

Abstract
For immune diagnostic purposes it would be critical to be able to distinguish between ongoing immune processes, such as active infections, and long-term immune memory, for example imprinted by infections that have been cleared a long time ago or by vaccinations. We tested the hypothesis that the secretion of Granzyme B, as detected in ex vivo ELISPOT assays, permits this distinction. We studied EBV-, flu- and CMV-specific CD8+ cells in healthy individuals, Vaccinia virus-reactive CD8+ cells in the course of vaccination, and HIV-specific CD8+ cells in HIV-infected individuals. Antigen-specific ex vivo GzB production was detected only transiently after Vaccinia immunization, and in HIV-infected individuals. Our data suggest that ex vivo ELISPOT measurements of granzyme B permit the identification of actively ongoing CD8+ cell responses – a notion that is pertinent to the immune diagnostic of infections, transplantation, allergies, autoimmune diseases, tumors, and vaccine development.
Keywords: cytokines, cytotoxicity, CD8+ T cells, ELISPOT, granzyme B, interferon-γ, immunodeficiency, memory, vaccination, viral infections
Introduction
CD8+ cells play a critical role in host defense [1-3]. Before the first encounter of their antigen CD8+ cells are naïve, occur in low clonal sizes, express cell surface markers characteristic of resting cells, and are not capable of exerting effector functions such as killing [4]. Shortly after an infection or vaccination the specific CD8+ cells are triggered to engage in an immune response. The antigen-specific cells undergo clonal expansion and acquire activation markers [5]. Such CD8+ cells that have recently encountered antigen in the context of a primary infection or vaccination are called effector cells. T cells that exert effector functions after secondary antigen encounter are referred to as effector memory cells [6-8]. Naïve CD8+ cells will proliferate vigorously but will not differentiate in lytic effector cells unless they also receive a “third signal” that can be provided by IL-12 [9, 10] or type I IFNs [11]. In the absence of such third signals, CD8+ cells will proliferate, express perforin but not Granzyme B (GzB) and will lack cytolytic functions [12]. If the immune response is successful and the antigen is cleared/controlled the CD8+ cells become quiescent, their clonal sizes drop, they reacquire a resting phenotype and lose the ability to engage in direct lytic effector functions. Such CD8+ cells that have encountered antigen long ago are called central memory cells [13]. Translating these notions into immune diagnostic considerations one might predict that high clonal sizes and the expression of activation/effector molecules would distinguish between CD8+ cells that are actively engaged in an immune response vs. resting memory cells. The clonal size, however, is not a reliable indicator of active immune processes -- frequencies of virus–specific CD8+ cells are frequently low in chronic viral infections [14-16]. When antigen persists, the antigen-specific CD8+ cell population undergoes exhaustion and the frequencies of the antigen-specific cells drop to barely detectable numbers as is typically seen in the chronic phase of many infections, including HIV, hepatitis B and C or tuberculosis [14, 17, 18]. In these diseases the frequencies of the pathogen-specific T cells can drop to levels so low that present technologies frequently have difficulties to discern uninfected or chronically infected individuals. The distinction between recently activated and long-term memory cells therefore needs to rely on techniques that not only permit the detection of rare antigen-specific cells, but in addition either delineate activation markers/effector molecules or functions of these cells.
Only recently activated CD8+ cells are capable of exerting perforin/GzB-mediated cytolytic functions [6]. Effector CD8+ cells store perforin and GzB molecules in their cytolytic granules, and upon antigen encounter, release these molecules towards the target cell [19-21]. Perforin molecules punch holes into the membrane of the target cell and GzB penetrates inducing apoptosis via activation of caspase pathways [21-23]. While resting CD8+ cells (naïve and central memory cells) have no cytolytic granules containing GzB, they start expressing granzyme within days after antigen encounter – provided they also receive “signal 3” [12, 23]. In the absence of ongoing antigen stimulation CD8+ cells stay GzB positive for approximately one month [24]. Due to ongoing stimulation with various environmental antigens up to 20% of all CD8+ cells in the PBMC of healthy individuals are GzB positive [24]. These notions have implications for the detection of antigen-specific CD8+ cells expressing GzB. Unlike with intracytoplasmic cytokine staining (ICS), where cytokine expression in the unstimulated T cells is very low and hence a small increase in the numbers of T cells expressing the cytokine after antigen stimulation permits the identification of the antigen-specific cells, for GzB the very high baseline expression in human CD8+ cells interferes with flow cytometry-based identification of the antigen-specific cells in a functional assay. Moreover, unlike for the ICS measurements, the stimulation of a granzyme positive CD8+ cell with antigen will not induce detectable additional synthesis of GzB, but to the contrary, will cause the CD8+ cells to release GzB from their lytic granules. In this degranulation process only a fraction of the GzB stored per cell is secreted (enabling the CD8+ cells to be “serial killers”) [22] which makes it challenging to detect the loss of GzB through degranulation by flow cytometry. In ELISPOT assays, on the contrary, the actually released GzB can be measured [25].
Based on the above considerations, tetramer/pentamer staining can be, and has been successfully combined with the detection of intracellularly stored GzB to identify in vivo activated antigen-specific CD8+ cells [26]. Since such effector memory cells have prestored GzB at isolation from the body, they should be able to release GzB upon direct ex vivo stimulation with the antigen. We set out to test this hypothesis. As a paradigm for recently activated effector memory cells, we studied Vaccinia-specific CD8+ cells after revaccination with this virus as well as HIV peptide-reactive CD8+ cells in HIV-infected subjects. As prototypic resting CD8+ memory cells, we tested EBV-, CMV- and flu peptide-reactive CD8+ cells in healthy individuals in whom these infections occurred in the indefinite past, and in whom there was no evidence for active infection.
Materials and Methods
Subjects, cell separation
Peripheral blood was obtained from female and male healthy human donors, 23–45 years of age. They were members of our laboratory or adjoining laboratories. High resolution HLA-typed PBMC were also purchased from Cellular Technology, Limited, CTL, Cleveland OH. Since most humans get exposed to flu, EBV and CMV early in life we assumed that the individuals in our test cohort that responded to such antigens in an ELISPOT assay have been exposed. This assumption should be valid since a clear correlation has been established between an individual's T cell response to viral recall antigen as measured by ELISPOT and a positive antibody titer [27, 28]. Moreover, ELISPOT assays have been shown to have high specificity in settings where antigen exposure affects only a fraction of the population, e.g. HIV infection [29], hepatitis C infection [14], and in settings of immunizations [30]. We therefore assume that positive ELISPOT results are specific and do not entail significant heterologous priming. We have several independent lines of evidence that acute flu, EBV and CMV infections did not occur in our cohort while the T cell data were obtained. First, the donors did not exhibit clinical signs of infections. Second, the frequencies of CD8+ cells specific for these viruses were found to be constant in several repeat experiments over an observation period of close to two years – infections should have resulted in increased frequencies. Third, the functional properties of the CD8+ cells specific for these viruses were constant over the observation period -- reactivation of these CD8+ cells by an acute infection should have resulted in conversion to a GzB positive phenotype.
Three volunteers who had been previously vaccinated with Vaccinia in childhood were revaccinated (using Dryvax®). The donors were bled on the day of the revaccination, prior to the revaccination (day 0), on days 14 and 105 thereafter. In addition, unvaccinated and childhood vaccinated donors were tested. The latter all showed the typical local lesion on the upper arm confirming the take of the prior vaccination. Blood was also obtained from HIV-infected patients from the Special Immunity Unit at the University Hospitals (Cleveland, OH). All HIV patients were under HAART therapy at the time of testing. All studies were performed under the approval of the Institutional Review Board for Human Investigation at the University Hospitals of Cleveland.
PBMC were isolated from heparinized blood by standard Ficoll-Hypaque density gradient centrifugation (using Isoprep, Robbins Scientific, Sunnyvale, CA). CD8+ T lymphocytes, CD4+ T lymphocytes and B lymphocytes were isolated from peripheral blood by negative selection using RosetteSepTM enrichment antibody cocktails according to the manufacturer's instructions (Stemcell Technologies, Vancouver, BC, Canada). The enrichment for the desired phenotype was >95% for CD4+ and CD8+ T lymphocytes and >81% for B lymphocytes as had been previously confirmed by FACS analysis.
Antigens
HLA-A2-restricted peptides of EBV (EBV early lytic protein BMLF1, peptide 259-267, GLCTLVAML; EBV LMP2A, CLGGLLTMV; EBV EBNA3a 325, FLRGRAYGL), CMV (pp65, peptide 495–503, NLVPMVATV; HCMV pp65 495, NLVPMVATV) and flu (Flu Mat 158, GILGFVFTL; Influenza M, ASCMGLIY) were purchased from Princeton Biomolecules (Langhorne, PA). An HIV-Peptide 9mer peptide (VIYQYMDDL, RT346-354 LAI) was used as unspecific control and was purchased from Interactiva (Ulm, Germany). The CEF Peptide Pool [31] containing 23 or 32 8-12mer peptides from CMV, EBV and Influenza as well as the HIV 1 Gag 20mer Peptides (∼1-480) were obtained through the NIH AIDS Research & Reference Reagent Program, Division of AIDS, NIAID, NIH. Single Peptides from the CEF Peptide Pool were purchased form Sigma-Genosys (Woodlands, TX). Mumps virus antigen was from Bio-Whittaker (Walkersville, MA), tetanus toxoid (TT) was a gift from Chiron Behringer GmbH & Co., Marburg, Germany. Purified protein derivative (PPD) prepared from human strains of Mycobacterium tuberculosis (MTB) was purchased from Evans Medical (Leatherhead, England), Candida Antigen was obtained from Greer (Lenoir, NC). Dust Mite Mix was from ALK Abello (Hørsholm, Denmark) and Vaccinia from WR/ABI (Advanced Biotechnologies Inc., Columbia, MD).
Cell culture and ELISPOT assays
When specified as “ex vivo”, PBMC were directly plated into a 24h ELISPOT assay within 5 h after blood draw. When specified, and as specified, PBMC were prestimulated with the nominal peptide for 3 to 8 days under sterile tissue culture conditions before the cells were transferred into an ELISPOT assay of 24 h duration. Cells were cultured in RPMI medium (Biowhittaker, Walkersville, MA) supplemented with 5% heat-inactivated ABO serum (Gemini Bioproducts, Calmasas, CA) and 1% L-glutamine (complete RPMI). If purified CD8+ T-cells were tested in an ELISPOT assay, autologous B cells were used as antigen presenting cells (APC). In the prestimulation cell cultures, purified CD8+ T-cells were supplemented with B cells as APC. Cells were cultured in the presence or absence of antigen challenge. After 3-6 days, the cells were harvested, washed and counted. The cells were then plated in regular 24 h ELISPOT assays.
Human ELISPOT assays were performed as described previously [32]. Briefly, ImmunoSpot M200 or P50 plates (Cellular Technology Ltd, Cleveland, OH) were coated overnight at 4 °C with the cytokine-specific capture antibody diluted in phosphate buffered saline (PBS) at concentrations specified below. The same concentrations were proven to be optimal for both the single color ELISPOT and the double color ELISPOT. Thus, for the double color ELISPOT, the two antibodies were added sequentially. Before plating of cells and antigens, the plates were blocked with bovine serum albumin (BSA) (10 g/l in PBS: PBS–BSA) for 1 h and washed 3x with PBS. PBMC were plated in complete RPMI medium at 2-4 × 105 cells per well (as specified in the table and figure legends). CD8+ T cells were plated at 105 cells per well with 25000 B lymphocytes as APCs. Pre-established optimal antigen concentrations were used for the ELISPOT assay [32]. All peptides were used at 20 µg/ml; Mumps antigen at a final dilution of 1:16. Tetanus Toxoid was used at 77 µg/ml, PPD at 20 µg/ml. Candida was used at 10 µg/ml and Dust mite was at 100 AU/ml. Vaccinia virus was used at 1 moi. We applied PHA at a final concentration of 10 µg/ml.
24 h ELISPOT assays were used for IFN-γ, GzB, IL-2, TNF-α and GM-CSF. For IL-4, IL-5 and IL-10 48 hr assays were performed. Thereafter, the plates were washed 3x with PBS, then 3x with PBS–Tween (0.025%), and the detection antibodies (in PBS–BSA–Tween) were added at previously established concentrations as indicated below. In the case of double color assays, the two antibodies were added simultaneously at the same concentrations as for the single color assays. After an overnight incubation at 4 °C, plates were washed 4x with PBS–Tween and the tertiary reagent was added in PBS–BSA for 2 h at room temperature. For the red system, Streptavidin-HRP conjugate was added at a 1:2000 dilution. For the blue system, Streptavidin-AP was added at a 1:1000 dilution. In the case of double color assays, we used Streptavidin-HRP conjugate at a 1:1000 dilution for the detection of biotin conjugated secondary antibodies and AP labeled anti-FITC antibodies at a dilution of 1:500 for the detection of FITC labeled secondary antibodies. All tertiary reagents were purchased from Dako (Carpenteria, CA). Afterwards, plates were washed 3x with PBS–Tween followed by multiple washes with PBS. The red spots were visualized using either HRP-substrate (Pierce Pharmaceuticals, Rockford, IL) or blue spots via nitriblue tetrazolium (NBT)/5-bromo-chloro-3-indoyl phosphate (BCIP) containing Vector Blue development solution (Vector Laboratories, Burlingame, CA). The Vector Blue development solution was prepared according to the manufacturer's instruction, filtered (0.22 µm) and added to the wells. A total of 200 µl of the development solution was plated per well. The reaction was stopped by rinsing with distilled water when spots became clearly visible macroscopically (10 to 45 min, dependent on the cytokine). When double color assays were performed, the blue color development was done first. The plates were then air-dried overnight before subjecting them to ELISPOT counting using an ImmunoSpot® Analyzer (Cellular Technology Ltd). Data are recorded as spot forming units (SFU) per well, i.e. the number of spots/well. To assess the amount of cytokine produced by single cells, we also measured the spot-size distribution, which is one of the built-in functions of the ImmunoSpot® software. For the ELISPOT assays, the following capture-antibodies were used: IFN-γ (2G1 from Endogen, Woburn, MA) at 3 µg/ml, GzB (GB-11 from BD Bioscience, San Jose, CA) at 5 µg/ml, IL-2 (5334.21 from R&D Systems, Minneapolis, MN) at 5 µg/ml, TNF-α (MAb1 from BD PharMingen, San Diego, CA) at 6 µg/ml, GM-CSF (BVD2-23B6 from BD PharMingen) at 6 µg/ml, IL-4 (8D4-8 from BD PharMingen) at 2 µg/ml, IL-5 (TRFK5 from BD PharMingen) at 4 µg/ml and IL-10 (JES3-9D7 from eBioscience, San Diego, CA) at 4 µg/ml. For the detection of single color ELISPOT assays we used biotinylated antibodies. In double color assays we used a FITC labeled IFN-γ detection antibody: IFN-γ (B133.5 from Endogen, biotinylated or FITC labeled in our laboratory) at 2 µg/ml, GzB (GB-10 from BD Bioscience, San Jose, CA) at 2 µg/ml, IL-2 (BG5 from Endogen) at 0.06 µg/ml, TNF-α (MAb11 from BD PharMingen, San Diego, CA) at 1.25 µg/ml, GM-CSF (BVD2-21C11 from BD PharMingen) at 2 µg/ml, IL-4 (MP4-25D2 from BD PharMingen) at 2 µg/ml, IL-5 (JES1-5A10 from BD PharMingen) at 4 µg/ml and IL-10 (JES3-12G8 from eBioscience) at 4 µg/ml.
Results
In HLA-A2 positive donors individual HLA-A2 restricted CEF peptides induce variable frequencies of IFN-γ producing CD8+ cells
In most healthy human donors infections with flu, EBV and CMV have occurred early in life. These infections have in common that – while the viruses are not necessarily cleared – they are readily controlled by the immune system and therefore the virus-specific memory cells might be considered resting. HLA-A2 restricted peptide determinants recognized by CD8+ cells have been well characterized for these viruses [31]. To study CD8+ cell responses to these three viruses PBMC from six HLA-A2 positive donors were tested for reactivity to five A2-restricted peptides of the above viruses in an IFN-γ ex vivo ELISPOT assay. In addition the CEF peptide pool was used. As can be seen in Table 1 all donors responded to the CEF peptide pool; however the extent to which the individual HLA-A2 peptides induced CD8+ cell responses was highly variable. For example, in donors 1 and 3 peptides EBV LMP2A (CLGGLLTMV) and HCMV pp65 495 (NLVPMVATV) activated a high number of peptide-specific CD8+ cells to produce IFN-γ, but these peptides did not activate CD8+ cells in donors 2, 4, 5 and 6. The latter donors however responded to other individual CEF peptides. These results were highly reproducible in repeat experiments with inter-assay variations less than 30% (data not shown). Cell separations confirmed that the peptide-induced IFN-γ producing cells were CD8+ cells (CD8 cell depletion abrogated IFN-γ production in the PBMC, CD4 cell depletion increased the frequencies of IFN-γ producing cells, see below). The data illustrate that even if donors are HLA-matched, and tested with corresponding “immune dominant” peptides, virus-specific memory cells frequently cannot be reliably detected in all individuals. The frequency of such peptide-reactive T cells apparently underlies considerable inter-individual variation being under the detection limit for a considerable fraction of the test subjects. However, when several peptides of the virus are tested using the CEF peptide pool, CD8+ cell memory is revealed. The data support the notion that in the human population peptide pools will more reliably detect CD8+ cell memory than individual peptides (whereby peptide pools are suitable for testing by ELISPOT and ICS, but not by tetramers).
Table 1.
HLA-A2-peptide induced direct ex vivo IFN-γ ELISPOT responses of HLA-A2 positive healthy donors' PBMCa
| Donors: | #1 | #2 | #3 | #4 | #5 | #6 |
|---|---|---|---|---|---|---|
| HLA-A allele “a” | 2-0201 | 2-0201 | 2-0201 | 2-0201 | 2-0201 | 2-0201 |
| HLA-A allele “b” | 33-3301 | 26-2601 | 2-0201 | 29-2902 | 24-2402 | 3-0301 |
| HLA-A2-restricted peptides: | ||||||
| Flu Mat 158 (GILGFVFTL) | 4 +/- 1 | 40+/- 12 | 72 +/- 38 | 28 +/- 4 | 33 +/- 1 | 4 +/- 0 |
| Flu A (FMYSDFHFI) | 24 +/- 10 | 37+/- 15 | 594 +/-142 | 32 +/- 6 | 32 +/- 3 | 6 +/- 0 |
| EBV LMP2A (CLGGLLTMV) | 139 +/- 15 | 3 +/- 1 | 779 +/-106 | 2 +/- 1 | 1 +/- 1 | 1 +/- 0 |
| EBV BMLF1259 (GLCTLVAML) | 13 +/- 1 | 12+/- 6 | 17 +/-3 | 7 +/- 1 | 4 +/- 1 | 35 +/- 11 |
| HCMV pp65 495 (NLVPMVATV) | 126 +/- 2 | 3 +/-1 | 762 +/-144 | 2 +/- 0 | 3 +/- 1 | 1 +/- 0 |
| CEF peptide pool (32 peptides) | 232 +/- 21 | 81 +/- 29 | 796 +/-60 | 273 +/-47 | 75 +/- 4 | 71 +/- 18 |
PBMC (2 × 105/ well) from the specified HLA-A2 positive donors (identified by high resolution typing) were cultured with the specified HLA-A2- or non-HLA-A2- restricted peptides (sequence specified, tested at 2 µg/ml) for 24 hr and the peptide-induced IFN-γ production was measured in a standard IFN-γ ELISPOT assay. CEF peptide pool (containing 32 peptides of EBV, CMV and flu) was also tested. In the negative control wells spot counts were smaller than 3 spots per well. The data are shown as mean IFN-γ SFU per 200,000 PBMC ± SD for triplicate wells for one experiment that has been reproduced twice with similar results – the inter-assay variation of peptide-induced responses was < 30%.
The extended secretion profile of CEF peptide-induced PBMC
HLA-A2 positive donor/peptide combinations that scored positive in the above screening (Table 1) were selected for the closer functional characterization of the peptide-specific CD8+ memory cells. The peptide-induced secretion of an extended panel of cytokines, i.e. IL-2, TNF-α, GM-CSF, IL-4, IL-5, IL-10 and GzB was measured in ex vivo ELISPOT assays, along with IFN-γ. We tested unseparated PBMC (as pertinent for clinical immune diagnostic) and purified CD8+ cells (verifying that the measured cytokine is indeed CD8+ cell derived). The results obtained for testing unseparated PBMC are summarized in Fig. 1a for EBV peptide BMLF1 259 and CMV pp65 peptide and are representative of all other individual CEF peptides tested, as well as for CEF peptide pool-induced responses. The CEF peptides that induced IFN-γ in the PBMC did not induce detectable GzB, IL-2, IL-4 or IL-5. Stimulation with these peptides induced increased numbers of TNF-α spots in most donors that responded with IFN-γ production to the peptide. IL-10 was induced by the peptides in the PBMC of several donors. With the exception of a single donor, the peptides did not induce GM-CSF in the PBMC. Therefore, in addition to IFN-γ production only TNF-α and IL-10 was reliably induced by CEF peptides in the PBMC.
Figure 1.
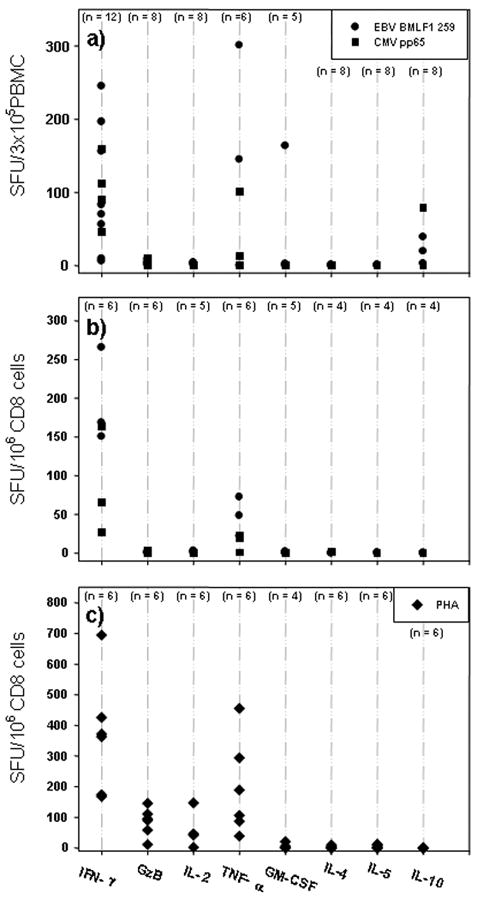
The cytokine signature of EBV- and CMV-specific CD8+ memory cells at isolation from healthy donors. Twenty nine donors were screened for CEF peptide reactivity. Of these subjects, donors were selected that are HLA-A2 positive and that showed an IFN-γ recall response to EBV BMLF1 259 and CMV pp 65 495, as specified by the solid circle or the solid square, respectively. (a) The peptide-induced secretion of an extended panel of cytokines (IL-2, TNF-α, GM-CSF, IL-4, IL-5, IL-10) and GzB was measured in ex vivo ELISPOT assays in freshly isolated PBMC. The medium subtracted mean spot number is shown for all cytokines. The number of donors tested for each cytokine and peptide was between 4 and 12, as specified. (b) The cytokine signature of EBV BMLF1 259 and CMV pp 65 495-specific purified CD8+ cells. CD8+ cells and B cells were purified by negative selection as specified in Materials and Methods. One hundred thousand CD8+ cells were plated with 50,000 B cells. The peptides are specified by the symbols as in Fig. 1a. (c) Purified CD8+ cells and B cells were stimulated with PHA. The legend to (a) and (b) applies.
The cytokine profile of purified CD8+ cells
Testing of cytokine induction in PBMC with class I restricted peptides is frequently done under the assumption that the induced cytokine is CD8+ cell derived. However, this notion is in need of experimental verification. Therefore we purified CD8+ cells from the PBMC of the above donors and tested these cells using B cells as APC. IFN-γ was readily induced in the purified CD8+ cells (Fig. 1b). The numbers of the IFN-γ spots generated by the purified CD8+ cells were compared to the numbers of spots induced in the PBMC. In average, 3.1 times the number of spots was induced using CD8+ cells compared to the respective PBMC (Fig. 2a). This result is consistent with CD8+ cells constituting ∼20-50% of the PBMC. The data suggest that all IFN-γ spots induced by the peptides in PBMC were CD8+ cell derived. Consistent with this notion, purified CD4+ cells did not respond to CEF peptides with IFN-γ spot formation (data not shown). The size of the spots induced in PBMC and CD8+ cells was comparable (Fig. 2c) further suggesting that the CEF peptide-induced IFN-γ is CD8+ cell derived.
Figure 2.
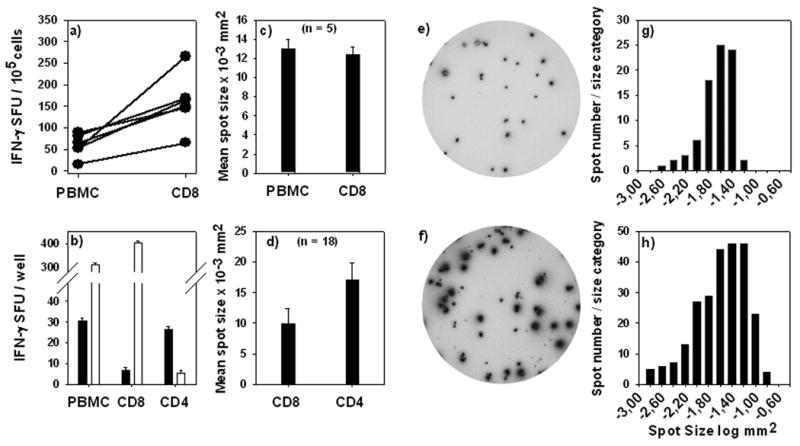
CD8+ cell-derived IFN-γ spots are smaller than CD4+ T cell-derived IFN-γ spots. (a) Spot numbers obtained for PBMC or purified CD8+ cells were normalized for 100,000 cells and corresponding data points are connected. (b) CEF-peptide induced IFN-γ spots are CD8+ cell derived while protein-antigen-induced IFN-γ spots are produced by CD4+ cells. PBMC, CD8+ and CD4+ enriched cells were tested for either mumps protein (solid bars) or EBV BMLF 1 259 peptide (open bars). Cell numbers of CD8+ and CD4+ cells were selected as specified in Materials and Methods matching the frequency of these cells in the PBMC. (c) Mean IFN-γ spot size induced by CEF peptides in PBMC vs. purified CD8+ cells. (d) Mean IFN-γ spot size produced by CD8+ and CD4+ cells. IFN-γ spot size distributions induced by the individual CEF peptides and the protein antigens mumps or candida were compared. The bars show the mean ± SD of spot sizes established in 18 donors for each purified cell type as indicated on the x-axis. A representative example of CEF-peptide-induced CD8+ cell derived IFN-γ spots is shown (e). Spot size distribution for such CD8 cell-derived spots from triplicate wells is shown in (g). A representative example of protein antigen-induced CD4 cell derived IFN-γ spots is shown in (f). The spot size distribution for triplicate wells of CD4 cell derived spots is shown in (h).
Unlike CEF peptides that induced CD8+ cells, protein antigens of mumps (Fig. 2b), Candida, PPD and tetanus toxoid (data not shown) triggered IFN-γ spot formation in CD4+ cells. CD8+ cells were found to produce less IFN-γ on a per cell basis than CD4+ cells. A representative example of IFN-γ spots produced by CD8+ cells is shown in Fig 2e. The spot size distribution for CD8+ cell derived spots is shown in Fig. 2g, which is characteristic for each of the CEF peptide-induced responses of 18 donors whose assay results were subject to detailed morphometric analysis. Parallel measurements of IFN-γ production by CD4+ cells yielded consistently larger spots, as shown by the example of the same donor (Fig. 2f and h vs. e and g) and on the cumulative results obtained for the 18 donors tested (Fig. 2d). CD8+ cells, therefore, produce less IFN-γ cytokine than CD4+ cells. This notion has to be accounted for when setting size gates in ELISPOT analysis.
Purified CD8+ cells produced TNF-α (Fig. 1b) although the spots were smaller, fainter, and lower in number than in PBMC (data not shown) suggesting that in addition to cognate TNF-α secretion by CD8+ cells there is bystander production of this cytokine when assays are performed with unseparated PBMC.
Except for IFN-γ and TNF-α no other cytokine (IL-2,-4, -5, -10, GM-CSF) or GzB was induced in purified CD8+ cells by the CEF peptides ex vivo (Fig. 1b). This outcome is surprising for IL-2 because central memory cells are thought to secrete IL-2 [33]. This finding is also surprising, because the notion is emerging that protective CD8+ cells are multi-cytokine producers, including the production of IL-2 [34]. Flu-specific CD8+ cells certainly qualify as protective CD8+ cells since they have cleared flu virus – yet they definitively did not produce IL-2. It should be noted that stimulation of purified CD4+ cells (or of CD4+ cells within PBMC) with protein antigens such as mumps, candida or PPD consistently yielded a high number of distinct IL-2 spots in the same assays and with cells of the same donors in whom the CEF peptides triggered vigorous IFN-γ production in CD8+ cells, but no detectable IL-2 (data not shown). Therefore, the lack of IL-2 detection when CD8+ cells are tested cannot be attributed to the insensitivity of the assay, but reflects CD8+ cell biology. While IL-2 can be commonly detected by intracytoplasmic staining in CD8+ cells, it seems to be biosynthesized, but not readily secreted by these cells. The IL-10 production induced by the peptides in PBMC (Fig.1a) was not detected in purified CD8+ cells (Fig. 1b), suggesting that this cytokine is produced by macrophages [32].
We stimulated purified CD8+ cells with the mitogen PHA to further verify the cytokine signature of CD8+ cells ex vivo (Fig. 1c). IFN-γ was the prevalently produced cytokine, TNF-α spots were also induced (but these TNF-α spots showed the aforementioned faint morphology). IL-4, IL-5, IL-10 and GM-CSF were not produced by CD8+ cells (but were abundantly generated in PHA stimulated PBMC, providing the positive control for the performance of the assay). Relative to IFN-γ, low frequencies of IL-2 and GzB producing CD8+ cells were detected after the mitogen stimulation.
IFN-γ positive, GzB negative CD8+ memory cells convert into GzB producing cells within 2 days after antigen stimulation
Freshly isolated CEF peptide-reactive CD8+ cells produced IFN-γ within 24 h after the antigen challenge, but did not produce GzB in the same time frame (see above). Purified CD8+ cells that responded with IFN-γ production to a peptide were stimulated with that peptide at isolation and kept in culture for up to 6 days. The production of IFN-γ and GzB was measured at days 2 (3), 4, and 6 as indicated. As shown in Fig. 3, such cells started producing GzB on day 2 with maximal GzB release on day 4, after which it declined. We tested the transferred cells with and without adding antigen again. The results were the same under both conditions. While the cells were thoroughly washed before the transfer to remove soluble peptide, washing apparently did not remove peptide that has already bound to MHC class I molecules. Thus, we attribute our finding to the transfer of peptide loaded APC.
Figure 3.
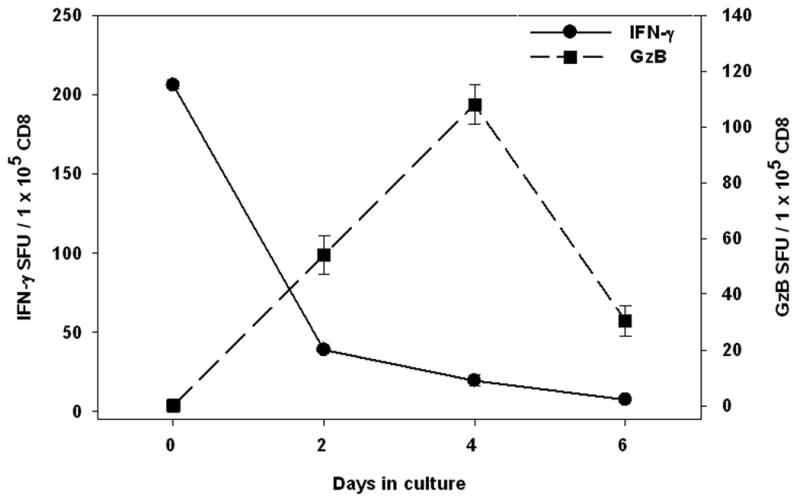
CD8+ memory cells that do not secrete GzB ex vivo start secreting it 2 - 4 days after restimulation. CD8+ cells and B cells were isolated from PBMC of healthy donors and cultured with an individual CEF peptide that induced an ex vivo IFN-γ response (here: EBV BMLF 1 259) or an A-2-restricted HIV control peptide (RT346-354 LAI – this peptide did not induce an ex vivo IFN-γ response). After culture for the specified duration in a tissue culture plate, the cells were harvested, counted, and cell numbers adjusted before transfer into IFN-γ or GzB pre-coated ELISPOT plates, followed by a 24 h single color ELISPOT test. The results for the IFN-γ assay correspond to the y-axis on the left, those of the GzB assay to the y-axis on the right. Mean and SD for duplicate wells are shown. Results are shown for one antigenic peptide (here: EBV BMLF 1 259) in one donor and are fully representative of six donors tested.
The induction of GzB in vitro was confined to in vivo primed memory cells because control peptides did not induce GzB during the 6 day culture (data not shown). As control peptides we used A2-restricted CEF peptides that did not induce IFN-γ in A-2 donors within the 24 h ex vivo culture. As an additional control peptide we used an A-2-restricted HIV peptide (RT346-354 LAI) [35] for these non HIV-infected healthy donors: also this peptide did not induce IFN-γ or GzB neither ex vivo, nor after up to 6 days of in vitro culture (data not shown).
Induction of GzB–producing CD8+ cells after vaccinia virus booster immunization
The data so far showed that CEF peptide-reactive long-term memory cells do not produce GzB ex vivo, which is consistent with the notion that these T cells are quiescent memory cells. To further test this notion, we performed IFN-γ and GzB ELISPOT assays to test for Vaccinia virus reactivity in individuals that had been vaccinated with the virus in childhood. In the majority of these individuals (10/11 individuals) weak but clear-cut IFN-γ responses were elicited ex vivo by Vaccinia virus while this virus did not induce IFN-γ production in non-vaccinated individuals (0/9 individuals). Vaccinia virus did not trigger GzB production ex vivo in any of these individuals (0/20). Three of these donors were revaccinated with Dryvax® through a classic intracutaneous inoculation and the formation of a local lesion in all three confirmed the take of vaccination. These donors were bled before the inoculation and tested in an IFN-γ and GzB ELISPOT assay (day 0 results). Low frequency IFN-γ spots, but no GzB spots were detected in all three donors (Fig. 4b and c). The donors were re-bled and tested on day 14. At this time the virus challenge not only induced a vigorous IFN-γ response, but also GzB production. When re-bled on day 105, the frequencies of Vaccinia-reactive IFN-γ producing cells declined, and at this late time point GzB production was no longer detectable. The original data for one of the donors are shown in Fig. 4a. The data suggest that only CD8+ cells that have been recently engaged in an immune response secrete GzB within 24 h ex vivo.
Figure 4.
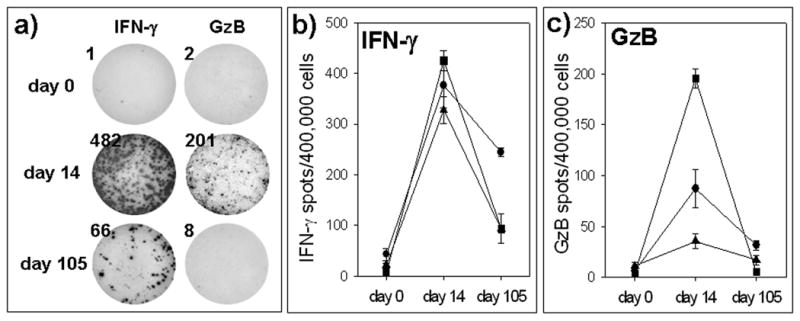
Detection of Vaccinia-specific CD8+ cells producing GzB post Vaccinia reimmunization. Three healthy volunteers who had been vaccinated in childhood were bled prior to revaccination with Dryvax® (day 0), and on days 14 and 105 after the revaccination. Ex vivo IFN-γ (a, b) or GzB (a, c) ELISPOT assays were performed with 400,000 PBMC per well, using Vaccinia virus (Vaccinia WR/ABI at 1 moi) for recall. For each time point and donor the mean spot numbers for triplicate wells are shown and connected by a line. (a) Original well images are shown for one of the donors, with analytes and time points specified.
HIV peptides induce GzB production in HIV-infected individuals directly ex vivo
In HIV infection the immune system does not succeed in controlling the virus. Therefore we postulated that CD8+ cells, which actively combat a virus in vivo at the time of their isolation, are in an activated/effector state and will secrete GzB within 24 h after the antigen challenge ex vivo. We tested this hypothesis by performing 24 h ex vivo GzB assays, in addition to IFN-γ measurements on PBMC of HIV-infected individuals (or healthy controls). These PBMC were challenged with a library of 49 20-mer peptides that cover the gag 1-480 sequence in steps of 5 amino acids. As shown in Figure 5a, the gag peptides induced IFN-γ production in HIV-infected individuals. Consistent with our previous observations [29] and those of others [36], different donors responded to different peptides (Fig. 5a) and no significant IFN-γ spot formation was induced in PBMC of healthy controls (data not shown). Because 20-mer peptides can trigger CD4+ cells in addition to CD8+ cells, we tested CD4 cell-depleted PBMC of HIV-infected subjects. An example of test results obtained with such CD4 cell-depleted PBMC is shown in Fig. 5c showing that these peptides stimulate CD8+ cells.
Figure 5.
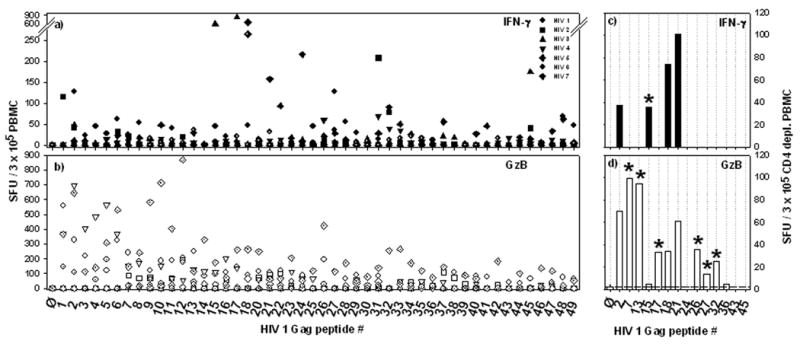
HIV peptides induce GzB secretion in PBMC of HIV-infected donors. GzB and IFN-γ secretion by CD8+ cells is dissociated in these donors. (a and b), PBMC were obtained from 7 HIV-infected individuals and tested in 24 h ex vivo IFN-γ (a) and GzB (b) ELISPOT assays. A peptide library covering HIV-1 gag peptides 1 – 480 was used for recall. The peptides were 20-mers and walked the sequence of gag in steps of 5 a.a. They were tested individually (as specified by the peptide serial numbers on the x-axis), in duplicate well. The means of the duplicates are shown. (c and d) Because 20 a.a. long peptides can induce CD4+ cells in addition to CD8+ cells, PBMC of HIV-infected subjects were CD4 cell-depleted before testing with the HIV peptide library in IFN-γ (c) and GzB (d) ELISPOT assays. Since all HIV+ donors had known low CD4+ cell count (approx. 150 CD4+ cells/µl), similar numbers of CD8+ T cells were used for antigen stimulation in both the total (a and b) and the CD4+ depleted (c and d) cell populations. Selected peptides were tested that previously scored positive/negative. An asterisk highlights peptides that either induced IFN-γ or GzB only.
In parallel to the IFN-γ measurements, GzB ELISPOT assays were performed on PBMC of HIV-infected donors (Fig. 5b). GzB producing cells could be detected in these 24 h ex vivo assays in frequencies similar and higher as compared to the numbers of IFN-γ producing cells. Gag peptides induced GzB spots in the CD4 cell-depleted PBMC of HIV-infected donors (Fig. 5d), but not in CD3 cell depleted PBMC (data not shown).
Similar to the healthy controls, CEF peptides induced IFN-γ production in HIV-infected individuals, but did not trigger the CEF-specific CD8+ cells to release GzB (data not shown). Therefore, only those CD8+ cells secrete GzB in HIV-infected individuals that are engaged in active host defense (the HIV-reactive CD8+ cells), but not the long term memory cells with third party antigen specificity (the CEF-reactive CD8+ cells).
Performing IFN-γ and GzB assays side-by-side on the same samples and testing for reactivity to the same HIV peptides permitted us to measure to what extent CD8+ cells in HIV co-express IFN-γ and GzB. Sixty nine percent of the gag peptides that induced GzB production in the PBMC (such cells are CD8+ cells) did not induce IFN-γ. Testing of CD4 cell depleted PBMC confirmed this observation. The peptides that induced GzB but no IFN-γ are highlighted by an asterisk in Figure 5d. A substantial proportion of HIV-specific CD8+ memory/effector cells, therefore, do not produce IFN-γ. Are these Tc2 cells? Looking at PBMC, the reverse was also frequently seen. Several peptides that induced IFN-γ did not induce GzB. Do CD8+ cells specific for these peptides target determinants that have mutated, and no longer maintain the CD8+ cells in an activated state?
Discussion
Resting CD8+ memory cells can be distinguished from recently activated CD8+ effector cells via cell surface marker expression [37-41], although there is some controversy how reliable the individual markers are delineating when the CD8+ cells last encountered antigen [42]. Tetramer/pentamer analysis excels in establishing such markers for peptide-specific CD8+ cells (except when multiple peptides are targeted, when multiple restriction elements need to be accounted for, and when the frequencies of the peptide-specific cells are low). Tetramer/pentamer-based studies, however, have the weakness that they provide limited functional information because TCR ligation with the tetramers/pentamers interferes with the T cell's functionality, typically inducing apoptosis in the T cells [43]. The weakness of ELISPOT assays is that they do not provide information on surface phenotypes -- but ELISPOT assays excel in assessing the functionality of T cells by measuring the actual release of cytokines and effector molecules after physiological stimulation. Linking functionality data (obtained by ELISPOT or ICS) with markers of resting memory cells is not straightforward, because the very stimulus used to detect the CD8+ cells in the functional assay leads to the activation of these cells and hence their conversion from a resting to an activated phenotype. Based on these considerations we did not seek to establish the complex relationship between phenotype and function, but focused on functions of CD8+ cells only.
The primary scope of our study was to determine whether the differential secretion of effector molecules permits to delineate CD8+ cells as resting memory cells vs. recently activated effector cells. The detection of antigen-induced IFN-γ production has been commonly used to monitor CD8+ cell immunity [44, 45], but IFN-γ is produced by resting and activated CD8+ cells alike not permitting conclusions on the antigen history of the cell [36, 44-48].
Our first set of experiments aimed at studying CD8+ cells that are specific for EBV, CMV and flu peptides. The common feature of these CD8+ cells in healthy subjects is their priming by viral infections at an earlier time point that cannot be exactly defined. However, these infections occurred in all likelihood more than several months, typically several years before the actual characterization of the specific CD8+ cells. While there have been reports suggesting that the different immunobiology of EBV, CMV and flu would differently affect CD8+ cells [49, 50] the memory cells specific for the three viruses showed indistinguishable patterns of cytokine production in all 10 healthy donors that we tested and also behaved identically with respect to the (lack of instant) secretion of GzB. Based on this uniform behavior of these CD8+ cells and on their lack of ex vivo GzB secretion, these CD8+ cells can be considered resting, long-term memory cells. We have extended the number of healthy donors whose PBMC have been tested for CEF-peptide induced IFN-γ and GzB to 29 to date. Consistent with the data shown here, only occasional donors were found to produce GzB upon CEF peptide stimulation, and when seen, the numbers of GzB producing cells were low, lower than for IFN-γ. It is possible that a recent flu infection or active EBV or CMV replication in these donors caused the rather atypical outcome. Unlike for IFN-γ assays, where the use of CEF peptides has become the gold standard for the positive control, for GzB ELISPOT assays these peptides do not seem to be suited as a reliable positive control – however, the mitogen PHA stimulation was found to consistently induce GzB secretion in PBMC.
There is evidence that CMV frequently persists in low viral load and subsequently maintains CMV-reactive CD8+ cells in an activated state, resulting in an activated phenotype and high clonal sizes [51]. When we stimulated PBMC with CMV peptides, we did not elicit GzB production in the CD8+ cells, although IFN-γ secretion was readily detectable – suggesting that even if the persisting antigen led to (partial) activation of these cells it did not result in the CD8+ cell transition into an effector cell that secretes GzB.
It is widely held that resting CD8+ memory (central memory cells) cells secrete IL-2 in addition to IFN-γ [33, 39]. While IFN-γ was readily induced by CEF peptides, none of the individual EBV, CMV and flu peptides, nor the pool of these peptides (CEF) [32] triggered significant IL-2 production in the CD8+ cells of all donors tested. IL-2 spots were either not induced at all, or, in the few instances when detected, they occurred in low numbers (Fig. 1) and in those cases the spot sizes (reflecting the per cell productivity) were orders of magnitudes smaller than the IL-2 spots generated by CD4+ cells. In contrast, protein antigens, including PPD, tetanus toxoid, mumps, and Candida induced IL-2 production by CD4+ cells resulting in large and distinct ELISPOTs (data not shown). Therefore, our IL-2 ELISPOT assay readily detects IL-2 producing cells, but resting CD8+ memory cells either do not release or produce IL-2 or produce it on a minimal scale relative to CD4+ cells. The co-expression of IL-2 with IFN-γ has been reported based on intracytoplasmic cytokine staining (ICS) [33, 52]. ICS and ELISPOT seem to suggest different cytokine profiles for resting CD8+ cells. The former measures intracytoplasmic cytokines in cells that have been treated with inhibitors of the Golgi apparatus and in addition in most instances with costimulatory anti-CD28 and anti-CD49d Abs. On the contrary, ELISPOT measures the actual secretion of the cytokine in unmanipulated cells. It is possible that the pharmacological treatment itself affects IL-2 gene expression, or, alternatively, the secretion of IL-2 might be regulated at a posttranslational level [53]. Other studies suggest a differential regulation of cytokine release dependent on the mode of stimulation [54-56]. Therefore, measurements of IL-2 in contrast to IFN-γ do not seem to be suited for the detection of long term memory CD8+ cells in healthy donors. At the basic immunology level, such EBV-, CMV- and flu-specific CD8+ cells in healthy donors do not qualify as “central memory cells” if IL-2 secretion is a defining parameter. Based on studies using flow cytometry it has been claimed that protective CD8+ cells are multi-cytokine producers, including the secretion of IL-2 [34]. Measuring the actually released IL-2, we have not found such CD8+ cells in the collective that we tested. Importantly, we have found that flu-reactive CD8+ memory cells, that have cleared the virus, and hence qualify as “protective CD8+ cells” showed the same secretory profile as CMV-reactive CD8+ cells that did not succeed in clearing the virus.
Neither the individual peptides, nor the CEF Peptide pool induced IL-4 or IL-5 in PBMC or purified CD8+ cells (Fig. 1a and b). These cytokines were not even induced upon the mitogen PHA stimulation in CD8+ cells (Fig. 1c). In contrast, using the same assays, we have found antigen or PHA stimulated CD4+ cells to secrete these cytokines [57, 58]. It has been reported that CD8+ cells, like CD4+ cells, can differentiate in type 2 cytokine producing cells, called [59]. Apparently, the natural infection by EBV, CMV or flu does not result in significant Tc2 differentiation. Moreover, the essentially undetectable numbers of PHA-inducible IL-4 and IL-5 producing CD8+ cells suggests that Tc2 cells occur at very low frequencies within the CD8+ cell pool of healthy individuals. IL-10 production was induced by several peptides in the PBMC of some donors (Fig. 1a). However, IL-10 was not elicited in the purified CD8+ cell fraction, and hence was not secreted by CD8+ cells. In a previous study we have reported that apparently antigen-induced IL-10 in PBMC is frequently not derived from T cells, but results from macrophage activation [32]. The CEF peptide-induced IL-10 production in PBMC also seems to fall in this category. IL-10 secretion was not induced in CD8+ cells even after PHA stimulation (but was readily seen in PBMC, controlling for the assay performance). Apparently, IL-10 secreting regulatory CD8+ cells also occur in very low frequencies within PBMC.
It has been suggested that CD8+ cells produce GM-CSF [60]. With the exception of one donor's PBMC response to a peptide, GM-CSF was not induced by the CEF peptides in PBMC and in purified CD8+ cells, and even PHA stimulation induced GM-CSF secreting cells only in low numbers (∼30 spots/100,000 CD8+ cells, data not shown). These results obtained by ELISPOT are clearly not a consequence of the assay insensitivity for detection because PHA stimulation of PBMC resulted in “too numerous to count” spots (data not shown).
TNF-α is produced by several cell types of the innate immune system but not by resting T cells [5, 8, 37, 61, 62] and generally not by B cells, although some studies suggest it might be secreted under certain conditions [63]. Frequently, high numbers of TNF-α “background” spots were seen in the medium control wells of PBMC cultures. The addition of antigenic peptide caused only minor increases in the overall spot number, resulting in low “stimulation indices” (antigen-induced spot numbers divided by spot counts in medium control wells). The spots in the medium control were clearly not CD8+ cell derived because purified CD8+ cells (plated with purified B cells as APC) did not produce TNF-α spots in the absence of antigen, but could be induced to produce TNF-α by the addition of the antigen. Thus, the CEF peptide-specific CD8+ cells (resting memory cells) were capable of producing TNF-α. However measuring the cognate CD8+ cell-derived cytokine required purifying the CD8+ cells and testing them on TNF-α negative APC (B cells). Unlike IFN-γ assays, in which very little to no background was seen, TNF-α assays performed on unseparated PBMC did not lend themselves well for monitoring antigen-specific CD8+ cells.
Resting memory CD8+ cells do not have preformed lytic granules containing GzB and it takes several days after reactivation for them to convert into effector cells that contain preformed granules [64]. Within 24 h activation culture CEF peptides (or the CEF peptide pool) rarely induced GzB ex vivo in the healthy donor collective we have studied. According to our results, resting CD8+ cells start secreting GzB 2-4 days after renewed antigen (Fig. 3) challenge. Thus, when freshly isolated PBMC are tested within a 24 h recall assay, resting CD8+ cells will not secrete GzB, while activated CD8+ cells will [41]. The GzB negative phenotype in the primary ELISPOT assay clearly delineates the CEF peptide-reactive CD8+ cells as resting memory cells as opposed to recently activated effector memory cells (whereby non-effector cells can have different activation stages and phenotypes) [49].
Except for GzB, the cytokine/effector molecule signature of CD8+ cells did not change during the 6 days of in vitro antigen stimulation. The major change was the induction of GzB secretion. These data suggested that GzB measurements would permit the distinction between CD8+ cells that have been recently activated in vivo and resting long-term memory cells. To address this hypothesis, we have studied GzB recall responses after Dryvax® immunization, and in HIV-infected individuals.
Supporting the hypothesis, in 24 h ex vivo assays, Vaccinia virus induced IFN-γ, but no GzB production in PBMC of long-term vaccinated individuals. After the reimmunization, however, on day 14, a vigorous GzB response was induced by Vaccinia virus recall in the PBMC. The GzB production was transient, because it became barely detectable by day 105. Using ELISPOT, these data confirm studies in which tetramers were used to track the vaccinia-specific CD8+ response in immunized individuals. One month after vaccinia immunization GzB containing tetramer positive CD8+ cells could be detected which declined within a year [24]. However, the ELISPOT approach should be a significant technological advance for immune diagnostic because it permits the detection of such cells in all humans (irrespective of HLA-type) and without the need to select peptides that are restricted by an allele, and without having to account for the different alleles present in each subject. This is a major advantage because – as shown in Table 1 and in Figure 5 – the peptide recognition patterns in infections are highly variable among different test subjects.
In subjects actively infected with HIV, viral peptides induced GzB production directly ex vivo (Fig. 5b). It is likely that these GzB producing CD8+ cells are effector cells that are actively engaged in combating the virus. Interestingly, in such HIV-infected individuals we detected CD8+ cells that produced IFN-γ, but no GzB (Fig. 5c and d). Based on their signature, these cells qualify as resting memory cells. The CEF peptide-reactive CD8+ cells showed the same phenotype in HIV-infected individuals. Because of the high mutation rate of HIV, it is tempting to postulate that the HIV peptides which induce IFN-γ, but no GzB delineate determinants of the virus that have mutated during the course of the infection. The original sequence (that is tested in the recall assay) has primed CD8+ cells at an earlier time point, inducing effector cells that were IFN-γ and GzB positive, and that then actively participated back in fighting the virus. However, if such determinants were silenced by mutations that led to peptide variants losing MHC-binding properties then the original peptide sequence would not be synthesized anymore. In the absence of stimulation by such peptides, the CD8+ cells that had targeted the original peptide will revert to the stage of resting memory cells and will produce IFN-γ, but no GzB. If this hypothesis is correct, peptides that induce ex vivo GzB in CD8+ cells delineate the determinants that are actively targeted at the time point of testing. Moreover, this hypothesis further predicts that measurements of GzB ex vivo permit monitoring whether a persisting virus such as HIV has been replicating within a time frame of approximately 100 days after which the CD8+ effector cells revert to a GzB negative phenotype. A renewed flare of virus replication will reactivate these resting memory cells and convert them to GzB positive effector memory cells. This notion may also be important for judging the impact of reimmunizations when immunity has already been established. Developing effective vaccines, e.g. for influenza or small pox continues to be high priority. In a clinical trial, the T cells of most patients will have been pre-primed by flu infection or by immunization with vaccinia virus. Such individuals will display IFN-γ producing (resting) CD8+ memory cells [65]. The impact of the newly administered vaccine could be judged by its ability to reactivate these memory cells and to convert them into GzB producing recently activated memory cells.
In summary, our data show that for the detection of resting CD8+ memory cells IFN-γ measurements are the most helpful. Such cells are GzB negative and they do not secrete the cytokines IL-2, IL-4, IL-5, IL-10 and GM-CSF. With respect to the above cytokine profile, activated CD8+ effector cells seem to be indistinguishable from CD8+ memory cells. However, CD8+ effector cells, unlike resting CD8+ memory cells, secrete GzB within 24 h after antigen challenge. Therefore, GzB measurements permit the identification of CD8+ cells that have undergone recent reactivation in vivo.
Acknowledgments
We thank Saada Eid for excellent technical assistance, and Drs J. Kennedy, D. D. Anthony and I. Moldovan for valuable discussions. This work was supported by NIH grant (AI-47756) to MTL and (RES425819) to PVL and by the NIAID contract NO1-AI-40098. TMN and SK are fellows of the Studienstiftung des deutschen Volkes.
Abbreviations
- Flu
influenza
- GzB
granzyme B
- ICS
intracellular cytokine staining
- PPD
Purified Protein Derivative
- TT
tetanus toxoid
- MTB
Mycobacterium tuberculosis
Footnotes
Disclosures PVL and MTL are founders of CTL, a Biotechnology company that specializes in ELISPOT assays.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Harty JT, Tvinnereim AR, White DW. CD8+ T cell effector mechanisms in resistance to infection. Annu Rev Immunol. 2000;18:275–308. doi: 10.1146/annurev.immunol.18.1.275. [DOI] [PubMed] [Google Scholar]
- 2.Ramshaw IA, Ramsay AJ, Karupiah G, Rolph MS, Mahalingam S, Ruby JC. Cytokines and immunity to viral infections. Immunol Rev. 1997;159:119–35. doi: 10.1111/j.1600-065x.1997.tb01011.x. [DOI] [PubMed] [Google Scholar]
- 3.Wong P, Pamer EG. CD8 T cell responses to infectious pathogens. Annu Rev Immunol. 2003;21:29–70. doi: 10.1146/annurev.immunol.21.120601.141114. [DOI] [PubMed] [Google Scholar]
- 4.Oehen S, Brduscha-Riem K. Differentiation of naive CTL to effector and memory CTL: correlation of effector function with phenotype and cell division. J Immunol. 1998;161:5338–46. [PubMed] [Google Scholar]
- 5.Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78:5535–45. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaech SM, Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol. 2001;2:415–22. doi: 10.1038/87720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takata H, Takiguchi M. Three memory subsets of human CD8+ T cells differently expressing three cytolytic effector molecules. J Immunol. 2006;177:4330–40. doi: 10.4049/jimmunol.177.7.4330. [DOI] [PubMed] [Google Scholar]
- 8.Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–34. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 9.Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med. 2003;197:1141–51. doi: 10.1084/jem.20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, Mescher MF. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol. 1999;162:3256–62. [PubMed] [Google Scholar]
- 11.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. 2005;174:4465–9. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 12.Curtsinger JM, Lins DC, Johnson CM, Mescher MF. Signal 3 tolerant CD8 T cells degranulate in response to antigen but lack granzyme B to mediate cytolysis. J Immunol. 2005;175:4392–9. doi: 10.4049/jimmunol.175.7.4392. [DOI] [PubMed] [Google Scholar]
- 13.Lanzavecchia A, Sallusto F. Understanding the generation and function of memory T cell subsets. Curr Opin Immunol. 2005;17:326–32. doi: 10.1016/j.coi.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 14.Anthony DD, Valdez H, Post AB, Carlson NL, Heeger PS, Lehmann PV. Comprehensive determinant mapping of the hepatitis C-specific CD8 cell repertoire reveals unpredicted immune hierarchy. Clin Immunol. 2002;103:264–76. doi: 10.1006/clim.2001.5193. [DOI] [PubMed] [Google Scholar]
- 15.Weekes MP, Wills MR, Sissons JG, Carmichael AJ. Large HIV-specific CD8 cytotoxic T-lymphocyte (CTL) clones reduce their overall size but maintain high frequencies of memory CTL following highly active antiretroviral therapy. Immunology. 2006;118:25–38. doi: 10.1111/j.1365-2567.2006.02334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitmire JK, Ahmed R. The economy of T-cell memory: CD4+ recession in times of CD8+ stability? Nat Med. 2001;7:892–3. doi: 10.1038/90923. [DOI] [PubMed] [Google Scholar]
- 17.Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science. 1999;286:958–61. doi: 10.1126/science.286.5441.958. [DOI] [PubMed] [Google Scholar]
- 18.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 19.Shresta S, Pham CT, Thomas DA, Graubert TA, Ley TJ. How do cytotoxic lymphocytes kill their targets? Curr Opin Immunol. 1998;10:581–7. doi: 10.1016/s0952-7915(98)80227-6. [DOI] [PubMed] [Google Scholar]
- 20.Trambas CM, Griffiths GM. Delivering the kiss of death. Nat Immunol. 2003;4:399–403. doi: 10.1038/ni0503-399. [DOI] [PubMed] [Google Scholar]
- 21.Trapani JA, Smyth MJ. Functional significance of the perforin/granzyme cell death pathway. Nat Rev Immunol. 2002;2:735–47. doi: 10.1038/nri911. [DOI] [PubMed] [Google Scholar]
- 22.Catalfamo M, Henkart PA. Perforin and the granule exocytosis cytotoxicity pathway. Curr Opin Immunol. 2003;15:522–7. doi: 10.1016/s0952-7915(03)00114-6. [DOI] [PubMed] [Google Scholar]
- 23.Trapani JA, Sutton VR. Granzyme B: pro-apoptotic, antiviral and antitumor functions. Curr Opin Immunol. 2003;15:533–43. doi: 10.1016/s0952-7915(03)00107-9. [DOI] [PubMed] [Google Scholar]
- 24.Rock MT, Yoder SM, Wright PF, Talbot TR, Edwards KM, Crowe JE., Jr Differential regulation of granzyme and perforin in effector and memory T cells following smallpox immunization. J Immunol. 2005;174:3757–64. doi: 10.4049/jimmunol.174.6.3757. [DOI] [PubMed] [Google Scholar]
- 25.Rininsland FH, Helms T, Asaad RJ, Boehm BO, Tary-Lehmann M. Granzyme B ELISPOT assay for ex vivo measurements of T cell immunity. J Immunol Methods. 2000;240:143–55. doi: 10.1016/s0022-1759(00)00191-5. [DOI] [PubMed] [Google Scholar]
- 26.Appay V, Rowland-Jones SL. The assessment of antigen-specific CD8+ T cells through the combination of MHC class I tetramer and intracellular staining. J Immunol Methods. 2002;268:9–19. doi: 10.1016/s0022-1759(02)00195-3. [DOI] [PubMed] [Google Scholar]
- 27.Frey SE, Newman FK, Yan L, Lottenbach KR, Belshe RB. Response to smallpox vaccine in persons immunized in the distant past. Jama. 2003;289:3295–9. doi: 10.1001/jama.289.24.3295. [DOI] [PubMed] [Google Scholar]
- 28.Hobeika AC, Morse MA, Osada T, Ghanayem M, Niedzwiecki D, Barrier R, Lyerly HK, Clay TM. Enumerating antigen-specific T-cell responses in peripheral blood: a comparison of peptide MHC Tetramer, ELISpot, and intracellular cytokine analysis. J Immunother (1997) 2005;28:63–72. doi: 10.1097/00002371-200501000-00008. [DOI] [PubMed] [Google Scholar]
- 29.Kleen TO, Asaad R, Landry SJ, Boehm BO, Tary-Lehmann M. Tc1 effector diversity shows dissociated expression of granzyme B and interferon-gamma in HIV infection. Aids. 2004;18:383–92. doi: 10.1097/00002030-200402200-00003. [DOI] [PubMed] [Google Scholar]
- 30.Kuerten S, Lichtenegger FS, Faas S, Angelov DN, Tary-Lehmann M, Lehmann PV. MBP-PLP fusion protein-induced EAE in C57BL/6 mice. J Neuroimmunol. 2006;177:99–111. doi: 10.1016/j.jneuroim.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 31.Currier JR, Kuta EG, Turk E, Earhart LB, Loomis-Price L, Janetzki S, Ferrari G, Birx DL, Cox JH. A panel of MHC class I restricted viral peptides for use as a quality control for vaccine trial ELISPOT assays. J Immunol Methods. 2002;260:157–72. doi: 10.1016/s0022-1759(01)00535-x. [DOI] [PubMed] [Google Scholar]
- 32.Guerkov RE, Targoni OS, Kreher CR, Boehm BO, Herrera MT, Tary-Lehmann M, Lehmann PV, Schwander SK. Detection of low-frequency antigen-specific IL-10-producing CD4(+) T cells via ELISPOT in PBMC: cognate vs. nonspecific production of the cytokine. J Immunol Methods. 2003;279:111–21. doi: 10.1016/s0022-1759(03)00240-0. [DOI] [PubMed] [Google Scholar]
- 33.Hamann D, Baars PA, Rep MH, Hooibrink B, Kerkhof-Garde SR, Klein MR, van Lier RA. Phenotypic and functional separation of memory and effector human CD8+ T cells. J Exp Med. 1997;186:1407–18. doi: 10.1084/jem.186.9.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Precopio ML, Betts MR, Parrino J, Price DA, Gostick E, Ambrozak DR, Asher TE, Douek DC, Harari A, Pantaleo G, Bailer R, Graham BS, Roederer M, Koup RA. Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8(+) T cell responses. J Exp Med. 2007;204:1405–16. doi: 10.1084/jem.20062363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harrer E, Harrer T, Barbosa P, Feinberg M, Johnson RP, Buchbinder S, Walker BD. Recognition of the highly conserved YMDD region in the human immunodeficiency virus type 1 reverse transcriptase by HLA-A2-restricted cytotoxic T lymphocytes from an asymptomatic long-term nonprogressor. J Infect Dis. 1996;173:476–9. doi: 10.1093/infdis/173.2.476. [DOI] [PubMed] [Google Scholar]
- 36.Addo MM, Yu XG, Rathod A, Cohen D, Eldridge RL, Strick D, Johnston MN, Corcoran C, Wurcel AG, Fitzpatrick CA, Feeney ME, Rodriguez WR, Basgoz N, Draenert R, Stone DR, Brander C, Goulder PJ, Rosenberg ES, Altfeld M, Walker BD. Comprehensive epitope analysis of human immunodeficiency virus type 1 (HIV-1)-specific T-cell responses directed against the entire expressed HIV-1 genome demonstrate broadly directed responses, but no correlation to viral load. J Virol. 2003;77:2081–92. doi: 10.1128/JVI.77.3.2081-2092.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ravkov EV, Myrick CM, Altman JD. Immediate early effector functions of virus-specific CD8+CCR7+ memory cells in humans defined by HLA and CC chemokine ligand 19 tetramers. J Immunol. 2003;170:2461–8. doi: 10.4049/jimmunol.170.5.2461. [DOI] [PubMed] [Google Scholar]
- 38.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 39.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–12. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 40.Tussey L, Speller S, Gallimore A, Vessey R. Functionally distinct CD8+ memory T cell subsets in persistent EBV infection are differentiated by migratory receptor expression. Eur J Immunol. 2000;30:1823–9. doi: 10.1002/1521-4141(200007)30:7<1823::AID-IMMU1823>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 41.Wolint P, Betts MR, Koup RA, Oxenius A. Immediate cytotoxicity but not degranulation distinguishes effector and memory subsets of CD8+ T cells. J Exp Med. 2004;199:925–36. doi: 10.1084/jem.20031799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rocha B, Tanchot C. CD8 T cell memory. Semin Immunol. 2004;16:305–14. doi: 10.1016/j.smim.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 43.Cebecauer M, Guillaume P, Hozak P, Mark S, Everett H, Schneider P, Luescher IF. Soluble MHC-peptide complexes induce rapid death of CD8+ CTL. J Immunol. 2005;174:6809–19. doi: 10.4049/jimmunol.174.11.6809. [DOI] [PubMed] [Google Scholar]
- 44.Kaufhold RM, Field JA, Caulfield MJ, Wang S, Joseph H, Wooters MA, Green T, Clark HF, Krah D, Smith JG. Memory T-cell response to rotavirus detected with a gamma interferon enzyme-linked immunospot assay. J Virol. 2005;79:5684–94. doi: 10.1128/JVI.79.9.5684-5694.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Terajima M, Cruz J, Raines G, Kilpatrick ED, Kennedy JS, Rothman AL, Ennis FA. Quantitation of CD8+ T cell responses to newly identified HLA-A*0201-restricted T cell epitopes conserved among vaccinia and variola (smallpox) viruses. J Exp Med. 2003;197:927–32. doi: 10.1084/jem.20022222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kristensen NN, Madsen AN, Thomsen AR, Christensen JP. Cytokine production by virus-specific CD8(+) T cells varies with activation state and localization, but not with TCR avidity. J Gen Virol. 2004;85:1703–12. doi: 10.1099/vir.0.79903-0. [DOI] [PubMed] [Google Scholar]
- 47.Slifka MK, Rodriguez F, Whitton JL. Rapid on/off cycling of cytokine production by virus-specific CD8+ T cells. Nature. 1999;401:76–9. doi: 10.1038/43454. [DOI] [PubMed] [Google Scholar]
- 48.Unsoeld H, Krautwald S, Voehringer D, Kunzendorf U, Pircher H. Cutting edge: CCR7+ and CCR7- memory T cells do not differ in immediate effector cell function. J Immunol. 2002;169:638–41. doi: 10.4049/jimmunol.169.2.638. [DOI] [PubMed] [Google Scholar]
- 49.Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, Ogg GS, King A, Lechner F, Spina CA, Little S, Havlir DV, Richman DD, Gruener N, Pape G, Waters A, Easterbrook P, Salio M, Cerundolo V, McMichael AJ, Rowland-Jones SL. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002;8:379–85. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- 50.Selin LK, Welsh RM. Plasticity of T cell memory responses to viruses. Immunity. 2004;20:5–16. doi: 10.1016/S1074-7613(03)00356-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klenerman P, Hill A. T cells and viral persistence: lessons from diverse infections. Nat Immunol. 2005;6:873–9. doi: 10.1038/ni1241. [DOI] [PubMed] [Google Scholar]
- 52.Sandberg JK, Fast NM, Nixon DF. Functional heterogeneity of cytokines and cytolytic effector molecules in human CD8+ T lymphocytes. J Immunol. 2001;167:181–7. doi: 10.4049/jimmunol.167.1.181. [DOI] [PubMed] [Google Scholar]
- 53.Gaffen SL, Liu KD. Overview of interleukin-2 function, production and clinical applications. Cytokine. 2004;28:109–23. doi: 10.1016/j.cyto.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 54.Beadling C, Slifka MK. Differential regulation of virus-specific T-cell effector functions following activation by peptide or innate cytokines. Blood. 2005;105:1179–86. doi: 10.1182/blood-2004-07-2833. [DOI] [PubMed] [Google Scholar]
- 55.Tough DF, Sun S, Sprent J. T cell stimulation in vivo by lipopolysaccharide (LPS) J Exp Med. 1997;185:2089–94. doi: 10.1084/jem.185.12.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tough DF, Zhang X, Sprent J. An IFN-gamma-dependent pathway controls stimulation of memory phenotype CD8+ T cell turnover in vivo by IL-12, IL-18, and IFN-gamma. J Immunol. 2001;166:6007–11. doi: 10.4049/jimmunol.166.10.6007. [DOI] [PubMed] [Google Scholar]
- 57.Helms T, Boehm BO, Asaad RJ, Trezza RP, Lehmann PV, Tary-Lehmann M. Direct visualization of cytokine-producing recall antigen-specific CD4 memory T cells in healthy individuals and HIV patients. J Immunol. 2000;164:3723–32. doi: 10.4049/jimmunol.164.7.3723. [DOI] [PubMed] [Google Scholar]
- 58.Kreher CR, Dittrich MT, Guerkov R, Boehm BO, Tary-Lehmann M. CD4+ and CD8+ cells in cryopreserved human PBMC maintain full functionality in cytokine ELISPOT assays. J Immunol Methods. 2003;278:79–93. doi: 10.1016/s0022-1759(03)00226-6. [DOI] [PubMed] [Google Scholar]
- 59.Woodland DL, Dutton RW. Heterogeneity of CD4(+) and CD8(+) T cells. Curr Opin Immunol. 2003;15:336–42. doi: 10.1016/s0952-7915(03)00037-2. [DOI] [PubMed] [Google Scholar]
- 60.Mosmann TR, Li L, Sad S. Functions of CD8 T-cell subsets secreting different cytokine patterns. Semin Immunol. 1997;9:87–92. doi: 10.1006/smim.1997.0065. [DOI] [PubMed] [Google Scholar]
- 61.Kaech SM, Ahmed R. Immunology. CD8 T cells remember with a little help. Science. 2003;300:263–5. doi: 10.1126/science.1084511. [DOI] [PubMed] [Google Scholar]
- 62.Wong GH, Goeddel DV. Tumour necrosis factors alpha and beta inhibit virus replication and synergize with interferons. Nature. 1986;323:819–22. doi: 10.1038/323819a0. [DOI] [PubMed] [Google Scholar]
- 63.Duddy ME, Alter A, Bar-Or A. Distinct profiles of human B cell effector cytokines: a role in immune regulation? J Immunol. 2004;172:3422–7. doi: 10.4049/jimmunol.172.6.3422. [DOI] [PubMed] [Google Scholar]
- 64.Bachmann MF, Barner M, Viola A, Kopf M. Distinct kinetics of cytokine production and cytolysis in effector and memory T cells after viral infection. Eur J Immunol. 1999;29:291–9. doi: 10.1002/(SICI)1521-4141(199901)29:01<291::AID-IMMU291>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 65.Kennedy JS, Frey SE, Yan L, Rothman AL, Cruz J, Newman FK, Orphin L, Belshe RB, Ennis FA. Induction of human T cell-mediated immune responses after primary and secondary smallpox vaccination. J Infect Dis. 2004;190:1286–94. doi: 10.1086/423848. [DOI] [PubMed] [Google Scholar]