

Abstract
Resin based materials are now widely used in dental restorations. While the use of these materials is aesthetically appealing in patients, they carry the risk of local and systemic adverse effects. The potential risks are direct damage to the cells and induction of immune-based hypersensitivity reactions. Dental Pulp Stromal Cells (DPSCs) and oral keratinocytes are the major cell types which may come in contact with dental resins such as 2-hydroxyethyl methacrylate (HEMA) after dental restorations. Here we show that N-acetyl cysteine (NAC) inhibits HEMA induced apoptotic cell death and restores the function of DPSCs and oral epithelial cells. NAC inhibits HEMA mediated toxicity through induction of differentiation in DPSCs since the genes for dentin sialoprotein (DSP), Osteopontin (OPN), Osteocalcin (OCN), and Alkaline Phosphatase (ALP) which are induced during differentiation are also induced by NAC. Unlike NAC, Vitamin E and C which known anti-oxidant compounds failed to prevent either HEMA mediated cell death or decrease in VEGF secretion by human DPSCs. More importantly, when added either alone or in combination with HEMA Vitamin E and Vitamin C did not increase the gene expression for OPN, and in addition Vitamin E inhibited the protective effect of NAC on DPSCs. NAC inhibited HEMA mediated decrease in NFκB activity thus, providing survival mechanism for the cells. Overall, the studies reported in this paper indicated that undifferentiated DPSCs have exquisite sensitivity to HEMA induced cell death, and their differentiation by NAC resulted in an increased NFκB activity which might have provided the basis for their increased protection from HEMA mediated functional loss and cell death.
Keywords: NAC, NFκB, Dental Pulp Stromal Cells, differentiation, VEGF
Introduction
Dental pulp contains a diverse population of the cells including odontoblasts, fibroblasts, immune cells, nerve cells and vascular cells. [1, 2]. Indeed, a population of putative post-natal stem cells within the dental pulp stromal cells (DPSCs) was found to have the capability to regenerate a dentin-pulp-like complex, which is composed of mineralized matrix with tubules lined with odontoblasts, and fibrous tissue containing blood vessels in an arrangement similar to the dentin-pulp complex found in normal human teeth [3, 4]. Differentiation of the pulp stromal cells into odontoblasts is essential as these cells are capable of laying secondary/reparative dentin which in turn is able to protect the pulp from further damage induced by a number of mechanisms including those induced by resin materials. Odontoblasts are highly specialized cells aligned in a single layer at the edge of the dental pulp and are responsible for secretion and mineralization of the fibrillar extracellular matrix of the dentin [4]. Odontoblasts are known to express specific proteins such as dentin sialoprotein (DSP), Osteopontin (OPN), Osteocalcin (OCN), type 1- collagen, Alkaline Phosphatase (ALP) and Dentin Matrix Protein-1 (DMP-1). [1, 2]. OPN which is an early stage osteoblastic marker is also expressed by odontoblasts since this is a component of their matrix.
Resin based materials are now widely used in dental practice. While the use of these materials is aesthetically appealing in patients, they carry the risk of local and systemic adverse effects. The potential risks are direct damage to the cells (cytotoxicity) and induction of immune-based hypersensitivity reactions. However, the mechanisms by which these materials cause adverse effects in patients have not been completely elucidated. Resin monomers such as HEMA (2-hydroxyethyl methacrylate) and TEGDMA (triethyleneglycol dimethacrylate) are shown to influence the differentiation of human pulp cells into odontoblasts [5]. We have shown previously that HEMA induces apoptosis in different cell types [6] and also in DPSCs and in odontoblast like cells. HEMA induced apoptosis has been linked to the decrease in intracellular glutathione (GSH) levels and the production of reactive oxygen species (ROS) in the cells. [7–11].
N-acetyl cysteine (NAC) is a membrane permeable aminothiol compound with diverse functions. NAC is shown to be the precursor of glutathiones (GSH) with a significant anti-oxidant effect. Although previously published reports on the function of NAC have largely been concentrated on its anti-oxidant effect, recent reports have underscored the significance of this compound in inhibition of proliferation and induction of differentiation [12–14]. NAC has also been regarded as an anti-inflammatory compound [15–17]. Therefore, NAC is shown to have a number of different functions depending on the nature of the cells and their stage of maturation. However, it seems one of the primary functions of NAC is inhibition of cell death. At present it is unclear how NAC protects the cells from cell death. However, previous work has attributed the protective effect of NAC in HEMA and TEGDMA mediated cell death to their anti-oxidant effect [18–20]. In contrast to these reports, our data suggests mechanisms for NAC function which are distinct from those shown previously and those mediated by other well established anti-oxidants such as Vitamin E and C [21].
In this paper we report that undifferentiated DPSCs have exquisite sensitivity to HEMA induced cell death, and their differentiation by NAC provides the basis for their increased protection from HEMA mediated functional loss and cell death. Increased differentiation triggered by NAC in DPSCs is paralleled with an increased induction of NFκB activity since this transcription factor is responsible for survival of the cells.
Materials and Methods
Cells and reagents
DMEM (Cellgro, VA) and RPMI were supplemented with 10 %FBS, 1% non-essential amino-acids, 1% sodium pyruvate, 1% streptomycin and 1%L-glutamine (Invitrogen; Carlsbad, CA) and used for cultures of DPSCs and UCLA-2 oral tumors respectively. β-glycerophosphate, ascorbic acid, dexamethasone, N-acetyl-cysteine (NAC), 2-hydroxyethyl methacrylate (HEMA) and HEPES were all purchased from Sigma (St. Louis, MO). The Fluorescein Isothiocyanate conjugated Annexin V/Propidium Iodide kit was purchased from Coulter Immunotech (Miami, FL). Recombinant IL-2 was purchased from the Chiron Corporation (Emeryville, CA). RNA extraction kit was purchased from Qiagen (Valencia, CA) and the reverse transcription kit was purchased from Invitrogen (Carlsbad, CA). All the PCR primers were obtained from Operon Biotechnologies (Huntsville, AL).
Dental pulp stromal cell cultures
DPSCs were obtained from both rat and human teeth. DPSCs from both species responded similarly to the effects of HEMA and NAC. Dental pulp was extracted from the upper and lower central incisors of 8-week old male Sprague-Dawley rats. The extracted tissue was treated with 0.1% collagenase (Sigma Aldrich, MO) and 0.25% trypsin EDTA (1mM) (Invitrogen, CA) at 37°C for 60 minutes to obtain single cell suspension of the pulp cells. Cells were then washed twice and re-suspended in complete medium (DMEM supplemented with 10% FBS, 1% antibiotic-antimycotic, 1% non-essential amino acids, 1% Na pyruvate). Dental pulp stromal cells were then cultured with ascorbic acid (50 μg/ml) and Na-β-glycerophosphate (10mM) (Sigma Aldrich, MO) with or without dexamethasone (10−8M) [22] to induce different degrees of differentiation as indicated in the result section. The cells were then cultured at 37°C in 5% CO2, and they were passaged and used in the experiments at 80% confluency.
Human dental pulp stromal cells were obtained from freshly extracted third molars and were cultured as described above [5].
Purification of Peripheral blood mononuclear cells (PBMCs)
All donors in this study provided informed consents, and protocols involving human subjects received UCLA Institutional Review Board approval (IRB # 97-10-049-14). PBMCs from the healthy donors were isolated as described before [23–25].
Determination of apoptosis
To determine the levels of apoptosis FITC-Annexin V and Propidium Iodide was used. The cells were treated HEMA and NAC, Vitamin C and Vitamin E as stated in the Results section, and they were washed twice and re-suspended in binding buffer containing FITC-Annexin V/Propidium iodide as suggested by the manufacturer. After 15 minutes of incubation on ice, Annexin V/PI stained cells were analyzed by flow cytometry. Flow cytometric analysis was performed using EPICs-ELITE flow cytometer (Coulter, Miami, FL). Dead cell fragments and debris were gated out by forward and side scatter analysis.
Alkaline Phosphatase (ALP) staining
Human DPSCs were treated with varying concentrations of HEMA, NAC and Vitamin E or co-cultured with and without untreated and IL-2 treated PBMCs as indicated in the result section. Cells were then washed twice with PBS and incubated with 120mM of Tris buffer (pH = 8.4) containing 0.9mM Napthol AS-M Phosphate and 1.8mM Fast Red TR (both purchased from Sigma, MO) for 30 minutes at 37 C. After 30 minute incubation, cells were washed three times with PBS and then fixed with 1ml cold ethanol (100%) for 30 minutes. The stained cultures were scanned using an Epson scanner 1250.
ELISA
Duo-set ELISA for measuring VEGF was purchased from R&D systems (Minneapolis, MN) and used according to the manufacturer’s recommendations. IFN-γ was measured as described previously [26].
3H Thymidine incorporation assay was performed as described previously [27]
Luciferase reporter assay
Human embryonic kidney (293T) cells, HEp2 oral epithelial cells and DPSCs were plated and maintained in DMEM medium supplemented with 10% FBS and 1% penicillin/streptomycin before transfection. Transfections were done using an NFκB Luciferase reporter vector [28] and Lipofectamine 2000 reagent (Invitrogen, CA) in Opti-MEM media (Invitrogen, CA) for 18 hours after which they were treated with HEMA, NAC and TNF-α as indicated in the Result section. The cells were then lysed with lysis buffer and the relative Luciferase activity was measured using the Luciferase assay reagent kit obtained from Promega (Madison, WI)
Reverse-transcriptase polymerase chain reaction (RT-PCR)
The rat and human DPSCs were cultured in a 6 well plate (2× 105/well) and treated as described in the result section for 18 hours, after which the cells were washed twice with PBS and then the total RNA was extracted using TRIzol (Invitrogen, CA) and a purification column (RNeasy, Qiagen, CA). Following DNAse I treatment, 0.5 μg of total RNA was incubated with MMLV reverse transcriptase (Invitrogen, CA) at 42 °C for an hour with oligodT primer (Invitrogen, CA). The PCR was performed using Platinum Taq DNA polymerase (Invitrogen, CA) to evaluate the gene expression for osteopontin, osteocalcin and dentin sialoprotein. GAPDH was used as the loading control. The resulting products were visualized with ethidium bromide staining on a 1.5% agarose gel. The primer design and PCR conditions are as follows:
| Rat primers | Forward | Reverse | Annealing Temperature | # of cycles |
|---|---|---|---|---|
| GAPDH | 5′-TGAAGGTCGGTGTCAACGGATTTGGC | 5′-CATGTAGGCCATGAGGTCCACCAC | 67 | 27 |
| Collagen-1 | 5′-GGCAACAGTCGATTCACC | 5′-AGGGCCAATGTCCATTCC | 58 | 28 |
| DSP | 5′-CGAGTCGATAGCCGTAGGAG | 5′-AACTCCACTCCCGTGTGTTC | 62 | 36 |
| Osteopontin | 5′–GATTATAGTGACACAGAC | 5′–AGCAGGAATACTAACTGC | 45 | 19 |
| Osteocalcin | 5′–GTCCCACACAGCAACTCG | 5′–CCAAAGCTGAAGCTGCCG | 61 | 25 |
| Human Primers | ||||
| GAPDH | 5′–CAGTCAGTTCTAGAATTCCATATG GGGAAGGTGAAGGTCGGAGTCAAC |
5′-GGTGGTCCAGGGATCGATCTC CTTGGAGGCCATGTGGGCCAT |
70 | 27 |
| Osteopontin | 5′-GGACAGCCAGGACTCCATTGA | 5′-CGTTTCATAACTGTCCTTCCCA | 71 | 35 |
| Osteocalcin | 5′-CAGAGTCCAGCAAAGGTGCAG | 5′-TCAGCCAACTCGTCACAGTCCG | 68 | 30 |
Statistical analysis
Unpaired two-tailed student t-test was performed for the statistical analysis. ANOVA with a Bonferroni post test was used to compare the different groups.
Results
Cell death induced in Dental Pulp Stromal Cells co-relates with the degree and stages of differentiation of the cells
The sensitivity to cell death in Dental Pulp Stromal Cells (DPSCs) were assessed in three different stages of cell growth and differentiation, 1-undifferentiated 2-differentiated in the presence of β-glycerophosphate and ascorbic acid and 3-differentiated in the presence of β-glycerophosphate, ascorbic acid and dexamethasone. These three stages of cell growth and differentiation can clearly be distinguished at the gene level using the gene expression for osteopontin (OPN) and osteocalcin (OCN) (Fig. 1A). Undifferentiated cells either did not express or expressed OPN or OCN at very low levels. Cells differentiated with β-glycerophosphate and ascorbic acid only increased OPN but not OCN whereas those differentiated with β-glycerophosphate, ascorbic acid and dexamethasone increased both the OPN and OCN (Fig. 1A).
Fig-1. Differentiation increases gene expression and survival in DPSCs.
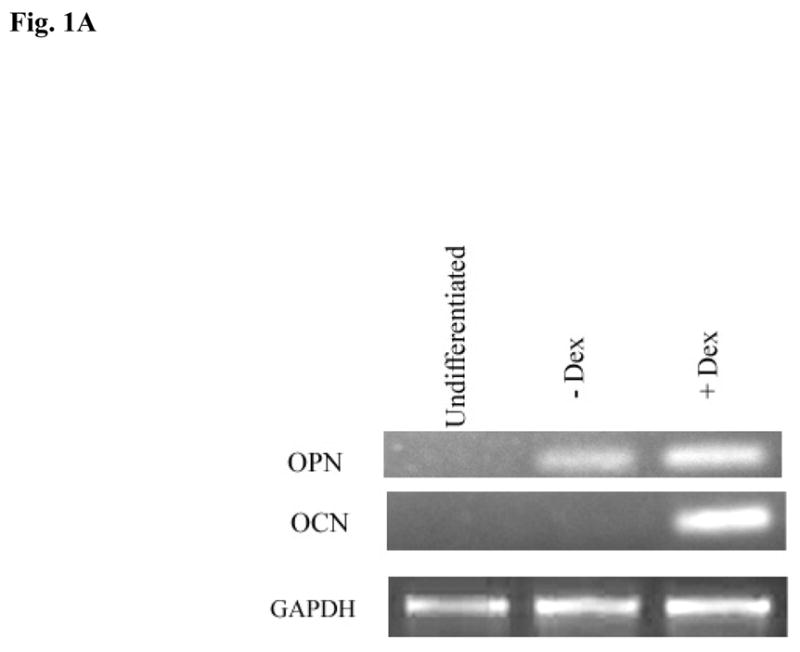
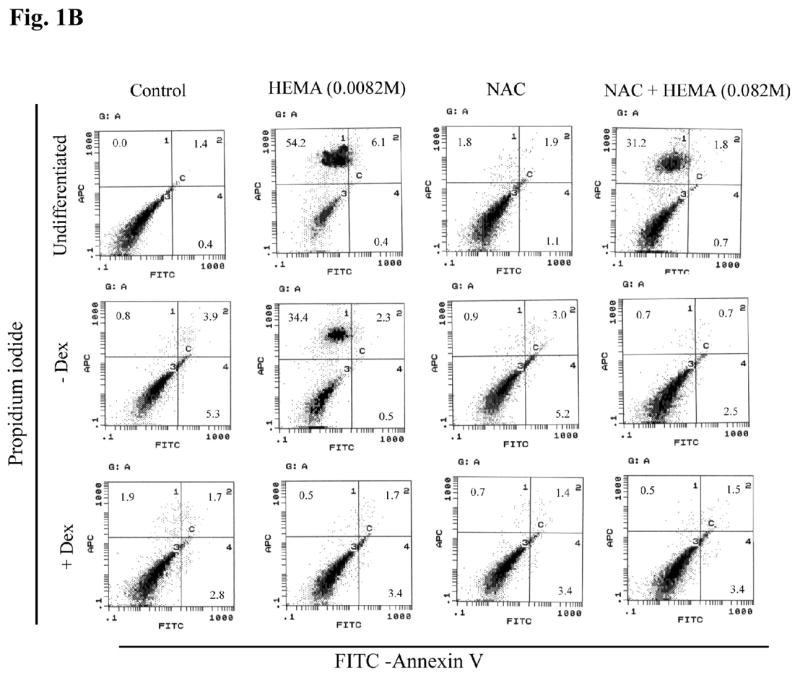
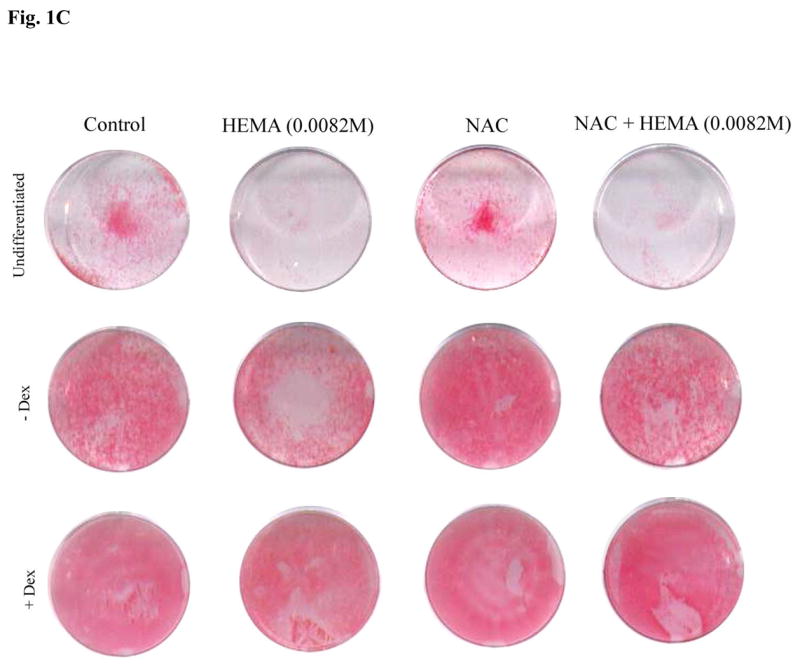
(A) Rat DPSCs (5×105/ml) were cultured in media alone (undifferentiated) or with the combination of β-glycerophosphate (10 mM) and ascorbic acid (50μg/ml) (-Dex), or with the combination of β-glycerophosphate (10 mM), ascorbic acid (50μg/ml) and Dexamethsone (10−8M) (+Dex). The levels of Osteopontin and Osteocalcin gene induction were determined by RT-PCR 14 days after the cells were seeded. GAPDH was used as the loading control. One of four independent experiments is shown in this figure (B) Rat DPSCs (1×106/ml) were cultured in media alone (undifferentiated) or with the combination of β-glycerophosphate (10 mM) and ascorbic acid (50μg/ml) (-Dex), or with the combination of β-glycerophosphate (10 mM), ascorbic acid (50μg/ml) and Dexamethsone (10−8M) (+Dex) for 14 days, after which they were treated with HEMA and NAC (20 mM) as indicated in the figure for 18 hours. The levels of cell death were assessed by FITC-Annexin V and Propidium Iodide (PI) staining. The numbers in each histogram are the percentages of cells positive for that quadrant. One of six independent experiments is shown in this figure (C) Rat DPSCs were treated as described in Fig. 1B and staining for ALP was carried out as described in the Materials and Methods section. One of six independent experiments is shown in this figure
Having established the differences in the gene induction patterns at different stages of differentiation, we then examined the ability of HEMA to induce cell death at different stages of differentiation. As shown in Fig. 1B the higher the stage of differentiation the lower were the levels of cell death. Indeed, undifferentiated DPSCs exhibited total of 60% cell death whereas β-glycerophosphate and ascorbic acid differentiated cells exhibited 37% and those treated with β-glycerophosphate, ascorbic acid and dexamethasone only exhibited 2.2% cell death. Therefore, the levels of differentiation correlated with the resistance of DPSCs to cell death. The different stages of differentiation are also marked by different levels of ALP staining in the presence and absence of HEMA treatment (Fig. 1C). Treatment of DPSCs at different stages of differentiation with N-acetyl cysteine (NAC) significantly blocked cell death mediated by HEMA (Fig. 1B) and increased the levels of ALP staining (Fig. 1C). However, decrease in HEMA mediated cell death by NAC was more apparent in differentiated than undifferentiated cells (Fig. 1B). Therefore, NAC may serve as an important agent of differentiation for DPSCs. Both rat and human DPSCs responded similarly to the effects of HEMA and NAC (please see below).
NAC prevents HEMA mediated decrease in VEGF secretion in DPSCs
DPSCs secrete Vascular Endothelial Growth Factor (VEGF) [29]. Therefore, to demonstrate that prevention of HEMA induced cell death by NAC has a significant functional consequences for DPSCs we measured secretion of VEGF when NAC was added with and without HEMA to DPSCs and to a primary oral keratinocyte tumor line (UCLA-2). The reasons we chose to concentrate on VEGF are the followings. First, VEGF is produced constitutively by UCLA-2 and DPSCs. Secondly, since naïve or IL-2 activated immune effectors do not produce measurable amounts of VEGF, their activating effect on increased release of VEGF by DPSCs can easily be investigated (please see Fig. 6B below). Lastly, the secreted levels of VEGF closely correlated with the viability and functional competency of UCLA-2 and HEp2 oral epithelial cells as well as DPSCs. Therefore, as shown in Table 1 and Fig. 6B, NAC significantly prevented HEMA induced decrease in VEGF secretion in both cell types. In addition, it is clear from this experiment that DPSCs have exquisite sensitivity to HEMA mediated effects since UCLA-2 exhibited no decrease in VEGF secretion at the lowest concentration of HEMA, whereas VEGF secretion remained significantly low in DPSCs at the same concentration when compared to control untreated cells (Table 1). Thus, prevention of cell death by NAC in DPSCs has also important functional consequences for the cells.
Fig-6. Increased Alkaline Phosphatase staining in DPSCs in the presence of naïve PBMCs and NAC.


(A) Human DPSCs (2.5×105/ml) cultured with the combination of β-glycerophosphate (10 mM) and ascorbic acid (50μg/ml) were washed extensively and co-cultured with naïve non activated PBMCs (PBMC to DPSC ratio 10:1) for 3 days after which they were stained for Alkaline Phosphatase activity. The density levels of Alkaline Phosphatase staining for each well were determined using Adobe Photoshop software. One of three independent experiments is shown in this figure. (*p≤ 0.0001 and **p≤ 0.0001, unpaired, two-tail t-test) (B) Human DPSCs were treated as described in Fig. 6A and co-cultured with naïve and IL-2 (1000u/ml) treated immune cells (PBMC to DPSC ratio 10:1) for 18 hours and the supernatants were removed and assayed for VEGF secretion using a sensitive and specific ELISA. One of three independent experiments is shown in this figure. (*p≤ 0.001, unpaired, two-tail) (C) Human DPSCs were treated as described in Fig. 6A and co-cultured with naïve and IL-2 (1000u/ml) treated immune cells (PBMC to DPSC ratio 10:1) for 18 hours and the supernatants were removed and assayed for IFN-γ secretion using a sensitive and specific ELISA. One of three independent experiments is shown in this figure. (*p≤ 0.001, unpaired, two-tail)
Table 1.
NAC prevents HEMA mediated decrease in VEGF secretion in DPSCs and UCLA-2 cells:
| VEGF (pg/ml) | ||
|---|---|---|
| Human DPSCs | UCLA -2 | |
| Control | 2263.9± 5.71 | 3195.7± 25.99 |
| HEMA (0.0164M) | 113.4± 17.1* | 180.2± 57.09* |
| HEMA (0.0082M) | 125.1± 8.24* | 319.6± 6.34* |
| HEMA (0.00164M) | 163.7± 3.8* | 3258.8± 15.89 |
| NAC | 1804.1± 18.04 | 2831.9± 70.37 |
| NAC +HEMA (0.0164M) | 1060.4± 12.04** | 1773.8± 90.02** |
| NAC + HEMA (0.0082M) | 1719.0± 6.34** | 2024.3± 31.7** |
| NAC + HEMA (0.00164M) | 2490.6± 46.91** | 3352.0± 18.38 |
Human DPSCs (2.5×105/ml) cultured with the combination of β-glycerophosphate (10 mM) and ascorbic acid (50μg/ml), and UCLA-2 (2.5×105/ml) oral tumors were treated with varying concentrations of HEMA as indicated in the table and NAC (20 mM), and after an overnight incubation the supernatants were removed and assayed for VEGF secretion using a sensitive and specific ELISA. One of three independent experiments is shown in this table. Results represent mean±sd of triplicates. Based on one way ANOVA
p ≤0.001 and
p≤0.001 was obtained for the differences between HEMA treated and untreated cells, and between NAC+HEMA treated cells and HEMA alone treated cells at the respective concentrations for human DPSCs and UCLA-2 cells.
Previous reports demonstrated an anti-proliferative effect for NAC, thus we determined the levels of thymidine incorporation in DPSCs and assessed the numbers of viable cells after treatment with and without NAC and HEMA. As shown in Fig. 2A we observed an increase rather than a decrease in 3H thymidine incorporation in DPSCs (Fig. 2A) as well as UCLA-2 cells (data not shown) when treated with NAC as compared to untreated cells. NAC also increased 3H thymidine incorporation significantly in cells treated with the combination of NAC and HEMA as compared to HEMA alone treated cells, although the extent of the inhibitory effect of NAC on HEMA mediated cell death was dependent on the dose of HEMA used (Fig. 2A). As expected, HEMA treatment caused a dose dependent inhibition in thymidine incorporation of the cells (Fig. 2A). In addition when the numbers of viable cells were determined in DPSCs an enhancement in the numbers of cells could be seen in samples treated with NAC (Fig. 2B). As expected HEMA treatment decreased the numbers of viable cells and the addition of NAC plus HEMA was able to increase the numbers of viable cells when compared to HEMA alone treated cells (Fig. 2B). The results indicated that NAC increased proliferation as well as the numbers of the viable cells when added to control untreated and HEMA treated cells.
Fig-2. NAC prevented HEMA mediated decrease in proliferation and cell numbers in DPSCs.

(A) Human DPSCs were treated with different concentrations of HEMA and NAC (20mM) as indicated in the figure for 72 hours after which the levels of 3H thymidine incorporation were determined in each sample using the β counter. One of two independent experiments is shown in this figure. Based on one way ANOVA the differences between the control group and those treated with HEMA and NAC are significant at p≤ 0.001 (B) Human DPSCs (2.5×105/ml) were treated with different concentrations of HEMA and NAC (20mM) as indicated in the figure for a period of 5 days after which they were trypsinized, and the cell numbers were determined in each sample. One of three independent experiments is shown in this figure. Based on one way ANOVA the differences between the control group and those treated with HEMA and NAC are significant at p≤ 0.001
Effect of NAC is distinct from that mediated by Vitamin E and C
Since NAC is considered as an important anti-oxidant agent we compared its effect to the well known anti-oxidant agents Vitamin E (Trolox) and Vitamin C. These two important anti-oxidants have previously been shown to inhibit HEMA mediated adverse effects [21]. Unlike NAC, Vitamin E failed to prevent either HEMA mediated cell death (Fig. 3A) or decrease in VEGF secretion by human DPSCs (Table 2). In addition, Vitamin E also inhibited the function of NAC on DPSCs since the addition of the combination of NAC and Vitamin E inhibited NAC mediated increase in VEGF secretion (Table 2). Unlike NAC, Vitamin E was unable to restore HEMA mediated decrease in Alkaline Phosphatase (ALP) staining in DPSCs (Fig. 3B). Moreover, the addition of vitamin C was unable to either prevent cell death (Fig. 4) or increase differentiation genes in DPSCs (Fig. 5). Therefore, the effect of NAC appears to be quite distinct from those mediated by Vitamin E and Vitamin C. These differences suggest that NAC may have an additional important function which may be distinct from that mediated by its well know anti-oxidant effect.
Fig-3. Vitamin E failed to prevent HEMA mediated cell death or HEMA induced decrease in ALP staining.
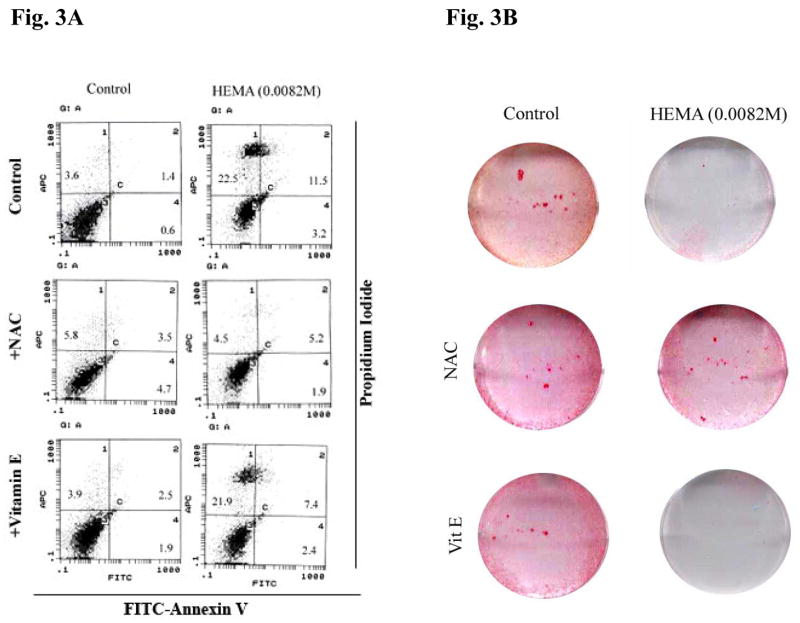
(A) Human DPSCs cultured with β-glycerophosphate (10 mM) and ascorbic acid (50μg/ml) (1×106/ml) were treated with HEMA (0.0082M), NAC (20 mM) and vitamin E (5 mM) for a period of 18 hours after which they were stained with FITC- Annexin V and Propidium Iodide and analyzed by EPICs ELITE flow cytometer. One of three independent experiments is shown in this figure. (B) Treatments were carried out as described in Fig. 3A, and the levels of Alkaline Phosphatase (ALP) staining were determined after 18 hours of incubation. One of three independent experiments is shown in this figure
Table 2.
Vitamin E failed to prevent HEMA mediated decrease in the levels of VEGF in human DPSCs:
| VEGF (pg/ml) | ||||
|---|---|---|---|---|
| Control | NAC | Vitamin E | NAC+ Vitamin E | |
| Control | 2263.9± 15.05 | 1532.4± 66.78 | 1966.2± 2.61 | 1472.6± 55.1 |
| HEMA (0.0164M) | 73.79± 9.28 | 1648.1± 45.08 | 63.16± 5.84 | 964.2± 37.1 |
| HEMA (0.0082M) | 72.6± 14.19 | 1477.5± 9.18 | 83.18± 4.10 | 1081.4± 6.68 |
| HEMA (0.00164M) | 57.26± 0.83 | 2123.9± 0.0 | 80.28± 0.0 | 871.5± 30.89 |
Human DPSCs (2.5×105/ml) cultured with the combination of β-glycerophosphate (10 mM) and ascorbic acid (50μg/ml), were treated with varying concentrations of HEMA as indicated in the table, Vitamin E (5 mM) and NAC (20 mM), and after an overnight incubation the supernatants were removed and assayed for VEGF secretion using a sensitive and specific ELISA. One of two independent experiments is shown in this table. Results represent mean±sd of triplicates. Based on one way ANOVA the differences between the control group and those treated with HEMA, NAC and Vitamin E are significant at p≤ 0.001
Fig-4. Vitamin C failed to prevent HEMA mediated cell death.
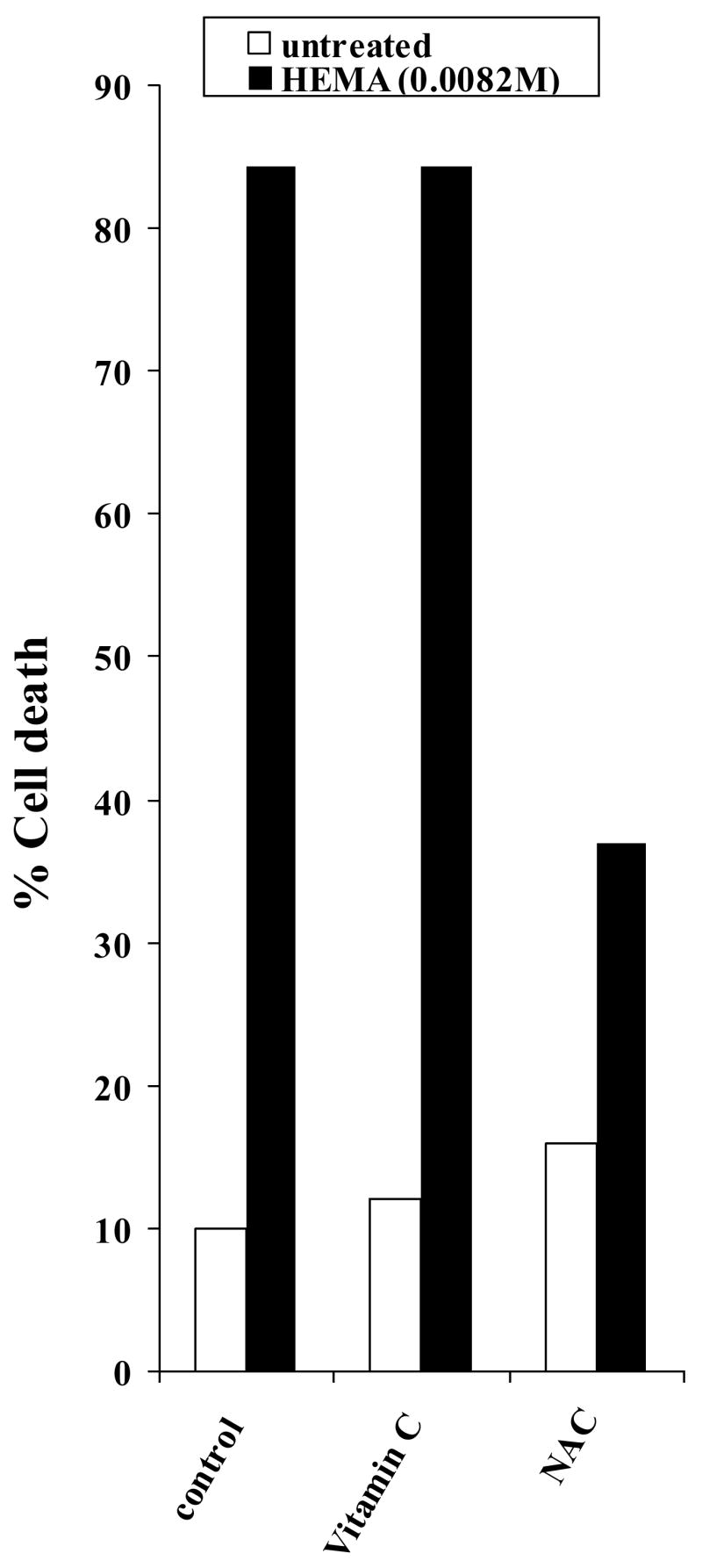
Undifferentiated DPSCs were either left untreated or treated with vitamin C (20 mM) or NAC (20mM) for 18 hours after which they were washed and stained with FITC- Annexin V and Propidium Iodide and analyzed by EPICs ELITE flow cytometer to determine the percentages of cell death in each sample. One of three independent experiments is shown in this figure
Fig-5. Increased induction of key differentiation genes in DPSCs by NAC but not by Vitamin E or C at different stages of differentiation.
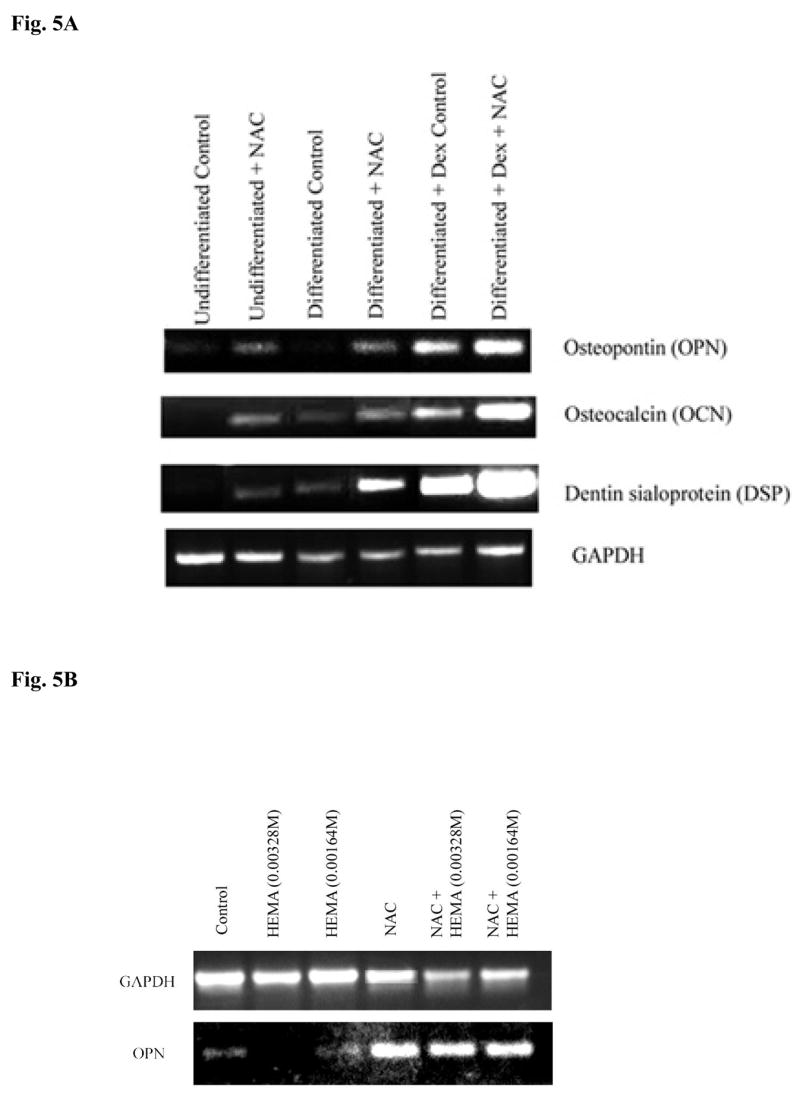
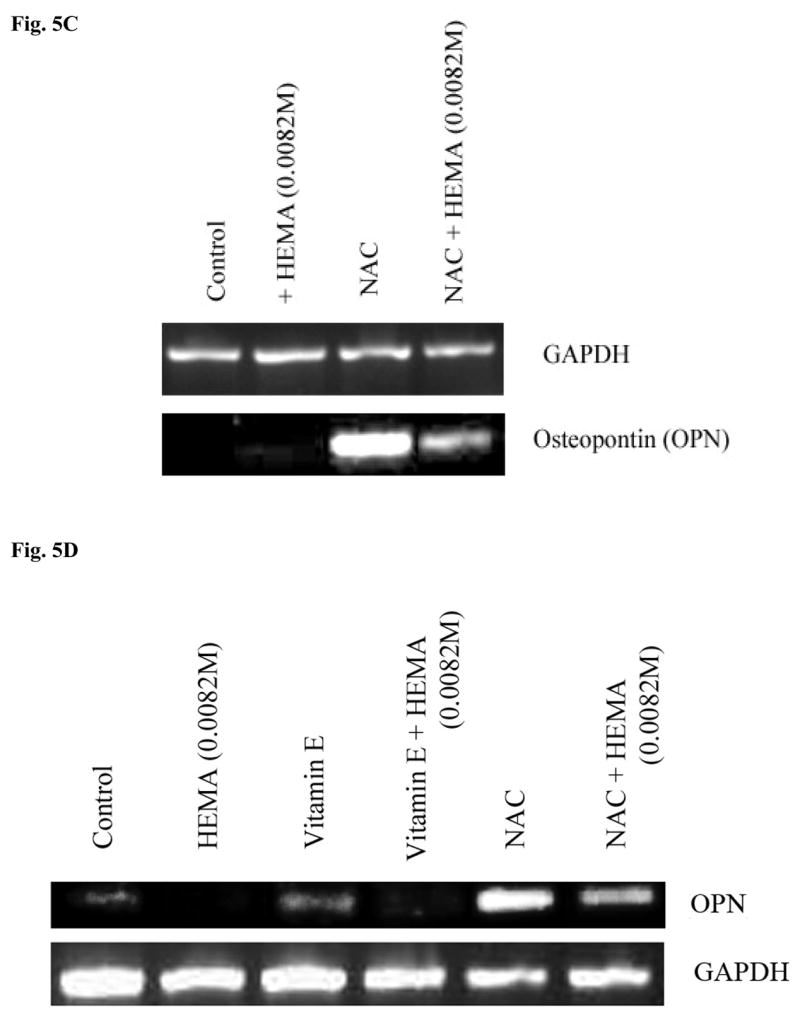
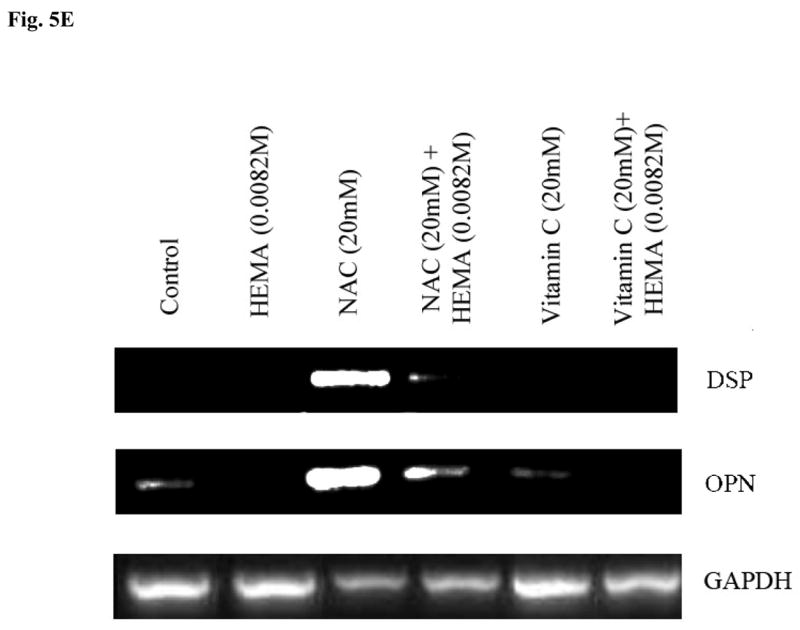
(A) Rat DPSCs (5×105/ml) were cultured in media alone (undifferentiated) or with the combination of β-glycerophosphate (10 mM) and ascorbic acid (50μg/ml) (Differentiated), or with the combination of β-glycerophosphate (10 mM), ascorbic acid (50μg/ml) and Dexamethsone (10−8M) (Differentiated+Dex) for 14 days after which the cells were treated with NAC (20 mM) overnight and the levels of Osteopontin, Osteocalcin and Dentin Sialoprotein gene induction were determined by RT-PCR. GAPDH was used as the loading control. One of three independent experiments is shown in this figure (B) Rat DPSCs (5×105/ml) cultured with the combination of β-glycerophosphate (10 mM) and ascorbic acid (50μg/ml) were treated with the varying concentrations of HEMA as indicated in the figure and NAC (20 mM) for 18 hours. Total RNA was then extracted, and the levels of ostepontin (OPN) were determined. GAPDH was used as the loading control. One of three independent experiments is shown in this figure. (C) Human DPSCs (5×105/ml) cultured with the combination of β-glycerophosphate (10 mM) and ascorbic acid (50μg/ml) were treated with HEMA as indicated in the figure and NAC (20 mM) for 18 hours. Total RNA was then extracted, and the levels of ostepontin (OPN) were determined. GAPDH was used as the loading control. One of three independent experiments is shown in this figure. (D) Rat DPSCs (5×105/ml) cultured with the combination of β-glycerophosphate (10 mM) and ascorbic acid (50μg/ml) were left untreated or treated with HEMA (0.0082M), NAC (20 mM), or Vitamin E (5 mM), or their combination as indicated in the figure for 18 hours. Total RNA was then extracted, and the levels of ostepontin (OPN) were determined. GAPDH was used as the loading control. One of three independent experiments is shown in this figure. (E) Rat DPSCs (5×105/ml) cultured in DMEM culture medium were either left untreated or treated with HEMA (0.0082M), NAC (20 mM), or Vitamin C (5 μg/ml), as indicated in the figure for 18 hours. Total RNA was then extracted, and the levels of ostepontin (OPN) were determined. GAPDH was used as the loading control. One of three independent experiments is shown in this figure.
Increased induction of key differentiation genes in DPSCs by NAC at different stages of differentiation
DPSCs were treated to induce different stages of differentiation, and the levels of OPN, OCN and Dentin Sialoprotein (DSP) gene expression were determined when NAC was also added to each sample. Undifferentiated DPSCs in the absence of NAC demonstrated no or low levels of differentiation gene expression (Fig. 5A). Treatment with NAC alone or its addition to the other differentiation agents substantially increased gene expression for OPN, OCN and DSP in DPSCs in a stepwise manner (Fig. 5A and 5B). Therefore, NAC not only can up-regulate gene expression on its own; it can also increase gene expression when added to other differentiation agents. In addition, even though the combination of HEMA and NAC treated cells exhibited lower levels of OPN gene expression when compared to NAC alone treated cells, the levels still remained higher and exceeded those seen by untreated cells (Figs. 5B,5C,5D and 5E). Increased gene expression for OPN was seen both in rat and human DPSCs treated with NAC (Figs. 5B, 5C and 5D). Furthermore, in agreement with the cell death data and ALP expression (Fig. 3 and 4), treatment with Vitamin E and Vitamin C, unlike NAC, failed to increase gene expression for OPN significantly (Fig. 5D and 5E). In addition, treatment with HEMA was inhibitory for the expression of OPN and unlike NAC the combination of vitamin E, vitamin E with HEMA and Vitamin C, Vitamin C with HEMA were unable to increase the expression of OPN (Fig. 5D and 5E).
Increased Alkaline Phosphatase staining in DPSCs in the presence of naïve PBMCs and NAC
Since inflamed dental pulp may contain immune effectors during the dental restoration procedures we determined the effect of peripheral blood mononuclear cells (PBMCs) on DPSCs when co-cultured with and without NAC. We determined the levels of ALP staining in untreated and NAC treated DPSCs co-cultured with naïve PBMCs. As shown in Fig. 6A, addition of PBMCs to DPSCs in the absence of NAC increased ALP staining in DPSCs when compared to non NAC treated DPSCs. Treatment with NAC increased the levels of ALP staining in DPSCs and the levels were further enhanced when naïve PBMCs were added to NAC treated DPSCs (Fig. 6A). Increased functional activation of DPSCs by naïve PBMCs was also observed when secretion of VEGF was determined in the co-cultures of DPSCs with PBMCs (Fig 6B). When IL-2 activated PBMCs were added to non NAC treated DPSCs, a 50% reduction in VEGF secretion could be seen compared to untreated PBMCs (Fig. 6B). IL-2 treated PBMCs mediated decrease in VEGF secretion by DPSCs was paralleled with a reduction in ALP staining of DPSCs (data not shown) as well as an increased secretion of IFN-γ in the co-cultures of DPSCs with PBMCs (Fig. 6C). Therefore, IL-2 activation of PBMCs is inhibitory for survival and function of DPSCs. In addition, when NAC was added to the co-cultures of IL-2 treated PBMCs with DPSCs the reduction in either ALP staining or VEGF secretion was lower when compared to co-cultures of naïve PBMCs with DPSCs in the absence of NAC (data not shown). The results indicated that naïve PBMCs have growth promoting effect on DPSCs whereas IL-2 activated PBMCs have growth inhibitory effect, and that NAC could contribute to growth promoting activity of PBMCs on DPSCs.
Increased function of nuclear NFκB by NAC
Since NFκB is an important inducer of cell survival and its activity increases during cellular differentiation, we next examined the effect of NAC in increasing NFκB function. 293T cells, as well as HEp2 oral epithelial cells and DPSCs were transfected with NFκB reporter vector and the transfected cells were treated with TNF-α, HEMA and NAC alone or in combinations as indicated in the figure 7, and the levels of NFκB activity were determined. Because of their excellent transfection efficiency 293T cells were used as a positive control, and demonstrated similar patterns of NFκB activation when compared to HEp2 oral epithelial cells or DPSCs, albeit the levels of NFκB activity differed between these cell types (Fig. 7). Since HEMA may come in contact with oral epithelial cells as well as DPSCs during dental restorations, HEp2 cells which had adequate transfection efficiency were also used in these experiments. DPSCs had very low transfection efficiency even when optimal conditions for transfection of primary cells were employed. NAC was able to significantly upregulate the NFκB activity in all the cells while HEMA inhibited NFκB activity compared to untreated controls (Fig. 7). NAC increased NFκB activation in HEMA-treated cells, but the levels of nuclear NFκB activity remained lower in cells treated with a combination of HEMA and NAC as compared to NAC alone. As expected, TNF-α was able to induce NFκB activity in all the cells. Although HEMA treatment decreased NFκB activity in TNF-α treated cells, the levels remained higher when compared to cells treated with HEMA alone. NAC, however, when added in combination with TNF-α had inhibitory effect on NFκB activity.
Fig-7. Increased function of nuclear NFκB by NAC in the presence and absence of PBMCs.
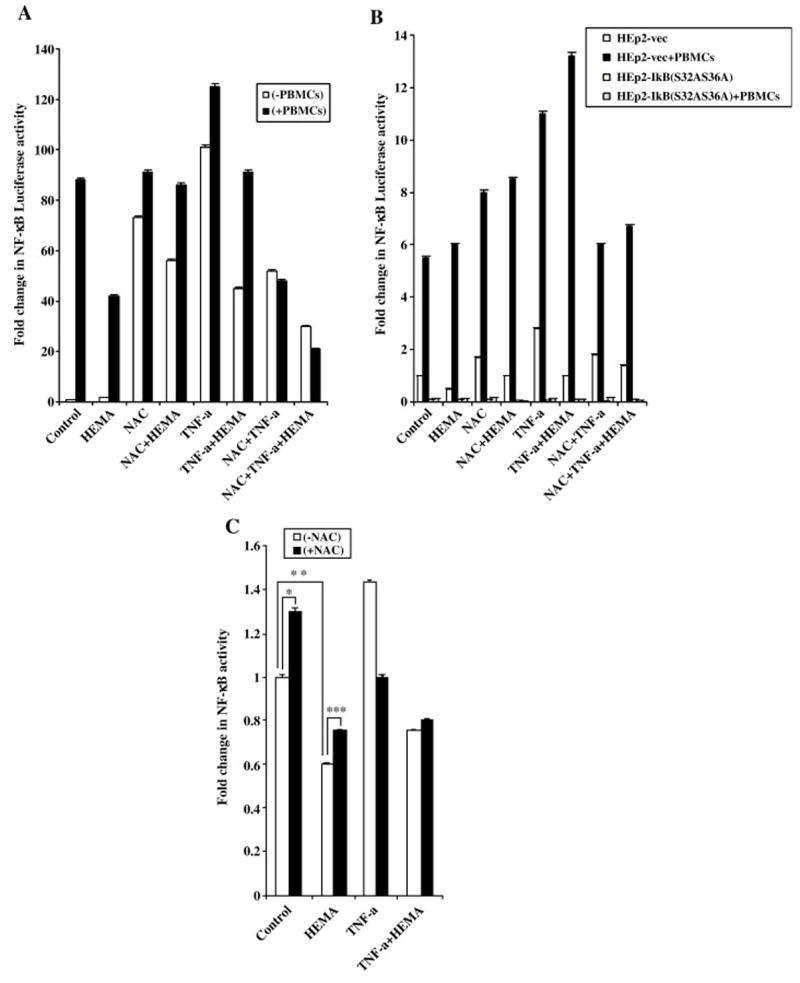

(A) 293T cells were transfected with the NFκB Luciferase reporter vector as described in the materials and methods section, and co-cultured with and without PBMCs (10:1 E:T ratio) before they were treated with HEMA (0.0082M), NAC (20mM) and TNF-α (20ng/ml) for 18 hours. The relative Luciferase activity was then determined in the lysates according to the manufacturer’s recommendation and fold change in luciferase activity were determined as compared to untreated cells. (Promega, Madison, WI). One of seven independent experiments is shown in this figure. Based on one way ANOVA the differences between the control group with PBMCs and those treated with HEMA, NAC and TNF-α are significant at p≤ 0.001based on ANOVA analysis. (B) Vector-alone or IκB(S32AS36A) transfected stable HEp2 oral epithelial cells were transfected with the NFκB Luciferase reporter vector as described in the materials and methods section, and then co-cultured with and without PBMCs before they were treated with HEMA (0.0082M), or NAC (20mM) or TNF-α (20ng/ml) and their combinations for 18 hours. The relative Luciferase activity was then determined in the lysates according to the manufacturer’s recommendation and fold change in luciferase activity were determined as compared to untreated cells. One of three independent experiments is shown in this figure. Based on one way ANOVA the differences between the Vector-alone control group and those treated with HEMA, NAC and TNF-α are significant at p≤ 0.001. (C) DPSCs were transfected with the NFκB Luciferase reporter vector as described in the materials and methods section, before they were treated with HEMA (0.0082M), or NAC (20mM) or TNF-α (20ng/ml) and their combinations for 18 hours. The relative Luciferase activity was then determined in the lysates according to the manufacturer’s recommendation and fold changes in luciferase activity were determined as compared to untreated cells. One of three independent experiments is shown in this figure. (***p≤ 0.001, **p≤ 0.0001 and *p≤ 0.001, unpaired, two-tail) (D) DPSCs were treated with HEMA (0.0082M), or NAC (20mM) or TNF-α (20ng/ml) and their combinations for 18 hours after which nuclear extracts were prepared and they were analyzed using western blot analysis. The specific antibody to p65 subunit of NFκB was used to determine the levels in each treated sample. The relative band densities were determined using Adobe Photoshop analysis software. One of two independent experiments is shown in this figure.
To examine the contributory role of PBMCs in increase activation of NFκB we measured NFκB activity when PBMCs were added to HEMA, NAC and TNF-α treated 293T and HEp2 cells. PBMCs induced NFκB activation in both cells significantly (Fig. 7A and 7B). PBMCs were able to increase NFκB in all treated cells substantially even though the patterns of activation remained similar to those described above in the presence of NAC, HEMA and TNF-α treatment. Blocking of NFκB with an IκB super-repressor abolished the activating effects of abovementioned treatments in the presence and absence of PBMCs. It is important to note that NAC had inhibitory effect on TNF-α treated cells as shown previously [30]. and in Fig. 7. Indeed, the pattern of NFκB activation under different treatments in both 293T and HEp2 cells resembled a bell shaped curve demonstrating increase with NAC or TNF-α but when added in combination, TNF-α and NAC had inhibitory effect on NFκB activity. However, when tested after a shorter incubation time period (3–4 hours), combinations of NAC and TNF-α had activating rather than inhibitory effect on NFκB activity in 293T cells indicating activation at earlier time points followed by a decline at 24 hours of treatment. Even though DPSCs had the least transfection efficiency when compared to 293T or HEp2 cells, the pattern of NFκB activation in DPSCs followed closely to those shown for 293T and HEp2 cells (Fig. 7C). NAC or TNF-α had enhancing effect on NFκB activity whereas HEMA was inhibitory for DPSCs. Both HEMA and NAC had inhibitory effect when added in combination with TNF-α. Similar profiles of NFκB activation was also observed when the levels of NFκB were determined in the nuclear extracts obtained from NAC, HEMA and TNF-α treated DPSCs (Fig. 7D). These data suggested that HEMA induced cell death could be mediated by the inhibition of NFκB.
Discussion
Differentiation status of the cells is important for their growth and survival. Here we report that the extent of differentiation significantly correlates with the ability to resist HEMA mediated cell death. Undifferentiated Dental pulp stromal cells have exquisite sensitivity to death inducing effect of HEMA. Once differentiated, depending on the stage of differentiation they gain step wise resistance to induction of cell death. For instance when DPSCs are treated with β-glycerophosphate and ascorbic acid an induction of osteopontin but not that of osteocalcin is observed and at this stage of differentiation a relatively higher resistance to cell death is observed when compared to undifferentiated DPSCs. However when dexamethasone is also supplemented in the presence of β-glycerophosphate and ascorbic acid the cells expressed both osteopontin and osteocalcin and exhibited the highest resistance to cell death mediated by HEMA when compared to the undifferentiated DPSCs or those differentiated in the absence of dexamethasone. This stepwise effect in resistance to cell death induced by HEMA is also evident when Alkaline Phosphatase (ALP) activity was determined in different stages of differentiation in DPSCs (Fig. 1C). Therefore the higher the stage of differentiation the higher is the resistance of the cells to death inducing signals.
N-acetyl cysteine (NAC) has long been considered as an antioxidant agent. However, recent work from our laboratory and those of the others established other important functions for NAC which could be the basis for its death inhibitory property [12, 13]. Here we demonstrate that the basis for cell death inhibitory function of NAC could be its capacity to induce NFκB, and modulate important differentiation and activation genes in DPSCs. NAC is capable of inducing a stepwise increase in osteopontin, osteocalcin and dentin sialoprotein depending on the stage of differentiation of the cells, and this correlated with significant inhibition of cell death at all stages of differentiation in DPSCs. It is important to note that we did not observe increased cell death by NAC when added to highly activated DPSCs since with the combined effects of all of the activating agents one might have expected to observe activation induced cell death. Therefore, the function of NAC may differ from other activating agents in that it is able to trigger activation in the absence of cell death. In addition, NAC mediated inhibition of cell death was more effective in differentiated than in undifferentiated cells. In this regard pre-treatment with NAC prior to HEMA treatment increased the effectiveness of NAC inhibition of cell death in undifferentiated DPSCs (data not shown). The inhibitory function of NAC on cell death was also seen when it was added to IL-2 activated immune effectors in the presence of their co-cultures with DPSCs. Therefore, on the basis of this function, NAC was suggested previously to be used as a therapeutic strategy in preventing depletion of CD4+ T cells in HIV + patients [31, 32]. At present it is not clear how NAC could in one hand induce differentiation and increase activation of the cells and on the other hand inhibit cell death since these two functions of cells namely cell death and activation are not mutually exclusive. Indeed, increased induction of cell death in highly differentiated cells is considered to be an important mechanism for guarding against possible cellular transformation. Whether NAC regulates differentiation through redox state of the cysteine-SH residues in protein tyrosine phosphorylation or tyrosine kinases needs further investigation.
We show that NAC activation mobilizes the transcription factor NFκB to the nucleus since we observed a significant rise in NFκB activity by NAC. This effect of NAC was also confirmed when NFκB activity was determined in a Western blot analysis of DPSCs treated with NAC (Fig. 7D). Since NFκB is a significant regulator of anti-apoptotic proteins, and it is also important for the differentiation of the cells it is likely that the inhibitory effect of NAC on cell death is partly due to the upregulation of this transcription factor. Indeed, when NFκB is inhibited by an IκB super repressor in oral keratinocytes the inhibitory effect of NAC on cell death was significantly abolished (manuscript submitted). Therefore, although we have also identified an NFκB independent pathway of prevention of cell death by NAC it appears that a large part of NAC’s protective effect on cell death is mediated by the activation of NFκB (manuscript submitted).
Unlike the previous reports we did not observe a significant decrease in cell proliferation or decrease in the numbers of the cells in the presence of NAC when compared to untreated control cells. [12, 13]. In contrast, significant increases in the cell proliferation and cell numbers was seen when NAC was added to untreated and HEMA treated cells when compared to HEMA alone treated cells. These results indicated that the increase in the cell numbers in HEMA and NAC treated cells could be due to both the inhibition of cell death as well as increase in proliferation of the surviving cells by NAC.
We show here that undifferentiated DPSCs have exquisite sensitivity to cell death when compared to their differentiated counterparts. Therefore, when exposed to restorative materials they are likely to become depleted first. Depending on the levels of exposure of dental pulp to restorative materials and their differentiation status different clinical pictures can be expected. In the worst case scenario, the pulp may become necrotic in which case the affected tooth will be lost under heavy exposure to dental resins. This may occur during pulp capping procedures and deep restorations where these restorative materials are either in direct contact with or in close proximity to the pulp. In case of an intermediate level of exposure to dental resins such that seen in many dental restorations, even though stem cells may become depleted, any remaining differentiated cells may provide certain levels of protection against HEMA induced effects. However, an immediate second exposure may be detrimental since it may cause the loss of the tooth.
It is important to note that a key anti-oxidant compound Vitamin E was found to contribute rather than inhibit HEMA mediated cell death when added to NAC. Moreover, no significant effect on prevention of cell death or induction of differentiation genes could be seen when vitamin C was added to DPSCs. Therefore, this may likely argue against a significant contribution of an anti-oxidant effect of NAC in inhibition of cell death. Indeed, the addition of Vitamin E to DPSCs not only did not change the course of HEMA mediated cell death, it also inhibited NAC mediated protection against loss of VEGF secretion induced by HEMA. More importantly, unlike NAC, Vitamin E was unable to increase gene expression for OPN when added alone or in combination with HEMA to DPSCs (Fig. 5D). Therefore, we propose that the effect of NAC on protection against HEMA mediated cell death is distinct from its well characterized anti-oxidant effect and relates to its enhancing effect on differentiation of the cells.
Similar to the effect of NAC, an increase in ALP staining was observed when PBMCs were co-cultured with DPSCs. Indeed, an enhancing effect on ALP staining could be obtained when NAC was added in the co-cultures of PBMCs with DPSCs. Increased ALP staining is considered to be a marker of increased differentiation in the cells [33–35]. Therefore, an important function of naïve immune effectors could be their enhancing effect on differentiation and survival of DPSCs. Increased differentiation and activation is further facilitated when NAC is added to the co-cultures of immune effectors with DPSCs. Therefore, not only NAC enhances differentiation and survival on its own, it can also enhance the effect mediated by immune effectors. In addition it can also protect the cells from adverse effects induced by the activated immune effectors. Indeed, when immune effectors were activated with IL-2 and added to DPSCs a significant decrease in ALP staining could be observed. Addition of NAC decreased the reduction in ALP staining by IL-2 activated immune effectors. Thus, NAC may not only increase the beneficial effect of immune effectors, it can also guard against adverse effects of activated immune effectors. Thus, one of the important functions of immune inflammatory cells in pulp is their enhancing effect on differentiation of DPSCs to withstand distinct environmental insults. In this regard most of the inflammatory cytokines induced in the co-cultures of naïve immune effectors with DPSCs or keratinocytes such as IL-6, IL-1β and TNF-α have all inducing effect on NFκB activation and this may in part be responsible for the increased survival of DPSCs and keratinocytes (data not shown) [26].
NAC is also regarded as an anti-inflammatory agent [15–17]. NAC can exert its anti-inflammatory effect at different stages of inflammation. NAC can prevent initiation of inflammation by preventing death of DPSCs since cell death is an important step in the recruitment of immune effectors [36]. Moreover, when even the immune cells gain access to the pulp area, they will eventually become inactivated and cleared since NAC will increase survival and activation of NFκB in DPSCs. This assumption is based on our previous observation that an increase in NFκB activation in keratinocytes induces immune inactivation by switching the balance towards an increased Th2 type cytokine such as IL-6 and eventual loss of the immune effectors [37, 38]. Needless to say, that cytokines such as IL-6, TNF-α or IL-1β can further increase NFκB activity in DPSCs perhaps causing faster resolution of inflammatory process.
Overall, the data presented in this paper underscores the significance of NAC in inducing differentiation of DPSCs and provides rationale for its inhibitory effect on HEMA mediated cell death. Figure 8 is a schematic diagram demonstrating the working model by which NAC may prevent cell death in DPSCs or oral keratinocytes. Therefore, to neutralize the toxic effects of dental materials NAC may be added immediately prior or along with the dental restorations to prevent their potential adverse effects.
Fig-8.
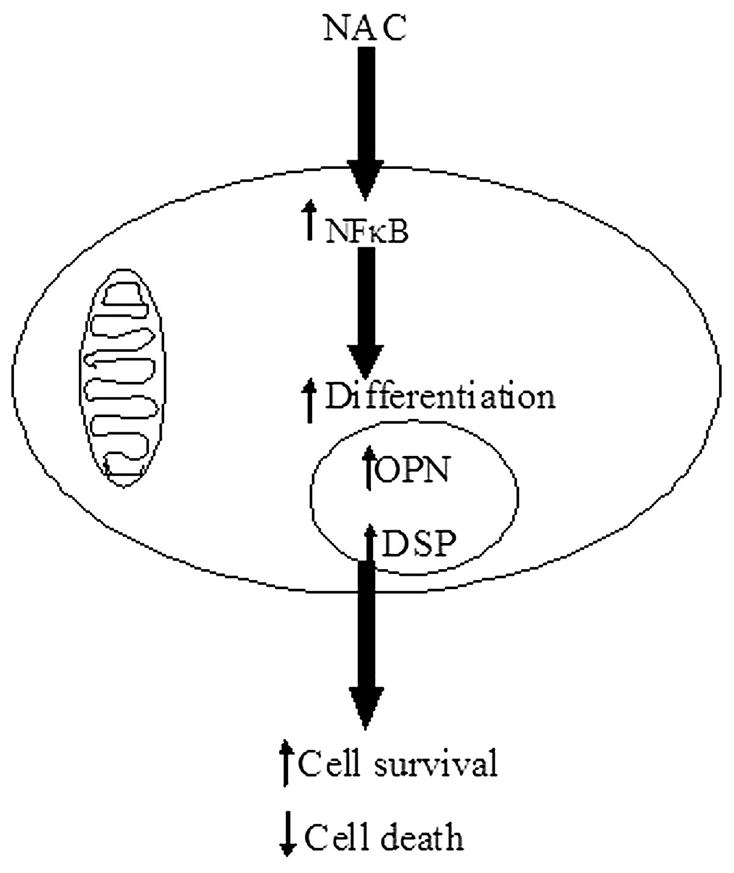
Schematic diagram representing the hypothetical model by which NAC may prevent cell death in DPSCs or oral keratinocytes
Acknowledgments
This work was supported by RO1-10331 from NIH-NIDCR
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bjorndal L, Mjor IA. Pulp-dentin biology in restorative dentistry. Part 4: Dental caries--characteristics of lesions and pulpal reactions. Quintessence Int. 2001;32:717–736. [PubMed] [Google Scholar]
- 2.Jontell M, Gunraj MN, Bergenholtz G. Immunocompetent cells in the normal dental pulp. J Dent Res. 1987;66:1149–1153. doi: 10.1177/00220345870660061101. [DOI] [PubMed] [Google Scholar]
- 3.Yu J, Deng Z, Shi J, Zhai H, Nie X, Zhuang H, Li Y, Jin Y. Differentiation of Dental Pulp Stem Cells into Regular-Shaped Dentin-Pulp Complex Induced by Tooth Germ Cell Conditioned Medium. Tissue Eng. 2006 doi: 10.1089/ten.2006.12.3097. [DOI] [PubMed] [Google Scholar]
- 4.Carda C, Peydro A. Ultrastructural patterns of human dentinal tubules, odontoblasts processes and nerve fibres. Tissue & cell. 2006;38:141–150. doi: 10.1016/j.tice.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 5.About I, Camps J, Mitsiadis TA, Bottero MJ, Butler W, Franquin JC. Influence of resinous monomers on the differentiation in vitro of human pulp cells into odontoblasts. J Biomed Mater Res. 2002;63:418–423. doi: 10.1002/jbm.10253. [DOI] [PubMed] [Google Scholar]
- 6.Paranjpe A, Bordador LC, Wang MY, Hume WR, Jewett A. Resin monomer 2-hydroxyethyl methacrylate (HEMA) is a potent inducer of apoptotic cell death in human and mouse cells. Journal of dental research. 2005;84:172–177. doi: 10.1177/154405910508400212. [DOI] [PubMed] [Google Scholar]
- 7.Stanislawski L, Daniau X, Lauti A, Goldberg M. Factors responsible for pulp cell cytotoxicity induced by resin-modified glass ionomer cements. J Biomed Mater Res. 1999;48:277–288. doi: 10.1002/(sici)1097-4636(1999)48:3<277::aid-jbm11>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 8.Stanislawski L, Lefeuvre M, Bourd K, Soheili-Majd E, Goldberg M, Perianin A. TEGDMA-induced toxicity in human fibroblasts is associated with early and drastic glutathione depletion with subsequent production of oxygen reactive species. J Biomed Mater Res A. 2003;66:476–482. doi: 10.1002/jbm.a.10600. [DOI] [PubMed] [Google Scholar]
- 9.Walther UI, Walther SC, Liebl B, Reichl FX, Kehe K, Nilius M, Hickel R. Cytotoxicity of ingredients of various dental materials and related compounds in L2- and A549 cells. J Biomed Mater Res. 2002;63:643–649. doi: 10.1002/jbm.10384. [DOI] [PubMed] [Google Scholar]
- 10.Hunag TH, Lii CK, Kao CT. Root canal sealers cause cytotoxicity and oxidative damage in hepatocytes. J Biomed Mater Res. 2001;54:390–395. doi: 10.1002/1097-4636(20010305)54:3<390::aid-jbm110>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 11.Stanislawski L, Soheili-Majd E, Perianin A, Goldberg M. Dental restorative biomaterials induce glutathione depletion in cultured human gingival fibroblast: protective effect of N-acetyl cysteine. J Biomed Mater Res. 2000;51:469–474. doi: 10.1002/1097-4636(20000905)51:3<469::aid-jbm22>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 12.Parasassi T, Brunelli R, Bracci-Laudiero L, Greco G, Gustafsson AC, Krasnowska EK, Lundeberg J, Lundeberg T, Pittaluga E, Romano MC, Serafino A. Differentiation of normal and cancer cells induced by sulfhydryl reduction: biochemical and molecular mechanisms. Cell Death Differ. 2005;12:1285–1296. doi: 10.1038/sj.cdd.4401663. [DOI] [PubMed] [Google Scholar]
- 13.Gustafsson AC, Kupershmidt I, Edlundh-Rose E, Greco G, Serafino A, Krasnowska EK, Lundeberg T, Bracci-Laudiero L, Romano MC, Parasassi T, Lundeberg J. Global gene expression analysis in time series following N-acetyl L-cysteine induced epithelial differentiation of human normal and cancer cells in vitro. BMC Cancer. 2005;5:75. doi: 10.1186/1471-2407-5-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edlundh-Rose E, Kupershmidt I, Gustafsson AC, Parasassi T, Serafino A, Bracci-Laudiero L, Greco G, Krasnowska EK, Romano MC, Lundeberg T, Nilsson P, Lundeberg J. Gene expression analysis of human epidermal keratinocytes after N-acetyl L-cysteine treatment demonstrates cell cycle arrest and increased differentiation. Pathobiology. 2005;72:203–212. doi: 10.1159/000086790. [DOI] [PubMed] [Google Scholar]
- 15.Gillissen A, Nowak D. Characterization of N-acetylcysteine and ambroxol in anti-oxidant therapy. Respiratory medicine. 1998;92:609–623. doi: 10.1016/s0954-6111(98)90506-6. [DOI] [PubMed] [Google Scholar]
- 16.Radomska-Lesniewska DM, Sadowska AM, Van Overveld FJ, Demkow U, Zielinski J, De Backer WA. Influence of N-acetylcysteine on ICAM-1 expression and IL-8 release from endothelial and epithelial cells. J Physiol Pharmacol. 2006;57(Suppl 4):325–334. [PubMed] [Google Scholar]
- 17.Mukherjee TK, Mishra AK, Mukhopadhyay S, Hoidal JR. High concentration of antioxidants N-acetylcysteine and mitoquinone-Q induces intercellular adhesion molecule 1 and oxidative stress by increasing intracellular glutathione. J Immunol. 2007;178:1835–1844. doi: 10.4049/jimmunol.178.3.1835. [DOI] [PubMed] [Google Scholar]
- 18.Schweikl H, Hartmann A, Hiller KA, Spagnuolo G, Bolay C, Brockhoff G, Schmalz G. Inhibition of TEGDMA and HEMA-induced genotoxicity and cell cycle arrest by N-acetylcysteine. Dent Mater. 2006 doi: 10.1016/j.dental.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 19.Lee DH, Lim BS, Lee YK, Ahn SJ, Yang HC. Involvement of oxidative stress in mutagenicity and apoptosis caused by dental resin monomers in cell cultures. Dent Mater. 2006;22:1086–1092. doi: 10.1016/j.dental.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 20.Spagnuolo G, D’Anto V, Cosentino C, Schmalz G, Schweikl H, Rengo S. Effect of N-acetyl-L-cysteine on ROS production and cell death caused by HEMA in human primary gingival fibroblasts. Biomaterials. 2006;27:1803–1809. doi: 10.1016/j.biomaterials.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 21.Walther UI, Siagian II, Walther SC, Reichl FX, Hickel R. Antioxidative vitamins decrease cytotoxicity of HEMA and TEGDMA in cultured cell lines. Archives of oral biology. 2004;49:125–131. doi: 10.1016/j.archoralbio.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura H, Saruwatari L, Aita H, Takeuchi K, Ogawa T. Molecular and biomechanical characterization of mineralized tissue by dental pulp cells on titanium. Journal of dental research. 2005;84:515–520. doi: 10.1177/154405910508400606. [DOI] [PubMed] [Google Scholar]
- 23.Wright S. Preparation for peripheral blood progenitor cell collection. Oncol Nurs Forum. 1998;25:471. [PubMed] [Google Scholar]
- 24.Jewett A, Bonavida B. Target-induced inactivation and cell death by apoptosis in a subset of human NK cells. J Immunol. 1996;156:907–915. [PubMed] [Google Scholar]
- 25.Jewett A, Bonavida B. Peripheral blood monocytes derived from HIV+ individuals mediate antibody-dependent cellular cytotoxicity (ADCC) Clin Immunol Immunopathol. 1990;54:192–199. doi: 10.1016/0090-1229(90)90081-z. [DOI] [PubMed] [Google Scholar]
- 26.Jewett A, Cacalano NA, Head C, Teruel A. Coengagement of CD16 and CD94 receptors mediates secretion of chemokines and induces apoptotic death of naive natural killer cells. Clin Cancer Res. 2006;12:1994–2003. doi: 10.1158/1078-0432.CCR-05-2306. [DOI] [PubMed] [Google Scholar]
- 27.Jewett A, Bonavida B. Interferon-alpha activates cytotoxic function but inhibits interleukin-2-mediated proliferation and tumor necrosis factor-alpha secretion by immature human natural killer cells. Journal of clinical immunology. 1995;15:35–44. doi: 10.1007/BF01489488. [DOI] [PubMed] [Google Scholar]
- 28.Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 29.Mantellini MG, Botero T, Yaman P, Dennison JB, Hanks CT, Nor JE. Adhesive resin and the hydrophilic monomer HEMA induce VEGF expression on dental pulp cells and macrophages. Dent Mater. 2006;22:434–440. doi: 10.1016/j.dental.2005.04.039. [DOI] [PubMed] [Google Scholar]
- 30.Araki S, Dobashi K, Kubo K, Yamamoto Y, Asayama K, Shirahata A. N-acetylcysteine attenuates TNF-alpha induced changes in secretion of interleukin-6, plasminogen activator inhibitor-1 and adiponectin from 3T3-L1 adipocytes. Life sciences. 2006;79:2405–2412. doi: 10.1016/j.lfs.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 31.Malorni W, Rivabene R, Lucia BM, Ferrara R, Mazzone AM, Cauda R, Paganelli R. The role of oxidative imbalance in progression to AIDS: effect of the thiol supplier N-acetylcysteine. AIDS research and human retroviruses. 1998;14:1589–1596. doi: 10.1089/aid.1998.14.1589. [DOI] [PubMed] [Google Scholar]
- 32.Spada C, Treitinger A, Reis M, Masokawa IY, Verdi JC, Luiz MC, Silveira MV, Michelon CM, Avila-Junior S, Gil DO, Ostrowskyl S. The effect of N-acetylcysteine supplementation upon viral load, CD4, CD8, total lymphocyte count and hematocrit in individuals undergoing antiretroviral treatment. Clin Chem Lab Med. 2002;40:452–455. doi: 10.1515/CCLM.2002.077. [DOI] [PubMed] [Google Scholar]
- 33.Thavarajah M, Evans DB, Kanis JA. Differentiation of heterogeneous phenotypes in human osteoblast cultures in response to 1,25-dihydroxyvitamin D3. Bone. 1993;14:763–767. doi: 10.1016/8756-3282(93)90208-r. [DOI] [PubMed] [Google Scholar]
- 34.Lee DH, Lim BS, Lee YK, Yang HC. Effects of hydrogen peroxide (H2O2) on alkaline phosphatase activity and matrix mineralization of odontoblast and osteoblast cell lines. Cell biology and toxicology. 2006;22:39–46. doi: 10.1007/s10565-006-0018-z. [DOI] [PubMed] [Google Scholar]
- 35.Alliot-Licht B, Bluteau G, Magne D, Lopez-Cazaux S, Lieubeau B, Daculsi G, Guicheux J. Dexamethasone stimulates differentiation of odontoblast-like cells in human dental pulp cultures. Cell and tissue research. 2005;321:391–400. doi: 10.1007/s00441-005-1115-7. [DOI] [PubMed] [Google Scholar]
- 36.Jewett A, Cacalano NA, Teruel A, Romero M, Rashedi M, Wang M, Nakamura H. Inhibition of nuclear factor kappa B (NFkappaB) activity in oral tumor cells prevents depletion of NK cells and increases their functional activation. Cancer Immunol Immunother. 2006;55:1052–1063. doi: 10.1007/s00262-005-0093-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jewett A, Head C, Cacalano NA. Emerging mechanisms of immunosuppression in oral cancers. Journal of dental research. 2006;85:1061–1073. doi: 10.1177/154405910608501201. [DOI] [PubMed] [Google Scholar]
- 38.Jewett A, Wang MY, Teruel A, Poupak Z, Bostanian Z, Park NH. Cytokine dependent inverse regulation of CD54 (ICAM1) and major histocompatibility complex class I antigens by nuclear factor kappaB in HEp2 tumor cell line: effect on the function of natural killer cells. Human immunology. 2003;64:505–520. doi: 10.1016/s0198-8859(03)00039-9. [DOI] [PubMed] [Google Scholar]