

Abstract
IKKβ-dependent NF-κB activation plays a key role in innate immunity and inflammation and inhibition of IKKβ has been considered as a likely anti-inflammatory therapy. Surprisingly, however, mice with a targeted IKKβ-deletion in myeloid cells are more susceptible to endotoxin-induced shock than control mice. Increased endotoxin susceptibility is associated with elevated plasma IL-1β as a result of increased pro-IL-1β processing, which was also seen upon bacterial infection. In macrophages enhanced pro-IL-1β processing depends on caspase-1 whose activation is inhibited by NF-κB-dependent gene products. In neutrophils, however, IL-1β secretion is caspase-1 independent and depends on serine proteases, whose activity is also inhibited by NF-κB gene products. Prolonged pharmacologic inhibition of IKKβ also augments IL-1β secretion upon endotoxin challenge. These results unravel a novel role for IKKβ-dependent NF-κB signaling in the negative control of IL-1β production and highlight potential complications of long-term IKKβ inhibition.
Introduction
Acute inflammation resulting in septic shock and multi-organ dysfunction (MOD) is a common cause of death in intensive care units (Hotchkiss and Karl, 2003). Sepsis is a systemic inflammatory response occurring after massive bacterial infection or severe trauma and in addition to septic shock and MOD can cause other complications such as adult respiratory distress syndrome (ARDS) (Cohen, 2002). Despite advances in antimicrobial treatment and supportive therapy, mortality remains high in septic patients (Hotchkiss and Karl, 2003).
During microbial infections, pathogen-associated molecular patterns (PAMP) are recognized by the host defense system, which mounts a protective response (Cohen, 2002). A similar response is triggered by endogenous mediators, which are released by necrotic cells during trauma (Karin et al., 2006). The initial inflammatory reaction is amplified and once amplification becomes excessive, shock and MOD can occur (Cohen, 2002). Myeloid cells are critical for PAMP recognition, initiation and amplification of the inflammatory cascade mainly through transcriptional regulation of genes encoding key inflammatory mediators, such as the cytokines TNF-α and IL-1β (Dinarello, 1997). In endotoxic shock TNF-α and IL-1β are released rapidly (30-90 min) (Dinarello, 1997) and activate a secondary inflammatory cascade, dependent on transcription factor NF-κB (Ghosh and Karin, 2002). More recently, a novel system termed the inflammasome, involved in post-transcriptional control of inflammation and innate immunity has been described (Martinon and Tschopp, 2004). The inflammasome system is based on ligand-dependent activation of caspase-1 and other pro-inflammatory caspases that carry out processing of cytoplasmatic pro-IL-1β and pro-IL-18 (Martinon and Tschopp, 2004). It is presently unclear whether the NF-κB and inflammasome systems interact, other than through the synthesis of pro-IL-1β, which is transcriptionally regulated by NF-κB.
One way to inhibit NF-κB activation is to use small molecule inhibitors of IKKβ (Karin et al., 2004), one of the two catalytic subunits of the IKK complex, that is critical for NF-κB activation during acute inflammation (Chen et al., 2003). IKK/NF-κB blockade has been proposed as a therapeutic modality for preventing mortality in septic shock and MOD (Zingarelli, 2005). However, there are certain potential complications associated with this approach, such as a marked increase in susceptibility to apoptosis (Chen et al., 2003; Kisseleva et al., 2006) or perhaps a failure to induce negative regulators, such as A20 (Lee et al., 2000). In addition, NF-κB plays a central role in activation of the host defense system (Li et al., 2002) and therefore IKK inhibition can increase susceptibility to infections. In fact, certain pathogens, such as Yersinia pestis, inhibit IKK activation to evade avoid host defense (Orth et al., 2000).
To examine whether IKKβ inhibition might prevent septic shock, we have conditionally disrupted the Ikkβ gene in myeloid cells, and subjected the resultant mice, termed IkkβΔmye, to endotoxin challenge. Surprisingly, IkkβΔmye mice were more susceptible to endotoxin-induced mortality and showed significantly increased levels of circulating IL-1β, due to enhanced pro-IL-1β processing. Enhanced IL-1β secretion in myeloid IKKβ-deficient mice was also seen upon bacterial infection. Prolonged pharmacological inhibition of IKKβ, which interferes with NF-κB activation in the whole animal, also increased lipopolysaccharide (LPS)-induced mortality and plasma IL-1β. Thus, our studies, which uncovered an unexpected role for IKKβ-driven NF-κB in the negative control of IL-1β secretion, strongly suggest that the therapeutic use of IKKβ inhibitors may result in unintended effects on the host response. Enhanced secretion of IL-1β and similarly-regulated cytokines by NF-κB deficient myeloid cells may have evolved as a protective strategy that provides innate immunity to microbes that produce NF-κB inhibitors.
Results
Mice lacking myeloid IKKβ are hypersusceptible to endotoxic shock
To examine the impact of myeloid-specific IKKβ inhibition on endotoxic shock, we crossed LysM-Cre mice (Clausen et al., 1999) to “floxed” Ikkβ mice to delete Ikkβ alleles in macrophages and neutrophils. The resulting mice, termed IkkβΔmye, lack IKKβ protein and kinase activity, resulting in defective NF-κB activation only in myeloid cells with no overt changes in histology of lymphoid organs and other sites rich in myeloid cells, such as the gastrointestinal tract and liver (Supplementary Figure 1) (Arkan et al., 2005; Greten et al., 2004). IkkβΔmye and IkkβF/F control mice were intraperitonealy (i.p.) challenged with a high dose of E. coli LPS (30 mg/kg) and monitored for survival. Unexpectedly, all IkkβΔmye mice died within 36 hrs, whereas 50% of IkkβF/F controls survived for >72 hrs (Figure 1A). Enhanced LPS toxicity in IkkβΔmye mice correlated with a marked increase in plasma IL-1β levels within the first 2 hrs (Figure 1B), and modestly elevated plasma TNF-α (Figure 1C), whereas there was no difference in plasma IL-6 (Figure 1D). To address whether increased IL-1β secretion was restricted to LPS challenge, we injected mice of either genotype with the TLR9 agonist CpG-ODN or Listeria monocytogenes. In both cases IkkβΔmye mice showed increased plasma IL-1β relative to IkkβF/F controls (Supplementary Figure 2), suggesting a general mechanism.
Figure 1. Increased endotoxin-induced mortaility is associated with elevated circulating IL-1β in IkkβΔmye mice.
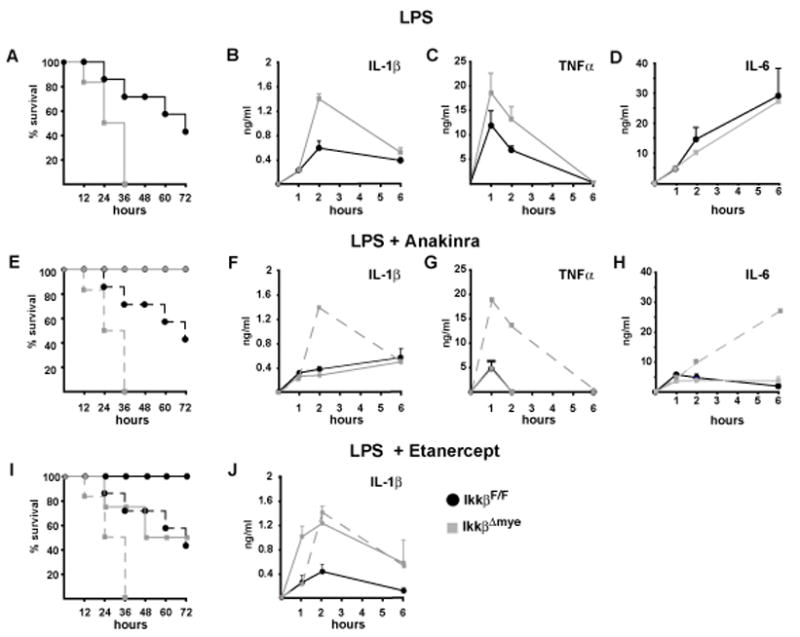
(A) Survival after endotoxin injection (30 mg/kg, E. coli O111:B4) of IkkβF/F (black) and IkkβΔmye mice (grey), (n = 4–6). (B) IL-1β, (C) TNF-α and (D) IL-6 plasma levels after LPS administration. (E, I) Survival of IkkβF/F (solid black lines) and IkkβΔmye mice (solid grey lines) after LPS administration in the presence of IL-1ra (E) and sTNFRII (I). Dashed lines represent survival without inhibitors. (F, J) IL-1β, (G) TNF-α and (H) IL-6 plasma levels in mice given LPS plus the indicated inhibitors. Dashed line represents respective plasma levels of IkkβΔmye mice without inhibitor. Data are averages of at least 4 animals per time point.
To determine whether enhanced LPS toxicity in IkkβΔmye mice was due to elevated IL-1β, we treated IkkβΔmye mice and IkkβF/F controls with recombinant IL-1 receptor (IL-1R) antagonist (IL-1ra, Anakinra) to block IL-1R activation. IL-1ra completely protected IkkβΔmye (and IkkβF/F) mice against LPS-induced mortality (Figure 1E), and decreased plasma levels of IL-1β, TNF-α and IL-6 (Figure 1 F, G, H). In comparison, inhibition of TNF-α signaling through administration of soluble TNF-α receptor (Etanercept) (Suffredini et al., 1995) was less efficient in reducing LPS toxicity in IkkβΔmye mice, although it completely protected IkkβF/F controls (Figure 1G). Unlike Anakinra, Etanercept did not reduce plasma IL-1β in IkkβΔmye mice (Figure 1H), suggesting that IL-1β was the key inflammatory mediator in LPS-challenged IkkβΔmye mice. In addition, the initial signaling by IL-1β may be responsible for further increase in secretion of IL-1β and other cytokines.
Because IKKβ is deleted in both macrophages and neutrophils in IkkβΔmye mice, we examined which cell type contributed to increased plasma IL-1β levels after LPS application. We used clodronate-containing liposomes (van Rooijen et al., 1997) and Gr-1 antibody (RB6-8C5) (Vassiloyanakopoulos et al., 1998) to deplete macrophages and neutrophils, respectively. Flow cytometry of splenocytes or peritoneal exudates, as well as immunohistochemical staining for the macrophage marker F4/80 in spleen and liver sections, confirmed efficient depletion of macrophages and Kupffer cells in IkkβΔmye and control mice without affecting Gr-1+ cells (Supplementary Figure 3). Macrophage depletion improved survival in LPS-treated IkkβΔmye mice (Figure 2A), and substantially decreased plasma IL-1β and TNF-α (Figure 2B, C). Successful neutrophil depletion by anti-Gr-1 administration, as confirmed on blood smears (data not shown), rendered IkkβΔmye mice significantly more resistant to LPS-induced toxicity than IkkβF/F controls (Figure 2D) and inhibited IL-1β release (Figure 2E), whereas the effect on plasma TNF-α was less pronounced (Figure 2F). Thus, both neutrophils and macrophages contribute to IL-1β release and mortality in LPS-challenged IkkβΔmye mice. Nonetheless, it should be recognized that the Gr-1 antibody used may also deplete Gr-1+CD11b+F4/80+ “inflammatory monocytes” that are also likely to contribute to IL-1β release (Taylor and Gordon, 2003)
Figure 2. Depletion of either macrophages or neutrophils improves survival in IkkβΔmye mice.
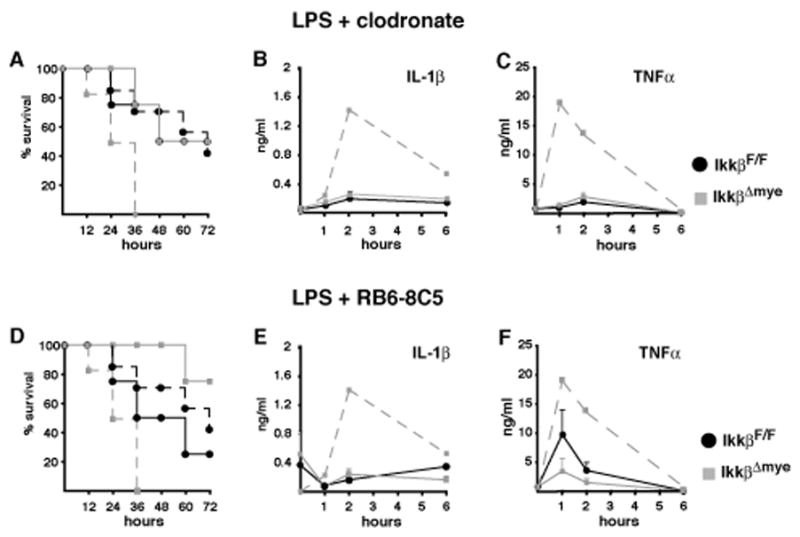
(A, D) Survival of IkkβF/F (black lines) and IkkβΔmye mice (grey lines) depleted of macrophages (A) or neutrophils (D). Dashed lines represent survival without depletion. (B, E) IL-1β and (C, F) TNF-α plasma levels after LPS administration to macrophage-depleted (B, C) or neutrophil-depleted (E, F) mice. Dashed line represents respective plasma levels in IkkβΔmye mice without depletion. Data are averages of at least 4 animals per time point.
The importance of IKKβ-deficient neutrophils for IL-1β release after endotoxin challenge was further examined in another mouse mutant, the previously characterized Mx1-Cre x IkkβF/F mouse, called IkkβΔ (Ruocco et al., 2005). In these mice induction of Mx1-Cre by systemic injection of the interferon (IFN) inducer poly-I:C (Kuhn et al., 1995) results in IKKβ deletion in IFN-responsive cells, including all myeloid cells (Maeda et al., 2005). IkkβΔ mice exhibited neutrophilia within the first 14 days after the first poly-IC injection, which was not only sustained over time but became more extensive (Figure 3A–D). This resembles observations made in mice transplanted with fetal liver cells from Ikkβ−/− or Rela−/− mice (Horwitz et al., 1997; Senftleben et al., 2001), indicating a role for NF-κB-dependent mechanisms in neutrophil homeostasis. Importantly, LPS administration to IkkβΔ mice caused 100% mortality within 24 hrs (Figure 3E), and increased plasma IL-1β dramatically, with a >40-fold increase over LPS-treated IkkβΔmye mice (compare Figure 1B and 3F), which do not display neutrophilia (data not shown). Yet, similar to IkkβΔmye mice, survival of IkkβΔ mice was markedly improved by inhibition of IL-1β with IL-1Ra (Supplementary Figure 4). These results support the important contribution made by neutrophils are to elevated plasma-IL-1β in endotoxin-challenged myeloid IKKβ-deficient mice and confirm that IL-1β is responsible for increased mortality in these mice.
Figure 3. IkkβΔ mice develop granulocytosis and show massively increased circulating IL-1β after endotoxin exposure.
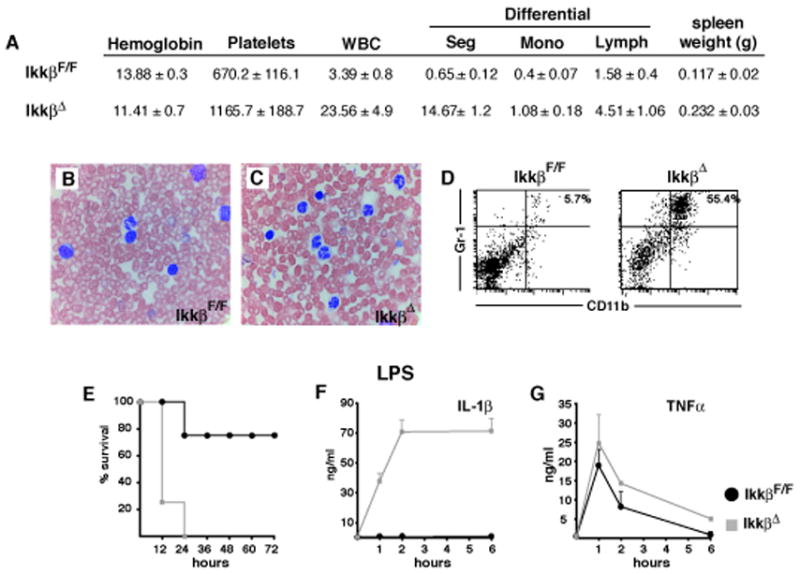
(A) Complete and differential blood counts and spleen weights of IkkβF/F and IkkβΔ mice. Data are average values for 5 mice of each genotype examined two weeks after a single poly-(I:C) injection. (B, C) Blood smears stained with Wright-Giemsa and (D) number of CD11b+/Gr-1+ cells in spleens of of IkkβF/F and IkkβΔ mice analyzed two weeks after poly-(I:C) injection. (E) Survival after endotoxin injection (20 mg/kg, E. coli O111:B4) of IkkβF/F (black) and IkkβΔ mice (grey), (n = 4–6). (F) IL-1β and (G) TNF-α plasma levels 1, 2 and 6 hrs after LPS administration.
Enhanced IL-1β release by IKKβ-deficient macrophages and neutrophils is mediated by distinct mechanisms
To investigate the mechanisms underlying the unexpected increase in plasma IL-1β and TNF-α, we prepared bone marrow-derived macrophages (BMDM) from IkkβΔ mice. These cells were highly compromised in LPS-induced NF-κB activation (Supplementary Figure 5) and showed decreased pro-IL-1β and TNF-α mRNA induction (Figure 4A), which was paralleled by reduced levels of pro-IL-1β and pro-TNF-α and decreased TNF-α release (Figure 4B, D). However, despite reduced pro-IL-1β expression LPS treatment of IkkβΔmacrophages resulted in more IL-1β secretion than the same treatment of NF-κB-competent IkkβF/F cells (Figure 4C). Immunoblot analysis revealed that the IL-1β secreted by IkkβΔmacrophages was properly processed (Figure 4D). Thus, IkkβΔmacrophages show the expected decrease in pro-IL-1β mRNA synthesis but due to very efficient pro-IL-1β processing they secrete much more IL-1β than control macrophages.
Figure 4. Increased IL-1β release correlates with elevated apoptosis of IKKβ-deficient macrophages and is inhibited by PAI-2.
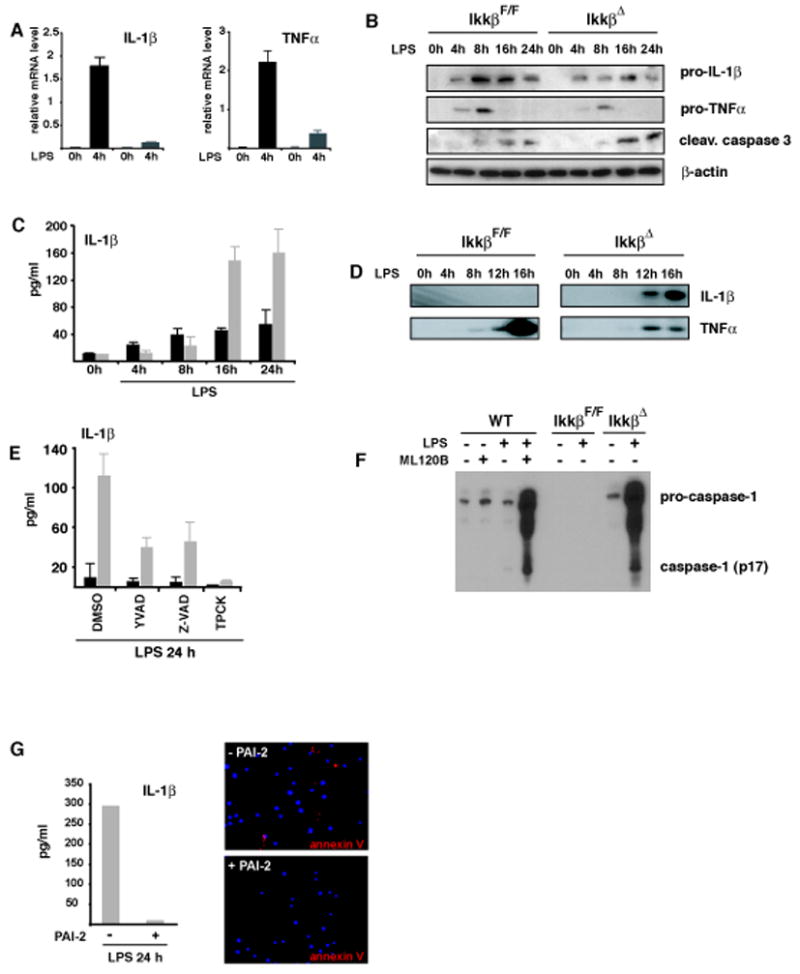
(A) Relative levels of IL-1β and TNF-α mRNA after incubation of IkkβF/F (black bars) and IkkβΔ (grey bars) BMDM with LPS (100 ng/ml). (B) Immunoblot analysis of intracellular pro-IL-1β, pro-TNF-α and cleaved caspase-3 in macrophages after LPS stimulation (100 ng/ml). (C) IL-1β levels in supernatants of IkkβF/F (black bars) and IkkβΔ (grey bars) macrophages after LPS stimulation. (D) Immunoblot analysis of processed IL-1β and TNF-α in supernatants of cultured macrophages. (E) IL-1β in supernatants of LPS stimulated IkkβF/F (black) and IkkβΔ (grey) macrophages in the presence of Ac-YVAD-cmk (100 μM), Z-VAD-fmk (10 μM) and TPCK (10 μM). Data are averages of at least three animals. (F) Loss of IKKβ activity enhances caspase-1 activation. WT or IKKβ-deficient macrophages were pretreated with ML120B (30 μM) or DMSO and either left unstimulated or incubated with LPS (100 ng/ml). After 22 hrs, culture supernatants were collected and analyzed by immunoblotting for secretion of activated caspase-1 (p17). The highest mobility represents uncleaved pro-caspase-1. (G) Reconstitution of IKKβ-deficient macrophages with PAI-2 blocks apoptosis and IL-1β release. Bone marrow of IkkβΔ mice was retrovirally transduced with EGFP-PAI-2. EGFP-positive cells were sorted, differentiated into macrophages and stimulated with LPS (100 ng/ml) for 24 hrs. Apoptosis was determined by annexin V staining and IL-1β levels in supernatants were determined by ELISA.
IKKβ-deficient macrophages are also more susceptible to LPS-induced apoptosis than normal cells (Park et al., 2005). To investigate whether increased macrophage apoptosis, which can result in caspase-1 activation, contributed to enhanced release of IL-1β, as proposed previously (Hogquist et al., 1991), we employed the pan-caspase inhibitor, Z-VAD-fmk, and the general serine protease inhibitor, TPCK, which inhibit apoptosis in IKKβ-deficient macrophages (Park et al., 2005). Both inhibitors were equally or more effective than the caspase-1 inhibitor Ac-YVAD-cmk, used as a positive control in these experiments, in inhibiting IL-1β secretion (Figure 4E), suggesting that enhanced IL-1β processing in IKKβ deficient macrophages is due to enhanced apoptosis and caspase-1 activation. Indeed, IkkβΔmye macrophages exhibited higher levels of activated caspase-3 (Figure 4B) and secreted more activated caspase-1 than control macrophages (Figure 4F), while expressing normal amounts of intracellular pro-caspase-1 (Supplementary Figure 6).
One of the NF-κB dependent anti-apoptotic genes in macrophages is the serpin plasminogen activator inhibitor 2 (PAI-2), which is also not expresse d in THP1 cells, a human myelomonocytic cell line in which LPS treatment alone (without co-treatment with ATP) results in IL-1β secretion (Martinon et al., 2002; Park et al., 2005). Reintroduction of PAI-2 into IkkβΔ macrophages inhibited LPS-induced apoptosis as judged by annexin V staining and completely blocked LPS-induced IL-1β secretion (Figure 4G). These results directly demonstrate that the product of a NF-κB regulated gene serves as a negative regulator of caspase-1-dependent IL-1β secretion. Recently, while this work was under revision, Bruey et al. demonstrated that another NF-κB-dependent anti-apoptotic gene product, Bcl-xL, is also a negative regulator of IL-1β secretion (Bruey et al. 2007).
We next turned to neutrophils to determine whether IL-1β processing is also enhanced in these cells in the absence of IKKβ. Thioglycollate-elicited peritoneal neutrophils were purified and examined for IL-1β and TNF-α expression and release. Immunoblot analysis corroborated efficient deletion of IKKβ in IkkβΔ neutrophils and inhibition of LPS-induced NF-κB activation (Supplementary Figure 7). To examine whether loss of IKKβ affects neutrophil function, we measured chemotaxis and bactericidal activity. Whereas chemotactic activity was increased in IkkβΔ neutrophils (Supplementary Figure 7D), we could not observe any differences in killing of E. coli within 8 hrs after infection (Supplementary Figure 7E). Interestingly, plasma IL-1β levels were also increased in E. coli infected IkkβΔ mice (Supplementary Figure 7). As in macrophages, we found reduced LPS-induced IL-1β and TNF-α mRNA in IkkβΔ neutrophils (Figure 5A). Intracellular pro-IL-1β levels were also downregulated (Figure 5B), to an even greater extent than in macrophages (Figure 4B). Nonetheless, supernatants of IkkβΔ neutrophils contained more immunoreactive IL-1β than those of IkkβF/F cells (Figure 5C), paralleling the results in macrophages. TNF-α secretion, however, was decreased as expected (Figure 5D).
Figure 5. Increased IL-1β release is independent of apoptosis in IKKβ-deficient neutrophils.
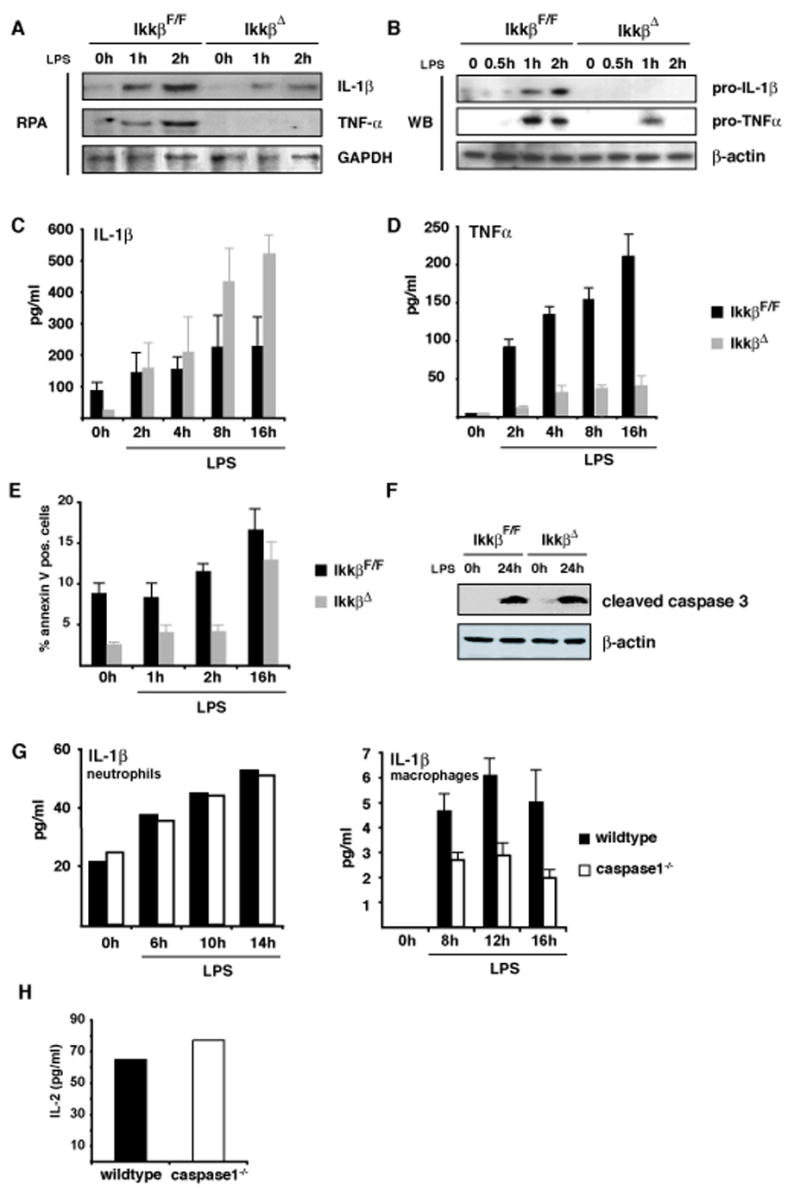
(A) Levels of IL-1β and TNF-α mRNAs in neutrophils after LPS stimulation were examined by RNase protection. (B) Immunoblot analysis of intracellular pro-IL-1β and pro-TNF-α in neutrophils after LPS stimulation. (C, D) IL-1β and TNF-α secretion by LPS-stimulated neutrophils determined by ELISA. (E) Number of annexin V positive Gr-1+ cells determined by flow cytometry. (F) Immunoblot analysis of cleaved caspase-3 in neutrophils after LPS stimulation (100 ng/ml). (G) IL-1β secretion by LPS-stimulated wt and caspase-1−/− macrophages and neutrophils. (H) Bioassay of supernatants of LPS-stimulated wt and caspase-1−/− neutrophils using EL4 cells.
To examine if increased IL-1β release by IKKβ-deficient neutrophils was also associated with enhanced apoptosis, we analyzed annexin V staining, which marks apoptotic cells, of Gr-1+-neutrophils before and after ex vivo LPS stimulation. In sharp contrast to macrophages, the number of apoptotic neutrophils was lower before and after LPS stimulation (especially at earlier time points) when cells were obtained from IkkβΔ mice instead of IkkβF/F mice (Figure 5E). In parallel, no difference in caspase-3 activation could be detected between IkkβF/F and IkkβΔ neutrophils 24 hrs after LPS addition (Figure 5F). Thus, in neutrophils, IKKβ deficiency attenuates apoptosis and may therefore account for the neutrophilia observed in IkkβΔ mice. Unlike macrophages in which LPS-induced IL-1β secretion was diminished in the absence of caspase-1, neutrophils from caspase-1−/− mice did not exhibit reduced bioactive IL-1β secretion after LPS stimulation (Figure 5G), as confirmed by an IL-1β bioassay (Figure 5H). Taken together, these data suggest that an IKKβ-regulated, caspase-1 independent mechanism of IL-1β processing accounts for IL-1β release by neutrophils.
Enhanced serine protease activity in IKKβ-deficient neutrophils
Proteinase 3 (PR3), neutrophil elastase (NE), and cathepsin G (CatG) are the main serine proteases of azurophilic granules of neutrophils (Pham, 2006), and may play a role in IL-1β and TNFα release (Coeshott et al., 1999). We therefore examined whether these enzymes are activated in IKKβ–deficient neutrophils and if their activation might be linked to IL-1β processing. MeOSuc-AAPV-pNA is a high affinity substrate for NE as well as murine PR3 (Wiesner et al., 2005), that can be used to measure the activity of NE and PR3 in mice. Hydrolysis of MeOSuc-AAPV-pNA was markedly elevated in lysates of IkkβΔ neutrophils even without LPS stimulation (Figure 6A). To distinguish between NE and PR3 we used recombinant secretory leukoprotease inhibitor (SLPI), an inhibitor of NE and CatG that does not block PR3 (Wiesner et al., 2005). SLPI blocked MeOSuc-AAPV-pNA hydrolysis by ~50% in lysates of IkkβΔ neutrophils, but a similar degree of inhibition was also seen in IkkβF/F control neutrophils, suggesting that NE activity is only partially responsible for elevated IL-1β processing in IKKβ-deficient neutrophils. The non-specific serine protease inhibitor, 3, 4-dichloroisocoumarin (DCIC), completely blocked protease activity, confirming the enzymatic specificity of the assay results (Figure 6A). To determine whether enhanced enzyme activity was due to increased expression, we examined PR3 protein levels, but found them to be only modestly (2-fold) upregulated in IkkβΔ neutrophils (Figure 6B). RNA analysis of PR3, NE and CatG confirmed this modest upregulation (Supplementary Figure 8). Because the two-fold increase in PR3 expression cannot account for the 5-fold increase in protease activity, PR3 enzymatic activity is also increased in IKKβ-deficient neutrophils. To determine which serpin could be responsible for this increased activity, we examined the RNA and protein expression of various serpins, some of which are known to be regulated by NF-κB. Interestingly, of these, serpinB1 (monocyte neutrophil elastase inhibitor, MNEI), showed an increased expression in IkkβΔ neutrophils, suggesting a compensatory mechanism. However, similarly to macrophages, PAI-2 expression was absent in IkkβΔ neutrophils, suggesting its involvement in PR3 regulation.
Figure 6. Elevated serine protease activity in IKKβ-deficient neutrophils accounts for enhanced IL-1β secretion.
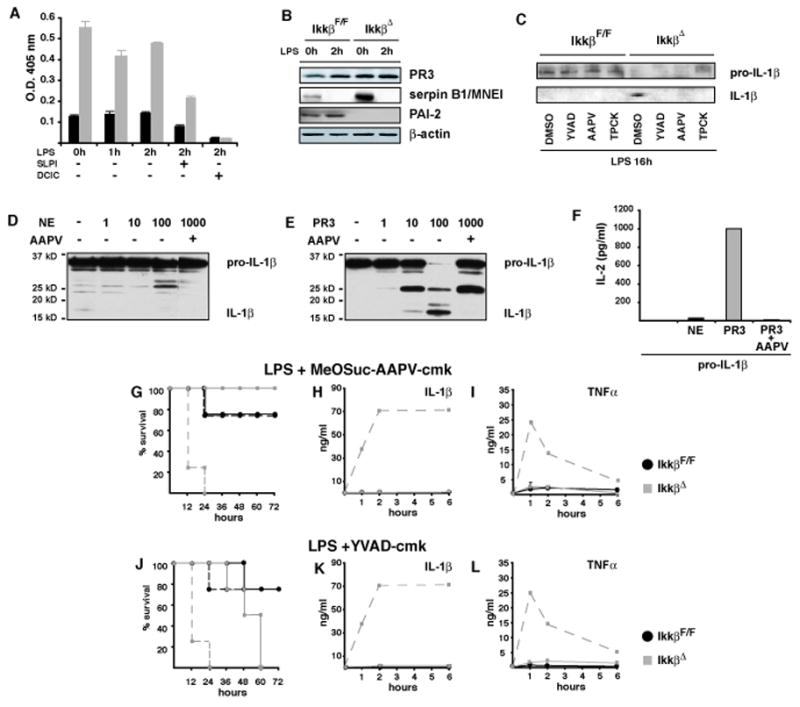
(A) Hydrolysis of MeOSuc-AAPV-pNA was used to determine serine protease activity in IkkβF/F (black bars) and IkkβΔ neutrophils (grey bars) treated with LPS and the protease inhibitors SLPI (1μg/ml) or 3, 4-DCIC (10 μM). (B) PR3, serpin B1/MNEI and PAI-2 expression in LPS–stimulated neutrophils determined by immunoblotting. (C) Immunoblot analysis of IL-1β expression in supernatants of neutrophils 16 hrs after LPS stimulation in the presence of Ac-YVAD-cmk (100 μM), MeOSuc-AAPV-cmk (500 μM) or TPCK (10 μM). (D, E) Immunoblot analysis of IL-1β produced in HEK293 cells after incubation with increasing amounts of purified (D) NE and (E) PR3 without or in the presence or AAPV. (F) Bioassay of extracts used in (D) and (E) in EL4 cells. (G–L) Survival of IkkβF/F mice (black lines) and IkkβΔ mice (grey lines) after pretreatment with the serine protease inhibitor MeOSuc-AAPV-cmk (1 mg/mouse) 1 hr before LPS application (G) or Ac-YVAD-cmk (J) and corresponding IL-1β (H, K) and TNF-α (I, J) plasma levels. Dashed lines in (G and J) represent survival without inhibitor and in (H, I) and (K, L) they represent plasma IL-1β and plasma TNF-α levels of IkkβΔmye mice without inhibitor, respectively.
We next determined whether PR3 or another serine protease is a major contributor to enhanced IL-1β processing in IKKβ deficient neutrophils. Neutrophils were treated or not with different protease inhibitors, stimulated with LPS, and IL-1β in culture supernatants was assayed. Immunoblot analysis revealed that in IKKβ-proficient neutrophils the majority of secreted IL-1β was unprocessed pro-IL-1β, whereas in supernatants of IkkβΔ neutrophils most of the immunoreactive IL-1β was the processed form, whose release was inhibited by the different protease inhibitors (Figure 6C). To confirm that NE and PR3 can process pro-IL-1β, we produced pro-IL-1β in HEK293 cells and incubated it with purified NE or PR3 (Figure 6D, E). PR3 efficiently and dose-dependently processed pro-IL-1β to IL-1β, and was clearly more potent than NE. Formation of mature bioactive IL-1β was confirmed by a bioassay (Figure 6F). Importantly, administration of MeOSuc-AAPV-cmk completely protected IkkβΔ mice from LPS-induced lethality (Figure 6G) and led to a dramatic decrease in plasma IL-1β and TNF-α (Figure 6H, I). Administration of α1-anti-trypsin, a potent inhibitor of extracellular but not intracellular NE and PR3, did not improve survival and did not decrease IL-1β plasma levels (data not shown). The caspase-1 inhibitor Ac-YVAD-cmk, was not as efficient in protecting IkkβΔ mice from LPS-induced lethality as MeOSuc-AAPV-cmk. Although MeOSuc-AAPV-cmk did not affect macrophage apoptosis, we cannot rule out certain off-target effects of this inhibitor and as discussed below the initial PR3 (or a different protease)-dependent IL-1β release by IKKβ-deficient neutrophils may trigger caspase-1-mediated IL-1β processing by IKKβ-deficient macrophages. Overall, increased intracellular serine protease activity, particularly PR3 and to a lesser extent NE, in IKKβ-deficient granulocytes can account for increased IL-1β release by these cells.
To evaluate whether PR3 activity is regulated directly by NF-κB, rather than other IKKβ functions (Wegener et al., 2006), we isolated neutrophils from Mx1-Cre x RelaF/F mice, called RelaΔ, which produce a RelA protein that lacks its nuclear localization signal (NLS) and part of the transactivation domain and are therefore functionally deficient for NF-κB p65/RelA (Algül et al., 2007). Like IkkβΔ neutrophils, RelaΔ neutrophils also displayed enhanced serine protease activity and continued to secrete IL-1β despite diminished pro-IL-1β expression levels (Supplementary Figure 9). These results indicate that defective NF-κB activation can account for increased serine protease activity and IL-1β secretion in IKKβ-deficient neutrophils.
Pharmacological IKKβ inhibition also results in neutrophilia and LPS-sensitivity
In IkkβΔmye and IkkβΔ mice, IKKβ is absent in specific cell types responsible for TNF-α and IL-1β production during endotoxin challenge. However, NF-κB is activated normally in other cells (Greten et al., 2004; Maeda et al., 2005). This situation may not fully mimic a potential therapeutic intervention with a systemically active inhibitor, which is likely to target all cells. We therefore, also employed a pharmacological approach, using the selective IKKβ inhibitor ML120B (Nagashima et al., 2006). ML120B effectively inhibited IKKβ and NF-κB activation in myeloid cells (Supplementary Figure 10), but its short-term administration to wild-type mice had little impact on LPS-induced mortality (Figure 7A). Plasma IL-1β levels were increased, whereas circulating TNF-α was dramatically decreased (Figure 7B, C).
Figure 7. Prolonged pharmacological inhibition of IKKβ enhances IL-1β secretion and endotoxic shock.
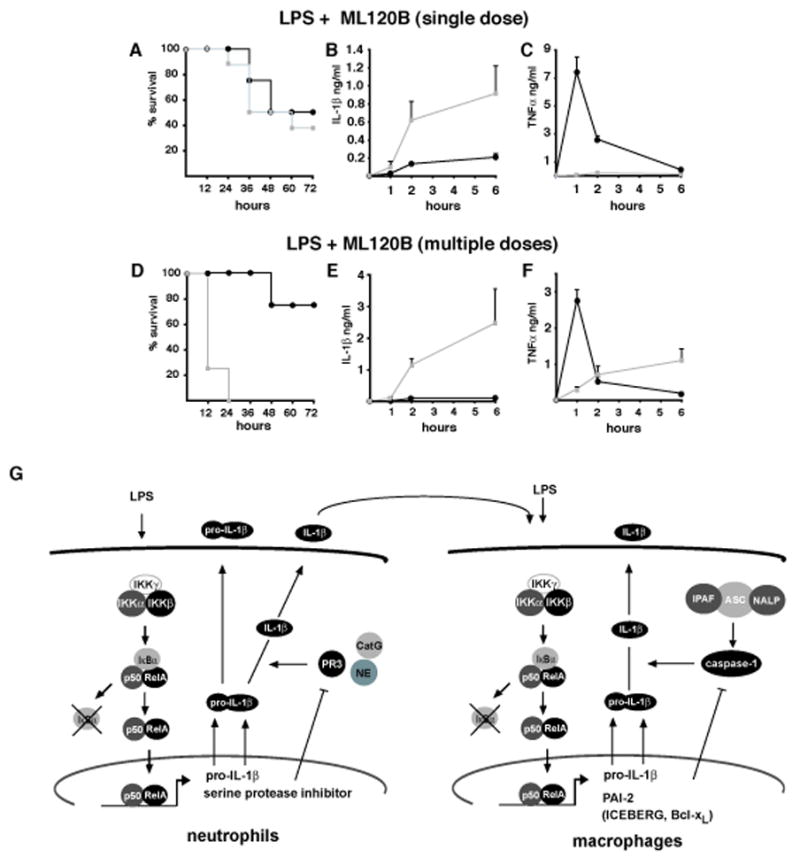
(A–C) Survival and corresponding IL-1β and TNF-α plasma levels of LPS-challenged wt mice given 300 mg/kg of the IKKβ inhibitor ML120B by oral gavage 1 hr prior to LPS administration (30 mg/kg). (D) Survival and corresponding IL-1β (E) and TNF-α (F) plasma levels in wt mice given 300 mg/kg ML120B by oral gavage twice daily for four days before LPS challenge. (G) Schematic representation of the proposed dual role of IKKβ-dependent NF-κB activation in regulation of IL-1β secretion by macrophages and neutrophils: in neutrophils, which appear to be a rapid but minor (albeit critical) source of IL-1β, IKKβ driven NF-κB positively regulates the transcription of pro-IL-1β mRNA and serine protease inhibitor genes whose products inhibit the activity of PR3, which can process pro-IL-1β. Secretion of biologically active IL-1β by neutrophils acts together with LPS to augment IL-1β secretion by macrophages, which represent the major source of this cytokine. In the macrophage, NF-κB controls pro-IL-1β mRNA synthesis as well as the expression of genes such as PAI-1, Bcl-xL and ICEBERG whose products inhibit caspase-1 activation.
Administration of ML120B for several days leads to granulocytosis (Nagashima et al., 2006), resembling the phenotype of IkkβΔ mice. We therefore examined whether repetitive ML120B treatment augmented LPS sensitivity. Wild-type mice were given ML120B twice daily (300 mg/kg each time) for 4 days and development of neutrophilia was confirmed in blood smears (data not shown). Similar to IkkβΔ mice, ML120B-treated animals were much more susceptible to LPS-induced mortality than untreated mice (Figure 7D). Plasma IL-1β was dramatically upregulated, whereas the initial LPS- induced surge in plasma TNF-α was suppressed (Figure 7E, F). Thus, prolonged IKKβ inhibition increases susceptibility to endotoxin-induced shock and mortality by enhancing IL-1β processing.
Discussion
IKKβ dependent NF-κB activation is considered to play a major role in the transcriptional control of acute and chronic inflammation (Bonizzi and Karin, 2004), suggesting that IKKβ inhibitors may be effective anti-inflammatory drugs (Karin et al., 2004). Unexpectedly, and counterintuitively, inhibition of this central pathway enhances susceptibility to endotoxin-induced shock and mortality by augmenting IL-1β processing and secretion. These findings stand in sharp contrast to the marked ability of IKKβ inhibition to prevent TNF-α expression and release, an endpoint that raised enthusiasm for targeting IKKβ in chronic inflammatory diseases, such as rheumatoid arthritis, and inflammation-induced bone loss (McIntyre et al., 2003; Ruocco et al., 2005). Thus, in addition to revealing a novel role for IKKβ dependent NF-κB activation as a negative regulator of pro-IL-1β processing, our results raise serious concerns about the long-term impact of IKKβ inhibition. However, it is possible that enhanced IL-1β processing may occur only upon acute septic infections, where, as discussed below, it is likely to play a protective role, and we suggest ways to avoid such complications. It should also be noted that partial and transient inhibition of IKKβ does not seem to be as problematic.
Prolonged inhibition of NF-κB, however, leads to enhanced processing of pro-IL-1β despite a profound transcriptional inhibition of NF-κB dependent gene expression including inhibition of IL-1β gene transcription. Whereas NF-κB positively regulates a major transcriptional control point in inflammation and innate immunity (Ghosh and Karin, 2002), the regulation of pro-IL-1β (and pro-IL-18) processing allows for an additional control of innate immunity and inflammation at the post-transcriptional level (Martinon and Tschopp, 2004). Regulation of IL-1β (and IL-18) secretion differs from that of most other cytokines, as IL-1β does not contain a signal peptide and its production and release depend on transcriptional and post-transcriptional processes (Dinarello, 2005; Martinon and Tschopp, 2004; Ogura et al., 2006). The majority of IL-1β induced upon endotoxin challenge in an NF-κB dependent manner remains in the cell as pro-IL-1β and the picogram amounts of IL-1β released by myeloid cells activated by LPS alone are small compared to nanogram amounts of TNF-α or IL-6. Whereas NF-κB is the main transcription factor controlling IL-1β gene induction, caspase-1 (also termed interleukin-1β-converting enzyme, ICE) is the major protease required for pro-IL-1β processing especially in macrophages (Kuida et al., 1995; Li et al., 1995). Akin to NF-κB whose activation depends on the signal-responsive IKK complex (Ghosh and Karin, 2002), also termed the signalsome (Mercurio et al., 1997), caspase-1 is regulated by the inflammasome, a group of related protein complexes which can respond to different ligands (Martinon and Tschopp, 2004; Ogura et al., 2006). However, several other proteases including NE, PR3, Cat G, chymase and chymotrypsin as well as certain matrix metalloproteinases, were shown to be capable of cleaving pro-IL-1β at critical sites involved in generation and secretion of the bioactive molecule (Black et al., 1988; Dinarello et al., 1986; Hazuda et al., 1990; Irmler et al., 1995; Mizutani et al., 1991). It was speculated that these enzymes, once secreted, cleave pro-IL-1β extracellularly thereby amplifying the inflammatory reaction (Fantuzzi et al., 1997). Our results strongly suggest that although the majority of IL-1β in mice lacking NF-κB activity in myeloid cells is derived from excessive caspase-1 activation in macrophages, neutrophils (and inflammatory monocytes) also play an important role in excessive IL-1β release, perhaps by augmenting its release from macrophages (Figure 7G). In neutrophils lacking IKKβ, IL-1β is secreted through a caspase-1-independent mechanism. In these cells intracellular serine proteases, especially PR3, are involved in IL-1β processing and secretion. In support of this, pretreatment of IkkβΔ mice with purified α1-anti-trypsin, which inhibits extracellular serine proteases (Liu et al., 2000), did not reduce IL-1β secretion, but a cell permeable peptide, specifically blocking NE and PR3, reduced IL-1β production substantially and protected IkkβΔ mice from endotoxin-induced death. Nonetheless, we cannot rule out off-target effects of this particular inhibitor, MeOSuc-AAPV-cmk, and a caspase-1 inhibitor, YVAD-cmk, also blocked excessive IL-1β secretion.
We tried to determine which cell type is responsible for enhanced IL-1β release after IKKβ or NF-κB depletion or inhibition in myeloid cells. We found that in LPS-challenged IkkβΔmye mice depletion of either neutrophils (and inflammatory monocytes) or macrophages was protective, suggesting critical involvement of all these cell types. Similar results were observed by the use of protease inhibitors. Both the caspase-1 inhibitor YVAD-cmk and the PR3 inhibitor MeOSuc-AAPV-cmk led to an almost complete inhibition of IL-1β secretion in IkkβΔ mice (Figure 6H, K). However, caspase-1 activity is required for IL-1β processing and secretion only in macrophages. In vitro, IKKβ-deficient macrophages secrete much more (7–10 fold) IL-1β per cell than IKKβ-deficient neutrophils after LPS stimulation. Furthermore, in vitro treatment with the IKKβ inhibitor readily augments caspase-1 activation and IL-1β secretion by LPS-treated macrophages (Figure 4F) and caspase-1 inhibition prevents IL-1β secretion. These results suggest that the major IL-1β producers under these conditions are the macrophages. Yet depletion of neutrophils (and inflammatory monocytes) via a strategy that leaves the macrophages intact also results in substantial inhibition of massive IL-1β secretion. To reconcile these results, we suggest that the initial and more rapid release of IL-1β by IKKβ-deficient neutrophils may augment the delayed and more substantial IL-1β secretion by IKKβ-deficient macrophages (Figure 7G). In support of this hypothesis, administration of an inhibitor of IL-1β signaling (IL-1Ra) also inhibits IL-1β secretion (Figure 1F).
It was previously found that caspase-1 activation is enhanced by macrophage apoptosis (Hogquist et al., 1991). Macrophage apoptosis is increased in the absence of NF-κB due to defective expression of A1/Bfl-1 and PAI-2, encoded by NF-κB target genes (Park et al., 2005). It is well established that in addition to these genes, NF-κB controls expression of genes that encode direct caspase inhibitors (Karin and Lin, 2002). Such molecules may inhibit caspase-1 activation directly and indirectly by suppressing macrophage apoptosis. In human cells, NF-κB may also regulate expression of ICEBERG, which appears to be a dedicated inhibitor of inflammasome activation (Humke et al., 2000). In addition, while this manuscript was under revision, it was shown that Bcl-xL, which is encoded by a NF-κB target gene and its relative Bcl-2 directly inhibit caspase-1 activation by NALP1 (Bruey et al., 2007). In neutrophils, instead of caspase-1, IL-1β production depends mainly on serine proteases, whose activity is also negatively regulated by NF-κB. It was documented that expression of several serine protease inhibitors (serpins), including PAI-2, MNEI and proteinase inhibitor 9 (PI-9) is transcriptionally activated by NF-κB (Kannan-Thulasiraman and Shapiro, 2002; Park et al., 2005; Zeng and Remold-O’Donnell, 2000). Of these, PAI-2 expression is dramatically reduced in IKKβ-deficient neutrophils and macrophages and restoration of PAI-2 expression in IKKβ-deficient macrophages inhibits LPS-induced IL-1β secretion. However, the direct target for PAI-2 involved in control of IL-1β processing is currently unknown.
Repetitive administration of a specific IKKβ inhibitor mimicked the effect of myeloid IKKβ deletion despite efficient inhibition of NF-κB mediated gene induction, including pro-TNF-α and pro-IL-1β synthesis. We suggest that the high stability of pro-IL-1β may account for its accumulation over time even when pro-IL-1β mRNA synthesis is inhibited. It is also plausible that the translational efficiency of pro-IL-1β mRNA may be increased once its levels drop below a certain threshold. In vitro, treatment of macrophages with the IKKβ inhibitor strongly augments LPS-induced caspase-1 activation and obliterates the need for a second signal, such as high concentrations of ATP.
Collectively, our results illustrate that quite unexpectedly IKKβ and NF-κB are also involved in negative regulation of inflammasome activation. It is well established that inflammation is a potentially dangerous, but protective response, that is controlled by numerous negative feedback mechanisms (Cohen, 2002; Karin et al., 2006). While many of these negative regulatory act transcriptionally by targeting NF-κB or IKK, our work reveals additional negative regulatory mechanisms that act post-transcriptionally through inhibition of pro-IL-1β processing. However, it is also possible that the major goal of the negative regulatory mechanisms we unraveled is to augment host defense rather than inhibit inflammation. As certain highly virulent pathogens have evolved the ability to inhibit NF-κB activation and thereby evade NF-κB dependent innate immunity (Orth et al., 2000), the augmented secretion of IL-1β by NF-κB-deficient myeloid cells may provide a compensatory mechanism that allows activation of an additional, NF-κB-independent, host defense response.
Undoubtedly, the mechanistic details of IL-1β production and its regulation by NF-κB also need to be assessed in normal human macrophages and neutrophils. Until then, our results raise serious concerns about possible complications associated with long-term inhibition of NF-κB. Although pharmacological inhibition is unlikely to be as complete as genetic disruption, it may, in addition to loss of innate immunity, augment acute inflammation associated with septic infections.
Materials and Methods
Mice
IkkβΔmye, IkkβΔ and IkkβF/F mice were described (Greten et al., 2004; Ruocco et al., 2005). To delete IKKβ in IkkβΔ mice, 250 μg poly(I:C) (Sigma) were injected i.p. two weeks before LPS administration or isolation of myeloid cells. Generation of RelaF/F mice is described elsewhere (Algül et al., 2007Algül et al., in press). Four to six mice of each genotype were i.p. injected with LPS (E. coli O111:B4, Sigma), CpG-DNA (ODN1668), E. coli (serotype O111:K58) or i.v. with Listeria monocytogenes wildtype (10403S). Etanercept (Enbrel®, Amgen), Ac-YVAD-cmk (Calbiochem), MeOSuc-AAPV-cmk (Calbiochem) were given i.p. 1 hr before LPS administration. Anakinra (Kineret®, Amgen) was injected subcutaneously every 4 hrs over a period of 12 hrs. ML120B (Nagashima et al., 2006a) was given by oral gavage either once before LPS or twice daily over four days. Clodronate-liposomes and RB6-8C5 were applied i.p. 24 hrs before LPS. Liposome-encapsulated clodronate (a kind gift of Roche Diagnostics GmbH, Mannheim, Germany) was prepared as previously described (Van Rooijen and Sanders, 1994).
Isolation of macrophages and neutrophils
BMDM were generated as described (Greten et al., 2004). To isolate neutrophils, mice were i.p. injected with 1 ml of 3% thioglycollate (DIFCO) and peritoneal cells were flushed out 3–5 hrs later. After blocking Fc-receptors with anti-CD16/32 (Pharmingen), cells were incubated with PE-labeled GR-1 antibody (Becton & Dickinson) and magnetically separated using anti-PE beads according to the manufacturer’s (Miltenyi Biotec) instructions. Retroviral reconstitution was performed essentially as described (Miething et al., 2007). Bone marrow was transduced with EGFP-PAI-2 retrovirus and EGFP positive cells were sorted by flow cytometry. EGFP+- and EGFP−-cells were differentiated into macrophages and stimulated with LPS.
Protein and RNA analysis
IL-1β and TNF-α in plasma and cell supernatants were measured using the DuoSet ELISA systems (R&D Systems) according to manufacturer’s instructions. RNA was extracted using Trizol (Invitrogen). RPA was performed as described (Park et al., 2005). cDNA synthesis, Real-Time PCR, immunoblot and EMSA were as described (Greten et al., 2004). Primer sequences are available upon request. Antibodies recognizing IL-1β, TNF-α (R&D Systems), IL-1β (for IP) and cleaved caspase 3 (Becton & Dickinson), β-actin (Sigma), IKKβ (UBI), RelA, IκBα, PR3 (Santa Cruz) were purchased from the indicated suppliers.
Protease activity determination and inhibition
Cell pellets were lysed and hydrolysis of MeOSuc-AAPV-pNA (Calbiochem) was determined in 0.05 M NaH2PO4, 0.1 M NaCl, 0.005 % Triton-X100 and 5 % DMSO at 405 nm. For inhibition experiments rhSLPI (R&D Systems) and 3, 4-dichloroisocoumarin (Calbiochem) were used.
In vitro processing of pro-IL-1β and IL-1β bioassay
HEK293 cells were transfected with pro-IL-1β expression plasmid and cells were lysed after 24 hrs. Lysates were incubated with purified NE and PR3 (Athens Research & Technology) for 30 min. IL-1β bioassay was performed using EL4-cells, which were stimulated for 24 hrs. IL-2 release by EL4 cells was measured by an ELISA (eBioscience) according to manufacturer’s instructions.
Chemotaxis assay
1×106 neutrophils were seeded in transwell plates and incubated in the presence of 10 μM fMLP in the lower chamber for 60 min after which cells in the lower chamber were counted.
Supplementary Material
Supplementary Figure 1: Deletion of IKKβ in myeloid cells does not lead to histological changes in IkkβΔmye mice
H&E stained sections of liver, lung, kidney and intestines from IkkβF/F and IkkβΔmye mice.
Supplementary Figure 2: Increased plasma IL-1β levels in IkkβΔmye mice after CpG-ODN challenge or Listeria monocytogenes infection
(A) Survival of IkkβF/F (black) and IkkβΔmye mice (grey) and corresponding IL-1β (B), TNF-α (C) and IL-6 (D) plasma levels after CpG-ODN administration (20 nmol/mouse). Data are averages of 4 animals per time point. Plasma levels of IL-1β (E), TNF-α (F) and IL-6 (G) in IkkβF/F (black) and IkkβΔmye (grey) mice after infection with Listeria monocytogenes (5×104 CFU i.v./mouse). Data are averages of at least 4 animals per time point. (H and I) Colony forming units (CFU) in spleens (H) and livers (I) of IkkβF/F (black) and IkkβΔmye mice (grey) measured 48 hrs after Listeria monocytogenes infection.
Supplementary Figure 3: Depletion of macrophages does not affect Gr-1+ cells
Forward- and side-scatter profiles of splenic Gr-1+ cells before and after depletion of macrophages in IkkβF/F and IkkβΔmye mice.
Supplementary Figure 4: Inhibition of IL-1β signaling improves survival in IkkβΔ mice after LPS challenge
(A) Survival of IkkβF/F (black) and IkkβΔ mice (grey) and corresponding IL-1β (B) and TNF-α (C) plasma levels after LPS administration (20 mg/kg) in the presence of Anakinra (50 mg/kg). Data are averages of at least 4 animals per time point. Dashed grey line in (A) represents survival in animals challenged with LPS without Anakinra and in (B and C) it represents plasma IL-1β (B) or TNF-α (C) level of IkkβΔ mice given LPS without Anakinra.
Supplementary Figure 5: IkkβΔ macrophages show more complete inhibition of NF-κB activation than IkkβΔmye macrophages
NF-κB DNA binding activity was measured by EMSA in extracts of LPS-stimulated IkkβF/F, IkkβΔmye and IkkβΔ macrophages.
Supplementary Figure 6: IkkβF/F and IkkβΔ macrophages do not show any differences in intracellular caspase-1 expression
Immunoblot analysis of caspase-1 expression in LPS-stimulated IkkβF/F and IkkβΔ macrophages.
Supplementary Figure 7: Characterization of IkkβΔ neutrophils
(A) Confirmation of IKKβ deletion by immunoblot analysis and (B) inhibition of NF-κB activation in response to LPS in IkkβΔ granulocytes. (C) Forward- and side-scatter profiles of IkkβF/F and IkkβΔ granulocytes do not show any differences. (D) Chemotaxis assay using IkkβF/F and IkkβΔ granulocytes incubated with or without 10 μM fMLP for 60 min. (E) Splenic CFU in IkkβF/F and IkkβΔ mice 8 hrs after infection with E. coli (2×108 cfu/mouse, i.p.) and corresponding plasma IL-1β levels (F) during infection. Results are averages for 4 mice per genotype.
Supplementary Figure 8: RNA expression levels of serine proteases and serpins in IkkβF/F and IkkβΔ granulocytes.
Real-Time PCR analysis of mRNA levels of the indicated serine protease and serpin genes in untreated and LPS-stimulated IkkβF/F and IkkβΔ granulocytes.
Supplementary Figure 9: Serine protease activity is increased in neutrophils of RelaΔ mice
(A) Increased MeOSuc-AAPV-pNA hydrolytic activity in neutrophils of RelaΔ mice. Substrate hydrolysis was measured using neutrophil lysates in the absence or presence of the protease inhibitor DCIC. (B) Successful deletion of full-length RelA in neutrophils of RelaΔ mice, reduced pro-IL-1β induction and PR3 expression as determined by immunoblot analysis. (C, D) IL-1β and TNF-α secretion by RelaF/F and RelaΔneutrophils after LPS stimulation as determined by an ELISA. Results shown are averages of three mice per genotype.
Supplementary Figure 10: Pharmacological inhibition of IKKβ in macrophages and neutrophils prevents NF-κB activition
Macrophages and neutrophils were incubated in the absence (DMSO) or presence of ML120B (30 μM) and stimulated with LPS (100 ng/ml). NF-κB activation was examined by EMSA.
Acknowledgments
We thank Rabea Konietschke, Birgit Wittig, Claudia Mugler and Yolanda Andersen for excellent technical assistance. We also thank Fabian Geisler for providing RelaΔ mice, Jürgen Ruland for providing EL-4 cells, Eileen Remold-O’Donnell for generously providing serpinB1/MNEI antibody, Vishva Dixit for generously providing caspase-1 antibody as well as Tim Sparwasser and Guy Salvesen for helpful discussions. This work was supported by grants from the Deutsche Forschungsgemeinschaft (Emmy-Noether-Program Gr1916/2-2), Deutsche Krebshilfe (106772), Fritz-Thyssen-Stiftung (10.05.2.168), and the Technical University Munich (KKF34-04) to F.R.G., as well as the National Institute of Health (AI56075, DK70867 to L.E., AI43477, AI061712, E06486, to M.K. and DK35108 to L.E. and M.K.). M.K. is an American Cancer Society Research Professor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- Black RA, Kronheim SR, Cantrell M, Deeley MC, March CJ, Prickett KS, Wignall J, Conlon PJ, Cosman D, Hopp TP, et al. Generation of biologically active interleukin-1 beta by proteolytic cleavage of the inactive precursor. J Biol Chem. 1988;263:9437–9442. [PubMed] [Google Scholar]
- Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Bruey JM, Bruey-Sedano N, Luciano F, Zhai D, Balpai R, Xu C, Kress CL, Bailly-Maitre B, Li X, Osterman A, et al. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell. 2007;129:45–56. doi: 10.1016/j.cell.2007.01.045. [DOI] [PubMed] [Google Scholar]
- Chen LW, Egan L, Li ZW, Greten FR, Kagnoff MF, Karin M. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med. 2003;9:575–581. doi: 10.1038/nm849. [DOI] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, Leimer AH, Cheronis J. Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc Natl Acad Sci U S A. 1999;96:6261–6266. doi: 10.1073/pnas.96.11.6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Proinflammatory and anti-inflammatory cytokines as mediators in the pathogenesis of septic shock. Chest. 1997;112:321S–329S. doi: 10.1378/chest.112.6_supplement.321s. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-1beta. Crit Care Med. 2005;33:S460–462. doi: 10.1097/01.ccm.0000185500.11080.91. [DOI] [PubMed] [Google Scholar]
- Dinarello CA, Cannon JG, Mier JW, Bernheim HA, LoPreste G, Lynn DL, Love RN, Webb AC, Auron PE, Reuben RC, et al. Multiple biological activities of human recombinant interleukin 1. J Clin Invest. 1986;77:1734–1739. doi: 10.1172/JCI112495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantuzzi G, Ku G, Harding MW, Livingston DJ, Sipe JD, Kuida K, Flavell RA, Dinarello CA. Response to local inflammation of IL-1 beta-converting enzyme- deficient mice. J Immunol. 1997;158:1818–1824. [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Hazuda DJ, Strickler J, Kueppers F, Simon PL, Young PR. Processing of precursor interleukin 1 beta and inflammatory disease. J Biol Chem. 1990;265:6318–6322. [PubMed] [Google Scholar]
- Hogquist KA, Nett MA, Unanue ER, Chaplin DD. Interleukin 1 is processed and released during apoptosis. Proc Natl Acad Sci U S A. 1991;88:8485–8489. doi: 10.1073/pnas.88.19.8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz BH, Scott ML, Cherry SR, Bronson RT, Baltimore D. Failure of lymphopoiesis after adoptive transfer of NF-kappaB-deficient fetal liver cells. Immunity. 1997;6:765–772. doi: 10.1016/s1074-7613(00)80451-3. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- Humke EW, Shriver SK, Starovasnik MA, Fairbrother WJ, Dixit VM. ICEBERG: a novel inhibitor of interleukin-1beta generation. Cell. 2000;103:99–111. doi: 10.1016/s0092-8674(00)00108-2. [DOI] [PubMed] [Google Scholar]
- Irmler M, Hertig S, MacDonald HR, Sadoul R, Becherer JD, Proudfoot A, Solari R, Tschopp J. Granzyme A is an interleukin 1 beta-converting enzyme. J Exp Med. 1995;181:1917–1922. doi: 10.1084/jem.181.5.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan-Thulasiraman P, Shapiro DJ. Modulators of inflammation use nuclear factor-kappa B and activator protein-1 sites to induce the caspase-1 and granzyme B inhibitor, proteinase inhibitor 9. J Biol Chem. 2002;277:41230–41239. doi: 10.1074/jbc.M200379200. [DOI] [PubMed] [Google Scholar]
- Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: a treasure trove for drug development. Nat Rev Drug Discov. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- Kisseleva T, Song L, Vorontchikhina M, Feirt N, Kitajewski J, Schindler C. NF-kappaB regulation of endothelial cell function during LPS-induced toxemia and cancer. J Clin Invest. 2006;116:2955–2963. doi: 10.1172/JCI27392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- Liu Z, Zhou X, Shapiro SD, Shipley JM, Twining SS, Diaz LA, Senior RM, Werb Z. The serpin alpha1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell. 2000;102:647–655. doi: 10.1016/s0092-8674(00)00087-8. [DOI] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561–574. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- McIntyre KW, Shuster DJ, Gillooly KM, Dambach DM, Pattoli MA, Lu P, Zhou XD, Qiu Y, Zusi FC, Burke JR. A highly selective inhibitor of I kappa B kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. Arthritis Rheum. 2003;48:2652–2659. doi: 10.1002/art.11131. [DOI] [PubMed] [Google Scholar]
- Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- Miething C, Grundler R, Mugler C, Brero S, Hoepfl J, Geigl J, Speicher MR, Ottmann O, Peschel C, Duyster J. Retroviral insertional mutagenesis identifies RUNX genes involved in chronic myeloid leukemia disease persistence under imatinib treatment. Proc Natl Acad Sci U S A. 2007;104:4594–4599. doi: 10.1073/pnas.0604716104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani H, Schechter N, Lazarus G, Black RA, Kupper TS. Rapid and specific conversion of precursor interleukin 1 beta (IL-1 beta) to an active IL-1 species by human mast cell chymase. J Exp Med. 1991;174:821–825. doi: 10.1084/jem.174.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima K, Sasseville VG, Wen D, Bielecki A, Yang H, Simpson C, Grant E, Hepperle M, Harriman G, Jaffee B, et al. Rapid TNFR1-dependent lymphocyte depletion in vivo with a selective chemical inhibitor of IKKbeta. Blood. 2006;107:4266–4273. doi: 10.1182/blood-2005-09-3852. [DOI] [PubMed] [Google Scholar]
- Ogura Y, Sutterwala FS, Flavell RA. The inflammasome: first line of the immune response to cell stress. Cell. 2006;126:659–662. doi: 10.1016/j.cell.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Orth K, Xu Z, Mudgett MB, Bao ZQ, Palmer LE, Bliska JB, Mangel WF, Staskawicz B, Dixon JE. Disruption of signaling by Yersinia effector YopJ, a ubiquitin-like protein protease. Science. 2000;290:1594–1597. doi: 10.1126/science.290.5496.1594. [DOI] [PubMed] [Google Scholar]
- Park JM, Greten FR, Wong A, Westrick RJ, Arthur JS, Otsu K, Hoffmann A, Montminy M, Karin M. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis--CREB and NF-kappaB as key regulators. Immunity. 2005;23:319–329. doi: 10.1016/j.immuni.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006;6:541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- Ruocco MG, Maeda S, Park JM, Lawrence T, Hsu LC, Cao Y, Schett G, Wagner EF, Karin M. I{kappa}B kinase (IKK){beta}, but not IKK{alpha}, is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J Exp Med. 2005;201:1677–1687. doi: 10.1084/jem.20042081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senftleben U, Li ZW, Baud V, Karin M. IKKbeta is essential for protecting T cells from TNFalpha-induced apoptosis. Immunity. 2001;14:217–230. doi: 10.1016/s1074-7613(01)00104-2. [DOI] [PubMed] [Google Scholar]
- Suffredini AF, Reda D, Banks SM, Tropea M, Agosti JM, Miller R. Effects of recombinant dimeric TNF receptor on human inflammatory responses following intravenous endotoxin administration. J Immunol. 1995;155:5038–5045. [PubMed] [Google Scholar]
- Taylor PR, Gordon S. Monocyte heterogeneity and innate immunity. Immunity. 2003;19:2–4. doi: 10.1016/s1074-7613(03)00178-x. [DOI] [PubMed] [Google Scholar]
- van Rooijen N, Bakker J, Sanders A. Transient suppression of macrophage functions by liposome-encapsulated drugs. Trends Biotechnol. 1997;15:178–185. doi: 10.1016/s0167-7799(97)01019-6. [DOI] [PubMed] [Google Scholar]
- Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Vassiloyanakopoulos AP, Okamoto S, Fierer J. The crucial role of polymorphonuclear leukocytes in resistance to Salmonella dublin infections in genetically susceptible and resistant mice. Proc Natl Acad Sci U S A. 1998;95:7676–7681. doi: 10.1073/pnas.95.13.7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener E, Oeckinghaus A, Papadopoulou N, Lavitas L, Schmidt-Supprian M, Ferch U, Mak TW, Ruland J, Heissmeyer V, Krappmann D. Essential role for IkappaB kinase beta in remodeling Carma1-Bcl10-Malt1 complexes upon T cell activation. Mol Cell. 2006;23:13–23. doi: 10.1016/j.molcel.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Wiesner O, Litwiller RD, Hummel AM, Viss MA, McDonald CJ, Jenne DE, Fass DN, Specks U. Differences between human proteinase 3 and neutrophil elastase and their murine homologues are relevant for murine model experiments. FEBS Lett. 2005;579:5305–5312. doi: 10.1016/j.febslet.2005.08.056. [DOI] [PubMed] [Google Scholar]
- Zeng W, Remold-O’Donnell E. Human monocyte/neutrophil elastase inhibitor (MNEI) is regulated by PU.1/Spi-1, Sp1, and NF-kappaB. J Cell Biochem. 2000;78:519–532. doi: 10.1002/1097-4644(20000915)78:4<519::aid-jcb1>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Zingarelli B. Nuclear factor-kappaB. Crit Care Med. 2005;33:S414–416. doi: 10.1097/01.ccm.0000186079.88909.94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Deletion of IKKβ in myeloid cells does not lead to histological changes in IkkβΔmye mice
H&E stained sections of liver, lung, kidney and intestines from IkkβF/F and IkkβΔmye mice.
Supplementary Figure 2: Increased plasma IL-1β levels in IkkβΔmye mice after CpG-ODN challenge or Listeria monocytogenes infection
(A) Survival of IkkβF/F (black) and IkkβΔmye mice (grey) and corresponding IL-1β (B), TNF-α (C) and IL-6 (D) plasma levels after CpG-ODN administration (20 nmol/mouse). Data are averages of 4 animals per time point. Plasma levels of IL-1β (E), TNF-α (F) and IL-6 (G) in IkkβF/F (black) and IkkβΔmye (grey) mice after infection with Listeria monocytogenes (5×104 CFU i.v./mouse). Data are averages of at least 4 animals per time point. (H and I) Colony forming units (CFU) in spleens (H) and livers (I) of IkkβF/F (black) and IkkβΔmye mice (grey) measured 48 hrs after Listeria monocytogenes infection.
Supplementary Figure 3: Depletion of macrophages does not affect Gr-1+ cells
Forward- and side-scatter profiles of splenic Gr-1+ cells before and after depletion of macrophages in IkkβF/F and IkkβΔmye mice.
Supplementary Figure 4: Inhibition of IL-1β signaling improves survival in IkkβΔ mice after LPS challenge
(A) Survival of IkkβF/F (black) and IkkβΔ mice (grey) and corresponding IL-1β (B) and TNF-α (C) plasma levels after LPS administration (20 mg/kg) in the presence of Anakinra (50 mg/kg). Data are averages of at least 4 animals per time point. Dashed grey line in (A) represents survival in animals challenged with LPS without Anakinra and in (B and C) it represents plasma IL-1β (B) or TNF-α (C) level of IkkβΔ mice given LPS without Anakinra.
Supplementary Figure 5: IkkβΔ macrophages show more complete inhibition of NF-κB activation than IkkβΔmye macrophages
NF-κB DNA binding activity was measured by EMSA in extracts of LPS-stimulated IkkβF/F, IkkβΔmye and IkkβΔ macrophages.
Supplementary Figure 6: IkkβF/F and IkkβΔ macrophages do not show any differences in intracellular caspase-1 expression
Immunoblot analysis of caspase-1 expression in LPS-stimulated IkkβF/F and IkkβΔ macrophages.
Supplementary Figure 7: Characterization of IkkβΔ neutrophils
(A) Confirmation of IKKβ deletion by immunoblot analysis and (B) inhibition of NF-κB activation in response to LPS in IkkβΔ granulocytes. (C) Forward- and side-scatter profiles of IkkβF/F and IkkβΔ granulocytes do not show any differences. (D) Chemotaxis assay using IkkβF/F and IkkβΔ granulocytes incubated with or without 10 μM fMLP for 60 min. (E) Splenic CFU in IkkβF/F and IkkβΔ mice 8 hrs after infection with E. coli (2×108 cfu/mouse, i.p.) and corresponding plasma IL-1β levels (F) during infection. Results are averages for 4 mice per genotype.
Supplementary Figure 8: RNA expression levels of serine proteases and serpins in IkkβF/F and IkkβΔ granulocytes.
Real-Time PCR analysis of mRNA levels of the indicated serine protease and serpin genes in untreated and LPS-stimulated IkkβF/F and IkkβΔ granulocytes.
Supplementary Figure 9: Serine protease activity is increased in neutrophils of RelaΔ mice
(A) Increased MeOSuc-AAPV-pNA hydrolytic activity in neutrophils of RelaΔ mice. Substrate hydrolysis was measured using neutrophil lysates in the absence or presence of the protease inhibitor DCIC. (B) Successful deletion of full-length RelA in neutrophils of RelaΔ mice, reduced pro-IL-1β induction and PR3 expression as determined by immunoblot analysis. (C, D) IL-1β and TNF-α secretion by RelaF/F and RelaΔneutrophils after LPS stimulation as determined by an ELISA. Results shown are averages of three mice per genotype.
Supplementary Figure 10: Pharmacological inhibition of IKKβ in macrophages and neutrophils prevents NF-κB activition
Macrophages and neutrophils were incubated in the absence (DMSO) or presence of ML120B (30 μM) and stimulated with LPS (100 ng/ml). NF-κB activation was examined by EMSA.