

Abstract
We studied secretory phospholipase A2 type IIA (sPLA2) activity toward phospholipids that are derivatized in the sn-1 position of the glycerol backbone. We explored what type of side group (small versus bulky groups, hydrophobic versus polar groups) can be introduced at the sn-1 position of the glycerol backbone of glycerophospholipids and at the same time be hydrolyzed by sPLA2. The biophysical characterization revealed that the modified phospholipids can form multilamellar vesicles, and several of the synthesized sn-1 functionalized phospholipids were hydrolyzed by sPLA2. Molecular dynamics simulations provided detailed insight on an atomic level that can explain the observed sPLA2 activity toward the different phospholipid analogs. The simulations revealed that, depending on the nature of the side chain located at the sn-1 position, the group may interfere with an incoming water molecule that acts as the nucleophile in the enzymatic reaction. The simulation results are in agreement with the experimentally observed sPLA2 activity toward the different phospholipid analogs.
INTRODUCTION
Phospholipase A2 (PLA2; EC 3.1.1.4) comprises a diverse superfamily of lipolytic enzymes that specifically cleave the sn-2 acyl ester bond of glycerophospholipids to produce free fatty acids and lysophospholipids (1–6). Over the past two decades, numerous PLA2s have been identified and characterized (7–9). According to their biochemical features, such as cellular localization, requirement of Ca2+, substrate specificity, and primary structure, these PLA2s are classified into several families, including low-molecular-weight secretory PLA2 (sPLA2), Ca2+-sensitive arachidonoyl-specific 85-kDa cytosolic PLA2 (cPLA2), and Ca2+-independent PLA2 (iPLA2). Secretory phospholipases are small enzymes (14–19 kDa) that are classified into at least four subgroups based on structural differences (10,11). They have several features that distinguish them from other PLA2 families, such as high-disulfide-bond content, a requirement for millimolar concentration of Ca2+ for catalysis, and a broad specificity for phospholipids with different polar headgroups and fatty acid chains (12). The activity of these lipolytic enzymes increases substantially when adsorbed onto a lipid-water interface (13–17)—a phenomenon known as interfacial activation (18). However, there are dramatically different affinities of the various sPLA2s for membrane surfaces containing different phospholipids (19,20). It has been suggested that this affinity is controlled by the difference in the amino acids located on the interfacial binding surface (interfacial specificity) rather than the substrate affinity being controlled by the active site residues (catalytic site specificity) (21–23). The interfacial binding specificity of sPLA2s has important physiological consequences. sPLA2 generally prefers membranes containing anionic lipids (20,24). Human sPLA2-IIA binds several orders of magnitude more tightly to anionic phospholipid membranes than to a membrane composed of zwitterionic (charge-neutral) phosphatidylcholine lipids (19,20,25). The specificity of the enzyme toward anionic membranes is driven by electrostatic forces (24,26,27), since the enzyme has a relatively large number of cationic residues, resulting in an isoelectric point for the enzyme that is larger than 10.5 (26,28). This electrostatic field provides nonspecific electrostatic interactions between the enzyme and membrane surface that consequently enhance binding to the anionic surface (1,16,18,27,29). Conversely, other sPLA2 types, such as those isolated from snake venom, lack the large number of cationic residues found in sPLA2-IIA (1,20). These species are able to bind and hydrolyze both zwitterionic and anionic lipid membranes, and do not exhibit the surface charge specificity of sPLA2-IIA (20,30). The ability of snake venom sPLA2 to also hydrolyze zwitterionic membranes has been connected to the presence of aromatic residues in the membrane putative interfacial binding surface (31). These aromatic residues are essential for the nonpolar interaction with the zwitterionic membrane (24,32), and they may interact with membranes in distinct modes that determine membrane-binding affinities and hence catalytic efficiency (32,33).
Members of the group II sPLA2s have drawn increased attention, since these enzymes have been associated with a wide variety of immune-mediated inflammatory pathologies in humans, ranging from systemic and acute inflammatory conditions to cancer (9,28,34–39). For instance, high levels of sPLA2 are associated with the onset of rheumatoid arthritis (40,41), allergic rhitinis (42,43), and septic shock (44,45). More recently, it was suggested that sPLA2-IIA has a central role in both tumor development and progression (46–49), since sPLA2-IIA occurs at elevated levels in cancer tissue. This observation provides an avenue for site-specific drug delivery by liposomal carriers, where the liposomes are degraded specifically in the tumor tissue by sPLA2 releasing antitumor drugs at the target site (48–50). Additionally, the fatty acid and lysophospholipid hydrolysis products generated by sPLA2 have been shown to display a synergistic effect as permeability enhancers for drug transport (2,48).
To further investigate the viability of using sPLA2-IIA as a trigger of targeted drug delivery systems, we are interested in new phospholipids with functionality in the sn-1 position. These lipid molecules should be hydrolyzed effectively by sPLA2-IIA and potentially serve as prodrugs. We demonstrated earlier that a relatively small hydrophobic side chain located at the sn-1 position is efficiently hydrolyzed by sPLA2-IIA (51).
The aim of this study was to explore further which type of side group (small versus bulky groups, hydrophobic versus polar) can be introduced at the sn-1 position of the glycerol backbone of glycerophospholipids. The insight gained from this study is essential for determining the structural criteria that allow sPLA2 to accommodate and tolerate phospholipids with sn-1 substitutions. Furthermore, this knowledge provides the basis for using the sn-1 position as a possible anchor point for drug conjugates or fluorescence probes. The latter can be used to probe interfacial properties of membranes. We have chosen an interdisciplinary approach that combines organic synthesis, biophysical characterization, and computer simulations to obtain a detailed understanding of structural properties in the substrate that determine sPLA2 activity. Here, we report the results of our biophysical and computational studies. The biophysical characterization was performed to determine whether the modified lipids can form multilamellar vesicles (MLVs) and be hydrolyzed by sPLA2. The experimentally determined sPLA2 activities toward the different phospholipids vary significantly and strongly depend on the type of substitution at the sn-1 position. These results were further supported by computer simulations that provide further insight into substrate recognition on a molecular level. Our results generally indicate that, independent of the polarity of the group substituted at the sn-1 position, limited space is available in the binding cleft of sPLA2. We could identify two essential contributions that may explain (in part) the difference in the experimentally observed sPLA2 activities against the synthesized substrates. First, specific lipid-protein interactions impair the catalytic reaction; and second, steric hindrance interferes with an incoming water molecule that acts as a nucleophile in the enzymatic reaction.
METHODS
Preparation of vesicles
Phospholipids were hydrated in an aqueous buffer (150 mM KCl, 10 mM HEPES, 30 μM CaCl2, 10 μM EDTA, pH = 7.5). MLVs formed spontaneously. To ensure complete hydration, the lipids were hydrated for 1 h at 65°C. During the hydration, the lipids were vortexed every 15 min. When small unilamellar vesicles (SUVs) were needed, the MLVs were sonicated for 1 h at 5°C above the lipid main-phase transition temperature. The total concentration of lipids in the buffer solutions was 2 mM.
Differential scanning calorimetry (DSC) and dynamic light scattering (DLS)
Differential scanning calorimetry (DSC) of 2 mM MLVs was performed by using a Microcal MC-2 (Northhampton, MA) ultrasensitive power compensating scanning calorimeter equipped with a nanovoltmeter. The scans were performed in the upscan mode at a scan rate of 10°C/h. An appropriate baseline was subtracted from the calorimeter curves to afford the melting enthalpies of the MLVs. Dynamic light scattering of the SUVs was performed on a Zetasizer Nano particle analyzer (ZS ZEN3600, Malvern Instruments, Westborough, MA).
Activity measurements
The conditions used to perform the sPLA2 activity measurements were as follows: 0.15 mM phospholipid as SUVs, 150 nM sPLA2, 0.15 M KCl, 30 μM CaCl2, 10 μM EDTA, 10 μM HEPES (pH 7.5). The catalytic reaction was initiated by addition of 8.9 μL of a 42-μM Agkistrodon piscivorus piscivorus snake venom sPLA2 stock solution to a 2.5-mL SUV suspension thermostated at the main phase-transition temperature of the vesicles before the addition of the enzyme. The time-dependent activity of sPLA2 was monitored from the changes in the 90° static light scattering giving information of changes in the lipid morphology as non-bilayer-forming lysophopholipids and fatty acids are generated. High-performance liquid chromatography (HPLC) quantification of the products from the enzymatic reaction was performed with a 5-μm diol column. Two different mobile phases (hereafter referred to as mobile phase A and mobile phase B) were used. Mobile phase A was a chloroform/methanol/25% ammonium hydroxide (800:195:5) solution, whereas mobile phase B was a chloroform/methanol/water/25% ammonium hydroxide (600:340:50:5) solution. The following gradients were used at a flow rate of 1 mL/min: 0–14 min, 100% A to 100% B using a linear gradient; 14–25 min, 100% B; 25–30 min, 100% B to 100% A using a linear gradient; 30–45 min, 100% A to regenerate. An evaporative light scattering detector was used for detection. Samples were collected at different time intervals by collecting 100 μL of the reaction mixture and rapidly mixing with a 0.5-mL chloroform/methanol/acetic acid (2:4:1) solution to quench the reaction. The solution was washed with 0.5 mL of water, and 50 μL of the organic phase was used for HPLC. Fluorescence measurements were performed using a DMX-1100 spectrofluorometer (SLM, Urbana, IL). Purified snake venom PLA2 was a generous gift from Dr. R. L. Biltonen (University of Virginia, Charlottesville, VA).
Molecular dynamics simulations
The crystal structures of European honeybee (Apis Mellifera) venoms sPLA2 complexed with the transition-state analog, 1-O-octyl-2-heptylphosphonyl-sn-glycero-3-phosphoethanolamine (diC8(2Ph)PE), resolved to 2.0 Å (15,52) and human PLA2-IIA complexed with 6-phenyl-4(R)-(7-phenyl-heptanoylamino)-hexanoic acid and resolved to 2.1 Å (53) were obtained from the Protein Data Bank (entry codes 1poc and 1kqu, respectively) (54).
The initial modeling step involved placing diC8(2Ph)PE into the binding cleft of hPLA2-IIA, which was done by 1), deleting the inhibitor in 1kqu (keeping the two calcium ions and water molecules in the structure); 2), deleting calcium ions and water molecules in 1poc; 3), aligning 1poc with 1kqu; and, finally, 4) deleting the European honeybee venom PLA2 structure. The structures for the different phospholipids (Fig. 1) were built from diC8(2Ph)PE using SPARTAN Version 1.0.2 (Wavefunction, Irvine, CA). The modified lipids were introduced in the protein structures by aligning the modified phospholipids with diC8(2Ph)PE in the enzyme-diC8(2Ph)PE structures and subsequently deleting diC8(2Ph)PE. Missing distance, angle, and torsion parameters for the sn-1 side chains were obtained from the CHARMM27 parameter set (55) describing similar atom types. The structures were solvated using the program SOLVATE (56). Eighteen water molecules were randomly replaced with chloride ions to neutralize the systems. The final systems contained ∼4900 water molecules, and the simulation cell dimensions were 52.7 × 51.7 × 67.3 Å3. For the molecular dynamics (MD) simulations, the software NAMD (57) was used with the Charmm27 all-hydrogens parameter set and with the TIP3 water model (55). Each complex was simulated at least three times starting from different initial conditions, which were obtained by different steps of energy minimization. The first energy minimization of the systems involved 1000 steps. For each subsequent simulation of a certain complex, the number of steps for energy minimization was increased by 250; that is, 1000, 1250, and 1500 steps were used for simulations of a complex that was repeated three times. The minimization procedure was followed by 100 ps of heating of the systems to 300 K. Each simulation was carried out for 10 ns in the NPT ensemble. A time step of 1 fs was used throughout all simulations. An isotropic constant ambient pressure of 1 atm was imposed using the Langevin piston method (58) with a damping coefficient of 5 ps−1, a piston period of 200 fs, and a decay of 500 fs. The particle mesh Ewald method was used for computation of the electrostatic forces (59,60). The grid spacing applied was ∼1.0 Å, and a fourth-order spline was used for the interpolation. The long-range part of the electrostatic forces was evaluated every fourth femtosecond. The van der Waals interactions were cut off at 12 Å using a switching function starting at 10 Å. Periodic boundary conditions were applied in x, y, and z directions. Examinations of the molecular structures and analyses of the trajectories were carried out using tcl-scripting within the graphical program Visualization Molecular Dynamics (61).
FIGURE 1.

Chemical structure of compounds A–H.
RESULTS AND DISCUSSION
The ability of secretory phospholipase A2 to hydrolyze a broad range of phospholipid analogs has been explored in developing new drug delivery systems (48,50). We previously demonstrated that sPLA2 can hydrolyze phospholipids with their headgroup in the sn-2 position and phospholipids with small substitutions in the sn-1 position (51,62). It is intriguing that sPLA2 is able to hydrolyze phospholipids with substitutions in the sn-1 position, since there is only limited space available in the binding cleft of sPLA2. To understand the structural restrictions for substitutions in the sn-1 position at a molecular level, we present results that are based on biophysical characterization of phospholipids and computer simulation of sPLA2-phospholipid analog complexes. The biophysical characterizations were performed to ensure that the phospholipid analogs formed vesicles and were hydrolyzed by sPLA2, whereas computer simulations were carried out to provide structural insight on an atomic level. We studied phospholipid analogs with sn-1 substitutes having different properties, i.e., the side chains have different size (bulkiness) and polarity. The chemical structures of the synthesized compounds are shown in Fig. 1, and the phospholipids contain hydroxy (compound A) methoxy (compound B), benzyloxy (compound C), allyl (compounds D and E), and ester (compounds F–H) groups. All compounds except compound B were synthesized, and all compounds were investigated in the computational study. The results obtained from simulations of sPLA2 in complex with its natural substrate (63) (hereafter referred to as “native”) are also included in the following discussion as a reference to the phospholipid analogs.
Differential scanning calorimetry
DSC scans were performed to verify that the phospholipid analogs formed MLVs and to ensure that the vesicles used in the activity measurement assay were at the main phase transition. All tested compounds showed a single peak in the thermograph with a main phase transition above 35°C. However, T1/2, which is a measure of the cooperativity of the phase transition, varies between the compounds (Table 1). For compounds C and E, T1/2 < 1°C, whereas for compounds A, D, and F–H, T1/2 > 1°C, with relatively broad tails indicating that this series of compounds may not form structural homogeneous MLVs. In particular, compounds A (T1/2 ≈ 3.7°C) and F (T1/2 ≈ 3.0°C) have relatively broad main phase-transition peaks, where the phase-transition enthalpy (ΔH) for compound F is approximately half the value observed for compounds C and E. These compounds have comparable ΔH (9.3–11.6 kJ/mole) and the same Tm (43.9°C) (Table 1). Larger variations are observed for the remaining phospholipid analogs. DSC of the SUVs showed the usually observed broadening of the main phase transition when MLVs are transformed into SUVs (64) (Fig. 2 D). Dynamic light scattering measurements of the different SUVs showed that the SUVs had an average diameter of 40–70 nm.
TABLE 1.
Differential calorimetry data of MLVs in the main phase transition for compounds A and C–H
| Compound | ΔH (kJ/mole) | Tm (°C) | T½ (°C) |
|---|---|---|---|
| A | 7.3 | 57.0 | 3.65 |
| C | 11.6 | 43.9 | 0.43 |
| D | 10.3 | 46.9 | 1.82 |
| E | 9.3 | 43.9 | 0.48 |
| F | 4.6 | 48.2 | 2.99 |
| G | 8.6 | 39.0 | 1.52 |
| H | 7.6 | 48.9 | 1.84 |
ΔH, melting enthalpy; T½, half-width; Tm, peak position.
FIGURE 2.

The typical appearance of a fluorescence measurement on (A) SUVs composed of phospholipids that are fairly good substrates for sPLA2, and (B) SUVs composed of phospholipids that are poor substrates for sPLA2. The upper lines show the intrinsic fluorescence from the enzyme, whereas the lower lines show the morphology changes of the systems. (C) Typical HPLC chromatogram illustrating the effect of sPLA2-catalyzed hydrolysis of a phospholipid from SUVs that is a fairly good substrate. The chromatograms show the amount of phospholipid before the addition of sPLA2 (solid line) and 1000 s (dashed line) and 6000 s (dotted line) after the addition of sPLA2. (D) Typical DSC scans for MLVs and SUVs. Scans are representative of all phospholipids studied.
Activity measurements
We used 90° light scattering to qualitatively determine sPLA2 activity toward the different compounds. This approach is only an indirect indication for sPLA2 activity, since it detects the morphology changes of the liposomes. These changes cannot be induced by the adsorption of the enzyme to the liposome surface, since the concentration of sPLA2 is significantly lower than the concentration of the liposomes. Hence, observed fluorescence changes are induced by hydrolysis. In this assay, SUVs consisting of compounds C, E, and G showed no effect upon sPLA2 addition, whereas morphology changes were detected for SUVs composed of compounds A, D, F, and H (Table 2). Fig. 2 A shows a typical appearance of a fluorescence measurement that is representative for SUVs composed of compounds A, D, F, or H (sPLA2 was added at 500 s to secure full equilibration of the systems). The lower curve monitoring the morphology changes of the system shows a period without any changes in the system followed by a sudden increase (“burst”) in fluorescence, indicating the breakdown of the SUVs due to sPLA2 hydrolysis.
TABLE 2.
Overview of the results obtained from fluorescence measurements and HPLC studies for compounds A and C–H
| Compound | Morphology changes observed by fluorescence | Activity observed by HPLC |
|---|---|---|
| A | + | + |
| C | − | − |
| D | + | + |
| E | − | − |
| F | + | + |
| G | − | − |
| H | + | + |
The intrinsic fluorescence from tryptophan in the enzyme shows a sudden sharp increase after addition of sPLA2 (Fig. 2 A, upper curve), which might be a result of sPLA2 immediately attaching to the surface of the negatively charged vesicles. During the experiment, the intrinsic fluorescence decreases, which is expected, since sPLA2 hydrolysis leads to the breakdown of SUVs, and consequently results in small aggregates to which sPLA2 binds less efficiently. Fig. 2 B shows a typical appearance of the fluorescence measurements representative for SUVs composed of compounds C, E, or G. No morphology changes of the systems (lower curve) are observed, indicating that these compounds are poor substrates for sPLA2. Furthermore, the intrinsic fluorescence from tryptophan of the enzyme (upper curve) shows no significant change after the addition of the enzyme. Again, the initial sharp increase might be a sign of instantaneous sPLA2 attachment to the surface of the SUVs. The fluorescence measurements were followed by HPLC studies to demonstrate the link between morphology changes and sPLA2 activity. Fig. 2 C shows an example of a typical HPLC measurement on systems in which fluorescence measurements indicate morphology changes. A substantial decrease in the amount of phospholipid in the sample before addition of sPLA2 (solid line) and 1000 s (dashed line) and 6000 s (dotted line) after addition of the enzyme is observed. Clearly, HPLC results confirm that the indirectly observed morphology changes are induced by sPLA2 hydrolysis of the phospholipids in the SUVs. The results obtained from the fluorescence measurements and the HPLC studies on compounds A and C–H are summarized in Table 2.
MD simulations
The biophysical characterization revealed that although the tested phospholipids can form MLVs, only some of the phospholipid analogs are relatively good substrates for sPLA2. To understand, on a structural level, the observed different sPLA2 activity toward the phospholipids, we applied computational methods. Initially, we applied simple docking of the phospholipid analogs to sPLA2. These results were not conclusive, and they indicated that all phospholipids fit into the binding cleft of sPLA2. We therefore applied MD simulations (65,66) to gain further insight into the mechanism(s) that could explain the experimentally observed activities. Enzymatic reaction depends on several factors, including binding of the enzyme to the membrane surface, membrane properties, formation of the Michaelis-Menten complex, etc. (18,66,67). Here, we only consider one step in the process, and have confined ourselves to a study, using classical mechanics, of the dynamics of substrate binding before the formation of the transition state. Our approach was motivated by the work of, e.g., Cho and co-workers (68) and Menger (69), who have challenged the classical hypothesis (70) that enzyme catalysis can only be improved by further stabilizing the transition state. The authors demonstrated that it may be at least as advantageous to consider ways of engineering the substrate, which could improve substrate binding, and/or product clearance (removal of product inhibition effects) (68–74).
It is well established that membrane properties affect enzyme activity (19), and that binding of some sPLA2 species to the membrane surface is governed by the interfacial charge distribution of the membrane (19,20,63). For instance, snake venom sPLA2 can hydrolyze both membranes consisting of zwitterionic phosphatidylcholine and those consisting of negatively charged phosphatidylglycerol (PG) phospholipids, whereas human sPLA2-IIA shows only activity toward membranes containing PG (1,16,19,24,27,29). This has been correlated to the binding surface of the enzyme. sPLA2-IIA has a relatively high positively charged binding surface (i-face) that makes critical contacts to negatively charged phospholipids (23,75). However, once bound to the surface, sPLA2-IIA can hydrolyze phosphocholine and PG phospholipids (20), indicating a more generic catalytic mechanism, which is not strongly dependent on the headgroup. Hence, it is expected that the results obtained in this study are relevant for snake venom sPLA2 as well as for human sPLA2-IIA. The series of compounds used in this study is only modified in the sn-1 position of PG, which means that measuring no activity cannot be due to the lack of binding of sPLA2 to the membrane surface, but has to be related to the catalytic reaction.
The catalytic mechanism of sPLA2 is schematically shown in Fig. 3, where the numbering corresponds to the numbering used for sPLA2-IIA. The catalytic site consists essentially of four components: a histidine (His47), an aspartate (Asp91), a calcium ion (cofactor), and a water molecule that acts as the nucleophile (15,76). The catalytic reaction is initiated by forming a Michaelis-Menten complex between the substrate and sPLA2. The complex is stabilized by Ca2+ that coordinates to the substrate via the carbonyl oxygen (O22) and phosphonate oxygen (O4), as well as to the protein via the backbone oxygen of Gly29 (O) and the two carboxylate oxygens of Asp48 (OD1 and OD2) (Fig. 4). Additionally, Lys62, which is highly conserved in the group II sPLA25, interacts with the sn-3 phosphate oxygen (1,77), thereby contributing to the stabilization of the complex (1,77). The reactive carbonyl carbon atom of the ester bond (Fig. 3, C21) is then attacked by a nucleophilic water molecule, leading to the formation of a tetrahedral intermediate (close to the transition state), which is stabilized by the backbone hydrogen of the NH group of Gly29 (oxyanion hole) and the calcium ion (15,76,78). During the formation of the intermediate phase, the hydrogen atom of the water molecule is transferred to His47, and thereby, the histidine imidazole ring becomes positively charged. The protonated His47 is stabilized by Asp91, which hydrogen-bonds to Tyr51 and Tyr66 (Fig. 3). The proton abstracted from His47 is ideally positioned to protonate the sn-2 oxygen in concert with the productive collapse of the tetrahedral intermediate (3,15,79). Molecular dynamics simulation is a classical approach that cannot account for transition state or processes involving bond breaking (66,71). We therefore focused on the Michaelis-Menten complex of the different compounds. One of the requirements for catalysis is that a stable complex can be formed and that the key distances (involving the Michaelis-Menten complex) are maintained. We have used these criteria to study the stability of the different complexes and to investigate whether that stability can be correlated to the observed sPLA2 activity toward the different compounds.
FIGURE 3.

Schematic representation of the catalytic mechanism of sPLA2 and key interactions that stabilize the Michaelis-Menten complex. Atom types given in parentheses refer to the Protein Data Bank nomenclature.
FIGURE 4.

RMSDs of Cα atoms with respect to the initial sPLA2 structure as a function of simulation time. (Inset) Comparison of the average residue fluctuations based on Cα atoms and averaged over the last 5 ns of the simulation (—) with the RMSD derived from the crystallograhic Bfactor: 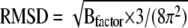 (·–·) (53,89). The data were extracted from a simulation of sPLA2 in complex with compound D and are representative for all simulations.
(·–·) (53,89). The data were extracted from a simulation of sPLA2 in complex with compound D and are representative for all simulations.
MD—protein stability
Simulations were performed at least three times for each complex, to verify the significance of the results (Table 3). For each simulation, the root mean-square deviation (RMSD) of Cα atoms with respect to the protein structure after minimization was calculated as a function of time to check the stability of the simulation. In Fig. 4, the time evolution of RMSD extracted from an sPLA2-compound D simulation is shown as an example. RMSD approaches a constant value of ∼1.7 Å within 3 ns. Similar results were obtained for the other simulations, and the average RMSDs calculated from the different simulations are summarized in Table 3. Variations are within the statistical uncertainties and the overall RMSD averaged over all RMSDs is 1.5 ± 0.1 Å. We also compared the fluctuations of Cα atoms with the crystallographically determined B-factors (53) (Fig. 4, inset). The fluctuations extracted from simulations resemble the B-factor, but have larger magnitudes in loop regions that are exposed to solvent. These deviations are not surprising, since these regions may be affected by crystal packing (71,80).
TABLE 3.
Summary of the simulations
| Compound | Number of simulations | Length of simulations (ns) | Root mean-square deviation mean ± SD (Å) |
|---|---|---|---|
| A | 3 | each 10 | 1.5 ± 0.1 |
| B | 3 | each 10 | 1.4 ± 0.1 |
| C | 3 | each 10 | 1.4 ± 0.1 |
| D | 3 | each 10 | 1.7 ± 0.1 |
| E | 4 | each 10 | 1.3 ± 0.1 |
| F | 3 | each 10 | 1.2 ± 0.1 |
| G | 4 | each 10 | 1.5 ± 0.1 |
| H | 3 | each 10 | 1.4 ± 0.1 |
| native | 6 | each 10 | 1.3 ± 0.1 |
The last column lists the average RMSD of Cα atoms with respect to the protein structure after minimization. Means and their standard deviations (SD) are based on a series of simulations of a particular complex:  “Native” refers to the natural substrate of sPLA2 ((R)-1,2-dipalmitoyl-glycero-3-phosphocholine) (63).
“Native” refers to the natural substrate of sPLA2 ((R)-1,2-dipalmitoyl-glycero-3-phosphocholine) (63).
MD—key distances
For the catalytic reaction to occur, a stable Michaelis-Menten complex has to form, which implies that distances stabilizing the complex are maintained (65,81). We therefore monitored distances involving the cofactor calcium (Ca2+ to Asp48(OD1), Asp48(OD2), Gly29(O), S(O22), S(O4) (Fig. 5, where atom types and S refer to the Protein Data Bank nomenclature and substrate, respectively) or residues in the vicinity of the substrate (Asp91(OD1)-Tyr51(OH), Asp91(OD2)-Tyr66(OH), His47(ND1)-S(C21), His47(HE2)-Asp91(OD1), and Gly29(O)-S(O22) (Fig. 6)). The data were extracted from an sPLA2-compound D simulation, and similar results were observed in all other simulations. To compare those, we have calculated mean distances for each complex averaged over time (the last 5 ns of the simulations) and over simulations of that complex. The average distances are listed in Table 4. For reference, the results for the sPLA2-native simulations are also included in the last column of Table 4. Distances extracted from simulations of different sPLA2-phospholipid complexes are within the statistical uncertainties and compare well to distances determined from the sPLA2-native simulations. Exceptions are particularly observed for distances Asp91–Tyr51 and Asp91–Tyr66, which are ∼1.2 Å shorter than those monitored in the sPLA2-native complex, indicating structural effects caused by the sn-1 substitute. The two tyrosines are not required to maintain activity, since mutants in which these two residues were mutated to phenylalanine had virtually unaltered catalytic performance (78). For the single mutant Asp→Asn, significant catalytic activity was retained. Both observations suggest that the hydrogen-bonding network plays only a minor role for the catalytic turnover. Variations in distance are also observed for His47(ND1)-S(C21) and S(O22)-Gly29(HN) in the different complexes. However, as shown in Fig. 7, these variations are within the statistical uncertainty when compared to the native phospholipid, and hence, we cannot attribute the lack of sPLA2 activity toward some of the phospholipid analogs to these variations.
FIGURE 5.

Time evolution of selected distances involving the cofactor calcium. Distances are given in the individual panels (A–D). S, substrate; atom types correspond to Protein Data Bank nomenclature (see also Fig. 4). The distances were extracted from a simulation of sPLA2 in complex with compound D and are representative for all simulations.
FIGURE 6.

Time evolution of selected distances between atoms that are close to the substrate. Distances are given in the individual panels. See Fig. 5 legend for more details.
TABLE 4.
Summary of the calculated distances between selected atoms that are involved in stabilization of the substrate or part of the catalytic device
| Compound | A | B | C | D | E | F | G | H | Native |
|---|---|---|---|---|---|---|---|---|---|
| Distances | Mean ± SD (Å) | Mean ± SD (Å) | Mean ± SD (Å) | Mean ± SD (Å) | Mean ± SD (Å) | Mean ± SD (Å) | Mean ± SD (Å) | Mean ± SD (Å) | Mean ± SD (Å) |
| D91(OD1)–Y51(OH) | 1.7 ± 0.2 | 1.7 ± 0.1 | 1.8 ± 0.2 | 1.8 ± 0.2 | 1.7 ± 0.1 | 1.7 ± 0.1 | 1.7 ± 0.1 | 1.7 ± 0.1 | 3.1 ± 0.3 |
| D91(OD2)–Y66(OH) | 1.7 ± 0.1 | 1.7 ± 0.1 | 1.7 ± 0.2 | 1.7 ± 0.1 | 1.7 ± 0.1 | 1.7 ± 0.1 | 1.7 ± 0.1 | 1.7 ± 0.1 | 2.7 ± 0.1 |
| H47(HE2)–D91(OD1) | 1.9 ± 0.2 | 1.9 ± 0.1 | 1.8 ± 0.2 | 1.8 ± 0.1 | 1.8 ± 0.2 | 1.9 ± 0.2 | 1.9 ± 0.2 | 1.9 ± 0.3 | 1.9 ± 0.3 |
| H47(ND1)–S(C21) | 5.2 ± 0.4 | 4.4 ± 0.3 | 4.5 ± 0.4 | 4.2 ± 0.3 | 4.8 ± 0.2 | 4.1 ± 0.4 | 4.8 ± 0.2 | 4.8 ± 0.4 | 4.5 ± 0.4 |
| Ca2+–D48(OD1) | 2.2 ± 0.1 | 2.2 ± 0.1 | 2.2 ± 0.1 | 2.2 ± 0.1 | 2.3 ± 0.3 | 2.2 ± 0.1 | 2.2 ± 0.1 | 2.2 ± 0.1 | 2.2 ± 0.2 |
| Ca2+–D48(OD2) | 2.4 ± 0.3 | 2.3 ± 0.2 | 2.3 ± 0.1 | 2.2 ± 0.1 | 2.5 ± 0.3 | 2.2 ± 0.1 | 2.2 ± 0.1 | 2.2 ± 0.1 | 2.2 ± 0.1 |
| Ca2+–G29(O) | 2.3 ± 0.1 | 2.3 ± 0.1 | 2.3 ± 0.1 | 2.4 ± 0.1 | 2.9 ± 0.3 | 2.3 ± 0.1 | 2.3 ± 0.1 | 2.3 ± 0.1 | 2.4 ± 0.2 |
| S(O22)–G29(HN) | 3.8 ± 0.9 | 3.0 ± 0.5 | 3.0 ± 0.5 | 3.6 ± 0.8 | 3.4 ± 0.7 | 2.6 ± 0.4 | 3.1 ± 0.5 | 2.6 ± 0.4 | 2.7 ± 0.5 |
| S(O22)–Ca2+ | 2.9 ± 0.5 | 2.4 ± 0.2 | 2.3 ± 0.2 | 2.3 ± 0.1 | 2.3 ± 0.1 | 2.4 ± 0.1 | 2.3 ± 0.1 | 2.3 ± 0.1 | 2.4 ± 0.2 |
| S(O4)–Ca2+ | 2.1 ± 0.1 | 2.1 ± 0.1 | 2.1 ± 0.1 | 2.1 ± 0.1 | 2.1 ± 0.1 | 2.1 ± 0.1 | 2.1 ± 0.1 | 2.1 ± 0.1 | 2.1 ± 0.1 |
FIGURE 7.

Average distances of His47(ND1)-S(C21) and S(O22)-Gly29(HN) extracted from the simulations of the different sPLA2-phospholipid complexes. Averages were calculated from a series of simulations of a particular complex, and only the last 5 ns of each simulation was used in the calculations. Dashed and dotted lines indicate the upper and lower error, based on the native substrate for His47(ND1)-S(C21) and S(O22)-Gly29(HN)), respectively. See Fig. 5 legend for more details.
MD—nucleophilic water molecule
We previously showed that for phospholipid analogs with their headgroup located in the sn-2 position, the enzymatic reaction is affected by the probability that a water molecule can reach the catalytic site (63). We speculated that a similar effect could be the reason for our observations in this study. We therefore monitored the movements of water molecules and registered when a water molecule reached the vicinity of His47(ND1) and S(C21) (Fig. 3) (63). These two residues will hereafter be referred to as H-S. An example of a water molecule trajectory monitored in an sPLA2-native simulation is shown in Fig. 8 A. Fig. 8 B displays images taken from the trajectory at different time intervals (t1 < t2 < t3) and shows the entering of a water molecule into the H-S region. From the data, we calculated the probability of water molecules being at a certain distance from H-S; the average relative water counts extracted from the simulations are shown in Fig. 9, and the data are provided in Table 5. There are significant differences and the probability of observing a water molecule at a certain distance from H-S depends on the compound. Within the time interval of the simulations, water molecules within 3.5 Å of H-S could be detected for compounds A, D, E, G, and the natural substrate. Water molecules within 4.5 Å of H-S could be observed for compounds B, F, and H, whereas no water molecules could be observed for compound C within this range. These findings relate partly to the observed sPLA2 activity, where we found that compounds A, D, F, and H serve as fairly good substrates for sPLA2. Our simulation results would suggest that compounds A, D, E, and G are relatively good substrates for sPLA2, whereas compounds B, F, and H are less efficiently hydrolyzed by sPLA2. Based on the relative water count, the activity can be ranked as follows: natural substrate ≈ A > G ≈ E > D > H ≈ F ≈ B. Besides compounds E and G, our findings agree well with the experimental data. To further elucidate the discrepancy observed for compounds E and G, we have followed the trajectories of water molecules and analyzed with which residues/atoms the water molecules interact as they approach H-S.
FIGURE 8.

(A) Trajectory of a water molecule monitored in an sPLA2-Native simulation. Distances are calculated with respect to the substrate S(C21) (upper) and His47(ND1) (lower). (B) Snapshots taken from the trajectory displayed in A. Images are taken at different time intervals (t1 < t2 < t3) and show the entrance of a water molecule into the H-S region. Note that, for clarity, only the water molecule that is entering the H-S region is shown.
FIGURE 9.

Relative water count extracted from the different simulations and averaged over a series of simulations of a particular complex. Before calculating the averages, the data extracted from each simulation were arbitrarily normalized by the water count at 6 Å. The numbers given in the graph belong to the third column and represent the ratio of the relative water count at 4.5 Å to the relative water count at 5.5 Å.
TABLE 5.
Relative water count extracted from the different simulations
| Compound | Relative water count at 3.5 Å (mean ± SD) | Relative water count at 4.5 Å (mean ± SD) | Relative water count at 5.5 Å (mean ± SD) |
|---|---|---|---|
| A | 0.02 ± 0.02 | 0.4 ± 0.3 | 0.6 ± 0.2 |
| B | 0 | 0.002 ± 0.003 | 0.3 ± 0.2 |
| C | 0 | 0 | 0.0002 ± 0.0005 |
| D | 0.0002 ± 0.0003 | 0.2 ± 0.3 | 0.6 ± 0.5 |
| E | 0.01 ± 0.02 | 0.2 ± 0.3 | 0.4 ± 0.3 |
| F | 0 | 0.004 ± 0.007 | 0.2 ± 0.3 |
| G | 0.1 ± 0.1 | 0.2 ± 0.3 | 0.3 ± 0.3 |
| H | 0 | 0.01 ± 0.02 | 0.3 ± 0.4 |
| Native | 0.1 ± 0.1 | 0.4 ± 0.2 | 0.7 ± 0.1 |
Before calculating the averages, the data were arbitrarily normalized by the water count at 6 Å, since there were significant differences in the water count within a series of simulations of a particular complex. See Table 3 note for more details.
MD—key structural interference
Despite the relatively high water count for compounds E and G, these phospholipid analogs are not hydrolyzed by sPLA2. Further analysis of the water trajectories provided additional information about the effect of the sn-1 side chain on the hydrolysis. The simulation revealed that the two methyl groups located at the first carbon of the sn-1 side chain cause steric hindrance such that water molecules cannot freely reach the H-S site. Fig. 10 displays snapshots of the active site of sPLA2 taken from sPLA2-compound D, sPLA2-compound E, and sPLA2-compound G simulations. The image taken from the sPLA2-compound D simulation is included as a reference. This compound is a relatively good substrate for sPLA2 and, as shown in Fig. 10, a water molecule can freely approach the H-S region. In contrast, for compounds E and G (Fig. 10, configuration 1), a water molecule can only be detected in the vicinity of His47 and cannot reach the H-S region. A methyl group located at the first carbon of the sn-1 chain blocks the water molecule from approaching the correct position (i.e., H-S region) such that it can act as the nucleophile in the enzymatic reaction. Furthermore, His47 cannot perfectly align in the dyad, due to the bulkiness of the methyl group. For compound G, a second configuration is observed where the carbonyl oxygen of the ester group coordinates to a water molecule that has entered the H-S region (Fig. 10, configuration 2). Here, strong coordination of the water molecule by the ester group prevents activation of the water molecule for the nucleophilic attack.
FIGURE 10.

Snapshots of the active site of sPLA2 taken from sPLA2-compound D, sPLA2-compound E, and sPLA2-compound G simulations. Secondary protein structure of the enzyme is shown in the ribbon mode. His48, the substrate, and a water molecule are shown in the liquorice and are colored according to atom type (H, white; N, blue; O, red; and P, gold). The cofactor calcium is shown in van der Waals and is colored blue.
CONCLUSION
We used biophysical techniques and MD simulations to understand, on a molecular level, the structural restrictions for substitutions in the sn-1 position. We studied phospholipid analogs with sn-1 substitutes of different properties, where size (i.e., bulkiness) and polarity are varied (Fig. 1). The biophysical characterization revealed that all phospholipid analogs form MLVs, but only compounds A, D, F, and H are potential good substrates for sPLA2.
Molecular dynamics simulations provided structural insight on an atomic level. Notwithstanding the clearly demonstrated importance of transition state studies (82–88), we suggest that the steps before the transition state can also critically determine the outcome of the reaction. Our simulations revealed that the difference observed in sPLA2 activity is caused by less efficient access of water molecules to the active site. We monitored the number of water molecules that enter the region between His47(ND1) and S(C21) (Fig. 3) for the different sPLA2-phospholipid complexes and found that the probability of a water molecule reaching the correct position such that hydrolysis can occur varies among the different phospholipid analogs. Based on the relative water count, the activity can be ranked as follows: natural substrate > A > D > H ≈ F. In agreement with experimental results, sPLA2 shows no activity toward compounds E and G, since the methyl group located at the first carbon of the sn-1 substitute (compound E) or the ester group (compound G) blocks water molecules from reaching the right position that would allow hydrolysis. The future plan for this work is to use the MD simulations to predict the ability of sPLA2 to hydrolyze a given substrate with more complicated residues in the sn-1 position. Consequently, our approach may be used in the design and development of new phospholipids with anticancer drugs linked to the sn-1 position.
Acknowledgments
Simulations were performed at the Danish Center for Scientific Computing at the University of Southern Denmark.
G.H.P. acknowledges financial support from the Danish National Research Foundation via a grant to the MEMPHYS-Center for Biomembrane Physics. L.L. is supported via a scholarship from the Technical University of Denmark. R.M. thanks the Lundbeck Foundation for financial support. The Center for Sustainable and Green Chemistry is supported by the Danish National Research Foundation.
Editor: Peter Tieleman.
References
- 1.Han, S. K., E. T. Yoon, D. L. Scott, P. B. Sigler, and W. Cho. 1997. Structural aspects of interfacial adsorption. A crystallographic and site-directed mutagenesis study of the phospholipase A2 from the venom of Agkistrodon piscivorus piscivorus. J. Biol. Chem. 272:3573–3582. [PubMed] [Google Scholar]
- 2.Mihelich, E. D., and R. W. Schevitz. 1999. Structure-based design of a new class of anti-inflammatory drugs: secretory phospholipase A2 inhibitors, SPI. Biochim. Biophys. Acta. 1441:223–228. [DOI] [PubMed] [Google Scholar]
- 3.Scott, D. L., and P. B. Sigler. 1994. Structure and catalytic mechanism of secretory phospholipase A2. Adv. Protein Chem. 45:53–88. [DOI] [PubMed] [Google Scholar]
- 4.Mayer, R. J., and L. A. Marshall. 1993. New insights on mammalian phospholipase A2(s); comparison of arachidonoyl-selective and -nonselective enzymes. FASEB J. 7:339–348. [DOI] [PubMed] [Google Scholar]
- 5.Kudo, I., M. Murakami, S. Hara, and K. Inoue. 1993. Mammalian non-pancreatic phospholipase A2. Biochim. Biophys. Acta. 1170:217–231. [DOI] [PubMed] [Google Scholar]
- 6.Gelb, M. H., M. K. Jain, A. M. Hanel, and O. G. Berg. 1995. Interfacial enzymology of glycerolipid hydrolases: lessons from secreted phospholipase A2. Annu. Rev. Biochem. 64:653–688. [DOI] [PubMed] [Google Scholar]
- 7.Dennis, E. A. 1994. Diversity of group types, regulation and function of phospholipase A2. J. Biol. Chem. 269:13057–13060. [PubMed] [Google Scholar]
- 8.Six, D. A., and E. A. Dennis. 2000. The expanding superfamily of phospholipase A2 enzymes: classification and characterization. Biochim. Biophys. Acta. 1488:1–19. [DOI] [PubMed] [Google Scholar]
- 9.Laye, J., and J. H. Gill. 2003. Phospholipase A2 expression in tumors: a target for therapeutic intervention? Drug Discov. Today. 8:710–716. [DOI] [PubMed] [Google Scholar]
- 10.Heinrikson, R. L., E. T. Krueger, and P. S. Keim. 1977. Amino acid sequence of phospholipase A2-alpha from the venom of Crotalus adamanteus. A new classification of phospholipases A2 based upon structural determinants. J. Biol. Chem. 252:4913–4921. [PubMed] [Google Scholar]
- 11.Davidson, F. F., and E. A. Dennis. 1990. Evolutionary relationships and implications for the regulation of phospholipase A2 from snake venom to human secreted forms. J. Mol. Evol. 31:228–238. [DOI] [PubMed] [Google Scholar]
- 12.Valentin, E., and G. Lambeau. 2000. Increasing molecular diversity of secreted phospholipase A2 and their receptors and binding proteins. Biochim. Biophys. Acta. 1488:59–70. [DOI] [PubMed] [Google Scholar]
- 13.Berg, O. G., M. H. Gelb, M.-D. Tsai, and M. K. Jain. 2001. Interfacial enzymology: The secreted phospholipase A2-paradigm. Chem. Rev. 101:2613–2653. [DOI] [PubMed] [Google Scholar]
- 14.Tatulian, S. A., R. L. Biltonen, and L. K. Tamm. 1997. Structural changes in a secretory phospholipase A2 induced by membrane binding: a clue to interfacial activation? J. Mol. Biol. 268:809–815. [DOI] [PubMed] [Google Scholar]
- 15.Scott, D. L., S. White, Z. Otwinowski, W. Yuan, M. H. Gelb, and P. B. Sigler. 1990. Interfacial catalysis: the mechanism of phospholipase A2. Science. 250:1541–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scott, D. L., A. M. Mandel, P. B. Sigler, and B. Honig. 1994. The electrostatic basis for the interfacial binding of secretory phosholipase A2. Biophys. J. 67:493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jain, M. K., G. Ranadive, B.-Z. Yu, and H. M. Verheij. 1991. Interfacial catalysis by phospholipase A2: monomeric enzyme is fully catalytically active at the bilayer interface. Biochemistry. 30:7330–7340. [DOI] [PubMed] [Google Scholar]
- 18.Peters, G. H., U. Dahmen-Levison, K. de Meijere, G. Brezesinski, S. Toxvaerd, A. Svendsen, and P. K. J. Kinnunen. 2000. Influence of surface properties of mix-monolayers on lipolytic hydrolysis. Langmuir. 16:2779–2788. [Google Scholar]
- 19.Mouritsen, O. G., T. L. Andresen, A. Halperin, P. L. Hansen, A. F. Jakobsen, U. B. Jensen, M. Ø. Jensen, K. Jørgensen, T. Kaasgaard, C. Leidy, A. C. Simonsen, G. H. Peters, and M. Weiss. 2006. Activation of interfacial enzymes at membrane surfaces. J. Phys. Condens. Matter. 18:1–12. [DOI] [PubMed] [Google Scholar]
- 20.Leidy, C., L. Linderoth, T. L. Andresen, O. G. Mouritsen, K. Jørgensen, and G. H. Peters. 2006. Domain-induced activation of human phospholipase A2 type IIA: local versus global lipid composition. Biophys. J. 90:3165–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singer, A. G., F. Ghomashchi, C. Le Calvez, J. Bollinger, S. Bezzine, M. Rouault, M. Sadilek, E. Nguyen, M. Lazdunski, G. Lambeau, and M. H. Gelb. 2002. Interfacial kinetic and binding properties of the complete set of human and mouse groups I, II, V, X, and XII secreted phospholipase A2. J. Biol. Chem. 277:48535–48549. [DOI] [PubMed] [Google Scholar]
- 22.Pan, Y. H., B.-Z. Yu, A. G. Singer, F. Ghomashchi, G. Lambeau, M. H. Gelb, M. K. Jain, and B. J. Bahnson. 2002. Crystal structure of human group X secreted phospholipase A2. J. Biol. Chem. 277:29086–29093. [DOI] [PubMed] [Google Scholar]
- 23.Bahnson, B. J. 2005. Structure, function and interfacial allosterism in phospholipase A2: insight from anion-assisted dimer. Arch. Biochem. Biophys. 433:96–106. [DOI] [PubMed] [Google Scholar]
- 24.Bezzine, S., J. G. Bollinger, A. G. Singer, S. L. Veatch, S. L. Keller, and M. H. Gelb. 2002. On the binding preference of human groups IIA and X phospholipases A2 for membranes with anionic phospholipids. J. Biol. Chem. 277:48523–48534. [DOI] [PubMed] [Google Scholar]
- 25.Leidy, C., O. G. Mouritsen, K. Jørgensen, and G. H. Peters. 2004. Evolution of a rippled membrane during phospholipase A2 hydrolysis studied by time-resolved AFM. Biophys. J. 87:408–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buckland, A. G., and D. C. Wilton. 2000. Anionic phospholipids, interfacial binding and the regulation of cell functions. Biochim. Biophys. Acta. 1483:199–216. [DOI] [PubMed] [Google Scholar]
- 27.Canaan, S., R. Nielsen, F. Ghomashchi, B. H. Robinson, and M. H. Gelb. 2002. Unusual mode of binding of human group IIA secreted phospholipase A2 to anionic interfaces as studied by continuous wave and time domain electron paramagnetic resonance spectroscopy. J. Biol. Chem. 277:30984–30990. [DOI] [PubMed] [Google Scholar]
- 28.Buckland, A. G., E. L. Heeley, and D. C. Wilton. 2000. Bacterial cell membrane hydrolysis by secreted phospholipases A2: a major physiological role of human group IIa sPLA2 involving both bacterial cell wall penetration and interfacial catalysis. Biochim. Biophys. Acta. 1484:195–206. [DOI] [PubMed] [Google Scholar]
- 29.Yu, B.-Z., M. J. Poi, U. A. Ramagopal, R. Jain, S. Ramakumar, O. G. Berg, M. D. Tsai, K. Sekar, and M. K. Jain. 2000. Structural basis of the anionic interface preference and kcat activation of pancreatic phospholipase A2. Biochemistry. 39:12312–12323. [DOI] [PubMed] [Google Scholar]
- 30.Gadd, M. E., and R. L. Biltonen. 2000. Characterization of the interaction of phospholipase A2 with phosphatidylcholine-phosphatidylglycerol mixed lipids. Biochemistry. 39:9623–9631. [DOI] [PubMed] [Google Scholar]
- 31.Gelb, M. H., W. Cho, and D. C. Wilton. 1999. Interfacial binding of secreted phospholipase A2: more than electrostatics and a major role for tryptophan. Curr. Opin. Struct. Biol. 9:428–432. [DOI] [PubMed] [Google Scholar]
- 32.Sumandea, M., S. Das, C. Sumandea, and W. Cho. 1999. Roles of aromatic residues in high interfacial activity of Naja naja atra phospholipase A2. Biochemistry. 38:16290–16297. [DOI] [PubMed] [Google Scholar]
- 33.Baker, S. F., R. Othman, and D. C. Wilton. 1998. Tryptophan-containing mutant of human (group IIa) secreted phospholipase A2 has a dramatically increased ability to hydrolyze phosphatidylcholine vesicles and cell membranes. Biochemistry. 37:13203–13211. [DOI] [PubMed] [Google Scholar]
- 34.Balsinde, J., M. A. Balboa, P. A. Insel, and E. A. Dennis. 1999. Regulation and inhibition of phospholipase A2. Annu. Rev. Pharmacol. Toxicol. 39:175–189. [DOI] [PubMed] [Google Scholar]
- 35.Thunnissen, M. M. G. M., E. Ab, K. H. Kalk, J. Drenth, B. W. Dijkstra, O. P. Kuipers, R. Dijkman, G. H. De Haas, and H. M. Verheij. 1990. X-ray structure of phospholipase A2 complexed with a substrate-derived inhibitor. Nature. 347:689–691. [DOI] [PubMed] [Google Scholar]
- 36.De Marino, V., M. Gentile, F. Granata, G. Marone, and M. Triggiani. 1999. Secretory phospholipase A2: a putative mediator of airway inflammation. Int. Arch. Allergy Immunol. 118:200–201. [DOI] [PubMed] [Google Scholar]
- 37.Denizot, Y., V. Truffinet, S. Bouvier, A. Gainant, P. Cubertafond, and M. Mathonnet. 2004. Elevated plasma phospholipase A2 and platelet-activating factor acetylhydrolase activity in colorectal cancer. Mediators Inflamm. 13:53–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murakami, M., Y. Nakatani, G. Atsumi, K. Inoue, and I. Kudo. 1997. Regulatory functions of phospholipase A2. Crit. Rev. Immunol. 17:225–283. [DOI] [PubMed] [Google Scholar]
- 39.Fuentes, L., M. Hernandez, M. L. Nieto, and M. S. Crespo. 2002. Biological effects of group IIA secreted phospholipase A 2. FEBS Lett. 531:7–11. [DOI] [PubMed] [Google Scholar]
- 40.Kramer, R. M., C. Hession, B. Johansen, G. Hayes, P. McGray, C. E. Pingchang, R. Tizard, and P. R. Blake. 1989. Structure and properties of human non-pancreatic phospholipase A2. J. Biol. Chem. 264:5768–5775. [PubMed] [Google Scholar]
- 41.Leistad, L., A. J. Feuerherm, M. Østensen, A. Faxvaag, and B. Johansen. 2004. Presence of secretory group IIa and V phospholipase A2 and cytosolic group IVa phospholipase A2 in chondrocytes from patients with rheumatoid arthritis. Clin. Chem. Lab. Med. 42:602–610. [DOI] [PubMed] [Google Scholar]
- 42.Stadel, J. M., K. Hoyle, R. M. Naclerio, A. Roshak, and F. H. Chilton. 1994. Characterization of phospholipase A2 from human nasal lavage. Am. J. Respir. Cell Mol. Biol. 11:108–113. [DOI] [PubMed] [Google Scholar]
- 43.Chilton, F. H., F. J. Averill, W. C. Hubbard, A. N. Fonteh, M. Triggiani, and M. C. Liu. 1996. Antigen-induced generation of lyso-phospholipids in human airways. J. Exp. Med. 183:2235–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Green, J.-L., G. M. Smith, R. Buchta, R. Lee, K. Y. Ho, I. A. Rajkovic, and K. F. Scott. 1991. Circulating phospholipase A2 activity associated with sepsis and septic shock is indistinguishable from that associated with rheumatoid arthritis. Inflammation. 15:355–367. [DOI] [PubMed] [Google Scholar]
- 45.Vadas, P. 1984. Elevated plasma phospholipase A2 levels: correlation with the hemodynamic and pulmonary changes in gram-negative septic shock. J. Lab. Clin. Med. 104:873–881. [PubMed] [Google Scholar]
- 46.Jiang, J., B. L. Neubauer, J. R. Graff, M. Chedid, J. E. Thomas, N. W. Roehm, S. Zhang, G. J. Eckert, M. O. Koch, J. N. Eble, and L. Cheng. 2002. Expression of group IIA secretory phospholipase A2 is elevated in prostatic intraepithelial neoplasia and adenocarcinoma. Am. J. Pathol. 160:667–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sved, P., K. F. Scott, D. McLeod, N. J. C. King, J. Singh, T. Tsatralis, B. Nikolov, J. Boulas, L. Nallan, M. H. Gelb, M. Sajinovic, G. G. Graham, P. J. Russell, and Q. Dong. 2004. Oncogenic action of secreted phospholipase A2 in prostate cancer. Cancer Res. 64:6934–6940. [DOI] [PubMed] [Google Scholar]
- 48.Andresen, T. L., S. S. Jensen, T. Kaasgaard, and K. Jørgensen. 2005. Triggered activation and release of liposomal prodrugs and drugs in cancer tissue by secretory phospholipase A2. Curr. Drug Deliv. 2:353–362. [DOI] [PubMed] [Google Scholar]
- 49.Andresen, T. L., S. S. Jensen, and K. Jørgensen. 2005. Advanced strategies in liposomal cancer therapy: problems and prospects of active and tumor specific drug release. Prog. Lipid Res. 44:68–97. [DOI] [PubMed] [Google Scholar]
- 50.Davidsen, J., K. Jørgensen, T. L. Andresen, and O. G. Mouritsen. 2003. Secreted phospholipase A2 as a new enzymatic trigger mechanism for localised liposomal drug release and absorption in diseased tissue. Biochim. Biophys. Acta. 1609:95–101. [DOI] [PubMed] [Google Scholar]
- 51.Linderoth, L., G. H. Peters, K. Jørgensen, R. Madsen, and T. L. Andresen. 2006. Synthesis of sn-1 functionalized phospholipids as substrates for secretory phospholipase A2. Chem. Phys. Lipids. 146:54–66. [DOI] [PubMed] [Google Scholar]
- 52.Scott, D. L., Z. Otwinowski, M. H. Gelb, and P. B. Sigler. 1990. Crystal-structure of bee-venom phospholipase A2 in a complex with a transition-state analogue. Science. 250:1563–1566. [DOI] [PubMed] [Google Scholar]
- 53.Hansford, K. A., R. C. Reid, C. I. Clark, J. D. A. Tyndall, M. W. Whitehouse, T. Guthrie, R. P. McGeary, K. Schafer, J. L. Martin, and D. P. Fairlie. 2003. D-tyrosine as a chiral precursor to potent inhibitors of human non-pancreatic secretory phospholipase A2 (IIA) with anti-inflammatory activity. ChemBioChem. 4:181–185. [DOI] [PubMed] [Google Scholar]
- 54.Bernstein, F. C., T. F. Koetzle, G. J. Williams, E. E. Meyer, M. D. Brice, J. R. Rodgers, O. Kennard, T. Shimanouchi, and M. Tasumi. 1977. The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 112:535–542. [DOI] [PubMed] [Google Scholar]
- 55.Jorgensen, W. L., J. Chandrasekhar, J. D. Medura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential models for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 56.Grubmuller, H. 1996. SOLVATE. Version 1.2. Theoretical Biophysics Group, Institute for Medical Optics, Ludwig-Maximilians University, Munich, Germany.
- 57.Kale, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, and K. Schulten. 1999. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 151:283–312. [Google Scholar]
- 58.Feller, S. E., Y. Zhang, R. W. Pastor, and B. R. Brooks. 1995. Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 103:4613–4621. [Google Scholar]
- 59.Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald: an N log(N) method for Ewald sums. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 60.Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- 61.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 62.Andresen, T. L., and K. Jørgensen. 2005. Synthesis and membrane behavior of a new class of unnatural phospholipid analogs useful as phospholipase A2 degradable liposomal drug carriers. Biochim. Biophys. Acta. 1669:1–7. [DOI] [PubMed] [Google Scholar]
- 63.Peters, G. H., M. S. Møller, K. Jørgensen, P. Rönnholm, M. Mikkelsen, and T. L. Andresen. 2007. Secretory phospholipase A2 hydrolysis of phospholipid analogues is dependent on water accessibility to the active site. J. Am. Chem. Soc. 129:5451–5461. [DOI] [PubMed] [Google Scholar]
- 64.Heimburg, T. 1998. Mechnical aspects of membrane thermodynamics. Estimation of the mechanical properties of lipid membranes close to the chain melting transition from calorimetry. Biochim. Biophys. Acta. 1415:147–162. [DOI] [PubMed] [Google Scholar]
- 65.Peters, G. H. 2004. Structure-based ligand design: from target protein to drug candidates. Rec. Res. Devel. Biophys. 3:501–526. [Google Scholar]
- 66.Peters, G. H. 2004. Computer imulations: a tool for investigating the function of complex biological macromolecules. In Enzyme Functionality: Design, Engineering, and Screening. A. Svendsen, editor. Marcel Dekker, New York. 97–147.
- 67.Peters, G. H. 2002. The dynamic response of a fungal lipase in the presence of charged surfactants. Colloids Surf. B Biointerfaces. 26:84–101. [Google Scholar]
- 68.Cho, Y. K., and D. B. Northrop. 1999. Effects of pressure on the kinetics of capture by yeast alcohol dehydrogenase. Biochemistry. 38:7470–7475. [DOI] [PubMed] [Google Scholar]
- 69.Menger, F. M. 1992. Analysis of ground-state and transition-state effects in enzyme catalysis. Biochemistry. 31:5368–5373. [DOI] [PubMed] [Google Scholar]
- 70.Pauling, L. 1948. The nature of forces between large molecules of biological interest. Nature. 161:707–709. [DOI] [PubMed] [Google Scholar]
- 71.Peters, G. H., and R. Bywater. 2002. Essential motions in a fungal lipase with bound substrate, covalently attached inhibitor and product. J. Mol. Recognit. 15:393–404. [DOI] [PubMed] [Google Scholar]
- 72.Peters, G. H., T. M. Frimurer, J. N. Andersen, and O. H. Olsen. 1999. Molecular dynamics simulations of protein-tyrosine phosphatase 1B. I. Ligand-induced changes in the protein motions. Biophys. J. 77:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peters, G. H., and R. Bywater. 2001. Influence of a lipid interface on protein dynamics in a fungal lipase. Biophys. J. 81:3052–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Peters, G. H., T. M. Frimurer, J. N. Andersen, and O. H. Olsen. 2000. Molecular dynamics simulations of protein-tyrosine phosphatase 1B. II. Substrate-enzyme interactions and dynamics. Biophys. J. 78:2191–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pan, Y. H., B.-Z. Yu, O. G. Berg, M. K. Jain, and B. J. Bahnson. 2002. Crystal structure of phospholipase A2 complex with the hydrolysis products of platelet activating factor: equilibrium binding of fatty acid and lysophospholipid-ether at the active site may be mutually exclusive. Biochemistry. 41:14790–14800. [DOI] [PubMed] [Google Scholar]
- 76.Janssen, M. J. W., W. A. E. C. van de Wiel, S. H. W. Beiboer, M. D. van Kampen, H. M. Verheij, A. J. Slotboom, and M. R. Egmond. 1999. Catalytic role of the active site histidine of porcine pancreatic phospholipase A2 probed by the variants H48Q, H48N and H48K. Protein Eng. 12:497–503. [DOI] [PubMed] [Google Scholar]
- 77.Scott, D. L., S. P. White, J. L. Browning, J. J. Rosa, M. H. Gelb, and P. B. Sigler. 1991. Structures of free and inhibited human secretory phospholipase A2 from inflammatory exudate. Science. 254:1007–1010. [DOI] [PubMed] [Google Scholar]
- 78.Sekar, K., B.-Z. Yu, J. Rogers, J. Lutton, X. Liu, X. Chen, M.-D. Tsai, M. K. Jain, and M. Sundaralingam. 1997. Phospholipase A2 engineering. Structural and functional roles of the highly conserved active site residue aspartate-99. Biochemistry. 36:3104–3114. [DOI] [PubMed] [Google Scholar]
- 79.White, S. P., D. L. Scott, Z. Otwinowski, M. H. Gelb, and P. B. Sigler. 1990. Crystal-structure of cobra-venom phospholipase A2 in a complex with a transition-state analog. Science. 250:1560–1563. [DOI] [PubMed] [Google Scholar]
- 80.Jørgensen, A. M., L. Tagmose, A. M. M. Jørgensen, K. P. Bøgesø, and G. H. Peters. 2007. Molecular dynamics simulations of Na+/Cl−-dependent neurotransmitter transporters in a model-membrane-aqueous system. ChemMedChem. 2:827–840. [DOI] [PubMed] [Google Scholar]
- 81.Peters, G. H., L. F. Iversen, H. S. Andersen, N. P. H. Møller, and O. H. Olsen. 2004. Residue 259 in protein-tyrosine phosphatase PTP1B and PTPα determines the flexibility of glutamine 262. Biochemistry. 43:18–28. [DOI] [PubMed] [Google Scholar]
- 82.Singh, U. C., and P. A. Kollman. 1986. A combined ab initio quantum mechanical and molecular mechanical method for carrying out simulations on complex molecular systems: applications to the CH3Cl+Cl− exchange reaction and gas phase protonation of polyethers. J. Comput. Chem. 7:718–730. [Google Scholar]
- 83.Burton, N. A., M. J. Harrison, J. C. Hart, I. H. Hillier, and D. W. Sheppard. 1998. Prediction of the mechanisms of enzyme-catalysed reactions using hybrid quantum mechanical/molecular mechanical methods. Faraday Discuss. 110:463–475. [DOI] [PubMed] [Google Scholar]
- 84.Rothlisberger, U., P. Carloni, K. Doclo, and M. Parrinello. 2000. A comparative study of galactose oxidase and active site analogs based on QM/MM Car-Parrinello simulations. J. Biol. Inorg. Chem. 5:236–250. [DOI] [PubMed] [Google Scholar]
- 85.Shokhen, M., and A. Albeck. 2000. Factors determining the relative stability of anionic tetrahedral complexes in serine protease catalysis and inhibition. Proteins Struct. Funct. Genet. 40:154–167. [DOI] [PubMed] [Google Scholar]
- 86.Bash, P. A., M. J. Field, R. C. Davenport, G. A. Petsko, D. Ringe, and M. Karplus. 1991. Computer simulation and analysis of the reaction pathway of triosephosphate isomerase. Biochemistry. 30:5826–5832. [DOI] [PubMed] [Google Scholar]
- 87.Åqvist, J., and A. Warshel. 1993. Simulation of enzyme-reactions using valence bond force-fields and other hybrid quantum-classical approaches. Chem. Rev. 93:2523–2544. [Google Scholar]
- 88.Feierberg, I., A. D. Cameron, and J. Åqvist. 1999. Energetics of the proposed rate-determining step of the glyoxalase I reaction. FEBS Lett. 453:90–94. [DOI] [PubMed] [Google Scholar]
- 89.Gargallo, R., B. Oliva, E. Querol, and F. X. Aviles. 2000. Effect of the reaction field electrostatic term on the molecular dynamics simulation of the activation domain of procarboxypeptidase B. Protein Eng. 13:21–26. [DOI] [PubMed] [Google Scholar]