardt LF. An alternatively spliced form of the nerve growth factor receptor TrkA confers an enhanced response to neurotrophin-3. Proc Natl Acad Sci USA. 1994;91:11133–11137. doi: 10.1073/pnas.91.23.11133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clary DO, Weskamp G, Austin LR, Reichardt LF. TrkA cross-linking mimics neuronal responses to nerve growth factor. Mol Biol Cell. 1994;5:549–563. doi: 10.1091/mbc.5.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cordon-Cardo C, Tapley P, Jing SQ, Nanduri V, O'Rourke E, Lamballe F, Kovary K, Klein R, Jones KR, Reichardt LF, Barbacid M. The trk tyrosine protein kinase mediates the mitogenic properties of nerve growth factor and neurotrophin-3. Cell. 1991;66:173–183. doi: 10.1016/0092-8674(91)90149-s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts-Meek S, Armanini MP, Ling LH, McMahon SB, Shelton DL, Levinson AD, Phillips HS. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 1994;76:1001–1011. doi: 10.1016/0092-8674(94)90378-6. [DOI] [PubMed] [Google Scholar]
- 17.Davies AM, Minichiello L, Klein R. Developmental changes in NT-3 signalliing via TrkA and TrkB in embryonic neurons. EMBO (Eur Mol Biol Organ) J. 1995;18:4482–4489. doi: 10.1002/j.1460-2075.1995.tb00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dechant G, Rodríguez-Tébar A, Kolbeck R, Barde YA. Specific high-affinity receptors for neurotrophin-3 on sympathetic neurons. J Neurosci. 1993;13:2610–2616. doi: 10.1523/JNEUROSCI.13-06-02610.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deckwerth TL, Johnson EM., Jr Neurotrophic factor deprivation-induced death. Ann NY Acad Sci. 1993;679:121–131. doi: 10.1111/j.1749-6632.1993.tb18293.x. [DOI] [PubMed] [Google Scholar]
- 21.DiCicco-Bloom E, Friedman WJ, Black IB. NT-3 stimulates sympathetic neuroblast proliferation by promoting precursor survival. Neuron. 1993;11:1101–1111. doi: 10.1016/0896-6273(93)90223-e. [DOI] [PubMed] [Google Scholar]
- 22.Dobrowsky RT, Werner MH, Castellino AM, Chao MV, Hannun YA. Activation of the sphingomyelin cycle through the low- affinity neurotrophin receptor. Science (Wash DC) 1994;265:1596–1599. doi: 10.1126/science.8079174. [DOI] [PubMed] [Google Scholar]
- 23.Dobrowsky RT, Jenkins GM, Hannun YA. Neurotrophins induce sphingomyelin hydrolysis: modulation by co-expression of p75 with Trk receptors. J Biol Chem. 1995;270:22135–22142. doi: 10.1074/jbc.270.38.22135. [DOI] [PubMed] [Google Scholar]
- 24.El-Shamy WM, Linnarsoon S, Lee K-F, Jaenisch R, Ernfors P. Prenatal and postnatal requirements of NT-3 for sympathetic neuroblast survival and innervation of specific targets. Development (Camb) 1996;122:491–500. doi: 10.1242/dev.122.2.491. [DOI] [PubMed] [Google Scholar]
- 25.Ernfors P, Lee KF, Kucera J, Jaenisch R. Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell. 1994;77:503–512. doi: 10.1016/0092-8674(94)90213-5. [DOI] [PubMed] [Google Scholar]
- 26.Farinas I, Jones KR, Backus C, Wang X-Y, Reichardt LF. Severe sensory and sympathetic deficits in mice lacking neurotrophin-3. Nature (Lond) 1994;369:658–661. doi: 10.1038/369658a0. [DOI] [PubMed] [Google Scholar]
- 27.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hallbook F, Ibáñez CF, Persson H. Evolutionary studies of the nerve growth factor family reveal a novel member abundantly expressed in xenopus ovary. Neuron. 1991;6:845–858. doi: 10.1016/0896-6273(91)90180-8. [DOI] [PubMed] [Google Scholar]
- 29.Hempstead BL, Rabin SJ, Kaplan L, Reid S, Parada LF, Kaplan DR. Overexpression of the trk tyrosine kinase rapidly accelerates nerve growth factor-induced differentiation. Neuron. 1992;9:883–896. doi: 10.1016/0896-6273(92)90241-5. [DOI] [PubMed] [Google Scholar]
- 30.Heumann R, Lindholm D, Bandtlow C, Meyer M, Radeke MJ, Misko TP, Shooter E, Thoenen H. Differential regulation of mRNA encoding nerve growth factor and its receptor in rat sciatic nerve during development, degeneration, and regeneration: role of macrophages. Proc Natl Acad Sci USA. 1987;84:8735–8739. doi: 10.1073/pnas.84.23.8735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ibáñez CF, Ebendal T, Barbany G, Murray-Rust J, Blundell TL, Persson H. Disruption of the low affinity receptor-binding site in NGF allows neuronal survival and differentiation by binding to the trk gene product. Cell. 1992;69:329–341. doi: 10.1016/0092-8674(92)90413-7. [DOI] [PubMed] [Google Scholar]
- 32.Ip NY, Stitt TN, Tapley P, Klein R, Glass DJ, Fandl J, Greene LA, Barbacid M, Yancopoulos GD. Similarities and differences in the way neurotrophins interact with the Trk receptors in neuronal and nonneuronal cells. Neuron. 1993;10:137–149. doi: 10.1016/0896-6273(93)90306-c. [DOI] [PubMed] [Google Scholar]
- 33.Kaplan DR, Hempstead BL, Martin-Zanca D, Chao MV, Parada LF. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- 34.Kaplan DR, Martin-Zanca D, Parada LF. Tryosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature (Lond) 1991;350:158–160. doi: 10.1038/350158a0. [DOI] [PubMed] [Google Scholar]
- 35.Kaplan DR, Matsumoto K, Lucarelli E, Thiele CJ. Induction of TrkB by retinoic acid mediates biologic responsiveness to BDNF and differentiation of human neuroblastoma cells. Neuron. 1993;11:321–331. doi: 10.1016/0896-6273(93)90187-v. [DOI] [PubMed] [Google Scholar]
- 36.Klein R, Jing S, Nanduri V, O'Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65:189–197. doi: 10.1016/0092-8674(91)90419-y. [DOI] [PubMed] [Google Scholar]
- 37.Klein R, Lamballe F, Bryant S, Barbacid M. The trkB tyrosine protein kinase is a receptor for neurotrophin-4. Neuron. 1992;8:947–956. doi: 10.1016/0896-6273(92)90209-v. [DOI] [PubMed] [Google Scholar]
- 38.Knusel B, Rabin SJ, Hefti F, Kaplan DR. Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J Neurosci. 1994;14:1542–1554. doi: 10.1523/JNEUROSCI.14-03-01542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kohn J, Toma JG, Kuchel GA, Miller FD. Differential localization of trkA and p75 neurotrophin receptors in postnatal and adult sympathetic neurons. Soc Neurosci Abstr. 1996;22:1009. [Google Scholar]
- 40.Lamballe F, Tapley P, Barbacid M. trkC encodes multiple neurotrophin-3 receptors with distinct biological properties and substrate specificities. EMBO (Eur Mol Biol Organ) J. 1993;12:3083–3094. doi: 10.1002/j.1460-2075.1993.tb05977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee K-F, Bachman K, Landis S, Jaenisch R. Dependence on p75 for innervation of some sympathetic targets. Science (Wash DC) 1994;263:1447–1449. doi: 10.1126/science.8128229. [DOI] [PubMed] [Google Scholar]
- 42.Leibrock J, Lottspeich F, Hohn A, Hofer M, Hengerer B, Masiakowski P, Thoenen H, Barde Y-A. Molecular cloning and expression of brain-derived neurotrophic factor. Nature (Lond) 1989;341:149–152. doi: 10.1038/341149a0. [DOI] [PubMed] [Google Scholar]
- 43.Levi-Montalcini R. The nerve growth factor 35 years later. Science (Wash DC) 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- 44.Levi-Montalcini R, Booker B. Excessive growth of the sympathetic ganglia evoked by a protein isolated from mouse salivary glands. Proc Natl Acad Sci USA. 1960;46:373–381. doi: 10.1073/pnas.46.3.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levi-Montalcini R, Booker B. Destruction of the sympathetic ganglia in mammals by an antiserum to a nerve growth protein. Proc Natl Acad Sci USA. 1960;46:381–390. doi: 10.1073/pnas.46.3.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma Y, Campenot RB, Miller FD. Concentration-dependent regulation of neuronal gene expression by nerve growth factor. J Cell Biol. 1992;117:135–141. doi: 10.1083/jcb.117.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 48.Mathew TC, Miller FD. Increased expression of Tα1 α-tubulin mRNA during collateral and NGF-induced sprouting of sympathetic neurons. Dev Biol. 1990;141:84–92. doi: 10.1016/0012-1606(90)90103-p. [DOI] [PubMed] [Google Scholar]
- 49.Meyer M, Matsuoka I, Wetmore C, Olson L, Thoenen H. Enhanced synthesis of brain-derived neurotrophic factor in the lesioned peripheral nerve: different mechanisms are responsible for the regulation of BDNF and NGF mRNA. J Cell Biol. 1992;119:45–54. doi: 10.1083/jcb.119.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller FD, Naus CCG, Durand M, Bloom FE, Milner RJ. Isotypes of α-tubulin are differentially regulated during neuronal maturation. J Cell Biol. 1987;105:3065–3073. doi: 10.1083/jcb.105.6.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller FD, Tetzlaff W, Bisby MA, Fawcett JW, Milner RJ. Rapid induction of the major embryonic α-tubulin mRNA Tα1, during nerve regeneration in adult rats. J Neurosci. 1989;9:1452–1463. doi: 10.1523/JNEUROSCI.09-04-01452.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller FD, Mathew TC, Toma JG. Regulation of nerve growth factor receptor gene expression by nerve growth factor in the developing peripheral nervous system. J Cell Biol. 1991;112:303–312. doi: 10.1083/jcb.112.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller FD, Speelman A, Mathew TC, Fabian J, Chang E, Pozniak C, Toma JG. Nerve growth factor derived from terminals selectively increases the ratio of p75 to trkA NGF receptors on mature sympathetic neurons. Dev Biol. 1994;161:206–217. doi: 10.1006/dbio.1994.1021. [DOI] [PubMed] [Google Scholar]
- 54.Purves, D. 1988. Body and Brain. A Trophic Theory of Neural Connections. Harvard University Press, Cambridge. [DOI] [PubMed]
- 55.Qui MS, Green SH. PC12 cell neuronal differentiation is associated with prolonged p21ras activity and consequent prolonged ERK activity. Neuron. 1992;9:705–717. doi: 10.1016/0896-6273(92)90033-a. [DOI] [PubMed] [Google Scholar]
- 56.Rodríguez-Tébar A, Dechant G, Barde YA. Binding of brainderived neurotrophic factor to the nerve growth factor receptor. Neuron. 1990;4:487–492. doi: 10.1016/0896-6273(90)90107-q. [DOI] [PubMed] [Google Scholar]
- 57.Rodríguez-Tébar A, Dechant G, Götz R, Barde YA. Binding of neurotrophin-3 to its neuronal receptors and interactions with nerve growth factor and brain-derived neurotrophic factor. EMBO (Eur Mol Biol Organ) J. 1992;11:917–922. doi: 10.1002/j.1460-2075.1992.tb05130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rydén M, Ibáñez CF. Binding of neurotrophin-3 to p75LNGFR, trkA and trkB mediated by a single functional epitope distinct from that recognized by trkC. J Biol Chem. 1996;271:5623–5627. doi: 10.1074/jbc.271.10.5623. [DOI] [PubMed] [Google Scholar]
- 59.Rydén M, Murray-Rust J, Glass D, Ilag LL, Trupp M, Yancopoulos GD, McDonald NQ, Ibáñez CF. Functional analysis of mutant neurotrophins deficient in low-affinity binding reveals a role for p75LNGFR in NT-4 signalling. EMBO (Eur Mol Biol Organ) J. 1995;14:1979–1990. doi: 10.1002/j.1460-2075.1995.tb07190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schecterson LC, Bothwell M. Novel roles for neurotrophins are suggested by BDNF and NT-3 mRNA expression in developing neurons. Neuron. 1992;9:449–463. doi: 10.1016/0896-6273(92)90183-e. [DOI] [PubMed] [Google Scholar]
- 61.Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature (Lond) 1994;368:246–248. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- 62.Snider WD. Nerve growth factor enhances dendritic arborization of sympathetic ganglion cells in developing mammals. J Neurosci. 1988;8:2628–2634. doi: 10.1523/JNEUROSCI.08-07-02628.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 64.Soppet D, Escandon E, Maragos J, Middlemas DS, Reid SW, Blair J, Burton LE, Stanton BR, Kaplan DR, Hunter T, et al. The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trkB tyrosine kinase receptor. Cell. 1991;65:895–903. doi: 10.1016/0092-8674(91)90396-g. [DOI] [PubMed] [Google Scholar]
- 65.Stephens RM, Loeb DM, Copeland TD, Pawson T, Greene LA, Kaplan DR. Trk receptors use redundant signal transduction pathways involving SHC and PLC-γ1 to mediate NGF responses. Neuron. 1994;12:691–705. doi: 10.1016/0896-6273(94)90223-2. [DOI] [PubMed] [Google Scholar]
- 66.Tessarollo L, Vogel KS, Palko ME, Reid SW, Parada LF. Targeted mutation in the neurotrophin-3 gene results in loss of muscle sensory neurons. Proc Natl Acad Sci USA. 1994;91:11844–11848. doi: 10.1073/pnas.91.25.11844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsoulfas P, Soppet D, Escandon E, Tessarollo L, MendozaRamirez J-L, Rosenthal A, Nikolics K, Parada LF. The rat trkC locus encodes multiple neurogenic receptors that exhibit differential response to neurotrophin-3 in PC12 cells. Neuron. 1993;10:975–990. doi: 10.1016/0896-6273(93)90212-a. [DOI] [PubMed] [Google Scholar]
- 68.Tsoulfas P, Stephens RM, Kaplan DR, Parada LF. TrkC isoforms with inserts in the kinase domain show impaired signaling responses. J Biol Chem. 1996;271:5691–5697. doi: 10.1074/jbc.271.10.5691. [DOI] [PubMed] [Google Scholar]
- 69.Valenzuela DM, Maisonpierre PC, Glass DJ, Rojas E, Nuñez L, Kong Y, Gies DR, Stitt TN, Ip NY, Yancopoulos GD. Alternative forms of rat trkC with different functional capabilities. Neuron. 1993;10:963–974. doi: 10.1016/0896-6273(93)90211-9. [DOI] [PubMed] [Google Scholar]
- 70.Verdi JM, Anderson DJ. Neurotrophins regulate sequential changes in neurotrophin receptor expression by sympthetic neuroblasts. Neuron. 1994;13:1359–1372. doi: 10.1016/0896-6273(94)90421-9. [DOI] [PubMed] [Google Scholar]
- 71.Verdi JM, Birren SJ, Ibáñez CF, Persson H, Kaplan DR, Benedetti M, Chao MV, Anderson DJ. p75LNGFRregulates Trk signal transduction and NGF-induced neuronal differentiation in MAH cells. Neuron. 1994;12:733–745. doi: 10.1016/0896-6273(94)90327-1. [DOI] [PubMed] [Google Scholar]
- 72.Wolf DE, McKinnon CA, Daou MC, Stephens RM, Kaplan DR, Ross AH. Interaction with trkA immobilizes gp75 in the high affinity nerve growth factor receptor complex. J Biol Chem. 1995;270:2133–2138. doi: 10.1074/jbc.270.5.2133. [DOI] [PubMed] [Google Scholar]
- 73.Wyatt S, Davies AM. Regulation of nerve growth factor receptor gene expression in sympathetic neurons during development. J Cell Biol. 1995;130:1435–1446. doi: 10.1083/jcb.130.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]