

Abstract
Background and Objectives
Mutations in the CARD15 gene affecting NOD2 function are susceptibility factors in Crohn’s disease. We set out to explore the mechanism of this susceptibility using mice that over-express NOD2.
Methods
Cellular and molecular responses of mice bearing a NOD2 transgene or administered plasmids that express wild-type and mutated NOD2 constructs were examined.
Results
In initial studies we showed that splenocytes from NOD2-transgenic mice as compared to littermate-controls (controls) exhibit greatly decreased IL-12p70 responses to peptidoglycan (PGN) a TLR2 ligand that contains MDP, but not other TLR ligands; in contrast, IL-12 responses to PAM3CSK4, a TLR2 ligand that does not contain MDP, were normal. Similarly, transgenic mice as compared to controls exhibited greatly decreased IL-12p40 responses to IP administration of PGN but not to LPS. In further studies we showed using EMSA that PGN-stimulated cells from transgenic mice exhibited greatly decreased p65 and c-Rel super-shifts, indicating decreased activation of NF-κB. Finally, in a series of studies on the effect of the NOD2 on susceptibility to induced colitis, we found that: 1) transgenic mice were highly resistant to induction of PGN-colitis and partially resistant to induction of TNBS-colitis; and 2) mice administered a plasmid expressing a wild-type NOD2 gene was completely resistant to TNBS-colitis whereas mice administered a plasmid expressing a NOD2 gene with the Crohn’s disease frame-shift mutation was only slightly resistant to TNBS-colitis.
Conclusion
these data offer strong new evidence that NOD2 mutations contribute to IBD by leading to excessive TLR2 cytokine responses.
Introduction
Approximately 15% of patients with Crohn’s disease bear homozygous or compound heterozygous mutations in the gene that encodes NOD2 (CARD15), an intra-cytoplasmic member of the family of proteins now known as the CLR or NLR proteins (1–4). These proteins are usually composed of a central NOD domain (nucleotide oligomerization domain) flanked on its C-terminal side by a LRR domain (leucine-rich repeat domain) that is capable of recognizing microbial components and on its N-terminal side by a CARD or pyrin domain that interacts with downstream molecules to bring about effector function (3–5). NOD2 has been shown to recognize muramyl dipeptide (MDP), a component of peptidoglycan (PGN), the latter itself a component of the bacterial wall of virtually all bacteria (6, 7).
Upon recognition of MDP, NOD2 engages a downstream molecule known as RICK (RIP2) via a CARD-CARD interaction (8). This leads to subsequent RICK-mediated ubiquitination of IKKγ (NEMO), the component of NF-κB that initiates NF-κB activation (9). Such MDP-induced NOD2 signaling is disturbed in Crohn’s disease because mutations in the LRR domain hinder recognition of MDP; thus, antigen-presenting cells (APCs) from these patients exhibit impaired MDP-induced activation of NF-κB (7). This raises the question of how mutations leading to loss of the ability to activate NF-κB can be a susceptibility factor in a disease in which increased activation of NF-κB is a necessary feature of the Th1/Th17 inflammation mediating the disease (10, 11).
One possible answer to this question was provided by recent studies of NOD2-deficient mice in which it was shown that NOD2-deficient APCs manifest increased NF-κB activation and IL-12 production upon exposure to PGN, the bacterial component that gives rise to MDP (12). In addition, it was shown that MDP could down-regulate the IL-12 response of normal APCs to PGN. On the basis of these findings the hypothesis was put forward that the mutations in NOD2 occurring in Crohn’s disease lead to loss of this down-regulatory function and hence an over-exuberant NF-κB-driven IL-12 response that is the hallmark of the disease.
In the present study we demonstrate that mice that over-express NOD2 due to the presence of a NOD2 transgene exhibit diminished PGN-driven IL-12 responses both in vitro and in vivo. In addition, we show that these mice are resistant to the induction of PGN-induced colitis and TNBS-colitis. Finally, we demonstrate that mice that over-expressing NOD2 due to administration of a plasmid that expresses NOD2 exhibits resistance to the induction of TNBS-colitis, whereas mice administered a plasmid with a NOD2 frame shift mutation such as that found in patients with Crohn’s disease exhibits only slight resistance to induction of TNBS-colitis. These findings support the view that mutations in NOD2 lead to Crohn’s disease because they cause a loss of the regulatory function of NOD2.
Material and Methods
Mice
Specific pathogen-free, 4–6-wk-old wild type or OT-II C57/BL6, or C57/BL10 mice were purchased from Jackson Laboratories (Bar Harbor). Transgenic mice bearing a NOD2 gene were developed at NIH as described below. Animal use adhered to National Institutes of Health Animal Care Guidelines.
Transgene Construction and Generation of Transgenic Mice
A pcDNA4HisMax plasmid containing a mouse NOD2 cDNA was subjected to BamH1 and XhoI digestion to obtain the NOD2 cDNA insert (13). The latter was blunt ended and sub-cloned into into a pCR Blunt II TOPO vector (invitrogen, Carlsbad, CA), and then cloned into pDOI-5 (7.2kb) vector at EcoRI site directly up-stream of a MHC class II promoter (14). Finally, a 6.8kb XbaI-BsaBI fragment of the MHC class II promoter linked to the NOD2 cDNA was microinjected to construct NOD2 transgenic mice. The NOD2Tg C57B6/129SV mouse founders were then backcrossed with C57/BL10 or C57/BL6 for 5–6 generations. The genotype of mouse off-spring was determined by PCR screening using primers NOD2(F):5′-agaagccctcctgcaggccctca-3′ and Rab-β-globin(R): 5′-ctcagtggtatttgtgagcca-3′.
Western blot analysis and Real Time RT-PCR analysis of NOD2 expression
MHC Class II+ or MHC class II- cells, CD11b+, and CD11c+ cells were isolated from the spleen by positive or negative selection using anti-mouse MHC Class II, CD11b, or CD11c microbeads (Miltenyi Biotech). Purified spleen cell populations were lysed in lysate buffer (Roche Applied Science), and 1% SDS for solubilization of Nonidet P-40-insoluble protein. Lysates were separated in NuPAGE 10% Bis-Tris Gel (Invitrogen) and transferred onto a nitrocellulose membrane. The membrane was then probed with rabbit anti-Nod2 Ab (Cayman Chemical) or rat anti-mouse NOD2 mAb (E-Biosciences, kindly provided by Dr. Peggy Lost) followed by HRP-conjugated anti-rabbit IgG or anti-rat IgG and then developed with a chemiluminescence detection system (Pierce). For real-time RT-PCR for NOD2 mRNA, total RNA was isolated from purified cell populations by RNeasy mini kit (Qiagen) and converted to cDNA with the High-Capacity cDNA Archive Kit (Applied Biosystems). cDNA (2μg of total RNA per sample) was added to the TaqMan® Universal Master Mix and analyzed in triplicate using an ABI PRISM 7700 unit (Applied Biosystems) with the Probe: 5′-(FAM) TGC TTT TTT GCC GCT TTC TAC TTG GCT (TAMRA)-3′, Forward primer: 5′-GAG TTC CTG CAC ATT ACC TTC CA-3′, and Reverse primer: 5′-AGA GGC CAC CGA TGT GTC AG-3′ (13). As an endogenous reference, primers and probe for glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) were included.
FACS analysis
Spleen cells were stained with FITC-conjugated mAbs (Pharmingen) and analyzed on FACSCalibur (BD Biosciences Immunocytometry Systems) with FlowJo software (FlowJo, LLC).
Cell cultures and cytokine assay
Splenocytes were cultured with LPS (1μg/ml, InvivoGen), PGN from Staphylococcus aureus (10μg/ml, Fluka), Pam3CSK4 (500ng/ml, InvivoGen), dsRNA (50μg/ml, InvivoGen), Loxoribine (100mM, invivoGen), CpG (1μM, InvivoGen) or MDP (at 10 or 100 μg/ml, Sigma) in RPMI1640 complete medium for 48 hrs as described previously(12, 15). Culture supernatants were assayed for IL-12p40, IL-12p70 and TNF using ELISA kits (Pharmingen and R&D systems). CD4+ T cells were isolated from the spleens of OT-II mice by anti-mouse CD4 microbeads (Miltenyi Biotech). By flow cytometry the purity of these cell populations were > 90%. OT-II CD4+ T cells (1×106/ml) were cultured with splenic CD11b+cells (1×106/ml) from Tg-BL/6 or Wt-BL/6mice loaded with OVA323–330 peptide (0.5μM) in RPMI1640 medium alone or in the presence of LPS, PGN, Pam3CSK4, CpG or MDP (15). Culture supernatants were harvested at 72hrs for determination of IFNγ production by ELISA (Pharmingen).
Systemic administration of PGN, LPS and Pam3CSK4
Wild-type or transgenic mice were administered 300μg of LPS (InvivoGen), PGN(Fluka) or 100μg Pam3CSK4 concomitantly with 100 μg of MDP (Sigma)) intravenously, and then serum samples were collected at 0, 2 and 5 h. Concentations of IL-12p40 and IL-6 in the serum samples were subsequently determined by ELISA kits (Pharmingen).
NF-κB activation assay
Nuclear extracts of splenic adherent cells stimulated for 2h with LPS or PGN were obtained using a nuclear extraction kit (Active Motif). Assay of NF-κB subunits in the extracts, was performed by EMSA as previously described (12). Anti-p65 or anti-c-Rel (Santa Cruz Biotechnology) were employed for super-shift analyses.
Induction of PGN-induced or TNBS-induced Colits and Culture of Lamina Propria Mononuclear Cells
PGN-colits was induced by intra-rectal administration of 1 mg of PGN (Lee Laboratories) of mice pre-administered 100 μl of 50% ethanol per rectum. TNBS-colits was induced by intrarectal administration of 3.75 mg of TNBS (Sigma) dissolved in 45% ethanol as previously described (16). Challenged mice were observed for 4–5 days and then sacrificed for histopathologic assessment and harvest of colonic LPMCs as previously described (16). LPMCs were cultured in 48-well plates (106 cells/ml) in wells coated with anti-CD3ε (10μg/ml, Pharmingen) and soluble anti-CD28 (1μg/ml, Pharmingen) for IFN-γ production, and with IFNγ (1000 units/ml, Pharmingen) and Staphylococcus aureus (1:10,000 Calbiochem) for IL-12p70 production. Culture supernatants were assayed by ELISA (Pharmingen).
Plasmids Expressing wild-type NOD2 or mutated NOD2 and their encapsulation for in vivo delivery
The pcDNA4HisMax plasmid containing a mouse NOD2 cDNA mentioned above served as the expression plasmid for wild-type NOD2. To obtain a mutated NOD2 plasmid, an Afl II mutation was introduced into this plasmid by the mutation primer 5′-CAGAAGCCCTCCTGCAGGCCCCTTAAGGGAACAGTGCCATTCTGGAG-3′ and its antisense primer using the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene). PCR with these primers introduced L980P followed by stop codon TAA in the mouse NOD2 cDNA that was equivalent to the human 3020insC Crohn disease frame-shift mutation (1, 2). For in vivo delivery of these plasmids, large amounts of plasmid was prepared using an endofree Plasmid Mega kit (Qiagen), and then packaged into a Hemagglutinin Virus Japan-Envelope vector using protamine sulfate (HVJ-E, GenomIdea, Osaka, Japan) according to the manufacturer’s instructions (16). Mice induced with TNBS-colitis were subjected to IP administration of 100 μg of plasmid DNA/mouse encapsulated in 5000 HAU of HVJ-E vector in 100μl of PBS at −1, 0 and +1 day of TNBS intrarectal challenge.
Results
Generation of NOD2-Transgenic Mice
To gain new insight into the function of NOD2 we probed responses of mice that over-express this protein due to the presence of a NOD2 transgene in antigen-presenting cells (APCs). The transgene construct that was used to generate the transgenic mice consisted of a mouse NOD2 cDNA under the control of the MHC class II promoter (see Methods) so that the transgenic mice obtained over-expressed NOD2 mainly in APCs. Thus, as shown in Figure 1A, studies of NOD2 expression using Western blotting with anti-NOD2 revealed that splenic MHC Class II+ and CD11b+ or CD11c+ cells (but not MHC Class II- cells) from hemizygous NOD2 transgenic (NOD2-Tg) mice contain greatly increased amounts of NOD2 as compared to extracts from littermate control wild type (Wt) mice and that the size of the transgenic NOD2 was the same as the size as endogenous NOD2 (which is only detectable in wild type CD11c+ cell lysates). In addition, in parallel studies shown in Figure 1B, quantitation of NOD2 mRNA by real-time PCR corroborated these results. Finally, it should be mentioned that NOD2-Tg mice on either a C57BL/6 or BL/10 background mice developed normally and, as shown in Supplemental Figure 1, exhibit normal cellular development.
Figure 1.
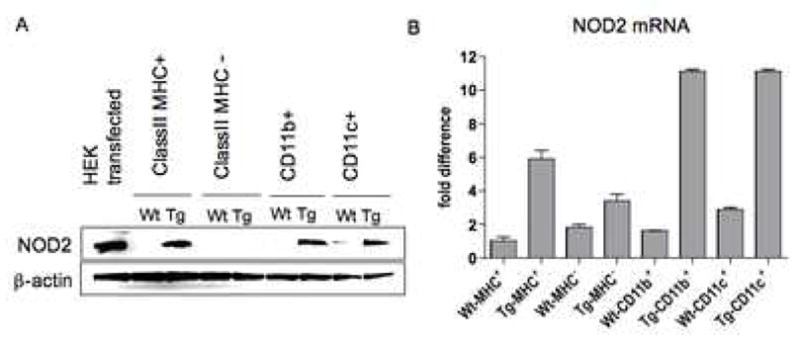
Expression of NOD2 in NOD2-Tg and littermate control (wild-type) mice. (A) Western blot analysis of NOD2 expression in spleen cell subpopulations from C57BL/6 NOD2-Tg and littermate control (Wt) mice. (B) Real time RT-PCR for quantitation of NOD2 mRNA in spleen cell subpopulations.
Splenocytes from NOD2-Tg mice exhibit enhanced MDP-mediated negative regulatory responses
In a previous study we demonstrated that MDP activation of NOD2 negatively regulates PGN-induced IL-12 production. To re-examine this in mice that over-express NOD2, we measured cytokine responses of splenocytes from NOD2-Tg and littermate control C57BL/6 (Wt-C57BL/6) mice after stimulation with a wide range of TLR ligands in the presence and absence of MDP. As shown in Fig. 2, panels A–B, there was a 5–10-fold reduction in PGN (TLR2)-induced IL-12p70 production by NOD2-Tg BL/6 splenocytes as compared to littermate control BL/6 splenocytes. In contrast, stimulation of cells from NOD2-Tg and littermate control mice with other TLR ligands led to similar levels of IL-12p70 production in the two groups. Comparable findings were obtained with respect to IL-12p40 production (data not shown). Finally, as shown in Fig. 2, panels C–D, an equivalent set of findings were found with NOD2-Tg BL/10 and littermate control splenocytes.
Figure 2.
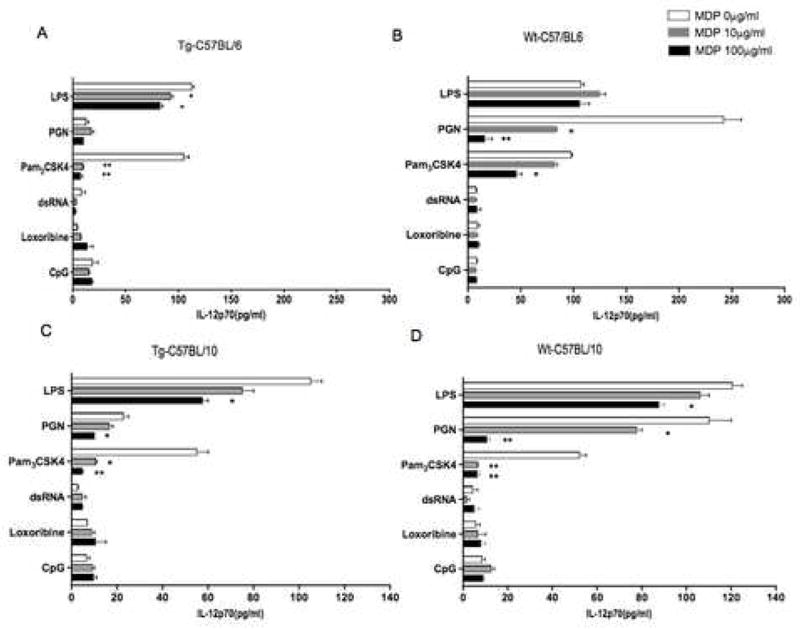
Splenocytes from C57BL/6 and C57BL/10 NOD2-Tg mice exhibit increased negative regulation of TLR2-induced IL-12p70 responses. (A, B) IL-12p70 production of splenocytes obtained from C57BL/6 NOD2-Tg and littermate-control (wild-type) mice; (C, D) IL-12p70 production of splenocytes obtained from C57BL/10 NOD2-Tg and littermate-control (wild-type) mice respectively. Total splenocytes were stimulated for 48h with LPS (1μg/m), PGN (10μg/ml), Pam3CSK4 (500ng/ml), dsRNA (25μg/ml), loxoribine (100μM), or CpG (1μM) in the absence or presence of MDP (0 μg/ml, 10μg/ml, or 100μg/ml); culture supernatants were then analyzed by ELISA. Each result presents the means ± SD of triplicate assays and is representative of three independent experiments. Statistical differences denoted by *p < 0.05, **p < 0.01 between the presence and absence of MDP.
Since PGN can activate both cell-surface TLR2 and intracellular NOD2 (the latter via MDP released by PGN degradation), it was possible that the decreased PGN response was due to negative regulation by MDP acting through NOD2. To examine this possibility more directly, we determined PGN- and Pam3CSK4-induced IL-12p70 responses in the presence of increasing concentrations of MDP. As shown in Figure 2, panels A–B, addition of MDP had a pronounced and dose-dependent down-regulatory effect on PGN-induced IL-12p70 production in cells from both NOD2-Tg and littermate control mice. This effect was more evident in cells from littermate control mice than in NOD2-Tg mice; however, this is probably due to the fact that cells from the NOD2-Tg mice had low baseline IL-12p70 production (i.e., IL-12 production in the absence of added MDP), probably because even the small amount of MDP released from PGN is sufficient for down-regulation in mice with NOD2 over-expression. Furthermore, in the case of Pam3CSK4, addition of MDP again led to down regulation of the IL-12p70 response and, as might be predicted, in this case the down regulation was more striking in NOD2-Tg mice than in littermate control mice. In contrast, MDP did not down-regulate responses to other TLR ligands. Finally, as shown in Figure 2, panels C–D, similar results were obtained with splenocytes from NOD2-Tg BL/10 mice, although in this case the differences between transgenic and littermate control mouse cells with respect to PAM3CSK4 were not as dramatic because the cells from control mice of this mouse strain also manifested a high degree of down-regulation with MDP.
Overall, this enhanced down-regulation of TLR2-induced IL-12p70 production by MDP in NOD2-Tg mice suggests a gene dose effect of NOD2 and supports the contention that NOD2 signaling leads to negative regulation of TLR2-induced IL-12 responses.
The above results obtained with whole splenocyte populations were most likely indicative of MHC class II+ cell (APC) responses within these populations. To examine this directly, we isolated CD11b+ dendritic cells from spleen populations with antibody-coated bead columns and then stimulated the cells with PGN or PGN plus MDP. In this case, the PGN used was a highly purified PGN free of lipotechoic acid shown in previous studies to not stimulate cells lacking TLR2. As shown in Supplemental Figure 2, CD11b+ cells from transgenic mice also exhibited decreased responses to PGN and cells from littermate control mice exhibited decreased responses upon addition of MDP.
Splenocytes from NOD2-Tg Mice do not display enhanced cytokine responses
Previous studies of responses of cells to MDP, conducted mainly in humans have shown that at least with respect to certain cytokines, MDP enhances rather than reduces responses to various TLR ligands (17–19). Thus, cells from NOD2-Tg might be expected to also exhibit enhanced responses in some instances. To explore this possibility we examined both TNF-α and IL-10 responses to various TLR ligands under the same conditions of IL-12 responses to TLR2 ligands. As shown in Supplemental Figure 3, panels A–B, NOD2-Tg cells displayed some enhancement of TNF-α responses following stimulation with TLR2 and TLR3 ligands, as compared to control cells but no enhancement of IL-10 responses. Furthermore, addition of MDP to the culture did not lead to significantly increased responses over baseline (no MDP) with either NOD2-Tg cells or control cells. Thus, over-expression of NOD2 did not cause further enhancement of previously noted increased responses induced by NOD2 signaling.
NOD2-Tg mice exhibit decreased in vivo IL-12 responses
To provide additional evidence that NOD2 signaling has a negative effect on IL-12 production mediated by PGN/TLR2, we measured the serum level of IL-12 in NOD2-Tg and littermate control (Wt) mice following intraperitoneal injection of PGN or LPS or injection of these ligands plus MDP. As shown in Figure 3A, the serum levels of both IL-12p40 and IL-6 were greatly reduced in NOD2-Tg mice as compared with those of littermate control mice when measured at the peak of their respective responses (either 2h or 5h after PGN injection). In littermate control mice the response of both cytokines was decreased by the co-administration of MDP whereas in NOD2-Tg mice co-administration caused no further decrease. In contrast, as shown in Figure 3B, the LPS-induced serum IL-12p40 and IL-6 responses in NOD2-Tg mice and littermate control mice were equivalent and no effect of co-administered MDP was seen. Similar studies were conducted to determine systemic responses to Pam3CSK4 and Pam3CSK4 plus MDP. In these studies IL-12p40 rather than IL-12p70 responses are reported as Pam3CSK4 did not induce a measurable IL-12p70 response. As shown in Figure 3C, while the Pam3CSK4 IL-12p40 response was not affected by the addition of MDP of littermate control mice, such addition did significantly lower the Pam3CSK4 IL-12p40 response in NOD2-Tg mice. These data show that activation of the NOD2 transgene leads to increased negative regulation of PGN/TLR2-mediated IL-12 responses in vivo as well as in vitro.
Figure 3.
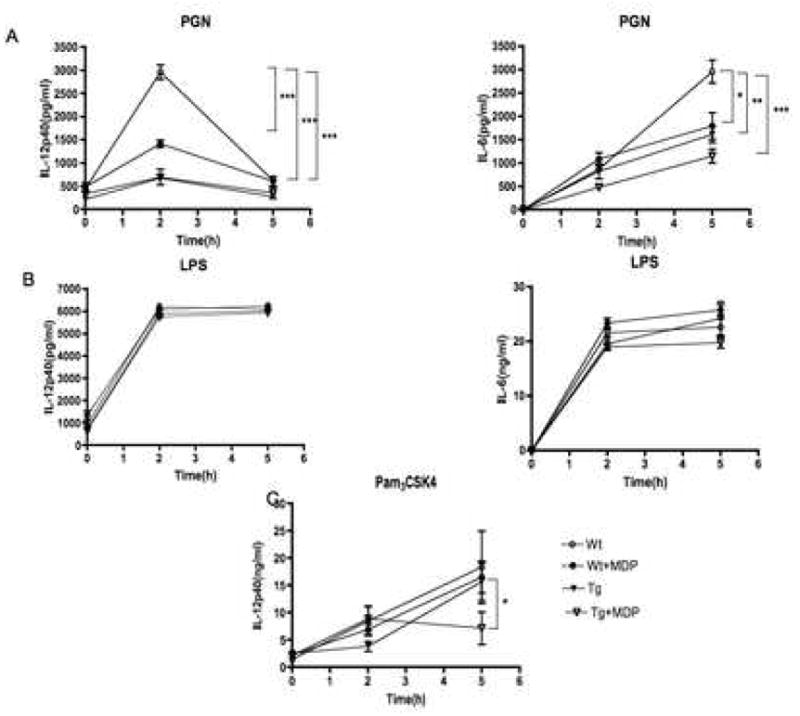
NOD2-Tg mice exhibit decreased serum IL-12p40 and IL-6 responses following systemic injection of PGN or PGN plus MDP. (A) IL-12p40 and IL-6 titers in sera after PGN (300μg) administration, (B) IL-12p40 and IL-6 titers in sera after LPS (300μg) administration, (C) IL-12p40 titers in sera after PAM3CSK4 administration with and without MDP (100μg) by intra-peritoneal injection of C57BL/6 NOD-2-Tg and littermate control (wild-type) BL/6 mice. Serum collected by intra-ocular bleeding was obtained at 0h, 2h and 5h after injection in 5 mice per each condition. Each result is representative of two independent studies. Statistical differences at peak levels were denoted by *, p <0.05; **, p <0.01 and ***, p <0.001.
T cell receptor (TCR)-Tg T cells exhibit a decreased antigen-specific Th1 cytokine response following activation by TLR2-stimulated NOD2 APCs
To study the effect of NOD2 over-expression on downstream antigen-specific T cell cytokine responses, we next determined the capacity of APCs from littermate control and NOD2-Tg mice to induce the differentiation of TCR-Tg T cells in the presence of antigen recognized by the T cells as well as a TLR ligand that provides co-stimulation of the APCs. In these studies we co-cultured CD4+ T cells isolated from OT-II transgenic mice (that recognize the peptide fragment 323–339 of ovalbumin (OVA323–339 peptide) in the context of IAb) with OVA323–339 peptide-pulsed splenic CD11b+ cells from NOD2 Tg mice or littermate control (Wt) mice in the presence of a variety of TLR ligands; then, after 72 h, we measured the level of IFN-γ in the culture supernatants. As shown in Fig. 4, OT-II CD4+ T cells exhibited no difference in IFN-γ production in the absence of TLR ligands whether OVA peptide was presented by NOD2-Tg APCs or littermate control APCs. In addition, OT-II CD4+ T cells exhibited enhanced IFN-γ production in cultures in which OVA-peptide was presented by either NOD2-Tg or littermate control APCs in the presence of the TLR ligands LPS or CpG as compared to cultures not containing TLR ligands. In contrast, OT-II CD4+ T cells manifested greatly decreased IFN-γ production when presented OVA-peptide by NOD2-Tg APCs cultured in the presence of PGN whereas robust IFN-γ production was seen in identical cultures containing littermate control APCs. In concert with this latter finding, while baseline IFN-γ responses of OT-II T cells to OVA peptide in the presence of Pam3CSK4 in this case was higher in the cultures containing transgenic APCs than in cultures containing WT APCs, IFN-γ responses of OT-II cells to OVA in the presence of both Pam3CSK4 and MDP were more profoundly reduced in the cultures containing transgenic APCs.
Figure 4.
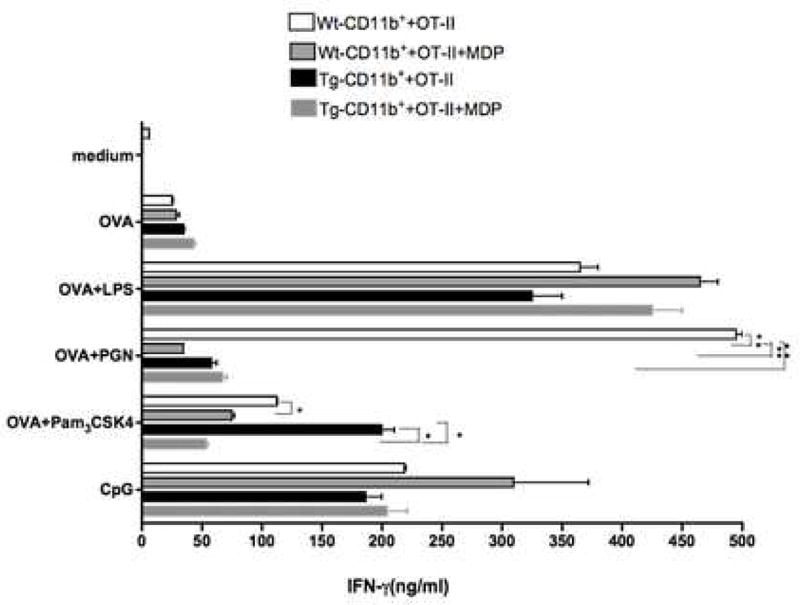
TCR-Tg T cells exhibit decreased antigen-specific Th1 responses when activated by TLR2-stimulated APCs from C57BL/6 NOD2-Tg mice. CD4+ T cells recognizing a peptide fragment of ovalbumin (OVA323–339 peptide, 0.5μM) were isolated from the spleens of OT-II TCR transgenic mice and then stimulated in vitro with OVA323–339 pulsed splenic CD11b+ cells from either NOD2-Tg or littermate control (wild-type) mice in the presence or absence of TLR ligands and MDP. The TLR ligands included LPS (1μg/m), PGN (10μg/ml), Pam3CSK4 (500ng/ml), CpG (1μM), and MDP (10μg/ml). Culture supernatants were harvested at 72 hrs and then subjected to ELISA for determination of cytokine levels. The data shown are mean values ± SD from three independent experiments. Statistical differences in the IFN-γ concentration between the NOD2-Tg groups and the littermate control groups were denoted by *, p < 0.05; **, p < 0.01.
NOD2 over-expression results in decreased NF-κB activation following stimulation by PGN
IL-12 production by APCs is dependent on activation and nuclear translocation of NF-κB components (20). Thus, the negative regulation of IL-12 occurring in APCs over-expressing NOD2 that are subjected to PGN stimulation implies that these cells manifest reduced nuclear translocation of NF-κB subunits during such stimulation. To explore this possibility we determined activation and nuclear translocation of NF-κB components by electrophoretic mobility shift assays (EMSAs) in cells stimulated by PGN and LPS under the same conditions used in the above in vitro cytokine production studies (see Methods). As shown in Fig. 5, there is no difference in the nuclear translocation of NF-κB components between NOD2-Tg and littermate control (Wt) APCs stimulated with LPS. In contrast, translocation of NF-κB was markedly suppressed in NOD2-Tg APCs but not in littermate control APCs stimulated by PGN. Furthermore, supershift assays using anti-p65 and anti-c-Rel Abs revealed that translocation of both p65 and c-Rel subunits of NF-κB is reduced in NOD2-Tg APCs stimulated with PGN as compared to littermate control (Wt) APCs stimulated in an identical fashion. Thus, these data indicate that down-regulation of IL-12 production by PGN-stimulated NOD2-Tg APCs is in fact associated with decreased activation and nuclear translocation of NF-κB components.
Figure 5.
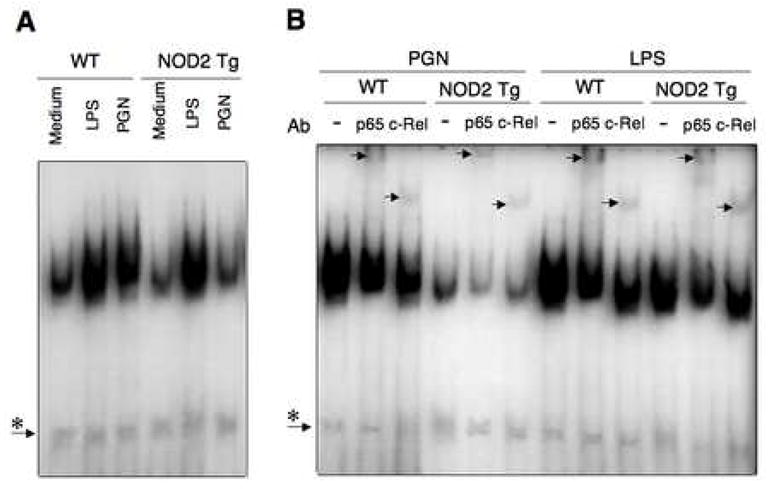
PGN stimulation of C57BL/6 NOD2-Tg splenic adherent cells results in down-regulated NF-κB signaling. (A) EMSA analysis of NF-κB with nuclear extracts from splenic adherent cells obtained from NOD2-Tg and littermate control (wild-type) mice stimulated for 2 hr with PGN (10mg/ml) or LPS (1mg/ml). (B) EMSA performed as in (A) including supershift analyses with anti-p65 and anti-c-Rel. Each lane in panel A and B is loaded with 5μg of nuclear extract; asterisks indicates non-specific band.
Mice that over-express NOD2 are resistant to the development of PGN- or TNBS-colitis
A prediction of the finding that NOD2 over-expression leads to down regulation of PGN-induced IL-12 production in vitro is that such over-expression would inhibit Th1 inflammation in vivo. To explore this possibility we determined if NOD2-Tg mice were resistant to the development of two forms of experimental colitis.
The first model used for this purpose took advantage of the fact that PGN itself can induce colitis (21, 22) and therefore we could test the effect of NOD2 over-expression on a colitis induced by a TLR ligand closely tied to NOD2-mediated negative regulation. In studies of PGN-colitis we administered 50% ethanol per rectum to both NOD2-Tg mice and littermate control mice alone or followed 8h later by soluble PGN (1mg) again per rectum and then characterized the ensuing colitis in the two mouse groups. As shown in Figure 6A, littermate control mice administered PGN experienced a severe and progressive weight loss beginning on day 1 after administration of PGN and continuing on subsequent days until day 3, at which time the mouse usually died. In contrast, NOD2-Tg mice administered PGN experienced only minimal weight loss at day 1 after administration of PGN and then rapidly regained lost weight so as to achieve a normal weight (compared to untreated littermate control mice) at day 3 after administration of PGN. This weight change in the NOD2-Tg animal that received PGN was not different from the mice that received ethanol alone (data not shown). As shown in Figures 6B and 6C these weight changes correlated with the histopathologic appearance of the bowel wall of mice in the various experimental groups as well as studies of IL-12p70 production ex vivo.
Figure 6.
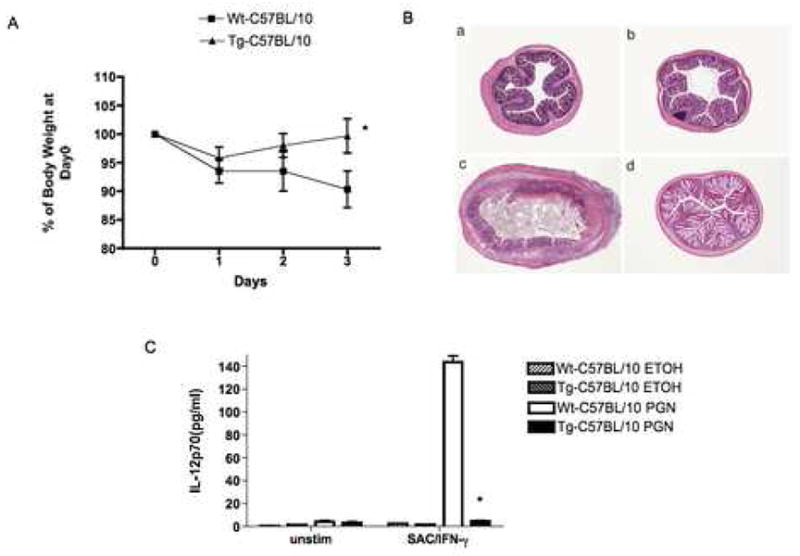
NOD2-Tg mice exhibit resistance to the induction of PGN-colits. C57BL/10 NOD2-Tg and littermate control (wild-type) mice were administered soluble PGN or ethanol alone (ETOH) per rectum. (A) Body weight changes (Mean± SD) of mice in each group (expressed as a percent of initial weight); on average, there were 10 mice in each group in two independent studies. Statistical differences between NOD-2 Tg and littermate control groups denoted by *, p < 0.05. (B) Representative H&E stained cross-sections of colons of mice at the time of sacrifice 3 days after PGN challenge (magnification: 25X); (a,c) littermate control mice administered ETOH alone or PGN respectively; (b,d) NOD2-Tg mice administered ETOH alone or PGN, respectively. (C) IL-12p70 production by LPMC obtained from mice 3 day after PGN or ETOH administration and cultured with Staphylococcus aureus (Cowan I) plus IFN-γ for 48h. Data are expressed as Mean ± SD from cells obtained from 10 mice in each group (derived from two separate studies). Statistical differences between NOD2-Tg and littermate control group administered PGN denoted by *, p < 0.05.
The second model of inflammation studied was trinitrobenzene sulfonic acid (TNBS)-colitis, a Th1 colitis that is initiated by TNP-substituted colonic proteins (11, 16). In studies of TNBS-colitis we administered intra-rectal TNBS (3.75 mg) in 45% ethanol or ethanol alone to C57BL/10 NOD2-Tg mice and littermate control mice and then characterized the colitis in these mouse groups. As shown in Figure 7A, whereas mice in both groups administered TNBS exhibited weight loss in the first two days after TNBS challenge, NOD2-Tg mice regained most of their initial weight in the final two days of study, whereas littermate control mice continued to lose weight. This weight change in the NOD2-Tg animal that received TNBS was not different from the mice that received ethanol alone (data not shown). As shown in Figure 7B and 7C this absence of weight loss in NOD2-Tg correlated with histopathologic changes and IL-12p70 production ex vivo. Thus, in parallel with their resistance to PGN-colitis, NOD2 mice also demonstrated a decreased susceptibility TNBS-colitis.
Figure 7.
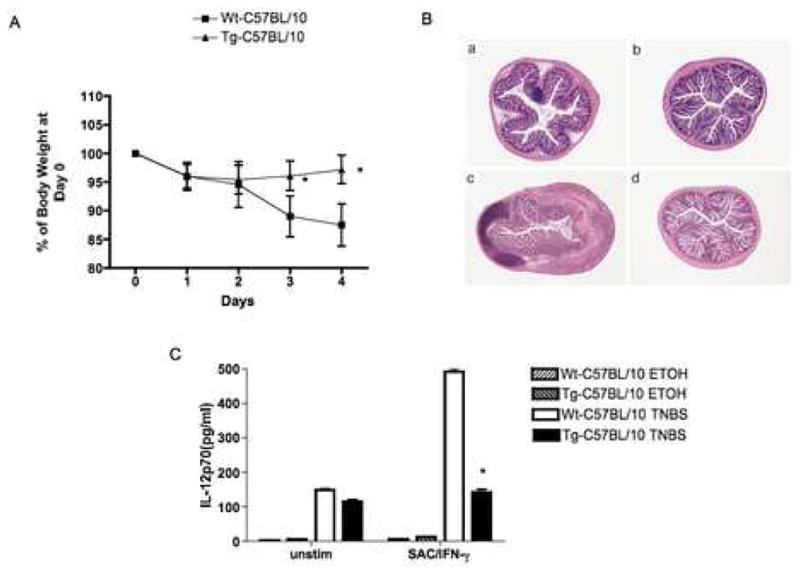
NOD2-Tg mice exhibit resistance to the induction of TNBS-colitis. C57BL/10 NOD2-Tg and littermate control (wild-type) mice were administered TNBS in ethanol or ethanol alone (ETOH) per rectum. (A) Body weight changes (Mean± SD) of mice in each group (expressed as a percent of initial weight); on average there were 10 mice in each group in two independent studies. Statistical differences between NOD2-Tg and littermate control group denoted by *, p < 0.05. (B) Representative H&E stained cross-sections of colons of mice at the time of sacrifice 4 days after TNBS challenge (magnification: 25X); (a,c) littermate control mice administered ETOH alone or TNBS respectively; (b,d) NOD2-Tg mice administered ETOH alone or TNBS respectively. (C) IL-12p70 production by LPMC obtained from mice 4 day after TNBS-colitis induction and cultured with Staphylococcus aureus (Cowan I) plus IFN-γ for 48h. Data are expressed as Mean ± SD from cells obtained from 10 mice in each group (derived from two separate studies). Statistical differences between NOD2-Tg and littermate control group administered TNBS denoted by *, p < 0.05.
Treatment of TNBS-colitis by administration of NOD2-expressing plasmids
In further in vivo studies exploring the effect of over-expression of NOD2 on the development of TNBS-colitis we determined the capacity of plasmids expressing NOD2 or NOD2 with a frame-shift mutation similar to the L1007fs mutation observed in patients with Crohn’s disease to prevent the development of this experimental colitis. Accordingly, plasmids of this type were constructed in which the wild type mouse NOD2 or mouse NOD2 with the mutation L980fs were expressed under a CMV promoter (see Methods). These plasmids as well as an “empty vector” control plasmid were encapsulated in an HVJ-E viral coat for efficient in vivo delivery and administered IP (100μg/mouse/dose) one day before, on the day of and one day after intra-rectal challenge with TNBS. As shown in Figure 8A, Western blot analysis of adherent spleen cells from mice administered plasmids exhibited robust expression of either wild type or L980 frame shift plasmid; furthermore, the level of expression of wild-type construct was considerably higher than in untransfected wild type cells (compared with Western blot shown in Figure 1A). As shown in Fig. 8B–D, administration of wild type NOD2-expressing plasmid led to prevention of TNBS-colitis as evaluated by weight loss, histopathology and in vitro production of both IL-12p70 and IFN-γ by stimulated cells from in mesenteric lymph node whereas administration of empty vector had no effect on the colitis. On the other hand, administration of a plasmid expressing a NOD2 with a frame-shift mutation in the NOD2 LRR had only a weak effect on the development of colitis by the same criteria. These studies thus show that over-expression of wild type NOD2 achieved by administration of a plasmid also has the capacity to prevent TNBS-colitis. The greater degree of prevention of colitis achieved with plasmid administration as compared to that in the transgenic mice is attributable to the fact that plasmid administration was accompanied by higher levels of ectopic NOD2 than the transgene. Furthermore, the partial down-regulation of the colitis obtained with the frameshift plasmid is attributable to the fact that the mutated molecule does retain some NOD2 function (2).
Figure 8.
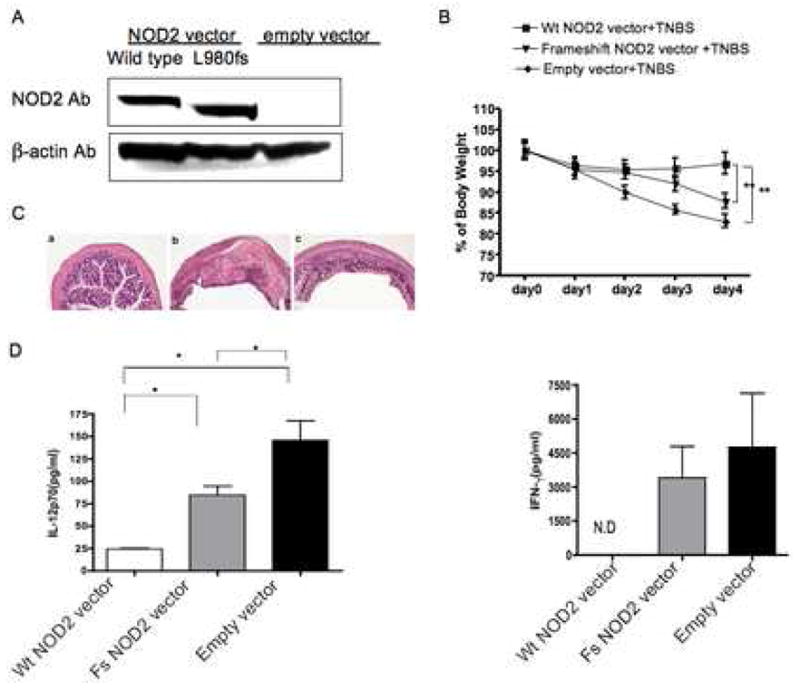
TNBS-colitis in wild-type mice administered an expression vector encoding normal NOD2 or NOD2 bearing a frame-shift mutation encapsulated in HVJ-E. (A) Western blot analysis of NOD2 expression in spleen adherent cells. (B) Body weight changes of mice in each group over the next four days (expressed as a percent of initial body weight); data from each group obtained from 10 mice per group drawn from two independent studies. Statistical differences between mice receiving a vector encoding normal NOD2 and NOD2 bearing a frame-shift mutation or empty vector denoted by **, p < 0.05. (C) Representative H & E stained cross-sections of colons of mice at the time of sacrifice 4 days after TNBS challenge (magnification: 25X); Mice administered: (a) vector encoding normal NOD2; (b) vector encoding frame shift NOD2; or (c) empty vector. (D) IL-12p70 and IFN-γ production by MLN obtained from mice 4 day after TNBS-colitis induction and cultured with Staphylococcus aureus (Cowan I) plus IFN-γ or anti-CD3/anti-CD28 for 48h. Data are expressed as Mean ± SD from cells obtained from 10 mice in each group (derived from two separate studies). Statistical differences between different groups denoted by *, p < 0.05.
Discussion
With this investigation of NOD2 transgenic mice we have provided a consistent set of data pointing to the view that MDP activation of NOD2 leads to a marked down-regulation of the TLR2-induced IL-12 responses. Thus, in in vitro studies, we demonstrated that cells from NOD2-Tg mice exhibit greatly diminished responses to PGN (a TLR2 ligand that generates MDP) in the absence of added MDP whereas their responses to Pam3CSK4 (a TLR ligands that does not generate MDP) were normal, but were greatly diminished in the presence of added MDP. Then, in in vivo studies we showed that colitis induced by intra-rectal administration of PGN, a substance that produces an intense and lethal colitis in littermate-control mice, was completely abrogated in NOD2-Tg mice. In addition, even a colitis induced by a stimulus not caused solely by TLR2 ligands, TNBS-colitis, was ameliorated in NOD2-Tg mice and prevented in wild-type mice administered a plasmid expressing NOD2. On the other hand, a plasmid with a frame-shift mutation such as that found in Crohn’s disease and expressing a truncated NOD2 had only a weak protective effect. Overall, these and other data provide strong evidence for the thesis that NOD2 exerts a negative effect on TLR2 responses; they thus lend further credence to the thesis that Crohn’s disease patients with NOD2 mutations are susceptible to disease because of loss of NOD2 regulatory function (3, 4).
In some prior studies of NOD2 function conducted (mainly) in humans it has been shown that MDP enhances cytokine production induced by several TLR ligands (17)-19). For instance, it has been shown that both human peripheral cells and peritoneal macrophages of mice manifest enhanced TNF-α, IL-10 or IL-1β responses to Pam3Cys4, and human cells manifest enhanced TNF-α responses to LPS, poly I:C, flagellin and CpG upon co-stimulation with MDP that is lost in patients with NOD2 mutations. In view of these findings it is notable that cells from transgenic mice with over-expression of NOD2 do not exhibit even more enhanced TNF-α and IL-10 responses to a variety of TLR ligands when co-stimulated with MDP. One possible explanation of this discrepancy is that the positive and negative effects of NOD2 are due to separate signaling pathways with differing kinetics. In this view, early after the initiation of NOD2 signaling one sees the combined effect of TLR signaling via the MyD88 pathway and NOD2 signaling via the RICK pathway that gives rise to enhanced cytokine production in the case of many cytokines. In contrast, later after the initiation of NOD2, a second type of NOD2 signaling supervenes that results in inhibition of responses, including those transiently enhanced initially. Additional studies of the combined effects of TLR/NOD2 signaling at several time points will be necessary to substantiate this possibility.
The resistance of C57BL/10 NOD2-Tg mice to induction of PGN-colitis was a particularly striking finding in this study given the intensity of the inflammation in comparable littermate-control mice. The fact that PGN induced a strong Th1 response in littermate control mice even though MDP generated from PGN could conceivably down-regulate the TLR2-mediated IL-12 response suggests that this regulatory system is not sufficiently robust in normal mice to prevent development of colitis. On the contrary, only when NOD2 is over-expressed does one see a sufficient down-regulation of the TLR2-mediated Th1 response to have an effect on colitis.
NOD2 over-expression also prevented the development of TNBS-colitis, i.e., a colitis that, unlike PGN-colitis, is not likely to be dependent solely on TLR2 signaling, but rather on a range of TLR signaling, including that of TLR4 and TLR9. This brings up the possibility that the down-regulation of NF-κB due to MDP stimulation of NOD2 could be affecting multiple TLR pathways in vivo, if not in vitro. In this context, it is possible that TLR2 (PGN) responses are more sensitive to NOD2 down-regulation than other forms of TLR stimulation, accounting for the in vitro findings and the fact that PGN-colitis was completely prevented in NOD2-Tg mice whereas TNBS-colitis was only partially prevented. Nevertheless, when there is even a greater degree of over-expression, as might be the case of the mice treated with a NOD2-expressing plasmid, one sees suppression of multiple TLR responses and therefore the complete suppression of TNBS-colitis. Regardless of the mechanism of suppression of TNBS-colitis by NOD2 over-expression it is noteworthy that NOD2 arising from a plasmid bearing a frame-shift mutation and thus giving rise to a similar truncated NOD2 protein found in Crohn’s disease with NOD2 mutations, had only a marginal ability to prevent TNBS-colitis.
The studies reported here bear on another view of how defects in NOD2 function lead to Crohn’s disease. This view is based on the fact that NOD2 is also expressed in specialized epithelial cells at the base of the intestinal crypts (Paneth cells) and that production of anti-bacterial polypeptides, α-defensins, by these cells may be partly dependent on NOD2 function (23, 24). Thus, NOD2 dysfunction could lead to undue expansion of intestinal bacteria in the intestinal crypts that, in turn, leads to excessive stimulation of the mucosal immune system and the production of Th1/Th17 pro-inflammatory cytokines characteristic of Crohn’s disease inflammation. Evidence for this view comes from a study of NOD2-deficient mice (distinct from those studied here) in which it was shown that the mice had decreased α-defensin production and oral administration of L. monocytogenes led to enhanced liver infection with this organism (25). In addition, there are data from patients with Crohn’s disease showing that decreased α-defensin production in patients with disease is more severe in patients with NOD2 mutations (24). These findings impact on the studies reported here because the NOD2 transgene was expressed under a MHC class II promoter and the NOD2 expressing plasmid was expressed under a CMV promoter and thus the over-expression could have also occurred in epithelial cells as well as in hematopoietic cells. It follows that the down regulation of responses and inflammation seen in our in vivo studies could be due, at least in part, to up-regulation of defensin production by epithelial cells by the exogenous NOD2. Two factors, however, render this possibility unlikely. The first is that cells from mice bearing the transgene manifest greatly reduced TLR2-induced IL-12 responses in vitro and in vivo that are independent of any effect of NOD2 on epithelial cell function. The second is that NOD2 over-expression leads to greatly increased resistance to the induction of rapidly induced colitides in which one intentionally disturbs the epithelial barrier (with ethanol) to initiate disease. However, to resolve this issue in a more definitive fashion, we are currently preparing NOD2 transgenic mice with a transgene construct driven by a CD11c promoter that will not be subject to epithelial cell expression of the transgene.
In summary, in the present work we have presented evidence that over-expression of NOD2 leads to decreased TLR2-driven IL-12 responses and protection from experimental colitis. These findings introduce the possibility of treating patients with agents that increase the expression of NOD2.
Supplementary Material
Figure 1. Flow cytometric studies of splenocyte subpopulation in NOD2-Tg and littermate control mice.
Figure 2. Responses of purified CD11b+ cells to purified PGN. CD 11b+ cells purified from whole splenocytes were cultured with LPS (1μg/ml), PGN (10 μg/ml), purified PGN (PPGN) (50 μg/ml) alone and in the presence of MDP (100 μg/ml). After 48 hours the supernatants were harvested and assayed for IL-12p40. Statistical differences denoted by *, p < 0.05, **, p < 0.01 between the presence and absence of MDP.
Figure 3. Splenocytes from NOD2-Tg mice exhibit normal regulation of TLR2-induced TNF-α and IL-10 responses. (A, B) TNF-α production and (C, D) IL-10 production of splenocytes obtained from C57BL/6 NOD2-Tg (A, C) and wild-type mice (B, D), respectively. Total splenocytes were stimulated for 48 h with LPS (1μg/m), PGN (10μg/ml), Pam3CSK4 (500ng/ml), dsRNA (25μg/ml), loxoribine (100μM), or CpG (1 μM) in the absence and presence of MDP (0 μg/ml, 10μg/ml, or 100μg/ml); culture supernatants were then subjected ELISA for determination of cytokine levels. Each result represents the mean ± SD of triplicate assays and is representative of three independent experiments.
Acknowledgments
Grant Support R03-015137 for JTR
We thank Dr. JianPing He (NIAID/NIH) for excellent technical help and Dr. Xiaowu Zhang (Cell Signaling Technology, Inc.) and Dr. Peggy Lost (E-Biosciences) for providing anti-NOD2 Abs.
Abbreviations used in this paper
- MDP
muramyl dipeptide
- Tg
Transgenic
- TLR
Toll-like receptor
- MDP
muramyl dipeptide
- PGN
peptidoglycan
- APCs
antigen-presenting cells
- TNBS
2,4,6-trinitribenzene sulfonic acid
- ELISA
enzyme-linked immunosorbent assay
- LPMC
lamina propria mononuclear cells
- HVJ-E
Hemagglutinin Virus Japan-Envelope
Footnotes
No conflicts of interest exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, Thomas G. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 2.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nunez G, Cho JH. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 3.Watanabe T, Kitani A, Strober W. NOD2 regulation of Toll-like receptor responses and the pathogenesis of Crohn’s disease. Gut. 2005;54:1515–1518. doi: 10.1136/gut.2005.071795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2 . Nat Rev Immunol. 2006;6:9–20. doi: 10.1038/nri1747. [DOI] [PubMed] [Google Scholar]
- 5.Inohara N, Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3:371–382. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- 6.Girardin SE, Travassos LH, Herve M, Blanot D, Boneca IG, Philpott DJ, Sansonetti PJ, Mengin-Lecreulx D. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem. 2003;278:41702–41708. doi: 10.1074/jbc.M307198200. [DOI] [PubMed] [Google Scholar]
- 7.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, Foster SJ, Moran AP, Fernandez-Luna JL, Nunez G. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 8.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 9.Abbott DW, Wilkins A, Asara JM, Cantley LC. The Crohn’s disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol. 2004;14:2217–2227. doi: 10.1016/j.cub.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 10.Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol. 2003;3:521–533. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- 11.Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549. doi: 10.1146/annurev.immunol.20.100301.064816. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–808. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- 13.Iwanaga Y, Davey MP, Martin TM, Planck SR, DePriest ML, Baugh MM, Suing CM, Rosenbaum JT. Cloning, sequencing and expression analysis of the mouse NOD2/CARD15 gene. Inflamm Res. 2003;52:272–276. doi: 10.1007/s00011-003-1170-z. [DOI] [PubMed] [Google Scholar]
- 14.Kouskoff V, Fehling HJ, Lemeur M, Benoist C, Mathis D. A vector driving the expression of foreign cDNAs in the MHC class II-positive cells of transgenic mice. J Immunol Methods. 1993;166:287–291. doi: 10.1016/0022-1759(93)90370-m. [DOI] [PubMed] [Google Scholar]
- 15.Watanabe T, Kitani A, Murray PJ, Wakatsuki Y, Fuss IJ, Strober W. Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity. 2006;25:473–485. doi: 10.1016/j.immuni.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 16.Fichtner-Feigl S, Fuss IJ, Preiss JC, Strober W, Kitani A. Treatment of murine Th1- and Th2-mediated inflammatory bowel disease with NF-kappa B decoy oligonucleotides. J Clin Invest. 2005;115:3057–3071. doi: 10.1172/JCI24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uehara A, Yang S, Fujimoto Y, Fukase K, Kusumoto S, Shibata K, Sugawara S, Takada H. Muramyldipeptide and diaminopimelic acid-containing desmuramylpeptides in combination with chemically synthesized Toll-like receptor agonists synergistically induced production of interleukin-8 in a NOD2-and NOD1-dependent manner, respectively, in human monocytic cells in culture. Cell Microbiol. 2005;7:53–61. doi: 10.1111/j.1462-5822.2004.00433.x. [DOI] [PubMed] [Google Scholar]
- 18.van Heel DA, Ghosh S, Butler M, Hunt KA, Lundberg AM, Ahmad T, McGovern DP, Onnie C, Negoro K, Goldthorpe S, Foxwell BM, Mathew CG, Forbes A, Jewell DP, Playford RJ. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn’s disease. Lancet. 2005;365:1794–1796. doi: 10.1016/S0140-6736(05)66582-8. [DOI] [PubMed] [Google Scholar]
- 19.Netea MG, Ferwerda G, de Jong DJ, Jansen T, Jacobs L, Kramer M, Naber TH, Drenth JP, Girardin SE, Kullberg BJ, Adema GJ, Van der Meer JW. Nucleotide-binding oligomerization domain-2 modulates specific TLR pathways for the induction of cytokine release. J Immunol. 2005;174:6518–6523. doi: 10.4049/jimmunol.174.10.6518. [DOI] [PubMed] [Google Scholar]
- 20.Hilliard BA, Mason N, Xu L, Sun J, Lamhamedi-Cherradi SE, Liou HC, Hunter C, Chen YH. Critical roles of c-Rel in autoimmune inflammation and helper T cell differentiation. J Clin Invest. 2002;110:843–850. doi: 10.1172/JCI15254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamada T, Sartor RB, Marshall S, Specian RD, Grisham MB. Mucosal injury and inflammation in a model of chronic granulomatous colitis in rats. Gastroenterology. 1993;104:759–771. doi: 10.1016/0016-5085(93)91011-6. [DOI] [PubMed] [Google Scholar]
- 22.Sartor RB, Bond TM, Schwab JH. Systemic uptake and intestinal inflammatory effects of luminal bacterial cell wall polymers in rats with acute colonic injury. Infect Immun. 1988;56:2101–2108. doi: 10.1128/iai.56.8.2101-2108.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ogura Y, Lala S, Xin W, Smith E, Dowds TA, Chen FF, Zimmermann E, Tretiakova M, Cho JH, Hart J, Greenson JK, Keshav S, Nunez G. Expression of NOD2 in Paneth cells: a possible link to Crohn’s ileitis. Gut. 2003;52:1591–1597. doi: 10.1136/gut.52.11.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, Feathers RW, Chu H, Lima H, Jr, Fellermann K, Ganz T, Stange EF, Bevins CL. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci U S A. 2005;102:18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307(5710):731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1. Flow cytometric studies of splenocyte subpopulation in NOD2-Tg and littermate control mice.
Figure 2. Responses of purified CD11b+ cells to purified PGN. CD 11b+ cells purified from whole splenocytes were cultured with LPS (1μg/ml), PGN (10 μg/ml), purified PGN (PPGN) (50 μg/ml) alone and in the presence of MDP (100 μg/ml). After 48 hours the supernatants were harvested and assayed for IL-12p40. Statistical differences denoted by *, p < 0.05, **, p < 0.01 between the presence and absence of MDP.
Figure 3. Splenocytes from NOD2-Tg mice exhibit normal regulation of TLR2-induced TNF-α and IL-10 responses. (A, B) TNF-α production and (C, D) IL-10 production of splenocytes obtained from C57BL/6 NOD2-Tg (A, C) and wild-type mice (B, D), respectively. Total splenocytes were stimulated for 48 h with LPS (1μg/m), PGN (10μg/ml), Pam3CSK4 (500ng/ml), dsRNA (25μg/ml), loxoribine (100μM), or CpG (1 μM) in the absence and presence of MDP (0 μg/ml, 10μg/ml, or 100μg/ml); culture supernatants were then subjected ELISA for determination of cytokine levels. Each result represents the mean ± SD of triplicate assays and is representative of three independent experiments.