

Abstract
The molecular neurobiology of depression begins with an idea: depressive disorders represent a family of related but distinct conditions. Different points of vulnerability in the brain may predispose to depressive disorders. For example, it is likely that the core pathophysiology of depression associated with early life adversity is different from non-trauma related disorders. In particular, the pathophysiology of trauma-related depression involves the hypothalamic-pituitary-adrenal (HPA) axis, although, in turn, this reflects underlying molecular etiologies. In particular, dysregulation of signal transduction mechanisms may be one site of origin. This may be the result of yet other processes such as the effects of reactive oxygen species (ROS) or genomic imprinting. Unraveling these complex causes may lead to novel treatments that can be used in a targeted fashion.
Keywords/phrases: Depression, molecular, signal transduction, gene expression, genomics
Unipolar depressive disorders encompass a range of features that strongly suggest a neurobiological substrate. These include symptoms such as include sleep and appetite disturbances (both up and down), loss of interest and pleasure, negative rumination, fatigue, and poor concentration, but also apparent abnormalities of the hypothalamic-pituitary-adrenal axis(1) or of neuroplasticity.(2) Moreover, depression appears to have genetic antecedents, which also point to a biological contribution to etiology.(3) However, the exact pathophysiology has been largely unknown until the last decade or so.
To begin, depression clearly is not one entity, but subsumes a range of causes that involve genetic contributors, along with early and later stress.(2;4) Therefore, the depressive spectrum include conditions related to early traumatic events (typically with early onset of depression as well), through episodes in the absence of early trauma that may or may not be precipitated by an acute stressor. Therefore, the heterogeneous nature of the condition must be taken into account in any search for a biological substrate.
Unfortunately, that is not usually the case. In fact, most studies simply take an unselected groups of “depressed” and “controls” for comparison. However, this is likely to contribute to type II error, since a particular biological finding may be applicable only to a subset of depressed patients. To illustrate this point, take a study from our own research laboratory (Figure 1).(5) In this study, protein kinase A (PKA) activity was determined in fibroblasts in culture from a group of depressed patients and controls. Panel A of Figure 1 shows the total group of depressed patients compared with normal controls. Clearly, some patients have reduced activity. However, there also is considerable overlap between depressed and control populations. Alternatively, a different picture emerges when the depressed population is broken down by subtype. The cells from patients with atypical features all fell within the control range. All but one melancholic patient were outside of the range, with the ones not subtyped as melancholic or atypical falling between. This indicates that a particular finding, low PKA activity, is characteristic of only one subset of depressed patients. Similar data in human post-mortem brain indicate that the reduced PKA activity may be associated with death by suicide.(6;7) Together, these results indicate that there will not be a single, unitary set of biological substrates that explain the full range of depressive disorders. Further, all contributors, such as genetic antecedents, early trauma, and recent stress, confer their own, perhaps unique, mechanisms of etiology
Figure 1.
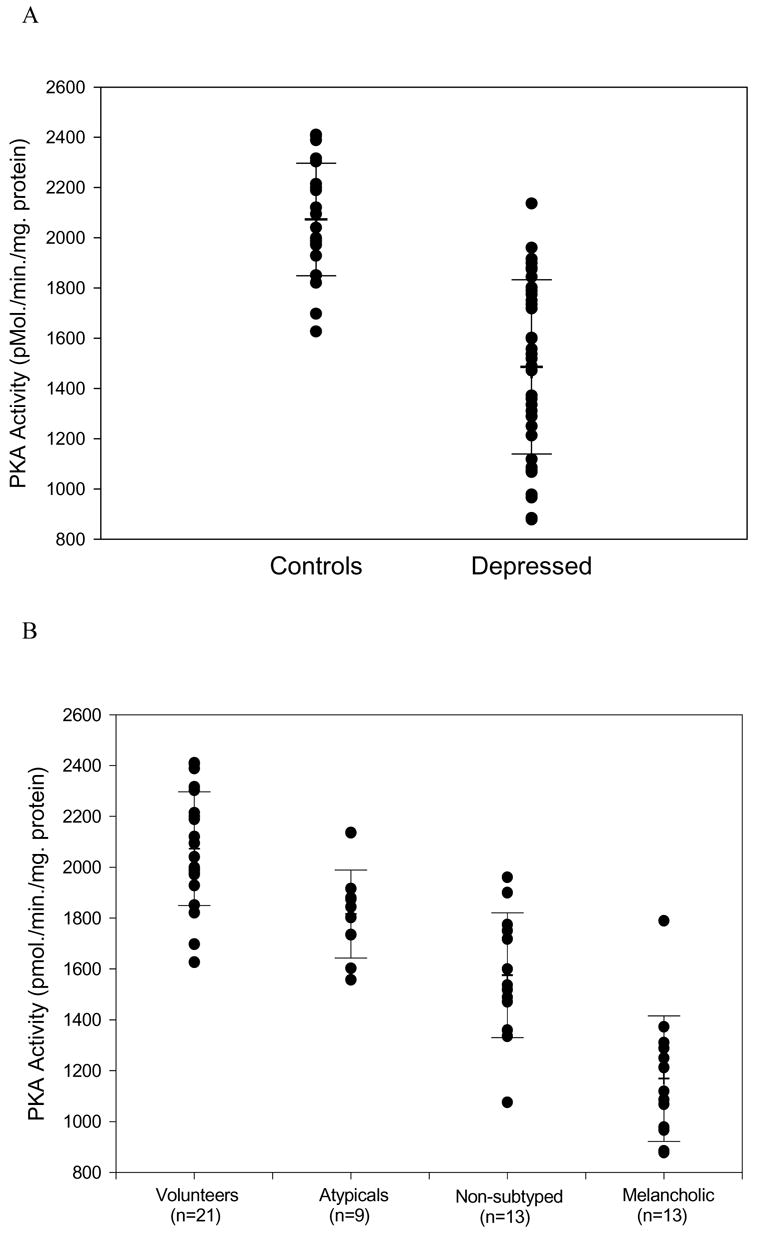
Protein kinase A activity in depressed patients and normal controls. Panel A: Activity in the total group, depressed and controls. Panel B: Activity in depressive subtypes and controls.(5)
The role of stress
That life stressors contribute in some fashion to depression is almost a truism and, essentially is an extension of what occurs normally. That is, adverse events confer negative mood states in normals. The search for physiological underpinnings, therefore, can be thought of as an extrapolation (and, hence, dysregulation) of normal responses, in many circumstances;(8) this will be discussed in greater detail later. The work of Kendler and colleagues(9;10) suggests that the both early and recent adversity contribute significantly to the potential for a depressive episode.(9;10) Moreover, early adverse events, particularly abuse, appear to confer heightened risk,(9;11–14) an issue that will resurface later in the discussion of cellular pathology.
The interplay of genetics and environment is highly complex and research in this area is in its infancy. Caspi et al.(15) have demonstrated a causal interaction between a specific polymorphic variant of the serotonin transporter (5HT-T), early and late adversity, and the occurrence of depression. This group investigated the contribution of a common and functional polymorphism of the promoter region of the 5HT-T gene (5HTTLPR). 5HT-T is the principal modular of synaptic serotonin (5HT) activity, and is blocked by serotonin selective reuptake inhibitors. This study evaluated 847 people followed over the course of two decades. The results suggested an interaction between the functional, so-called “short” (s) variant of 5HTTLPR with both early and late adversity. For example, stressful events were tabulated for the period between ages 21 and 26. There was no relationship between the s variant and the occurrence of depression in participants who reported no major stressors during this period. However, the occurrence of stressors conferred increased risk primarily in persons carrying the s allele. Moreover, there was an environmental stress “dose” effect – i.e., more stressful events produced higher risk. As well, there was a “gene dose” effect in which the ss carriers had the highest risk, the “long-long” (ll) the lowest, and the ls falling in between. There also was a relationship between early traumas and risk for depression that was modulated by genotype. Specifically, persons with no history of early maltreatment did not show any association between transporter alleles and depression. However, in those who did have early maltreatment, the risk segregated according to allelic status, with the ss having the highest risk. Again, there was both a “stress dose” and “gene dose” effect. These results were confirmed in a recent study with a new cohort.(16) These results suggest a strong interaction between genetic diathesis (and, hence, biological substrate) with both early and recent stressors. Although this is a complicating element, in fact it opens particular avenues of investigation.
Stress and the hypothalamic pituitary adrenal axis dysfunction: Risk for depression
Abnormalities of the HPA axis in depressed patients are well-described.(1) However, overt dysregulation is found only in a subset of depressed patients. As articulated by Nemeroff and colleagues,(17;18) depression and early trauma converge at the level of HPA axis regulation. Adversity that is early in life, severe, and prolonged, appears to contribute significantly both to subsequent risk for anxiety and depressive disorders, and HPA dysfunction. Traumas such as physical or sexual abuse or the loss of a parent that occur during critical periods of development result in a permanent alteration of stress reactivity in the CNS. Actually, from a teleological perspective, the maintenance of an activated stress response system following chronic or severe stress makes adaptive sense. The concept being that high threat intensity would result in a persistently elevated “alert” status.
Persistent enhancement of stress reactivity could explain the findings of heightened HPA axis activity in some depressed patients including elevated peripheral cortisol(1;19;20) and central corticotrophin releasing hormone (CRH).(21;22) The interplay of genetic predisposition and early adversity would then lead to a vulnerable phenotype. Moreover, this is likely to result in heightened stress reactivity by at least two mechanisms. The first would be the biological diathesis. The second would be a pattern of maladaptive responses to stressors. For example, withdrawal from a stressor would be highly adaptive in the case of early abuse. However, that same behavior, manifested in adult life, would be equally maladptive. These, then, would combine to elevate risk for depression.
Alternatively, the fact that not all depressed persons have been abused in childhood would explain why only a subset shows HPA activation. The etiological pathways and the subcellular dysfunction associated with it could, then, differ in depressed persons without history of early adversity.
Intracellular signal transduction
A number of studies implicate intracellular signal transduction in the pathophysiology of depression. One key set of mechanisms involve phosphorylation enzymes, including protein kinases A (PKA) and C (PKC) (Figure 2). The binding of a transmitter with a g-protein linked receptor activates the coupling of g-proteins (Gs and Gq) with second messenger enzymes such adelylate cyclase (AC) or phospholipase C (PLC). These, in turn, catalyze the formation of the second messengers cyclic AMP and diacylglycerol (DAG). These, in turn, bind to PKA and PKC respectively, which facilitates phosphorylation by these enzymes.(23)
Figure 2.
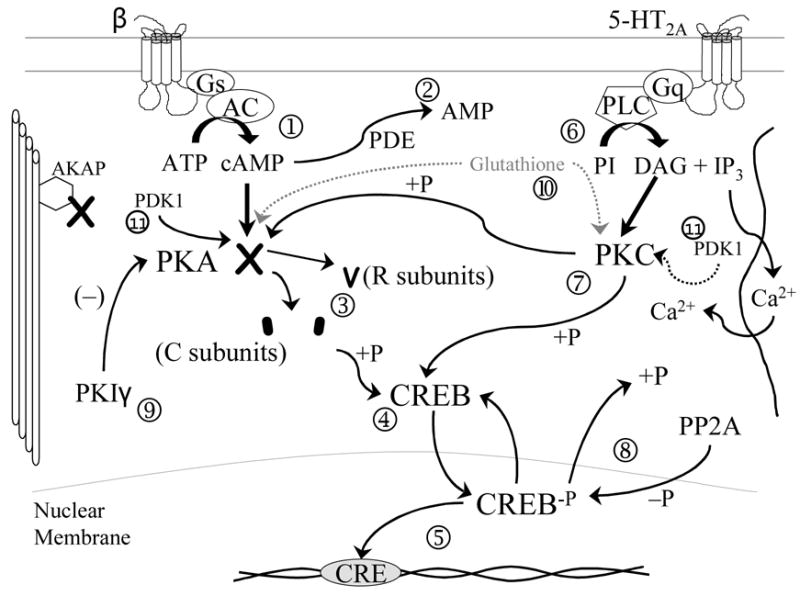
Representative intracellular g-protein coupled signal transduction cascades. Subheading: Cell surface receptors, such as norepinephrine ß receptors, are coupled to stimulatory G (Gs) proteins. On transmitter binding, Gs activates adenylate cyclase (AC), catalyzing the formation of cyclic AMP from ATP ➀. Cyclic AMP is degraded to AMP by phosphodiesterases (PDE), which inactivate the cyclic AMP signal ➁. Cyclic AMP binds to the regulator subunits (R) of protein kinase A (PKA), resulting in a conformational change and the release of two catalytic subunits (C) ➂. The C subunits then are capable of phosphorylating serine and threonine residues on target polypeptides. One such protein is cyclic AMP response element binding protein (CREB), a transcriptional factor ➃. Phosphorylation of CREB results in translocation of CREB-P to the nucleus and subsequent binding to CRE-containing gene promoter regions activating expression ➄. A parallel pathway, exemplified by the 5-HT2A transductional cascade, couples to Gq (another stimulatory G protein), activating phospholipase C (PLC) catalyzing the conversion of phosphotidylinositol (PI) to diacylglycerol (DAG) and inositol triphosphate (IP3) ➅. IP3 mobilizes Ca2+ release from intracellular stores. DAG binds to protein kinase C resulting in phosphorylation of target proteins; these include both CREB and PKA ➆. The phosphorylation of PKA regulates the activity of the enzyme. Phosphorylated substrates are subsequently dephosphorylated by protein phosphatase 2A ➇. On dephosphorylation, CREB returns to the cytosol. Another regulator of PKA activity is protein kinase inhibitor (PKI); in the case of fibroblasts, this is the PKIγ isoform ➈. Both PKA and PKC can be glutathionylated, protecting them from oxidative degradation ➉. The activation loops of both enzymes are phosphorylated by phosphoinositide dependent kinase 1 (PDK1)
Protein kinases are critical elements of stimulus-response coupling.(24) One critical effect is the subsequent phosphorylation of the transcriptional factor cyclic AMP response element binding protein (CREB). CREB phosphorylation is linked to both norepinephrine- (NE) and serotonin- (5-HT) linked cascades and may represent a common target of action of more noradrenergic and serotonergic antidepressants.(24) Phosphorylated CREB binds to cyclic AMP response element (CRE) in the promoter region of genes, which regulates gene expression.(25) This represents an integrative set of mechanisms in which antidepressants acting via either NE or 5-HT can target a common set of genes and their respective protein products.
A variety of research has implicated deficiencies in PKA and PKC in a subset of depressives.(26) For example, our research group has shown that, relative to normal controls, certain depressed patients have deficient PKA and PKC protein levels,(27;28) lower binding of cyclic AMP to PKA,(29) reduced phosphorylation of CREB,(29) and altered gene expression patterns;(30) these findings have also been shown in both peripheral and post-mortem brain tissues by other groups.(6;7;7;31–39) This decrease in the activity of these two key enzymes would be expected to alter the expression of genes that contain CRE elements in their promoters; these would include key proteins that regulate the stress response in brain, including brain derived neurotrophic factor (BDNF),(40–42) the BDNF receptor trk-b,(43) and glucocorticoid receptors (GR).(44) Moreover, GR functions as a transcriptional factor and regulates the expression of other genes, specifically exerting an inhibitory effect on corticotrophin releasing hormone (CRH).(45) Hence, reduced activity of these key enzymes could enhance stress reactivity via altered regulation of the expression of specific genes. Important elements of stress regulation could, then, be vulnerable under demand conditions. In fact, the knockout of PKA and PKC protein isoforms in mice have demonstrated a variety of cellular effects,(46;47) most particularly hippocampal neuroplasticity,(46;48) which has been implicated in depression.(49)
Mechanisms for altered protein activity
Although the evidence for altered kinase activity seems sound, the exact mechanisms for inducing these effects are unknown. One possible avenue is via protein oxidation. The oxidation-reduction (redox) potential is altered by reactive oxygen species (ROS), which are formed by a variety of factors, including inflammatory cytokines.(50) Altered redox potential has been shown to affect the activity of kinases.(50) Glutathione, an intracellular antioxidant, is involved in the protection of proteins against oxidative stress,(50–52) by binding to redox sensitive amino acids (particularly cysteines). Recently, glutathionylation has been described as an important regulator of PKA degradation,(53) and could be involved in the availability of PKA proteins.
As noted, proinflammatory cytokines are involved in the genesis of ROS and thereby affect oxidation and, hence, degradation of proteins. Altered cytokine activity is well-described in depression,(54) and antidepressant drugs have been shown to regulate the expression of specific cytokines, in particular, inhibiting pro-inflammatory cytokines that are involved in enhancing ROS.(55) Curiously, this appears to be mediated via kinase-related pathways.(56) Maes et al. showed that the inhibitory effects of fluoxetine on tumor necrosis factor alpha are mediated, in part, by PKA. This suggests an interactive system in which kinase dependent pathways may modulate cytokine expression, and pro-inflammatory cytokines may affect kinase-dependent pathways via protein oxidation.
Another key element in regulation of stress-responsivity is epigenetic regulation of gene expression. DNA does not exist in an unregulated, “open” fashion. DNA is associated with histone proteins which determine gene expression to a significant extent(57–59) A number of protein modification processes, including phosphorylation, acetylation, methylation, ubiquitination, and ribosylation, can influence these proteins similarly to other protein elements in cells.(59;60) Furthermore, direct methylation of DNA sequences, particularly promoter regions, also exerts major influences on activity. Methylation is a key determinant of DNA expression. This occurs on cytosines of the dinucleotide CpG via DNA methyltransferases.(59;60) These mechanisms, termed epigenetic regulation, represent inducible and, potentially, reversible phenomena that can be modified by environmental factors. These effects may, then, mediate gene-environment interactions and determine both short- and longer-term responses to environmental cues.(59;60)
This concept was tested recently in an elegant set of experiments from the laboratory of Dr. Michael Meaney.(61–67) Rat mothers periodically engage in licking and grooming (LG) and arched-back nursing (ABN) of their pups. However, they show a range of such responses, with some mothers showing high- and others low-LG-ABN and others demonstrating LG-ABN. Low-LG-ABN was associated with a marked elevation in methylation of exon 17 of the GR promoter region relative to high-LG-ABN, an effect that persisted into adult life. The cross-fostering of pups reversed this pattern, indicating that maternal behavior was the mediator. This increased promoter methylation resulted in decreased GR expression in brain, and a heightened corticosterone response to stress. These data indicate a causal relationship between maternal behavior and subsequent stress reactivity that is mediated via methylation of a specific gene.
Summary
Depression is a condition with a complex biological pattern of etiology. We have seen how environmental stressors modulate subsequent vulnerability to depression. In particular, early adversity appears to induce heightened reactivity to stress via several possible mechanisms, both biological and psychological. This increased reactivity results in an enhancement of biological stress-response mechanisms, especially the HPA axis. Regulators of this system, particularly signal transduction pathways involving PKA and PKC, may be important in the regulation of key genes in this system including genes for GR, BDNF, and trkb. This system is potentially vulnerable to ROS and, therefore, indirectly to the effects of cytokines. Finally, some of these effects may be controlled by chemical modification of DNA, specifically, methylation of promoters or other gene regions. This is a mechanism by which long-term biological change can be induced via environmental stressors.
The brain is homeostatic; therefore, it is possible that alterations at multiple points in this system may induce dysregulation and, as a result, vulnerability to stress. Therefore, a person may be vulnerable to depression, which may be a final common “pathway” for this family of conditions. However, people may vary considerably with regard to the locus of the problem. Therefore, for example, functional variants in a set of genes might predispose some people, while others may have epigenetic imprinting, and yet others with different causes. Although complicated, it is not impossible to unravel this mix. In fact, this could lead to the development of novel interventions that could target specific points of vulnerability, allowing an improved matching of patient to treatment based on differential abnormalities at the cellular level.
Footnotes
Supported in part by NIMH grants MH073630, MH01741, and MH52339.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Gillespie CF, Nemeroff CB. Hypercortisolemia and depression. Psychosom Med. 2005;67 (Suppl 1):S26–S28. doi: 10.1097/01.psy.0000163456.22154.d2. [DOI] [PubMed] [Google Scholar]
- 2.Hayley S, Poulter MO, Merali Z, Anisman H. The pathogenesis of clinical depression: Stressor- and cytokine-induced alterations of neuroplasticity. Neuroscience. 2005;135(3):659–678. doi: 10.1016/j.neuroscience.2005.03.051. [DOI] [PubMed] [Google Scholar]
- 3.Kendler KS, Davis CG, Kessler RC. The familial aggregation of common psychiatric and substance use disorders in the National Comorbidity Survey: a family history study. Br J Psychiatry. 1997;170:541–548. doi: 10.1192/bjp.170.6.541. [DOI] [PubMed] [Google Scholar]
- 4.Kendler KS, Kessler RC, Walters EE, MacLean C, Neale MC, Heath AC, et al. Stressful life events, genetic liability, and onset of an episode of major depression in women. Am J Psychiatry. 1995 Jun;153:833–842. doi: 10.1176/ajp.152.6.833. [DOI] [PubMed] [Google Scholar]
- 5.Shelton RC, Manier DH, Peterson CS, Ellis TC, Sulser F. Cyclic AMP-dependent protein kinase in subtypes of major depression and normal volunteers. Int J Neuropsychopharmcol. 1999;2(3):187–192. doi: 10.1017/S1461145799001509. [DOI] [PubMed] [Google Scholar]
- 6.Dwivedi Y, Conley RR, Roberts RC, Tamminga CA, Pandey GN. [(3)H]cAMP binding sites and protein kinase a activity in the prefrontal cortex of suicide victims. Am J Psychiatry. 2002;159(1):66–73. doi: 10.1176/appi.ajp.159.1.66. [DOI] [PubMed] [Google Scholar]
- 7.Dwivedi Y, Rizavi HS, Shukla PK, Lyons J, Faludi G, Palkovits M, et al. Protein kinase A in postmortem brain of depressed suicide victims: altered expression of specific regulatory and catalytic subunits. Biol Psychiatry. 2004;55(3):234–243. doi: 10.1016/j.biopsych.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Shelton RC. Intracellular mechanisms of antidepressant drug action. Harv Rev Psychiatry. 2000;8(4):161–174. [PubMed] [Google Scholar]
- 9.Kendler KS, Gardner CO, Prescott CA. Toward a comprehensive developmental model for major depression in women. Am J Psychiatry. 2002;159(7):1133–1145. doi: 10.1176/appi.ajp.159.7.1133. [DOI] [PubMed] [Google Scholar]
- 10.Kendler KS, Gardner CO, Prescott CA. Toward a comprehensive developmental model for major depression in men. Am J Psychiatry. 2006;163(1):115–124. doi: 10.1176/appi.ajp.163.1.115. [DOI] [PubMed] [Google Scholar]
- 11.Kendler KS, Kuhn JW, Prescott CA. Childhood sexual abuse, stressful life events and risk for major depression in women. Psychol Med. 2004;34(8):1475–1482. doi: 10.1017/s003329170400265x. [DOI] [PubMed] [Google Scholar]
- 12.Kendler KS, Sheth K, Gardner CO, Prescott CA. Childhood parental loss and risk for first-onset of major depression and alcohol dependence: the time-decay of risk and sex differences. Psychol Med. 2002;32(7):1187–1194. doi: 10.1017/s0033291702006219. [DOI] [PubMed] [Google Scholar]
- 13.Kendler KS, Thornton LM, Gardner CO. Genetic risk, number of previous depressive episodes, and stressful life events in predicting onset of major depression. Am J Psychiatry. 2001;158(4):582–586. doi: 10.1176/appi.ajp.158.4.582. [DOI] [PubMed] [Google Scholar]
- 14.Kendler KS, Thornton LM, Gardner CO. Stressful life events and previous episodes in the etiology of major depression in women: an evaluation of the "kindling" hypothesis. Am J Psychiatry. 2000;157(8):1243–1251. doi: 10.1176/appi.ajp.157.8.1243. [DOI] [PubMed] [Google Scholar]
- 15.Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301(5631):386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 16.Kendler KS, Kuhn JW, Vittum J, Prescott CA, Riley B. The interaction of stressful life events and a serotonin transporter polymorphism in the prediction of episodes of major depression: a replication. Arch Gen Psychiatry. 2005;62(5):529–535. doi: 10.1001/archpsyc.62.5.529. [DOI] [PubMed] [Google Scholar]
- 17.Heim C, Plotsky PM, Nemeroff CB. Importance of studying the contributions of early adverse experience to neurobiological findings in depression. Neuropsychopharmacology. 2004;29(4):641–648. doi: 10.1038/sj.npp.1300397. [DOI] [PubMed] [Google Scholar]
- 18.Nemeroff CB. Neurobiological consequences of childhood trauma. J Clin Psychiatry. 2004;65 (Suppl 1):18–28. [PubMed] [Google Scholar]
- 19.Wong M-L, Kling MA, Munson PJ. Pronounced and sustained central hypernoradrenergic function in mahor depression with melancholic features:relation to hypercortisolism and corticotrophin-releasing hormone. PNAS 97. 2000:325–330. doi: 10.1073/pnas.97.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gold PW, Drevets WC, Charney DS. New insights into the role of cortisol and the glucocorticoid receptor in severe depression. Biol Psychiatry. 2002;52(5):381–385. doi: 10.1016/s0006-3223(02)01480-4. [DOI] [PubMed] [Google Scholar]
- 21.Heuser I, Bissette G, Dettling M, Schweiger U, Gotthardt U, Schmider J, et al. Cerebrospinal fluid concentrations of corticotropin-releasing hormone, vasopressin, and somatostatin in depressed patients and healthy controls: response to amitriptyline treatment. Depress Anxiety. 1998;8(2):71–79. [PubMed] [Google Scholar]
- 22.Banki CM, Karmacsi L, Bissette G, Nemeroff CB. CSF corticotropin-releasing hormone and somatostatin in major depression: response to antidepressant treatment and relapse. Eur Neuropsychopharmacol. 1992;2(2):107–113. doi: 10.1016/0924-977x(92)90019-5. [DOI] [PubMed] [Google Scholar]
- 23.Nestler EJ, Greengard P. Protein Phosphorylation in the Nervous System. New York: John Wiley & Sons; 1984. [Google Scholar]
- 24.Hyman SE, Nestler EJ. Initiation and adaptation: a paradigm for understanding psychotropic drug action. Am J Psychiatry. 1996;153(2):151–162. doi: 10.1176/ajp.153.2.151. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto KK, Gonzalez GA, Biggs WH, III, Montminy MR. Phosphorylation-induced binding and transcriptional efficacy of nuclear factor CREB. Nature. 1988;334(6182):494–498. doi: 10.1038/334494a0. [DOI] [PubMed] [Google Scholar]
- 26.Pandey GN, Dwivedi Y. Focus on protein kinase A and protein kinase C, critical components of signal transduction system, in mood disorders and suicide. Int J Neuropsychopharmacol. 2005;8:1–4. doi: 10.1017/S1461145704004936. [DOI] [PubMed] [Google Scholar]
- 27.Akin D, Manier DH, Sanders-Bush E, Shelton RC. Decreased serotonin 5-HT2A receptor-stimulated phosphoinositide signaling in fibroblasts from melancholic depressed patients. Neuropsychopharmacology. 2004;29:2081–2087. doi: 10.1038/sj.npp.1300505. [DOI] [PubMed] [Google Scholar]
- 28.Akin D, Manier DH, Sanders-Bush E, Shelton RC. Signal transduction abnormalities in melancholic depression. Int J Neuropsychopharmacol. 2005;8:5–16. doi: 10.1017/S146114570400478X. [DOI] [PubMed] [Google Scholar]
- 29.Manier DH, Shelton RC, Ellis TC, Peterson CS, Eiring A, Sulser F. Human fibroblasts as a relevant model to study signal transduction in affective disorders. J Affect Disord. 2000;61(1–2):51–58. doi: 10.1016/s0165-0327(99)00190-1. [DOI] [PubMed] [Google Scholar]
- 30.Shelton RC, Liang S, Liang P, Chakrabarti A, Manier DH, Sulser F. Differential expression of pentraxin 3 in fibroblasts from patients with major depression. Neuropsychopharmacol. 2003 doi: 10.1038/sj.npp.1300307. in press. [DOI] [PubMed] [Google Scholar]
- 31.Pandey GN, Dwivedi Y, Pandey SC, Conley RR, Roberts RC, Tamminga CA. Protein kinase C in the postmortem brain of teenage suicide victims. Neurosci Lett. 1997;228(2):111–114. doi: 10.1016/s0304-3940(97)00378-9. [DOI] [PubMed] [Google Scholar]
- 32.Pandey GN, Dwivedi Y, Kumari R, Janicak PG. Protein kinase C in platelets of depressed patients. Biol Psychiatry. 1998;44:909–911. doi: 10.1016/s0006-3223(97)00535-0. [DOI] [PubMed] [Google Scholar]
- 33.Pandey GN, Dwivedi Y, SridharaRao J, Ren X, Janicak PG, Sharma R. Protein kinase C and phospholipase C activity and expression of their specific isozymes is decreased and expression of MARCKS is increased in platelets of bipolar but not in unipolar patients. Neuropsychopharmacology. 2002;26(2):216–228. doi: 10.1016/S0893-133X(01)00327-X. [DOI] [PubMed] [Google Scholar]
- 34.Dwivedi Y, Rao JS, Rizavi HS, Kotowski J, Conley RR, Roberts RC, et al. Abnormal expression and functional characteristics of cyclic adenosine monophosphate response element binding protein in postmortem brain of suicide subjects. Arch Gen Psychiatry. 2003;60(3):273–282. doi: 10.1001/archpsyc.60.3.273. [DOI] [PubMed] [Google Scholar]
- 35.Pandey GN, Dwivedi Y, Ren X, Janicak PG. American College of Neuropsychopharmacology, Annual Meeting. 2003. Decreased Protein Expression and CRE - DNA Binding Activity of Cyclic Adenosine Monophosphate Response Element Binding Protein in Neutrophil of Depressed Patients. Ref Type: Generic. [Google Scholar]
- 36.Hrdina P, Faludi G, Li Q, Bendotti C, Tekes K, Sotonyi P, et al. Growth-associated protein (GAP-43), its mRNA, and protein kinase C (PKC) isoenzymes in brain regions of depressed suicides. Molecular Psychiatry. 1998;3(5):411–418. doi: 10.1038/sj.mp.4000435. [DOI] [PubMed] [Google Scholar]
- 37.Coull MA, Lowther S, Katona CL, Horton RW. Altered brain protein kinase C in depression: a post-mortem study. Eur Neuropsychopharmacol. 2000;10(4):283–288. doi: 10.1016/s0924-977x(00)00084-5. [DOI] [PubMed] [Google Scholar]
- 38.Perez J, Tardito D, Racagni G, Smeraldi E, Zanardi R. Protein kinase A and Rap1 levels in platelets of untreated patients with major depression. Mol Psychiatry. 2001;6(1):44–49. doi: 10.1038/sj.mp.4000795. [DOI] [PubMed] [Google Scholar]
- 39.Perez J, Tardito D, Racagni G, Smeraldi E, Zanardi R. cAMP signaling pathway in depressed patients with psychotic features. Mol Psychiatry. 2002;7(2):208–212. doi: 10.1038/sj.mp.4000969. [DOI] [PubMed] [Google Scholar]
- 40.Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20(4):727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- 41.Meller R, Babity JM, Grahame-Smith DG. 5-HT2A receptor activation leads to increased BDNF mRNA expression in C6 glioma cells. NeuroMolecular Medicine. 2002;1(3):197–205. doi: 10.1385/NMM:1:3:197. [DOI] [PubMed] [Google Scholar]
- 42.Karege F, Schwald M, El Kouaissi R. Drug-induced decrease of protein kinase a activity reveals alteration in BDNF expression of bipolar affective disorder. Neuropsychopharmacology. 2004;29(4):805–12. doi: 10.1038/sj.npp.1300384. [DOI] [PubMed] [Google Scholar]
- 43.Deogracias R, Espliguero G, Iglesias T, Rodriguez-Pena A. Expression of the neurotrophin receptor trkB is regulated by the cAMP/CREB pathway in neurons. Mol Cell Neurosci. 2004;26(3):470–480. doi: 10.1016/j.mcn.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 44.Barrett TJ, Vedeckis WV. Occupancy and composition of proteins bound to the AP-1 sites in the glucocorticoid receptor and c-jun promoters after glucocorticoid treatment and in different cell types. Recept Signal Transduct. 1996;6(3–4):179–193. [PubMed] [Google Scholar]
- 45.Malkoski SP, Dorin RI. Composite glucocorticoid regulation at a functionally defined negative glucocorticoid response element of the human corticotropin-releasing hormone gene. Mol Endocrinol. 1999;13(10):1629–1644. doi: 10.1210/mend.13.10.0351. [DOI] [PubMed] [Google Scholar]
- 46.Qi M, Zhuo M, Skalhegg BS, Brandon EP, Kandel ER, McKnight GS, et al. Impaired hippocampal plasticity in mice lacking the Cbeta1 catalytic subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1996;93(4):1571–1576. doi: 10.1073/pnas.93.4.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, et al. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol. 2000;279(3):L429–L438. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 48.Nguyen PV, Woo NH. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol. 2003;71(6):401–437. doi: 10.1016/j.pneurobio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 49.Duman RS. Synaptic plasticity and mood disorders. Mol Psychiatry. 2002;7(Suppl 1):S29–34. S29–S34. doi: 10.1038/sj.mp.4001016. [DOI] [PubMed] [Google Scholar]
- 50.Adler V, Yin Z, Tew KD, Ronai Z. Role of redox potential and reactive oxygen species in stress signaling. Oncogene. 1999;18(45):6104–6111. doi: 10.1038/sj.onc.1203128. [DOI] [PubMed] [Google Scholar]
- 51.Dickinson DA, Moellering DR, Iles KE, Patel RP, Levonen AL, Wigley A, et al. Cytoprotection against oxidative stress and the regulation of glutathione synthesis. Biol Chem. 2003;384(4):527–537. doi: 10.1515/BC.2003.061. [DOI] [PubMed] [Google Scholar]
- 52.Dickinson DA, Forman HJ. Glutathione in defense and signaling: lessons from a small thiol. Ann N Y Acad Sci. 2002;973:488–504. doi: 10.1111/j.1749-6632.2002.tb04690.x. [DOI] [PubMed] [Google Scholar]
- 53.Humphries KM, Juliano C, Taylor SS. Regulation of cAMP-dependent protein kinase activity by glutathionylation. J Biol Chem. 2002;277(45):43505–43511. doi: 10.1074/jbc.M207088200. [DOI] [PubMed] [Google Scholar]
- 54.Licinio J, Wong ML. The role of inflammatory mediators in the biology of major depression: central nervous system cytokines modulate the biological substrate of depressive symptoms, regulate stress-responsive systems, and contribute to neurotoxicity and neuroprotection. Molecular Psychiatry. 1999;4(4):317–327. doi: 10.1038/sj.mp.4000586. [Review] [92 refs] [DOI] [PubMed] [Google Scholar]
- 55.Maes M. The immunoregulatory effects of antidepressants. Hum Psychopharmacol. 2001;16(1):95–103. doi: 10.1002/hup.191. [DOI] [PubMed] [Google Scholar]
- 56.Maes M, Kenis G, Kubera M, De Baets M, Steinbusch H, Bosmans E. The negative immunoregulatory effects of fluoxetine in relation to the cAMP-dependent PKA pathway. Int Immunopharmacol. 2005;5(3):609–618. doi: 10.1016/j.intimp.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 57.Jenuwein T, Allis CD. Translating the Histone Code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 58.Jenuwein T, Allis CD. Translating the histone code. [see comment] Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [Review] [94 refs] [DOI] [PubMed] [Google Scholar]
- 59.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 60.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genetics. 2003;33:Suppl-54. doi: 10.1038/ng1089. [Review] [186 refs] [DOI] [PubMed] [Google Scholar]
- 61.Champagne FA, Weaver IC, Diorio J, Dymov S, Szyf M, Meaney MJ. Maternal care associated with methylation of the estrogen receptor-alpha1b promoter and estrogen receptor-alpha expression in the medial preoptic area of female offspring. Endocrinology. 2006 Jun;147:2909–2915. doi: 10.1210/en.2005-1119. [DOI] [PubMed] [Google Scholar]
- 62.Weaver IC, Champagne FA, Brown SE, Dymov S, Sharma S, Meaney MJ, et al. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J Neurosci. 2005;1925:11045–11054. doi: 10.1523/JNEUROSCI.3652-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Szyf M, Weaver IC, Champagne FA, Diorio J, Meaney MJ. Maternal programming of steroid receptor expression and phenotype through DNA methylation in the rat. Front Neuroendocrinol. 2005;1926:139–162. doi: 10.1016/j.yfrne.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 64.Meaney MJ, Szyf M. Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues Clin Neurosci. 2005;2007:103–123. doi: 10.31887/DCNS.2005.7.2/mmeaney. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fish EW, Shahrokh D, Bagot R, Caldji C, Bredy T, Szyf M, et al. Epigenetic Programming of Stress Responses through Variations in Maternal Care. Ann N Y Acad Sci. 2004 Dec;1036:167–180. doi: 10.1196/annals.1330.011. [DOI] [PubMed] [Google Scholar]
- 66.Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;2007:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 67.Weaver IC, Szyf M, Meaney MJ. From maternal care to gene expression: DNA methylation and the maternal programming of stress responses. Endocr Res. 2002;1928:699. doi: 10.1081/erc-120016989. [DOI] [PubMed] [Google Scholar]