

Abstract
2-Amino-3,8-dimethylimidazo-[4,5-f]quinoxaline (MeIQx) is one of the most potent and abundant mutagens in the western diet. Bioactivation includes N-hydroxylation catalyzed by cytochrome P450s followed by O-acetylation catalyzed by N-acetyltransferase 2 (NAT2). Nucleotide excision repair-deficient chinese hamster ovary (CHO) cells were constructed by stable transfection of human cytochrome P4501A1 (CYP1A1) and a single copy of either NAT2*4 (rapid acetylator) or NAT2*5B (slow acetylator) alleles. CYP1A1 and NAT2 catalytic activities were undetectable in untransfected CHO cell lines. CYP1A1 activity did not differ significantly (p > 0.05) among the CYP1A1-transfected cell lines. Cells transfected with NAT2*4 had significantly higher levels of sulfamethazine N-acetyltransferase (p = 0.0001) and N-hydroxy-MeIQx O-acetyltransferase (p = 0.0093) catalytic activity than cells transfected with NAT2*5B. Only cells transfected with both CYP1A1 and NAT2*4 showed concentration-dependent cytotoxicity and hypoxanthine phosphoribosyl transferase (hprt) mutagenesis following MeIQx treatment. dG-C8-MeIQx was the primary DNA adduct formed and levels were dose-dependent in each cell line and in the order: untransfected < transfected with CYP1A1 < transfected with CYP1A1 & NAT2*5B < transfected with CYP1A1 & NAT2*4. MeIQx DNA adduct levels were significantly higher (p < 0.001) in CYP1A1/NAT2*4 than CYP1A1/NAT2*5B cells at all concentrations of MeIQx tested. MeIQx-induced DNA adduct levels correlated very highly (r2 = 0.88) with MeIQx-induced mutants. These results strongly support extrahepatic activation of MeIQx by CYP1A1 and a robust effect of human NAT2 genetic polymorphism on MeIQx –induced DNA adducts and mutagenesis. The results provide laboratory-based support for epidemiological studies reporting higher frequency of heterocyclic amine-related cancers in rapid NAT2 acetylators.
Introduction
Heterocyclic amine carcinogens are produced by condensation of creatinine with amino acids and are often generated during high temperature cooking of meats (1). 2-Amino-3,8-dimethylimidazo-[4,5-f]quinoxaline (MeIQx) is one of the most potent and abundant mutagens formed during high temperature cooking of meats prevalent in the western diet. In three large cohort studies, estimated mean daily intake of MeIQx ranged from 33 to 45 ng/day (2). MeIQx is one of four heterocyclic amine carcinogens designated as “reasonably anticipated to be a human carcinogen” (3). The target organ specificity of MeIQx differs with species and gender, but it has been associated with liver tumors, lung tumors, lymphoma and leukemia in mice (4) and with liver, zymbal-gland, skin, and clitoral-gland tumors in rats (5). Case control studies suggest that MeIQx may increase the risk of colorectal adenoma (6,7), lung cancer (8), and breast cancer (9) in humans.
MeIQx-induced DNA adduct formation and mutagenesis require metabolic activation, and one important pathway includes N-hydroxylation catalyzed by cytochrome P450s (10–12). Although many heterocyclic amine carcinogens undergo N-hydroxylation by CYP1A2, this isoform is primarily restricted to the liver. Metabolic activation of MeIQx by extrahepatic cytochrome P450 isoforms, such as CYP1A1, also has been reported (13). N-hydroxy-MeIQx can be further O-acetylated by N-acetyltransferase 2 (NAT2) to the acetoxy-derivative that is highly unstable, leading to electrophilic intermediates that are mutagenic (14) and form DNA adducts (15).
MeIQx-induced DNA adducts primarily form at the C8 position of deoxyguanosine (16) and dG-C8-MeIQx adducts have been identified in human tissues (17). Bulky DNA adducts such as these are recognized by the nucleotide excision repair (NER) pathway (18). MeIQx-induced DNA adducts have been measured using high performance liquid chromatography-tandem mass spectrometry (LC-MS-MS) technology (19).
Since heterocyclic amines require metabolic activation to exert their carcinogenic effects, human metabolic polymorphisms in the enzymes catalyzing the activation and/or detoxification pathways of carcinogen metabolism may account for differences in susceptibility to carcinogens between individuals (10). Humans exhibit genetic polymorphism in NAT2 resulting in rapid and slow acetylator phenotypes. The NAT2*4 allele is associated with rapid acetylator phenotype, whereas the NAT2*5B allele is associated with slow acetylator phenotype (20,21). Relative to the NAT2*4 reference allele, NAT2*5B possesses three single nucleotide polymorphisms in the NAT2 coding region: T341C (I114T), C481T (silent) and A803G (K268R) (20,22). Epidemiological studies suggest a role for NAT2 genetic polymorphism in susceptibility to various cancers related to heterocyclic amine exposures but conflicting results suggest the need for laboratory-based experiments to support the biological plausibility and the conclusions inferred from these epidemiological studies (20,22).
Since O-acetylation catalyzed by NAT2 is an activation pathway, we hypothesized that MeIQx-induced DNA damage and subsequent mutagenesis would be greater in rapid than slow NAT2 acetylators. Therefore, we constructed NER-deficient chinese hamster ovary (CHO) cells transfected with human CYP1A1, human NAT2*4 (rapid acetylator allele) or human NAT2*5B (slow acetylator allele) to test this hypothesis.
Materials and Methods
Cell Culture
The UV5-CHO cell line, a NER-deficient derivative of the AA8 line, was obtained from the ATCC (Catalog number: CRL-1865). UV5-CHO is hypersensitive to bulky adduct mutagens and belongs to the excision repair cross complementation group 2. All cells were grown in alpha-modified minimal essential medium (Cambrex, Walkersville, MD) without L-glutamine, ribosides and deoxyribosides supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), 100 U/ml penicillin, 100 U/ml streptomycin (Cambrex) and 2 mM L-glutamine (Cambrex) at 37 °C in 5% CO2. Media were supplemented with appropriate selective agents to maintain stable transfectants. All transfection techniques were according to instructions provided with the Effectene Transfection Reagent kit (Qiagen, Valencia, CA). Successful transfections were validated as described below.
Construction of UV5/CHO Cells Expressing CYP1A1 and Human NAT2*4 or NAT2*5B
The procedures used were slightly modified from those recently reported (23). Briefly, the pFRT/lacZeo plasmid (Invitrogen, Carlsbad, CA) was transfected into NER-deficient UV5 cell lines to generate a UV5 cell line containing an integrated FRT site (UV5FRT). Clones possessing a single integrated FRT site were identified by PCR and Southern blot analysis as previously described (23). Plasmid pUV1 containing human NADPH-cytochrome P450 reductase gene (POR) was kindly provided by Dr. Frank Gonzalez (National Cancer Institute, Bethesda, MD). Total RNA was isolated from human fibroblasts previously transfected with human CYP1A1 genomic DNA (24). An aliquot (1.5 μg) of total RNA was reverse-transcribed into cDNA using Advantage RT-for-PCR kit (BD Bioscience Clontech, Palo Alto, CA) in a reaction mixture (20 μl) containing 50 mM Tris-HCl (pH 8.3), 75 mM KCl, 3mM MgCl2, 2 mM dNTPs, Recombinant RNase inhibitor (20 unit), 1 μM Oligo(dT)18 primer and MMLV Reverse transcriptase (100 unit). The open reading frame of human CYP1A1 was amplified by PCR using an aliquot (3 μl) of the cDNA solution as template. The forward and reverse primers for human CYP1A1 amplification were 5′-actgatgctagcatgcttttcccaatctccatgt-3′ and 5′-actgatacgcgtctaagagcgcagctgcattt-3′, respectively. The cDNA of human POR was amplified by PCR directly from plasmid pUV1 using primers 5′-atctagtagcggccgcctagctccacacgtccaggg-3′ and 5′-actagttctagaatgggagactcccacgtgga-3′. The RT-PCR products were purified by electrophoresis in a 0.6% agarose gel.
Both purified PCR products were digested and ligated into similarly treated pIRES vector, and transformed into DH5α competent cells as described by the manufacturer (Life Technologies, Rockville, MD). The cDNA sequence of human CYP1A1 and POR were confirmed by automated DNA sequencing using the Big Dye Terminator kit (Applied Biosystems). The pIRES plasmid containing cDNAs of human CYP1A1 and POR was transfected into the newly established UV5FRT cell line using the Effectene Transfection Reagent kit (Qiagen). Transfected cells were selected by resistance to 500 μg/ml geneticin (G418). Those UV5 cells expressing a single FRT site, CYP1A1, and POR are referred to as UV5/CYP1A1 cells throughout the manuscript. These colonies were expanded and intact geneticin-resistant cells were assayed for CYP1A1 activity by measuring 7-ethoxyresorufin O-deethylase (EROD) activity as previously described (23). Cell lines with similar levels of EROD activity were selected for additional transfection with either NAT2*4 or NAT2*5B.
The open reading frames of NAT2*4 and NAT2*5B were amplified by PCR, digested with NheI and XhoI (New England Biolabs, Ipswich, MA) and inserted into the similarly prepared pcDNA5/FRT vector (Invitrogen) as previously described (23). The identity of the NAT2*4 or NAT2*5B gene was confirmed by automated DNA sequencing using the Big Dye Terminator kit as previously described (23). The pcDNA5/FRT plasmid containing human NAT2*4 or NAT2*5B was co-transfected with pOG44, a Flp recombinase expression plasmid, into UV5FRT cells. Integration of the pcDNA5/FRT construct into the FRT site was confirmed by PCR as previously described (23).
N-and O-Acetyltransferase Assays
N-acetyltransferase activities were measured with the human NAT2-selective substrate sulfamethazine (SMZ) using high performance liquid chromatography (HPLC) to separate N-acetyl-SMZ product from SMZ substrate as described previously (25). N-hydroxy-MeIQx O-acetyltransferase assays were determined by HPLC using modifications of an assay recently described (26). Briefly, reaction mixtures containing equal amounts of cell lysate protein, 1 mg/ml deoxyguanosine, 100 μM N-hydroxy-MeIQx (Toronto Research Chemicals, North York, Canada), and 1 mM acetyl coenzyme A were incubated at 37°C for 10 min and stopped by the addition of water saturated ethyl acetate. The reactions were centrifuged for 10 min and the organic phase was transferred, evaporated to dryness, and resuspended in 100 μl of 10% acetonitrile. HPLC separation was achieved using a gradient of 85:15 sodium perchlorate pH 2.5:acetonitrile to 0:100 sodium perchlorate pH 2.5:acetonitrile over 10 min. The retention time of dG-C8-MeIQx was 15.6 min. Baseline measurements using extracts of UV5 and UV5/CYP1A1 cells were subtracted from measurements in the NAT2*4-and NAT2*5B-transfected CHO cell lines.
Cytotoxicity and Mutagenesis
Assays for cytotoxicity and mutagenesis were previously described (23). Briefly, cells were grown for 12 doublings, with selective agents in complete HAT medium (30 μM hypoxanthine, 0.1 μM aminopterin, and 30 μM thymidine). Cells were plated at a density of 5 × 105 cells/T-25 flask and incubated for 24 h, after which media were changed and the cells treated for 48 h with various concentrations of MeIQx (Toronto Research Chemicals, North York, Ontario, Canada) or vehicle control (0.5% DMSO). Survival was determined by colony forming assay and expressed as percent of vehicle control. The remaining cells were replated and subcultured. After 7 days of growth, cultures were plated for cloning efficiency in complete media and for mutations in complete medium containing 40μM 6-thioguanine (Sigma, St. Louis, MO). Dishes were seeded with 1 × 105 cells/100 mm dish (10 replicates) and incubated for 7 days; cloning efficiency dishes were seeded with 100 cells/well/6-well plate in triplicate and incubated for 6 days.
Identification and Quantitation of MeIQx-DNA Adducts
Cells grown in 15 cm plates were treated with MeIQx as described above for the cytotoxicity and mutagenesis assays. Cells were harvested after 48 h of treatment and DNA was extracted and quantified as previously described (23). dG-C8-MeIQx and dG-C8-MeIQx-D3 adduct standards (>95% purity) were kindly provided by Dr. Rob Turesky (Wadsworth Center, New York State Department of Health, Albany, NY). Details of their synthesis and spectral analysis have been published previously (19). Several standard DNA digestion methods were compared prior to selecting the most efficient and optimal digestion protocol for recovery of dG-C8-MeIQx. One-tenth volumes each of proteinase K solution (20 mg/mL) and 10% SDS were added to the cell lysate, and the mixture was incubated at 37°C for 60 min. One volume of phenol equilibrated with 10 mM Tris HCl (pH 8.0), was added to the mixture, which was then vortexed and centrifuged at 3600 × g for 15 min. The aqueous layer was removed and added to 1 volume of phenol:chloroform:isoamyl alcohol (25:24:1) saturated with 10 mM Tris HCl (pH 8.0), which was vortexed and centrifuged. The aqueous layer was removed and added to 1 volume of cold (−20°C) isopropanol, and the mixture was vortexed and centrifuged. The DNA pellet was washed with 70% ethanol and redissolved in 5 mM Tris (pH 7.4) containing 1 mM CaCl2, 1 mM ZnCl2, and 10 mM MgCl2. DNA was quantified by A260. DNA quality was monitored by A260/280 and was consistently above 1.9. DNA samples (200 μg) spiked with one nanogram (3.3 adducts per 106 DNA bases) deuterated internal standard (dG-C8-MeIQx-D3) were digested at 37°C with 10 U of DNAse I (US Biological, Swampscott, MA) for 1 h followed by 5 U of micrococcal nuclease (Sigma), 5 U of nuclease P1 (US Biological), 0.01 U of spleen phosphodiesterase (Sigma), and 0.01 U of snake venom phosphodiesterase (Sigma) for 6 h followed by 5 U of alkaline phosphatase (Sigma) overnight. Two volumes of acetonitrile were added to the digest, which was then filtered and concentrated to 100 μl in a speed vacuum.
Samples were subjected to HPLC and introduced into a Micromass Quattro LC triple quadrupole mass spectrometer using a custom-built nanospray as previously described (23). Samples were loaded onto a Inertsil C18 precolumn (5 mm × 300 μm i.d., 5 μm, LC Packings) using Perkin Elmer ABI 140D syringe pumps and a Hewlett Packard 1100 Series autosampler. Binary gradient HPLC separation was as described previously (23). Solvent A was water with 0.05% formic acid, and solvent B was acetonitrile with 0.05% formic acid. Loading conditions were 5% solvent B for 15 min at a flow rate of 20 μl/min. During sample loading, the capillary column was equilibrated at 5% solvent B. After 15 min of on-line sample clean-up, a switching valve was used to back flush the analyte onto the Intersil C18 capillary column (15 cm × 75 μm i.d., 5 μm, LC Packings). The sample was eluted using Hewlett Packard 1100 Series binary pumps set at a flow rate of 0.4 ml/min equipped with a pre-column flow splitter having a fixed ratio flow split of 2000:1. Gradient conditions were 5% B to 60% B over 5 min; 60% B for 10 min; 60% B to 90% B over 5 min; 90% B for 5 min; 90% B to 5% B over 10 min; 5% B for 5 min. During sample elution onto the analytical column, the precolumn was washed using the following gradient conditions: 5% B to 90% B over 10 min; 90% B for 10 min; 90% B to 5% B over 10 min; 5% B for 10 min.
Samples were introduced into a Micromass Quattro LC triple quadrupole mass spectrometer using a custom-built nanospray with 20 μm i.d. fused silica tubing inserted through 125 μm i.d. PEEK tubing. All samples were analyzed using the positive ionization mode. Capillary voltages, cone voltages, collision energies, and collision gas (argon) pressures were optimized by monitoring ion intensities while infusing 10 ng/μL of standards in 5% aqueous acetonitrile, 0.05% formic acid at a flow rate of 0.5 μL/min. Electrospray ionization was set at 2.5 kV and cone voltages were 25 V. Argon was used as the collision gas and set at 0.0002 mBar. Collision energies were 50 V for adduct characterization and 25 V for quantitation. Multiple reaction monitoring (MRM) (dwell time 0.5 sec; span 0.4 Daltons) was used to measure the [M+H]+ to [(M-116) + H]+ (loss of deoxyribose) mass transition. The dG-C8-MeIQx adduct was monitored using the transition from m/z 479 to m/z 363 and the deuterated internal standard (dG-C8-MeIQx-D3) was monitored using the transition from m/z 482 to m/z 366.
Results
Human CYP1A1 Expression
The transfection of CYP1A1 was confirmed by measurement of CYP1A1 catalytic activity. The CYP1A1-, CYP1A1/NAT2*5B and CYP1A1/NAT2*4-transfected cell lines catalyzed EROD activity at rates that did not differ significantly (p>0.05) from each other (Fig. 1). CYP1A1 EROD catalytic activity was not detected in the untransfected UV5 cells.
Figure 1.
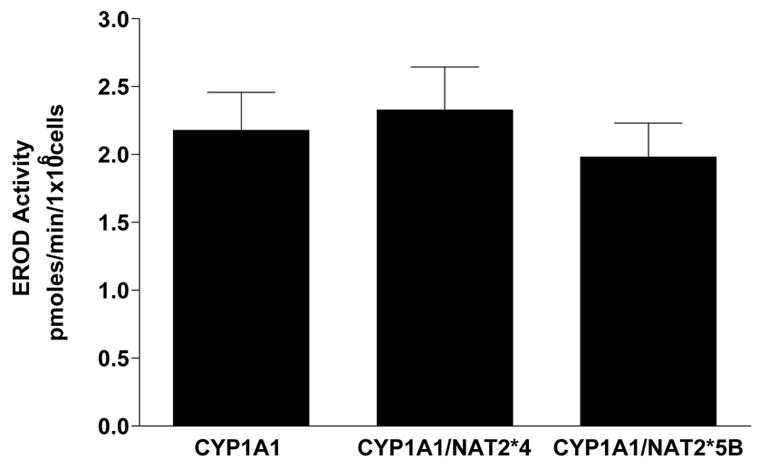
EROD activities in UV5 CHO cell lines. Each bar represents Mean ± S.E.M. for three experiments. EROD activity in UV5 cells was below the level of detection (< 0.2 pmoles/min/million cells). No significant (p>0.05) differences in EROD activity among the CYP1A1-transfected CHO cells were observed.
Human NAT2 Expression
Transfection of NAT2*4 or NAT2*5B to the single FRT site, verified by two separate PCRs, was confirmed by measurement of human NAT2-specific mRNA and catalytic activity. Human NAT2-specific mRNA was not detected in UV5 or UV5/CYP1A1 cells, and did not differ statistically (p>0.05) between UV5/CYP1A1/NAT2*4 and UV5/CYP1A1/NAT2*5B cells (data not shown). SMZ N-acetyltransferase catalytic activity was over 20-fold greater (p=0.0001) in the UV5/CYP1A1/NAT2*4 cell line than the UV5/CYP1A1/NAT2*5B cell line (Fig. 2). SMZ N-acetyltransferase catalytic activity was not detected in the UV5 or UV5/CYP1A1 cell lines (<20 pmole/min/mg). Cell lysates from UV5 and each of the transfected CHO cell lines were tested for their capacity to activate N-OH-MeIQx to form dG-C8-MeIQx adducts. The N-hydroxy-MeIQx OAT activity in UV5/CYP1A1/NAT2*4 cells was about 4.5-fold higher (p=0.0093) than in the UV5/CYP1A1/NAT2*5B cell line (Fig. 2). Low but detectable levels of N-hydroxy-MeIQx activation were detected in the UV5 and the UV5/CYP1A1 cell lines that were subtracted from the experimental measurements in the NAT2-transfected cells.
Figure 2.

SMZ N-acetyltransferase (top panel) and N-hydroxy-MeIQx O-acetyltransferase (bottom) activities in cell lysates of CYP1A1/NAT2*4- and CYP1A1/NAT2*5B-transfected CHO cells. Activity in the UV5 and UV5/CYP1A1 cell lines were below the level of detection for SMZ NAT (< 20 pmol/min/mg). Low but detectable levels of N-hydroxy-MeIQx activation detected in the UV5 and the UV5/CYP1A1 cell lines were subtracted from the experimental measurements in the NAT2-transfected cells. *Both SMZ NAT and N-hydroxy-MeIQx OAT activities were significantly (p < 0.01) higher in CYP1A1/NAT2*4- than CYP1A1/NAT2*5B-transfected CHO cells.
MeIQx-induced Cytotoxicity and Mutagenesis
Only the UV5/CYP1A1/NAT2*4 and UV5/CYP1A1/NAT2*5B cell lines showed concentration-dependent cytotoxicity (Fig 3) following MeIQx treatment. Dose-dependent cytotoxicity from MeIQx was greater in the UV5/CYP1A1/NAT2*4 than in the UV5/CYP1A1/NAT2*5B cell line. Only the UV5/CYP1A1/NAT2*4 cell lines showed dose-dependent MeIQx-induced hprt mutants (Fig 4).
Figure 3.
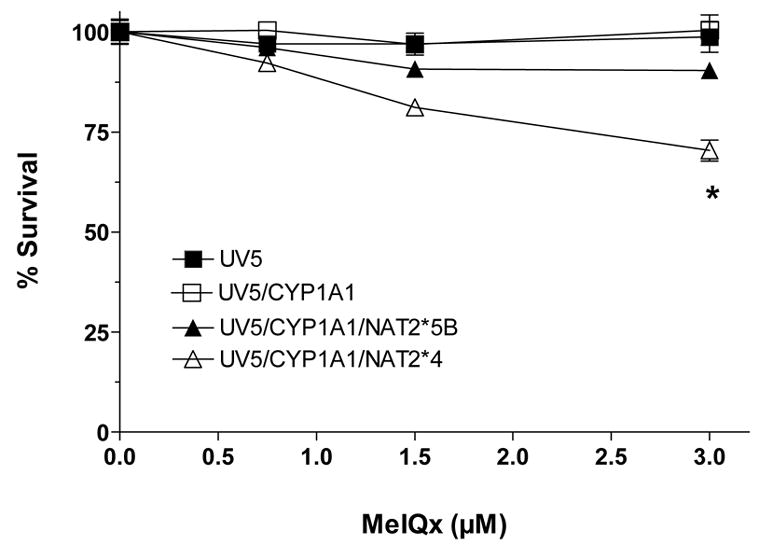
MeIQx-induced cytotoxicity in UV5 CHO cell lines. Percent survival on the ordinate is plotted versus MeIQx treatment concentration on the abscissa. Each data point represents Mean ± S.E.M. for three experiments. MeIQx-induced cytotoxicity was significantly greater (p<0.05) in CYP1A1/NAT2*4- than CYP1A1/NAT2*5B-transfected CHO cells at 3 μM.
Figure 4.
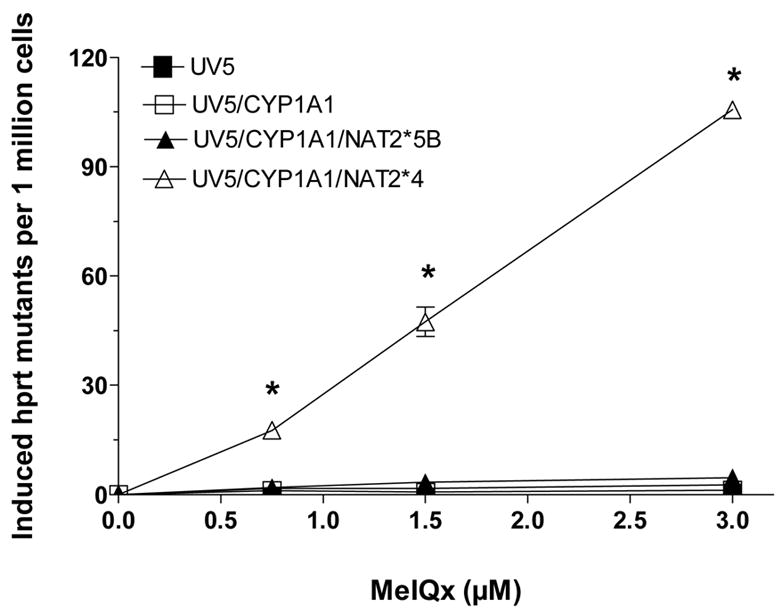
MeIQx-induced hprt mutants in UV5 CHO cell lines. MeIQx-induced hprt mutants are plotted on the ordinate versus MeIQx treatment concentration on the abscissa. Each data point represents Mean ± S.E.M. for three experiments. *MeIQx-induced hprt mutants were concentration dependent only in the CYP1A1/NAT2*4- transfected CHO cells.
Identification and Quantitation of DNA Adducts
The deoxyguanosine (dG)-C8-MeIQx standard was characterized by HPLC-tandem mass spectrometry (LC-MS/MS) and used to verify the identity of DNA adducts formed in vitro. With the low energy MRM mass spectrometry scanning conditions, one principal adduct had a molecular weight of 479, corresponding to dG-C8-MeIQx or 482 corresponding to dG-C8-MeIQx-D3 (Figure 5). Identical patterns of fragmentation were observed for dG-C8-MeIQx and dG-C8-MeIQx-D3, except that the latter were shifted by 3 Da (Figure 5). One adduct (dG-C8-MeIQx) was identified in CHO cells incubated with MeIQx (Figure 6). As shown in Figure 7, dG-C8-MeIQx DNA adduct levels were dose-dependent only in the CYP1A1/NAT2*4 and CYP1A1/NAT2*5B cell lines. MeIQx DNA adduct levels were consistently in the order: CYP1A1 < CYP1A1/NAT2*5B < CYP1A1/NAT2*4, and these differences were significant (p<0.0001) at all MeIQx concentrations. MeIQx DNA adduct levels in the CYP1A1/NAT2*4 cells were substantially and significantly (p < 0.001) higher than in the CYP1A1/NAT2*5B cells at all concentrations. The number of MeIQx-induced hprt mutants correlated very highly (r2 = 0.88) with the number of MeIQx-DNA adducts in each of the cell lines.
Figure 5.
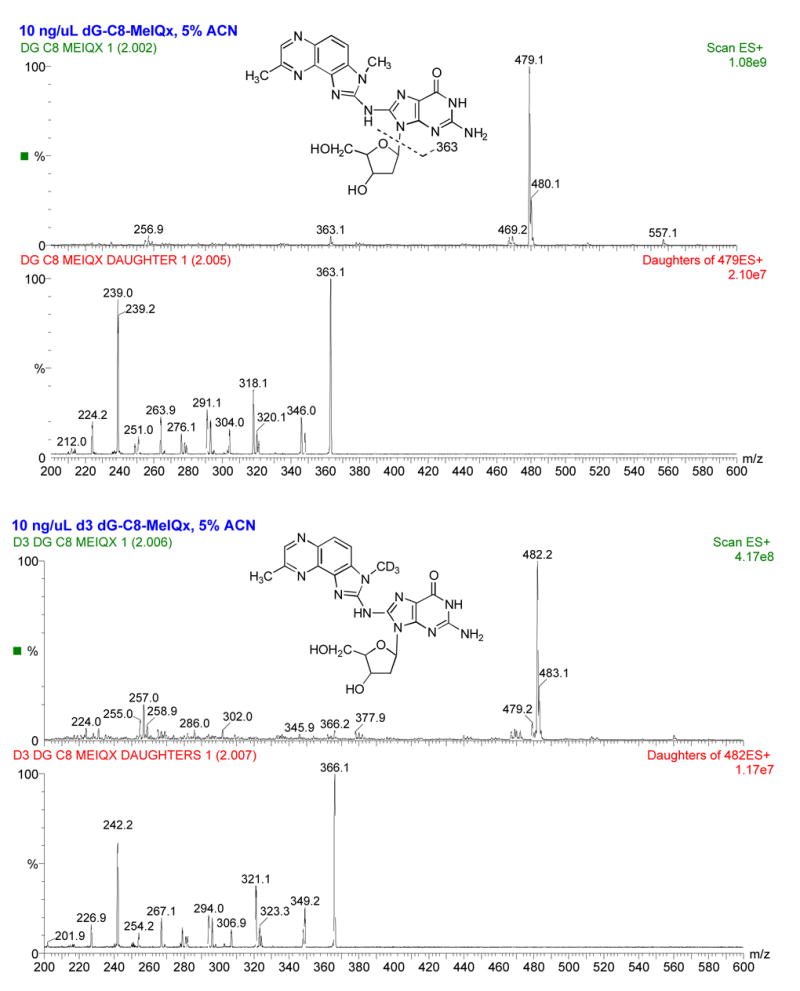
Structure and electrospray ionization spectra of dG-C8-MeIQx (top two panels) and dG-C8-MeIQx-D3 (bottom two panels) showing [M+H]+ = 479 and 482, respectively. Collision induced dissociation fragmentation of dG-C8-MeIQx and d3 dG-C8-MeIQx at −50 V collision energy shows fragmentation of the MeIQx and guanosine moieties dominated by the major fragment produced from loss of deoxyribose: the aglycone ion of m/z 363 or 366 for dG-C8-MeIQx and dG-C8-MeIQx-D3, respectively. The collision energy was −25 V for MRM transitions used during quantitation.
Figure 6.

Chromatograms of representative HPLC-tandem mass spectrometry traces of dG-C8-MeIQx (top panel) and dG-C8-MeIQx-D3 (bottom panel) of DNA from CHO cells treated with MeIQx and spiked with dG-C8-MeIQx-D3. Elution time in minutes is plotted on the abscissae. Multiple reaction monitoring (MRM) was used to measure the [M+H]+ to [(M-116) + H]+ (loss of deoxyribose) mass transition. The dG-C8-MeIQx adduct was monitored using the transition from m/z 479 to m/z 363 (top) and the deuterated internal standard (dG-C8-MeIQx-D3) was monitored using the transition from m/z 482 to m/z 366 (bottom).
Figure 7.

MeIQx-induced dG-C8-MeIQx adduct levels in UV5 CHO cell lines. Adduct levels are plotted on the ordinate versus MeIQx treatment concentration on the abscissa. Each data point represents Mean ± S.E.M. for three experiments (the S.E.M. sometimes falls within the symbol). *dG-C8-MeIQx adducts were significantly higher (p<0.001) in CYP1A1/NAT2*4- than CYP1A1/NAT2*5B-transfected CHO cells at all concentrations tested.
Discussion
Previous studies have suggested that rapid acetylator NAT2 phenotype increases colorectal (27–31), breast (32,33), and lung (34) cancer risk in individuals exposed to heterocyclic amine carcinogens. Based on food frequency questionnaires, increased risk for colorectal cancer in rapid NAT2 acetylators was most significant when stratified specifically for MeIQx intake (29). The biological basis for the hypothesis that rapid NAT2 acetylators are predisposed to MeIQx-induced cancers is greater metabolic activation of the N-hydroxy metabolites via O-acetylation in rapid acetylators. Mechanistic support for this hypothesis derives from previous studies showing 1) greater metabolic activation of heterocyclic amines in mammary epithelial cells derived from rapid acetylators (35), 2) MeIQx caused greater cytotoxicity and mutagenicity in chinese hamster CHL cells transfected with human CYP1A2 and NAT2 (36), and 3) structurally related heterocyclic amine carcinogen 2-amino-3-methylimidazo[4,5-f] quinoline caused greater cytotoxicity and mutagenicity in DNA repair deficient CHO cells transfected with rat CYP1A2 and human NAT2 (37).
Cancers such as colorectal, breast, and lung that are related to heterocyclic amine carcinogen exposures may require metabolic activation within the target organ. In extrahepatic tissues, N-hydroxylation of MeIQx in humans is more likely to be catalyzed by CYP1A1 rather than CYP1A2, and a recent study suggested that combinations of CYP1A1 and NAT2 may increase risk of colorectal cancer from heterocyclic amines (38). Thus, our study focused on the effect of CYP1A1 in combination with rapid or slow acetylator NAT2 on mutagenesis and DNA adduct formation from MeIQx. Whereas the previous studies have shown that transfection of human NAT2 increases MeIQx-induced cytotoxicity and mutagenesis, the focus of our hypothesis was on comparisons between rapid and slow acetylator NAT2 phenotypes.
Our studies strongly suggest that metabolic activation of MeIQx can be catalyzed by CYP1A1, but that mutagenesis and DNA adduct formation requires further metabolic activation by NAT2. Indeed, MeIQx-induced mutagenesis and DNA adduct formation were clearly concentration dependent in the rapid acetylator UV5/CYP1A1/NAT2*4 cell line. MeIQx-induced mutagenesis and DNA adduct formation was negligible in the other cell lines, although DNA adduct formation was concentration-dependent and slightly higher in the slow acetylator UV5/CYP1A1/NAT2*5B cell line than in the UV5 and UV5/CYP1A1 cell lines. Thus, these results confirm that metabolic O-acetylation by NAT2 substantially enhances DNA adduct formation and mutagenesis and that rapid acetylator NAT2 catalyzes this activation to a substantially greater extent than slow acetylator NAT2. The primary DNA adduct formed in our UV5/CYP1A1/NAT2*4 CHO cells was dG-C8-MeIQx, which is the primary adduct that has been identified in human tissues (17, 39). The product ion fragmentation patterns for dG-C8-MeIQx and dG-C8-MeIQx-D3 were remarkably similar to those reported previously (19). The dG-C8-MeIQx adduct quantity correlated very highly (r2 = 0.88) with hprt mutant frequency across all cell lines and MeIQx concentration levels.
These findings provide laboratory support for the epidemiological studies cited above reporting higher frequencies of colorectal, breast, and lung cancers related to heterocyclic amine exposures in human rapid NAT2 acetylators. It should be noted that some N-hydroxy-heterocyclic amine carcinogens such as N-hydroxy-2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine also have been shown to be activated by other phase II enzymes such as sulfotransferases (40), but these enzymes were not the focus of our study and were not transfected into the CHO cells used here. In conclusion, our results strongly support extrahepatic activation of MeIQx by CYP1A1 and a robust effect of human NAT2 genetic polymorphism on MeIQx –induced mutagenesis and DNA adduct levels. The differences in N-hydroxy-MeIQx O-acetyltransferase catalytic activity observed between the CYP1A1/NAT2*4- and CYP1A1/NAT2*5B-transfected CHO cells lines reflected differences in MeIQx-induced DNA adduct formation and mutagenesis. The effect of NAT2 phenotype on MeIQx-induced DNA adduct formation and mutagenesis correlated very highly further suggesting that N-hydroxylation followed by NAT2-catalyzed O-acetylation is an important pathway in the metabolic activation of MeIQx. Although MeIQx activation undoubtedly involves multiple enzymatic pathways (11), our results provide laboratory-based support for epidemiological studies reporting higher frequency of heterocyclic amine-related cancers in rapid NAT2 acetylators.
Acknowledgments
We thank Dr. Robert Turesky for his very kind donation of dG-C8-MeIQx and dG-C8-MeIQx-D3 adduct standards and Dr. Frank Gonzalez for kind donation of plasmid pUV1 containing POR.
Footnotes
Financial Support: United States Public Health Service grant R01-CA34627 from the National Cancer Institute.
References
- 1.Keating GA, Bogen KT. Estimates of heterocyclic amine intake in the US population. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;802:127–33. doi: 10.1016/j.jchromb.2003.10.047. [DOI] [PubMed] [Google Scholar]
- 2.Byrne C, Sinha R, Platz EA, et al. Predictors of dietary heterocyclic amine intake in three prospective cohorts. Cancer Epidemiol Biomarkers Prev. 1998;7:523–9. [PubMed] [Google Scholar]
- 3.National Toxicology Program. Report on Carcinogenesis. 11. U.S. Department of Health and Human Services, Public Health Service, Research; Triangle Park, NC: 2005. [Google Scholar]
- 4.Ohgaki H, Hasegawa H, Suenaga M, Sato S, Takayama S, Sugimura T. Carcinogenicity in mice of a mutagenic compound, 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline (MeIQx) from cooked foods. Carcinogenesis. 1987;8:665–8. doi: 10.1093/carcin/8.5.665. [DOI] [PubMed] [Google Scholar]
- 5.Kato T, Ohgaki H, Hasegawa H, Sato S, Takayama S, Sugimura T. Carcinogenicity in rats of a mutagenic compound, 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline. Carcinogenesis. 1988;9:71–3. doi: 10.1093/carcin/9.1.71. [DOI] [PubMed] [Google Scholar]
- 6.Sinha R, Kulldorff M, Chow WH, Denobile J, Rothman N. Dietary intake of heterocyclic amines, meat-derived mutagenic activity, and risk of colorectal adenomas. Cancer Epidemiol Biomarkers Prev. 2001;10:559–62. [PubMed] [Google Scholar]
- 7.Ishibe N, Sinha R, Hein DW, et al. Genetic polymorphisms in heterocyclic amine metabolism and risk of colorectal adenomas. Pharmacogenetics. 2002;12:145–50. doi: 10.1097/00008571-200203000-00008. [DOI] [PubMed] [Google Scholar]
- 8.Sinha R, Kulldorff M, Swanson CA, Curtin J, Brownson RC, Alavanja MC. Dietary heterocyclic amines and the risk of lung cancer among Missouri women. Cancer Res. 2000;60:3753–6. [PubMed] [Google Scholar]
- 9.De Stefani E, Ronco A, Mendilaharsu M, Guidobono M, Deneo-Pellegrini H. Meat intake, heterocyclic amines, and risk of breast cancer: a case-control study in Uruguay. Cancer Epidemiol Biomarkers Prev. 1997;6:573–81. [PubMed] [Google Scholar]
- 10.Turesky RJ. The role of genetic polymorphisms in metabolism of carcinogenic heterocyclic aromatic amines. Curr Drug Metab. 2004;5:169–80. doi: 10.2174/1389200043489036. [DOI] [PubMed] [Google Scholar]
- 11.Turesky RJ. Formation and biochemistry of carcinogenic heterocyclic aromatic amines in cooked meats. Toxicol Lett. 2007;168:219–27. doi: 10.1016/j.toxlet.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 12.Kim D, Guengerich FP. Cytochrome P450 activation of arylamines and heterocyclic amines. Annu Rev Pharmacol Toxicol. 2005;45:27–49. doi: 10.1146/annurev.pharmtox.45.120403.100010. [DOI] [PubMed] [Google Scholar]
- 13.Josephy PD, Batty SM, Boverhof DR. Recombinant human P450 forms 1A1, 1A2, and 1B1 catalyze the bioactivation of heterocyclic amine mutagens in Escherichia coli lacZ strains. Environ Mol Mutagen. 2001;38:12–8. doi: 10.1002/em.1045. [DOI] [PubMed] [Google Scholar]
- 14.Wild D, Feser W, Michel S, Lord HL, Josephy PD. Metabolic activation of heterocyclic aromatic amines catalyzed by human arylamine N-acetyltransferase isozymes (NAT1 and NAT2) expressed in Salmonella typhimurium. Carcinogenesis. 1995;16:643–8. doi: 10.1093/carcin/16.3.643. [DOI] [PubMed] [Google Scholar]
- 15.Hein DW, Fretland AJ, Doll MA. Effects of single nucleotide polymorphisms in human N-acetyltransferase 2 on metabolic activation (O-acetylation) of heterocyclic amine carcinogens. Int J Cancer. 2006;119:1208–11. doi: 10.1002/ijc.21957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turesky RJ, Rossi SC, Welti DH, Lay JO, Jr, Kadlubar FF. Characterization of DNA adducts formed in vitro by reaction of N-hydroxy-2-amino-3-methylimidazo[4,5-f]quinoline and N-hydroxy-2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline at the C-8 and N2 atoms of guanine. Chem Res Toxicol. 1992;5:479–90. doi: 10.1021/tx00028a005. [DOI] [PubMed] [Google Scholar]
- 17.Turteltaub KW, Mauthe RJ, Dingley KH, et al. MeIQx-DNA adduct formation in rodent and human tissues at low doses. Mutat Res. 1997;376:243–52. doi: 10.1016/s0027-5107(97)00049-3. [DOI] [PubMed] [Google Scholar]
- 18.Schut HA, Snyderwine EG. DNA adducts of heterocyclic amine food mutagens: implications for mutagenesis and carcinogenesis. Carcinogenesis. 1999;20:353–68. doi: 10.1093/carcin/20.3.353. [DOI] [PubMed] [Google Scholar]
- 19.Paehler A, Richoz J, Soglia J, Vouros P, Turesky RJ. Analysis and quantification of DNA adducts of 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline in liver of rats by liquid chromatography/electrospray tandem mass spectrometry. Chem Res Toxicol. 2002;15:551–61. doi: 10.1021/tx010178e. [DOI] [PubMed] [Google Scholar]
- 20.Hein DW. Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutat Res. 2002;506–507:65–77. doi: 10.1016/s0027-5107(02)00153-7. [DOI] [PubMed] [Google Scholar]
- 21.Hein DW. N-acetyltransferase 2 genetic polymorphism: effects of carcinogen and haplotype on urinary bladder cancer risk. Oncogene. 2006;25:1649–58. doi: 10.1038/sj.onc.1209374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hein DW, Doll MA, Fretland AJ, et al. Molecular genetics and epidemiology of the NAT1 and NAT2 acetylation polymorphisms. Cancer Epidemiol Biomarkers Prev. 2000;9:29–42. [PubMed] [Google Scholar]
- 23.Metry KJ, Zhao S, Neale JR, et al. 2-amino-1-methyl-6-phenylimidazo [4,5-b] pyridine-induced DNA adducts and genotoxicity in chinese hamster ovary (CHO) cells expressing human CYP1A2 and rapid or slow acetylator N-acetyltransferase 2. Mol Carcinog. 2007;46:553–63. doi: 10.1002/mc.20302. Epub Feb 12, 2007. [DOI] [PubMed] [Google Scholar]
- 24.States JC, Quan T, Hines RN, Novak RF, Runge-Morris M. Expression of human cytochrome P450 1A1 in DNA repair deficient and proficient human fibroblasts stably transformed with an inducible expression vector. Carcinogenesis. 1993;14:1643–9. doi: 10.1093/carcin/14.8.1643. [DOI] [PubMed] [Google Scholar]
- 25.Leff MA, Epstein PN, Doll MA, et al. Prostate-specific human N-acetyltransferase 2 (NAT2) expression in the mouse. J Pharmacol Exp Ther. 1999;290:182–7. [PubMed] [Google Scholar]
- 26.Hein DW, Doll MA, Nerland DE, Fretland AJ. Tissue distribution of N-acetyltransferase 1 and 2 catalyzing the N-acetylation of 4-aminobiphenyl and O-acetylation of N-hydroxy-4-aminobiphenyl in the congenic rapid and slow acetylator Syrian hamster. Mol Carcinog. 2006;45:230–8. doi: 10.1002/mc.20164. [DOI] [PubMed] [Google Scholar]
- 27.Lang NP, Butler MA, Massengill J, et al. Rapid metabolic phenotypes for acetyltransferase and cytochrome P4501A2 and putative exposure to food-borne heterocyclic amines increase the risk for colorectal cancer or polyps. Cancer Epidemiol Biomarkers Prev. 1994;3:675–82. [PubMed] [Google Scholar]
- 28.Le Marchand L, Hankin JH, Wilkens LR, et al. Combined effects of well-done red meat, smoking, and rapid N-acetyltransferase 2 and CYP1A2 phenotypes in increasing colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2001;10:1259–66. [PubMed] [Google Scholar]
- 29.Le Marchand L, Hankin JH, Pierce LM, et al. Well-done red meat, metabolic phenotypes and colorectal cancer in Hawaii. Mutat Res. 2002;506–507:205–14. doi: 10.1016/s0027-5107(02)00167-7. [DOI] [PubMed] [Google Scholar]
- 30.Lilla C, Verla-Tebit E, Risch A, et al. Effect of NAT1 and NAT2 genetic polymorphisms on colorectal cancer risk associated with exposure to tobacco smoke and meat consumption. Cancer Epidemiol Biomarkers Prev. 2006;15:99–107. doi: 10.1158/1055-9965.EPI-05-0618. [DOI] [PubMed] [Google Scholar]
- 31.Ognjanovic S, Yamamoto J, Maskarinec G, Le Marchand L. NAT2, meat consumption and colorectal cancer incidence: an ecological study among 27 countries. Cancer Causes Control. 2006;17:1175–82. doi: 10.1007/s10552-006-0061-3. [DOI] [PubMed] [Google Scholar]
- 32.Deitz AC, Zheng W, Leff MA, et al. N-Acetyltransferase-2 genetic polymorphism, well-done meat intake, and breast cancer risk among postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2000;9:905–10. [PubMed] [Google Scholar]
- 33.Gallicchio L, McSorley MA, Newschaffer CJ, et al. Flame-broiled food, NAT2 acetylator phenotype, and breast cancer risk among women with benign breast disease. Breast Cancer Res Treat. 2006;99:229–33. doi: 10.1007/s10549-006-9203-2. [DOI] [PubMed] [Google Scholar]
- 34.Chiou HL, Wu MF, Chien WP, et al. NAT2 fast acetylator genotype is associated with an increased risk of lung cancer among never-smoking women in Taiwan. Cancer Lett. 2005;223:93–101. doi: 10.1016/j.canlet.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 35.Stone EM, Williams JA, Grover PL, Gusterson BA, Phillips DH. Interindividual variation in the metabolic activation of heterocyclic amines and their N-hydroxy derivatives in primary cultures of human mammary epithelial cells. Carcinogenesis. 1998;19:873–9. doi: 10.1093/carcin/19.5.873. [DOI] [PubMed] [Google Scholar]
- 36.Yanagawa Y, Sawada M, Deguchi T, Gonzalez FJ, Kamataki T. Stable expression of human CYP1A2 and N-acetyltransferases in Chinese hamster CHL cells: mutagenic activation of 2-amino-3-methylimidazo[4,5-f]quinoline and 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline. Cancer Res. 1994;54:3422–7. [PubMed] [Google Scholar]
- 37.Wu RW, Tucker JD, Sorensen KJ, Thompson LH, Felton JS. Differential effect of acetyltransferase expression on the genotoxicity of heterocyclic amines in CHO cells. Mutat Res. 1997;390:93–103. doi: 10.1016/s0165-1218(97)00005-0. [DOI] [PubMed] [Google Scholar]
- 38.Murtaugh MA, Sweeney C, Ma KN, Caan BJ, Slattery ML. The CYP1A1 genotype may alter the association of meat consumption patterns and preparation with the risk of colorectal cancer in men and women. J Nutr. 2005;135:179–86. doi: 10.1093/jn/135.2.179. [DOI] [PubMed] [Google Scholar]
- 39.Totsuka Y, Fukutome K, Takahashi M, et al. Presence of N2-(deoxyguanosin-8-yl)-2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline (dG-C8-MeIQx) in human tissues. Carcinogenesis. 1996;17:1029–34. doi: 10.1093/carcin/17.5.1029. [DOI] [PubMed] [Google Scholar]
- 40.Wu RW, Panteleakos FN, Kadkhodayan S, Bolton-Grob R, McManus ME, Felton JS. Genetically modified Chinese hamster ovary cells for investigating sulfotransferase-mediated cytotoxicity and mutation by 2-amino-1-methyl-6- phenylimidazo[4,5-b]pyridine. Environ Mol Mutagen. 2000;35:57–65. [PubMed] [Google Scholar]