

Abstract
Kinesin and myosin have been proposed to transport intracellular organelles and vesicles to the cell periphery in several cell systems. However, there has been little direct observation of the role of these motor proteins in the delivery of vesicles during regulated exocytosis in intact cells. Using a confocal microscope, we triggered local bursts of Ca2+-regulated exocytosis by wounding the cell membrane and visualized the resulting individual exocytotic events in real time. Different temporal phases of the exocytosis burst were distinguished by their sensitivities to reagents targeting different motor proteins. The function blocking antikinesin antibody SUK4 as well as the stalk-tail fragment of kinesin heavy chain specifically inhibited a slow phase, while butanedione monoxime, a myosin ATPase inhibitor, inhibited both the slow and fast phases. The blockage of Ca2+/calmodulin-dependent protein kinase II with autoinhibitory peptide also inhibited the slow and fast phases, consistent with disruption of a myosin-actin– dependent step of vesicle recruitment. Membrane resealing after wounding was also inhibited by these reagents. Our direct observations provide evidence that in intact living cells, kinesin and myosin motors may mediate two sequential transport steps that recruit vesicles to the release sites of Ca2+-regulated exocytosis, although the identity of the responsible myosin isoform is not yet known. They also indicate the existence of three semistable vesicular pools along this regulated membrane trafficking pathway. In addition, our results provide in vivo evidence for the cargo-binding function of the kinesin heavy chain tail domain.
To dock and fuse with the plasma membrane in response to localized calcium influx, vesicles for Ca2+-regulated exocytosis must first be recruited to the cell surface from intracellular pools. Although much has been learned about the molecular mechanisms of the docking and fusion reactions of regulated exocytosis (Sudhof et al., 1993; Bennett and Scheller, 1994; De Camilli, 1995), the few studies of the vesicle recruitment process have focused on membrane recycled from endocytosis (Betz and Bewick, 1992; Ryan et al., 1993; Betz and Wu, 1995), and as yet there has been little direct in vivo observation of the roles of motor proteins in recruiting vesicles to exocytotic sites (Scholey, 1996; Vallee and Sheetz, 1996).
The motor protein kinesin is a good candidate for part of the transport machinery in the pathway of regulated exocytosis. Kinesin has been demonstrated to move along microtubule tracks towards the plus end by hydrolyzing ATP in several in vitro assays (Goldstein, 1993; Bloom and Endow, 1995). It has also been shown to associate with vesicle and organelle membranes in different cell types (Bloom and Endow, 1995). Several antikinesin antibodies were able to inhibit fast axonal transport (Vale et al., 1985b ; Brady et al., 1990), the centrifugal migration of pigment granules (Rodionov et al., 1991), and the formation of tubular lysosomal structure (Hollenbeck and Swanson, 1990). Mutations in Drosophila kinesin impaired the transport of membrane proteins to their appropriate cellular locations (Saxton et al., 1991; Gho et al., 1992). Based on these findings, it has been widely predicted that this motor protein will be shown to play an essential role in transporting vesicles to sites of Ca2+-regulated exocytosis.
A kinesin holoenzyme is composed of two identical heavy chains and two light chains. The kinesin heavy chain (KHC)1 consists of an amino-terminal globular head domain that is linked to the carboxyl-terminal tail domain through a stalk region that dimerizes two KHCs to form the kinesin motor (Fig. 1) (Yang et al., 1989). The KHC head domain is highly conserved among different kinesin-related proteins (KRPs) and has been shown to be responsible for ATP hydrolysis and force generation (Yang et al., 1990). The tail domain is more variable and is thought to be important for kinesin cargo binding (Hirokawa et al., 1989; Bloom and Endow, 1995). This was further supported by in vitro observations that the bacterially expressed stalk-tail fragment, but not the stalk fragment of sea urchin KHC, was able to bind microsomal membranes isolated from sea urchin eggs in a saturable manner and compete with native kinesin for membrane binding (Skoufias et al., 1994). However, an in vivo demonstration has been difficult because the in vivo function of sea urchin kinesin was not known.
Figure 1.

Primary sequence of KHC. Arrows indicate approximate sites for antibody recognition. Numbers refer to KHC amino acid sequence number starting from the amino terminus. The “Stalk-Tail” and “Stalk” are the two KHC fragments used in this experiment.
The actin-based motor myosin is another candidate that may drive vesicle recruitment in regulated exocytotic pathways (Fath and Burgess, 1994; Hasson and Mooseker, 1995; Langford, 1995). Evidence that some of the myosin isoforms may power membrane transport came from several systems including yeast (Johnston et al., 1991; Drubin and Nelson, 1996), algae (Adams and Pollard, 1986; Grolig et al., 1988), squid axoplasm (Kuznetsov et al., 1992), and polarized epithelial cells (Fath et al., 1994). There was also evidence for the presence of both microtubule- and actin-based motors on the same membranous organelles (Fath et al., 1994). However, with one exception, there has been no in vivo demonstration for the role of myosin in regulated exocytosis, and the exact interrelationship between the microtubule-based and actin-based systems has yet to be elucidated (Langford, 1995). The one exception is a study in which a smooth muscle antimyosin II antibody microinjected into presynaptic neurons inhibited synaptic transmission (Mochida et al., 1994). Transmission was also inhibited in a dose-dependent manner by two inhibitors of myosin light chain kinase. The identity of the neuronal myosin subtype affected in this study remains an open question since other studies have failed to find myosin II presynaptically (Miller et al., 1992).
Ca2+/calmodulin-dependent protein kinase II (CaM kinase II) has been implicated in the regulation of exocytosis at an actin binding step (Ceccaldi et al., 1995). CaM kinase II injected presynaptically in squid giant synapse was found to facilitate transmitter release (Llinas et al., 1991). Additionally, extracellular application of synthetic peptide inhibitors of CaM kinase II preferentially suppressed the phosphorylation of synapsin I at the CaM kinase II–specific site and decreased excitatory synaptic responses elicited in the CA1 region of hippocampal slices (Waxham et al., 1993). Recently, Ceccaldi showed that the presence of dephosphorylated synapsin I is necessary for synaptic vesicles to bind actin, and the effect is abolished upon its phosphorylation by CaM kinase II (Ceccaldi et al., 1995). Therefore, CaM kinase II is postulated to play an essential role in recruitment of vesicles since its action would be required to free vesicles from the actin-bound pool.
Using a confocal microscope, we were able to visualize and quantify individual exocytotic events in real time by triggering local bursts of regulated exocytosis in early sea urchin embryonic cells by wounding the cell membrane with a laser beam and inducing Ca2+ influx through the wound (Bi et al., 1995; Terasaki, 1995). This system allowed us to directly investigate the in vivo roles of protein motors in the recruitment of vesicles to exocytotic sites and address several related issues. By analyzing the temporal patterns of exocytosis when the functions of kinesin, myosin, and CaM kinase were inhibited, we concluded that exocytotic vesicles were recruited by a two-step transport mechanism mediated by kinesin and myosin sequentially. Our results also revealed distinct intermediate vesicle pools along the transport pathway. In addition, our results demonstrated the in vivo cargo binding function of the KHC tail domain.
Materials and Methods
Materials
For isolation and purification of bacterially expressed KHC fragments, P11 cellulose phosphate cation exchanger was obtained from Whatman International Ltd. (Maidstone, Kent, UK). DEAE Bio-Gel A (100–200 mesh) anion exchange resin was obtained from Bio-Rad Laboratories (Hercules, CA). All other reagents were from Sigma Chemical Co. (St. Louis, MO) unless otherwise specified. SUK4 and SUK2 monoclonal antibody stocks were purified IgG isolated on protein A columns at 4.5 and 3 mg/ml in PBS. They were mixed with Fura-2 salt and aspartate buffer (containing 100 mM potassium aspartate, 20 mM Hepes, pH 7.2) to make the injection solutions that contained 2.7 mg/ml of either antibody.
2, 3-butanedione monoxime (BDM, from Sigma Chemical Co.) was dissolved into natural sea water to make a fresh stock of 0.5 M on the same day of the experiment. It was then added into sea water to reach required final concentrations. Washes were performed by replacing the extracellular BDM solution with fresh natural sea water at least three times in 5 min. Both the powder and solutions of BDM were kept in dark to avoid light inactivation.
CaM kinase peptides CaMK(273-302) and CaMK(284-302) were synthesized using an automated synthesizer (Applied Biosystems, Inc., Foster City, CA), purified by HPLC, and assessed by sequencing. CaMK(273-302) and CamK(284-302) peptide stocks were 2.5 and 5 mM in aspartate buffer (containing 100 mM potassium aspartate, 20 mM Hepes, pH 7.2). They were mixed with Fura-2 potassium salt to make the injection solutions that contained 0.25 and 0.5 mM of either peptide.
Isolation and Purification of Bacterially Expressed KHC Fragments
Previously, two Escherichia coli bacterial strains were transformed with T7 vectors containing partial inserts of sea urchin conventional kinesin heavy chain: one 1.4-kb insert encoded the tail and a portion of the stalk (amino acids 579–1031) and one 0.8-kb insert encoded a portion of the stalk only (amino acids 579–858), as described (Skoufias et al., 1994). Isolation of the KHC fragments was performed as previously described (Skoufias et al., 1994) with the following modifications. Frozen transformed cell stocks were grown up in Luria Broth containing 50 μg/ml each of ampicillin and chloramphenicol. Bacteria were induced, harvested, washed as before, and then lysed by sonication followed by three rounds of freezing and thawing in lysis buffer as described (Skoufias et al., 1994). Stalk and stalk-tail fragments were isolated by ion exchange chromatography in 20 mM Tris, pH 8, 6 M urea, 1 mM DTT and eluted as before, except DEAE Bio-Gel A was substituted for DE52 DEAE. Column elutates were dialyzed sequentially with 1 liter of 3 and 1.5 M urea in 20 mM Tris, pH 8, 1 mM EGTA, 1 mM EDTA, 150 mM NaCl, 1 mM MgSO4, 2 mM DTT, plus protease inhibitors (0.1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 2 μg/ml aprotinin, 20 μg/ml benzamidine, and 1 mg/ml TAME). Final purification was performed by sedimentation through a 1–15% sucrose density gradient in the same buffer with 1.5 M urea. Peak gradient samples were pooled and dialyzed sequentially against 1 liter each of 1.5, 0.75, and 0 M urea in aspartate buffer that contained 100 mM l-aspartic acid, 20 mM Hepes, pH 7.2, plus protease inhibitors. Final concentrations of KHC stalk fragment was 2.7 mg/ml = 80 μM; stalk-tail fragment was 0.75 mg/ml = 14.5 μM.
Cells, Microinjection, and Test of Resealing
Lytechinus pictus sea urchins (from Marinus Inc., Long Beach, CA) were handled as previously described (Bi et al., 1995). Microinjections and resealing tests were performed as described (Steinhardt et al., 1994; Bi et al., 1995). Briefly, eggs and sperm were obtained by injection of 0.55 M KCl into the sea urchin interperitoneal cavity. Sperm was collected dry and stored at 4°C for up to 1 wk. Eggs were collected in natural sea water, dejellied by passing through 80-μm Nitex mesh (Tetko Inc., Depew, NY), washed three times in natural sea water, and were allowed to settle onto glass coverslips coated with 10 mg/ml poly-dl-lysine (Sigma Chemical Co.). Kinesin reagents or buffer were pressure-injected into unfertilized or newly fertilized sea urchin eggs. All injection solutions contained 2.5 mM Fura-2 salt (Molecular Probes, Eugene, OR) as a fluorescent marker for injected cells. Injection volume was about 5% of cell volume. For tests of resealing, injected cells were wounded in natural sea water with a glass micropipette that penetrated the plasma membrane and was withdrawn immediately after wounding. The size of this wounding is similar to that produced by a typical microinjection. Resealing was monitored by photometric measurement of Fura-2 emission fluorescence at 510 nm after alternate excitation with 357- and 385-nm ultraviolet light (Steinhardt et al., 1994) and subsequently confirmed visually.
Confocal Microscopy
Confocal microscopy was performed using a confocal microscope (model MRC-600; Bio-Rad Labs) equipped with a 40× Neofluor objective (NA = 1.3, oil immersion; Carl Zeiss, Inc., Thornwood, NY), a 15 mW argon ion laser, and a UV epifluorescence system (Carl Zeiss, Inc.). For microscopy, eggs settled on poly-lysine–coated coverslips were injected within 5 min after fertilization and were allowed to develop to two-cell stage (∼2 h at 16.5°C) before being mounted into a handmade chamber that held ∼150 μl of natural sea water containing 100 μM rhodamine dextran (3,000 molecular weight; Molecular Probes). Confocal experiments were performed at 20°C. Two- to four-cell stage embryos with relatively flat surfaces adhering to the coverslip were selected for experiments. Injected embryos could be easily identified by their Fura-2 fluorescence with UV epifluorescence filters (360 nm excitation and >400 nm emission).
During each imaging series, the cell was first wounded by “parking” the confocal beam at maximum power (neutral density filter setting at ND 0) focused at a selected position on the cell surface for 10–25 s to trigger local exocytosis. Neutral density filter setting ND 3 was then used for subsequent imaging to attenuate the beam power and reduce cell damage. The confocal scanning plane was focused just inside the cell surface to visualize the formation of bright fluorescent “disks.” These disks have been shown to be exocytotic vesicle pockets filled with extracellular fluorescent dextrans, based on their rapid formation and their sensitivity to proteolytic neurotoxins that specifically target the exocytosis machinery (Bi et al., 1995). Cells were scanned continuously (1 frame/s), and image frames were collected and stored when new exocytotic events occurred or after a period of time (∼20 s) without new events. A semiautomatic macro program was written to perform these operations. Images were processed by NIH Image (Bethesda, MD) and Adobe Photoshop (San Jose, CA). Quantification was performed manually during playback of recorded image series by direct counting the number of new exocytotic events in each frame.
Results
A Slow Phase of Ca2+-triggered Exocytosis Requires a Functional Kinesin Motor
We used the laser beam of a confocal microscope to wound the membranes of embryonic sea urchin cells so that Ca2+ influx through the wound would trigger a burst of local exocytosis (Bi et al., 1995). By setting the confocal scanning plane right underneath the cell surface, individual exocytotic events could be visualized as bright disks in the confocal images (Bi et al., 1995; Terasaki, 1995). In Fig. 2, each newly emerged bright disk indicated by an arrow represents a single newly exocytosed vesicle. Under normal conditions, the exocytotic burst usually lasts for about a minute and ceases after the wound reseals, Ca2+ influx stops, and intracellular free Ca2+ levels return to normal (Fig. 2 A). To investigate the possible transport role of the motor protein kinesin (Scholey et al., 1985; Vale et al., 1985a ) in this exocytotic pathway, we injected cells with SUK4, a monoclonal antibody targeting the motor domain of KHC (Fig. 1) (Wright et al., 1991). This antibody had been shown to block the kinesin-mediated microtubule motility in vitro (Ingold et al., 1988) and to inhibit exocytosis-dependent membrane resealing (Steinhardt et al., 1994). Surprisingly, the early exocytotic events were virtually unaffected by the antibody injection (5 to 10 s in Fig. 2 B). However, the later exocytotic events were inhibited (Fig. 2 B, 26 s and later). As a control, some cells were injected with another antikinesin antibody, SUK2, that targets the kinesin stalk domain (Fig. 1) (Wright et al., 1991) and does not interfere with the motor function (Ingold et al., 1988) or membrane resealing (Steinhardt et al., 1994) at similar intracellular concentrations. These cells exhibited a normal pattern of exocytosis in response to Ca2+ influx through the laser wound (Fig. 2 C).
Figure 2.
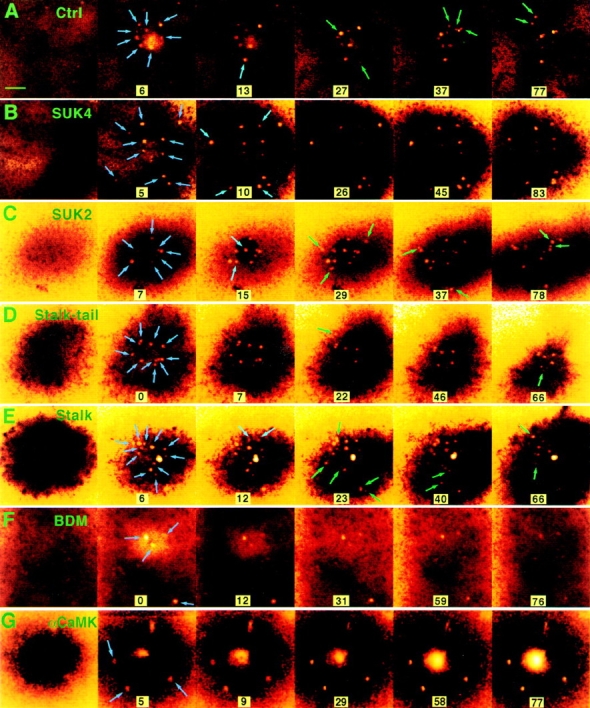
Effect of reagents targeted against motor proteins on Ca2+-regulated exocytosis induced by laser wounding. The pseudocolor pictures are confocal fluorescence images of extracellular rhodamine dextran showing the exocytotic pockets into confocal focal plane just under the plasma membrane (Bi et al., 1995). Exocytotic events are visualized as the appearance of bright disks (0.5–1 μm diameter) against the dark intracellular background indicated by arrows. Blue arrows indicate early events, while green arrows indicate later events. (A) The exocytotic pattern of a sea urchin embryonic cell in natural sea water. In other series, the embryos were previously injected with (B) SUK4, (C) SUK2 antikinesin, (D) stalk-tail fragment, and (E) stalk fragment of KHC and were kept in natural sea water. In series F, the embryo was treated with 50 mM BDM. In series G, the embryo was injected with CaMK(273-302), an autoinhibitory peptide from the regulatory domain of CaM kinase type II. The large central stain in G is dye entering the unhealed wound. The first frame in each series was collected before wounding. Time 0 is defined as the moment immediately after wounding. The unit for time labels is seconds. Arrows indicate new exocytotic events that occurred since the previous frame. Bar, 5 μm.
Fig. 3 A shows examples of the quantified time course of exocytosis after individual membrane wounds. Fig. 3 B averages the results from 17 or more experiments and reveals two distinct phases in the temporal pattern of this regulated exocytosis: a slow phase that is inhibited by SUK4 and a fast one not affected by this function-blocking antibody. Although we can not exclude the possibility that kinesin may play some role in the organization of intracellular structure, which may in turn affect the temporal pattern of exocytosis, it appears more likely that this slow phase simply represents a kinesin-mediated step in the regulated exocytosis pathway, the step of vesicle recruitment on microtubules directed toward the release site of membrane wounding and Ca2+ influx. Furthermore, since conventional kinesin is currently the only known KRP that is recognized by SUK4, we suspect that this most abundant KRP is responsible for the transport of vesicles in this ubiquitous regulated exocytosis pathway.
Figure 3.

Specific inhibition of a slow phase of Ca2+-regulated exocytosis by function-blocking antikinesin antibody SUK4. A quantifies cumulative number of exocytotic events in individual cells from typical experiments. The control cell (Ctrl) was not injected with any reagent. B summarizes the average number of exocytotic events that occurred within different time ranges from n experiments. n = 37 for Ctrl, 17 for SUK4, and 23 for SUK2. A biphasic temporal pattern of exocytosis is seen by comparing the SUK4 injection data with the Ctrl or SUK2 injection data. The slow phase (after 16 s), but not the fast phase (0–15 s) of exocytosis is inhibited by SUK4 antikinesin. Error bars are standard errors.
Kinesin Mediates Vesicle Transport for Ca2+-regulated Exocytosis through Its Cargo-binding Tail Domain
To confirm that the effect of SUK4 was due to inhibition of kinesin-mediated vesicle transport and to further identify the molecular identity of the responsible KRP isoform, we generated the stalk-tail and stalk only fragments of KHC expressed in bacteria (Fig. 1). Here the term “kinesin” refers to the conventional kinesin, as opposed to other KRPs. In an in vitro assay, this stalk-tail fragment, but not the stalk fragment, effectively competed with kinesin for vesicle binding (Skoufias et al., 1994). If the KHC tail domain is responsible for vesicular cargo binding in vivo, as has been postulated, introduction of the stalk-tail fragment, but not the stalk alone into the cell, should arrest kinesin-mediated transport by competition for the binding of cargo receptors. If kinesin, but not other KRPs, does mediate vesicle recruitment for regulated exocytosis, this arrest of transport should alter the temporal pattern of exocytosis, as did the SUK4 antibody.
We injected the cells with the stalk-tail fragment at ∼650 nM intracellular concentration. This is 32-fold greater than the K d of stalk-tail binding to microsomes (Skoufias et al., 1994). These cells indeed showed a block in the slow phase of exocytosis similar to that seen in cells injected with SUK4 (Figs. 2 D and 4). In contrast, stalk fragment injection (intracellular concentration ∼2.4 μM) had no effect on exocytosis (Fig. 2 E and 4). Because the tail domain is less conserved among different KRPs, the stalk-tail competition for putative vesicle receptors should be very specific. It is therefore highly likely that conventional kinesin is the protein motor that drives vesicle recruitment in Ca2+-regulated exocytosis. In addition, this result also provides direct in vivo evidence for the postulated vesicular cargo binding function of the kinesin tail domain.
Figure 4.

Specific inhibition of the slow phase of Ca2+-regulated exocytosis by KHC stalk-tail fragment. (A) Typical examples of the cumulative number of exocytotic events in individual cells. (B) The average number of exocytotic events that occurred within different time ranges from n experiments. n = 37 for Ctrl, 27 for Stalk-tail, and 33 for Stalk.
A Myosin Motor May Mediate Vesicle Transport at a Step Downstream to Kinesin Transport
Members of the myosin family have also been implicated in membranous organelle transport in several systems (Adams and Pollard, 1986; Grolig et al., 1988; Kuznetsov et al., 1992). Does it play any role in the regulated exocytotic pathway? If yes, what is the relationship between the kinesin–microtubule and actomyosin transport systems? To address these questions, we treated the cells with BDM, a known reversible inhibitor of myosin ATPase that has been shown to block myosin-dependent cell spreading in epithelial cells (Cramer and Mitchison, 1995) and retrograde F-actin flow in neuronal growth cones (Lin et al., 1996) but has not been found to affect kinesin ATPase activity (Cramer and Mitchison, 1995). BDM is at present the only potent general inhibitor of myosin function available. In the absence of knowledge of specific myosin isoforms and in the absence of more specifically targeted reagents, BDM is as close as one can get now to implicating myosin in a functional assay. BDM is fully reversible in functional studies of myosin. Exocytosis was reversibly inhibited by 50 mM BDM (Figs. 2 F and 5). In contrast to the inhibition by SUK4 and KHC stalk-tail, BDM inhibited exocytotic events from both the slow and the fast phase (Fig. 5). This is particularly obvious when comparing the exocytosis under these conditions within the 6–15-s range (Fig. 3–5). The onset of BDM effect on exocytosis pattern took about 10 min. After that, the exocytosis pattern was stable for at least 1 h, until BDM was washed away. Exocytosis recovered to its normal pattern within 15 min after BDM was washed away (Fig. 5).
Figure 5.

Reversible inhibition of both the slow and the fast phases of exocytosis by BDM. The cells were in 50 mM BDM for 10–60 min. For “Wash” experiments, cells were in 50 mM BDM for at least 45 min and were then transferred to BDM-free sea water for at least 15 min before imaging. (A) Quantified examples of individual experiments under different conditions. (B) Average number of exocytotic events that occurred within different time ranges from n experiments. n = 37 for Ctrl, 17 for BDM, and 8 for Wash.
To make sure that the BDM effect was not due to any inhibition of the vesicle fusion step of exocytosis, we tested exocytosis of previously docked cortical granules found at the plasma membrane in unfertilized sea urchin eggs at the same BDM concentration. Exocytosis of these vesicles was not affected. (Nine out of nine cells tested showed normal exocytosis in response to membrane wounding; data not shown.) This is also consistent with the fact that in embryonic cells, BDM inhibited later events (>5 s after wounding) to a much higher degree than the initial burst (Fig. 5). The initial burst of exocytosis is most likely from previously docked vesicles. Therefore, we believe that BDM affected the regulated exocytosis at a step upstream of the final fusion event. Furthermore, this step, possibly a myosin-mediated transport step, appears to be downstream of the kinesin transport step, because in addition to the slow phase of exocytosis, BDM also inhibited the fast phase of exocytosis that was not affected by our blocks of kinesin function.
To obtain further evidence consistent with an actin-based step in vesicle recruitment, we investigated the possible role of CaM kinase II by injecting cells with CaMK(273-302), an autoinhibitory peptide from the regulatory domain of CaM kinase. Although CaM kinase II is a multifunctional enzyme and could have multiple targets, we examined its specific effect on vesicle recruitment to sites of Ca2+-regulated exocytosis. CaMK(273-302) had been previously shown to inhibit a CaM kinase II–specific Ca2+/calmodulin-dependent phosphorylation in cell extracts of Lytechinus pictus (Baitinger et al., 1990) and to inhibit exocytosis-dependent membrane resealing (Steinhardt et al., 1994). Figs. 3 G and 6 A show typical examples of cumulative exocytotic events. Fig. 6 B averages the results from 18 or more experiments and reveals that both fast and slow vesicle exocytosis have been inhibited by CaMK(273-302). CaMK(273-302) did not block exocytosis completely, which is consistent with its action on CaM kinase II in vitro (Baitinger et al., 1990). To demonstrate that the inhibition was not on the fusion of vesicles, we tested the effect of CaMK(273-302) on laser wound–triggered exocytosis of previously docked cortical granules found at the plasma membrane in unfertilized sea urchin eggs. Exocytosis of these vesicles was not affected (four of four cells tested). As a control, some cells were injected with the shorter peptide, CaMK(284-302), which does not inhibit Ca2+/calmodulin-dependent phosphorylation in vitro (Baitinger et al., 1990) or membrane resealing (Steinhardt et al., 1994). Those cells exocytosed normally in response to Ca2+ influx through the laser wound (Fig. 6). It appears that CaMK(273-302) and BDM have similar effects on the recruitment of vesicles, a result consistent with a block of the late step of the vesicle transportation process from the actin matrix to the plasma membrane.
Figure 6.

Inhibition of both the slow and the fast phases of exocytosis by CaMK(273-302), an autoinhibitory peptide from the regulatory domain of CaM kinase type II. Control peptide CaMK(284-302) did not inhibit exocytosis. (A) Typical examples of cumulative number of exocytotic events in individual cells. (B) The average number of exocytotic events that occurred within different time ranges from n experiments. n = 18 for Ctrl, 39 for CaMK(273-302), and 22 for CaMK(284-302).
Three Semistable Vesicle Pools in the Pathway of Regulated Exocytosis
One interesting feature in the above results is that blocking either motor system could not completely block injury-induced regulated exocytosis. When kinesin transport was blocked, only the slow phase of exocytosis was inhibited. The fast phase was virtually intact (Figs. 3 and 4). With BDM inhibition of putative myosin transport, a significant portion of the very fast events still persisted (Fig. 5). If the vesicle recruitment process is a simple chain of constitutive sequential steps and the regulation is only at the final exocytosis step, a blockade at any point in the chain should similarly inhibit the whole process. However, the above experiments showed distinct temporal patterns when different motors were blocked, indicating the existence of semistable intermediate vesicle pools that may be regulated separately in the membrane transport pathway.
Quantification of the temporal patterns of exocytosis under different conditions (Figs. 3–5) allows us to mathematically separate these vesicle pools. As shown in Figure 7 A, an “Immediate” pool of vesicles could be defined as those that can exocytose in the presence of BDM because they respond to Ca2+ influx most rapidly (within a few seconds) and do not seem to depend on either myosin or kinesin transport mechanisms. By subtracting the myosin- independent (BDM-insensitive) component from the kinesin-independent exocytosis (average of SUK4 and stalk-tail data), a “Fast” pool could be defined as vesicles whose exocytosis are myosin-dependent but kinesin independent. Exocytosis of vesicles from this fast pool usually took place within 10 s after Ca2+ influx. Finally, a “Slow” pool could be isolated by subtracting the kinesin-independent exocytosis data (summation of immediate and the fast pools) from the average of control experiments including the control, SUK2, and Stalk data from Figs. 3–5. Apparently, vesicles in this slow pool require normal levels of kinesin and myosin transport for their final exocytosis. These vesicles take a much longer time to exocytose (peaking at 30 s) than those in the first two pools, presumably because of the delay in long-distance transport by the kinesin–microtubule system, as will be discussed later.
Motor-driven Vesicle Recruitment Is Essential for Cell Membrane Resealing
Although the main objective of this study was to directly observe motor-driven vesicle recruitment steps in a regulated pathway in vivo, we also investigated the role of these steps in cell membrane resealing (Steinhardt et al., 1994; Bi et al., 1995; Miyake and McNeil, 1995). After injection into the cell, the stalk-tail fragment of KHC inhibited membrane resealing in sea urchin embryonic cells or activated eggs (Table I) when tested by mechanical wounding with a glass needle, as did the SUK4 antibody (Steinhardt et al., 1994). Stalk injection had no significant effect on resealing (Table I). As expected, BDM treatment also inhibited membrane resealing in activated eggs or embryonic cells but not in unfertilized eggs, which all have thousands of cortical vesicles already docked at their plasma membrane (Table II). We also found that resealing in activated eggs and embryonic cells was inhibited by cytoskeleton-disrupting drugs. (Nine out of nine cells tested failed to reseal after 0.5 h in 25–30 μM nocodozole, which disrupts microtubules; 10 out of 10 cells failed to reseal after 0.5 h in 10–20 μM cytochalasin D or B, which both disrupt actin filaments.) This is consistent with the function of microtubules and actin filaments as tracks for appropriate protein motors. Therefore, both kinesin–microtubule and actomyosin transport systems appeared to be not only important for the ubiquitous process of Ca2+-regulated exocytosis but were also essential for normal cell membrane resealing.
Table I.
Effect of KHC fragments on Cell Membrane Resealing in Activated Eggs of L. pictus
| Percent resealing (n) | ||||||
|---|---|---|---|---|---|---|
| Reagents injected | 10–30 min | 31–60 min | 61–120 min | |||
| Stalk-tail | 100 (13) | 0 (17) | 0 (9) | |||
| Stalk | 100 (13) | 100 (17) | 100 (11) | |||
Embryonic cells were injected with fragments of KHC and were then wounded by micropipette penetration at different times after injection. Wounding and resealing were monitored both by Fura-2 fluorescence and by visual inspection (Steinhardt et al., 1994; Bi et al., 1995). Stalk-tail injection completely inhibited resealing after a delay of 30 min. Injection of stalk fragment had no effect on resealing. n is the total number of cells tested (same for Table II).
Table II.
Effect of BDM on Cell Membrane Resealing in Activated Eggs of L. pictus
| Concentration of BDM | Percent resealing (n) | |||||||
|---|---|---|---|---|---|---|---|---|
| 1–20 min | 21–40 min | 41–60 min | Wash | |||||
| 20 | 100 (15) | 100 (22) | 100 (28) | – | ||||
| 40 | 100 (25) | 59 (22) | 0 (17) | – | ||||
| 50 | 74 (27) | 10 (30) | 0 (42) | 100 (16) | ||||
Cells were treated with BDM and then wounded to test for resealing at different times. At high concentration of BDM (50 mM), cells showed failure to reseal as early as 8 min after BDM addition. The inhibition of resealing was completely reversed after BDM was washed away. (Cells were tested 15–45 min after BDM washout.) In parallel experiments, resealing in unfertilized sea urchin eggs was not affected by 50 mM BDM (100% normal resealing 21–70 minutes after BDM addition, n = 15).
Discussion
During the past few years, a great deal has been learned about vesicle docking and fusion mechanisms (Sudhof et al., 1993; Bennett and Scheller, 1994; De Camilli, 1995) in Ca2+-regulated exocytosis. However, much less is known about the recruitment of vesicles in the regulated exocytotic pathway (Scholey, 1996; Vallee and Sheetz, 1996). In these experiments, the distinct temporal patterns of inhibition of exocytosis by reagents targeting two different motor systems are consistent with two sequential motor-driven recruitment steps for vesicle exocytosis—a kinesin-mediated transport step followed by an actomyosin-based step. Fig. 7 B summarizes the essential features of this transport/exocytosis pathway. This picture is also consistent with the fact that astral microtubules extend from the cell center to the cell periphery, while short actin filaments form networks near the cell membrane (Hollenbeck and Cande, 1985), and with the coexistence of kinesin–microtubule and actomyosin motor systems on the same organelles (Fath et al., 1994; Morris and Hollenbeck, 1995). Similar mechanisms have been postulated in other systems based on elegant in vitro observations (Fath et al., 1994; Allan, 1995; Langford, 1995). Our results, however, provide in vivo evidence for this postulated two-step pathway in general vesicular trafficking. Furthermore, our results also indicate that the recruitment of an average vesicle may need about 20 s of kinesin transport and about 5–10 s of myosin transport. Translating into spatial measures, these numbers correspond to about 20 μm of microtubule track and about 5–10 μm of actin filament track, assuming the average motor speeds are both around 1 μm/s (Vale et al., 1985b ; Porter et al., 1987; Langford et al., 1994; Bearer et al., 1996). Considering that the cell size is ∼40 μm in diameter for a four-cell stage embryo, there is enough time to recruit vesicles from any intracellular location to the cell surface.
Figure 7.

Three distinct vesicle pools and two-step vesicular recruitment for Ca2+-regulated exocytosis. (A) Three vesicle pools and their temporal distribution of exocytosis rates (number of exocytotic events per unit time) based on the data shown in Figs. 3–5. The “Immediate” pool of vesicles (squares) are BDM insensitive (and also kinesin reagents insensitive) and are therefore not dependent of either kinesin or myosin transport mechanism. The “Fast” pool (diamonds) is myosin dependent but kinesin independent. It was calculated by subtracting the myosin-independent (BDM-insensitive) component from the kinesin-independent exocytosis (average of SUK4 and Stalk-tail data). The “Slow” pool (circles) is kinesin dependent and was obtained by subtracting the kinesin-independent exocytosis from the average of control, SUK2, and Stalk data. (B) The proposed relative distribution of different vesicle pools and the kinesin- and myosin-mediated transport mechanisms.
Conventional kinesin is the most abundant member in the large family of KRPs. In sea urchin embryonic cells, there is at least 10-fold more kinesin than the next most abundant KRP, kinesin-2. (Scholey, J., unpublished observation). Conventional kinesin associates with the vesicles in the mitotic spindle asters (Wright et al., 1991) but is apparently not essential for mitosis (Wright et al., 1993). Because SUK4 is specific for kinesin and does not cross-react with any other known KRPs, and more importantly, because the KHC tail domain is variable among different KRPs, the inhibition of exocytosis by SUK4 and KHC stalk-tail domain strongly suggests that the conventional kinesin, rather than any other KRPs, is the protein motor for vesicle transport in regulated exocytosis. In addition, these results clearly demonstrate that the variable tail domain is responsible for in vivo vesicle cargo binding, as has been suggested based on in vitro observations (Skoufias et al., 1994; Bloom and Endow, 1995; Vallee and Sheetz, 1996). Kinectin, an integral membrane protein of the endoplasmic reticulum, has been identified as a membrane anchor protein for kinesin in the chick brain (Kumar et al., 1995). A similar protein may be responsible for the KHC tail binding in our system.
The myosin motor protein family consists of many structural and functional distinct isoforms (Hasson and Mooseker, 1995). Which isoforms may be involved in vesicular transport is still an open question (Hasson and Mooseker, 1995). BDM inhibits many myosin isoforms and could also have other effects. Possible complications from BDM effects on membrane conductance do not play a role in our preparation since Ca2+ influx is directly from a membrane wound. By having the advantage of direct visualization of regulated exocytotic events, our system provides a relatively simple in vivo assay that may help more explicitly identify the myosins responsible for vesicle recruitment when more specific reagents become available.
The inhibition of CaM kinase II has a similar effect on exocytosis as BDM, and this encourages us to believe both are affecting an actin-based step in vesicle delivery. However, the inhibitory effects we observed on exocytosis, while consistent with interruption of vesicle transport at the myosin-actin–dependent step, cannot be conclusive. CaM kinase II has been shown to be able to phosphorylate a number of targets in docking/fusion: synaptobrevin, synaptotagmin, and rabphilin-3A (Popoli, 1993; Fykse et al., 1995; Nielander et al., 1995; Hirling and Scheller, 1996; Popoli et al., 1997). Any of these proteins might conceivably be a target, and their phosphorylation by CaM kinase II may yet be shown to play a regulatory role. However, at the moment, there is no evidence suggesting that these phosphorylations must precede successful docking and fusion. More telling, in our results when we specifically tested the block of CaM kinase on previously docked cortical granules, it had no effect on the fusion step of exocytosis. The most abundant synaptic phosphoprotein, synapsin and its homologues, are more likely to be the targets affected by CaM kinase II in vesicle recruitment for exocytosis. Double knockouts of synapsin I and II exhibited decreased posttetanic potentiation and severe synaptic depression upon repetitive stimulation (Rosahl et al., 1995). Peptide inhibitors of CaM kinase II preferentially suppressed the phosphorylation of synapsin I at the CaM kinase II–specific site and decreased excitatory synaptic responses elicited in the CA1 region of hippocampal slices (Waxham et al., 1993). These inhibitory effects on exocytosis could be explained by deficient recruitment of vesicles to the active zone, in line with the need for phosphorylation of synapsin by CaM kinase II to free vesicles from actin (Ceccaldi et al., 1995).
The three vesicle pools identified by the action of blockers of kinesin function and BDM are closely related to the two-step transport mechanism. As shown in Fig. 7, the immediate pool represents vesicles that can exocytose when transport mechanisms are blocked. These vesicles must have passed the recruitment steps and are functionally docked on the plasma membrane, ready to fuse in response to Ca2+ influx. The fast pool, on the other hand, should consist of vesicles that have just passed the kinesin transport step but still require myosin-mediated transport before docking and fusion. The slow pool is the most upstream in this trafficking pathway. These vesicles must be first transported via a kinesin–microtubule system and then via an actomyosin system before approaching the plasma membrane. Recent studies on the nerve termini have defined vesicle pools using different criteria (Borges et al., 1995; Pieribone et al., 1995; Rosahl et al., 1995; Stevens and Tsujimoto, 1995; Rosenmund and Stevens, 1996). The time constant for replenishment of the readily releasable pool at nerve termini is also about 10 s (Stevens and Tsujimoto, 1995). It will be interesting to know how much of the vesicle recruitment scheme described above is applicable to neuronal transmission. One study found that a smooth muscle antimyosin II antibody microinjected into presynaptic neurons inhibited synaptic transmission; however, the antibody fraction with this activity is no longer available (Mochida et al., 1994).
The fast pool, which we are labeling myosin dependent, must be relatively stable in the absence of Ca2+ influx because it would otherwise have been depleted when kinesin transport was blocked. Therefore, the myosin-mediated transport is not constitutively active but is probably turned up by intracellular Ca2+ elevation. This may be accomplished, at least in part, by the activation of CaM kinase II since BDM and CaM kinase II autoinhibitory peptide disrupt at the fast pool step. Likewise, the kinesin-mediated transport should also be activated by Ca2+ influx through the wounding because otherwise the vesicle pools after the kinesin transport step would have accumulated and would therefore be much larger in the control cases than in the cases with SUK4 or KHC stalk-tail injection. The nature and kinetics of the regulation for either transport system are not clear. However, Ca2+ ions appear to play a key role in both steps because it is probably the first signal the cell can receive after membrane wounding. This is also consistent with the observation of multiple calcium-dependent processes related to secretion in bovine chromaffin cells (Neher and Zucker, 1993).
Finally, this study adds to our knowledge of the process that reseals disrupted cell membrane. Recent experiments have indicated that Ca2+-regulated exocytosis is a ubiquitous process in many cell types (Dan and Poo, 1992; Steinhardt et al., 1994; Bi et al., 1995; Girod et al., 1995; Coorssen et al., 1996; Rodriguez et al., 1997) and is essential for cell membrane resealing (Steinhardt et al., 1994; Bi et al., 1995; Miyake and McNeil, 1995). Although the detailed physical picture of the resealing process is not clear, it has been demonstrated that the addition of membrane lipids proximal to the wound site by vesicle exocytosis is necessary for the final “closure” of wounded membrane (Bi et al., 1995). The inhibition of membrane repair by block of CaM kinase II (Steinhardt et al., 1994) is now directly correlated with the inhibition of exocytosis. In addition, consistent with our earlier results (Steinhardt et al., 1994), we found that cells could not reseal when their kinesin transport was inhibited (Table I), even though the early phases of exocytosis were intact under the same conditions (Figs. 2–4). It therefore appears that exocytosis must be sustained and prolonged by the active transport of vesicles to successfully reseal a disrupted membrane and ensure cell survival.
Acknowledgments
We thank A. Tieu and D. MacDermed for technical assistance.
This work was supported by the Committee on Research of the University of California, Berkeley, the National Institutes of Health, and the American Cancer Society.
Abbreviations used in this paper
- BDM
2,3-butanedione monoxime
- CaM kinase II
Ca2+/calmodulin-dependent protein kinase II
- KHC
kinesin heavy chain
- KRP
kinesin-related protein
Footnotes
Address all correspondence to Richard A. Steinhardt, Department of Molecular and Cell Biology, University of California, Berkeley, CA 94720-3200. Tel.: (510) 642-3517. Fax: (510) 643-6791. E-mail: rick@mendel.berkeley.edu
Guo-Qiang Bi's current address is Department of Biology, University of California at San Diego, La Jolla, CA 92093-0357.
References
- Adams RJ, Pollard TD. Propulsion of organelles isolated from Acanthamoeba along actin filaments by myosin-I. Nature (Lond) 1986;322:754–756. doi: 10.1038/322754a0. [DOI] [PubMed] [Google Scholar]
- Allan V. Membrane traffic motors. FEBS Lett. 1995;369:101–106. doi: 10.1016/0014-5793(95)00615-g. [DOI] [PubMed] [Google Scholar]
- Baitinger C, Alderton J, Poenie M, Schulman H, Steinhardt RA. Multifunctional Ca2+/calmodulin-dependent protein kinase is necessary for nuclear envelope breakdown. J Cell Biol. 1990;111:1763–1773. doi: 10.1083/jcb.111.5.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearer EL, DeGiorgis JA, Medeiros NA, Reese TS. Actin-based motility of isolated axoplasmic organelles. Cell Motil Cytoskel. 1996;33:106–114. doi: 10.1002/cm.970330202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MK, Scheller RH. Molecular correlates of synaptic vesicle docking and fusion. Curr Opin Neurobiol. 1994;4:324–329. doi: 10.1016/0959-4388(94)90092-2. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science (Wash DC) 1992;255:200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Wu LG. Synaptic transmission. Kinetics of synaptic-vesicle recycling. Curr Biol. 1995;5:1098–1101. doi: 10.1016/s0960-9822(95)00220-x. [DOI] [PubMed] [Google Scholar]
- Bi GQ, Alderton JM, Steinhardt RA. Calcium-regulated exocytosis is required for cell membrane resealing. J Cell Biol. 1995;131:1747–1758. doi: 10.1083/jcb.131.6.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom GS, Endow SA. Motor proteins 1: kinesins. Protein Profile. 1995;2:1105–1171. [PubMed] [Google Scholar]
- Borges S, Gleason E, Turelli M, Wilson M. The kinetics of quantal transmitter release from retinal amacrine cells. Proc Natl Acad Sci USA. 1995;92:6896–6900. doi: 10.1073/pnas.92.15.6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady ST, Pfister KK, Bloom GS. A monoclonal antibody against kinesin inhibits both anterograde and retrograde fast axonal transport in squid axoplasm. Proc Natl Acad Sci USA. 1990;87:1061–1065. doi: 10.1073/pnas.87.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi PE, Grohovaz F, Benfenati F, Chieregatti E, Greengard P, Valtorta F. Dephosphorylated synapsin I anchors synaptic vesicles to actin cytoskeleton: an analysis by videomicroscopy. J Cell Biol. 1995;128:905–912. doi: 10.1083/jcb.128.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coorssen JR, Schmitt H, Almers W. Ca2+triggers massive exocytosis in Chinese hamster ovary cells. EMBO (Eur Mol Biol Organ) J. 1996;15:3787–3791. [PMC free article] [PubMed] [Google Scholar]
- Cramer LP, Mitchison TJ. Myosin is involved in postmitotic cell spreading. J Cell Biol. 1995;131:179–189. doi: 10.1083/jcb.131.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Quantal transmitter secretion from myocytes loaded with acetylcholine. Nature (Lond) 1992;359:733–736. doi: 10.1038/359733a0. [DOI] [PubMed] [Google Scholar]
- De Camilli P. The eighth Datta Lecture. Molecular mechanisms in synaptic vesicle recycling. FEBS Lett. 1995;369:3–12. doi: 10.1016/0014-5793(95)00739-v. [DOI] [PubMed] [Google Scholar]
- Drubin DG, Nelson WJ. Origins of cell polarity. Cell. 1996;84:335–344. doi: 10.1016/s0092-8674(00)81278-7. [DOI] [PubMed] [Google Scholar]
- Fath KR, Burgess DR. Membrane motility mediated by unconventional myosin. Curr Opin Cell Biol. 1994;6:131–135. doi: 10.1016/0955-0674(94)90126-0. [DOI] [PubMed] [Google Scholar]
- Fath KR, Trimbur GM, Burgess DR. Molecular motors are differentially distributed on Golgi membranes from polarized epithelial cells. J Cell Biol. 1994;126:661–675. doi: 10.1083/jcb.126.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fykse EM, Li C, Sudhof TC. Phosphorylation of rabphilin-3A by Ca2+/calmodulin- and cAMP-dependent protein kinases in vitro. J Neurosci. 1995;15:2385–2395. doi: 10.1523/JNEUROSCI.15-03-02385.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gho M, McDonald K, Ganetzky B, Saxton WM. Effects of kinesin mutations on neuronal functions. Science (Wash DC) 1992;258:313–316. doi: 10.1126/science.1384131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girod R, Popov S, Alder J, Zheng JQ, Lohof A, Poo MM. Spontaneous quantal transmitter secretion from myocytes and fibroblasts: comparison with neuronal secretion. J Neurosci. 1995;15:2826–2838. doi: 10.1523/JNEUROSCI.15-04-02826.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LS. With apologies to Scheherazade: tails of 1001 kinesin motors. Annu Rev Genet. 1993;27:319–351. doi: 10.1146/annurev.ge.27.120193.001535. [DOI] [PubMed] [Google Scholar]
- Grolig F, Williamson RE, Parke J, Miller C, Anderton BH. Myosin and Ca2+-sensitive streaming in the alga Chara: detection of two polypeptides reacting with a monoclonal anti-myosin and their localization in the streaming endoplasm. Eur J Cell Biol. 1988;47:22–31. [PubMed] [Google Scholar]
- Hasson T, Mooseker MS. Molecular motors, membrane movements and physiology: emerging roles for myosins. Curr Opin Cell Biol. 1995;7:587–594. doi: 10.1016/0955-0674(95)80017-4. [DOI] [PubMed] [Google Scholar]
- Hirling H, Scheller RH. Phosphorylation of synaptic vesicle proteins: modulation of the alpha SNAP interaction with the core complex. Proc Natl Acad Sci USA. 1996;93:11945–11949. doi: 10.1073/pnas.93.21.11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Pfister KK, Yorifuji H, Wagner MC, Brady ST, Bloom GS. Submolecular domains of bovine brain kinesin identified by electron microscopy and monoclonal antibody decoration. Cell. 1989;56:867–878. doi: 10.1016/0092-8674(89)90691-0. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Cande WZ. Microtubule distribution and reorganization in the first cell cycle of fertilized eggs of Lytechinus pictus. . Eur J Cell Biol. 1985;37:140–148. [PubMed] [Google Scholar]
- Hollenbeck PJ, Swanson JA. Radial extension of macrophage tubular lysosomes supported by kinesin. Nature (Lond) 1990;346:864–866. doi: 10.1038/346864a0. [DOI] [PubMed] [Google Scholar]
- Ingold AL, Cohn SA, Scholey JM. Inhibition of kinesin-driven microtubule motility by monoclonal antibodies to kinesin heavy chains. J Cell Biol. 1988;107:2657–2667. doi: 10.1083/jcb.107.6.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston GC, Prendergast JA, Singer RA. The Saccharomyces cerevisiaeMYO2 gene encodes an essential myosin for vectorial transport of vesicles. J Cell Biol. 1991;113:539–551. doi: 10.1083/jcb.113.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J, Yu H, Sheetz MP. Kinectin, an essential anchor for kinesin-driven vesicle motility. Science (Wash DC) 1995;267:1834–1837. doi: 10.1126/science.7892610. [DOI] [PubMed] [Google Scholar]
- Kuznetsov SA, Langford GM, Weiss DG. Actin-dependent organelle movement in squid axoplasm. Nature (Lond) 1992;356:722–725. doi: 10.1038/356722a0. [DOI] [PubMed] [Google Scholar]
- Langford GM. Actin- and microtubule-dependent organelle motors: interrelationships between the two motility systems. Curr Opin Cell Biol. 1995;7:82–88. doi: 10.1016/0955-0674(95)80048-4. [DOI] [PubMed] [Google Scholar]
- Langford GM, Kuznetsov SA, Johnson D, Cohen DL, Weiss DG. Movement of axoplasmic organelles on actin filaments assembled on acrosomal processes: evidence for a barbed-end-directed organelle motor. J Cell Sci. 1994;107:2291–2298. doi: 10.1242/jcs.107.8.2291. [DOI] [PubMed] [Google Scholar]
- Lin CH, Espreafico EM, Mooseker MS, Forscher P. Myosin drives retrograde F-actin flow in neuronal growth cones. Neuron. 1996;16:769–782. doi: 10.1016/s0896-6273(00)80097-5. [DOI] [PubMed] [Google Scholar]
- Llinas R, Gruner JA, Sugimori M, McGuinness TL, Greengard P. Regulation by synapsin I and Ca(2+)-calmodulin-dependent protein kinase II of the transmitter release in squid giant synapse. J Physiol. 1991;436:257–282. doi: 10.1113/jphysiol.1991.sp018549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M, Bower E, Levitt P, Li D, Chantler PD. Myosin II distribution in neurons is consistent with a role in growth cone motility but not synaptic vesicle mobilization. Neuron. 1992;8:25–44. doi: 10.1016/0896-6273(92)90106-n. [DOI] [PubMed] [Google Scholar]
- Miyake K, McNeil PL. Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J Cell Biol. 1995;131:1737–1745. doi: 10.1083/jcb.131.6.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida S, Kobayashi H, Matsuda Y, Yuda Y, Muramoto K, Nonomura Y. Myosin II is involved in transmitter release at synapses formed between rat sympathetic neurons in culture. Neuron. 1994;13:1131–1142. doi: 10.1016/0896-6273(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J Cell Biol. 1995;131:1315–1326. doi: 10.1083/jcb.131.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Zucker RS. Multiple calcium-dependent processes related to secretion in bovine chromaffin cells. Neuron. 1993;10:21–30. doi: 10.1016/0896-6273(93)90238-m. [DOI] [PubMed] [Google Scholar]
- Nielander HB, Onofri F, Valtorta F, Schiavo G, Montecucco C, Greengard P, Benfenati F. Phosphorylation of VAMP/synaptobrevin in synaptic vesicles by endogenous protein kinases. J Neurochem. 1995;65:1712–1720. doi: 10.1046/j.1471-4159.1995.65041712.x. [DOI] [PubMed] [Google Scholar]
- Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ, Greengard P. Distinct pools of synaptic vesicles in neurotransmitter release [see comments] Nature (Lond) 1995;375:493–497. doi: 10.1038/375493a0. [DOI] [PubMed] [Google Scholar]
- Popoli M. Synaptotagmin is endogenously phosphorylated by Ca2+/calmodulin protein kinase II in synaptic vesicles. FEBS Lett. 1993;317:85–88. doi: 10.1016/0014-5793(93)81496-m. [DOI] [PubMed] [Google Scholar]
- Popoli M, Venegoni A, Vocaturo C, Buffa L, Perez J, Smeraldi E, Racagni G. Long-term blockade of serotonin reuptake affects synaptotagmin phosphorylation in the hippocampus. Mol Pharm. 1997;51:19–26. doi: 10.1124/mol.51.1.19. [DOI] [PubMed] [Google Scholar]
- Porter ME, Scholey JM, Stemple DL, Vigers GP, Vale RD, Sheetz MP, McIntosh JR. Characterization of the microtubule movement produced by sea urchin egg kinesin. J Biol Chem. 1987;262:2794–2802. [PubMed] [Google Scholar]
- Rodriguez A, Webster P, Ortego J, Andrews NW. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol. 1997;137:93–104. doi: 10.1083/jcb.137.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov VI, Gyoeva FK, Gelfand VI. Kinesin is responsible for centrifugal movement of pigment granules in melanophores. Proc Natl Acad Sci USA. 1991;88:4956–4960. doi: 10.1073/pnas.88.11.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Sudhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation [see comments] Nature (Lond) 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Reuter H, Wendland B, Schweizer FE, Tsien RW, Smith SJ. The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron. 1993;11:713–724. doi: 10.1016/0896-6273(93)90081-2. [DOI] [PubMed] [Google Scholar]
- Saxton WM, Hicks J, Goldstein LS, Raff EC. Kinesin heavy chain is essential for viability and neuromuscular functions in Drosophila, but mutants show no defects in mitosis. Cell. 1991;64:1093–1102. doi: 10.1016/0092-8674(91)90264-y. [DOI] [PubMed] [Google Scholar]
- Scholey JM. Kinesin-II, a membrane traffic motor in axons, axonemes, and spindles. J Cell Biol. 1996;133:1–4. doi: 10.1083/jcb.133.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholey JM, Porter ME, Grissom PM, McIntosh JR. Identification of kinesin in sea urchin eggs, and evidence for its localization in the mitotic spindle. Nature (Lond) 1985;318:483–486. doi: 10.1038/318483a0. [DOI] [PubMed] [Google Scholar]
- Skoufias DA, Cole DG, Wedaman KP, Scholey JM. The carboxyl-terminal domain of kinesin heavy chain is important for membrane binding. J Biol Chem. 1994;269:1477–1485. [PubMed] [Google Scholar]
- Steinhardt RA, Bi G, Alderton JM. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science (Wash DC) 1994;263:390–393. doi: 10.1126/science.7904084. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Tsujimoto T. Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proc Natl Acad Sci USA. 1995;92:846–849. doi: 10.1073/pnas.92.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, De Camilli P, Niemann H, Jahn R. Membrane fusion machinery: insights from synaptic proteins. Cell. 1993;75:1–4. [PubMed] [Google Scholar]
- Terasaki M. Visualization of exocytosis during sea urchin egg fertilization using confocal microscopy. J Cell Sci. 1995;108:2293–2300. doi: 10.1242/jcs.108.6.2293. [DOI] [PubMed] [Google Scholar]
- Vale RD, Reese TS, Sheetz MP. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 1985a;42:39–50. doi: 10.1016/s0092-8674(85)80099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Schnapp BJ, Mitchison T, Steuer E, Reese TS, Sheetz MP. Different axoplasmic proteins generate movement in opposite directions along microtubules in vitro. Cell. 1985b;43:623–632. doi: 10.1016/0092-8674(85)90234-x. [DOI] [PubMed] [Google Scholar]
- Vallee RB, Sheetz MP. Targeting of motor proteins. Science (Wash DC) 1996;271:1539–1544. doi: 10.1126/science.271.5255.1539. [DOI] [PubMed] [Google Scholar]
- Waxham MN, Malenka RC, Kelly PT, Mauk MD. Calcium/ calmodulin-dependent protein kinase II regulates hippocampal synaptic transmission. Brain Res. 1993;609:1–8. doi: 10.1016/0006-8993(93)90847-g. [DOI] [PubMed] [Google Scholar]
- Wright BD, Henson JH, Wedaman KP, Willy PJ, Morand JN, Scholey JM. Subcellular localization and sequence of sea urchin kinesin heavy chain: evidence for its association with membranes in the mitotic apparatus and interphase cytoplasm [published erratum appears 114:following 863] J Cell Biol. 1991;113:817–833. doi: 10.1083/jcb.113.4.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright BD, Terasaki M, Scholey JM. Roles of kinesin and kinesin-like proteins in sea urchin embryonic cell division: evaluation using antibody microinjection. J Cell Biol. 1993;123:681–689. doi: 10.1083/jcb.123.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JT, Laymon RA, Goldstein LS. A three-domain structure of kinesin heavy chain revealed by DNA sequence and microtubule binding analyses. Cell. 1989;56:879–889. doi: 10.1016/0092-8674(89)90692-2. [DOI] [PubMed] [Google Scholar]
- Yang JT, Saxton WM, Stewart RJ, Raff EC, Goldstein LS. Evidence that the head of kinesin is sufficient for force generation and motility in vitro. Science (Wash DC) 1990;249:42–47. doi: 10.1126/science.2142332. [DOI] [PubMed] [Google Scholar]