

Abstract
A number of mechanistic and predictive genetic regulatory networks (GRNs) comprising dozens of genes have already been characterized at the level of cis-regulatory interactions. Reconstructions of networks of 100’s to 1000’s of genes and their interactions are currently underway. Understanding the organizational and functional principles underlying these networks is probably the single greatest challenge facing genomics today. We review the current approaches to deciphering large-scale GRNs and discuss some of their limitations. We then propose a bottom-up approach in which large-scale GRNs are first organized in terms of functionally distinct GRN building blocks of one or a few genes. Biological processes may then be viewed as the outcome of functional interactions among these simple, well-characterized functional building blocks. We describe several putative GRN functional building blocks and show that they can be located within GRNs on the basis of their interaction topology and additional, simple and experimentally testable constraints.
Keywords: Genetic regulatory networks, systems biology, transcriptional regulation, visualization
INTRODUCTION
The availability of a large number of annotated genomes has shown that metazoan complexity arises primarily from more complex regulatory interactions among genes and their products [1]. At the same time, high throughput technologies delineate the location, abundance, state and interactions of genes and their products with increasing resolution in terms of cell types, time points and conditions (see for example [2, 3]). A recent phenomenon accompanying the reconstruction of large-scale molecular interaction maps is the publication of often unfathomable large network diagrams of the type shown in Fig. (1) (zoom-in view in upper section). The inclusion of such figures (especially given the tight space constraints of most journals) is perplexing, since the figures can provide no insights by themselves. For such figures to be comprehensible, it is necessary to superimpose one or more layers of functional abstraction on top of the low-level interactions that comprise these very complex networks, as discussed extensively in Chapter 4 of [4].
Fig. (1).
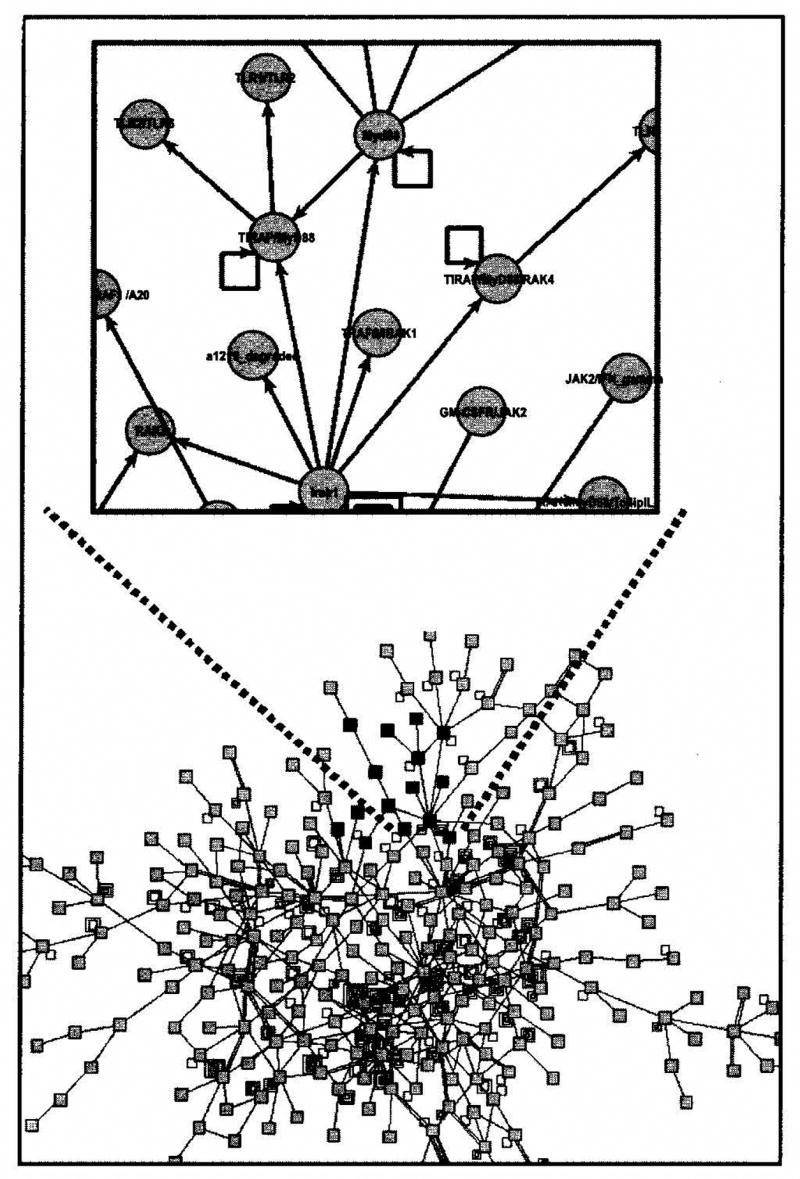
Example of a large-scale genetic regulatory network (GRN) diagram viewed as a homogenous, automatically laid out graph of interacting genes and proteins. Zoomed in view of a portion of the network is shown in the inset. Lack of space, time and functional information makes the network diagram difficult to decipher.
INTERACTION CLUSTERS PROVIDE A HIGHER LEVEL OF ABSTRACTION
In the area of genetic regulatory networks (GRNs), a number of different ways of clustering the components of large-scale molecular interaction networks into distinct groups have been proposed. These include batteries of co-expressed genes [5], sets of highly interacting genes [6], genes sharing a common Gene Ontology annotation or known pathway, statistically over-represented topological motifs [7, 8] and functional building blocks [9] (for a discussion of potential functional building blocks in protein interaction networks, see [10]). Indeed, current systems biology network documentation and analysis tools such as Cytoscape (http://Cytoscape.org) implement many of the above mentioned methods and use ‘guilt by association’ to decompose a large-scale network map into a set of interacting functional modules such as “lipid metabolism” and “DNA repair” (see for example [11, 12]). These approaches greatly aid in the understanding of large-scale datasets and have provided significant insights into cellular organization and the regulatory interactions among different cellular processes. However, since the subsystems delineated in this way tend to comprise dozens of interacting genes and gene products, their mechanisms of action can be difficult to understand.
HIGHER-LEVEL ABSTRACTIONS NEED A FUNCTIONAL BASIS
Figs. (2) and (3) show two different views of the widely studied galactose utilization module in yeast [13, 14]. Fig. (2) was derived manually from the literature [15]. Accompanying this figure are biochemical and mathematical descriptions, which uniquely and unambiguously define the nature of the interactions within this system and the resulting dynamic behavior. Fig. (3) is an example of a system reconstructed automatically from high-throughput data. It was reconstructed by statistically merging data from 18 different global datasets (see [12] for details). The resulting network map was then clustered using the above mentioned ‘guilt-by-association’ approach. Because of the nature of the data they are derived from, the interactions indicated in Fig. (3) are only qualitative.
Fig. (2).

The CRN regulating galactose uptake in yeast. The constitutively expressed transcription activator Gal4p binds to the promoters of GAL genes. In the absence of galactose, the represser Gal80p blocks the activation domain of Gal4p. In the presence of intracellular galactose. the signal transducer Gal3p is activated, which sequesters Gal80p in the cytoplasm, leading to transcription of the GAL genes. The signal transducer gal3 and the represser gal80 are basally expressed in non-induced, non-glucose-repressed conditions. Lines terminating in arrowheads denote positive regulation. Lines terminating in bars denote repression. Gene symbols denote transcription. For enzymatic interactions, the relevant enzymes arc indicated next to each line. The double ellipses denote Gal4p dimers. See main text for references.
Fig. (3).
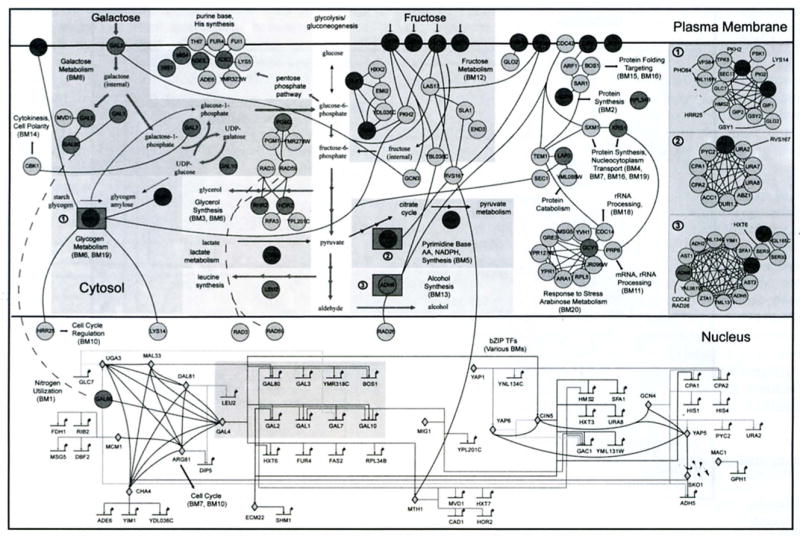
The yeast galactose utilization network derived from high-throughput global assays. Shaded background boxes in the cytosol delineate the known metabolic pathways. Transcription factors in the nucleus are represented by diamonds (genes within the grey box are the regulatory genes in Fig. 2). All other proteins (circles) arc located according to their sub-cellular localizations (plasma membrane, cytosol and nucleus). Colors (not shown) can be used to indicate increase, decrease and no significant change in gene expression when the carbon source is changed from raffinose to galactose (see (12] for color). The three numbered squares shown at the right represent complexes. Numbers preceded with BM mark functionally clustered biomodules. Short black arrows indicate communication between the modules.
Each interaction in Fig. (2) is known at the biochemical level and the module as a whole can be and has been, modeled and studied dynamically. The system behavior under different cellular conditions can be and has been, predicted and experimentally verified (see for example [14]). By comparison, the network in Fig. (3) includes many interactions about which little is known. Thus, its behavior under novel conditions can only be guessed at. On the other hand, Fig. (3) includes many important interactions between the core galactose pathway and other cellular processes such as cell cycle regulation and fructose metabolism. These are however not apparent in Fig. (2). To understand galactose uptake in yeast, we need to be able to decompose the network of Fig. (3) into sub-modules with specific functional roles of the type in Fig. (2). We argue that the best way to do this is to start at the bottom, characterizing functional building blocks of one or few genes. We could then look for distinct functional groupings of these functional building blocks (or sub-circuits, as described in [4]) and build a hierarchical library of functional sub-system archetypes underlying all GRNs.
THE SAME TOPOLOGICAL MOTIF CAN IMPLEMENT SEVERAL DIFFERENT FUNCTIONS
A key step towards the identification of GRN functional building blocks was the discovery of statistically surprising recurring topological motifs in GRNs. These were first found in E. coli [7] and subsequently in yeast and human cells [16, 17]. Instances of these motifs can also be seen in GRNs from several other species, including Ciona [18], Drosophila [19], sea urchin [4, 20] and C. elegans [3]. Examples of topological motifs include autoregulation, feed-forward loops, densely overlapping regulons (transcription factors with overlapping target genes) and bi-fans (two factors that co-regulate target genes). It has been tempting to consider these topological motifs as promising candidates for describing and understanding GRNs at a higher level of abstraction.
Some of the simpler topological motifs easily map to a particular function. For example, a positively auto-regulating gene can be quickly identified as a likely candidate for a latch that continues to drive a regulatory circuit after the initial signal has been removed. However not every positive auto-regulator can be guaranteed to have this property, but the great majority will. For the equally simple case of a negatively auto-regulated gene, the situation is much more complex. Depending on the kinetic parameters of the system, such a simple circuit can be tuned to provide a regulated activity level, a single pulse, a rapid response, or a long lasting oscillation. Similarly, the ‘densely overlapping regulon’ motif has connectivity suggestive of a battery of co-regulated genes. On the other hand, the irregular pattern of transcription factor binding sites on the target genes allows the possibility that distinct combinations of inputs may regulate each gene differently.
Thus, the same topological motif can imply radically different functions. In general, detailed experimental data are needed to correctly assign a function to most topological motifs. The converse is also true: the same function may arise from a variety of different network topologies. For example, GRN sub-circuits with the functionality of a latch may contain one [16] or multiple genes (e.g. the sea urchin blimp1/otx/gatae, see Fig. (6) and [20]) that participate to implement the latch.
Fig. (6).
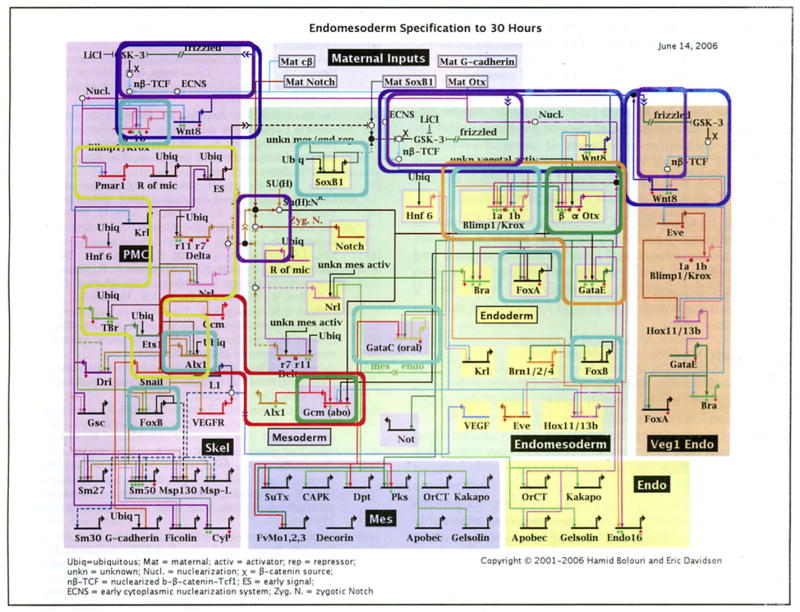
Occurrences of putative functional building blocks in the GRN underlying endomesoderm specification in the embryo of the sea urchin Strongylocentrotus purpuratus. Each occurrence is indicated by a rounded bounding box. Box colors identify different types of functional building blocks. Green: single-gene intra-cellular positive feedback latches. Orange: a multi-gene intra-cellular positive feedback latch. Dark blue: inter-cellular positive feedback latch (the community effect) mediated by Wnt8 signaling. Cyan: instances of negative auto-regulation. foxA expression is oscillatory (Oliver! P, Tu Q, Walton K, Davidson EH and McClay DR, manuscript in preparation). The expression patterns of the other auto-repressive genes are consistent with the ‘single pulse’ functional building block. Purple: signal mediated toggle switches mediated via β-catenin and TCF/LEF in Wnt signaling and Su(H) in Notch signaling. Red: the alx1-gcm mutual exclusion operator. Alx1 is on in the PMC domain and off in the mesoderm. Gcm is on in the mesoderm and off in the PMC. Yellow: the pmar1 gradient detection/analogue to digital switch. Pmar1 represses an uncharacterized repressor which in turn represses es, delta, nrs, alx1, tbr and ets1.
Each short horizontal line from which a bent arrow extends to indicate transcription represents the cis-regulatory element that is responsible for expression of the gene named in the domain shown. The arrows and barred lines indicate the normal function of the gene (activation or repression) as deduced from changes in transcript levels in perturbation experiments. Red diamonds indicate that a cis-regulatory element, which controls relevant expression has been isolated. Green diamonds indicate reported experimental evidence validates expected target site. The rectangles in the lower tier of the diagram show downstream differentiation genes. Dashed lines indicate inferred or possibly indirect relationships. Arrows inserted in arrow tails indicate intercellular signaling interactions. Large open ovals represent cytoplasmic biochemical interactions at the protein level. Sec [4] for a detailed description.
Another complication in assigning unique functions to topological motifs is the context in which the constituent genes operate. For example, dorsal, twist and snail form a so-called feedforward loop (FFL) motif during dorsal/ventral (D/V) patterning in Drosophila. High levels of Dorsal trigger the expression of twist and both in turn trigger the expression of the snail represser [19]. Dorsal, Twist and Snail then jointly regulate several downstream genes. In E. coli, the FFL motif has previously been shown to act to accelerate transcriptional response [21] or filter noise spikes [21, 22]. However, the operational context of the D/V FFL motif is that there is a gradient of Dorsal concentration along the D/V axis. Twist and Snail also diffuse, but to differing extents. Furthermore, different dorsal binding sites have different binding affinities [23] and the activator twist and the represser snail compete for the same binding site on some of the target genes [19]. These features, which are not apparent in the topological motif, mean that this particular instance of a feedforward motif acts as a spatial analog-to-digital converter, which translates a single Dorsal concentration gradient into different but highly specific patterns of gene activation in different nuclei along the D/V axis. Topological graphs do not show the (on or off DNA) regulatory interactions among the inputs incident on a target gene. Consequently, assigning a function to a topological motif requires additional time course, spatial and/or interaction data.
SOME CANDIDATE FUNCTIONAL BLOCKS
By comparing known regulatory interactions in different pathways, cells and organisms, it is possible to identify some candidate conserved functional building blocks (as discussed extensively in Chapter 4 of [4]). Fig. (4) illustrates some examples (see [9] for an earlier version). For simplicity, the figure shows most of these functional blocks in the style of the simplest corresponding network motifs. However, our definition of these blocks is based on function and not topology. Thus, in building block (A), the positive feedback reinforcement loop can involve one, two, or more genes and be intra- or inter-cellular, as shown schematically. Similarly, functional building block (B) represents all classes of negative auto-regulation. It may involve one or more genes and signaling between one or more cells. The key characteristic of building block (C) is mutual repression between two genes. Again, specific instances of this type of interaction may be implemented via intermediary genes, or via intercellular signaling. For the remaining candidate building blocks, we do not have candidate multi-gene or multi-cell versions, but expect that they will be discovered.
Fig. (4).
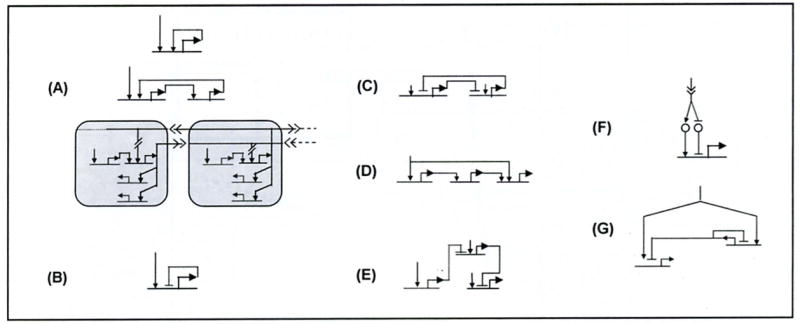
Some proposed GRN functional building blocks. See text for detailed descriptions.
The function assigned to each of these simple building blocks has been characterized experimentally in at least one organism and hypothesized in additional organisms. Some of the circuits are capable of multiple functions depending on the biochemical kinetics of their components. In the next section, we give an example of how mathematical analysis can impose specific, experimentally testable constraints for each function. Using these constraints, a given network map (and associated expression data) can either be immediately decomposed into specific functional building blocks, or specific experiments can be proposed to enable such a functional decomposition. Below, we summarize the functional characteristics of each of these candidate functional building blocks.
-
Building block (A) implements a latch that once activated, turns fully on and remains on. It has been observed in sea urchin embryos [20] and is also thought to occur in E.coli [7], yeast [8], human LS cells [16] and mouse pancreatic B-cells [24] (see [4], pi77 for a discussion). An interesting potential instance of the positive feedback functional building block appears to underlie cardiac development [1]. As Davidson & Erwin [1] show, the positive feedback loop is conserved in heart development across multiple phyla even though both the topology and the member genes have diverged (see also [4], p218).
The inter-cellular version of this positive feedback function serves as the underlying control mechanism behind the community effect [25]. Here the emphasis is on promoting a common fate lock-in among neighboring cells. Wnt signaling in sea urchin endomesoderm specification and eFGF signaling in Xenopus myogenesis [26] and notochord development [27] use this building block.
The negative auto-regulation in building block (B) can imply one of four possible functions depending on kinetic parameter values: (1) A single pulse element. This function turns a long lasting trigger (a step function) into a transient pulse of transcriptional activity. It was identified in Drosophila embryos in a series of ground breaking papers during the 1970s and 1980s by Ashburner and colleagues [28]. The stress-inducible gene ATF3 functions similarly in a variety of human cell lines [29]; (2) A rapid-response element. This function was first discovered as a topological motif in E. coli [7] and then experimentally and theoretically analyzed by Alon and colleagues [30]; (3) A ‘programmable’ homeostatically-fixed expression level. Gsc in the Xenopus dorsal mesoderm implements this function, i.e. it negatively auto-regulates itself to maintain a fixed expression level [31]; and (4) Sustained oscillations. An example occurrence of this function is the oscillatory expression of hes-1 in a variety of mammalian cells (see [32] for experimental verification, [33] for theoretical analysis).
The mutual exclusion functional block in (C) can be implemented with two mutually repressing genes, each activated by different inputs. Only one of the two genes can be active in any one cell at a given time. This building block can be observed in sea urchin endomesoderm specification (alx and gem mutually repress, see Fig. (6) and private communications Oliveri P. and Davidson EH). It also occurs in the Drosophila segment polarity network where en and sip exclude one another, see [34]. Just as the intra-cellular latch functional block (B) has an inter-cellular signaling equivalent, an inter-cellular version of the mutual exclusion block in (C) has also been proposed: Notch mediated lateral specification in mouse pancreas development [35] (reviewed in [36]).
The spike filter functional block shown in (D) is an example of a functional block built upon feedforward loop (FFL) topology. This building block occurs in E. coli and has been functionally characterized in a synthetically constructed circuit by Alon el al. [22, 37].
The functional building block (E) is a gradient-detection or analog-to-digital converter similar in function to the example described above in the D/V patterning network in Drosophila (interactions of dorsal, twist and snail). Pmar1 and its downstream targets in the sea urchin primary mesenchyme cells are examples of this functional block ([38, 39], also private communications Oliver! P. and Davidson EH; see Fig. (6)). The ability of this building block to act as a highly non-linear switch was functionally characterized in a synthetically constructed circuit by Weiss and colleagues [40].
The signal-mediated toggle switch (F) is conserved in Notch and Wnt signaling across several phyla, as discussed by Barolo and Posakony [41]. Here, a factor that acts as a signal-mediated transcriptional activator (suppressor of hairless for Notch and TCF/LEF for Wnt signaling), will act to repress its target genes in the absence of signaling (see [42] and [43] respectively). The result is that downstream genes are either fully on, or fully off (see also [44]).
The homeostatic switch in (G) is a surprising functional variant of the FFL topology. In this sub-system however, the intermediate gene first activates and then resets (deactivates) a set of downstream genes. It was recently discovered in mouse macrophages [45].
FUNCTIONAL BLOCKS MUST BE ROBUST TO VARIATIONS IN KINETIC PARAMETERS
The network building blocks highlighted above have been shown to be capable of the function described in each of the specified cases. This is a necessary but not a sufficient condition. Ideally, a given functional building block should behave in a predictable manner, regardless of inevitable variations in the kinetic parameters of the component interactions (in a single cell over time and from cell to cell). As with the example of heart development [1], we may also expect a functional building block to perform the same function when implemented with orthologous components and topologies. In contrast, simple synthetic genetic circuits are not robust to such variations and perform very differently and unpredictably from cell to cell [46].
In many cases, an analysis of the plausible ranges of kinetic parameters within which a network performs a specified function can be used to identify plausible network blocks [45, 47]. In addition to the size of the operating region, its shape (whether it is convex or concave) and the rate at which performance degrades with parameter changes can be used to pinpoint sensitivities in a model. Analysis of sensitivity to kinetic parameters can be used to impose specific biological/biochemical constraints that a topological network block must meet to be capable of a hypothesized function. An excellent example of this is described by Cherry and Adler [48]. As Cherry and Adler show, a pair of mutually repressing genes can only act as a (bistable) mutual exclusion operator if the repressive interactions of the two genes are higher than first order, for example via dimerization of the regulatory gene products, or the presence of multiple cooperating cis-regulatory binding sites on each gene. Fig. (5), which is adapted from [48], shows that the kinetic parameter region within which mutually repressing genes will act as a bistable mutual exclusion switch covers a large portion of the plausible parameter ranges for such a system. It also shows that greater cooperativity leads to more robust switches. Together these figures can be used to check the plausibility of a given mutual repression motif as a mutual exclusion functional block. Similar analyses can be performed on the other functional building blocks discussed and can lead to similar experimentally testable constraints that topological observations must meet in order to be considered a functional building block.
GRNs CAN BE DESCRIBED IN TERMS OF HIERARCHIES OF FUNCTIONAL ABSTRACTIONS
Functional building blocks of the type described here can be thought of as an intermediate layer of abstraction between the molecular interaction map at the most detailed level and a “process diagram” summarizing the key processes that define a system behavior at a very abstract level (see [49]). By analogy to engineered systems, one may envisage a number of additional layers of abstraction between the building block and process layers. For example, functional building blocks may be grouped together to define modules with specific higher level functions. The extent to which such intermediate level groupings (functional modules) will turn out to be preserved and the number of layers of abstraction that describe a system in the most informative manner can only be determined when more GRNs have been characterized. For a comprehensive discussion of GRN organization and conserved GRN components at different levels of abstraction, see chapter 4 of [4].
FUNCTIONAL BLOCKS ARE FLUID AND CONTEXT-DEPENDENT
In contrast to engineered systems, where each system component is designed to be part of one functional module and is expected to perform a single function at all times, genes and their products take part in many different processes depending on time, cell types and environmental stimuli. Thus, the functional building blocks in a network will tend to form, execute their function and then disband. Components of these functional blocks will be reused in different functional blocks. It also seems likely that a single gene (or its product) may simultaneously take part in more than one functional block. These considerations suggest that static network diagrams and network maps that show only a single copy of each molecular species may not have sufficient explanatory power (see for example the time-space GRN visualizations offered by the BioTapestry software [50], http://www.BioTapestry.org). Even when considering a single cell type at a particular time, multiple, dynamic representations of processes, functional blocks and molecular species will be needed to provide multiple functional views of the same network at the same time, with each view providing a different organizational perspective.
DELINEATING FUNCTIONAL BLOCKS REQUIRES FUNCTIONAL DATA
As illustrated above, topology alone is not sufficient to understand the behavior of genetic regulatory networks. Even at the level of the simplest network building blocks, nonlinear regulatory interactions lead to multiple possible behaviors for a given topological motif. Additional information can be used to first predict, then iteratively refine potential functions of a given topological motif. Below is an example procedure.
for each cell type of interest, determine the time-course of expression of each component gene. Are the expression patterns consistent with a theoretically-predicted function for the motif?
if post-transcriptional modifications are known to regulate the transcription factors involved, does available data support the hypothesized regulatory functions within the block?
for each of the above genes, determine the cis-regulatory inputs active in the cell type and time-period of interest. Are these inputs necessary and sufficient to explain the output activity of each gene in the block? Are the roles assigned to the inputs (transcription factors) and their interactions consistent with the literature?
When the answer to any of the above questions is no, the implication is that one or more of our assumptions about the system requires further elaboration or is considered incorrect. This leads to new, experimentally-testable hypotheses about the system and further rounds of model refinement. When the answers to all of the above questions are positive, the outcome is a specific, mechanistic and causal model of function and behavior of the network motif considered. The validity of the model can then be verified by additional perturbation analyses. A validated functional model can be further refined by more detailed studies of the biochemical kinetics of its component interactions. It can also be further characterized in terms of the interactions of the modeled functional block with other functional blocks within the cell and their collective role in larger cellular processes.
It should be noted that the demonstration that a given set of genes collectively perform a particular function, does not preclude additional functional roles for the same genes and interactions. Indeed, known functional building blocks often have multiple functional roles (consider for example how a door handle is commonly used to both free the latch and also to pull the door open).
CONCLUSION
As shown in Fig. (6), the simple functional building bocks described herein densely cover almost the entire known sea urchin endomesoderm specification network. The functions of many of these instances have been characterized experimentally and concur with our description above (see http://www.its.caltech.edu/~mirsky/pubs.htm for full list of papers). For others that have not been characterized yet, the assigned function in Fig. (3) explains the observed patterns of gene activity correctly. One may therefore reasonably hope that the use of functional building blocks will provide a useful means of deciphering the behavior of other GRNs currently under reconstruction.
The candidate functional network building blocks described above are at this point supported by limited experimental evidence. Further investigation is needed to verify and refine the functions assigned to these minimal modules. As more networks involved in different cellular processes and different species are delineated, many more candidate conserved functional blocks may be discovered. Each of these will need to be subjected to rigorous experimental and theoretical analysis to verify that it can plausibly perform its assumed function given natural variations in kinetic rates and perhaps, even using different components.
Fig. (5).
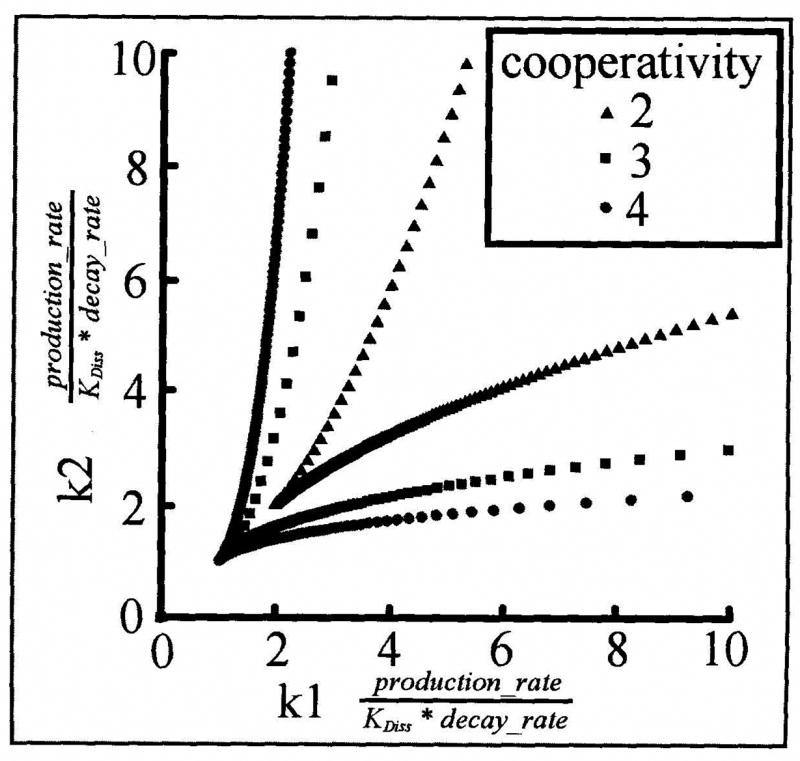
Parameter values for which a pair of mutually repressing genes will act as a mutual exclusion switch (adapted from [48]). So long as the repressive interactions of the two genes are greater than first order, a large range of parameter values (between the two dotted boundaries marked) results in robust switch-like behavior. The example boundaries shown are calculated assuming a Hill-function for the repressive interactions. In this case, the Hill coefficient stands in for cooperative binding on DNA and/or cooperative complex formation prior to DNA binding. Boundaries for Hill coefficients of 2, 3 and 4 are shown. The larger the degree of cooperativity, the more robust the mutual exclusion switch (larger area enclosed by the dotted boundaries).
Acknowledgments
We thank Eric H. Davidson for many helpful discussions. This work was supported in part by NIGMS grant GM061006.
References
- 1.Davidson EH, Erwin DH. Gene regulatory networks and the evolution of animal body plans. Science. 2006;311:796–800. doi: 10.1126/science.1113832. [DOI] [PubMed] [Google Scholar]
- 2.Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, Vidalain PO, Han JD, Chesneau A, Hao T, Goldberg DS, Li N, Martinez M, Rual JF, Lamesch P, Xu L, Tewari M, Wong SL, Zhang LV, Berriz GF, Jacotot L, Vaglio P, Reboul J, Hirozane–Kishikawa T, Li Q, Gabel HW, Elewa A, Baumgartner B, Rose DJ, Yu H, Bosak S, Sequerra R, Fraser A, Mango SE, Saxton WM, Strome S, Van Den Heuvel S, Piano F, Vandenhaute J, Sardet C, Gerstein M, Doucette-Stamm L, Gunsalus KC, Harper JW, Cusick ME, Roth FP, Hill DE, Vidal M. A map of the interactome network of the metazoan C. elegans. Science. 2004;303:540–3. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deplancke B, Mukhopadhyay A, Ao W, Elewa AM, Grove CA, Martinez NJ, Sequerra R, Doucette-Stamm L, Reece-Hoyes JS, Hope IA, Tissenbaum HA, Mango SE, Walhout AJ. A gene-centered C. elegans protein-DNA interaction network. Cell. 2006;125:1193–205. doi: 10.1016/j.cell.2006.04.038. [DOI] [PubMed] [Google Scholar]
- 4.Davidson EH. The Regulatory Genome: Gene Regulatory Networks in Development and Evolution. Elsevier; 2006. [Google Scholar]
- 5.Ihmels J, Bergmann S, Barkai N. Defining transcription modules using large-scale gene expression data. Bioinformatics. 2004;20:1993–2003. doi: 10.1093/bioinformatics/bth166. [DOI] [PubMed] [Google Scholar]
- 6.Rives AW, Galitski T. Modular organization of cellular networks. Proc Natl Acad Sci USA. 2003;100:1128–33. doi: 10.1073/pnas.0237338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen-Orr SS, Milo R, Mangan S, Alon U. Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 2002;31:64–8. doi: 10.1038/ng881. [DOI] [PubMed] [Google Scholar]
- 8.Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, Zeitlinger J, Jennings EG, Murray HL, Gordon DB, Ren B, Wyrick JJ, Tagne JB, Volkert TL, Fraenkel E, Gifford DK, Young RA. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 9.Bolouri H, Davidson EH. Modeling transcriptional regulatory networks. Bioessays. 2002;24:1118–29. doi: 10.1002/bies.10189. [DOI] [PubMed] [Google Scholar]
- 10.Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr Opin Cell Biol. 2003;15:221–31. doi: 10.1016/s0955-0674(03)00017-6. [DOI] [PubMed] [Google Scholar]
- 11.Hwang D, Rust AG, Ramsey S, Smith JJ, Leslie DM, Weston AD, de Atauri P, Aitchison JD, Hood L, Siegel AF, Bolouri H. A data integration methodology for systems biology. Proc Natl Acad Sci USA. 2005;102:17296–301. doi: 10.1073/pnas.0508647102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hwang D, Smith JJ, Leslie DM, Weston AD, Rust AG, Ramsey S, de Atauri P, Siegel AF, Bolouri H, Aitchison JD, Hood L. A data integration methodology for systems biology: experimental verification. Proc Natl Acad Sci USA. 2005;102:17302–7. doi: 10.1073/pnas.0508649102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ideker T, Thorsson V, Ranish JA, Christmas R, Buhler J, Eng JK, Bumgarner R, Goodlett DR, Aebersold R, Hood L. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 2001;292:929–34. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- 14.Acar M, Becskei A, van Oudenaarden A. Enhancement of cellular memory by reducing stochastic transitions. Nature. 2005;435:228–32. doi: 10.1038/nature03524. [DOI] [PubMed] [Google Scholar]
- 15.de Atuari POD, Ramsey S, Bolouri H. Evolution of design principles in biochemcial networks. IEE Syst Biol. 2004;1:28–40. doi: 10.1049/sb:20045013. [DOI] [PubMed] [Google Scholar]
- 16.Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, Young RA. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–56. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, Jennings EG, Zeitlinger J, Pokholok DK, Kellis M, Rolfe PA, Takusagawa KT, Lander ES, Gifford DK, Fraenkel E, Young RA. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imai KS, Levine M, Satoh N, Satou Y. Regulatory blueprint for a chordate embryo. Science. 2006;312:1183–7. doi: 10.1126/science.1123404. [DOI] [PubMed] [Google Scholar]
- 19.Stathopoulos A, Levine M. Genomic regulatory networks and animal development. Dev Cell. 2005;9:449–62. doi: 10.1016/j.devcel.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 20.Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, Minokawa T, Amore G, Hinman V, Arenas-Mena C, Otim O, Brown CT, Livi CB, Lee PY, Revilla R, Rust AG, Pan Z, Schilstra MJ, Clarke PJ, Arnone MI, Rowen L, Cameron RA, McClay DR, Hood L, Bolouri H. A genomic regulatory network for development. Science. 2002;295:1669–78. doi: 10.1126/science.1069883. [DOI] [PubMed] [Google Scholar]
- 21.Mangan S, Itzkovitz S, Zaslaver A, Alon U. The incoherent feed-forward loop accelerates the response-time of the gal system of Escherichia coli. J Mol Biol. 2006;356:1073–81. doi: 10.1016/j.jmb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Mangan S, Alon U. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci USA. 2003;100:11980–5. doi: 10.1073/pnas.2133841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papatsenko D, Levine M. Quantitative analysis of binding motifs mediating diverse spatial readouts of the Dorsal gradient in the Drosophila embryo. Proc Natl Acad Sci USA. 2005;102:4966–71. doi: 10.1073/pnas.0409414102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Servitja JM, Ferrer J. Transcriptional Networks controlling pancreatic development and beta cell function. Diabetologica. 2004;47:597–613. doi: 10.1007/s00125-004-1368-9. [DOI] [PubMed] [Google Scholar]
- 25.Gurdon JB. A community effect in animal development. Nature. 1988;336:772–4. doi: 10.1038/336772a0. [DOI] [PubMed] [Google Scholar]
- 26.Standley HJ, Zorn AM, Gurdon JB. eFGF and its mode of action in the community effect during Xenopus myogenesis. Development. 2001;128:1347–57. doi: 10.1242/dev.128.8.1347. [DOI] [PubMed] [Google Scholar]
- 27.Weston MJD, Kato K, Gurdon JB. A community effect is required for amphibian notochord differentiation. Development Genes and Evolution. 1994;203:250–3. doi: 10.1007/BF00360520. [DOI] [PubMed] [Google Scholar]
- 28.Ashburner M. Puffs, genes and hormones revisited. Cell. 1990;61:1–3. doi: 10.1016/0092-8674(90)90205-s. [DOI] [PubMed] [Google Scholar]
- 29.Wolfgang CD, Liang G, Okamoto Y, Allen AE, Hai T. Transcriptional autorepression of the stress-inducible gene ATF3. J Biol Chem. 2000;275:16865–70. doi: 10.1074/jbc.M909637199. [DOI] [PubMed] [Google Scholar]
- 30.Rosenfeld N, Elowitz MB, Alon U. Negative autoregulation speeds the response times of transcription networks. J Mol Biol. 2002;323:785–93. doi: 10.1016/s0022-2836(02)00994-4. [DOI] [PubMed] [Google Scholar]
- 31.Koide T, Hayata T, Cho KW. Xenopus as a model system to study transcriptional regulatory networks. Proc Natl Acad Sci USA. 2005;102:4943–8. doi: 10.1073/pnas.0408125102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirata H, Yoshiura S, Ohtsuka T, Bessho Y, Harada T, Yoshikawa K, Kageyama R. Oscillatory expression of the bHLH factor Hes1 regulated by a negative feedback loop. Science. 2002;298:840–3. doi: 10.1126/science.1074560. [DOI] [PubMed] [Google Scholar]
- 33.Monk NA. Oscillatory expression of Hes1, p53 and NF-kappaB driven by transcriptional time delays. Curr Biol. 2003;13:1409–13. doi: 10.1016/s0960-9822(03)00494-9. [DOI] [PubMed] [Google Scholar]
- 34.Alexandre C, Vincent JP. Requirements for transcriptional repression and activation by Engrailed in Drosophila embryos. Development. 2003;130:729–39. doi: 10.1242/dev.00286. [DOI] [PubMed] [Google Scholar]
- 35.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;254:770–6. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 36.Jensen J. Gene regulatory factors in pancreatic development. Dev Dyn. 2004;229:176–200. doi: 10.1002/dvdy.10460. [DOI] [PubMed] [Google Scholar]
- 37.Dekel E, Mangan S, Alon U. Environmental selection of the feed-forward loop circuit in gene-regulation networks. Phys Biol. 2005;2:81–8. doi: 10.1088/1478-3975/2/2/001. [DOI] [PubMed] [Google Scholar]
- 38.Oliveri P, Carrick DM, Davidson EH. A regulatory gene network that directs micromere specification in the sea urchin embryo. Dev Biol. 2002;246:209–28. doi: 10.1006/dbio.2002.0627. [DOI] [PubMed] [Google Scholar]
- 39.Oliveri P, Davidson EH, McClay DR. Activation of pmar1 controls specification of micromeres in the sea urchin embryo. Dev Biol. 2003;258:32–43. doi: 10.1016/s0012-1606(03)00108-8. [DOI] [PubMed] [Google Scholar]
- 40.Hooshangi S, Thiberge S, Weiss R. Ultrasensitivity and noise propagation in a synthetic transcriptional cascade. Proc Natl Acad Sci USA. 2005;102:3581–6. doi: 10.1073/pnas.0408507102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barolo S, Posakony JW. Three habits of highly effective signaling pathways: principles of transcriptional control by developmental cell signaling. Genes Dev. 2002;16:1167–81. doi: 10.1101/gad.976502. [DOI] [PubMed] [Google Scholar]
- 42.Morel V, Schweisguth F. Repression by suppressor of hairless and activation by Notch are required to define a single row of single-minded expressing cells in the Drosophila embryo. Genes Dev. 2000;14:377–88. [PMC free article] [PubMed] [Google Scholar]
- 43.Daniels DL, Weis WI. Beta-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat Struct Mol Biol. 2005;12:364–71. doi: 10.1038/nsmb912. [DOI] [PubMed] [Google Scholar]
- 44.Istrail S, Davidson EH. Logic functions of the genomic cis-regulatory code. Proc Natl Acad Sci USA. 2005;102:4954–9. doi: 10.1073/pnas.0409624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gilchrist M, Thorsson V, Li B, Rust AG, Korb M, Kennedy K, Hai T, Bolouri H, Aderem A. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441:173–8. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- 46.Guet CC, Elowitz MB, Hsing W, Leibler S. Combinatorial synthesis of genetic networks. Science. 2002;296:1466–70. doi: 10.1126/science.1067407. [DOI] [PubMed] [Google Scholar]
- 47.Morohashi M, Winn AE, Borisuk MT, Bolouri H, Doyle J, Kitano H. Robustness as a measure of plausibility in models of biochemical networks. J Theor Biol. 2002;216:19–30. doi: 10.1006/jtbi.2002.2537. [DOI] [PubMed] [Google Scholar]
- 48.Cherry JL, Adler FR. How to make a biological switch. J Theor Biol. 2000;203:117–33. doi: 10.1006/jtbi.2000.1068. [DOI] [PubMed] [Google Scholar]
- 49.Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, Minokawa T, Amore G, Hinman V, Arenas-Mena C, Otim O, Brown CT, Livi CB, Lee PY, Revilla R, Schilstra MJ, Clarke PJ, Rust AG, Pan Z, Arnone MI, Rowen L, Cameron RA, McClay DR, Hood L, Bolouri H. A provisional regulatory gene network for specification of endomesoderm in the sea urchin embryo. Dev Biol. 2002;246:162–90. doi: 10.1006/dbio.2002.0635. [DOI] [PubMed] [Google Scholar]
- 50.Longabaugh WJR, Davidson EH, Bolouri H. Computational representation of developmental genetic regulatory networks. Dev Biol. 2005;283:1–16. doi: 10.1016/j.ydbio.2005.04.023. [DOI] [PubMed] [Google Scholar]