

Abstract
A systematic study of α-iodination and subsequent Suzuki-Miyaura couplings between non-attenuated enaminones and a wide range of aromatic boronic acids is reported. The microwave-assisted variant of this transformation furnished the α-arylenaminones in significantly shorter reaction times and slightly improved yields as compared to conventional heating.
Recently we reported a new method for the preparation of mono and bicyclic enaminones.1 This procedure allows direct access to a variety of substituted, six membered vinylogous amides. Although compounds such as these have been studied, it is our view that they remain underutilized in natural product and chemical library synthesis.2,3 To increase their synthetic value we became interested in employing them as coupling partners in the Suzuki-Miyaura reaction (Figure 1).4,5
Figure 1.
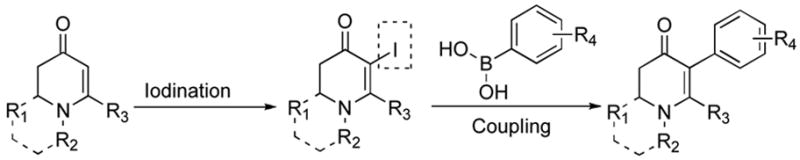
Proposed route to α-aryl enaminones.
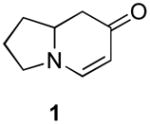
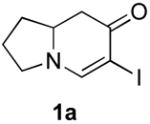
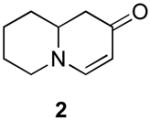
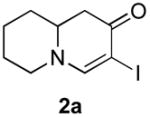
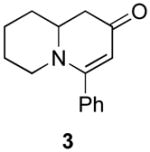
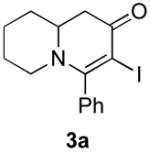
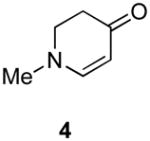
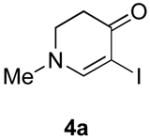
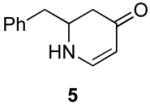
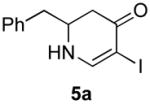
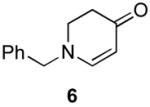
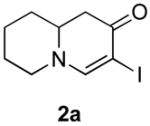
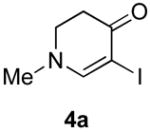
Our strategy relied upon the unique ambident nature of the enaminone to first function as a nucleophile (α-iodination) and then as an electrophile (oxidative insertion). While the α-iodination of unprotected E-enaminones is well known,6 the same transformation with unprotected Z-enaminones has only been recently reported.4,7 Conditions for the α-iodination of enones first developed by Johnson,8 as well as modifications thereof,6a,b were investigated on a series of vinylogous amides, which were prepared according to our previously reported procedure (Table 1).1 Gratifyingly, installation of iodine was achieved in near quantitative yield under the standard Johnson conditions (Table 1, conditions A).9 Furthermore, the modified Johnson conditions reported by Kim et al. (Method B) proceeded smoothly on both bicyclic (1–3) and monocyclic (4–6) systems.10 It is notable that tertiary Z-enaminones (1–4 and 6) posed no problems despite a report that this reaction does not proceed on tertiary E-enaminones.6a In addition, the use of I2 and DMAP also provided the desired iodinated enaminones in excellent yields (Method C).6b,11 Furthermore, a tolerance for substitution adjacent to the ring fused nitrogen (3a) as well as a secondary vinylogous amide nitrogen (5a) was demonstrated.
Table 1.
α-Iodination of enaminones.
| Enaminone | Conditions | α-Iodo enaminone | Yield (%)a |
|---|---|---|---|
|
A. pyridine/CCl4, I2 |
|
99 |
| B. CH2Cl2, I2, then NEt3 | 95 | ||
|
B. CH2Cl2, I2, then NEt3 |
|
87 |
| C. CH2Cl2, I2, DMAP | 97 | ||
|
B. CH2Cl2, I2, then NEt3 |
|
85 |
|
B. CH2Cl2, I2, then NEt3 |
|
88 |
|
B. CH2Cl2, I2, then NEt3 |
|
85 |
|
C. CH2Cl2, I2, DMAP |
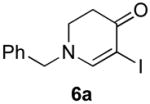
|
94 |
Isolated yield
With the α-iodoenaminones in hand, we next turned to their coupling with boronic acids. A survey of the literature reveals that coupling reactions have been achieved on α-halo vinylogous amides with success.13 However, in most cases, the resonance contribution of the enaminone nitrogen is attenuated via an electron withdrawing protecting group.12 Without this protecting group the resonance contribution of the nitrogen is greatly enhanced. In turn, this electron rich system is less willing to undergo oxidative insertion, often the rate limiting step in this coupling process, leading to poor conversion and low yields.4a This limits the use of Suzuki coupling reactions in these systems to those that do not contain tertiary aliphatic substitution. This is unfortunate, given the common occurrence of this structural feature in alkaloid natural products.13
A variety of conditions were explored with enaminone 1a and 4-methoxyphenylboronic acid to determine optimal conditions for coupling (Table 2). In all cases Pd(PPh3)4 was employed as the palladium source and heating was required for conversion. In addition, it was critical that the solvent mixtures be carefully deoxygenated. The best conditions were obtained when a mixture of dioxane and water was used as solvent and Ba(OH)2 employed as base (Table 2, entry 6).14 Although pleased with the results of our optimization, the reaction times were less than ideal. Microwave irradiation has been used to decrease reaction times and in some cases increase reaction yields.5 Thus microwave conditions were applied to this system and a dramatic rate enhancement was noted (Table 2, entry 7, 14–20 h reduced to 15 min) along with a modest increase in yield.15
Table 2.
Optimization of coupling reaction between enaminone 1a and 4-methoxyphenylboronic acid.a
| Entry | Base | Solvent(s), Temperature | Reaction Time | Yield %b |
|---|---|---|---|---|
| 1 | CaCO3 | DMF/H2O (3/1), 150 °C | 20 h | 0 |
| 2 | Na2CO3 | DME/H2O (1/1), 100 °C | 20 h | 13 |
| 3 | Na2CO3 | toluene, 110 °C | 20 h | 23 |
| 4 | Na2CO3 | toluene/EtOH (1/1), 110 °C | 20 h | 25 |
| 5 | CsF | MeCN/H2O (1/1), 100 °C | 14 h | 27 |
| 6 | Ba(OH)2 | dioxane/H2O (3/1), 110 °C | 14 h | 65 |
| 7c | Ba(OH)2 | dioxane/H2O (3/1), 150 °C MWc | 15 min | 70 |
Compound 1a (0.10 mmol) was dissolved in degassed solvent mixture (0.25–0.5 M) under argon and 4-methoxyphenylboronic acid (0.17 mmol), base (0.20 mmol) and Pd(PPh3)4 (0.02 mmol) added.
Isolated yield.
Carried out employing microwave irradiation (150 °C, 15 min).
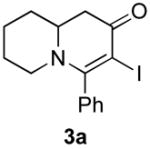
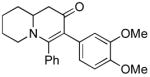
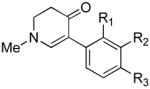
The scope of the reaction was next investigated under the optimized microwave conditions using the enaminones and boronic acid coupling partners shown in Table 3. In general, electron rich boronic acids participated well in the coupling reaction while electron poor coupling partners delivered moderate yields (1b–1h and 4b–4h). Employing an unprotected phenolic boronic acid resulted in a poor yield of enaminones 1g and 4g. Both the 5/6 (1a) and 6/6 (2a and 3a) enaminone systems couple effectively as do monocyclic enaminones (4a–6a). It should be pointed out that even the sterically encumbered substrate 3a underwent coupling under these conditions. In addition it should be noted that a free NH was tolerated (5a).
Table 3.
Cross coupling of iodoenaminones and arylboronic acids.a
| α-Iodo enaminone | Product | Substitution (R1, R2, R3) | Yield (%)b |
|---|---|---|---|
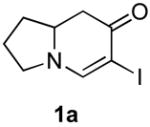
|
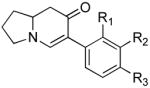
|
1b (H, H, OMe) | 70 |
| 1c (H, H, OBn) | 71 | ||
| 1d (H, NO2, H) | 57 | ||
| 1e (Cl, Cl, H) | 60 | ||
| 1f (H, H, H) | 65 | ||
| 1g (H, H, OH) | 45 | ||
| 1h (H, OMe, OMe) | 75c | ||
|

|
2b (as shown) | 72 |
|
|
3b (as shown) | 60 |
|
|
4b (H, H, OMe) | 60 |
| 4c (H, H, OBn) | 71 | ||
| 4d (H, NO2, H) | 62 | ||
| 4e (Cl, Cl, H) | 50 | ||
| 4f (H, H, H) | 55 | ||
| 4g (H, H, OH) | 36 | ||
| 4h (H, OMe, OMe) | 68 | ||
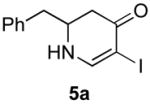
|
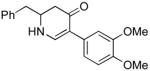
|
5b (as shown) | 70 |
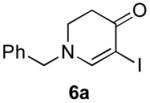
|
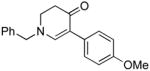
|
6b (as shown) | 69 |
In summary, we have demonstrated a significant improvement in the technology available for preparing α-iodoenaminones and coupling them with arylboronic acids. The use of microwave irradiation proved advantageous in the coupling process. The scope of both the enaminone and boronic acid partners demonstrated versatility. Ongoing studies in these labs are directed toward further method development upon these vinylogous amide scaffolds as well as applications in natural product and diversity-oriented synthesis (DOS).
Acknowledgments
We thank the National Institutes of Health for their generous support of our programs (NIH CA90602, NIH GM076302, and NIH GM069663). B.J.T. was supported by the Department of Defense breast cancer predoctoral fellowship (DAMD17-02-1-0435).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Turunen BJ, Georg GI. J Am Chem Soc. 2006;128:8702. doi: 10.1021/ja0609046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For recent reviews, see: Negri G, Kascheres C, Kascheres AJ. J Heterocycl Chem. 2004;41:461.Elassar AZA, El-Khair AA. Tetrahedron. 2003;59:8463.
- 3.Michael JP, De Koning CB, Gravestock D, Hosken GD, Howard AS, Jungmann CM, Krause RWM, Parsons AS, Pelly SC, Stanbury TV. Pure Appl Chem. 1999;71:979. [Google Scholar]; (b) Comins DL. J Heterocycl Chem. 1999;36:1491. [Google Scholar]
- 4.During the course of our investigation an example of iodination and Suzuki-Miyaura coupling of an N-arabinosyl dehydropiperidinone appeared: Kranke B, Kunz H. Can J Chem. 2006;84:625.Kranke B, Kunz H. Org Biomol Chem. 2007;5:349. doi: 10.1039/b615113b.
- 5.(a) Suzuki A. J Organomet Chem. 1999;576:147. [Google Scholar]; (b) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]; (c) Fitzmaurice RJ, Etheridge ZC, Jumel E, Woolfson DN, Caddick S. Chem Commun. 2006:4814. doi: 10.1039/b610734f. [DOI] [PubMed] [Google Scholar]; (d) Kappe OC. Angew Chem, Int Ed. 2004;43:6250. doi: 10.1002/anie.200400655. [DOI] [PubMed] [Google Scholar]; (e) Kellogg RM. Chemtracts. 2004;17:451. [Google Scholar]; (f) Song YS, Kim BT, Heo JN. Tetrahedron Lett. 2005;46:5987. [Google Scholar]; (g) Song YS, Lee YJ, Kim BT, Heo JN. Tetrahedron Lett. 2006;47:7427. [Google Scholar]
- 6.(a) Kim JM, Na JE, Kim JN. Tetrahedron Lett. 2003;44:6317. [Google Scholar]; (b) Ohshima T, Xu Y, Takita R, Shibasaki M. Tetrahedron. 2004;60:9569. [Google Scholar]; (c) Sha CK, Shen CY, Jean TS, Chiu RT, Tseng WH. Tetrahedron Lett. 1993;34:7641. [Google Scholar]; (d) Sha CK, Santhosh KC, Tseng CT, Lin CT. J Chem Soc, Chem Commun. 1998:397. [Google Scholar]; (e) Campos PJ, Arranz J, Rodriguez MA. Tetrahedron Lett. 1997;38:8397. [Google Scholar]; (f) Matsuo K, Ishida S, Takuno Y. Chem Pharm Bull. 1994;42:1149. doi: 10.1248/cpb.42.1325. [DOI] [PubMed] [Google Scholar]; (g) Kordik CP, Reitz AB. J Org Chem. 1996;61:5644. [Google Scholar]; (h) Ramesh NG, Heijne EH, Klunder AJH, Zwanenburg B. Tetrahedron Lett. 1998;39:3295. [Google Scholar]; (i) Ramesh NG, Heijne EH, Klunder AJH, Zwanenburg B. Tetrahedron. 2002;58:1361. [Google Scholar]; (j) Papoutsis I, Spyroudis S, Varvoglis A, Callies JA, Zhdankin VV. Tetrahedron Lett. 1997;38:8401. [Google Scholar]
- 7.For an example of a bromodesilylation on an unprotected enaminone see: Comins DL, Chen X, Morgan LA. J Org Chem. 1997;62:7435. doi: 10.1021/jo9711495.
- 8.Johnson CR, Adams JP, Braun MP, Senanayake CBW, Wovkulich PM, Uskokovic MR. Tetrahedron Lett. 1992;33:917. [Google Scholar]
- 9.Table 1, method A: 6-Iodo-2,3,8,8a-tetrahydroindolizin-7(1H)-one (1a). Enaminone 1 (760 mg, 5.54 mmol) was dissolved in CCl4/pyridine (1:1, 10 mL) and cooled to 0 °C. To this mixture was added iodine (3.52 g, 13.9 mmol) in CCl4/pyridine (1:1, 2 mL) dropwise. The reaction was then allowed to warm to rt. After 5 h this solution was concentrated into a brown oil under reduced pressure and purified via flash column chromatography (silica gel, 1% Et3N in EtOAc) to provide 1.45 g of the title compound (99%) as a yellow solid (mp = 97–98 °C: 1H NMR (500 MHz, CDCl3) δ 1.62–1.72 (m, 1H) 1.90–2.10 (m, 1H), 2.07–2.17 (m, 1H), 2.22–2.23 (m, 1H), 2.43 (dd, J = 16.2 Hz, 16.2 Hz, 1H), 2.74 (dd, J = 15.9 Hz, 4.7 Hz, 1H), 3.59–3.66 (m, 2H), 3.81–3.93 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 24.6, 32.4, 40.3, 49.8, 58.3, 59.9, 154.7, 185.3; IR (neat) 2970, 2872, 1620, 1559, 1296, 1223 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C8H11INO: 263.9885, found 263.9886.
- 10.Iodination Method B: 6-Iodo-2,3,8,8a-tetrahydroindolizin-7(1H)-one (1a). To a stirred solution of enaminone 1 (137 mg, 1.0 mmol) and iodine (254 mg, 1.0 mmol) in CH2Cl2 (3 ml) was added Et3N (140 μl, 1.0 mmol) at rt. After 5 min this solution was concentrated into a brown oil under reduced pressure then purified via flash chromatography (silica gel, 1% Et3N in EtOAc) to provide the desired α-iodoenaminone 1a (95%).
- 11.Iodination Method C: 3-Iodo-7,8,9a-tetrahydro-1H-quinolizin-2(6H)-one (2a). A solution of I2 in CH2Cl2 (210 mg, 0.83 mmol, 0.05M) was added dropwise over 20 min to a solution of enaminone 2 (104 mg, 0.69 mmol) and DMAP (171 mg, 1.38 mmol) in CH2Cl2 (15 mL) at rt. After stirring overnight at the same temperature, the reaction mixture was quenched by the addition of water and extracted with CH2Cl2 (2×). The combined organic layers were washed with saturated aq. NH4Cl, water, saturated aq. Na2S2O3, water, brine, dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography (silica gel, 50% EtOAc in hexanes) to afford iodide 2a (186 mg, 97%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 1.30–1.57 (m, 3H), 1.69–1.83 (m, 3H), 2.42 (dd, 1H, J = 13.2 Hz), 2.70 (dd, 1H, J = 5.4 Hz), 3.04 (td, 1H , J = 3 Hz, 3.36–3.43 (m, 2H), 7.34 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 22.91, 25.53, 31.55, 41.94, 53.17, 57,26, 62.71, 159.48, 186.03; IR (neat) 2924, 2853, 1630, 1564, 1461, 1318, 1136 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C9H13INO : 278.0036, found 278.0038.
- 12.(a) Comins DL, Joseph SP, Chen X. Tetrahedron Lett. 1995;36:9141. [Google Scholar]; (b) Felpin FX. J Org Chem. 2005;70:8575. doi: 10.1021/jo051501b. [DOI] [PubMed] [Google Scholar]
- 13.Cordell GA. The Alkaloids: Chemistry and Biology. 2003;60 doi: 10.1016/s1099-4831(07)00010-7. [DOI] [PubMed] [Google Scholar]
- 14.Conventional heating method: 6-(3,4-Dimethoxyphenyl)-2,3,8,8a-tetrahydroindolizin-7(1H)-one (1h). α-Iodoenaminone 1a (240 mg, 0.91 mmol) was dissolved in degassed dioxane:water (3:1, 4 mL). The 3,4-dimethoxyphenylboronic acid (282 mg, 1.55 mmol), barium hydroxide (345 mg, 1.82 mmol), and Pd(PPh3)4 (210 mg, 0.182 mmol) were added sequentially. This mixture, equipped with reflux condenser, was heated to 110 °C for 18 h. The reaction was allowed to cool to rt, concentrated under reduced pressure, dissolved in CH2Cl2, filtered, dried over Na2SO4, and purified via flash column chromatography (silica gel, 1% Et3N in EtOAc) to provide 161 mg of the title compound (65%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 1.65–1.80 (m, 1H) 1.91–2.07 (m, 1H), 2.08–2.18 (m, 1H), 2.26–2.39 (m, 1H), 2.45–2.60 (m, 2H), 3.55–3.63 (m, 2H), 3.83–3.88 (m, 1H), 3.86 (s, 3H), 3.88 (s, 3H), 6.80–6.86 (m, 2H), 7.09 (s, 1H), 7.42 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 24.8, 33.4, 42.7, 50.2, 56.2, 56.3, 58.2, 110.0, 111.5, 112.0, 119.7, 129.0, 130.3, 147.4, 148.9, 149.3, 189.5; IR (neat) 3381, 2935, 2835, 1609, 1514, 1407, 1250 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C16H20NO3 : 274.1443, found 274.1441.
- 15.Microwave irradiation method: 6-(3,4-Dimethoxyphenyl)-2,3,8,8a-tetrahydroindolizin-7(1H)-one (1h). α-Iodo enaminone 1a (240 mg, 0.91 mmol) was dissolved in degassed dioxane:water (3:1, 4 mL). The 3,4-dimethoxyphenylboronic acid (282 mg, 1.55 mmol), barium hydroxide (345 mg, 1.82 mmol), and Pd(PPh3)4 (210 mg, 0.182 mmol) were added sequentially. This mixture was heated under microwave irradiation at 150 °C for 1 h. The reaction was allowed to cool to rt, concentrated under reduced pressure, dissolved in CH2Cl2, filtered, dried over Na2SO4, and purified via flash column chromatography (silica gel, 1% Et3N in EtOAc) to provide 187 mg of the title compound (75%).