

Abstract
Many secreted bioactive signaling molecules, including the yeast mating pheromones a-factor and α-factor, are initially synthesized as precursors requiring multiple intracellular processing enzymes to generate their mature forms. To identify new gene products involved in the biogenesis of a-factor in Saccharomyces cerevisiae, we carried out a screen for MAT a-specific, mating-defective mutants. We have identified a new mutant, ste24, in addition to previously known sterile mutants. During its biogenesis in a wild-type strain, the a-factor precursor undergoes a series of COOH-terminal CAAX modifications, two sequential NH2-terminal cleavage events, and export from the cell. Identification of the a-factor biosynthetic intermediate that accumulates in the ste24 mutant revealed that STE24 is required for the first NH2-terminal proteolytic processing event within the a-factor precursor, which takes place after COOH-terminal CAAX modification is complete. The STE24 gene product contains multiple predicted membrane spans, a zinc metalloprotease motif (HEXXH), and a COOH-terminal ER retrieval signal (KKXX). The HEXXH protease motif is critical for STE24 activity, since STE24 fails to function when conserved residues within this motif are mutated. The identification of Ste24p homologues in a diverse group of organisms, including Escherichia coli, Schizosaccharomyces pombe, Haemophilus influenzae, and Homo sapiens, indicates that Ste24p has been highly conserved throughout evolution. Ste24p and the proteins related to it define a new subfamily of proteins that are likely to function as intracellular, membrane-associated zinc metalloproteases.
The precursors of eukaryotic secreted hormones, neuropeptides, and other signaling molecules undergo a variety of posttranslational maturation events, including proteolytic processing and chemical modification, which result in biologically active molecules that are released from the cell. In Saccharomyces cerevisiae, the mating pheromones, a-factor and α-factor, are secreted by haploid cells of the opposite mating type, MAT a and MATα, respectively, and function to initiate mating responses that lead to the formation of MAT a/MATα diploids. Both pheromones are synthesized as precursors that undergo an ordered set of maturation events (Sprague and Thorner, 1992). In the case of α-factor, these events are well understood, and include signal sequence cleavage, glycosylation, and proteolytic processing by three peptidases (Kex2p, Kex1p, and Ste13p) during the transit of α-factor through the classical secretory pathway (Fuller et al., 1988). The identification of yeast Kex2 as a serine protease acting at paired basic residues of pro–α-factor (Julius et al., 1984) paved the way for the discovery of the homologous mammalian neuroendocrine processing enzymes furin and the proprotein convertases (Seidah and Chretien, 1994; Bruzzaniti et al., 1996; Seidah et al., 1996). In contrast to our extensive knowledge about the enzymes that execute α-factor processing, the identification of enzymes involved in a-factor maturation has not yet been completed.
The mating pheromone a-factor is encoded by two similar and functionally redundant genes, MFA1 and MFA2 (Brake et al., 1985; Michaelis and Herskowitz, 1988). The a-factor precursors encoded by these genes (36 and 38 amino acids long, respectively) are ultimately processed to the mature, bioactive form of a-factor, which is 12 amino acids long and contains a farnesylated and carboxylmethylated COOH-terminal cysteine residue (Anderegg et al., 1988). A schematic view of the a-factor precursor encoded by MFA1 is shown at the top of Fig. 1. Flanking the “mature” region of the precursor is an “NH2-terminal extension” and a COOH-terminal “CAAX” motif (C is cysteine, A is aliphatic, and X can be various amino acids; Zhang and Casey, 1996). As indicated, the biogenesis of a-factor can be viewed as comprising three separate general stages: COOH-terminal modification, NH2-terminal processing, and export (Chen et al., 1997). During COOH-terminal modification, the CAAX motif of the a-factor precursor is first prenylated, the AAX residues (VIA, for a-factor) are proteolysed, and the newly exposed cysteine residue is carboxylmethylated. Subsequently, the COOH-terminally modified a-factor precursor undergoes NH2-terminal processing, which consists of two distinct and sequential cleavage steps, P1→ P2 cleavage occurring between residues T7 and A8, and P2→ M cleavage occurring between residues N21 and Y22 (Chen, 1993; Chen et al., 1997). Finally, fully matured a-factor is exported out of the cell by a nonclassical export mechanism that involves the ATP-binding cassette transporter Ste6p. The CAAX motif is known to signal COOH-terminal prenylation, proteolysis, and methylation of a number of key eukaryotic proteins, most notably the Ras proteins (Powers et al., 1986; Zhang and Casey, 1996). To date, however, a-factor is the only known example of a prenylated and methylated molecule that is secreted from the cell.
Figure 1.

Model for the pathway of a-factor biogenesis. The a-factor biosynthetic intermediates and the components of the a-factor biogenesis machinery are shown (see the text for more information). Several of the a-factor intermediates can be directly visualized by SDS-PAGE and are designated P0, P1, P2, and M (Chen et al., 1997). The a-factor precursor contains an NH2-terminal extension, a mature portion, and a COOH-terminal CAAX motif, as indicated at top. During a-factor biogenesis, the unmodified a-factor precursor (P0) undergoes COOH-terminal modification (prenylation, proteolytic cleavage of AAX, and carboxylmethylation) to yield the fully COOH-terminally modified species P1. Next, NH2-terminal proteolytic processing occurs in two distinct steps, the first (P1→ P2) cleavage removing seven residues from the NH2-terminal extension to yield the P2 species, and the second (P2→ M) cleavage generating mature a-factor, which is exported from the cell. Question marks indicate that the corresponding component has not yet been genetically identified.
Over the past years, several components that mediate distinct events of a-factor biogenesis have been identified (see Fig. 1), either as mutants with a MAT a-specific sterile phenotype, or as mutants exhibiting decreased activity of a hyperactive Ras allele. The α and β subunits of the Ras and a-factor farnesyltransferase are encoded by the RAM2 and RAM1 genes, respectively, which have homologous mammalian counterparts (Powers et al., 1986; Goodman et al., 1990; Schafer et al., 1990; He et al., 1991; Chen et al., 1991a ,b). The carboxyl methyltransferase that methylates both a-factor and Ras is encoded by the STE14 gene (Hrycyna and Clarke, 1990; Hrycyna et al., 1991; Sapperstein et al., 1994); a mammalian version of Ste14p is biochemically, but not genetically, well characterized (Stephenson and Clarke, 1990; Tan et al., 1991; Pillinger et al., 1994). In the case of NH2-terminal processing, a homologue of the human insulin-degrading enzyme (IDE)1 family of proteases, encoded by the AXL1 gene, has recently been shown to be responsible for the second (P2→ M) cleavage of the a-factor precursor (Adames et al., 1995). AXL1 also appears to play a key role in the presumably distinct and unrelated process of bud site selection in haploid cells (Fujita et al., 1994). Finally, the export of a-factor is mediated by the product of the STE6 gene, an ATP-binding cassette protein that is closely related to the mammalian multidrug resistance protein (Mdr1; Kuchler et al., 1989; McGrath and Varshavsky, 1989; Michaelis, 1993).
From the above description, it is clear that many gene products implicated in a-factor biogenesis are now known. These provide important tools for studying the cell biology and biochemistry of a-factor maturation and export. However, at least two genes encoding proteases involved in the biogenesis of a-factor are still missing. One of these is the CAAX protease, which has been biochemically characterized, but remains to be genetically identified (Ashby et al., 1992; Hrycyna and Clarke, 1992). Another is the protease(s) required for the first (P1→ P2) cleavage step within the NH2-terminal extension of the a-factor precursor. The identification of novel a-factor biogenesis genes is the goal of the present study.
Here we describe the isolation and characterization of a new sterile mutant and identification of the corresponding gene, designated STE24. We demonstrate that STE24 is required for the first (P1→ P2) NH2-terminal proteolytic cleavage event during biogenesis of the a-factor precursor, as indicated in Fig. 1. STE24 encodes a protein with several distinctive features, including multiple predicted transmembrane spans, a COOH-terminal ER retrieval signal, and a zinc metalloprotease motif. We show by mutational analysis that the metalloprotease motif is essential for Ste24p functional activity. Taken together, these results suggest that Ste24p itself may directly mediate the P1→ P2 NH2-terminal proteolytic cleavage of the a-factor precursor; this cleavage event may occur on the cytosolic face of either the ER or another organellar membrane. Ste24p is the founding member of a family of membraneassociated zinc metalloproteases that exhibit striking evolutionary conservation from bacteria to mammals.
Materials and Methods
Strains, Media, and Growth Conditions
The yeast strains used in this study are listed in Table I. SM1058 was formerly designated EG123 (Michaelis and Herskowitz, 1988). All strains are isogenic to SM1058, with the exception of the mating and halo testers SM1067, SM1068, and SM1086. The FUS1-lacZ strain used for the mutant screen was generated as follows: the FUS1-lacZ integration plasmid pGA1716 (Rhodes et al., 1990) was kindly provided by B. Errede (University of North Carolina, Chapel Hill, NC). The plasmid pGA1716 was linearized by digestion with ApaI and integrated into the ura3 locus of the mata1 − strain SM1061. Two independent transformants were confirmed for FUS1-lacZ induction upon α-factor treatment, and were designated SM3038 and SM3039.
Table I.
Yeast Strains Used in This Study
| Strain | Genotype | Reference | ||
|---|---|---|---|---|
| SM1058 | MAT a leu2 ura3 trp1 his4 can1 | Michaelis and Herskowitz (1988) | ||
| SM1059 | MATα leu2 ura3 trp1 his4 can1 | Michaelis and Herskowitz (1988) | ||
| SM1060 | MAT a/MATα leu2/leu2 ura3/ura3 trp1/trp1 his4/his4 can1/can1 | Michaelis and Herskowitz (1988) | ||
| SM1061 | mata1− leu2 ura3 trp1 his4 can1 | This laboratory | ||
| SM1067 | MAT a lys1 cry1 | Michaelis and Herskowitz (1988) | ||
| SM1068 | MATα lys1 | Michaelis and Herskowitz (1988) | ||
| SM1086 | MATα ss2-1 his6 met1 can1 cyh2 | Michaelis and Herskowitz (1988) | ||
| SM1853 | MATa ste14::URA3 leu2 ura3 trp1 his4 can1 [pCEN TRP1 MFA1] | This laboratory | ||
| SM1871 | MAT a ste14::TRP1 leu2 ura3 trp1 his4 can1[pCEN URA3 MFA1] | This laboratory | ||
| SM2059 | MAT a mfa1::LEU2 mfa2::lacZ leu2 ura3 trp1 his4 can1 [pCEN URA3 mfa1-A8G] | This laboratory | ||
| SM3038 | mata1 − leu2 trp1 his4 can1 URA3::FUS1-lacZ | This study | ||
| SM3039 | mata1 − leu2 trp1 his4 can1 URA3::FUS1-lacZ | This study | ||
| SM3040 | MAT a leu2 ura3 trp1 can1 | This laboratory | ||
| SM3042 | MATα leu2 ura3 trp1 can1 | This laboratory | ||
| SM3044 | mata1 − ste24-1 leu2 trp1 his4 can1 URA3::FUS1-lacZ | This study | ||
| SM3060 | MAT a ste24-1 leu2 ura3 trp1 can1 | This study | ||
| SM3061 | mata1 − axl1-2 leu2 trp1 his4 can1 URA3::FUS1-lacZ | This study | ||
| SM3062 | mata1 − axl1-2 leu2 trp1 his4 can1 URA3::FUS1-lacZ | This study | ||
| SM3063 | mata1 − axl1-4 leu2 trp1 his4 can1 URA3::FUS1-lacZ | This study | ||
| SM3064 | mata1 − ste14-11 leu2 trp1 his4 can1 URA3::FUS1-lacZ | This study | ||
| SM3069 | SM3060 [pCEN URA3] | Transformant of SM3060 with pRS316 | ||
| SM3071 | SM3060 [pCEN URA3 STE24::HA] | Transformant of SM3060 with pSM1107 | ||
| SM3072 | SM3060 [pCEN URA3 ste24-H297A::HA] | Transformant of SM3060 with pSM1103 | ||
| SM3073 | SM3060 [pCEN URA3 ste244-E298A::HA] | Transformant of SM3060 with pSM1104 | ||
| SM3074 | SM3060 [pCEN URA3 ste244-E298D::HA] | Transformant of SM3060 with pSM1105 | ||
| SM3082 | SM3060 [pCEN LEU2 STE24] | Transformant of SM3060 with pSM1098 | ||
| SM3083 | MAT a ste24-1 leu2 ura3 trp1 can1 | This study | ||
| SM3088 | mata1 −/MAT a ste24-1/STE24 his4/HIS4 leu2/leu2 ura3/ura3 trp1/trp1 can1/can1 | This study | ||
| SM3089 | mata1 −/MAT a his4/HIS4 leu2/leu2 ura3/ura3 trp1/trp1 can1/can1 | This study | ||
| SM3095 | MAT a/MATα ste24Δ::LEU2/STE24 leu2/leu2 ura3/ura3 trp1/trp1 his4/his4 can1/can1 | This study | ||
| SM3103 | MAT a ste24Δ::LEU2 leu2 ura3 trp1 his4 can1 | Progeny of SM3095 | ||
| SM3104 | SM3039 [pCEN TRP1 MFA1] | Transformant of SM3039 with pSM464 | ||
| SM3105 | SM3044 [pCEN TRP1 MFA1] | Transformant of SM3044 with pSM464 | ||
| SM3106 | SM3061 [pCEN TRP1 MFA1] | Transformant of SM3061 with pSM464 | ||
| SM3107 | SM3062 [pCEN TRP1 MFA1] | Transformant of SM3062 with pSM464 | ||
| SM3108 | SM3063 [pCEN TRP1 MFA1] | Transformant of SM3063 with pSM464 | ||
| SM3286 | SM3103 [pCEN URA3 MFA1] | Transformant of SM3103 with pSM233 | ||
| SM3309 | SM3060 [pCEN URA3 MFA1] | Transformant of SM3060 with pSM322 | ||
| SM3310 | SM1058 [pCEN URA3 MFA1] | Transformant of SM1058 with pSM233 |
Transformation of yeast was performed either by the lithium acetate method (Ito et al., 1983) or by the method of Elble (1992). In the latter case, 10 μl of 1 M DTT was added to each 500-μl transformation to increase the transformation efficiency. Yeast cultures were grown at 30°C in complete media (YEPD), SC drop-out media, or SD minimal media, which were prepared as described previously (Michaelis and Herskowitz, 1988).
Mutant Isolation and Screen
To find new genes required for the biogenesis of a-factor, we carried out the multistep screen schematized in Fig. 2 to identify sterile mutants in a mata1 − strain background. The parental strains SM3038 and SM3039 contain a FUS1-lacZ reporter gene to facilitate discrimination between mutants involved in the pheromone signal response versus mutants involved in the pheromone production. Two methods of mutagenesis (chemical and insertional) were used. For chemical mutagenesis, strains SM3038 and SM3039 were grown overnight at 30°C and 108 cells were harvested by centrifugation. Cells were washed twice with sterile water, resuspended in 0.1 M potassium phosphate buffer (pH 8.0), and mutagenized with 3% ethylmethanesulfonate (EMS) to 50–65% survival. After shaking for 1 h at room temperature, the cells were washed with 5% sodium thiosulfate, transferred to a new tube, and washed twice with the same solution. Cells were resuspended in 1 ml of YEPD, diluted to 103 cells/ml in YEPD, a 0.2- ml aliquot of cells was plated onto YEPD plates, and these were grown for 2–3 d at 30°C. The colonies were screened for mating defects by replica plate mating onto a lawn of the MATα tester strain SM1068 on SD plates containing 0.05% YEPD. We screened ∼40,000 colonies, of which 530 exhibited less efficient mating than the parental strain.
Figure 2.
Scheme of the screen for MAT a-specific sterile mutants. Details of this screen are described in the text.
To perform insertion mutagenesis, we used a yeast genomic library that had been mutagenized in Escherichia coli by random insertion with a mini-Tn3 element containing the lacZ fusion gene and the yeast-selectable marker LEU2 (Burns et al., 1994), which was kindly provided by M. Snyder (Yale University, New Haven, CT). The population of mutagenized yeast DNA inserts were released en masse from the vector by digestion with NotI and transformed into yeast strains SM3038 and SM3039 to generate a library of gene replacements. About 25,000 transformants selected on SC-Leu plates were pooled together, plated on YEPD at a density of ∼250 colonies per plate, and subjected to a mating test. We screened ∼30,000 colonies, of which 63 showed a down-mater phenotype.
To rescue plasmids from cells mutagenized by random mini-Tn3 lacZ insertion so that we could analyze chromosomal sequences flanking the insertion element, we essentially followed the method described by Burns et al. (1994). To integrate the YIp5 plasmid (Botstein et al., 1979) into the AmpR gene located within the mini-Tn3 lacZ insertion, we first selected for a ura3 − derivative using 5-fluoro-orotic acid (5-FOA) (Boeke et al., 1984), which selects for loss of the wild-type URA3 gene that is adjacent to the FUS1-lacZ construct in our parental background. This procedure was carried out for the 11 mutants we obtained that were complemented by the AXL1 gene. From DNA sequencing data and Southern blotting analysis, these 11 mutants could be classified into three new alleles; axl1-2 represents an insertion at codon 867, axl1-3 an insertion at 1167, and axl1-4 contains a complete deletion of the AXL1 gene together with flanking sequences, and presumably arose during an aberrant Tn3 insertion event in E. coli.
Plate Assay for FUS1-lacZ Induction
Patches of cells grown on YEPD plates were replica plated to X-gal indicator plates (Rose et al., 1990) with and without 0.1 μg/ml α-factor. After a 4-h incubation at 30°C, colonies were exposed to chloroform vapor for 10 min and were incubated at 37°C overnight. Colonies of cells that were normal for their mating response expressed lacZ upon α-factor treatment and turned blue on plates with, but not without, α-factor.
Cloning and Sequencing
Before cloning the STE24 gene, it was necessary to determine whether or not the ste24-1 mutation was recessive. Generating a diploid in which MAT a mating could be tested required several steps. The strain SM1061 (mata1 STE24) was first transformed with a plasmid carrying MATα and URA3, and this transformant was crossed with SM3083 (MAT a ste24-1). From the resultant diploid, Ura− cells that had lost the MATα plasmid were recovered on 5-FOA, yielding SM3088 (MAT a/mata1 STE24/ste24-1), which is heterozygous for STE24. SM3088 could be tested for its ability to mate to MATα cells, and was found to mate equally as well as the homozygous strain SM3089 (MAT a/mata1 STE24/STE24), indicating that the ste24-1 mutation is recessive.
The STE24 gene was cloned by complementation of the mating defect associated with the ste24-1 allele. Strain SM3083 (MAT a ste24-1) was transformed with a yeast genomic library constructed in the CEN LEU2 vector p366, which was provided by P. Hieter (Johns Hopkins University, Baltimore, MD). Approximately 42,000 transformants were tested for the mating activity on SD plates containing 0.05% YEPD (see mating assay below). Five transformants exhibited a higher efficiency of mating than the original ste24-1 mutant.
Two of the five plasmids, pSM1054 and pSM1058, conferred a wildtype level of mating to the ste24-1 mutant and contained a common ∼10kb fragment. We subcloned three BamHI fragments (1.7, 2.5, or 5.9 kb in size) from pSM1054 into pRS304 (TRP1; Sikorski and Hieter, 1989) to generate pSM1089, pSM1062, or pSM1061, respectively, and sequenced one end of each fragment. Comparison of these sequences and yeast genome database revealed that this ∼10-kb DNA fragment is located on the right arm of chromosome X. Several DNA fragments (see Plasmid Constructions) were subcloned and tested for complementation activity of ste24-1. The subcloning studies revealed that open reading frame (ORF) YJR117w encodes the STE24 gene. We resequenced a 1.7-kb DNA region of pSM1064 (see below) containing the entire STE24 gene to confirm its sequence.
The remaining three clones obtained from the library conferred lower than the wild-type level of mating activity to the ste24-1 mutant and were shown to contain an overlapping (∼6 kb) DNA fragment. A 4.3-kb BamHI-BamHI fragment from one of these plasmids, pSM1057, was subcloned into the BamHI site of the plasmid pRS304 to generate pSM1088. Sequence analysis of one end of this 4.3-kb fragment revealed that the library insert contains the STE4 gene. We constructed pSM1090, which contains solely the STE4 gene, by subcloning of the 2.3-kb BamHI-MluI fragment of pSM1088 into the plasmid pRS315 (CEN ARS LEU2; Sikorski and Hieter, 1989). We also constructed pSM1091, which contains a truncated STE4 gene plus the remaining region of the overlapping fragment among the three clones, by subcloning the 5.8-kb BglII-BglII fragment of pSM1057 into pRS315. These plasmids were tested for the complementation of the mating defect of ste24-1. Plasmid pSM1090 conferred mating activity to the ste24-1 mutant while pSM1091 did not. Thus, we concluded that the complementing activity of these three clones is associated with the STE4 gene. STE4 encodes the β subunit of the heterotrimeric G protein involved in the pheromone signaling pathway. Overexpression of the STE4 gene is known to cause wild-type cells to undergo G1 cell cycle arrest (Cole et al., 1990; Nomoto et al., 1990; Whiteway et al., 1990). Thus, even when present on a low copy–number vector, a single extra copy of the STE4 gene activates the signal transduction pathway, compensating in part for the mating defect of ste24-1, presumably by increasing the level of a-factor production.
Plasmid Constructions
The integration plasmids pSM1061 and pSM1062 were constructed by subcloning the 5.9- and 2.5-kb BamHI-BamHI fragments from pSM1054 into the BamHI site of the integration plasmid pRS304, respectively. Both plasmids were digested with BglII, located in the middle of the cloned genomic fragment, and transformed into the wild-type strain SM1059.
The plasmids used in the complementation tests to identify the STE24 ORF (see Fig. 7) were constructed as follows: pSM1054 was digested with SphI and religated to produce pSM1063. pSM1065 and pSM1066 were constructed by subcloning the 4.3-kb EcoRV-EcoRV fragment and the 8.5-kb ScaI-EcoRV fragment from pSM1054 into the SmaI site of the plasmid pRS315, respectively. Plasmid pSM1067 was constructed by cloning the 7.6-kb XhoI-EcoRV fragment from pSM1054 between the XhoI-SmaI sites of pRS315. pSM1067 was digested with XhoI and HpaI, and the resulting 2.7-kb fragment was subcloned between the XhoI-SmaI sites of pRS315 to produce pSM1069. pSM1070 was constructed by subcloning the 3.4-kb HpaI-SacI fragment from pSM1067 between the SmaI-SacI sites of pRS315. The 5.9-kb BamHI-BamHI fragment from pSM1054 was cloned into the pRS315 to generate pSM1064. pSM1071 was constructed by cloning the 2.9-kb XbaI-XbaI fragment containing the 2.9-kb XbaI-BamHI fragment of the genome into the XbaI site of pRS315. pSM1064 was digested with BglII and religated to generate pSM1068.
Figure 7.

Complementation analysis to identify the STE24 gene and map of the disruption construct used to generate the ste24Δ::LEU2 null allele. A map of the cloned DNA that complements ste24-1 is shown. Boxes containing arrows indicate the ORFs; the database designations are shown above their respective boxes. Shaded boxes below the map show subclones tested for complementation. The ability of DNA fragments to complement the mating- defective phenotype in strain SM3060 (ste24-1) is indicated at the right. Shown above the map is the γ-disruption vector used to construct the chromosomal ste24Δ::LEU2 allele. Restriction sites are as follows: Bg, BglII; HI, BamHI; Kp, KpnI; P, PstI; RV, EcoRV; Sa, SacI; Sc, ScaI; Sp, SphI; Xa, XbaI; Xh, XhoI.
The plasmid pSM1072 was used for γ gene disruption (Sikorski and Hieter, 1989) of STE24, and it generates a chromosomal null allele in which nearly the entire coding sequence of STE24 (codons 1–444) is deleted. pSM1072 was constructed as follows: The 1.5-kb SphI-PstI fragment and the 2.6-kb SphI-XbaI fragment were prepared from pSM1064 and were ligated into the integration plasmid pRS305 (LEU2; Sikorski and Hieter, 1989) linearized with XbaI and PstI. pSM1072 was linearized with SphI and transformed into the diploid wild-type strain SM1060 (see Fig. 7).
Epitope Tagging and Oligonucleotide-directed Mutagenesis of STE24
We constructed a derivative of STE24 that contains a triply iterated HA epitope tag at the NH2 terminus of STE24. To do this, a new NotI site was first created immediately after the initiating methionine. To disrupt the NotI site of the polylinker, the plasmid pSM1069 (STE24 CEN LEU2) was digested with NotI, treated with Klenow, and religated. The resulting plasmid was designated pSM1092. An ∼350-bp portion of the STE24 5′ sequence was amplified by PCR using the primers oSM219 and oSM218. oSM219 is 5′-AAACTGCAGGCCTTTATTCatgAGCGGCCGCTTTGATCTTAAGACG-3′ (PstI and NotI sites are in italics and bold, respectively; the STE24 start codon is in lowercase letters), and oSM218, which is complementary to the sequence 13 bp downstream of the internal NdeI site of the STE24, is 5′-CATAAACTCTGTGCGACAG-3′. The amplified DNA fragment was digested with PstI and NdeI, and was cloned between the PstI-NdeI sites of pSM1092, yielding pSM1095. pSM1095 was linearized at the newly introduced NotI site and ligated with a 111-bp fragment from pSM491 encoding three copies of the HA epitope (Berkower et al., 1994) to generate pSM1097 (STE24::HA CEN LEU2), which contains the HA epitope in the sense orientation. pSM1098 (STE24::HAreverse CEN LEU2), which contains the HA epitope in the opposite orientation, was also derived from pSM1095. The orientations of the HA epitope in pSM1097 and pSM1098 were confirmed by restriction enzyme digestion and sequencing. We also constructed pSM1107 (STE24::HA CEN URA3) by subcloning the entire DNA insert of pSM1097 containing STE24::HA into pRS316 (CEN ARS URA3; Sikorski and Hieter, 1989).
Oligonucleotide-directed PCR mutagenesis of the HEXXH motif in STE24 was carried out essentially by the method described by Higuchi et al. (1988). First, two DNA fragments were amplified by PCR. One is a 192-bp fragment just upstream of the mutation site in STE24 and was obtained using oligonucleotides oSM259 and oSM260 as the NH2- and COOH-terminal primers. The other is an ∼220-bp fragment containing the mutated sequence as the NH2-terminal primer (oSM261, oSM262, and oSM263 for the H297A, E298A, and the E298D substitutions, respectively) and oSM225 as the COOH-terminal primer. These two PCR products were purified from an agarose gel and used as templates for the second PCR reaction, in which oligonucleotides oSM259 and oSM225 were used to generate a 393-bp DNA fragment. This second PCR product was digested with XbaI and EcoRI and used to replace the XbaI-EcoRI region of the wild-type STE24::HA. The mutations were confirmed by nucleotide sequencing. The sequence of the primers used for these mutageneses are as follows: oSM259, 5′-ATCTATTGAAAGTTTGGC-3′, oSM260, 5′-GCCAAAACAGCCGTAATTTC-3′, oSM261, 5′-ATTACGGCTGTTTTGGCAGCTGAAATCGG-3′, oSM262, 5′-TTACGGCTGTTTTGGCCCATGCGATCGGTC-3′, oSM263, 5′-TTACGGCTGTTTTGGCCCATGATATCGGTC-3′, and oSM225, 5′-ATAAATCCAATGATAATG-3′. The CEN URA plasmids containing ste24-H297A::HA, ste24-E298A::HA, and ste24-E298D::HA were designated pSM1103, pSM1104, and pSM1105, respectively.
Metabolic Labeling and Immunoprecipitation of a-Factor
Metabolic labeling and immunoprecipitation of a-factor were essentially carried out as described previously (Chen et al., 1997). To label intracellular a-factor, cells were grown in SD medium with appropriate supplements to OD600 0.7, harvested, and for each separate labeling, 5 OD600 U were resuspended in 0.5 ml of growth medium in a polypropylene tube. Labeling was carried out for 5 min with 150 μCi of [35S]cysteine. The chase was initiated by the addition of 10 μl of 1 M nonradioactive cysteine, and was terminated by the addition of an equal volume of cold azide stop mix (40 mM methionine, 40 mM cysteine, 20 mM NaN3, 500 μg/ml BSA). Intracellular (I) and extracellular (E) fractions were prepared and subjected to immunoprecipitation with a-factor antiserum 9-137, as described previously (Chen et al., 1997). Immunoprecipitates were electrophoresed in a 16% SDS-PAGE gel, followed by fluorography or phosphoimager analysis.
Assay for the Methylation of a-Factor
Cells were grown and harvested as described above. 5 OD600 U resuspended in 0.25 ml of growth medium were double labeled with 150 μCi of [35S]cysteine and 50 μCi S-adenosyl-l-[3H-methyl]methionine (15 Ci/mmol; Amersham, Arlington Heights, IL) for 6 min and terminated by addition of 0.25 ml ice-cold azide stop mix. Cells were pelleted by centrifugation, washed once with H2O, resuspended in 0.5 ml of breaking buffer (50 mM potassium phosphate buffer, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 1 mM β-mercaptoethanol, 1 mM PMSF) and lysed using glass beads at 4°C. The cell lysate was separated from the beads by centrifugation and transferred to a new tube. The beads were washed twice with 0.5 ml breaking buffer, and the two washes were pooled together with the original cell lysate. The a-factor species present in the combined lysate were immunoprecipitated with a-factor antiserum 9-137 and the immunoprecipitate was recovered from protein A–Sepharose beads by boiling in 15 μl of 2× SDS sample buffer. Recovered immunoprecipitates were subjected to SDS-PAGE. After fixation by 20% TCA for 7 min, gels were dried under a vacuum at 80°C and subjected to autoradiography.
Radiolabeled methyl esters were assayed by the method described by Xie et al. (1990) and Hrycyna et al. (1991). The region of the dried gel containing the P1 species was excised and mixed with 150 μl 1 M NaOH in a polypropylene microcentrifuge tube. The tube was placed in a 20-ml scintillation vial containing 5 ml scintillation fluid (Bio-Safe II; Research Products International Corp., Mt. Prospect, IL) and capped, allowing the volatile [3H]methanol released by base cleavage of methyl ester to diffuse into the scintillation fluid. After 24 h at 37°C, the microfuge tube was removed, and the radioactivity that had been transferred by diffusion to the scintillation fluid as [3H]methanol was assayed in a liquid scintillation counter. Total 35S radioactivity incorporation was assayed by adding 1 ml SOLVABLE (NEN Research Products, Boston, MA), a tissue solubilizer, to each of the microcentrifuge tubes containing the gel slices and allowing them to incubate for 6 h at 65°C. The contents of the tube were mixed with 10 ml of scintillation fluid (Bio-Safe II) and 100 μl of glacial acetic acid, and were subsequently counted in a liquid scintillation counter. The extent of a-factor methylation in each strain was assessed by calculating the 3H cpm/35S cpm ratio of the P1 gel band. This value was set at 100% for the wild-type strain.
Immunoblotting Analysis
2 OD600 U of cells growing logarithmically in SD dropout medium were harvested, washed once with sterile water, and resuspended in 1 ml of sterile water. Cells were lysed in 0.24 M NaOH/1% β-mercaptoethanol, and proteins were precipitated with 6% TCA on ice and resuspended in sample buffer (3.5% SDS, 14% glycerol, 80 mM Tris base, 80 mM Tris pH 8, 8 mM EDTA, 120 mM DTT, 0.01% bromophenol blue). After incubation at 65°C for 5 min, the samples were subjected to SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was incubated in blocking reagent (Boehringer Mannheim, Indianapolis, IN) diluted 10% in TBST (20 mM Tris-Cl, pH 7.6, 137 mM NaCl, and 0.1% Tween 20) at room temperature for 30 min, and then incubated with the anti-HA mAb 12CA5 (Babco, Richmond, CA) diluted 1:10,000 in TBST containing 5% blocking reagent (Boehringer Mannheim) for 2 h at room temperature. After three washes with TBST for 5 min each, the membrane was incubated with anti–mouse peroxide-linked secondary antibody (Amersham) diluted 1:10,000 in TBST containing 5% blocking reagent. After three final washes with TBST, the membrane was developed using the chemiluminescence detection kit (Boehringer Mannheim).
Mating and Halo Assay
Quantitative mating assays were performed by the plate mating procedure, essentially as described previously. (Michaelis and Herskowitz, 1988). Mutants to be tested were grown to saturation in YEPD medium and serially diluted in 10-fold increments into YEPD. A 0.1-ml aliquot of each dilution was spread on an SD plate together with ∼107 cells of the MATα mating tester SM1068 in 0.1 ml YEPD. Plating the cells in a small amount of YEPD provides essential nutrients that allow haploids to form microcolonies that can contact one another, thereby increasing their opportunity to mate. All mutant MAT a strains tested are Trp−, and the MATα mating tester is Lys−. Diploids resulting from mating are prototrophic and, hence, can form colonies on SD. After 3 d, diploids were counted and normalized to the total number of MAT a cells plated. The efficiency of diploid formation for each mutant is expressed as a percentage of that of the wild-type strain (100%).
Patch halo and mating assays were carried out essentially as described previously (Michaelis and Herskowitz, 1988). MAT a strains to be tested were spread in patches on YEPD or SD dropout plates, and were incubated at 30°C for 1–2 d. For the halo assay, these master patches were replica plated onto YEPD plates spread with the MATα halo tester SM1086 and incubated for 24 h. For the patch mating assay, the master patches were replica plated onto an SC minimal plate containing a small amount of YEPD, which had been spread with a lawn of the MATα mating tester SM1068 and incubated at 30°C for 2 d. The “stringency” of patch matings could be adjusted by varying the amount of YEPD (0.05%, stringent; 1%, permissive). To test mating of the MAT a strain, SM1067 was used as the MAT a mating tester.
Results
A Screen for MATa-specific Sterile Mutants Defective in a-Factor Biogenesis
While certain enzymes involved in a-factor biogenesis are known, there are clearly missing components for which the corresponding genes have not yet been identified. To identify additional genes required for a-factor maturation, we performed a multistep screen to isolate mutants that showed a MAT a-specific reduction in mating efficiency (Fig. 2). In addition to mutants specifically defective in a-factor production, we expected that mutants defective in pheromone-mediated signal transduction would represent a large proportion of our initial isolates. To eliminate this class of mutants, our parental strain contained the pheromone-inducible reporter construct (FUS1-lacZ; Rhodes, et al., 1990), which provided an assay for assessing whether signal transduction was normal or reduced. An additional feature of our parental strain was a mata1 mutation; a mata1 mutant mates like a MAT a wild-type strain, but has the advantage that it can be converted to a strain with MATα properties by transformation with a MATα plasmid. This feature enabled us to easily test our MAT a mating-defective mutants for their MATα mating properties.
Two different methods were used for mutagenesis: chemical mutagenesis by EMS, and insertional mutagenesis by the integration of a random Tn3-lacZ insertion element into the yeast genome, as described by Burns et al. (1994). An important feature of our screen is that we sought not only strong, but also weak down-maters, based on the hypothesis that mutants exhibiting a significant amount of residual mating (and thus scoring as only weak down-maters) may have been overlooked in previous screens. For this purpose, we performed mating tests under stringent conditions, rather than the permissive conditions ordinarily used in mating tests (Sprague, 1991). As shown in Fig. 3 A, we can distinguish a mating defect for an axl1 mutant under stringent conditions, but not while using standard permissive conditions.
Figure 3.
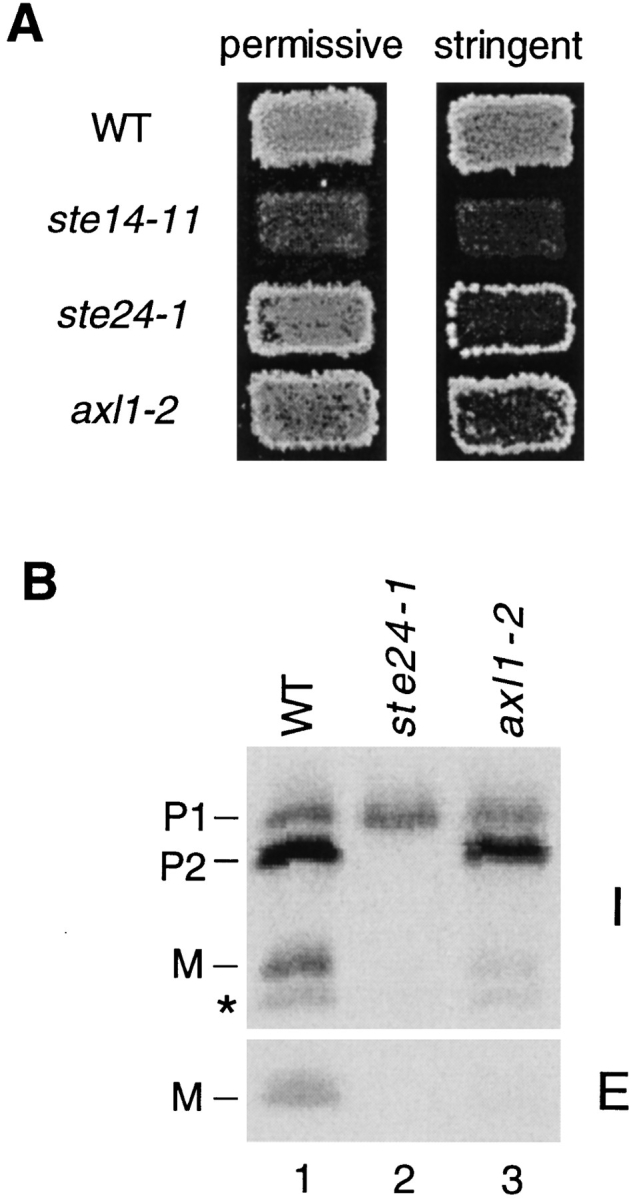
Mating and a-factor biogenesis phenotypes of the axl1-2 and ste24-1 mutants. (A) Patch mating assay. Patches of the indicated MAT a strains were replica-plated onto a lawn of the MATα mating tester stain SM1068 on an SD plate containing 1% (permissive) or 0.05% (stringent) YEPD. Plates were incubated at 30°C for 2 d. Strains tested are wild-type (SM3039), ste14-11 (SM3064), ste24-1 (SM3044), and axl1-2 (SM3061); these three mutants were obtained in this study (see Materials and Methods). (B) Immunoprecipitation of a-factor. To examine their a-factor biogenesis profiles, wild-type and mutant cells were metabolically labeled for 5 min with [35S]cysteine and the label was chased for 15 min. Intracellular and extracellular fractions were separated, subjected to immunoprecipitation with a-factor antiserum, and the fate of a-factor was analyzed by SDS-PAGE, followed by autoradiography, as described in Materials and Methods. The intracellular precursor species (P1 and P2) and mature a-factor are indicated. The band below the mature M species marked with an asterisk (*) is an a-factor–related peptide (AFRP) that does not appear on all gels (Chen, 1993); AFRP will be described elsewhere (Chen, P., and S. Michaelis, manuscript in preparation). Strains shown are wild type (SM3104), ste24-1 (SM3105), and axl1-2 (SM3106).
We screened a total of 65,000 mutagenized colonies obtained from either the EMS (40,000) or random insertion (25,000) mutagenesis procedures, and we identified 593 down-maters by replica plate mating tests. The down-maters were tested for FUS1-lacZ induction, yielding mutants (247) that showed normal expression of lacZ upon α-factor induction, ruling out the possibility that their reduced mating was caused by a defect in their mating response pathway. A subset of these (124) were transformed with plasmids harboring the MATα gene to test their mating activity, as MATα cells, and the majority (73) were found to mate normally with the MAT a tester strain SM1067. Of those, mutants exhibiting a reduced a-factor halo (44) were selected as biogenesis-defective candidates and were transformed with plasmids carrying genes previously known to be involved in a-factor processing. Of the 44 mutants tested, 28 were complemented by known genes: 12 by STE6, 5 by STE14, and 11 by AXL1, but none by either RAM1 or RAM2.
The remaining 16 mutants showed relatively weak mating defects in comparison to the ste6 and ste14 mutants. Nine of the mutants, which exhibited the most obvious mating defect by patch mating under stringent conditions, plus one of the axl1 mutants, were directly examined for an a-factor processing defect. After metabolic labeling of cells followed by immunoprecipitation with a-factor antibodies, a-factor biosynthetic intermediates can be directly visualized by an SDS-PAGE (Chen et al., 1997). As shown in Fig. 3 B, three major intracellular species of a-factor are apparent in the wild-type strain (lane 1): the COOH-terminally modified full-length precursor (P1), the NH2-terminally cleaved precursor (P2), in which seven amino acids have been removed from the NH2-terminal extension, and mature a-factor (M). For a schematic diagram of these species, see Fig. 1. Mature a-factor is also present in the extracellular culture fluid. In contrast to the wild-type pattern (Fig. 3 B, lane 1), one of the axl1 mutants identified in our screen (Fig. 3 B, lane 3) exhibits a defect in P2→ M processing, so that less intracellular and extracellular mature a-factor is present in comparison to wild-type, as previously reported by Adames et al. (1995). Interestingly, one of the new mutants, designated ste24, which arose from the EMS mutagenesis, also showed a strong defect in a-factor processing (Fig. 3 B, lane 2). In addition to a lack of mature a-factor, the P2 precursor species is also absent. This strain showed a significant mating defect under stringent testing conditions (Fig. 3 A). Since none of the remaining eight mutants exhibited an a-factor biogenesis defect by this assay (data not shown), they were not pursued further.
To determine whether the defect in SM3044 was caused by a single mutation, this strain was transformed with a plasmid (pSM857) that carries the MATα gene and a LEU2 marker, crossed to a wild-type MAT a strain (SM3040), sporulated, and the resultant diploid was subjected to tetrad analysis. Progeny were grown in YEPD overnight to allow plasmid loss, screened for Leu− segregants, and these were tested for mating activity to a MATα mating tester. Of 15 tetrads examined, the mating defect segregated 2:2 in all cases, indicating that it is caused by a single mutation. We also demonstrated this mutation is recessive, as described in Materials and Methods. The mutant was designated ste24-1 and was further analyzed below.
The ste24 Mutant Is Defective in a-Factor Processing
To examine the processing of a-factor in a ste24 mutant over an extended period of time, we performed pulsechase experiments. The ste24-1 mutant (SM3044) and the wild-type strain (SM3039) were transformed with plasmids carrying the MFA1 gene to facilitate detection of a-factor. Cells were pulse labeled with [35S]cysteine for 5 min, label was chased for varying lengths of time, and intracellular (I) and extracellular (E) fractions were prepared and subjected to immunoprecipitation with a-factor antiserum. As shown in Fig. 4, in wild-type cells, a portion of labeled a-factor was already converted to mature by the time the pulse was complete (the 0 time point in Fig. 4, lane 1); a significant amount of mature a-factor appears in the extracellular fraction by the end of the 5 min-chase (Fig. 4, lane 2), and export is essentially complete by 15 min (Fig. 4, lanes 3 and 4), as we have previously observed in a wildtype strain (Chen et al., 1997). By contrast, in a ste24 mutant, neither the P2 precursor nor the mature form of a-factor can be detected, even after 30 min (Fig. 4, lanes 5–8). Instead, only a single species that comigrates with the fully COOH-terminally modified precursor P1 is evident, suggesting that the ste24 mutant is defective in the P1→ P2 processing step within the NH2-terminal extension of a-factor.
Figure 4.

Processing of the a-factor precursor in wild-type and ste24 mutant cells. Cells were pulse labeled with [35S]cysteine for 5 min and chased for the indicated times. Intracellular and extracellular fractions were separated and subjected to immunoprecipitation, electrophoresis, and autoradiography. Positions of the P1, P2, and M forms of a-factor are indicated. Strains are wild type (SM3104) and ste24-1 (SM3105).
The ste24 mutant Is Defective in the First Cleavage within the NH2-terminal Extension of a-Factor
If the a-factor maturation defect in the ste24 mutant is in the P1→ P2 step of NH2-terminal processing, then the COOHterminal modification of a-factor should be unaffected in the ste24 mutant (see Fig. 1). Because methylation is obligatorily the last step in the COOH-terminal modification of CAAX proteins, methylation can be used as a marker to signify the completion of the preceding steps of prenylation and AAX proteolysis (Hrycyna and Clarke, 1992; Ashby and Rine, 1995; Hrycyna et al., 1995). Accordingly, to more precisely determine which step of a-factor biogenesis is defective in the ste24 mutant, we asked whether the a-factor intermediate that accumulates in theste24 mutant strain is methylated.
We examined the methylation status of a-factor biosynthetic intermediates in a wild-type strain, two ste24 mutants (ste24-1 and ste24Δ::LEU2, see below), and as a negative control in a ste14 mutant defective for the carboxyl methyltransferase enzyme itself. Cells were double labeled with the methyl group donor [3H-methyl]-S-adenosylmethionine and the protein label [35S]cysteine, extracts were prepared, and immunoprecipitation with a-factor antiserum was carried out. The immunoprecipitates were subjected to SDS-PAGE and autoradiography. Subsequently, the P1 species of the wild-type and ste14 strains and the corresponding species that accumulate in the ste24 strains were excised from the gel. These species were assayed for total 35S-labeled protein by direct scintillation counting and for the presence of 3H-labeled methyl ester groups by the vapor phase equilibrium assay (Xie et al., 1990). This assay specifically detects ester-linked methyl groups, which are liberated as [3H]methanol after base hydrolysis (see Materials and Methods). The level of 3H counts relative to 35S counts indicates the extent of methylation of the species of a-factor being analyzed. As shown in Fig. 5, the level of a-factor methylation in two different ste24 mutants is equivalent to (or possibly even slightly higher than) the wild-type strain. This is in contrast to the ste14 mutant, which is completely deficient in methylation. These results indicated that in an ste24 mutant, the COOH-terminal modification of a-factor has been completed. Thus, we concluded that the a-factor biosynthetic intermediate that accumulates in the ste24 mutant is indeed P1, which has undergone COOH-terminal modification, and that the ste24-1 mutant is deficient in carrying out the first NH2terminal cleavage step of a-factor biogenesis.
Figure 5.
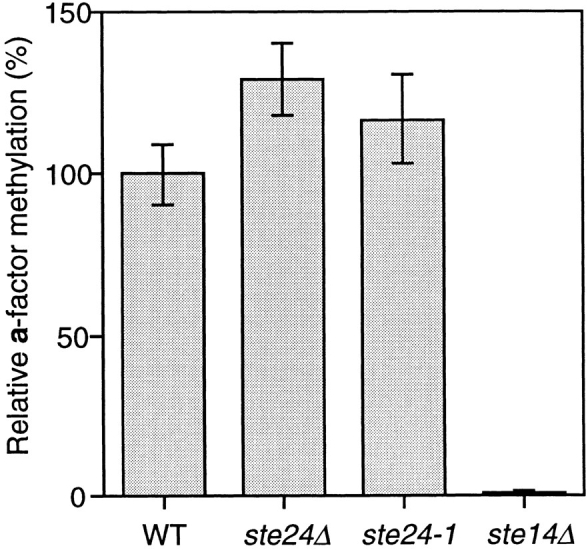
Methylation of a-factor in wild-type and ste24 mutant cells. Cells were double-labeled with [35S]cysteine and S-adenosyl-l-[3H-methyl]methionine. Total cell extracts were prepared, immunoprecipitated with a-factor antiserum, and subjected to SDS-PAGE. The P1 species was excised from the gel. P1 was assayed for base-volatile [3H]methanol counts, reflecting its level of methylesterification and for [35S]cysteine incorporation to measure the total amount of labeled P1 that is present, as described in the Materials and Methods. The extent of P1 methylation in each strain was assessed by calculating the 3H cpm/35S cpm ratio. The level of P1 methylation in the mutant strains relative to wild type (100%) is indicated. Mean and standard error for three or four experiments is shown. Strains are wild type (SM3310), ste24-1 (SM3309), ste24Δ (SM3286), and ste14Δ (SM1871).
A ste24 Mutant and an a-Factor P1→ P2 Cleavage Site Mutant Exhibit the Same Defect in a-Factor Processing
Our laboratory has generated a large collection of a-factor mutants, several of which have alterations in the NH2-terminal extension of MFA1 in residues adjacent to the P1→ P2 cleavage site (Nouvet, F., A. Kistler, and S. Michaelis, manuscript in preparation). We compared the a-factor processing of one such a-factor mutant, mfa1- A8G (Fig. 6 A), which is expressed in a wild-type (STE24) strain, to that of wild-type MFA1 expressed in a ste24Δ mutant, by a pulse-chase immunoprecipitation experiment. As can be seen in Fig. 6 B, the mfa1-A8G mutant, although slightly “leaky,” showed essentially the same deficiency in P1→ P2 conversion and exhibited the same stable accumulation of P1 as did the ste24 mutant. These results indicated that the mfa1-A8G a-factor “substrate” mutant and the ste24 “machinery” mutant are impaired at the same processing step.
Figure 6.

Comparison of a-factor processing in ste24 mutant cells and in cells bearing an mfa1 mutant plasmid defective in P1→ P2 cleavage. (A) Position of the mfa1-A8G mutation in a-factor. The mfa1-A8G mutation and the site of P1→ P2 cleavage are indicated. (B) Processing of a-factor in ste24 mutant cells and in a strain expressing the a-factor mutant mfa1-A8G. Cells were pulse labeled with [35S]cysteine, and the label was chased for the indicated times. Intracellular and extracellular fractions were separated, subjected to immunoprecipitation, electrophoresis, and phosphoimager analysis. Positions of the P1, P2, and M forms of a-factor are indicated. Strains are wild type (SM3310), ste24Δ (SM3286), and mfa1A8G (SM2059).
Cloning the STE24 Gene
To clone STE24, the ste24-1 mutant (SM3083) was transformed with a CEN-based yeast genomic library, and transformants were tested for their mating activity. Of 42,000 transformants screened, five showed complementation of the ste24 mating defect. Three of these complemented only partially and were shown to harbor the STE4 gene (see Materials and Methods). The remaining two transformants fully complemented the mating defect of the ste24-1 mutant. The plasmids that were recovered from these strains contained a common ∼10-kb fragment. We confirmed that one of them (pSM1054) could restore the a-factor processing defect of ste24-1 by a-factor immunoprecipitation experiments (data not shown). To determine whether this insert contained the STE24 structural gene, we integrated TRP1-marked integrating plasmids bearing a portion of the cloned DNA (a 5.9-kb BamHI fragment [pSM1061] or a 2.5-kb BamHI fragment [pSM1062]) into the genome of a wild-type strain (SM1059). The resultant strain was crossed with a ste24-1 trp1 strain (SM3060) and the diploid was sporulated. Tetrad analysis revealed that TRP1 + and ste24-1 showed complete linkage in 24 tetrads that were tested. Therefore, we concluded that the cloned gene is allelic to the ste24-1 locus.
We sequenced ∼150 bp from three different regions of the ∼10-kb library insert carrying STE24 (see Fig. 7, arrow) and searched for these DNA sequences in the S. cerevisiae genome database. The database search revealed that the 10-kb segment of DNA encoding STE24 is located on chromosome X and contains eight ORFs. Subcloning studies (see Fig. 7) indicated that a restriction fragment containing ORF YJR117w complemented the ste24-1 mutant. We have therefore designated ORF YJR117w as STE24.
The STE24 Gene Product Contains Multiple Predicted Membrane Spans, a Metalloprotease Motif, and an ER Retrieval Signal; Ste24p Defines a Novel Family of Putative Membrane Metalloproteases
The STE24 gene has the potential to encode a 453–amino acid protein whose predicted molecular mass is 52.3 kD (Fig. 8, A and B). Hydropathy analysis of Ste24p predicts that it contains multiple membrane spanning segments (possibly six), suggesting that Ste24p is an integral membrane protein (Fig. 8, A and B; see also Fig. 9 C). Interestingly, we found that the Ste24p sequence contains two additional distinctive sequences: a metalloprotease consensus motif, HEXXH (Jongeneel et al., 1989; residues 297–301), and a COOH-terminal ER retrieval signal, KKXX (Jackson et al., 1990, 1993; residues 450–453).
Figure 8.

Ste24p predicted sequence and structure. (A) The amino acid sequence of Ste24p. Putative transmembrane segments predicted by hydropathy analysis (Kyte and Doolittle, 1982) are above the line. The zinc metalloprotease and di-lysine consensus motifs are shown in black and gray boxes, respectively. These sequence data are available from GenBank/EMBL/DDBJ under accession No. U77137. (B) Schematic structure of Ste24p. The hatched boxes show predicted transmembrane domains. The relative position of the zinc metalloprotease (HEXXH) and dilysine (KKXX) motifs are indicated. Circles indicate potential sites of N-glycosylation, based on the presence of an NXS/T consensus motif. Because the presence of a di-lysine retrieval signal suggests that the COOH terminus of Ste24p is cytosolically disposed, the sites rendered in light gray are predicted to be cytosolically oriented and thus are unlikely to be glycosylated.
Figure 9.
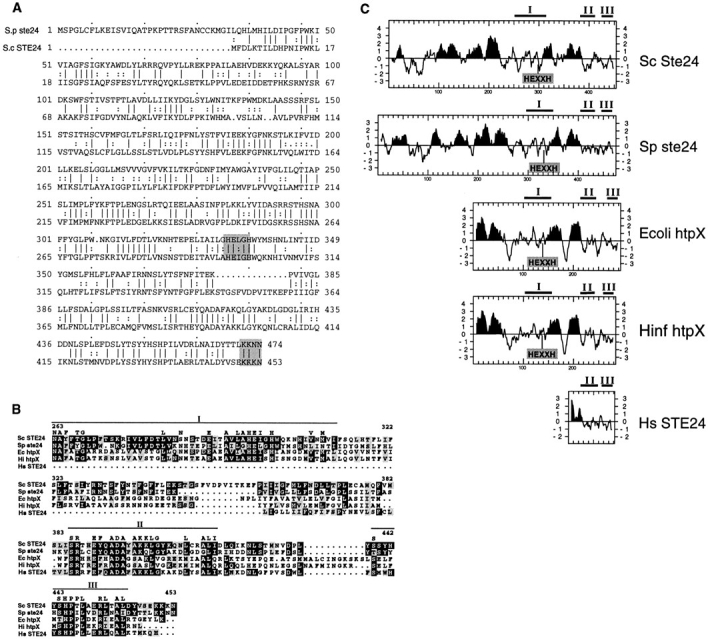
Comparison of Ste24p with related proteins in other organisms. (A) The amino acid sequence of S. cerevisiae Ste24p (Sc Ste24) is compared with the S. pombe homologue (YAN5, protein accession number Q10071; here designated Sp ste24) using the Bestfit program (GCG software package). Identity is indicated by a line between the sequences and conserved changes are indicated by two dots (two corresponding bases in a codon). Gaps are shown as dots within the sequence. The zinc metalloprotease motif (HEXXH) and ER retrieval signal (KKXX) are shown in gray boxes. The Sc Ste24p and Sp ste24 proteins are 41.3% identical at the amino acid level. (B) Alignment of the COOH-terminal region of Ste24p and related sequences from other species. The COOH-terminal region of Ste24p homologues from five organisms are aligned by the Blosum-62 amino acid similarity matrix. Bars mark highly conserved subdomains (I, II, and III). Black boxes denote amino acid identity with S. cerevisiae Ste24p, and gray boxes denote amino acid similarity with Ste24p. Residues that are identical between more than two proteins in these subdomains are indicated above. The amino acid numbers of Sc Ste24p are shown. The accession numbers of E. coli htpX protein and H. influenzae htpX homologue protein are P23894 and P44840, respectively. The partial H. sapiens homologue (Hs STE24) sequence was assembled from the expressed sequence tag database sequences (Z43273, T35312, R54272, N76181, F11310, and T12172). (C) Comparison of hydropathy plots of Ste24p and related proteins. The algorithm of Kyte and Doolittle (1982) was used to generate hydropathy profiles for Ste24p homologues, with a window of 11 amino acids. Potential transmembrane regions are in black. The highly conserved subdomains shown in B are indicated above the boxes. The position of the zinc metalloprotease motif (HEXXH) is indicated.
A homology search of the GenBank/EMBL/DDBJ data base using the BLAST algorithm (Altschul et al., 1990) identified the uncharacterized Schizosaccharomyces pombe protein YAN5 (accession No. Q10071) as a close homologue of Ste24p. The Ste24p and YAN5 proteins share significant homology throughout their entire length (41% identity and 63% similarity). Like Ste24p, YAN5 (here designated Sp ste24) contains a metalloprotease motif, a COOH-terminal ER retrieval signal, and multiple membrane spans (Fig. 9, A and C).
Our BLAST search also revealed other proteins related to Ste24p. One is the E. coli 32-kD transmembrane protein HtpX (Kornitzer et al., 1991), whose expression is induced by temperature upshift. The overexpression of a truncated form of the HtpX protein in bacterial cells causes a higher than wild-type rate of degradation of abnormal proteins (Kornitzer et al., 1991). A Haemophilus influenzae htpX homologue, which was identified by DNA sequence analysis (Fleischman et al., 1995), also shows high degree of similarity to Ste24p. Finally, Ste24p is homologous to at least a segment of a human protein (designated here as Hs STE24) identified in the expressed sequence tag (EST) database (Bassett et al., 1995; Boguski et al., 1994).
Fig. 9 B shows an alignment of a portion of the COOHterminal S. cerevisiae Ste24p sequence with that of related proteins from other organisms. There are three highly conserved domains (designated I, II, and III) within the region shown. These regions are not present in other zinc metalloproteases, and thus distinguish Ste24p and its related proteins as a distinctive metalloprotease subfamily. Hydropathy plots suggest that each member of this gene family has multiple predicted transmembrane domains, although this cannot be determined for Hs STE24, since only a portion of its sequence is available (Fig. 9 C). Importantly, the HEXXH motif is in an analogous location relative to the last two potential membrane spans for each member of the Ste24p family.
STE24 Is a Nonessential Gene
The phenotype of an ste24 null mutation was examined by constructing a MAT a/MATα diploid strain heterozygous for a deletion that removes nearly the entire STE24 ORF (Fig. 7, ste24Δ::LEU2). Sporulation of this strain (SM3095) gave rise to four viable spores per tetrad, indicating that STE24 is not an essential gene. We tested mating activity and a-factor biogenesis in strains that contained the ste24Δ::LEU2 null allele. The mating efficiency of the ste24Δ null mutant (SM3103) was essentially the same as that of the ste24-1 mutant (SM3060); both exhibit a mating efficiency of ∼5% that of a wild-type strain, as assessed by quantitative mating. Pulse-chase immunoprecipitation experiments of a-factor demonstrated that the ste24Δ null mutant exhibited the same gel pattern as ste24-1, indicative of a P1→ P2 cleavage defect (see Fig. 6 B). Aside from an a-factor processing defect, we have not detected other obvious phenotypes for the ste24Δ mutant: either at 30°C or 37°C, the ste24Δ mutant grew at the same rate as the wild type, and the MATα ste24Δ mutant showed the same mating efficiency as the wild-type MATα strain.
Since STE24 is required for the biogenesis of a-factor, we examined whether STE24 is expressed in MAT a and MATα haploid cells and in MAT a/MATα diploids. To detect Ste24p, we have constructed an HA epitope–tagged derivative of STE24 in a CEN vector (pSM1107), as described in Materials and Methods. Total cell extracts prepared from MAT a haploid (SM1058), MATα haploid (SM1059), and MAT a/MATα diploid (SM1060) strains harboring the HA-tagged STE24 plasmid were analyzed by Western blotting with an anti-HA mAb (12CA5). Each strain showed an equivalent signal, indicating that STE24 is expressed in all cell types (data not shown).
The Zinc Metalloprotease Motif, HEXXH, Is Essential for Ste24p Activity
Mutational studies with zinc metalloproteases have identified a key sequence motif (HEXXH) that contains two invariant histidine residues and a glutamate. Substitution mutations that alter either of the histidine residues cause a loss of proteolytic activity, as well as a defect in metal binding (Medina et al., 1991). The glutamate residue is also important for proteolysis. To examine the importance of the zinc metalloprotease motif for Ste24p activity, we carried out PCR-mediated, site-directed mutagenesis to generate three HEXXH motif mutations in Ste24p (see Materials and Methods). These correspond to substitutions of the first histidine residue, ste24-H297A, and of the glutamate residue, ste24-E298A and ste24-E298D. All three mutants, even the conservative substitution E298D, lost the ability to complement the mating-defective phenotype of ste24-1 (Fig. 10 A). To confirm that the loss of function of these mutants is not caused by reduced expression or stability of Ste24p, we examined the steady-state level of the mutant proteins by Western blotting. As shown in Fig. 10 B, the mutant proteins were detected at the same level as wild-type Ste24-HAp. These results suggest that the function of Ste24p in a-factor processing depends on its zinc metalloprotease activity.
Figure 10.

Phenotype of ste24 HEXXH metalloprotease motif mutants. (A) Patch mating test of ste24 metalloprotease domain mutants. Patches of ste24-1 transformants harboring plasmids with the indicated HA epitope–tagged versions of wild-type and mutant STE24 were replica plate mated to a lawn of the MAT a mating tester strain SM1068 on an SD plate containing 0.05% YEPD. Strains are SM3069 (vector, pRS316), SM3071 (STE24::HA, pSM1107), SM3072 (ste24-H297A::HA, pSM1103), SM3073 (ste24E298A::HA, pSM1104), and SM3074 (ste24-E298D, pSM1105). (B) Steady-state levels of wild-type and mutant Ste24 proteins. Crude extracts prepared from strains bearing the indicated mutations were analyzed by immunoblotting with anti-HA 12CA5 antibodies. Extract from 0.5 OD600 U of cells was loaded in each lane. Lane 1, SM3082 (STE24 tagged in the reverse orientation, pSM1098); lane 2, SM3071 (STE24::HA, pSM1107); lane 3, SM3072 (ste24-H297A::HA, pSM1103); lane 4, SM3073 (ste24E298A::HA, pSM1104); and lane 5, SM3074 (ste24-E298D, pSM1105).
Discussion
STE24 Is Required for the First NH2-terminal Processing Step of a-Factor Biogenesis
In this study, we report the identification of a new gene, STE24, that is required for the biogenesis of a-factor in S. cerevisiae. The ste24-1 mutant was isolated in a screen for MAT a-specific sterile mutants, which identified both known (ste6, ste14, axl1) and new (ste24-1) mutants. During biogenesis, the a-factor precursor undergoes COOHterminal modification (including prenylation, proteolytic cleavage, and methylation) and NH2-terminal processing (involving two distinct proteolytic cleavage steps) to yield mature a-factor (Fig. 1; Chen et al., 1997). Our results here suggest that Ste24p is required for the first (P1→ P2) of the two NH2-terminal processing steps, since we observe the accumulation in the ste24-1 mutant strain of the a-factor biosynthetic intermediate P1, which has completed COOHterminal modification, but contains an intact NH2-terminal extension. Cloning and sequence analysis reveal that Ste24p contains multiple membrane spans, a di-lysine ER retrieval motif (discussed below), and importantly, a zinc metalloprotease motif, HEXXH. We demonstrate that the protease motif is essential for the function of Ste24p, since mutations that alter either of two highly conserved residues within this region (H297A, E298A, and E298D) result in the loss of complementation of the mating defect associated with ste24-1. These results support the view that Ste24p exerts its biological function as a membrane-associated protease, acting either directly or indirectly to carry out endoproteolytic cleavage between residues T7 and A8 of P1. A membrane location for this a-factor NH2-terminal protease is consistent with our previous observation that all COOH-terminally prenylated a-factor intermediates, including P1, are membrane associated (Chen, 1993; Chen et al., 1997).
Another protease involved in a-factor biogenesis, Axl1p, was recently identified in a screen for sterile mutants similar to ours that was carried out by Adames et al. (1995); at least three distinct alleles of AXL1 were also obtained in the present study (see Fig. 3 and Materials and Methods). In an axl1 mutant, the second (P2→ M) NH2-terminal proteolytic processing step in a-factor maturation is impaired. AXL1 encodes a putative zinc metalloprotease that, unlike Ste24p, belongs to the IDE subfamily of metalloproteases that has the HXXEH sequence instead of HEXXH (Fujita et al., 1994). Together, the two distinct proteases Ste24p and Axl1p can account for the two-step removal of the NH2-terminal extension of the a-factor precursor.
Interestingly, the Ste24p- and Axl1p-dependent proteolytic cleavages occur sequentially, in an obligatorily ordered fashion, as indicated by two separate findings. First, a ste24 mutation blocks not only the first (P1→ P2) NH2terminal cleavage of the a-factor precursor, but also the subsequent Axl1-dependent, NH2-terminal cleavage step. Second, when the STE24-dependent (P1→ P2) cleavage of a-factor is prevented by the mutation of a residue adjacent to the P1→ P2 processing site (mfa1-A8G), the AXL1- dependent cleavage is also blocked (Fig. 6; Nouvet, F., A. Kistler, and S. Michaelis, manuscript in preparation). It is likely that there are interesting cell biological or enzymological reasons to account for why the STE24-dependent processing of a-factor is indispensable for AXL1-dependent processing. For instance, it is possible that the two processing steps occur in distinct cellular locations and that the initial cleavage event is necessary to expose a targeting signal within a-factor. Alternatively, only the P2 form of a-factor may be recognized as a substrate by AXL1-dependent processing machinery. The in vitro reconstitution of a-factor NH2-terminal processing will be important for examining the interrelationship between these two processing steps and ultimately for establishing whether Ste24p and Axl1p act directly to cleave the a-factor NH2-terminal extension, or if they do so indirectly by activating another protease(s).
It is also notable that the COOH-terminal prenylation of a-factor appears to be obligatory for NH2-terminal processing, as evidenced by the finding that in ram1, ram2, or a-factor CAAX mutants, the NH2-terminal cleavage of a-factor cannot proceed (He et al., 1991; Chen et al., 1997). This may reflect the fact that prenylation is required for a-factor to become membrane localized, and thus to engage the STE24-dependent machinery in the membrane. Alternatively the COOH-terminal prenyl group could directly serve as a recognition determinant for the a-factor NH2-terminal processing enzyme(s). Unlike prenylation, carboxylmethylation is dispensable for the NH2-terminal processing of a-factor (Sapperstein et al., 1994).
Whereas Ste24p and Axl1p are both required for a-factor processing, these two gene products may also act independently to carry out additional but distinct cellular functions. Axl1p is expressed in haploid cells, but not in diploid cells (Fujita et al., 1994). In addition to its role in the NH2terminal processing of a-factor, Axl1p is also critical for axial budding in haploid yeast, although its protease domain appears to be dispensable for the latter function (Adames et al., 1995). Ste24p, on the other hand, does not play a role in budding, since both ste24-1 and ste24Δ haploids exhibit the normal axial budding pattern (data not shown). In addition, Ste24p is expressed in both haploid and diploid cell types, as assessed by Western analysis of HA-tagged Ste24p; thus, if it does have a cellular function in addition to a-factor processing, this function is not likely to be restricted to haploid cells. It should be noted that a recent report suggests that the product of AFC1, a gene that is identical to STE24, may play a role in the COOHterminal AAX processing of farnesylated proteins (Boyartchuk, V.L., M.N. Ashby, and J. Rine. 1996. Yeast Genetics and Molecular Biology Meeting Abstracts). While it may be that Ste24p could provide a minor contribution to AAX proteolysis of a-factor, the results we present here indicate that its major role for the biogenesis of the a-factor precursor is in NH2-terminal processing. Aside from the a-factor processing defect, we have not detected other obvious phenotypes for the ste24Δ mutant.
Ste24p Is Likely to be Associated with Intracellular Membranes
The deduced amino acid sequence of STE24 reveals that in addition to its HEXXH protease domain, Ste24p contains multiple predicted transmembrane spans and a COOH-terminal di-lysine motif, KKXX. Considerable evidence indicates that the di-lysine motif serves as an ER retrieval sequence for integral membrane proteins. First, KKXX is present at the COOH terminus of several mammalian and yeast ER membrane resident proteins and, when mutated, results in their aberrant localization (Jackson et al., 1993; Letourneur et al., 1994). In addition, the di-lysine motif from one of these proteins (Wbp1) was shown to be sufficient, when affixed to the COOH terminus of the plasma membrane protein Ste2p, to cause Ste2p to become ER localized (Letourneur et al., 1994). It is noteworthy, however, that a recently discovered yeast protein, Emp47p, which also terminates with a di-lysine motif, is Golgi localized, suggesting that additional signals within a protein can modulate or override the KKXX information (Schroder et al., 1995). Clearly, in the case of Ste24p, further analyses, including immunofluorescence and subcellular fractionation, will be required to establish its localization pattern within the cell. Preliminary results with the HA epitope–tagged Ste24p construct suggest it is an ER membrane protein (Fujimura-Kamada, K., and S. Michaelis, unpublished data). Defining the pattern of Ste24p trafficking and localization will further contribute to clarifying the general role of di-lysine motifs as targeting signals.
The biogenesis of a-factor appears to be intimately associated with membranes, since the prenylated a-factor biosynthetic intermediates P1, P2, and M are all membrane bound (Chen et al., 1997). It has not yet been established, however, which cellular membranes serve as sites for the a-factor modification and processing reactions. Since Ste24p, together with Axl1, are poised to function in the “middle” of the a-factor biogenesis pathway, acting after completion of COOH-terminal CAAX modification and before the Ste6p-mediated export step, knowing the location(s) of Ste24p and Axl1p would immediately help to restrict our views as to whether all or some of these steps occur within a multicomponent a-factor processing complex, or whether they are carried out separately, in distinct cellular locations.
Our model for the topology of Ste24p predicts that it contains six membrane spans and that the HEXXH metalloprotease motif and the KKXX motif both lie in cytosolic domains (between spans 4 and 5, and in the COOH-terminal tail, respectively). Our topology model is based on the knowledge that the KKXX sequence is recognized by the coatomer protein complex COPI, which resides in the cytoplasm (Cosson and Letourneur, 1994). Hence, to exert its role as a signal for COPI-mediated retrieval, the Ste24p di-lysine motif would need to be cytosolically oriented. Because two membrane spans are predicted to lie between the di-lysine and protease motifs, the HEXXH protease active site is also likely to be cytosolically disposed. Thus, it seems most likely that Ste24p exerts its proteolytic function for the NH2-terminal processing of a-factor at the cytosolic face of an organellar membrane, most likely the ER membrane.
A New Mutant Hunt for MATa-specific Sterile Mutants; a-Factor Biogenesis Components Have Been Previously Missed because of Functional Redundancy
Previous mutant hunts aimed at isolating MAT a-specific sterile mutants have identified a number of a-factor biogenesis components, including STE6, STE14, and RAM1 (MacKay and Manney, 1974; Blair, 1979; Rine, 1979; Wilson and Herskowitz, 1987; Sprague and Thorner, 1992). It is significant, however, that certain components have been elusive. In particular, until quite recently, there were no candidate genes for either of the NH2-terminal processing enzymes or for the COOH-terminal AAX protease. A common reason that a gene may be missed in a genetic screen is functional redundancy. For a pair of genes that can carry out the same process, mutating only one partner of the pair is likely to produce only a subtle phenotype that could be easily overlooked. For instance, the functionally redundant a-factor precursor structural genes MFA1 and MFA2 were not identified in screens for MAT asterile mutants, but instead, were obtained from a plasmid library based on the known amino acid sequence of a-factor (Michaelis and Herskowitz, 1988); indeed, deletion of either of these genes results in only a slight diminution of a-factor activity, whereas deletion of both results in a completely a-factorless strain that is sterile.
A main feature of our screen for new sterile mutants is that we paid particular attention to weak sterile mutants with a subtle phenotype, in contrast to previous screens in which only strong sterile mutants were sought. The ste24Δ mutant exhibits significant residual mating; and as expected, a low level of mature a-factor is revealed by overexposure of a-factor gels (data not shown). These observations imply that another gene product, yet to be identified, is able to carry out P1→ P2 processing of a-factor at low efficiency. Such a partial redundancy is observed between Axl1p and the Axl1p homologue, Ste23p; whereas the axl1Δ mutant exhibits low but residual mating, deletion of both genes (axl1Δste23Δ) leads to nearly complete sterility (Adames et al., 1995). Although a search of the yeast database does not reveal additional STE24 homologues, it is certainly possible that a protease(s) that is not homologous to Ste24p may provide a functionally redundant function capable of mediating NH2-terminal processing of the a-factor precursor.
Ste24p Defines a New and Ubiquitous Subfamily of Membrane-associated Metalloproteases
A search of the genome database reveals Ste24p homologues in diverse organisms, including bacteria, yeast, and mammals, which are indicative of a remarkable degree of evolutionary conservation (Fig. 9). The S. pombe homologue (Sp ste24) shares a high level of homology with S. cerevisiae Ste24p throughout its entirety (41% identity and 63% similarity). Significantly, Sp ste24 contains both the HEXXH protease motif and the COOHterminal di-lysine motif. S. pombe cells of the Minus mating type secrete a mating pheromone, M-factor, whose chemical modifications (including prenylation and methylation) and overall gene structure are similar to that of a-factor (reviewed by Nielsen and Davey, 1995). Thus, it is likely that the biogenesis of a-factor and M-factor will be mediated by similar cellular machinery. Based on the similarity between Sc Ste24p and Sp ste24, it is reasonable to predict that Sp ste24 is involved in the biogenesis of S. pombe M-factor.
In addition to the existence of an S. pombe homologue, Escherichia coli, H. influenzae, and Homo sapiens also encode proteins that are highly related to S. cerevisiae Ste24p (Fig. 9). Although only a small portion from the COOHterminal end of the human gene (Hs STE24) has been sequenced, it exhibits a high degree of similarity to Sc Ste24p (45% identity and 63% similarity) in the sequenced portion. It is also notable that Hs STE24 has a COOH-terminal sequence that resembles, but is distinct from, the known di-lysine motifs KKXX and KXKXX. Hs STE24 has two lysines, which are positioned three and six residues from the COOH terminus. A lysine at the −3 position has been shown to play a critical role in the ER retention of a protein with a canonical KKXX motif (Jackson et al., 1990). The remaining proteins related to Ste24p, for which the complete sequence is known, all contain multiple predicted transmembrane spans and an HEXXH motif that resides in an analogous position relative to the two COOH-terminal membrane spans.
In recent years, a number of zinc metalloproteases possessing a motif identical or related to HEXXH, have been identified. These have been classified into several subfamilies, based on sequence comparison (Jiang and Bond, 1992; Hooper, 1994). One of these families is the IDE subfamily, which contains the “inverted motif” HXXEH, and also includes Axl1p and Ste23p. Another newly appreciated subfamily of zinc metalloproteases is the AAA subfamily, whose members (e.g., Escherichia coli FtsH and S. cerevisiae Yme1p) contain an HEXXH motif and an ATP-binding domain, and are associated with membranes (Tomoyasu et al., 1995; Weber et al., 1996). By sequence comparison, the Ste24p-related proteins do not belong to this or any of the previously defined subfamilies, but instead represent a new subfamily, that is defined by highly conserved boxes (I, II, and III) at their COOH terminus (Fig. 9 B). Our mutagenesis studies of the metalloprotease motif provides indirect evidence that Ste24p functions as a protease. Moreover, for one member of the Ste24p subfamily, HtpX, it has been reported that the overexpression of a truncated form of the protein, which still contains the HEXXH motif, leads to an increase in the degradation of abnormal proteins (Kornitzer et al., 1991. Thus, whereas the Ste24prelated proteins have not yet been proven biochemically to be zinc metalloproteases, these observations suggest that they are indeed highly likely to function as proteases. Our discovery that Ste24p homologues are present in organisms as diverse as bacteria and mammals suggests that proteins of this subfamily exist ubiquitously. It will be of great interest to learn if, in addition to yeast a-factor, members of the Ste24p subfamily are involved in the posttranslational modification of other secreted signaling molecules.
Acknowledgments
We thank B. Errede for generously providing FUS1-lacZ plasmid, M. Snyder for the libraries for insertional mutagenesis, P. Hieter for providing genomic libraries, and N.L. Craig, D. Loayza, W.K. Schmidt, A. Tam, and S.K. Sapperstein for comments on this manuscript. K.F.-K. would like to thank Y. Kamada for helpful discussion.
This work was supported by a National Institutes of Health grant (GM41223) to S. Michaelis.
Abbreviations used in this paper
- E
extracellular
- EMS
ethylmethanesulfonate
- I
intracellular
- IDE
insulin-degrading enzyme
- M
mature species of a-factor
- ORF
open reading frame
- P1
fully COOH-terminally modified precursor species of a-factor
- P2
fully COOH-terminally modified, partially NH2-terminally cleaved precursor species of a-factor
- 3-FOA
5-fluoro-orotic acid
Footnotes
Please address all correspondence to S. Michaelis, Department of Cell Biology and Anatomy, The Johns Hopkins University School of Medicine, 725 North Wolfe Street, Baltimore, MD 21205. Tel.: (410) 955-8286. Fax: (410) 955-4129. E-mail: susan_michaelis@qmail.bs.jhu.edu
References
- Adames N, Blundell K, Ashby MN, Boone C. Role of yeast insulin-degrading enzyme homologues in propheromone processing and bud site selection. Science (Wash DC) 1995;270:464–467. doi: 10.1126/science.270.5235.464. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Anderegg RJ, Betz R, Carr SA, Crabb JW, Duntze W. Structure of Saccharomyces cerevisiae mating hormone a-factor. Identification of S-farnesyl cysteine as a structural component. J Biol Chem. 1988;263:18236–18240. [PubMed] [Google Scholar]
- Ashby MN, King DS, Rine J. Endoproteolytic processing of a farnesylated peptide in vitro. Proc Natl Acad Sci USA. 1992;89:4613–4617. doi: 10.1073/pnas.89.10.4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby MN, Rine J. Ras and a-factor converting enzyme. Methods Enzymol. 1995;250:235–250. doi: 10.1016/0076-6879(95)50076-6. [DOI] [PubMed] [Google Scholar]
- Bassett DE, Jr, Boguski MS, Spencer F, Reeves R, Goebl M, Hieter P. Comparative genomics, genome cross-referencing and XREFdb. Trends Genet. 1995;11:372–373. doi: 10.1016/s0168-9525(00)89109-x. [DOI] [PubMed] [Google Scholar]
- Berkower C, Loayza D, Michaelis S. Metabolic instability and constitutive endocytosis of STE6, the a-factor transporter of Saccharomyces cerevisiae. . Mol Biol Cell. 1994;5:1185–1198. doi: 10.1091/mbc.5.11.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair, L.C. 1979. Genetic analysis of mating type switching in yeast. (Ph.D. thesis. University of Oregon, Eugene, OR).
- Boeke JD, Lacroute F, Fink GR. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoroorotic acid resistance. Mol Gen Genet. 1984;197:345–346. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- Boguski MS, Tolstoshev CM, Bassett JDE. Access to dbEST is available through the World Wide Web. Science (Wash DC) 1994;265:1993–1994. [Google Scholar]
- Botstein D, Falco SC, Stewart SE, Brennan M, Scherer S, Stinchcomb DT, Struhl K, Davis RW. Sterile host yeast (SHY): a eukaryotic system of biological containment for recombinant DNA experiments. Gene (Amst) 1979;8:17–24. doi: 10.1016/0378-1119(79)90004-0. [DOI] [PubMed] [Google Scholar]
- Brake, A.J., C. Brenner, R. Najarian, P. Laybourn, and J. Merryweather. 1985. Structure of genes encoding precursors of the yeast peptide mating pheromone a -factor. In Protein Transport and Secretion. M.J. Gething, editor. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 103–108.
- Bruzzaniti A, Goodge K, Jay P, Taviaux SA, Lam MH, Berta P, Martin TJ, Moseley JM, Gillespie MT. PC8, a new member of the convertase family. Biochem J. 1996;314:727–731. doi: 10.1042/bj3140727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns N, Grimwade B, Ross-Madonald PB, Choi E-Y, Finberg K, Roeder GS, Snyder M. Large-scale analysis of gene expression, protein localization, and gene disruption in Saccharomyces cerevisiae. . Genes Dev. 1994;8:1087–1105. doi: 10.1101/gad.8.9.1087. [DOI] [PubMed] [Google Scholar]
- Chen, P. 1993. Biogenesis of the S. cerevisiae mating pheromone a-factor. (Ph.D. thesis. Johns Hopkins School of Medicine, Baltimore, MD). 251 pp.
- Chen P, Sapperstein SK, Choi JD, Michaelis S. Biogenesis of the Saccharomyces cerevisiae mating pheromone a-factor. J Cell Biol. 1997;136:251–269. doi: 10.1083/jcb.136.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WJ, Andres DA, Goldstein JL, Brown MS. Cloning and expression of a cDNA encoding the alpha subunit of rat p21ras protein farnesyltransferase. Proc Natl Acad Sci USA. 1991a;88:11368–11372. doi: 10.1073/pnas.88.24.11368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WJ, Andres DA, Goldstein JL, Russell DW, Brown MS. cDNA cloning and expression of the peptide-binding beta subunit of rat p21ras farnesyltransferase, the counterpart of yeast DPR1/RAM1. . Cell. 1991b;66:327–334. doi: 10.1016/0092-8674(91)90622-6. [DOI] [PubMed] [Google Scholar]
- Cole GM, Stone DE, Reed SI. Stoichiometry of G protein subunits affects the Saccharomyces cerevisiaemating pheromone signal transduction pathway. Mol Cell Biol. 1990;10:510–517. doi: 10.1128/mcb.10.2.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosson P, Letourneur F. Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science (Wash DC) 1994;263:1629–1631. doi: 10.1126/science.8128252. [DOI] [PubMed] [Google Scholar]
- Elble R. A simple and efficient procedure for transformation of yeast. BioTechniques. 1992;13:53–65. [PubMed] [Google Scholar]
- Fleischman RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb J-F, Dougherty BA, Merrick JM, et al. Whole-genome random sequencing and assembly of Haemophilus influenzaeRd. Science (Wash DC) 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- Fujita A, Oka C, Arikawa Y, Katagai T, Tonouchi A, Kuhara S, Misumi Y. A yeast gene necessary for bud-site selection encodes a protein similar to insulin-degrading enzymes. Nature (Lond) 1994;372:567–570. doi: 10.1038/372567a0. [DOI] [PubMed] [Google Scholar]
- Fuller RS, Sterne RE, Thorner J. Enzymes required for yeast prohormone processing. Annu Rev Physiol. 1988;50:345–362. doi: 10.1146/annurev.ph.50.030188.002021. [DOI] [PubMed] [Google Scholar]
- Goodman LE, Judd SR, Farnsworth CC, Powers S, Gelb MH, Glomset JA, Tamanoi F. Mutants of Saccharomyces cerevisiaedefective in the farnesylation of Ras proteins. Proc Natl Acad Sci USA. 1990;87:9665–9669. doi: 10.1073/pnas.87.24.9665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Chen P, Chen S-Y, Vancura KL, Michaelis S, Powers S. RAM2, an essential gene of yeast, and RAM1 encode the two polypeptide components of the farnesyltransferase that prenylates a-factor and Ras proteins. Proc Natl Acad Sci USA. 1991;88:11373–11377. doi: 10.1073/pnas.88.24.11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi R, Krummel B, Saiki RK. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper NM. Families of zinc metalloproteases. FEBS Lett. 1994;354:1–6. doi: 10.1016/0014-5793(94)01079-x. [DOI] [PubMed] [Google Scholar]
- Hrycyna CA, Clarke S. Farnesyl cysteine C-terminal methyltransferase activity is dependent upon the STE14 gene product in Saccharomyces cerevisiae. . Mol Cell Biol. 1990;10:5071–5076. doi: 10.1128/mcb.10.10.5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrycyna CA, Sapperstein SK, Clarke S, Michaelis S. The Saccharomyces cerevisiae STE14 gene encodes a methyltransferase that mediates C-terminal methylation of a-factor and RAS proteins. EMBO (Eur Mol Biol Organ) J. 1991;10:1699–1709. doi: 10.1002/j.1460-2075.1991.tb07694.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrycyna CA, Clarke S. Maturation of isoprenylated proteins in Saccharomyces cerevisiae. . J Biol Chem. 1992;267:10457–10464. [PubMed] [Google Scholar]
- Hrycyna CA, Wait SJ, Backlund PS, Michaelis S. Use of the yeast STE14 methyltransferase, expressed as a TrpE-STE14 fusion protein in Escherichia coli, for in vitro carboxylmethylation of isoprenylated polypeptides. Methods Enzymol. 1995;250:251–256. doi: 10.1016/0076-6879(95)50077-4. [DOI] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MR, Nilsson T, Peterson PA. Identification of a consensus motif for the retention of transmembrane proteins in the endoplasmic reticulum. EMBO (Eur Mol Biol Organ) J. 1990;9:3153–3162. doi: 10.1002/j.1460-2075.1990.tb07513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MR, Nilsson T, Peterson PA. Retrieval of transmembrane proteins to the endoplasmic reticulum. J Cell Biol. 1993;121:317–333. doi: 10.1083/jcb.121.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Bond JS. Families of metalloendopeptidases and their relationship. FEBS Lett. 1992;242:110–114. doi: 10.1016/0014-5793(92)80916-5. [DOI] [PubMed] [Google Scholar]
- Jongeneel CV, Bouvier J, Bairoch A. A unique signature identifies a family of zinc-dependent metallopeptidases. FEBS Lett. 1989;242:211–214. doi: 10.1016/0014-5793(89)80471-5. [DOI] [PubMed] [Google Scholar]
- Julius D, Brake A, Blair L, Kunisawa R, Thorner J. Isolation of the putative structural gene for the lysine-arginine-cleaving endo-peptidase required for processing of yeast prepro-α-factor. Cell. 1984;37:1075–1089. doi: 10.1016/0092-8674(84)90442-2. [DOI] [PubMed] [Google Scholar]
- Kornitzer D, Teff D, Altuvia S, Oppenheim AB. Isolation, characterization, and sequence of an Escherichia coli heat shock gene, htpx. . J Bacteriol. 1991;173:2944–2953. doi: 10.1128/jb.173.9.2944-2953.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchler K, Sterne RE, Thorner J. Saccharomyces cerevisiae STE6gene product: a novel pathway for protein export in eukaryotic cells. EMBO (Eur Mol Biol Organ) J. 1989;8:3973–3984. doi: 10.1002/j.1460-2075.1989.tb08580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Letourneur F, Gaynor EC, Hennecke S, Demolliere C, Duden R, Emr SD, Riezman H, Cosson P. Coatomer is essential for retrieval of dilysine-tagged proteins to the ER. Cell. 1994;79:1199–1207. doi: 10.1016/0092-8674(94)90011-6. [DOI] [PubMed] [Google Scholar]
- MacKay V, Manney TR. Mutations affecting sexual conjugation and related processes in Saccharomyces cerevisiae.I. Isolation and phenotypic characterization of nonmating mutants. Genetics. 1974;76:255–271. doi: 10.1093/genetics/76.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JP, Varshavsky A. The yeast STE6gene encodes a homologue of the mammalian multidrug resistance P-glycoprotein. Nature (Lond) 1989;340:400–404. doi: 10.1038/340400a0. [DOI] [PubMed] [Google Scholar]
- Medina JF, Wetterholm A, Radmark O, Shapiro R, Haeggstrom JZ, Vallee BL, Samuelsson B. Leukotriene A4 hydrolase: determination of the three zinc-binding ligands by site-directed mutagenesis and zinc analysis. Proc Natl Acad Sci USA. 1991;88:7620–7624. doi: 10.1073/pnas.88.17.7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis S. STE6, the yeast a-factor transporter. Semin Cell Biol. 1993;4:17–27. doi: 10.1006/scel.1993.1003. [DOI] [PubMed] [Google Scholar]
- Michaelis S, Herskowitz I. The a-factor pheromone of Saccharomyces cerevisiaeis essential for mating. Mol Cell Biol. 1988;8:1309–1318. doi: 10.1128/mcb.8.3.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen O, Davey J. Pheromone communication in the fission yeast Schizosaccharomyces pombe. . Semin Cell Biol. 1995;6:95–104. doi: 10.1016/1043-4682(95)90006-3. [DOI] [PubMed] [Google Scholar]
- Nomoto S, Nakayama N, Arai K, Matsumoto K. Regulation of the yeast pheromone response pathway by G protein subunits. EMBO (Eur Mol Biol Organ) J. 1990;9:691–696. doi: 10.1002/j.1460-2075.1990.tb08161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillinger MH, Volker C, Stock JB, Weissmann G, Philips MR. Characterization of a plasma membrane-associated prenylcysteine-directed alpha carboxyl methyltransferase in human neurophils. J Biol Chem. 1994;269:1485–1492. [PubMed] [Google Scholar]
- Powers S, Michaelis S, Broek D, Santa S, Anna, Field J, Herskowitz I, Wigler M. RAM, a gene of yeast required for a functional modification of RAS proteins and for production of mating pheromone. Cell. 1986;47:413–422. doi: 10.1016/0092-8674(86)90598-2. [DOI] [PubMed] [Google Scholar]
- Rhodes N, Connell L, Errede B. STE11is a protein kinase required for cell-type-specific transcription and signal transduction in yeast. Genes Dev. 1990;4:1862–1874. doi: 10.1101/gad.4.11.1862. [DOI] [PubMed] [Google Scholar]
- Rine, J.D. 1979. Regulation and transposition of cryptic mating type genes in Saccharomyces cerevisiae. Ph.D. thesis, University of Oregon, Eugene, OR.
- Rose, M.D., F. Winston, and P. Hieter. 1990. Methods in Yeast Genetics. A Laboratory Course Manual 1. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 181–182.
- Sapperstein S, Berkower C, Michaelis S. Nucleotide sequence of the yeast STE14 gene, which encodes the RAS and a-factor isoprenylcysteine methyltransferase, and demonstration of its essential role in a-factor export. Mol Cell Biol. 1994;14:1438–1449. doi: 10.1128/mcb.14.2.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer WR, Trueblood CE, Yang CC, Mayer MP, Rosenberg S, Poulter CD, Kim SH, Rine J. Enzymatic coupling of cholesterol intermediates to a mating pheromone precursor and to the ras protein. Science (Wash DC) 1990;249:1133–1139. doi: 10.1126/science.2204115. [DOI] [PubMed] [Google Scholar]
- Schroder S, Schimmoller F, Singer-Kruger B, Riezman H. The Golgi-localization of yeast Emp47p depends on its di-lysine motif but is not affected by the ret1-1mutation in alpha-COP. J Cell Biol. 1995;131:895–912. doi: 10.1083/jcb.131.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidah NG, Chretien M. Pro-protein convertases of subtilisin/kexin family. Methods Enzymol. 1994;244:175–188. doi: 10.1016/0076-6879(94)44015-8. [DOI] [PubMed] [Google Scholar]
- Seidah NG, Hamelin J, Mamarbachi M, Dong W, Tardos H, Mbikay M, Chretien M, Day R. cDNA structure, tissue distribution, and chromosomal localization of rat PC7, a novel mammalian proprotein convertase closest to yeast kexin-like proteinases. Proc Natl Acad Sci USA. 1996;93:3388–3393. doi: 10.1073/pnas.93.8.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague, G.F., and J.W. Thorner. 1992. Pheromone Response and Signal Transduction during the Mating Process of Saccharomyces cerevisiae The Molecular and Cellular Biology of the Yeast Saccharomyces: Gene Expression. E.W. Jones, J.R. Pringle, and J.R. Broach, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 657–744.
- Sprague JGF. Assay of yeast mating reaction. Methods Enzymol. 1991;194:77–93. doi: 10.1016/0076-6879(91)94008-z. [DOI] [PubMed] [Google Scholar]
- Stephenson RC, Clarke S. Identification of a C-terminal protein carboxyl methyltransferase in rat liver membranes utilizing a synthetic farnesyl cysteine-containing peptide substrate. J Biol Chem. 1990;265:16248–16254. [PubMed] [Google Scholar]
- Tan EW, Perez-Sala D, Canada FJ, Rando RR. Identifying the recognition unit for G protein methylation. J Biol Chem. 1991;266:10719–10722. [PubMed] [Google Scholar]
- Tomoyasu T, Gamer J, Bukau B, Kanemori M, Mori H, Rutman AJ, Oppenheim AB, Yura T, Yamanaka K, Niki H, Hiraga S, Ogura T. Escherichia coli FtsH is a membrane-bound, ATP-dependent protease which degrades the heat-shock transcription factor σ32 . EMBO (Eur Mol Biol Organ) J. 1995;14:2551–2560. doi: 10.1002/j.1460-2075.1995.tb07253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber ER, Hanekamp T, Thorsness PE. Biochemical and functional analysis of the YME1 gene product, an ATP and zinc-dependent mitochondrial protease from S. cerevisiae. . Mol Biol Cell. 1996;7:307–317. doi: 10.1091/mbc.7.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteway M, Hougan L, Thomas DY. Overexpression of the STE4 gene leads to mating response in haploid Saccharomyces cerevisiae. . Mol Cell Biol. 1990;10:217–222. doi: 10.1128/mcb.10.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson K, Herskowitz I. STE16, a new gene required for pheromone production by a cells of Saccharomyces cerevisiae. . Genetics. 1987;115:441–449. doi: 10.1093/genetics/115.3.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Yamane H, Stephenson R, Ong O, Fung BK-K, Clarke S. Analysis of prenylated carboxyl-terminal cysteine methylesters in proteins. METHODS (a companion to Methods Enzymol) 1990;1:270–282. [Google Scholar]
- Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Ann Rev Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]