

Abstract
The site-specific solvation of the photoexcited protochlorophyllide a (Pchlide a) in methanol solvent was investigated using the time-dependent density functional theory method for the first time to our knowledge. The intermolecular site-specific coordination and hydrogen-bonding interactions between Pchlide a and methanol molecules play a very important role in the steady-state and time-resolved spectra. All the calculated absorption and fluorescence spectra of the isolated Pchlide a and its coordinated and hydrogen-bonded complexes with methanol demonstrate that the novel fluorescence shoulder at ∼690 nm of Pchlide a in methanol should be ascribed to the coordinated and hydrogen-bonded Pchlide a-(MeOH)4 complex. This coordinated and hydrogen-bonded complex can also account for the intermediate state found in the time-resolved spectroscopic studies. Herein, we have theoretically confirmed that the intermolecular coordination and hydrogen bonds between Pchlide a and methanol molecules can be strengthened in the electronically excited state of Pchlide a. Furthermore, the site-specific solvation of the photoexcited Pchlide a can be induced by the intermolecular coordination and hydrogen-bond strengthening upon photoexcitation. Then the hydrogen-bonded intermediate state is formed in 22–27 ps timescales after the site-specific solvation. All the steady-state and time-resolved spectral features of Pchlide a in different solvents can be explained by the formation of this hydrogen-bonded intermediate state after the site-specific solvation, which is induced by the coordination and hydrogen-bond strengthening.
INTRODUCTION
Protochlorophyllide a (Pchlide a) is a precursor in the biosynthesis of chlorophyll, which is a sort of ubiquitous pigment of photosynthesis in plants, green algae, and cyanobacteria (1–9). Pchlide a belongs to the plant tetrapyrroles, which are of significant biological importance (1). The chemical structure of Pchlide a is a Mg-tetrapyrrole made up of four pyrrole-type rings linked together with four methane bridges (2,3). In the biosynthetic pathway of chlorophyll, Pchlide a is synthesized from 5-aminolevulinic acid in a sequence of enzyme-catalyzed reactions. In a consequent step, Pchlide a is reduced at the C17/C18 double bond to yield the chlorine macrocycle of chlorophyllide a (4–6). The reduction of Pchlide a is catalyzed by two distinct enzymes, the light-dependent and light-independent NADPH/protochlorophyllide oxidoreductase (POR) (7–9). Both of these enzymes are widely distributed among phototrophic organisms. Especially, the light-dependent POR enzyme is not only of importance as a key regulator of chlorophyll synthesis but also is one of only two enzymes in which catalytic activity is initiated by the absorption of light (8,9). Until now, we have had little knowledge about the molecular structure of Pchlide-POR-NADPH complexes, as well as about the nature of the pigment-surroundings interaction.
Many experimental and theoretical methods have been performed to investigate the biological macromolecules and their interactions with their surroundings (10–30). The requirement for light makes the POR an attractive model for studying the primary events of an enzymatic reaction in real time by using time-resolved spectroscopy (7–9). Recently, the first femtosecond study, performed by van Stokkum et al., on the ultrafast reaction dynamics in the POR enzyme, a ternary complex formed by the apoprotein, the substrate Pchlide a, and the coenzyme NADPH, has been reported (10). The experimental results were interpreted in terms of a complex mechanism with two parallel reactions leading to the formation of chlorophyllide a on the timescale of 3 ps and 400 ps, respectively (10). In addition, many spectroscopic studies on the enzymatic catalysis and protein-folding dynamics have been perfectly carried out by Callender et al. (11–14). One can also note that many quantum mechanics and quantum chemistry methods have been developed and applied to the investigations of the biological macromolecules (16–30). It has been demonstrated that these theoretical calculations are very useful for understanding the complex structure and dynamics of the biological macromolecules.
To facilitate understanding for the complex photochemical reaction in the POR enzyme, Dietzek et al. have investigated the excited-state dynamics of the substrate Pchlide a in solution, separated from the POR apoprotein, using time-resolved absorption and fluorescence spectroscopy (31–33). To mimic the different environmental conditions in the oxidoreductase complex, a variety of solvents were chosen in the femtosecond time-resolved absorption experiments. They demonstrated that the excited state dynamics of Pchlide a strongly depends on the solvent polarity (31,32). In polar solvents, such as methanol and acetonitrile, the excited state relaxation dynamics is multiexponential with three distinguishable timescales of 4.0–4.5 ps for vibrational relaxation and vibrational energy redistribution of the initially excited S1 state; 22–27 ps for the formation of an intermediate state, most likely with a charge transfer character; and 200 ps for the decay of this intermediate state back to the ground state (32). In nonpolar solvent cyclohexane, only the 4.5 ps relaxational process can be observed (32). In addition to polarity, the viscosity of solvent can also affect the excited state relaxation processes. Upon increasing the viscosity by adding glycerol to a methanolic solution of Pchlide a, two of the former relaxation processes are found to be decelerated (31,32). This means that not only is the vibrational cooling of the S1 state slowed in the more viscous surrounding, but the formation rate of the intermediate state with charge transfer character can also be reduced, suggesting that nuclear motions along the reaction coordinate are involved in the charge transfer (31–33). Consequently, the formation of the intermediate state may be related to the dynamic solvation process of Pchlide a in the S1 state.
The steady-state spectral properties of Pchlide a in various solvents have been extensively investigated in previous work (34–44). Determination of the spectral properties for Pchlide a in different solvents would be helpful to understand the properties and function of Pchlide a in vivo (34). In all the investigated solvents, the absorption spectra have a shape characteristic of chlorophyll and its derivatives in the monomeric form, consisting of Soret and Qy bands that are accompanied with satellite bands of lower intensity on the high energy side (34–37). The observed absorption maxima are within the ranges of 432.5–451 nm and 624–633 nm for Soret and Qy bands, respectively. For Pchlide a in nonpolar benzene solvent, the absorption maxima for Soret and Qy bands are 442 and 632.5 nm, respectively (38–44). For Pchlide a in polar protic methanol solvent, the absorption maxima for Soret and Qy bands are 434 and 629 nm, respectively. Moreover, the fluorescence spectrum of Pchlide a in methanol exhibits a strong S1→S0 band centered at 641 nm (31–33,44). The Stokes shift of Pchlide a in the investigated solvents shows a slight increase for increasing solvent orientation polarizability. This indicates that the spectral properties of Pchlide a have low sensitivity to the nonspecific solvation (42–44). Thus, the differences for fluorescence emission spectra of Pchlide a in various solvents are mainly due to the site-specific solvation between solutes and solvents (44). Moreover, it can be found that the fluorescence lifetimes of Pchlide a in more polar solvents are shortened, which may result from the site-specific solute-solvent interactions (42). Hydrogen bonding between solvent and solute molecules can cause a decrease of fluorescence lifetime, which can be observed when comparing the lifetimes for acetone and ethanol solvents, as well as for methanol and acetonitrile solvents (44). The fluorescence lifetime of Pchlide a is shorter in pyridine than in tetrahydrofuran, which may reflect a different character of Mg ligation in these solvents. This means that the fluorescence lifetime can also be influenced by the coordination bonding between Pchlide a and polar solvents (44). It should be noted that there was a novel shoulder at ∼690 nm in the fluorescence spectra of Pchlide a in polar solvents. It has been excluded that the shoulder is formed due to the Pchlide a aggregations, since no changes characteristic of Pchlide a aggregation are observed in the absorption and fluorescence spectra for the pigment concentration used (31–33,44). As a result, the novel fluorescence shoulder at 690 nm may be correlated with the site-specific solvation of the photoexcited Pchlide a in polar solvents by the site-specific coordination and hydrogen bonding. In addition, the fluorescence shoulder may also be associated with the formation of the intermediate state.
In this work, we theoretically investigated the structure and dynamics of the site-specific solvation for the Pchlide a in polar protic methanol solvent in the electronically excited state using the time-dependent density functional theory (TDDFT) method. The TDDFT method has been confirmed as a very useful and reliable tool to study the excited states of large molecules (45–54). The intermolecular coordination and hydrogen-bonding interactions between Pchlide a and methanol molecules were discussed in detail. The geometric and the electronic structures of the coordinated and hydrogen-bonded Pchlide a-(MeOH)n complexes were calculated in both the ground and excited states. It would be helpful for understanding the steady-state and time-resolved spectral features of Pchlide a in the polar protic methanol solvent. It was demonstrated that the fluorescence shoulder at 690 nm can be ascribed to the coordinated and hydrogen-bonded complex, which is formed due to the site-specific solvation in the electronically excited state of the Pchlide a molecule through the intermolecular coordination and hydrogen bonding. Furthermore, we also theoretically demonstrated that the intermolecular coordination and hydrogen bonds between Pchlide a and methanol molecules can be strengthened in the electronically excited state of Pchlide a. As a result, the site-specific solvation of photoexcited Pchlide a in methanol could be induced by the excited-state coordination and hydrogen-bond strengthening.
THEORY AND METHODS
In this work, the generalized gradient approximation (GGA) for the exchange correlation potential (BP86) was employed both in the density functional theory (DFT) calculation for ground state and TDDFT calculation for the electronically excited state (55–60). The resolution-of-the-identity (RI) approximation was also used to improve the efficiency without sacrificing the accuracy of the results (56–58). The triple-ζ valence quality with one set of polarization functions (TZVP) was chosen as basis sets and the corresponding auxiliary basis sets for the RI approximation throughout (59). Fine quadrature grids of size 4 were also employed. All the electronic structure calculations were carried out using the TURBOMOLE program suite (60).
RESULTS AND DISCUSSION
To delineate the detailed aspects of site-specific interactions between Pchlide a and methanol molecules in solution, we have been motivated to present a coordinated and hydrogen-bonded Pchlide a-(MeOH)n complex. As we know, the three C=O groups in Pchlide a are good sites which can be responsible for the formation of three hydrogen bonds C=O···H–O between Pchlide a and methanol molecules (48–51). Moreover, it has been reported that chlorophyll a can be coordinated with one water molecule between Mg atom in chlorophyll a and O atom in water molecule (40–44). Similarly, Pchlide a can be coordinated with one methanol molecule between the Mg atom in Pchlide a and the O atom in the methanol molecule. As a result, four methanol molecules should be included in the hydrogen-bonded Pchlide a-(MeOH)n complex. In Fig. 1, the fully optimized geometric conformations of isolated Pchlide a and the coordinated and hydrogen-bonded Pchlide a-(MeOH)4 complex are shown. A strong coordination bond (CB) Mg-O with a bond length of 2.105 Å can be formed between Pchlide a and methanol molecules.
FIGURE 1.

Geometric structures of Pchlide a (A) and its hydrogen-bonded complex with methanol (B). Nitrogen and magnesium atoms and some important carbon atoms are labeled. Dotted lines denote the intermolecular hydrogen bonds. CB, coordination bond; HB, hydrogen bond.
At the same time, this coordinated methanol molecule and its adjacent C=O group can form a hydrogen-bonding chain by bridging another methanol molecule. Herein, the hydrogen bonds O–H···O and C=O···H are denoted HB-I and HB-II, respectively (Fig. 1 B). In our optimized conformation of the coordinated and hydrogen-bonded Pchlide a-(MeOH)4 complex, the bond lengths of the HB-I and HB-II are calculated to be 1.637 and 1.833 Å, respectively. In addition, the angles formed by the HB-I and HB-II are calculated to be 177° and 166°, respectively. One can also see that both the methyl groups of the two methanol molecules reside out of the plane of the Pchlide a molecule. At the site of the C13 carbonyl group, a hydrogen bond C=O···H with a bond length of 1.860 Å and a bond angle of 174° can be formed between Pchlide a and the methanol molecule. At the same time, another relatively weak hydrogen bond C–H···O with a bond length of 2.337 Å and a bond angle of 176° can also be formed between this methanol molecule and the adjacent methyl group in Pchlide a. The hydrogen bonds C=O···H and C–H···O are marked here as HB-III and HB-IV, respectively. Similarly, hydrogen bonds C=O···H and O–H···O can be formed between one methanol molecule and Pchlide a at the site of C17. The bond length and angle of the hydrogen bond O–H···O, here denoted HB-V, are calculated to be 1.670 Å and 159°, respectively; whereas the other hydrogen bond C=O···H, here denoted HB-VI, is calculated to have a bond length of 1.914 Å and a bond angle of 139°.
In our theoretical investigation, some other hydrogen-bonded Pchlide a-(MeOH)n (n < 4) complexes have also been considered. Herein, we denote the complex formed by Pchlide a and the coordinated methanol molecule as Pchlide a-MeOH. The Pchlide a-MeOH dimer and the methanol molecule bonded by hydrogen bonds HB-I and HB-II can form a hydrogen-bonded trimer, which we denote here Pchlide a-(MeOH)2. The hydrogen-bonded complex formed by Pchlide a-(MeOH)2 and the methanol molecule which is bonded by hydrogen bonds HB-V and HB-VI are denoted Pchlide a-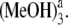 Similarly, Pchlide a-
Similarly, Pchlide a-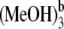 refers to the hydrogen-bonded complex formed by Pchlide a-(MeOH)2 and the methanol molecule which is bonded by hydrogen bonds HB-III and HB-IV. All these coordinated and hydrogen-bonded complexes as well as the isolated Pchlide a molecule have also been fully optimized. Moreover, the binding energies of the intermolecular coordination and hydrogen bonds between Pchlide a and methanol molecules are calculated. The calculated binding energy of CB is 56.42 kJ/mol. The total binding energy of hydrogen bonds HB-I and HB-II is calculated to be 68.83 kJ/mol, whereas the calculated total binding energy of hydrogen bonds HB-III and HB-IV is as small as 25.41 kJ/mol. In addition, the total binding energy of hydrogen bonds HB-V and HB-VI is calculated to be 49.76 kJ/mol.
refers to the hydrogen-bonded complex formed by Pchlide a-(MeOH)2 and the methanol molecule which is bonded by hydrogen bonds HB-III and HB-IV. All these coordinated and hydrogen-bonded complexes as well as the isolated Pchlide a molecule have also been fully optimized. Moreover, the binding energies of the intermolecular coordination and hydrogen bonds between Pchlide a and methanol molecules are calculated. The calculated binding energy of CB is 56.42 kJ/mol. The total binding energy of hydrogen bonds HB-I and HB-II is calculated to be 68.83 kJ/mol, whereas the calculated total binding energy of hydrogen bonds HB-III and HB-IV is as small as 25.41 kJ/mol. In addition, the total binding energy of hydrogen bonds HB-V and HB-VI is calculated to be 49.76 kJ/mol.
The electronic excitation energies and corresponding oscillator strengths for the low-lying singlet excited states of the coordinated and hydrogen-bonded Pchlide a-(MeOH)n (n ≤ 4) complexes as well as the isolated Pchlide a are presented in Table 1. All the energy levels of the Qy (S1) and the Soret bands are calculated here. It is characteristic that the energy levels of all the complexes are red shifted compared to those of isolated Pchlide a because of the intermolecular coordination and hydrogen-bonding interactions. Moreover, it can be found that both the electronic excitation energies and corresponding oscillator strengths of the hydrogen-bonded Pchlide a-(MeOH)2 are nearly the same as that of the hydrogen-bonded Pchlide a-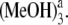 Similarly, the hydrogen-bonded Pchlide a-(MeOH)4 and the Pchlide a-
Similarly, the hydrogen-bonded Pchlide a-(MeOH)4 and the Pchlide a-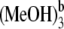 complexes also have nearly the same electronic spectra. Therefore, this indicates that the strong hydrogen bonds HB-V and HB-VI cannot significantly influence the electronic spectra of the hydrogen-bonded Pchlide a-(MeOH)n complexes. This may be because the hydrogen bonds HB-V and HB-VI are far away from the conjugated moiety of the Pchlide a molecule.
complexes also have nearly the same electronic spectra. Therefore, this indicates that the strong hydrogen bonds HB-V and HB-VI cannot significantly influence the electronic spectra of the hydrogen-bonded Pchlide a-(MeOH)n complexes. This may be because the hydrogen bonds HB-V and HB-VI are far away from the conjugated moiety of the Pchlide a molecule.
TABLE 1.
Calculated electronic excitation energies (in nm) and corresponding oscillator strengths (in the parentheses) of the low-lying electronically excited states for isolated Pchlide a and the hydrogen-bonded Pchlide a-(MeOH)1,2,3,4 complexes at the level of TD-RI-BP86 with the basis set TZVP
| Pchlide a | Pchlide a-MeOH | Pchlide a-(MeOH)2 | Pchlide a-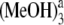
|
Pchlide a-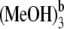
|
Pchlide a-(MeOH)4 | |
|---|---|---|---|---|---|---|
| S1:Abs. | 615(0.014) | 631(0.015) | 647(0.018) | 647(0.018) | 667(0.020) | 668(0.020) |
| Flu. | H→L 72.9% | H→L 76.7% | H→L 79.8% | H→L 79.9% | H→L 82.9% | H→L 83.1% |
| 632(0.023) | 651(0.021) | 673(0.022) | 674(0.023) | 707(0.022) | 708(0.022) | |
| S2 | 601(0.033) | 606(0.032) | 612(0.037) | 612(0.037) | 620(0.051) | 620(0.052) |
| S3 | 517(0.017) | 529(0.015) | 541(0.013) | 540(0.013) | 559(0.002) | 559(0.001) |
| S4 | 501(0.000) | 490(0.001) | 496(0.003) | 495(0.003) | 558(0.008) | 557(0.009) |
| S5 | 478(0.006) | 483(0.008) | 487(0.076) | 487(0.079) | 510(0.001) | 509(0.001) |
| S6 | 471(0.094) | 479(0.075) | 465(0.016) | 466(0.012) | 497(0.063) | 497(0.064) |
| S7 | 457(0.004) | 459(0.057) | 464(0.025) | 463(0.029) | 475(0.001) | 476(0.000) |
| S8 | 450(0.099) | 446(0.004) | 451(0.276) | 451(0.265) | 467(0.048) | 467(0.069) |
| S9 | 436(0.113) | 442(0.294) | 437(0.006) | 438(0.021) | 460(0.230) | 461(0.194) |
| S10 | 431(0.017) | 432(0.003) | 430(0.116) | 430(0.110) | 444(0.048) | 445(0.066) |
| S11 | 421(0.408) | 425(0.213) | 423(0.202) | 423(0.202) | 435(0.017) | 435(0.012) |
| S12 | 413(0.201) | 418(0.263) | 420(0.017) | 420(0.020) | 433(0.032) | 433(0.026) |
| S13 | 403(0.111) | 404(0.170) | 407(0.280) | 407(0.264) | 423(0.192) | 424(0.193) |
| S14 | 400(0.080) | 394(0.103) | 397(0.119) | 397(0.121) | 409(0.363) | 409(0.362) |
| S15 | 393(0.127) | 386(0.303) | 388(0.245) | 388(0.268) | 398(0.142) | 398(0.127) |
To distinctly present the electronic spectra, we simulate all the calculated absorption spectra of the isolated Pchlide a and the coordinated and hydrogen-bonded Pchlide a-(MeOH)n (n ≤ 4) complexes and show them in Fig. 2. In this work, only the methanol molecules in the first solvation shell which are directly coordinated and hydrogen bonded with Pchlide a are involved without consideration of the bulk effect of the outer solvation shells, since only the methanol molecules in the inner solvation shell of Pchlide a can be attributed to the site-specific solvation of the photoexcited Pchlide a. For comparison, the experimental values for the Qy (S1) and the Soret bands are also presented here. One can find that the calculated Soret band for all the complexes is at ∼434 nm, which is in good agreement with the experimental spectra. From Table 1, it has been found that the calculated Qy (S1) band of the isolated Pchlide a is at 615 nm, whereas this band of the coordinated and hydrogen-bonded complexes are calculated to be at the range from 631 to 668 nm. Since only one peak in the experimental Qy (S1) absorption band is located at 629 nm (31–33), it can be concluded that this peak is mainly ascribed to the absorption of the isolated Pchlide a, whereas the absorption of the coordinated and hydrogen-bonded complexes in this band are relatively weak. Thus, no evident absorption peak attributed to the coordinated and hydrogen-bonded Pchlide a-(MeOH)n complexes can be found in the experimental Qy (S1) band. At the same time, it is also demonstrated that the effect of bulk methanol solvent cannot significantly influence the absorption spectra of Pchlide a in methanol.
FIGURE 2.

Calculated absorption (solid lines) and fluorescence spectra (dotted lines) of isolated Pchlide a and its various hydrogen-bonded Pchlide a-(MeOH)1,2,3,4 complexes. Bold arrows indicate the order with increasing the number of MeOH molecules. Herein, Pchlide a-MeOH refers to the complex formed by Pchlide a and the coordinated methanol. The Pchlide a-MeOH dimer and the methanol hydrogen bonded by HB-I and HB-II form the hydrogen-bonded trimer Pchlide a-(MeOH)2. The hydrogen-bonded complex formed by Pchlide a-(MeOH)2 and the methanol which is hydrogen bonded by HB-V and HB-VI is denoted Pchlide a-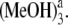 Pchlide a-
Pchlide a-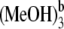 refers to the hydrogen-bonded complex formed by Pchlide a-(MeOH)2 and the methanol molecule which is bonded by hydrogen bonds HB-III and HB-IV. The vertical lines show the absorption and fluorescence peaks in the experiments.
refers to the hydrogen-bonded complex formed by Pchlide a-(MeOH)2 and the methanol molecule which is bonded by hydrogen bonds HB-III and HB-IV. The vertical lines show the absorption and fluorescence peaks in the experiments.
After excitation at 627 nm, two fluorescence components can be found to be at 641 and 690 nm in the experimental fluorescence spectra (31–33). As discussed above, it has been excluded that the shoulder at 690 nm in the fluorescence spectra is due to the Pchlide a aggregations (31–33,44). The novel fluorescence shoulder may be associated with the coordinated and hydrogen-bonded Pchlide a-(MeOH)n complexes after the site-specific solvation in the electronically excited state of Pchlide a in methanol. All the geometric optimizations of the S1 state for the isolated Pchlide a and the Pchlide a-(MeOH)n complexes have been performed using the TDDFT method. The calculated fluorescence emission energies are also listed in Table 1. At the same time, the calculated fluorescence spectra for the isolated Pchlide a and all the complexes are also shown in Fig. 2.
The calculated fluorescence of isolated Pchlide a is at 632 nm, which coincides with the fluorescence maximum at 641 nm in experiments. In addition, the fluorescence energies of the coordinated and hydrogen-bonded complexes are calculated to be in the range from 651 to 708 nm. It is evident that the experimental shoulder at 690 nm is located in this range. Thus, it can be demonstrated that the fluorescence maximum at 641 nm in experiments originates from the isolated Pchlide a, whereas the fluorescence shoulder at ∼690 nm of the Pchlide a in methanol solvent should be ascribed to the coordinated and hydrogen-bonded Pchlide a-(MeOH)4 complex. Furthermore, one can find that the site-specifically coordinated and hydrogen-bonded methanol molecules cause very large spectral shifts. However, the bulk effect of the outer solvation shell can only induce a slight spectral shift (42–44). So the site-specific solvation plays a more important role for the fluorescence spectra of the photoexcited Pchlide a in methanol than the nonspecific polar solvation.
Consequently, a dynamic equilibrium between the isolated Pchlide a and its site-specifically solvated forms in both the ground state and excited state can be given from the steady-state absorption and fluorescence spectral analysis (31–33,52). The Pchlide a in methanol solvent is located in the equilibrium between the isolated Pchlide a and the weak coordinated and hydrogen-bonded forms in the ground state. Thus, the equilibrium remains strongly in favor of the isolated Pchlide a in the ground state. So only the absorption peak which is ascribed to the isolated Pchlide a can be distinctly seen in the absorption spectra. However, the equilibrium may be changed and in favor of the coordinated and hydrogen-bonded forms in the electronically excited state. Therefore, the fluorescence shoulder attributed to the coordinated and hydrogen-bonded complexes can be distinctly found in the fluorescence spectra. The change of the dynamic equilibrium in the electronically excited state relative to that in the ground state should be the result of the site-specific solvation of the photoexcited Pchlide a in methanol.
According to the orbital transition contributions to the electronically excited state listed in Table 1, we know that the S1 state of the isolated Pchlide a and all its coordinated and hydrogen-bonded complexes in a dominative manner corresponds to the orbital transition from highest occupied molecular orbital (HOMO) to lowest unoccupied molecular orbital (LUMO). Thus, only the HOMO and LUMO of the isolated Pchlide a and all the complexes are listed in Table 2. For the isolated Pchlide a, it is clear that the electron density of the HOMO is localized on the porphyrin macrocycle, whereas the electron density of the LUMO is delocalized over the porphyrin moiety. So it is confirmed that the S1 state of the isolated Pchlide a molecule is of intramolecular charge transfer (ICT) character. The charge can be transferred from the porphyrin macrocycle to the cyclopentanone ring, in particular the carbonyl group of this ring, as well as the ethylene group at the site of C3.
TABLE 2.
Frontier MOs of isolated Pchlide a and its complexes calculated at the level of RI-BP86 with the basis set TZVP
 |
It should be noted that both the HOMO and LUMO are nearly unaffected by the polar protic methanol molecules in the coordinated and the hydrogen-bonded Pchlide a-(MeOH)n complexes. They are also of the same ICT character from the porphyrin macrocycle to the cyclopentanone ring and the ethylene group at the site of C3. This is not in accordance with the proposed ICT nature of the intermediate state found by Dietzek et al. in their time-resolved spectroscopic experiments. They believed that the intermediate state of the ICT nature is only formed for Pchlide a in the polar solvents, and no ICT state can be found for Pchlide a in the nonpolar solvents (31–33). It is evidently not so since the S1 state of the isolated Pchlide a is of ICT character. Therefore, the intermediate state formed in 22–27 ps timescales for Pchlide a in polar solvents should not be only assigned an ICT state. Moreover, the time constant for the formation of the intermediate state is significantly affected by the solvent viscosity (32). As a result, the formation of the intermediate state should be associated with the excited-state solvation process for the photoexcited Pchlide a in methanol solvents.
The calculated binding energies for the intermolecular coordination and hydrogen bonds in the coordinated and hydrogen-bonded Pchlide a-(MeOH)4 complex in different electronic states at the level of (TD)-RI-BP86 with the basis set TZVP are listed in Table 3. Our calculations at this level give high accuracy results and the error of the calculated binding energy will be within 1∼3 kJ/mol (61–63). The total binding energy of the two hydrogen bonds formed by one methanol molecule is calculated. One can find that the binding energy of the coordination bond CB is increased from the 56.42 kJ/mol in the ground state to the 61.76 kJ/mol in the S1 state. The total binding energy of the hydrogen bonds HB-I and HB-II is 68.83 kJ/mol in ground state, whereas it is increased to 73.91 kJ/mol in the S1 state. In addition, the total binding energy of the relatively weak hydrogen bonds HB-III and HB-IV is also increased from the 25.41 kJ/mol in ground state to the 32.09 kJ/mol in the S1 state. It should be noted that the strong hydrogen bonds HB-V and HB-VI are nearly unchanged upon photoexcitation to the S1 state.
TABLE 3.
Calculated binding energies (in kJ/mol) of the coordination bond (CB) and various hydrogen bonds (HB) in different electronic states at the level of TD-RI-BP86 with the basis set TZVP
| CB | HB-I + HB-II | HB-III + HB-IV | HB-V + HB-VI | |
|---|---|---|---|---|
| S0 | 56.42 | 68.83 | 25.41 | 49.76 |
| S1 | 61.76 | 73.91 | 32.09 | 50.28 |
From the calculated fluorescence emission energies of complexes listed in Table 1, one can also find that both the fluorescence emission energies and corresponding oscillator strengths of the hydrogen-bonded Pchlide a-(MeOH)2 and Pchlide 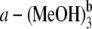 complexes are nearly the same as those of the hydrogen-bonded Pchlide
complexes are nearly the same as those of the hydrogen-bonded Pchlide 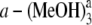 and Pchlide a-(MeOH)4 complexes, respectively. Thus, it can be concluded that the fluorescence emission energies of coordinated and hydrogen-bonded Pchlide a-(MeOH)n complexes are significantly influenced by the change of the intermolecular coordination and hydrogen bonds in the electronically excited state. So the hydrogen bonds HB-V and HB-VI cannot strongly affect the fluorescence spectra of the hydrogen-bonded Pchlide a-(MeOH)n complexes, since they are nearly unchanged in the S1 state of the Pchlide a-(MeOH)4 complex as discussed above. In Table 4, the bond lengths of the intermolecular coordination and the hydrogen bonds in ground and excited states are listed. It can be seen that with the increasing of the binding energies in the S1 state compared to those in ground state, the bond lengths of the coordination bond CB and the hydrogen bonds HB-I, HB-II, HB-III, and HB-IV are correspondingly shortened, whereas the hydrogen-bond lengths of HB-V and HB-VI remain unchanged upon photoexcitation to the S1 state, which coincides with the unchanged hydrogen-bond binding energies.
and Pchlide a-(MeOH)4 complexes, respectively. Thus, it can be concluded that the fluorescence emission energies of coordinated and hydrogen-bonded Pchlide a-(MeOH)n complexes are significantly influenced by the change of the intermolecular coordination and hydrogen bonds in the electronically excited state. So the hydrogen bonds HB-V and HB-VI cannot strongly affect the fluorescence spectra of the hydrogen-bonded Pchlide a-(MeOH)n complexes, since they are nearly unchanged in the S1 state of the Pchlide a-(MeOH)4 complex as discussed above. In Table 4, the bond lengths of the intermolecular coordination and the hydrogen bonds in ground and excited states are listed. It can be seen that with the increasing of the binding energies in the S1 state compared to those in ground state, the bond lengths of the coordination bond CB and the hydrogen bonds HB-I, HB-II, HB-III, and HB-IV are correspondingly shortened, whereas the hydrogen-bond lengths of HB-V and HB-VI remain unchanged upon photoexcitation to the S1 state, which coincides with the unchanged hydrogen-bond binding energies.
TABLE 4.
Calculated bond lengths (in Å) of the coordination bond (CB) and various hydrogen bonds (HB) in different electronic states of Pchlide a-(MeOH)4 complex
| CB | HB-I | HB-II | HB-III | HB-IV | HB-V | HB-VI | |
|---|---|---|---|---|---|---|---|
| S0 | 2.105 | 1.637 | 1.833 | 1.860 | 2.337 | 1.670 | 1.914 |
| S1 | 2.092 | 1.607 | 1.775 | 1.789 | 2.314 | 1.669 | 1.915 |
The intermolecular coordination and hydrogen-bond strengthening in the electronically excited states can be followed by the rearrangement of the coordinated and hydrogen-bonded solvent molecules. As a consequence, the site-specific solvation could be induced by the intermolecular coordination and hydrogen-bond strengthening upon photoexcitation to the S1 state of Pchlide a in methanol solvent. All the steady-state and time-resolved spectral features of Pchlide a in different solvents can be explained by the site-specific solvation which is induced by the excited-state coordination and hydrogen-bond strengthening. As discussed above, Pchlide a in polar solvents is located in the equilibrium between the free and the weakly solvated forms in the ground state. So this ground-state equilibrium remains markedly in favor of the free form. Only one peak ascribed to the absorption of the free Pchlide a is found in the experimental Qy (S1) absorption band (26).
Upon photoexcitation, the relatively weak intermolecular coordination and hydrogen bonds between Pchlide a and polar solvents in ground state can be strengthened in the electronically excited state. Therefore, after 4.0–4.5 ps for the ultrafast vibrational relaxation and vibrational energy redistribution of the initially excited S1 state, a strongly coordinated and hydrogen-bonded intermediate state is formed by the site-specific solvation which is induced by the intermolecular coordination and hydrogen-bond strengthening. The site-specific solvation occurs in 22–27 ps timescales, which is dependent on the viscosity of the polar solvents. The excited-state equilibrium will become strongly in favor of the coordinated and hydrogen-bonded forms. As a result, the fluorescence shoulder at ∼690 nm can be distinctly observed in the fluorescence spectra for Pchlide a in polar solvents. After site-specific solvation, the hydrogen-bonded intermediate state decays back to the ground state through the radiative deactivation within ∼200 ps (31–33). For the case of the Pchlide a in nonpolar solvents, no coordinated and hydrogen-bonded intermediate state can be formed by the site-specific solvation induced by the intermolecular coordination and hydrogen-bond strengthening. Therefore, only the vibrational relaxation process taking place on a 4.5 ps timescale can be observed for the photoexcited Pchlide a in the nonpolar solvents.
CONCLUSIONS
The TDDFT method was performed to investigate the site-specific solvation of the photoexcited Pchlide a in the polar protic methanol solvent. The intermolecular site-specific coordination and hydrogen-bonding interactions between Pchlide a and methanol molecules play a very important role in the steady-state and time-resolved spectra. All the absorption and fluorescence spectra of the isolated Pchlide a and its coordinated and hydrogen-bonded Pchlide a-(MeOH)n (n ≤ 4) complexes are calculated. We have theoretically demonstrated that only the peak in the experimental Qy (S1) absorption band located at 629 nm and the fluorescence maximum at 641 nm can be mainly ascribed to the absorption and emission of the isolated Pchlide a. Furthermore, the novel fluorescence shoulder at ∼690 nm of the Pchlide a in methanol solvent can be ascribed to the coordinated and hydrogen-bonded Pchlide a-(MeOH)4 complex. According to our frontier molecular orbitals (MOs) analysis, it is confirmed that the S1 state of both the isolated Pchlide a and its coordinated and hydrogen-bonded complexes are of the same ICT character. This is not in accordance with the fact that the intermediate state of an ICT nature can only be formed for Pchlide a in the polar solvents, whereas no ICT state can be found for Pchlide a in the nonpolar solvents. Therefore, the intermediate state formed in 22–27 ps timescales for the photoexcited Pchlide a in polar solvents should not be assigned only the ICT state. We think that it is associated with the excited-state solvation for the photoexcited Pchlide a in polar solvents, since the time constant for the formation of the intermediate state is significantly affected by solvent viscosities.
In this work, we have theoretically demonstrated that the intermolecular coordination and hydrogen bonds between Pchlide a and methanol molecules can be strengthened in the electronically excited state of Pchlide a according to the calculated binding energies and bond lengths for these bonds in different electronic states. The site-specific solvation of the photoexcited Pchlide a can be induced by the intermolecular coordination and hydrogen-bond strengthening upon photoexcitation. All the steady-state and time-resolved spectral features of Pchlide a in different solvents can be interpreted by the site-specific solvation induced by the excited-state coordination and hydrogen-bond strengthening. Due to the site-specific solvation, the dynamic equilibrium in the electronically excited state of Pchlide a will be changed and becomes in favor of the hydrogen-bonded form. Consequently, the formation of the hydrogen-bonded intermediate state for the photoexcited Pchlide a in methanol can be induced by the hydrogen-bond strengthening after the site-specific solvation taking place in the 22–27 ps timescales.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 20373071 and No. 20333050).
Editor: Steven D. Schwartz.
References
- 1.Suzuki, J. Y., D. W. Bolivar, and C. E. Bauer. 1997. Genetic analysis of chlorophyll biosynthesis. Annu. Rev. Genet. 31:61–89. [DOI] [PubMed] [Google Scholar]
- 2.Lebedev, N., and M. P. Timko. 1998. Protochlorophyllide photoreduction. Photosynth. Res. 58:5–23. [Google Scholar]
- 3.Bollivar, D. W. 2006. Recent advances in chlorophyll biosynthesis. Photosynth. Res. 90:173–194. [DOI] [PubMed] [Google Scholar]
- 4.Cornah, J. E., M. J. Terry, and A. G. Smith. 2003. Green or red: what stops the traffic in the tetrapyrrole pathway? Trends Plant Sci. 8:224–230. [DOI] [PubMed] [Google Scholar]
- 5.Eckhardt, U., B. Grimm, and S. Hörtensteiner. 2004. Recent advances in chlorophyll biosynthesis and breakdown in higher plants. Plant Mol. Biol. 56:1–14. [DOI] [PubMed] [Google Scholar]
- 6.Moulin, M., and A. G. Smith. 2005. Regulation of tetrapyrrole biosynthesis in higher plants. Biochem. Soc. Trans. 33:737–742. [DOI] [PubMed] [Google Scholar]
- 7.Schoefs, B., and F. Franck. 2003. Protochlorophyllide reduction: mechanisms and evolution. Photochem. Photobiol. 78:543–557. [DOI] [PubMed] [Google Scholar]
- 8.Masuda, T., and K. Takamiya. 2004. Novel insights into the enzymology, regulation and physiological functions of light-dependent protochlorophyllide oxidoreductase in angiosperms. Photosynth. Res. 81:1–29. [DOI] [PubMed] [Google Scholar]
- 9.Yang, J., and Q. Cheng. 2004. Origin and evolution of the light-dependent protochlorophyllide oxidoreductase (LPOR) genes. Plant Biol. 6:537–544. [DOI] [PubMed] [Google Scholar]
- 10.Heyes, D. J., C. N. Hunter, I. H. M. van Stokkum, and R. van Grondelle. 2003. Ultrafast enzymatic reaction dynamics in protochlorophyllide oxidoreductase. Nat. Struct. Biol. 10:491–492. [DOI] [PubMed] [Google Scholar]
- 11.Deng, H., R. Callender, and E. Howell. 2001. Vibrational structure of dihydrofolate bound to R67 dihydrofolate reductase. J. Biol. Chem. 276:48956–48960. [DOI] [PubMed] [Google Scholar]
- 12.Cheng, H., I. Nikolic-Hughes, J. H. H. Wang, H. Deng, P. J. O'Brien, L. Wu, Z. Y. Zhang, D. Herschlag, and R. Callender. 2002. Environmental effects on phosphoryl group bonding probed by vibrational spectroscopy: implications for understanding phosphoryl transfer and enzymatic catalysis. J. Am. Chem. Soc. 124:11295–11306. [DOI] [PubMed] [Google Scholar]
- 13.McClendon, S., N. Zhadin, and R. Callender. 2005. The approach to the Michaelis complex in lactate dehydrogenase: the substrate binding pathway. Biophys. J. 89:2024–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Callender, R., and R. B. Dyer. 2006. Advances in time-resolved approaches to characterize the dynamical nature of enzymatic catalysis. Chem. Rev. 106:3031–3042. [DOI] [PubMed] [Google Scholar]
- 15.Zuo, P., B. X. Li, X. H. Zhao, Y. S. Wu, X. C. Ai, J. P. Zhang, L. B. Li, and T. Y. Kuang. 2006. Ultrafast carotenoid-to-chlorophyll singlet energy transfer in the cytochrome b(6)f complex from Bryopsis corticulans. Biophys. J. 90:4145–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang, Y. L., and X. C. Hu. 2002. Quantum chemistry study of π–π stacking interactions of the bacteriochlorophyll dimer in the photosynthetic reaction center of Rhodobacter sphaeroides. J. Chem. Phys. 117:1–4. [Google Scholar]
- 17.Wang, Y. L., and X. C. Hu. 2002. A quantum chemistry study of binding carotenoids in the bacterial light-harvesting complexes. J. Am. Chem. Soc. 124:8445–8451. [DOI] [PubMed] [Google Scholar]
- 18.Mao, L. S., Y. L. Wang, and X. C. Hu. 2003. π–π stacking interactions in the peridinin-chlorophyll-protein of Amphidinium carterae. J. Phys. Chem. B. 107:3963–3971. [Google Scholar]
- 19.Wang, Y. L., L. S. Mao, and X. C. Hu. 2004. Insight into the structural role of carotenoids in the Photosystem I: a quantum chemical analysis. Biophys. J. 86:3097–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang, D. W., and J. Z. H. Zhang. 2004. Full ab initio computation of protein-water interaction energies. J. Theor. Comput. Chem. 3:43–49. [Google Scholar]
- 21.Mei, Y., E. L. Wu, K. L. Han, and J. Z. H. Zhang. 2006. Treating hydrogen bonding in ab initio calculation of biopolymers. Int. J. Quantum Chem. 106:1267–1276. [Google Scholar]
- 22.Mei, Y., C. G. Ji, and J. Z. H. Zhang. 2006. A new quantum method for electrostatic solvation energy of protein. J. Chem. Phys. 125:094906. [DOI] [PubMed] [Google Scholar]
- 23.York, D. M., T. S. Lee, and W. T. Yang. 1996. Quantum mechanical study of aqueous polarization effects on biological macromolecules. J. Am. Chem. Soc. 118:10940–10941. [Google Scholar]
- 24.York, D. M., T. S. Lee, and W. T. Yang. 1998. Quantum mechanical treatment of biological macromolecules in solution using linear-scaling electronic structure methods. Phys. Rev. Lett. 80:5011–5014. [Google Scholar]
- 25.Hori, T., H. Takahashi, M. Nakano, T. Nitta, and W. T. Yang. 2006. A QM/MM study combined with the theory of energy representation: solvation free energies for anti/syn acetic acids in aqueous solution. Chem. Phys. Lett. 419:240–244. [Google Scholar]
- 26.Karmacharya, R., D. Antoniou, and S. D. Schwartz. 2001. Nonequilibrium solvation and the quantum Kramers problem: proton transfer in aqueous glycine. J. Phys. Chem. A. 105:2563–2567. [Google Scholar]
- 27.Nunez, S., D. Antoniou, V. L. Schramm, and S. D. Schwartz. 2004. Promoting vibrations in human purine nucleoside phosphorylase. A molecular dynamics and hybrid quantum mechanical/molecular mechanical study. J. Am. Chem. Soc. 126:15720–15729. [DOI] [PubMed] [Google Scholar]
- 28.Basner, J. E., and S. D. Schwartz. 2005. How enzyme dynamics helps catalyze a reaction in atomic detail: a transition path sampling study. J. Am. Chem. Soc. 127:13822–13831. [DOI] [PubMed] [Google Scholar]
- 29.Antoniou, D., J. Basner, S. Nunez, and S. D. Schwartz. 2006. Computational and theoretical methods to explore the relation between enzyme dynamics and catalysis. Chem. Rev. 106:3170–3187. [DOI] [PubMed] [Google Scholar]
- 30.Exequiel, J. R., T. Pineda, R. Callender, and S. D. Schwartz. 2007. Ligand binding and protein dynamics in lactate dehydrogenase. Biophys. J. 10.1529/biophysj.107.106146. [DOI] [PMC free article] [PubMed]
- 31.Dietzek, B., R. Maksimenka, T. Siebert, E. Birckner, W. Kiefer, J. Popp, G. Hermann, and M. Schmitt. 2004. Excited-state processes in protochlorophyllide a: a femtosecond time-resolved absorption study. Chem. Phys. Lett. 397:110–115. [DOI] [PubMed] [Google Scholar]
- 32.Dietzek, B., W. Kiefer, J. Popp, G. Hermann, and M. Schmitt. 2006. Solvent effects on the excited-state processes of protochlorophyllide: a femtosecond time-resolved absorption study. J. Phys. Chem. B. 110:4399–4406. [DOI] [PubMed] [Google Scholar]
- 33.Dietzek, B., W. Kiefer, A. Yartsev, V. Sundström, P. Schellenberg, P. Grigaravicius, G. Hermann, J. Popp, and M. Schmitt. 2006. The excited-state chemistry of protochlorophyllide a: a time-resolved fluorescence study. ChemPhysChem. 7:1727–1733. [DOI] [PubMed] [Google Scholar]
- 34.El Hamouri, B., M. Brouers, and C. Sironval. 1981. Pathway from photoinactive P633–628 protochlorophyllide to the P696–682 chlorophyllide in cucumber etioplast suspensions. Plant Sci. Lett. 21:375–379. [Google Scholar]
- 35.Ryderg, M., and C. Sundqvist. 1982. Spectral forms of protochlorophyllide in prolamellar bodies and prothylakoids fractionated from wheat etioplasts. Physiol. Plant. 56:133–138. [Google Scholar]
- 36.Böddi, B., A. Lindsten, M. Ryderg, and C. Sundqvist. 1989. On the aggregational states of protochlorophyllide and its protein complexes in wheat etioplasts. Physiol. Plant. 76:135–143. [Google Scholar]
- 37.Franck, F., B. Bereza, and B. Böddi. 1999. Protochlorophyllide-NADP+ and protochlorophyllide-NADPH complexes and their regeneration after flash illumination in leaves and etioplast membranes of dark-grown wheat. Photosynth. Res. 59:53–61. [Google Scholar]
- 38.Mysliwa-Kurdziel, B., F. Franck, and K. Strzalka. 1999. Analysis of fluorescence lifetime of protochlorophyllide and chlorophyllide in isolated etioplast membranes measured from multifrequency cross-correlation phase fluorometry. Photochem. Photobiol. 70:616–623. [Google Scholar]
- 39.Shipman, L. I., T. M. Cotton, J. R. Norris, and J. J. Katz. 1976. An analysis of the visible absorption spectrum of chlorophyll a monomer, dimer, and oligomers in solution. J. Am. Chem. Soc. 98:8222–8230. [DOI] [PubMed] [Google Scholar]
- 40.Renge, I., and R. Avarmaa. 1985. Specific solvation of chlorophyll a: solvent nucleophility, hydrogen bonding and steric effects on absorption spectra. Photochem. Photobiol. 42:253–260. [Google Scholar]
- 41.Krawczyk, S. 1989. The effects of hydrogen bonding and coordination interaction in visible absorption and vibrational spectra of chlorophyll a. Biochim. Biophys. Acta. 976:140–149. [Google Scholar]
- 42.Limantara, L., S. Sakamoto, Y. Koyama, and H. Nagae. 1997. Effects of nonpolar and polar solvents on the Qx and Qy energies of bacteriochlorophyll a and bacteriopheophytin a. Photochem. Photobiol. 65:330–337. [Google Scholar]
- 43.Vladkova, R. 2000. Chlorophyll a self-assembly in polar solvent-water mixtures. Photochem. Photobiol. 71:71–83. [DOI] [PubMed] [Google Scholar]
- 44.Mysliwa-Kurdziel, B., J. Kruk, and K. Strzalka. 2004. Fluorescence lifetimes and spectral properties of protochlorophyllide in organic solvents in relation to the respective parameters in vivo. Photochem. Photobiol. 79:62–67. [PubMed] [Google Scholar]
- 45.Sobolewski, A. L., and W. Domcke. 1999. Photophysics of malonaldehyde: an ab initio study. J. Phys. Chem. A. 103:4494–4504. [Google Scholar]
- 46.Sobolewski, A. L., and W. Domcke. 1999. Ab initio investigations on the photophysics of indole. Chem. Phys. Lett. 315:293–298. [Google Scholar]
- 47.Sobolewski, A. L., and W. Domcke. 2000. Photoejection of electrons from pyrrole into an aqueous environment: ab initio results on pyrrole-water clusters. Chem. Phys. Lett. 321:479–484. [Google Scholar]
- 48.Sudholt, W., A. Staib, A. L. Sobolewski, and W. Domcke. 2000. Molecular-dynamics simulations of solvent effects in the intramolecular charge transfer of 4-(N,N-dimethylamino)benzonitrile. Phys. Chem. Chem. Phys. 2:4341–4353. [Google Scholar]
- 49.Sobolewski, A. L., and W. Domcke. 2004. Intramolecular hydrogen bonding in the S1(π-π*) excited state of anthranilic acid and salicylic acid: TDDFT calculation of excited-state geometries and infrared spectra. J. Phys. Chem. A. 108:10917–10922. [Google Scholar]
- 50.Sobolewski, A. L., W. Domcke, and C. Hattig. 2006. Photophysics of organic photostabilizers. Ab initio study of the excited-state deactivation mechanisms of 2-(2′-hydroxyphenyl)benzotriazole. J. Phys. Chem. A. 110:6301–6306. [DOI] [PubMed] [Google Scholar]
- 51.Zhao, G.-J., and K.-L. Han. 2007. Early time hydrogen-bonding dynamics of photoexcited coumarin 102 in hydrogen-donating solvents: theoretical study. J. Phys. Chem. A. 111:2469–2474. [DOI] [PubMed] [Google Scholar]
- 52.Zhao, G.-J., and K.-L. Han. 2007. Ultrafast hydrogen bond strengthening of the photoexcited fluorenone in alcohols for facilitating the fluorescence quenching. J. Phys. Chem. A. 10.1021/jp0719659. [DOI] [PubMed]
- 53.Zhao, G.-J., J.-Y. Liu, L.-C. Zhou, and K.-L. Han. 2007. Site-selective photoinduced electron transfer from alcoholic solvents to the chromophore facilitated by hydrogen bonding: a new fluorescence quenching mechanism. J. Phys. Chem. B. 111:8940–8945. [DOI] [PubMed] [Google Scholar]
- 54.Zhao, G.-J., and K.-L. Han. 2007. Novel infrared spectra for intermolecular dihydrogen bonding of the phenol-borane-trimethylamine complex in electronically excited state. J. Chem. Phys. 127:024306. [DOI] [PubMed] [Google Scholar]
- 55.Furche, F., and R. Ahlrichs. 2002. Adiabatic time-dependent density functional methods for excited state properties. J. Chem. Phys. 117:7433–7447. [Google Scholar]
- 56.Whitten, J. L. 1973. Coulombic potential energy integrals and approximations. J. Chem. Phys. 58:4496–4501. [Google Scholar]
- 57.Dunlap, B. I., J. W. D. Conolly, and J. R. Sabin. 1979. On some approximations in applications of Xα theory. J. Chem. Phys. 71:3396–3402. [Google Scholar]
- 58.Vahtras, O., J. E. Almlöf, and M. W. Feyereisen. 1993. Integral approximations for LCAO-SCF calculations. Chem. Phys. Lett. 213:514–518. [Google Scholar]
- 59.Schäfer, A., C. Huber, and R. Ahlrichs. 1994. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 100:5829–5835. [Google Scholar]
- 60.Ahlrichs, R., M. Bär, H. Horn, and C. Kölmel. 1989. Electronic structure calculations on workstation computers: the program system turbomole. Chem. Phys. Lett. 162:165–169. [Google Scholar]
- 61.Neugebauer, J., and M. Reiher. 2004. Vibrational center-ligand coupling in transition metal complexes. J. Comput. Chem. 25:587–597. [DOI] [PubMed] [Google Scholar]
- 62.Weigend, F. 2002. A fully direct RI-HF algorithm: implementation, optimised auxiliary basis sets, demonstration of accuracy and efficiency. Phys. Chem. Chem. Phys. 4:4285–4291. [Google Scholar]
- 63.Jacquemin, D., E. A. Perpète, G. Scalmani, M. J. Frisch, X. Assfeld, I. Ciofini, and C. Adamo. 2006. Time-dependent density functional theory investigation of the absorption, fluorescence, and phosphorescence spectra of solvated coumarins. J. Chem. Phys. 125:164324. [DOI] [PubMed] [Google Scholar]