

Abstract
Pyruvate dehydrogenase kinase 2 (PDHK2) is a unique mitochondrial protein kinase that regulates the activity of the pyruvate dehydrogenase multienzyme complex (PDC). PDHK2 is an integral component of PDC tightly bound to the inner lipoyl-bearing domains (L2) of the dihydrolipoyl transacetylase component (E2) of PDC. This association has been reported to bring about an up to 10-fold increase in kinase activity. Despite the central role played by E2 in the maintenance of PDHK2 functionality in the PDC-bound state, the molecular mechanisms responsible for the recognition of L2 by PDHK2 and for the E2-dependent PDHK2 activation are largely unknown. In this study, we used a combination of molecular modeling and site-directed mutagenesis to identify the amino acid residues essential for the interaction between PDHK2 and L2 and for the activation of PDHK2 by E2. On the basis of the results of site-directed mutagenesis, it appears that a number of PDHK2 residues located in its R domain (P22, L23, F28, F31, F44, L45, and L160) and in the so-called “cross arm” structure (K368, R372, and K391) are critical in determining the strength of the interaction between PDHK2 and L2. The residues of L2 essential for recognition by PDHK2 include L140, K173, I176, E179, and to a lesser extent D164, D172, and A174. Importantly, certain PDHK2 residues forming interfaces with L2, i.e., K17, P22, F31, F44, R372, and K391, are also critical for the maintenance of enhanced PDHK2 activity in the E2-bound state. Finally, evidence that the blood glucose-lowering compound AZD7545 disrupts the interactions between PDHK2 and L2 and thereby inhibits PDHK2 activity is presented.
Oxidative decarboxylation of pyruvate catalyzed by the mitochondrial pyruvate dehydrogenase complex (PDC)1 serves as an important metabolic link that connects glycolysis and the citric acid cycle. It is generally believed that the pyruvate dehydrogenase reaction is the rate-limiting step in the aerobic stage of oxidation of carbohydrate fuels (1). The activity of PDC is regulated through a reversible phosphorylation (inactivation)–dephosphorylation (reactivation) cycle catalyzed by a dedicated pyruvate dehydrogenase kinase (PDHK) and pyruvate dehydrogenase phosphatase (PDP), respectively (2). Under most circumstances, both PDHK and PDP are continuously active, maintaining a certain phosphorylation state or activity state of PDC. Under normal feeding conditions, the activity state of PDC in different tissues varies on average from 20 to 50% (1). Under starvation, the activity state of PDC decreases dramatically and approaches approximately 1–2% of the total activity (1). This is thought to be a mechanism for conservation of carbohydrate fuels for brain and some other tissues. Importantly, changes in the activity state of PDC comparable to those observed under starvation also occur in diabetes. However, in diabetes, this is an unwanted effect because it prevents the disposal of excessive carbohydrates.
PDC is a large multienzyme complex built of 30 copies of pyruvate dehydrogenase (E1, heterotetramer with an α2β2 composition), 60 copies of dihydrolipoyl transacetylase (E2), and six copies of dihydrolipoyl dehydrogenase (E3, α2 dimer) (3). The entire complex is assembled around the 60-mer of E2 with other components being attached to the E2 core. PDHK is an integral part of PDC (two to three PDHK dimers per complex) (4). A growing body of evidence strongly suggests that PDHK is attached to so-called inner lipoyl-bearing domains of E2 (L2) (5–7). Roche et al. (8) proposed that L2 attachment facilitates the movement of the PDHK molecule around the E2 core, thereby providing PDHK with access to numerous copies of the E1 component that serves as a PDHK substrate. Inactivation of E1 occurs as a result of phosphorylation of three serine residues (S264 or site 1, S271 or site 2, and S203 or site 3) located on the α chain of E1 (9). Although all three phosphorylation sites were shown to be inactivating (10, 11), the regulation of PDC in vivo largely correlates with phosphorylation of site 1 (12), which is the site most rapidly modified by PDHK (10, 11). Under starvation and in diabetes, all three sites of E1 become heavily phosphorylated, reflecting an increase in intramitochondrial concentrations of NADH and acetyl-CoA, which promote PDHK activity, as well as an increase in the intramitochondrial concentration of the PDHK protein, which comes about as a result of de novo synthesis (1, 2).
A strong correlation between the PDHK activity and phosphorylation state of PDC in diabetes makes PDHK an attractive drug target. In mammals, there are four biochemically and genetically distinctive forms of PDHK (PDHK1, PDHK2, PDHK3, and PDHK4) (13, 14). Two of these forms, i.e., PDHK2 and PDHK4, have been implicated in misregulation of PDC in diabetes (15–17). PDHK2 is the most common form of PDHK in mammalian tissues (13). It displays brisk responses to all major effectors of PDHK activity, such as pyruvate, thiamine pyrophosphate, CoA, NAD+, acetyl-CoA, and NADH (18). In contrast, PDHK4 appears to be a specialized, inducible enzyme, which is produced in response to high concentrations of circulating fatty acids, glucocorticoids, thyroids, and retinoic acid, and is overexpressed in animal models of diabetes (15, 19–22). The contribution of PDHK2 to the hyperphosphorylation of PDC in diabetes is further supported by recent data presented by Mayers and colleagues (23). The authors have found that a PDHK2-specific inhibitor (AZD7545) significantly elevates PDC activity in muscle of obese, insulin-resistant, Zucker rats and, at the same time, markedly improves the postprandial blood glucose profile (23). Considering that AZD7545 inhibits PDHK2 activity in “a non-ATP competitive manner”, Morrell and co-authors (24) proposed that AZD7545 blocks an interaction of PDHK2 with the E2 component. To date, the molecular mechanism responsible for the recognition of lipoamide as well as of the entire L2 by PDHK2 is largely unknown. To identify the amino acid residues contributing to the recognition of lipoamide, L2, and AZD7545 by PDHK2, we conducted an alanine-scanning mutagenesis of rat isozyme. Here, we report the first data on the mechanism of recognition of lipoamide, L2, and AZD7545 by PDHK2.
EXPERIMENTAL PROCEDURES
Vector Construction and Protein Expression
Construction of the expression vectors for human E1, the human E2–E3BP subcomplex, Escherichia coli lipoyl-protein ligase A, human His6-L2 (amino acids Ser 127–Ile 214), human GST-L2 (amino acids Ser 127–Ile 214), and rat PDHK2 was described elsewhere (10, 18, 25–27). Mutagenesis was conducted on previously described pPDHK2 and pGST-L2 vectors using appropriate oligonucleotide primers (18, 27). Reactions were carried out using the ExSite site-directed mutagenesis kit (Stratagene, La Jolla, CA) essentially as recommended by the manufacturer. The presence of mutations and the fidelity of the rest of the DNAs were confirmed by sequencing (28). General conditions for the expression of E1, the E2–E3BP subcomplex, PDHK2, His6-L2, and GST-L2 were described previously (18, 25, 27). PDHK2 carrying various point mutations was expressed following the established protocol (18). The plasmid directing the synthesis of molecular chaperonins GroEL and GroES was obtained as a generous gift from A. Gatenby at DuPont Central Research and Development (Wilmington, DE). Purification of E1, the E2–E3BP subcomplex, PDHK2, His6-L2, and GST-L2 was described elsewhere (10, 18, 25, 27). The protein composition of each protein preparation was evaluated by SDS–PAGE analysis. Gels were stained with Coomassie R250. All preparations used in this study were more than 90% pure.
Circular Dichroism (CD) Spectroscopy
CD spectra for wild-type and mutant PDHK2 proteins were recorded in quartz cells with a 1 mm light path at 30 °C using a Jasco (Easton, MD) J815 spectrometer. Protein samples (1.0 mg/mL) were prepared in 20 mM potassium phosphate buffer (pH 7.5). Protein concentrations were determined on the basis of the 280 nm absorption. Recordings were made from 260 to 185 nm at 1 nm resolution. CD spectra were obtained by averaging at least three accumulations. Spectra were smoothed and corrected for buffer blanks. The molar ellipticity values were calculated according to the expression [Θ] = (Θ/10) × (112.95/lc), where Θ is the ellipticity in millidegrees, 112.95 is the mean residues molecular weight in grams per mole, l is the path length in centimeters, and c is the protein concentration in grams per liter. The calculations of the secondary structure content of PDHK2 and its variants were carried out using SELCON (29).
PDHK2 Pulldown on GST-L2 Protein
Pulldown experiments were performed as described elsewhere (27). Briefly, 50 μL of a 50% (v/v) slurry of glutathione–Sepharose beads was placed in a Spin-X microcentrifuge filter device with a pore diameter of 0.22 μm (Corning Inc., Corning, NY). Beads were washed three times with buffer A [25 mM Tris-HCl buffer (pH 8.0), 0.1 mM EDTA, 2.5 mM MgCl2, 0.1 M KCl, 5 mM dithiothreitol, 1% (v/v) glycerol, and 0.1 mg/mL BSA]. Equilibrated beads were incubated with an appropriate GST-L2 protein (0.25 mg/mL) in 400 μL of buffer A for 10 min. GST-L2-decorated glutathione–Sepharose beads were washed and incubated with an appropriate PDHK2 mutant protein (0.1 mg/mL) prepared in 400 μL of buffer A for 10 min. After incubation, unbound PDHK2 was removed by centrifugation for 1 min at 6000g followed by three consecutive washes in buffer A. To elute bound proteins, 0.1 mL of buffer A supplemented with 10 mM reduced glutathione was added to beads and beads were incubated for 5 min at room temperature, followed by centrifugation at 6000g for 1 min. Free and bound PDHK2 were analyzed using SDS–PAGE. Gels were stained with Coomassie R250. Stained gels were analyzed by scanning densitometry. Scans were quantified using the UN-SCAN-IT automated digitizing system (Silk Scientific, Inc., Orem, UT).
Isothermal Titration Calorimetry (ITC)
ITC experiments were carried out essentially as described previously (27) using a MicroCal (Northampton, MA) VP-ITC microcalorimeter. All titrations were performed at 30 °C. In a typical L2 binding measurement, the concentration of PDHK2 in the calorimeter cell was 10 μM, and the concentration of L2 in the injection syringe was 250 μM with 240 s spacing between injections (5 μL of L2 per injection). The heat change accompanying the addition of the buffer to PDHK2 and the heat of dilution of the ligands were subtracted from the raw results. The equilibrium association constant of L2 (KA) and the molar heat of binding (ΔH) were obtained by nonlinear least-squares fitting of experimental data using a single-site binding model of the Origin software package (version 7.0) provided with the instrument (OriginLab, Northampton, MA). The affinity of L2 for PDHK2 is given as the dissociation constant (KD = 1/KA). Higher protein concentrations were used in the case of weak binding to achieve an accurate determination of binding parameters. For ITC measurements, GST-L2 proteins were treated essentially as described by Liu and colleagues (30).
Tryptophan Fluorescence Quenching
Measurements of tryptophan fluorescence quenching were carried out essentially as described in ref 31. Briefly, steady state fluorescence spectra of PDHK2 were recorded at 25 °C in 50 mM potassium phosphate buffer (pH 7.5) containing 0.25 mM EDTA and 2.0 mM MgCl2, using a Cary Eclipse fluorescence spectrophotometer (Varian Inc., Palo Alto, CA). Samples were excited at 290 nm (5 nm slit), and emission spectra were recorded from 320 to 420 nm (5 nm slit). The titration experiments were conducted in standard (1 cm × 1 cm) cells containing protein samples in an initial volume of 2.0 mL. The appropriate ligands, i.e., ADP or DCA, were added in 2 or 10 μL increments with constant stirring. Wild-type and mutant PDHK2 proteins were used at concentrations of 0.2–0.5 μM. Titrations with AMP or acetic acid were performed as controls. The concentrations of ADP and AMP were evaluated on the basis of their absorbance coefficient (ε = 1.54 × 104 M−1 cm−1) at 259 nm.
Acetylation of L2’s
The rate of acetylation of unaltered and mutant GST-L2 proteins was determined following the procedure described by Liu and co-authors (32) with minor modifications. Prior to the experiment, highly purified E1 and an appropriate GST-L2 protein were desalted in 50 mM KH2PO4 (pH 7.5) and 0.25 mM EDTA. The E1 solution was supplemented with BSA (7.5 mg/mL) made in the same buffer. The reaction mixtures (final volume of 30 μL) containing 50 mM KH2PO4 (pH 7.5), 0.1 mM EDTA, 1 mM MgCl2, 0.2 mM TPP, 20–50 μM GST-L2, 50 ng of E1, and 1.5 mg/mL BSA were equilibrated at 30 °C for 60 s. After equilibration, reactions were initiated by the addition of [2-14C]pyruvate (25–30 cpm/pmol) to a final concentration of 120 μM. We terminated reactions after 1 or 30 min by quenching 10 μL aliquots onto P81 filter paper discs (Whatman Inc., Florham Park, NJ) presoaked in 1 M acetic acid. Filters were washed five to six times for 30 min in 1 M acetic acid followed by two 5 min consecutive washes in ethanol and ethyl ether. Retained radioactivity was determined by liquid scintillation counting. A negative control (without GST-L2) was used to assess nonspecific incorporation.
Phosphorylation Assay
General conditions for the phosphorylation assay based on incorporation of [32P]phosphate from [γ-32P]ATP into the α chain of the E1 component were described previously (10). Briefly, reaction mixtures were set up at 37 °C in a final volume of 50 μL containing 20 mM Tris-HCl (pH 7.8), 5 mM MgCl2, 50 mM KCl, 5 mM DTT, 2.0% (v/v) ethylene glycol, 0.5 mg/mL E1, 20 μg/mL kinase, and 0.1 mM [γ-32P]ATP (specific radioactivity of 100–200 cpm/pmol). In some experiments, reaction mixtures were supplemented with the E2–E3BP complex used at final concentration of 0.5 mg/mL. Phosphorylation reactions were initiated by the addition of ATP after equilibration at 37 °C for 60 s. ATP was added in a volume of 1/10 of the total reaction volume. After incubation for 1 min, 40 μL aliquots were quenched on Whatman 3MM filters presoaked in a solution of 20% (w/v) trichloroacetic acid, 50 mM sodium pyrophosphate, and 50 mM ATP. After extensive washing, the protein-bound radioactivity was determined by liquid scintillation counting. A negative control (without PDHK2) was used to assess nonspecific incorporation. All assays were conducted in triplicate.
Other Procedures
SDS–PAGE was carried out according to the method of Laemmli (33). Protein concentrations were determined according to the method of Lowry (34) with bovine serum albumin as a standard. The extent of L2 lipoylation was examined following the procedure described by Quinn and colleagues (35); the lipoate content of all L2 constructs was greater than 90%.
RESULTS AND DISCUSSION
Characterization of PDHK2 Proteins with Point Mutations in the Putative L2-Binding Site
Despite the central role played by L2 in the maintenance of PDHK2 functionality in the PDC-bound state, the molecular mechanism responsible for the recognition of L2 by PDHK2 is largely unknown. Recently, Kato and colleagues (36) reported the crystal structure of an enzyme related to PDHK2, PDHK3, in a complex with L2. According to this structure, the centerpiece of the L2-binding site on PDHK3 is a lipoyl-binding cavity located on the edge of the so-called B domain (36) [R domain in PDHK2 (37)]. The inner lining of this cavity is made of many conserved hydrophobic residues. The second prominent interface is built of the amino acids furnished by the amino terminus of the B domain, as well as the amino acids of so-called “cross arm” (37), which are almost immediately adjacent to the kinase or K domain of PDHK3 (36). Finally, the last prominent interface is formed by the amino acids of the far carboxyl terminus of the cross arm coming from the neighboring protomer of the PDHK3 dimer (36). Considering that PDHK2 and PDHK3 are structurally related (36–38), it is likely that PDHK2 binds L2 in a similar fashion. To identify the amino acid residues of PDHK2 that might form an interface with L2, the model of the PDHK2–L2 complex was built on the basis of the published structure of the PDHK3–L2 complex [PDB entry 1y8n (36)] using the homology modeling program Swiss-Model (39). As illustrated in Figure 1, this model predicted that the side chains of L23, F28, F31, F44, L45, L160, I167, and F168 of PDHK2 may form the lining of a putative lipoyl-binding cavity (Figure 1B). Amino acids K17, P22, Q47, K368, and R372 may contribute to the second interface, and amino acids E389 and K391 may be a part of the interface between the carboxyl end of the cross arm and the tip of L2 (Figure 1A).
Figure 1.

Recognition of L2 by PDHK2. (A) Space filling representation of the PDHK2 dimer viewed down the lipoyl-bearing domain-binding site. The model of the PDHK2–L2 complex was based on the published structure of the PDHK3–L2 complex [PDB entry 1y8n (36)] using the homology modeling program Swiss-Model (39). PDHK2 subunit I is colored yellow. PDHK2 subunit II is colored green. Amino acid residues of PDHK2 located within contact distance of L2 in the PDHK2–L2 complex are highlighted: basic residues are colored blue, acidic residues red, and neutral residues sand. (B) Space filling representation of the R domain of PDHK2 subunit II viewed down the lipoate-binding cavity. Lipoate residues (LPA) are shown as a stick model. Hydrophobic residues lining up the lipoate-binding cavity are the color of sand. Residues L160, I167, and F168 located inside of the lipoate-binding cavity are not shown. (C) Space filling representation of L2 based on 1y8n coordinates (36). Amino acid residues of L2 located within contact distance of PDHK2 in the PDHK2–L2 complex are highlighted: basic residues are colored blue, acidic residues red, and neutral residues sand. The graphics were generated using PyMOL, version 0.98.
To investigate the functional significance of the amino acid residues identified through the modeling experiments, we carried out alanine-scanning mutagenesis (Table 1). Mutagenized cDNAs were expressed in bacteria. All of them directed an abundant synthesis of corresponding protein products. However, cellular fractionation revealed that two PDHK2 proteins (PDHK2-L23A and PDHK2-F28A) were present almost exclusively in inclusion bodies, indicating the existence of potential folding and/or stability problems. Nevertheless, we were successful in isolating small quantities of these kinases, which were sufficient for the initial characterization. The rest of the mutant PDHK2 proteins were isolated in great quantities using metal affinity chromatography (18).
Table 1.
Properties of PDHK2 Proteins Carrying Point Mutations within a Putative Lipoyl-Bearing Domain-Binding Site
| PDHK2 | dissociation constant for ADP (μM)a | dissociation constant for DCA (μM)a | E1 kinase activity (nmol min−1 mg−1)b | E2-dependent activation (x-fold)b |
|---|---|---|---|---|
| wild-type | 7.5 ± 0.8 | 160 ± 19 | 31 ± 6 | 4.3 ± 0.4 |
| K17A | 8.6 ± 0.4 | 190 ± 35 | 33 ± 1 | 1.7 ± 0.5 |
| P22A | 6.0 ± 0.2 | 190 ± 18 | 26 ± 4 | 2.4 ± 0.5 |
| L23A | 7.4 ± 1.2 | 220 ± 25 | 4.8 ± 0.7 | 12 ± 5 |
| F28A | 11 ± 2 | not applicable | 2.5 ± 1.2 | 10 ± 1 |
| F31A | 5.2 ± 0.3 | 260 ± 13 | 36 ± 6 | 1.8 ± 0.2 |
| F44A | 9.7 ± 0.4 | 210 ± 15 | 20 ± 3 | 1.7 ± 0.1 |
| L45A | 6.8 ± 0.8 | 160 ± 16 | 16 ± 2 | 4.3 ± 0.9 |
| Q47A | 5.9 ± 0.2 | 190 ± 29 | 41 ± 7 | 3.3 ± 0.3 |
| L160A | 2.2 ± 0.3 | 250 ± 17 | 11 ± 1 | 4.2 ± 0.3 |
| I167A | 7.7 ± 0.3 | 110 ± 10 | 15 ± 4 | 5.7 ± 0.4 |
| F168A | 3.4 ± 0.1 | 220 ± 16 | 16 ± 2 | 4.2 ± 0.2 |
| K368A | 5.1 ± 0.7 | 160 ± 10 | 24 ± 4 | 4.1 ± 0.8 |
| R372A | 4.1 ± 0.4 | 170 ± 23 | 35 ± 3 | 2.0 ± 0.1 |
| E389A | 5.5 ± 0.7 | 190 ± 18 | 25 ± 3 | 4.0 ± 0.9 |
| K391A | 8.8 ± 0.8 | 150 ± 17 | 27 ± 6 | 2.8 ± 0.4 |
Determined by intrinsic tryptophan fluorescence quenching as described in Experimental Procedures. Intrinsic tryptophan fluorescence quenching data were processed as described by Hiromasa and colleagues (40). Binding constants represent means ± the standard deviation for three to five independent determinations.
Determined on the basis of the incorporation of [32P]phosphate into E1 during 1 min of the reaction as described previously (18). Data are expressed as means ± the standard deviation for six to nine independent determinations.
To identify any significant folding problems, wild-type and mutant PDHK2 proteins were characterized using CD spectroscopy. The far-UV spectra of all PDHK2 proteins exhibited a characteristic negative band with double minima at 208 and 222 nm and a large positive peak at approximately 195 nm (data not shown), which is typical of proteins containing a large proportion of α-helical secondary structure (40). Importantly, there were no appreciable changes in the CD spectra of different mutant PDHK2 proteins, suggesting that mutations did not cause gross changes in PDHK2 structure.
To investigate this issue further, wild-type and mutant PDHK2 proteins were analyzed for their ability to bind the nucleotide and dichloroacetate (DCA). On the basis of the structural studies, PDHK2 consists of two domains almost equal in length: the amino-terminal R domain assembled as a four-helix bundle and the carboxy-terminal K domain folded as a mixed α/β sandwich (37, 38). The DCA-binding site is located in the middle of the R domain (37), while the nucleotide-binding site is positioned in the middle of the K domain (37, 38). The nucleotide and DCA form multiple contacts with the amino acids of R and K domains, making DCA and ADP binding assays extremely valuable tools for the assessment of the overall integrity of these domains. In addition, nucleotide binding is also a powerful tool for the assessment of the integrity of communications between the K and R domains, because binding of ADP to the K domain of one protomer causes fluorescence quenching of W383 (31), which is buried deeply in the R domain of the neighboring protomer (36, 37). As summarized in Table 1, with the exception of PDHK2-F28A, it was found that all PDHK2 proteins were capable of ADP and DCA binding, suggesting that their R and K domains were properly folded, which would be expected considering that the majority of side chains targeted for mutagenesis are solvent-exposed. PDHK2-F28A was the only protein that appeared to be defective in DCA binding, which might be an indication of a partial unfolding in its R domain. In contrast, PDHK2-L23A behaved reasonably well in both DCA and ADP binding assays despite the fact that PDHK2-L23A was clearly less stable in solution than other PDHK2 variants.
The ability of wild-type and mutant PDHK2 proteins to catalyze the phosphorylation reaction was assessed using the standard kinase assay with free E1 as a substrate (10). All PDHK2 variants were found to be catalytically active (Table 1). Most of the mutant kinases phosphorylated E1 at a rate of 30–100% of that of the wild-type enzyme. The overall rate of the E1 kinase reaction observed in these experiments was typical of the values reported previously for the rat PDHK2 isozyme (18). Two PDHK2 variants, i.e., PDHK2-L23A and PDHK2-F28A, displayed significantly reduced E1 kinase activities. As discussed above, this could be due in part to potential folding problems, lower stability, or both. Usually, the addition of E2 to the phosphorylation cocktail is associated with a significant increase in PDHK2 activity (26, 27). This phenomenon was rationalized in terms of (1) colocalization and mutual orientation of E1 and PDHK2 caused by their binding to E2, (2) E2-facilitated access of PDHK2 to the multiple copies of E1 attached to the E2 core, and (3) E2-dependent activation of PDHK2 (8). Of 15 PDHK2 variants analyzed in this study, six variants exhibited a significantly weakened response to the addition of E2 (Table 1). Two of those variants (PDHK2-F31A and PDHK2-F44A) carried amino acid substitutions in the lining of the putative lipoyl-binding cavity. Three other variants (PDHK2-K17A, PDHK2-P22A, and PDHK2-R372A) carried the substitutions of amino acids contributing to the second interface between PDHK2 and L2, while the last one (PDHK2-K391A) had a mutation in the third interface. Taken together, these results are consistent with the interpretation that PDHK2 residues K17, P22, F31, F44, R372, and K391 play an important role in the recognition of L2, in the maintenance of the PDHK2-enhanced activity in the E2-bound state, or in both. In marked contrast, PDHK2-L23A and PDHK2-F28A proteins displayed a great increase in the level of E2-dependent activation (Table 1). On the basis of this observation alone, it would be attractive to propose that the side chains of PDHK2 L23 and F28 amino acid residues work as negative modulators of PDHK2 activity in the complex-bound form. However, this interpretation should be taken with caution considering that these variants were significantly less stable than wild-type PDHK2, and therefore, a greater response to the addition of E2 observed with PDHK2-L23A and PDHK2-F28A could be explained at least in part by the E2-dependent stabilization of PDHK2-L23A and PDHK2-F28A rather than by the E2-dependent activation.
Binding of Wild-Type and Mutant PDHK2 Proteins to L2
Several lines of evidence strongly suggest that L2’s serve as principal docking sites for the PDHK2 molecule on the surface of the E2 core (5–7). Thus, it would be expected that at least some of the side chains identified above as essential for the E2-dependent activation of PDHK2 are also contributing to the binding of L2. To examine this idea, we employed a GST-L2-based pulldown assay that was described previously (27). In this assay, GST-L2 constructs adsorbed on glutathione–Sepharose are used as bait for PDHK2 molecules to isolate PDHK2–L2 complexes. In GST-L2 pulldown assay, five PDHK2 variants (PDHK2-L23A, PDHK2-F28A, PDHK2-F31A, PDHK2-F44A, and PDHK2-R372A) exhibited a greatly reduced ability to bind L2 (Figure 2A and Table 2). The other six kinases (PDHK2-P22A, PDHK2-L45A, PDHK2-L160A, PDHK2-F168A, PDHK2-K368A, and PDHK2-K391A) exhibited a significant reduction in the level of L2 binding. Finally, PDHK2-K17A, PDHK2-Q47A, PDHK2-I167A, and PDHK2-E389A bound GST-L2 in a manner essentially similar to that of the wild-type protein (Figure 2A and Table 2).
Figure 2.
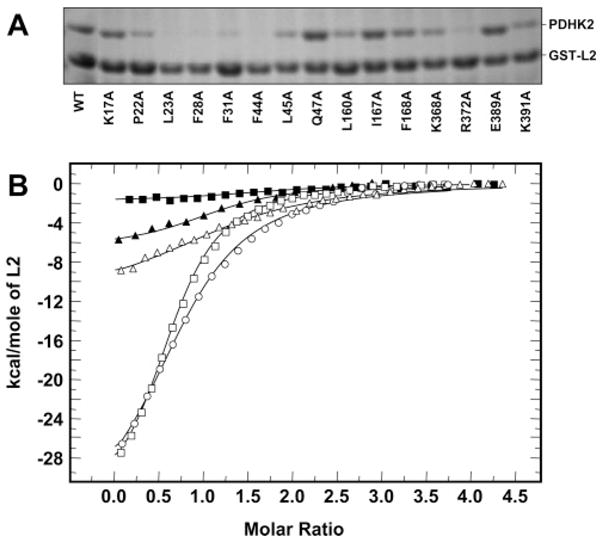
Binding of wild-type and mutant PDHK2 proteins to the unaltered L2. (A) SDS–PAGE analysis of wild-type and mutant PDHK2 proteins pulled down using an unaltered GST-L2 construct. Pulldown experiments were carried out as described previously (27). Gels were stained with Coomassie R250. Shown are representative data from four experiments. (B) Binding isotherms of L2 and PDHK2. ITC measurements were performed at 30 °C in a VP-ITC microcalorimeter (MicroCal). Unaltered L2 (250 μM) in the syringe was injected into the reaction cell containing 10 μM wild-type or mutant PDHK2. The molar ratio represents L2 monomer to PDHK2 dimer. Solid lines depict the least-square fitting curves obtained using a single-site L2 binding model. Fittings were made using Origin, version 7.0. Data for binding of wild-type PDHK2 are shown as empty circles. Data for binding of PDHK2-K17A, PDHK2-Q47A, PDHK2-R372A, and PDHK2-K391A proteins are shown as empty triangles, empty squares, filled squares, and filled triangles, respectively.
Table 2.
Binding of Wild-Type and Mutant PDHK2 Proteins to the Unaltered L2 Domain
| GST-L2 pulldown
|
ITC with L2
|
||
|---|---|---|---|
| PDHK2 | PDHK2 binding (%)a | KD (μM) | ΔH (kcal/mol) |
| wild-type | 100 ± 9b | 8.3 ± 0.3c | −46 ± 1 |
| K17A | 110 ± 20 | 26 ± 2 | −12 ± 1 |
| P22A | 45 ± 14 | NMd | – |
| L23A | 10 ± 6 | NDe | – |
| F28A | 10 ± 5 | NDe | – |
| F31A | 17 ± 6 | NMd | – |
| F44A | 7 ± 1 | NMd | – |
| L45A | 47 ± 11 | NMd | – |
| Q47A | 130 ± 30 | 3.0 ± 0.1 | −34 ± 1 |
| L160A | 48 ± 4 | NMd | – |
| I167A | 110 ± 5 | 13 ± 1 | −24 ± 1 |
| F168A | 65 ± 8 | NMd | – |
| K368A | 52 ± 14 | 64 ± 6f | −12 ± 1 |
| R372A | 9 ± 3 | NMd | – |
| E389A | 100 ± 7 | 7.8 ± 0.3 | −24 ± 1 |
| K391A | 62 ± 3 | 78 ± 3f | −4.2 ± 0.1 |
As discussed in Experimental Procedures, fractions containing free and GST-L2-bound PDHK2 variants were separated via SDS–PAGE. Gels were stained with Coomassie R250. Stained gels were analyzed by scanning densitometry. Scans were quantified using an UN-SCAN-IT automated digitizing system (Silk Scientific, Inc.). Data represent the ratio between GST-L2 protein and corresponding PDHK2 protein. The ratio of wild-type PDHK2 to GST-L2 was taken to be 100%.
Results are expressed as means ± the standard deviation of at least three experiments.
Results are expressed as means ± the standard deviation of three experiments conducted with different preparations of proteins. ITC measurements were performed at 30 °C in a VP-ITC microcalorimeter (MicroCal). Unaltered L2 domain (250 μM) in the syringe was injected into the reaction cell containing 10 μM wild-type or mutant PDHK2. Dissociation constants and enthalpy changes were obtained using Origin, version 7.0, supplied by manufacturer.
Not measurable due to the insufficient amount of differential heat produced in the binding reaction.
Not done due to the limited yields of PDHK2-L23A and PDHK2-F28A proteins.
To achieve an accurate determination of binding parameters, measurements were conducted using 20 μM PDHK2-K368A or PDHK2-K391A and 400 μM L2 in the syringe.
To further investigate the contributions of the 15 PDHK2 residues mentioned above to the interaction with L2, we performed isothermal titration calorimetry studies. In these experiments, the wild-type and mutant PDHK2 proteins were titrated with monomeric L2 essentially as described previously (27). The representative titrations for wild-type PDHK2, PDHK2-K17A, PDHK2-Q47A, PDHK2-R372A, and PDHK2-K391A are shown in Figure 2B. Table 2 summarizes the enthalpy changes and the binding constants obtained by deconvolution of the total binding isotherms. In agreement with the previously reported data (27), wild-type PDHK2 bound L2 with a dissociation constant of approximately 8.3 μM. PDHK2-Q47A displayed a somewhat greater affinity for L2 (dissociation constant of approximately 3.0 μM), while PDHK2-E389A bound L2 in a manner similar to that of the wild-type kinase (dissociation constant of approximately 7.8 μM). Titrations employing PDHK2-I167A, PDHK2-K17A, PDHK2-K368A, and PDHK2-K391A proteins yielded higher dissociation constants of approximately 13.5, 26.3, 64.1, and 77.5 μM, respectively (Table 2). Binding of L2 to PDHK2-P22A, PDHK2-F31A, PDHK2-F44A, PDHK2-L45A, PDHK2-L160A, PDHK2-K168A, and PDHK2-R372A gave a very weak heat signal, which, in conjunction with GST-L2 pulldown experiments, suggested that the binding affinities of these PDHK2 variants for L2 were very weak. Low heat production along with limitations in PDHK2 solubility (27) prevented us from more accurate determination of the thermodynamic parameters of these proteins. On the basis of the sensitivity of ITC, it seems reasonable to propose that the affinity of PDHK2-P22A, PDHK2-F31A, PDHK2-F44A, PDHK2-L45A, PDHK2-L160A, PDHK2-K168A, and PDHK2-R372A proteins for L2 decreased at least 10-fold. Thus, despite the somewhat different nature of the ligand employed in ITC and in pulldown experiments (41), both approaches demonstrated that a number of PDHK2 amino acid residues, i.e., K17, P22, F31, F44, L45, L160, F168, K368, R372, and K391, are essential for L2 binding.
To better appreciate the significance of ITC and GST-L2 pulldown experiments, these data have to be considered in relation to the outcome of E2-dependent PDHK2 activation assay. When the results of the latter assay are taken into consideration, PDHK2 variants can be divided into three major categories: those that display a reduction in the level of L2 binding without an appreciable change or even an increase in their level of activation by E2 (PDHK2-L23A, PDHK2-F28A, PDHK2-L45A, PDHK2-L160A, and PDHK2-K368A); those that show a correlation between L2 binding and E2-dependent activation (PDHK2-P22A, PDHK2-F31A, PDHK2-F44A, PDHK2-R372A, and PDHK2-K391A); and, finally, one variant (PDHK2-K17A) that binds L2 only slightly worse than wild-type PDHK2 but has a level of activation significantly reduced by the E2 component. The behavior of kinases in the first group appears to be almost identical to the behavior of PDHK2 carrying certain very short truncations at the carboxyl terminus (27). The truncated kinases also showed a great decrease in the level of L2 binding without an appreciable decrease in the level of E2-dependent activation or E2 binding (27). This behavior can be understood in the context of the entire complex. On the surface of the E2 core, PDHK2 is exposed to a very high local concentration of lipoyl domains that well exceeds 1 mM (8). The affinity of PDHK2 for L2 is approximately 8.3 μM. Thus, even if a mutation decreases the affinity of PDHK2 for L2 by 1 order of magnitude, which can be readily detected by a pulldown assay or ITC, this kinase will still be able to show an E2-dependent activation after its integration into the structure of the multienzyme complex. On the basis of this consideration, it seems reasonable to propose that the side chains of L23, F28, L45, L160, and K368 contribute to the binding of L2. Their alteration by site-directed mutagenesis causes a significant decrease in the affinity of PDHK2 for L2. However, this decrease in the level of L2 binding is not accompanied by a decrease in the level of E2-dependent activation, because it is not great enough to prevent the genetically altered PDHK2 from being integrated into the complex. In this respect, PDHK2-L23A and PDHK2-F28A variants appear to be mechanistically similar to PDHK2-L45A, PDHK2-L160A, and PDHK2-K368A. However, their responses to the addition of E2 are amplified because of their instability in the free form in solution. In marked contrast, the outcome of experiments with PDHK2-P22A, PDHK2-F31A, PDHK2-F44A, PDHK2-R372A, and PDHK2-K391A cannot be explained by the reduction in their ability to bind L2, because these kinases display an altered response to E2 as well. This suggests that, in addition to their participation in L2 binding, the side chains of P22, F31, F44, R372, and K391 also serve as positive modulators of PDHK2 activity in the PDC-bound state. This idea is further exemplified by the behavior of the PDHK2-K17A variant, which shows that K17 has no great importance for L2 binding but plays a significant role in E2-dependent stimulation of PDHK2 activity. Taken together, these data strongly suggest that certain amino acid residues located in the L2-binding pocket of PDHK2 are critical for the maintenance of the enhanced kinase activity in a complex-bound state rather than for L2 binding per se.
Binding of L2 Mutants to Wild-Type PDHK2
On the basis of the modeling experiments, the aliphatic part of the PDHK2 K17 side chain is stacked against the side chains of L140 and I176 of L2, with the ε-amino group exposed to the highly acidic cluster made of the side chains of E162, D164, E179, E182, and E183 of L2 (Figure 1A,C). The side chains of PDHK2 L23, F28, F31, F44, L45, and L160 form an interface with the lipoamide (Figure 1B). PDHK2 K368 and R372 are located in the vicinity of the acidic cluster formed by E162, D164, E179, E182, and E183 of L2 (Figure 1A). Finally, PDHK2 E389 and K391 are located close to the tip of L2 which carries the lipoylation site, i.e., L2 K173, and, therefore, may participate in the electrostatic interactions with L2 R196 and L2 D172 as proposed by Kato and colleagues (36) for PDHK3 (Figure 1A,C).
To better understand the roles of L2 amino acids in PDHK2 binding, we subjected L2 to alanine-scanning mutagenesis (aliphatic A174 had been converted into the hydrophilic seryl residue). Genetically altered L2’s were expressed in bacteria as fusions with GST and purified on glutathione–Sepharose following the standard protocol (27). Their lipoylation state was evaluated after thrombin cleavage using a gel-shift assay (35). On the basis of this assay, all but L2-K173A variants were lipoylated in a manner similar to that of unaltered L2 (data not shown).
As shown in Figure 3A and Table 3, when subjected to a GST-L2 pulldown assay with wild-type PDHK2 as a catch, four variants displayed either no appreciable PDHK2 binding or a greatly reduced level of PDHK2 binding, i.e., L2-L140A, L2-K173A, L2-I176A, and L2-E179A. Three other variants (L2-D164A, L2-D172A, and L2-A174S) showed a small but statistically significant reduction in the level of PDHK2 binding. Finally, L2-E162A, L2-E182A, L2-E183A, and L2-R196A mutants bound PDHK2 in a manner similar to that of unaltered L2.
Figure 3.
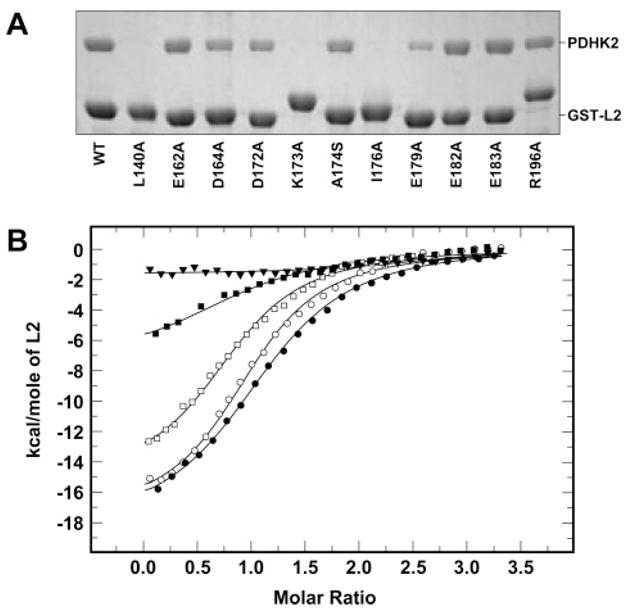
Binding of unaltered and mutant L2 proteins to wild-type PDHK2. (A) SDS–PAGE analysis of wild-type PDHK2 pulled down using unaltered and mutant GST-L2 proteins. Pulldown experiments were carried out as described previously (27). Gels were stained with Coomassie R250. Shown are representative data of five experiments. (B) Binding isotherms of L2 and PDHK2. ITC measurements were performed at 30 °C in a VP-ITC microcalorimeter (MicroCal). Unaltered or mutant L2 (250 μM) in the syringe was injected into the reaction cell containing 10 μM wild-type PDHK2. The molar ratio represents L2 monomer to PDHK2 dimer. Solid lines depict the least-square fitting curves obtained using a single-site L2 binding model. Fittings were made using Origin, version 7.0. Data for binding of unaltered L2 are shown as empty circles. Data for binding of L2-E162A, L2-D164A, L2-K173A, and L2-E183A proteins are shown as empty squares, filled squares, filled triangles, and filled circles, respectively.
Table 3.
Binding of Unaltered and Mutant L2 Proteins to Wild-Type PDHK2
| GST-L2 pulldown
|
ITC with L2
|
||
|---|---|---|---|
| L2 | PDHK2 binding (%)a | KD (μM) | ΔH (kcal/mol) |
| unaltered | 100 ± 5b | 8.6 ± 0.4d | −27 ± 1 |
| L140A | NMc | NMc | – |
| E162A | 93 ± 4 | 7.8 ± 0.1 | −19 ± 1 |
| D164A | 55 ± 3 | 44 ± 12e | −8.6 ± 0.9 |
| D172A | 66 ± 3 | 21 ± 3 | −11 ± 1 |
| K173A | NMc | NMc | – |
| A174S | 69 ± 4 | NDf | – |
| I176A | NMc | NMc | – |
| E179A | 28 ± 2 | NDf | – |
| E182A | 100 ± 5 | 6.8 ± 0.9 | −27 ± 1 |
| E183A | 98 ± 5 | 4.3 ± 0.2 | −19 ± 1 |
| R196A | 79 ± 3 | 13 ± 1 | −21 ± 1 |
As discussed in Experimental Procedures, fractions containing free and GST-L2-bound PDHK2 were separated via SDS–PAGE. Gels were stained with Coomassie R250. Stained gels were analyzed by scanning densitometry. Scans were quantified using an UN-SCAN-IT automated digitizing system (Silk Scientific, Inc.). Data represent the ratio between GST-L2 protein and PDHK2 protein. The ratio of PDHK2 to the unaltered GST-L2 construct was taken to be 100%.
Results are expressed as means ± the standard deviation of at least three experiments.
Not measurable.
Results are expressed as means ± the standard deviation of three experiments conducted with different preparations of proteins. ITC measurements were performed at 30 °C in a VP-ITC microcalorimeter (MicroCal). Unaltered or mutant L2 protein (250 μM) in the syringe was injected into the reaction cell containing 10 μM wild-type PDHK2. Dissociation constants and enthalpy changes were obtained using Origin, version 7.0, supplied by manufacturer.
To achieve an accurate determination of binding parameters, measurements were conducted using 20 μM PDHK2 and 400 μM L2-D164A in the syringe.
Not done due to the limited yields of L2-A174S and L2-E179A variants when prepared following the procedure described by Liu et al. (30).
To determine the contributions of the side chains listed above to the binding affinity of L2, thermodynamics of the interaction of wild-type PDHK2 with unaltered and mutant L2s were characterized using ITC (Figure 3B and Table 3). For these experiments, the monomeric L2’s were released from the corresponding GST-L2 constructs by the thrombin cleavage essentially as described by Liu and co-authors (30). In general, we found that binding of L2 made from GST fusion proteins tended to generate less molecular heat than that of monomeric L2 used in the experiments described in Figure 2B and Table 2. This was due presumably to the difference in the length of the amino-terminal spacer produced by the thrombin cleavage of GST-L2 constructs. On the other hand, the binding constants for both types of L2’s were remarkably similar, 8.3 and 8.6 μM (Tables 2 and 3, respectively). Overall, the analysis of the data presented in Table 3 shows that the effect of E162A, E182A, E183A, and R196A mutations on binding of L2 to the wild-type PDHK2 was rather minor with binding constants ranging form 4.4 to 13.5 μM. The substitutions of either D164 or D172 had a greater impact on the interaction of L2 with PDHK2. The extent of binding of the D164A mutant decreased by approximately 2.5-fold, while the extent of binding of the D172A mutant decreased by approximately 5-fold. In contrast, L2-L140A, L2-K173A, and L2-I176A proteins failed to generate molecular heat sufficient for accurate measurements, which in conjunction with the pulldown experiment indicates that binding of L2-L140A, L2-K173A, and L2-I176A proteins was so weak that it was beyond the range of detection by the assay. Finally, the yields of L2-A174A and L2-E179A proteins generated by thrombin cleavage were too prohibitively low to be used in ITC experiments.
Thus, in agreement with the earlier studies (8, 26), both types of assays strongly suggest that lipoamide itself is the major factor determining the strength of binding of PDHK2 to L2 as evidenced by the results with the lipoylation-deficient L2-K173A variant. Previously, L140 and E179 of L2 were shown to be critical for PDHK3 binding (8, 42). Our data demonstrate that these residues are also important for PDHK2 binding. In addition, we found that another branched chain amino acid residue of L2 juxtapositioned to L140, i.e., I176, is critical for PDHK2 binding. It is currently unknown whether I176 is essential for PDHK3 binding. Interestingly, both L2-L140A and L2-I176A variants displayed greatly reduced activities in the E1-mediated acetylation assay (data not shown), suggesting that E1 and PDHK2 might recognize similar binding determinants of L2. On the basis of our modeling experiments, the side chains of L140 and I176 of L2 are stacked against the aliphatic part of the K17 side chain of PDHK2 and, therefore, create a hydrophobic patch around K17, which might be critical for the maintenance of enhanced PDHK2 activity in the E2-bound state. Alternatively, L140 and I176 might be essential for the maintenance of L2 in its native conformation, while their substitution might cause L2 to assume an altered conformation with a reduced affinity for PDHK2 as well as E1. Kato and colleagues (36) reported that L2 D172 forms a salt bridge with PDHK3 R397, which is an equivalent of K391 in PDHK2. Therefore, it is feasible that the D172/K391 pair plays a similar role in PDHK2 binding. In agreement with this idea, we have found that the substitution of K391 for alanine causes a significant decrease in the affinity of PDHK2 for L2. Surprisingly, the alteration of the L2 D172 side chain by site-directed mutagenesis had a rather small effect on PDHK2 binding, suggesting that the charges of either K391 or D172 are not essential for the interaction between PDHK2 and L2. It seems reasonable to propose that PDHK2 K391 contributes to the binding of L2 primarily through the hydrophobic interactions between the aliphatic part of the K391 side chain and the prosthetic group. Another ionic interaction between PDHK3 E395 (E389 in PDHK2) and L2 R196 thought to be important for the interaction between L2 and PDHK3 (8, 35) appears to be inconsequential for PDHK2 binding. Finally, our data suggest that the acidic cluster located at the base of L2 is essential for PDHK2 binding due presumably to the ionic interactions with PDHK2 K368 and/or R372. Importantly, L2 E179 appears to be the only acidic residue critical for PDHK2 binding. Another acidic residue, i.e., L2 E162, which, in addition to E179, was implicated in PDHK3 binding (8), does not appreciably contribute to the binding of PDHK2. Thus, several amino acid residues of L2 (E162, D172, A174, and R196), which were reported to be essential for PDHK3 binding on the basis of site-directed mutagenesis (8) and structural data (35), have little if any effect on PDHK2 binding, strongly suggesting that individual isozymes might recognize L2 somewhat differently.
Effect of AZD7545 on PDHK2
As discussed in the introductory section, AZD7545 is thought to inhibit PDHK2 activity by displacing the kinase molecule from the complex-bound state (24). On the basis of this idea, it would be expected that the effect of AZD7545 on PDHK2 activity has to be E2-dependent. To explore this hypothesis, we investigated the PDHK2 inhibition pattern by AZD7545 using free or E2-bound E1 as a substrate (Figure 4A). We found that, when E2-bound E1 was used as a substrate, AZD7545 effectively inhibited PDHK2, which is in agreement with the earlier results (24). AZD7545 worked as a partial inhibitor, causing a concentration-dependent decrease in PDHK2 activity that reached approximately 20–25% of the initial activity at an AZD7545 concentration of 100 μM [Figure 4A (●)]. In marked contrast, when free E1 was used as a PDHK2 substrate, there was no appreciable decrease in the kinase activity over a wide range of AZD7545 concentrations (from 10−9 to 10−4 M) [Figure 4A (○)]. In fact, with free E1, we consistently observed a slight increase in kinase activity. Furthermore, when similar experiments were conducted using highly diluted PDC components, i.e., under conditions that promote dissociation of PDHK2 from PDC (43), the effect of AZD7545 on PDHK2 activity was proportionally smaller than that observed with concentrated components (data not shown). Taken together, these data strongly suggest that the effect of AZD7545 on PDHK2 activity is E2-dependent and, therefore, provide the first experimental support for the hypothesis proposed by Morrell and colleagues (24).
Figure 4.

Effect of AZD7545 on wild-type PDHK2. (A) Effects of AZD7545 on PDHK2 activity determined with free E1 (○) or E2-bound E1 (●) as a substrate. The activity of PDHK2 was determined on the basis of the incorporation of [32P]phosphate into E1 during 1 min of the reaction as described previously (18). Each data point represents the mean ± the standard deviation for three to five independent determinations. (B) Effect of AZD7545 on binding of PDHK2 to the unaltered GST-L2 protein. The left panel depicts experiments carried out with vehicle alone. The right panel shows the experiment carried out with 6 μM AZD7545 added to the binding mixture. Shown are representative data from four experiments. (C) Displacement of PDHK2 from the PDHK2–GST-L2 complex by AZD7545. Preformed PDHK2–GST-L2 complexes were incubated in binding buffer with vehicle (left) or 6 μM AZD7545 (right). Protein contents of free (F) and bound (B) fractions were analyzed using SDS–PAGE. Gels were stained with Coomassie R250. Shown are representative data from four experiments. AZD7545 was added from the stock solution made in dimethyl sulfoxide (DMSO). The final concentration of DMSO in the binding mixture was less than 1% (v/v).
PDHK2 is docked to PDC through L2’s furnished by the E2 component (5–7). Therefore, if AZD7545 acts by displacing PDHK2 from PDC, it has to interfere with L2 binding by PDHK2. This idea was explored using a GST-L2-based pulldown assay. As shown in Figure 4B (left panel), in the assay that utilizes 80 μg of PDHK2 and 200 μg of GST-L2 construct, approximately half of the PDHK2 protein was recovered in the L2-bound form. Similar experiments conducted with AZD7545 added directly to the binding mixture at a final concentration of 6 μM yielded all PDHK2 in the free form (Figure 4B, right panel), which is consistent with the interpretation that AZD7545 can prevent PDHK2 from binding to L2. Several lines of evidence suggest that, in vivo, a significant amount of PDHK2 protein exists as an integral component of PDC, being exposed to the very high local concentration of lipoyl domains (>1 mM) (8, 41). Thus, to be able to inhibit PDHK2 activity through the displacement mechanism, AZD7545 should be capable of releasing PDHK2 from the L2-bound state. To examine this idea, PDHK2 was pre-adsorbed on immobilized GST-L2, and the excess of PDHK2 was removed by extensive washing (Figure 4C). PDHK2-L2 complexes were incubated in the binding buffer containing vehicle alone (Figure 4C, left panel) or in the binding buffer containing 6 μM AZD7545 added in vehicle solution (Figure 4C, right panel). After incubation, beads were extensively washed in the binding buffer to remove free PDHK2. SDS–PAGE analysis of the resulting fractions revealed that AZD7545 was very effective in promoting the release of L2-bound PDHK2. These data along with the results of direct competition experiments summarized in Figure 4B strongly suggest that AZD7545 acts as a functional antagonist of L2.
Although data presented in Figure 4 support the hypothesis that AZD7545 acts as an L2 antagonist, they do not answer the question of whether AZD7545 directly competes with L2 for the same binding site or whether AZD7545 binding leads to a conformational change that promotes the release of the bound L2. To explore these hypotheses, we examined the effect of AZD7545 on the activities of PDHK2 variants carrying point mutations in the L2-binding site. Considering that, in our hands, AZD7545 inhibited only the complex-bound form of PDHK2 (Figure 4A), these experiments were conducted with the E2-bound form of E1 as a substrate. Of 15 PDHK2 variants characterized in this study, two (PDHK2-F31A and PDHK2-F44A) displayed a significant reduction in the effect of AZD7545 (Figure 5A). In fact, PDHK2-F44A showed little if any response to the addition of the inhibitor at a concentration as high as 10−4 M, suggesting that a specific mutation in the L2-binding site of PDHK2 can greatly suppress the effect of AZD7545 on the kinase activity. Importantly, this mutation disrupts the effect of AZD7545 without any appreciable influence on PDHK2 protein folding as evidenced by CD spectroscopy (Figure 5B). PDHK2 F31 and F44 are highly conserved residues that, on the basis of the modeling experiments, form the inner lining of the lipoamide-binding cavity. Thus, it is likely that AZD7545 mimics the lipoate prosthetic group and exerts its inhibitory effect on kinase activity by competing for the lipoamide-binding cavity of PDHK2.
Figure 5.
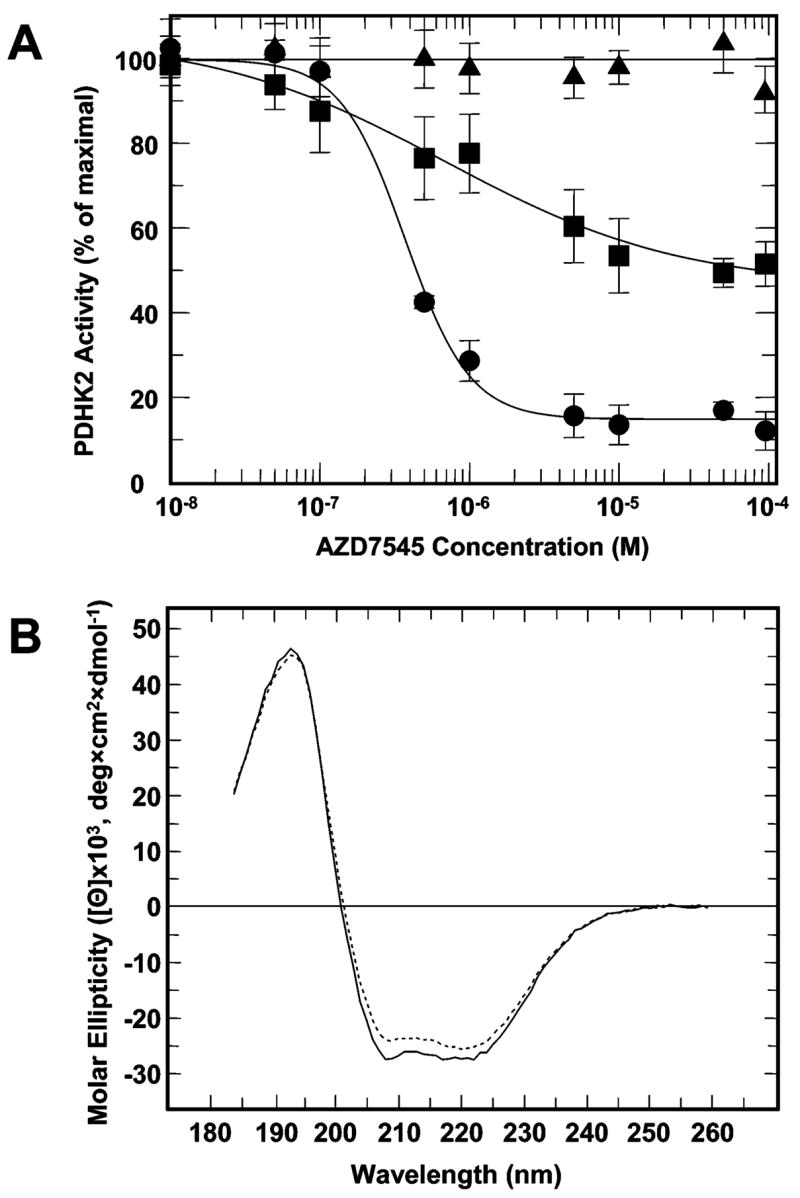
Effect of AZD7545 on the activity of wild-type and mutant PDHK2. (A) The effect of AZD7545 on the activity of wild-type PDHK2 is shown with filled circles. The effects of AZD7545 on the activities of PDHK2-F31A and PDHK2-F44A are shown with filled rectangles and filled triangles, respectively. The activity of PDHK2 was determined on the basis of the incorporation of [32P]phosphate into the E2/E3BP-bound E1 subunit. Kinetic data were fitted and analyzed using GraFit, version 5.0. Each data point represents the mean ± the standard deviation for three to five independent determinations. AZD7545 was added from the stock solutions made in DMSO. The final concentration of DMSO in reaction mixtures was 1% (v/v). (B) CD spectra of wild-type PDHK2 (–) and PDHK2-F44A (- - -) proteins. CD spectra were recorded using a Jasco J815 spectrometer (Jasco Inc., Easton, MD) as described in Experimental Procedures.
Acknowledgments
We are thankful to Drs. Rachel Mayers and Elaine Kilgour at AstraZeneca (Mereside, Alderley Park, Macclesfield, Cheshire, U.K.) for the generous gift of the AZD7545 sample. We also thank Dr. Michael J. Jablonsky (Department of Chemistry, University of Alabama, Birmingham, AL) for his help with CD spectroscopy.
Footnotes
This work was supported by Grant GM51262 from the U.S. Public Health Service.
Abbreviations: PDC, pyruvate dehydrogenase complex; PDHK, pyruvate dehydrogenase kinase; PDHK1–PDHK4, isozymes 1–4 of pyruvate dehydrogenase kinase, respectively; E1, pyruvate dehydrogenase component of PDC; E2, dihydrolipoyl transacetylase component of PDC; E3, dihydrolipoyl dehydrogenase component of PDC; E3BP, E3-binding protein component of PDC; L2, inner lipoyl-bearing domain of dihydrolipoyl transacetylase; DCA, dichloroacetate; LPA, lipoic acid; SDS–PAGE, polyacrylamide gel electrophoresis in the presence of sodium dodecyl sulfate; DTT, dithiothreitol; ITC, isothermal titration calorimetry; DMSO, dimethyl sulfoxide.
References
- 1.Randle PJ. Metabolic fuel selection: General integration at the whole-body level. Proc Nutr Soc. 1995;54:317–327. doi: 10.1079/pns19950057. [DOI] [PubMed] [Google Scholar]
- 2.Holness MJ, Sugden MC. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans. 2003;31:1143–1151. doi: 10.1042/bst0311143. [DOI] [PubMed] [Google Scholar]
- 3.Zhou ZH, McCarthy DB, O’Connor CM, Reed LJ, Stoops JK. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc Natl Acad Sci USA. 2001;98:14802–14807. doi: 10.1073/pnas.011597698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel MS, Roche TE. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 1990;4:3224–3233. doi: 10.1096/fasebj.4.14.2227213. [DOI] [PubMed] [Google Scholar]
- 5.Ravindran S, Radke GA, Guest JR, Roche TE. Lipoyl domain-based mechanism for the integrated feedback control of the pyruvate dehydrogenase complex by enhancement of pyruvate dehydrogenase kinase activity. J Biol Chem. 1996;271:653–662. doi: 10.1074/jbc.271.2.653. [DOI] [PubMed] [Google Scholar]
- 6.Yang D, Gong X, Yakhnin A, Roche TE. Requirements for the adaptor protein role of dihydrolipoyl acetyltransferase in the up-regulated function of the pyruvate dehydrogenase kinase and pyruvate dehydrogenase phosphatase. J Biol Chem. 1998;273:14130–14137. doi: 10.1074/jbc.273.23.14130. [DOI] [PubMed] [Google Scholar]
- 7.Tuganova A, Popov KM. Role of protein-protein interactions in the regulation of pyruvate dehydrogenase kinase activity. Biochem J. 2005;387:147–153. doi: 10.1042/BJ20040805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roche TE, Baker JC, Yan X, Hiromasa Y, Gong X, Peng T, Dong J, Turkan A, Kasten SA. Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog Nucleic Acid Res Mol Biol. 2001;70:33–75. doi: 10.1016/s0079-6603(01)70013-x. [DOI] [PubMed] [Google Scholar]
- 9.Yeaman SJ, Hutcheson ET, Roche TE, Pettit FH, Brown JR, Reed LJ, Watson DC, Dixon GH. Sites of phosphorylation on pyruvate dehydrogenase from bovine kidney and heart. Biochemistry. 1978;17:2364–2370. doi: 10.1021/bi00605a017. [DOI] [PubMed] [Google Scholar]
- 10.Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J. 2001;358:69–77. doi: 10.1042/0264-6021:3580069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korotchkina LG, Patel MS. Probing the mechanism of inactivation of human pyruvate dehydrogenase by phosphorylation of three sites. J Biol Chem. 2001;276:5731–5738. doi: 10.1074/jbc.M007558200. [DOI] [PubMed] [Google Scholar]
- 12.Sale GJ, Randle PJ. Role of individual phosphorylation sites in inactivation of pyruvate dehydrogenase complex in rat heart mitochondria. Biochem J. 1982;203:99–108. doi: 10.1042/bj2030099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gudi R, Bowker-Kinley MM, Kedishvili NY, Zhao Y, Popov KM. Diversity of the pyruvate dehydrogenase kinase gene family in humans. J Biol Chem. 1995;270:28989–28994. doi: 10.1074/jbc.270.48.28989. [DOI] [PubMed] [Google Scholar]
- 14.Rowles J, Scherer SW, Xi T, Majer M, Nickle DC, Rommens JM, Popov KM, Harris RA, Riebow NL, Xia J, Tsui LC, Bogardus C, Prochazka M. Cloning and characterization of PDK4 on 7q21.3 encoding a fourth pyruvate dehydrogenase kinase isoenzyme in human. J Biol Chem. 1996;271:22376–22382. doi: 10.1074/jbc.271.37.22376. [DOI] [PubMed] [Google Scholar]
- 15.Wu P, Sato J, Zhao Y, Jaskiewicz J, Popov KM, Harris RA. Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase isoenzyme 4 in rat heart. Biochem J. 1998;329:197–201. doi: 10.1042/bj3290197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugden MC, Holness MJ. Therapeutic potential of the mammalian pyruvate dehydrogenase kinases in the prevention of hyperglycaemia. Curr Drug Targets: Immune, Endocr Metab Disord. 2002;2:151–165. [PubMed] [Google Scholar]
- 17.Bajotto G, Murakami T, Nagasaki M, Qin B, Matsuo Y, Maeda K, Ohashi M, Oshida Y, Sato Y, Shimomura Y. Increased expression of hepatic pyruvate dehydrogenase kinases 2 and 4 in young and middle-aged Otsuka Long-Evans Tokushima Fatty rats: Induction by elevated levels of free fatty acids. Metabolism. 2006;55:317–323. doi: 10.1016/j.metabol.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 18.Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998;329:191–196. doi: 10.1042/bj3290191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holness MJ, Kraus A, Harris RA, Sugden MC. Targeted upregulation of pyruvate dehydrogenase kinase (PDK)-4 in slow-twitch skeletal muscle underlies the stable modification of the regulatory characteristics of PDK induced by high-fat feeding. Diabetes. 2000;49:775–781. doi: 10.2337/diabetes.49.5.775. [DOI] [PubMed] [Google Scholar]
- 20.Huang B, Wu P, Bowker-Kinley MM, Harris RA. Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptor-α ligands, glucocorticoids, and insulin. Diabetes. 2002;51:276–283. doi: 10.2337/diabetes.51.2.276. [DOI] [PubMed] [Google Scholar]
- 21.Holness MJ, Bulmer K, Smith ND, Sugden MC. Investigation of potential mechanisms regulating protein expression of hepatic pyruvate dehydrogenase kinase isoforms 2 and 4 by fatty acids and thyroid hormone. Biochem J. 2003;369:687–695. doi: 10.1042/BJ20021509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwon HS, Huang B, Ho-Jeoung N, Wu P, Steussy CN, Harris RA. Retinoic acids and trichostatin A (TSA), a histone deacetylase inhibitor, induce human pyruvate dehydrogenase kinase 4 (PDK4) gene expression. Biochim Biophys Acta. 2006;1759:141–151. doi: 10.1016/j.bbaexp.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 23.Mayers RM, Butlin RJ, Kilgour E, Leighton B, Martin D, Myatt J, Orme JP, Holloway BR. AZD7545, a novel inhibitor of pyruvate dehydrogenase kinase 2 (PDHK2), activates pyruvate dehydrogenase in vivo and improves blood glucose control in obese (fa/fa) Zucker rats. Biochem Soc Trans. 2003;31:1165–1167. doi: 10.1042/bst0311165. [DOI] [PubMed] [Google Scholar]
- 24.Morrell JA, Orme J, Butlin RJ, Roche TE, Mayers RM, Kilgour E. AZD7545 is a selective inhibitor of pyruvate dehydrogenase kinase 2. Biochem Soc Trans. 2003;31:1168–1170. doi: 10.1042/bst0311168. [DOI] [PubMed] [Google Scholar]
- 25.Harris RA, Bowker-Kinley MM, Wu P, Jeng J, Popov KM. Dihydrolipoamide dehydrogenase-binding protein of the human pyruvate dehydrogenase complex. DNA-derived amino acid sequence, expression, and reconstitution of the pyruvate dehydrogenase complex. J Biol Chem. 1997;272:19746–19751. doi: 10.1074/jbc.272.32.19746. [DOI] [PubMed] [Google Scholar]
- 26.Tuganova A, Boulatnikov I, Popov KM. Interaction between the individual isoenzymes of pyruvate dehydrogenase kinase and the inner lipoyl-bearing domain of transacetylase component of pyruvate dehydrogenase complex. Biochem J. 2002;366:129–136. doi: 10.1042/BJ20020301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klyuyeva A, Tuganova A, Popov KM. The carboxy-terminal tail of pyruvate dehydrogenase kinase 2 is required for the kinase activity. Biochemistry. 2005;44:13573–13582. doi: 10.1021/bi0505868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sreerama N, Woody RW. A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal Biochem. 1993;209:32–44. doi: 10.1006/abio.1993.1079. [DOI] [PubMed] [Google Scholar]
- 30.Liu S, Baker JC, Andrews PC, Roche TE. Recombinant expression and evaluation of the lipoyl domains of the dihydrolipoyl acetyltransferase component of the human pyruvate dehydrogenase complex. Arch Biochem Biophys. 1995;316:926–940. doi: 10.1006/abbi.1995.1124. [DOI] [PubMed] [Google Scholar]
- 31.Hiromasa Y, Hu L, Roche TE. Ligand-induced effects on pyruvate dehydrogenase kinase isoform 2. J Biol Chem. 2006;281:12568–12579. doi: 10.1074/jbc.M513514200. [DOI] [PubMed] [Google Scholar]
- 32.Liu S, Gong X, Yan X, Peng T, Baker JC, Li L, Robben PM, Ravindran S, Andersson LA, Cole AB, Roche TE. Reaction mechanism for mammalian pyruvate dehydrogenase using natural lipoyl domain substrates. Arch Biochem Biophys. 2001;386:123–135. doi: 10.1006/abbi.2000.2222. [DOI] [PubMed] [Google Scholar]
- 33.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 35.Quinn J, Diamond AG, Masters AK, Brookfield DE, Wallis NG, Yeaman SJ. Expression and lipoylation in Escherichia coli of the inner lipoyl domain of the E2 component of the human pyruvate dehydrogenase complex. Biochem J. 1993;289:81–85. doi: 10.1042/bj2890081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kato M, Chuang JL, Tso S-C, Wynn RM, Chuang DT. Crystal structure of pyruvate dehydrogenase kinase 3 bound to lipoyl domain 2 of human pyruvate dehydrogenase complex. EMBO J. 2005;24:1763–1774. doi: 10.1038/sj.emboj.7600663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knoechel TR, Tucker AD, Robinson CM, Phillips C, Taylor W, Bungay PJ, Kasten SA, Roche TE, Brown DG. Regulatory roles of the N-terminal domain based on crystal structures of human pyruvate dehydrogenase kinase 2 containing physiological and synthetic ligands. Biochemistry. 2006;45:402–415. doi: 10.1021/bi051402s. [DOI] [PubMed] [Google Scholar]
- 38.Steussy CN, Popov KM, Bowker-Kinley MM, Sloan RB, Jr, Harris RA, Hamilton JA. Structure of pyruvate dehydrogenase kinase. Novel folding pattern for a serine protein kinase. J Biol Chem. 2001;276:37443–37450. doi: 10.1074/jbc.M104285200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holzwarth G, Doty P. The ultraviolet circular dichroism of polypeptides. J Am Chem Soc. 1965;87:218–228. doi: 10.1021/ja01080a015. [DOI] [PubMed] [Google Scholar]
- 41.Baker JC, Yan X, Peng T, Kasten S, Roche TE. Marked differences between two isoforms of human pyruvate dehydrogenase kinase. J Biol Chem. 2000;275:15773–15781. doi: 10.1074/jbc.M909488199. [DOI] [PubMed] [Google Scholar]
- 42.Tso SC, Kato M, Chuang JL, Chuang DT. Structural Determinants for Cross-talk between Pyruvate Dehydrogenase Kinase 3 and Lipoyl Domain 2 of the Human Pyruvate Dehydrogenase Complex. J Biol Chem. 2006;281:27197–27204. doi: 10.1074/jbc.M604339200. [DOI] [PubMed] [Google Scholar]
- 43.Hiromasa Y, Roche TE. Facilitated interaction between the pyruvate dehydrogenase kinase isoform 2 and the dihydrolipoyl acetyltransferase. J Biol Chem. 2003;278:33681–33693. doi: 10.1074/jbc.M212733200. [DOI] [PubMed] [Google Scholar]