

Abstract
Successful zygote formation during yeast mating requires cell fusion of the two haploid mating partners. To ensure that cells do not lyse as they remodel their cell wall, the fusion event is both temporally and spatially regulated: the cell wall is degraded only after cell–cell contact and only in the region of cell–cell contact. To understand how cell fusion is regulated, we identified mutants defective in cell fusion based upon their defect in mating to a fus1 fus2 strain (Chenevert, J., N. Valtz, and I. Herskowitz. 1994. Genetics 136:1287–1297). Two of these cell fusion mutants are defective in the FPS1 gene, which codes for a glycerol facilitator (Luyten, K., J. Albertyn, W.F. Skibbe, B.A. Prior, J. Ramos, J.M. Thevelein, and S. Hohmann. 1995. EMBO [Eur. Mol. Biol. Organ.] J. 14:1360–1371). To determine whether inability to maintain osmotic balance accounts for the defect in cell fusion in these mutants, we analyzed the behavior of an fps1Δ mutant with reduced intracellular glycerol levels because of a defect in the glycerol-3-phosphate dehydrogenase (GPD1) gene (Albertyn, J., S. Hohmann, J.M. Thevelein, and B.A. Prior. 1994. Mol. Cell. Biol. 14:4135– 4144): deletion of GPD1 partially suppressed the cell fusion defect of fps1 mutants. In contrast, overexpression of GPD1 exacerbated the defect. The fusion defect could also be partially suppressed by 1 M sorbitol. These observations indicate that the fusion defect of fps1 mutants results from inability to regulate osmotic balance and provide evidence that the osmotic state of the cell can regulate fusion. We have also observed that mutants expressing hyperactive protein kinase C exhibit a cell fusion defect similar to that of fps1 mutants. We propose that Pkc1p regulates cell fusion in response to osmotic disequilibrium. Unlike fps1 mutants, fus1 and fus2 mutants are not influenced by expression of GPD1 or by 1 M sorbitol. Their fusion defect is thus unlikely to result from altered osmotic balance.
The joining of two cells occurs during certain specialized cell–cell interactions such as sperm–egg fusion during fertilization, myoblast fusion during myotube formation, and gamete fusion during yeast mating. Intercellular fusion requires successful completion of a number of different events, the molecular details of which are poorly understood. The interacting cells must first recognize and adhere to each other. Extracellular material separating the interacting cells must then be removed. The zona pellucida surrounding the egg, extracellular matrix components separating myoblasts, and cell wall material separating haploid yeast cells must be removed to place the plasma membranes of the interacting cells into apposition. Finally, the plasma membranes of the two cells fuse, forming a single heterokaryon which can then undergo fusion of intracellular organelles.
The mating pathway of Saccharomyces cerevisiae culminates in the fusion of two haploid cells of opposite mating type (a and α) into an a/α diploid zygote. The events leading up to cell–cell contact are well characterized. Haploid cells secrete peptide pheromones (a-factor by a cells and α-factor by α cells) that are important for intercellular recognition and for preparing cells for fusion. These pheromones activate a G protein–coupled receptor on the surface of the opposite mating partner, which in turn activates a mitogen-activated protein (MAP)1 kinase cascade, inducing a morphological response (shmoo formation), cell cycle arrest, and transcriptional induction (for reviews see Kurjan, 1992; Sprague and Thorner, 1992; Bardwell et al., 1994; Herskowitz, 1995). The mating pheromones prepare cells to fuse by inducing expression and localization of fusion components. In particular, synthesis of Fus1p and Fus2p, proteins required for cell fusion, is induced by pheromone (Trueheart et al., 1987; McCaffrey et al., 1987; Elion et al., 1995). These proteins are localized to the region of future cell contact (Trueheart et al., 1987; Elion et al., 1995). Cells polarize the actin cytoskeleton and secretory apparatus toward their selected mating partner by detecting a pheromone gradient (Jackson and Hartwell, 1990; Madden and Snyder, 1992; Segall, 1993). As a result, new membrane and cell wall material is deposited at the site of future cell contact (Field and Schekman, 1980; Adams and Pringle, 1984; Novick and Botstein, 1985; Hasek et al., 1987; Read et al., 1992), which may be important for localized cell wall modifications (Lipke et al., 1976; Tkacz and MacKay, 1979; Schekman and Brawley, 1979; Baba et al., 1989) and targeting of the fusion machinery.
Although pheromones activate cells for fusion, cell wall degradation does not begin until the mating partners contact each other. Initially, cell surface agglutinins mediate attachment of the mating partners (Lipke and Kurjan, 1992), which is reversible by sonication. The cell walls then become irreversibly attached. Once cell–cell contact occurs, a thinning of the cell wall is observed that begins in the center of the region of cell contact and proceeds toward the edges (Osumi et al., 1974). Cell wall degradation and remodeling normally occur quickly, so that few cells in a population of mating cells are adhered but not fused (Trueheart et al., 1987). In mutants defective in cell fusion, zygote formation is blocked after the cells have adhered but before the intervening wall has been degraded, producing a dumbbell-shaped structure called a prezygote. The persistence of the cell wall in these mutants creates a physical barrier between mating partners, preventing cytoplasmic mixing and nuclear fusion (Trueheart et al., 1987; McCaffrey et al., 1987).
Products of the FUS1-FUS3, FUS5-FUS8, and CEF1 genes are required for cell fusion (Trueheart et al., 1987; McCaffrey et al., 1987; Elion et al., 1990, 1995; Kurihara et al., 1994; Elia and Marsh, 1996). Fus1p is a transmembrane protein with an intracellular SH3 domain (Trueheart et al., 1987; Trueheart and Fink, 1989). Fus2p has no similarity to known proteins (Elion et al., 1995). Despite their lack of homology, Fus1p and Fus2p have overlapping functions, as overexpression of one can partially suppress loss of the other. Absence of both proteins results in a synthetic fusion defect. In addition, the fusion defect is greatly enhanced when both mating partners are mutant, suggesting that at least some activities required for cell fusion can be provided by either partner (Trueheart et al., 1987). Unlike FUS1 and FUS2, which are specifically required for cell fusion, the other FUS genes have additional functions during mating. For instance, FUS3, whose role in cell fusion is unknown, encodes a MAP kinase that functions in the pheromone response pathway (Elion et al., 1990). FUS5, FUS8, and CEF1 genes correspond to AXL1, RAM1, and STE6, respectively. These genes were previously identified for their role in a-factor production (Adames et al., 1995; Powers et al., 1986; Kuchler et al., 1989), suggesting that high levels of pheromone may play a role in cell fusion (Elia and Marsh, 1996; Brizzio et al., 1996). It is also possible that these proteins (e.g., Ste6p, the a-factor transporter) are required for cell fusion independently of their role in pheromone production or secretion (Elia and Marsh, 1996). Whether the products of these genes play direct roles in cell fusion or are involved in regulating fusion is unclear.
Cell fusion requires that cell contact be sensed and that the cell surface be remodeled in response to this contact. To prevent cell lysis and to maintain cell integrity, cell wall degradation must be highly regulated, occurring only after cell–cell contact and only in the region of cell–cell contact. We show that the glycerol facilitator, Fps1p, is required for cell fusion. Our studies on Fps1p provide evidence that the osmotic state of the cell regulates cell fusion. We present additional studies suggesting that protein kinase C, previously recognized for its role in osmotic regulation (Davenport et al., 1995), negatively regulates cell fusion, further linking osmosensing pathways to regulation of cell fusion.
Materials and Methods
Yeast Strains and Media
Yeast strains are described in Table I. Standard yeast growth conditions and genetic manipulations are described in Rose et al. (1990). Cells were grown at 30°C in yeast extract/peptone/dextrose medium unless otherwise noted. DNA manipulations were performed as described in Sambrook et al. (1989).
Table I.
Yeast Strains and Plasmids Used in This Study
| Strain | Genotype | Source | ||
|---|---|---|---|---|
| IH2350 | MATα ura 3-52 his4-34 trp1 Δ1 | IH collection | ||
| IH2351* | MATα ura 3-52 trp1Δ1 fus1Δ1 fus2Δ3 | IH collection | ||
| IH2353* | MATa ura 3-52 trp1Δ1 fus1Δ1 fus2Δ3 | IH collection | ||
| IH3160 | MATa | IH collection | ||
| IH3179 | MATa fps1-1 | IH collection | ||
| IH3182 | MATa fps1-2 | IH collection | ||
| IH3186 | MATα | This study | ||
| IH3190 | MATα trp1Δ99 | This study | ||
| IH3194 | MATα trp1Δ99 leu2Δ1 | This study | ||
| IH3196 | MATa leu2Δ1 | This study | ||
| JP52 | MATa fus1Δ::TRP1 trp1Δ99 leu2Δ1 | This study | ||
| JP147 | MATa fps1Δ::URA3 leu2Δ1 | This study | ||
| JP150 | MATa FPS1::FPS1-URA3 | This study | ||
| JP153 | MATa ura3-52::URA3 | This study | ||
| JP154 | MATa fps1-1::FPS1-URA3 | This study | ||
| JP157 | MATa fps1-1 ura3-52::URA3 | This study | ||
| JP158 | MATa fps1-2::FPS1-URA3 | This study | ||
| JP161 | MATa fps1-2 ura3-52::URA3 | This study | ||
| JP163 | MATa fps1Δ:: LEU2Δ1 | This study | ||
| JP165 | MATa fps1Δ:: URA3 gpd1Δ::TRP1 leu2Δ1 trp1Δ99 | This study | ||
| JP168 | MATa gpd1Δ::TRP1 leu2Δ1 trp1Δ99 | This study | ||
| JP226 | MATα fps1Δ::URA3 trp1Δ99 | This study | ||
| JP233 | MATα fps1Δ::URA3 gpd1Δ::TRP1 trp1Δ99 | This study | ||
| JP236 | MATα gpd1Δ::TRP1 trp1Δ99 | This study | ||
| JP257 | MATα fus2Δ::URA3 trp1Δ99 | This study | ||
| JP285 | MATa fus1Δ::TPR1 gpd1Δ::TRP1 trp1Δ99 | This study | ||
| JP287 | MATα fus2Δ::URA3 gpd1Δ:: TRP1 trp1Δ99 | This study | ||
| JP317 | MATa PKC1::PKC1-R398P-URA3 leu2Δ1 | This study | ||
| JP325‡ | MATa fps1Δ::LEU2 | This study | ||
| JP326‡ | MATa mpk1Δ:::TRP1 | This study | ||
| JP327‡ | MATα fps1Δ::LEU2 mpk1Δ::TRP1 | This study | ||
| JP328‡ | MATα | This study | ||
| Strains containing plasmids: | ||||
| JP199 | IH3196 containing YEpGPD1 | |||
| JP200 | JP163 containing YEpGPD1 | |||
| JP300 | IH3196 containing pJP67 | |||
| JP301 | IH3196 containing YCp50 | |||
| JP333 | IH3194 containing pJW192 | |||
| JP400 | IH3196 containing YCplac195 | |||
| JP401 | JP163 containing YCplac195 | |||
| Plasmid name | Description | Source | ||
| pDL242 | pGAL1[PKC1-R398A] (pBM743) | D. Levin (Watanabe et al., 1994) | ||
| pDL293 | pGAL1[PKC1::HA] (pBM743) | D. Levin (Watanabe et al., 1994) | ||
| pDL295 | pGAL1[PKC1-K853R::HA] (pBM743) | D. Levin (Watanabe et al., 1994) | ||
| pJP2 | fus1Δ::TRP1 | This study | ||
| pJP30 | FPS1-URA3 (pRS306)§ | This study | ||
| pJP31 | fps1Δ::LEU2 | This study | ||
| pJP52 | fps1Δ::URA3 | This study | ||
| pUCgpd1Δ::TRP1 | gpd1Δ::TRP1 | S. Hohmann (Albertyn et al., 1994 b) | ||
| YEpGPD1 | 2μ GPD1 (YEplac195)§ | S. Hohmann (Albertyn et al., 1994 b) | ||
| pJP67 (YCp50-DS1) | PKC1-R398P (YCp50)§ | Y. Takai (Nonaka et al., 1995) | ||
| pJP72 | PKC1-R398P (pRS306)§ | This study | ||
| pJW192 | RAS2-GFP | J. Whistler | ||
| pKOFUS2 | fus2Δ::URA3 | IH collection |
Isogenic to IH2350.
Isogenic derivatives in the EG123 strain background, whose full genotype is trp1 ura3 his4 leu2 can1; all other strains are isogenic derivatives of IH3160, whose full genotype is MATa ade2-101 ura3-52 met1-1 HMLa HMRa.
pRS306, YCplac195, and YCp50 are described in Guthrie and Fink (1991). IH, I. Herskowitz.
Yeast Plasmids and Transformations
YEpGPD1 is a 2 μ URA3 plasmid (derived from YEplac195) containing the GPD1 gene, as described in Albertyn et al. (1994 b) (kindly provided by S. Hohmann, Katholieke University, Leuven, Belgium). pJP67 (YCp50-DS1) is a YCp50-derived plasmid containing the PKC1-R398P allele as described in Nonaka et al. (1995) (kindly provided by Y. Takai, Osaka University, Osaka, Japan). The 4.3-kb SphI fragment containing PKC1-R398P from pJP67 was cloned into BamHI and SalI sites of pRS306 to generate pJP72. This plasmid was used to integrate PKC1-R398P at its genomic locus, generating strain JP317. Plasmids containing PKC1 under control of the GAL1 promoter are pDL242 (pGAL1[PKC1-R398A]), pDL293 (pGAL1[PKC1::HA]), and pDL295 (pGAL1[PKC1-K853R:: HA]) as described in Watanabe et al. (1994). They were kindly provided by D. Levin (Johns Hopkins University, Baltimore, MD). pJW192 codes for a RAS2–green fluorescent protein (GFP) fusion protein under control of the GPD promoter on a 2μ TRP1 marked plasmid (kindly provided by J. Whistler, University of California, Berkeley). Yeast transformations were performed by the lithium acetate method (Ito et al., 1983).
Strain Construction
The GPD1 gene was deleted from strains using pUCgpd1Δ::TRP1, a construct designed to replace GPD1 with TRP1 as described in Albertyn et al. (1994 b) (kindly provided by S. Hohmann). Strains were confirmed to be gpd1Δ by their sensitivity to high osmolarity media and by PCR analysis. fus1Δ strains were constructed using pJP2, which contains a substitution of the FUS1 open reading frame (ORF) by TRP1. This plasmid was generated by cloning the 1.9-kb PstI–KpnI fragment containing FUS1 into the PstI–KpnI sites of pBluescript KS+. The FUS1 ORF was removed by cloning a ClaI–HincII fragment containing TRP1 into the AccI–HincII sites of FUS1. fus2Δ strains were generated using pKOFUS2, a plasmid in which the 1.6-kb HindIII fragment containing FUS2 was replaced by the 1.1-kb HindIII fragment containing URA3. fus1Δ and fus2Δ strains were confirmed by their defective mating and by PCR analysis. fps1Δ deletion strains were generated using either pJP31 or pJP52. pJP31 replaces FPS1 with LEU2; pJP52 replaces FPS1 with URA3 (see Fig. 1 b). The BglII– HindIII fragment containing FPS1 was cloned into the BamHI–HindIII sites of pUC18. To generate pJP31, LEU2 was removed from pUC18-LEU2 (Herskowitz collection), in which LEU2 is cloned into the SalI site of pUC18. A PstI–XbaI fragment, containing LEU2, was cloned into the NsiI–AccI site of FPS1. pJP52 was constructed from a plasmid in which a 111-nucleotide fragment was inserted at the stop codon of FPS1, generating a BamHI site 70 nucleotides 3′ to the stop codon. This plasmid was cut with BamHI and NsiI and a BamHI–NsiI fragment containing URA3 was inserted. The URA3 fragment was obtained from pSM32 (Herskowitz collection), a pUC18 plasmid containing URA3. fps1Δ strains were confirmed by defective mating and by PCR analysis.
Figure 1.

Morphological phenotype of cell fusion mutants. (a) Wild-type (IH3160), (b) M8 (IH3179), (c) M11 (IH3182), (d) fps1Δ (JP147), and (e) PKC1-R398P (JP317) strains were mated on filters to wild-type strain IH3186 (two left columns) or to wild-type strain JP333 carrying the RAS2–GPF fusion plasmid (two right columns) as described in Materials and Methods. Nuclei were visualized by DAPI staining (second column). RAS2–GFP was visualized by fluorescence microscopy (fourth column).
Mating Assays
Quantitative mating was as described in Valtz and Herskowitz (1996) except that 6 × 106 cells of each mating partner were mixed. Mating assays scored microscopically were performed by mixing equal numbers of log phase a and α cells (6 × 106), collecting cells on 0.45-μm filters (Millipore Corp., Bedford, MA), and incubating on YEPD plates for ∼4 h at 30°C. Cells were resuspended in 5 ml 70% ethanol by vortexing, washed, and resuspended in 50% glycerol + 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI; Sigma Chemical Co., St. Louis, MO). Samples were sonicated and viewed with a microscope at 100× (Axioskop; Carl Zeiss, Inc., Thornwood, NY). Percentage of prezygotes was defined as prezygotes/(prezygotes + zygotes). At least 100 partnered cells (zygotes + prezygotes) were counted per sample. Numbers represent the average of at least three experiments unless otherwise noted. Assays in which one partner contained the RAS2-GFP plasmid were performed in essentially the same manner, except that strains were grown in SD-TRP (Rose et al., 1990) to select for the plasmid. In this case, cells were resuspended from the filter into 5 ml YEPD by vortexing, sonicated, and viewed with a microscope at 100× (BX50; Olympus Corp., Lake Success, NY). Mating assays scored on plates were performed by spreading a lawn of 9 × 106 log phase fus1 fus2 mating tester cells on a YEPD plate. Patches of cells grown on YEPD (or on minimal media to select for plasmids) were replica plated to the lawn. After incubation at 30°C for 3.5–4.5 h, plates were replica plated to media selective for growth of diploids. For cells expressing PKC1 alleles under control of the GAL1 promoter (pDL242, pDL293, pDL295), patches were grown on raffinose-URA plates to select for the plasmid but maintain the GAL1 promoter inactive. They were replica plated to a lawn of an MATα fus1 fus2 strain at 30°C, on either YEPD or YEPGalactose. Cells were allowed to mate for 3.75 h and then replica plated to select for diploids in the presence of glucose.
Cloning of FPS1
FPS1 was cloned by complementation of the mating defect of the M8 mutant (Chenevert et al., 1994). Five plasmids that rescued the mating defect were isolated from a 2μ YEp24-derived library (Carlson and Botstein, 1982) from 22,000 transformants screened. All contained a common 6.5-kb overlapping DNA segment. An EcoRI–SalI fragment was subcloned into pRS316, a CEN-ARS vector, and deletion analysis was performed to identify the complementing ORF (see Fig. 2 c). A plasmid containing a 2.2-kb XhoI–HindIII fragment (pJP27) fully restored mating to the M8 and M11 mutants. The 2.4-kb BglII–HindIII segment containing FPS1 was cloned into pRS306, a URA3-marked, integrating vector to form pJP30, which was able to restore mating and fusion when integrated at the FPS1 genomic locus. A wild-type strain was transformed with the integrating plasmid, marking the FPS1 locus with URA3 (JP150), and then crossed to M8 and M11 mutants, which were ura3. 12 of 13 complete tetrads from the M8 cross showed 2:2 segregation of Ura3+:Ura3− and Mating+:Mating− phenotypes. One tetrad showed 1:3 segregation of Ura3+:Ura3− and 2:2 segregation of Mating+:Mating−, presumably because of gene conversion at URA3. 10 complete tetrads from the M11 cross showed 2:2 segregation of Ura3 and mating phenotypes. In all spores, the Ura3+ phenotype cosegregated with mating proficiency, indicating that the mutations responsible for the mating defects of M8 and M11 are linked to FPS1.
Figure 2.

Restriction map (a), disruption constructs (b), and deletion analysis (c) of FPS1 region. The ability of each plasmid to complement the mating defect of the M8 and M11 mutants is indicated on the right: +, complementation; −, no complementation. pJP are plasmids carrying the indicated segments. Restriction enzymes: S, SalI; B, BamHI; Bg, BglII; Xh, XhoI; N, NsiI; K, KpnI; H, HindIII; X, XbaI; E, EcoRI. Additional information is given in Materials and Methods and in the text.
Analysis of fps1Δ mpk1Δ Double Mutants
Segregants obtained by crossing an fps1Δ strain with an mpk1Δ strain were allowed to germinate on YEPD containing 1 M sorbitol at 25°C. Cells were passaged on YEPD (except the mpk1Δ fps1Δ double mutant, which was grown on YEPD + 1 M sorbitol), streaked for single colonies on either YEPD or YEPD + 1 M sorbitol, and grown for 2 d at 25°C.
Assaying of Intracellular Glycerol
Intracellular glycerol was assayed enzymatically using a glycerol determination kit (Boehringer Mannheim Biochemicals, Indianapolis, IN) essentially as described (Albertyn et al., 1994a ). Cells were grown to mid-log phase in YEPD unless selecting for plasmids, in which case they were grown in SD-URA (Rose et al., 1990). From each culture, three 10-ml aliquots were separately filtered onto 0.45-μm filters (Millipore Corp.), washed quickly with 5 ml ice-cold YEPD, and resuspended in 2 ml 0.5 M Tris-HCl, pH 7.5, by vortexing. Samples were heated to 95°C for 10 min, and cell debris was removed by centrifugation. Aliquots were pooled, and glycerol determinations were performed as per manufacturer's specifications. Protein concentrations were determined using the Bio-Rad protein assay (Bio Rad Laboratories, Hercules, CA). Glycerol concentrations were normalized to total protein. Fold increase in glycerol concentrations was determined by normalizing to the level of intracellular glycerol in wild-type cells. Data represent the average of at least three experiments.
Results
Identification of Mutants Defective in Cell Fusion
Mutants defective in cell fusion were identified based upon their defect in mating to a fus1 fus2 mutant (Chenevert et al., 1994). fus1 fus2 double mutants are mildly compromised in mating to a wild-type strain but are severely defective in mating to fus1 or fus2 strains (Trueheart et al., 1987; Elion et al., 1995). Hence, a screen to find mutants defective in mating to a fus1 fus2 strain should identify mutants defective in FUS1, FUS2, and other genes required for fusion. To determine if any of the mutants were defective in FUS1 or FUS2, plasmids containing these genes were tested for their ability to restore mating to the mutants, which identified one mutant as defective in FUS2 (Chenevert et al., 1994).
To identify additional cell fusion mutants, mutants exhibiting a mating defect but normal shmoo morphology (class 4 mutants in Chenevert et al., 1994) were examined microscopically to ascertain if prezygotes accumulated when the mutants were mated to a wild-type strain. Prezygotes were scored as structures in which the nuclei of mating partners remained unfused, as evidenced by two distinct DAPI staining structures, and in which a septum was visible between adherent mating partners. 9 of 13 mutants exhibited increased frequency of prezygotes, indicative of a fusion defect (data not shown). The mutants were of two classes: five were a cell specific and four were non–cell type specific, exhibiting mating defects as both a and α cells. Two mutants of the latter class, designated M8 and M11 (B6 and J10, respectively, in Chenevert et al., 1994), were further characterized and are described here.
M8 and M11 exhibit normal pheromone signaling and events leading up to cell–cell contact. They produce and respond to pheromone normally, as assayed by cell cycle arrest and shmoo formation (Chenevert et al., 1994; Philips, J., unpublished observations). We observed that, in matings between M8 or M11 mutants and a wild-type partner, the percentage of partnered cells ([prezgotes + zygotes]/total cells) was normal, suggesting that the defect in mating is not due to inability of the partners to find or adhere to each other. Fig. 1 illustrates the aberrant morphological structures that accumulated in mating mixes in which one partner is M8 or M11. More than 30% of the partnered cells were prezygotes (Fig. 1, b and c). In contrast, in mating reactions between wild-type partners (Fig. 1 a), <1% of the partnered cells were prezygotes. Further evidence that cell fusion was blocked before plasma membrane fusion was obtained by mating cells to a partner that produces a RAS2–GFP fusion protein (kindly provided by J. Whistler), which localizes green fluorescence around the periphery of the cell (Whistler, J., and J. Rine, personal communication). In mating reactions containing wild-type cells, zygotes showed the green fluorescent signal throughout the entire zygote (Fig. 1 a). In contrast, when mutants were mated to the wild-type partner containing RAS2– GFP, prezygotes were found in which the green fluorescent signal remained restricted to one cell, indicating a failure of plasma membrane fusion and cytoplasmic mixing (Fig. 1, b and c). We conclude that M8 and M11 mutants are defective in cell fusion but are normal for earlier events of mating.
M8 and M11 Are Defective in the FPS1 Gene
To clone the gene defective in the M8 mutant, the strain was transformed with a high copy YEp24-derived library, and 22,000 transformants were screened for ability to mate with a fus1 fus2 strain. Five plasmids were identified that restored mating. All contained a common 6.5-kb overlapping DNA segment. This segment was subcloned into pRS316, a CEN-ARS vector, and further subcloning was performed to identify the minimal fragment capable of complementation (Fig. 2 c). A 2.2-kb XhoI–HindIII fragment (pJP27) restored mating to the M8 and M11 mutants. This fragment contains the FPS1 ORF, which fully restored mating and cell fusion to M8 and M11 when integrated at its genomic locus (Fig. 3 and data not shown). The mutation responsible for the mating defect of M8 and M11 was demonstrated to be allelic to FPS1 by following the segregation of M8 and M11 in crosses where the FPS1 locus was marked (see Materials and Methods). The plasmid complementation and segregation analyses indicate that the mating defect of M8 and M11 strains is due to mutations in FPS1, which are designated fps1-1 and fps1-2, respectively.
Figure 3.

The FPS1 gene complements the mating defect of M8 and M11. (Right) WT (JP150), M8 (JP154), and M11 (JP158), all carrying FPS1 plasmid pJP30 at the FPS1 genomic locus; (left) WT (JP153), M8 (JP157), and M11 (JP161), all carrying the vector alone (pRS306). Strains were mated to a MATα fus1 fus2 strain (IH2351) as described in Materials and Methods.
Mating Defect of fps1Δ Mutants
Analysis of strains deleted for FPS1 confirmed that Fps1p is required for cell fusion. FPS1 was deleted from the genome by replacing the FPS1 coding sequence with either LEU2 or URA3 (Fig. 2 b; see Materials and Methods). fps1Δ strains, like fps1-1 and fps1-2 mutants, were defective in mating to fus1 fus2 strains (Fig. 4) and accumulated prezygotes that were morphologically indistinguishable from those observed in mating reactions with fps1-1 and fps1-2 mutants (Fig. 1 d). In mating mixes of fps1Δ or fps1-2 mutants to a wild-type partner, 30–45% of partnered cells were prezygotes. To determine if the fps1-1 allele was quantitatively similar to the fps1Δ allele, FPS1 was deleted in two different fps1-1 strains. These fps1Δ deletion strains behaved like the fps1-1 parent strains with respect to the percentage of prezygotes accumulated (data not shown). Thus, fps1-1 and fps1-2 appear to be null alleles of FPS1. FPS1 deletion mutants in the EG123 strain background have a similar defect in cell fusion (data not shown).
Figure 4.
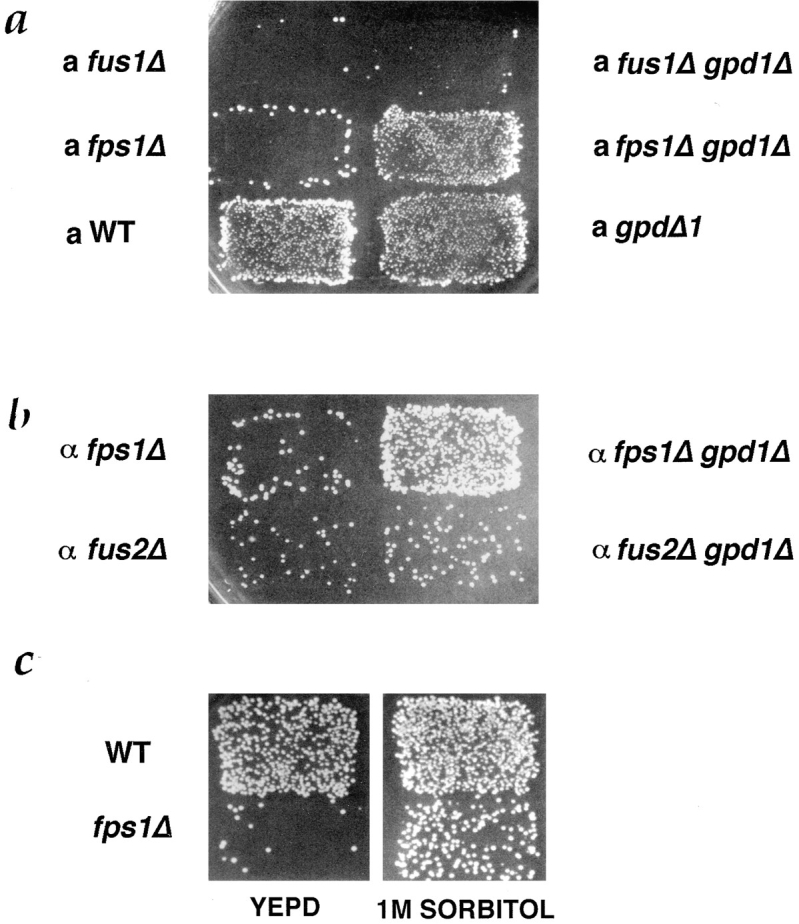
Suppression of mating defect of fps1Δ by gpd1Δ and 1 M sorbitol. (a) Patches of MAT a strains, WT (IH3160), fps1Δ (JP147), fus1Δ (JP52) (left) and gpd1Δ (JP168), fps1Δ gpd1Δ (JP165), and fus1Δ gpd1Δ (JP285) (right), were mated to a MATα fus1 fus2 strain (IH2351) as described in Materials and Methods. (b) Patches of MATα strains, fps1Δ (JP226) and fus2Δ (JP257) (left) and fps1Δ gpd1Δ (JP233) and fus2Δ gpd1Δ (JP287) (right), were mated to a MATa fus1 fus2 strain (IH2353) as described in Materials and Methods. (c) Patches of MATa strains, WT (IH3196), and fps1Δ (JP147), were mated on YEPD (left) or YEPD containing 1 M sorbitol (right) to a MATα fus1 fus2 strain (IH2351).
Quantitative mating assays demonstrated that, like fus1 fus2 strains, fps1Δ strains were mildly defective in mating, exhibiting a two- to threefold decrease in diploid formation (Table II). Unlike fus1 fus2 mutants, which exhibit an enhanced defect when both mating partners are mutant (Trueheart et al., 1987; McCaffrey et al., 1987; Elion et al., 1995), fps1Δ mutants did not mate significantly worse or accumulate more prezygotes when mated to an fps1Δ partner than to a wild-type partner. In fact, when fps1Δ mutants are mated to wild-type cells, they yielded 61 ± 17% or 32 ± 5% prezygotes, depending upon the mating type of the mutant, whereas matings between two fps1Δ mutants yield 18 ± 9% prezygotes. Thus, unlike fus1 and fus2 mutants, matings between fps1Δ mutants may exhibit some suppression of the fusion defect. In contrast, fps1Δ mutants mated to fus1 fus2 mutants at a much lower efficiency compared with wild type, a decrease of ∼25-fold (Table II).
Table II.
Quantitative Mating Defect of fps1Δ Mutants
| a strain | α strain | Mating efficiency | Prezygotes | |||
|---|---|---|---|---|---|---|
| % | % | |||||
| 1. WT (IH3196) | WT (IH3190) | 100 | 0.8 ± 0.2 | |||
| 2. fps1Δ (JP147) | WT (IH3190) | 46 ± 10 | 61 ± 17 | |||
| 3. WT (IH3196) | fps1Δ (JP226) | 39 ± 8 | 32 ± 5 | |||
| 4. fps1Δ (JP147) | fps1Δ (JP226) | 32 ± 15 | 18 ± 9 | |||
| 5. WT (IH3196) | WT (IH2350) | 100 | ND | |||
| 6. WT (IH3196) | fus1 fus2 (IH2351) | 35 ± 10 | ND | |||
| 7. fps1Δ (JP147) | fus1 fus2 (IH2351) | 1.4 ± 1.0 | ND |
Mating efficiencies were calculated as described in Material and Methods. Mating efficiencies were normalized to the isogenic wild-type mating. For lines 2–4, the values were normalized to the efficiency of IH3196 × IH3190 mating (line 1). Values are the means of at least four experiments ± SD. For lines 6 and 7, the values were normalized to the efficiency of IH3196 × IH2350 mating (line 5) and are the mean of three experiments ± SD. Percentage of prezygotes represents the number of prezygotes/(zygotes + prezygotes). At least 100 partnered cells were counted for each experiment. Values are the means of three experiments ± SD.
Glycerol Accumulation in fps1Δ Mutants Is Correlated with Defective Cell Fusion
Fps1p is a member of the major intrinsic protein family, an evolutionarily conserved family of channel proteins that transport small molecules (for review see Reizer et al., 1993). It is most similar to the Escherichia coli glycerol facilitator, GlpF, and has been proposed to function as a glycerol transporter in yeast (Luyten et al., 1995). Consistent with its role as a glycerol transporter, fps1Δ mutants contain approximately twofold more intracellular glycerol than wild-type strains, suggesting that they are defective in glycerol efflux (Luyten et al., 1995). We observed a similar increase in intracellular glycerol in our fps1Δ mutant (Table III).
Table III.
Intracellular Glycerol Concentrations in fps1Δ Strains
| Strain | Fold change in intracellular glycerol | |
|---|---|---|
| WT | 1.0 | |
| fps1Δ | 2.1 ± 0.2 | |
| gpd1Δ | 1.3 ± 0.3 | |
| fps1Δ gpd1Δ | 1.1 ± 0.2 | |
| WT + vector | 1.0 | |
| WT + 2μ GPD1 | 2.2 ± 0.3 | |
| fps1Δ + vector | 2.4 ± 0.5 | |
| fps1Δ + 2μ GPD1 | 4.2 ± 0.9 |
Strains were WT (IH3196), fps1Δ (JP147), gpd1Δ (JP168), and fps1Δ gpd1Δ (JP165). Fold change in intracellular glycerol was determined by normalizing to the glycerol level of the WT strain (n = 5). Strains containing plasmids were WT + vector (JP400), WT + YEpGPD1 (JP199), fps1Δ + vector (JP401), and fps1Δ + YEpGPD1 (JP200). Fold change in intracellular glycerol was determined by normalizing to WT + vector (n = 3). Glycerol concentrations were determined as described in Materials and Methods. Values are the means ± SD.
To ascertain whether the defect in cell fusion of fps1Δ strains results from elevated levels of intracellular glycerol, we determined whether reducing intracellular glycerol levels restored cell fusion. We reduced intracellular glycerol levels by deleting the GPD1 gene, which encodes the NADH-dependent glycerol-3-phosphate dehydrogenase (GPDH) required for the first step of glycerol biosynthesis (Gancedo et al., 1968). Although yeast cells have a second NADH-dependent GPDH (Gpd2p), Gpd1p is responsible for ∼95% of the NADH-dependent GPDH activity in the cell (Albertyn et al., 1994 b). We found that the level of intracellular glycerol in the fps1Δ GPD1 mutant was 2.1-fold higher than in the wild-type strain (FPS1 GPD1) and was only 1.1-fold higher than wild-type strains in the fps1Δ gpd1Δ mutant (Table III).
We next determined whether this decrease in intracellular glycerol level in fps1Δ gpd1Δ mutants suppressed the mating defect. We found that deletion of GPD1 suppressed the mating defect of fps1Δ strains, whereas deletion of GPD1 in FPS1 strains had no effect on mating (Fig. 4). To determine if gpd1Δ suppressed the cell fusion defect of fps1Δ strains, we microscopically assayed the double mutant for accumulation of prezygotes: deletion of GPD1 in an fps1Δ mutant decreased the number of prezygotes from 42% to 14% (Table IV). Again, we saw little effect of the GPD1 deletion in an FPS1 strain. Thus, deletion of GPD1 partially restored both intracellular glycerol levels and mating to an fps1Δ mutant.
Table IV.
Effect of Altering GPD1 Expression on the Defect in Cell Fusion of fps1Δ Mutants
| Percentage of prezygotes in matings to * | ||||||
|---|---|---|---|---|---|---|
| Genotype | α GPD1 | α gpd1Δ | ||||
| a WT | 0.75 ± 0.55 | 0.75 ± 0.55 | ||||
| a gpd1Δ | 1.50 ± 0.75 | 1.75 ± 0.55 | ||||
| a fps1Δ | 42.3 ± 6.7 | 55.5 ± 4.6 | ||||
| a fps1Δ gpd1Δ | 14.0 ± 2.5 | 26.0 ± 7.2 | ||||
| α WT + vector | α WT + 2μ GPD1 | |||||
| a WT + vector | 2 | <1 | ||||
| a WT + 2μ GPD1 | 5 | <1 | ||||
| a fps1Δ + vector | 20 | 7 | ||||
| a fps1Δ + 2μ GPD1 | 84 | 33 | ||||
In the top four lines, α strains were WT (IH3186) and gpd1Δ (JP236). a strains were WT (IH3196), gpd1Δ (JP168), fps1Δ (JP147), and fps1Δ gpd1Δ (JP165). At least 100 partnered cells were counted for each experiment. Values are the means of four experiments ± SD. In the bottom four lines the α strain was IH3190. a strains were WT + vector (JP400), WT + YEpGPD1 (JP199), fps1Δ + vector (JP401), and fps1Δ + YEpGPD1 (JP200). More than 100 partnered cells were counted per sample. Data are from one representative experiment.
Percentage of prezygotes represents the number of prezygotes/(zygotes + prezygotes).
If increased intracellular glycerol is responsible for the cell fusion defect of fps1Δ mutants, we hypothesized that this defect would be exacerbated by further increasing intracellular glycerol levels. Overexpression of GPD1 in fps1Δ strains raises intracellular glycerol levels (Luyten et al., 1995). We observed that overexpression of GPD1 in fps1Δ mutant strain JP200 led to a 1.8-fold increase in glycerol levels in comparison with the fps1Δ strain carrying a control plasmid (JP401) (Table III, seventh and eighth lines). We observed that the glycerol-overproducing strain JP200 exhibited an enhanced defect in cell fusion, producing 84% prezygotes in comparison with strain JP401, which yielded 20% prezygotes when mated to a wild-type strain (Table IV). No significant increase in prezygotes was observed in the FPS1 strain carrying the GPD1 plasmid. We also examined the ability of these strains to mate with partners that overexpress GPD1. Once again, we observed that mating with JP200 yielded nearly fivefold more prezygotes than JP401: 33% vs 7% (Table IV, second column, seventh and eight lines). Overexpression of GPD1 increased the levels of intracellular glycerol and exacerbated the defect in cell fusion of fps1Δ mutants. We conclude that the defect in cell fusion in fps1Δ mutants correlates with increased levels of intracellular glycerol.
In contrast with what was observed when GPD1 was overexpressed in the fps1Δ mutant, we found that when GPD1 was overexpressed in the mating partner there was a decrease in prezygotes formed, from 20 to 7% for the fps1Δ strain carrying the control plasmid (Table IV, seventh line) and from 84 to 33% for the fps1Δ strain carrying the GPD1 plasmid (Table IV, eighth line). One explanation for this improvement in fusion is that overexpression of GPD1 in the mating partner led to increased extracellular glycerol, which restored osmotic balance to the fps1Δ mutant. Consistent with the idea that glycerol produced by the mating partner may osmotically stabilize the fps1Δ mutant, we found that deletion of GPD1 in the partner somewhat exacerbated the defect of the fps1Δ mutant. fps1Δ mutants mated to GPD1 strains produced 42% prezygotes, which increased to 55% when mated to a gpd1Δ partner (Table IV, third line). Additionally, fps1Δ gpd1Δ mutants mated to GPD1 strains produced 14% prezygotes, which increased to 26% when mated to a gpd1Δ partner (Table IV, fourth line).
High Osmolarity Partially Suppresses the Cell Fusion Defect of fps1Δ Mutants
The correlation between increased intracellular glycerol levels and a defect in cell fusion suggests at least two possible causes of the cell fusion defect. Intracellular glycerol itself may inhibit cell fusion. Another possibility is that an imbalance between intracellular and extracellular solute levels inhibits fusion. To distinguish between these possibilities, we determined whether restoration of osmotic balance by 1 M sorbitol could suppress the cell fusion defect of fps1Δ mutants. If elevated intracellular glycerol per se inhibits cell fusion, then 1 M sorbitol should either have no effect or may exacerbate the cell fusion defect of fps1Δ mutants, since 1 M sorbitol induces GPD1 expression (Hirayama et al., 1995). If the mating defect is due to osmotic imbalance, then mating in the presence of 1 M sorbitol may suppress the defect. We observed the latter: the mating defect of fps1Δ mutants was partially alleviated by 1 M sorbitol (Fig. 4 c). Because this mating assay involves mating to a fus1 fus2 strain, it was possible that the improvement in mating resulted from an effect of the 1 M sorbitol on the fus1 fus2 partner rather than on the fps1Δ partner. To address this issue, we examined the effect of 1 M sorbitol on fps1Δ mutants mated to a wild-type strain. We found that the fusion defect was suppressed in the presence of 1 M sorbitol; the number of prezygotes declined from 37 to 10% (Table V). Moreover, the presence of 1 M sorbitol did not improve mating by fus1 fus2 mutants (data not shown) or result in decreased levels of prezygotes (Table V). We conclude that the mating defect of fps1Δ mutants can be suppressed by restoring osmotic balance to these cells.
Table V.
1 M Sorbitol Partially Suppresses the Cell Fusion Defect of fps1Δ Strains
| Percentage of prezygotes* in mating on YEPD | ||||
|---|---|---|---|---|
| Genotypes | Unsupplemented | + 1 M sorbitol | ||
| a WT | 0.3 ± 0.4 | 1 ± 0.7 | ||
| a fps1Δ | 37 ± 4 | 10 ± 2 | ||
| a fus1Δ | 12 ± 3 | 14 ± 2 | ||
| α fus2Δ | 7 ± 1 | 15 ± 2 | ||
| α WT | 1.7 ± 0.4 | 4.0 ± 3.1 | ||
| α fus1 fus2 | 58.3 ± 19.0 | 90 ± 5.5 | ||
Cells were mated either on YEPD or YEPD supplemented with 1 M sorbitol. a strains were WT (IH3196), fps1Δ (JP147), and fus1Δ (JP52); they were mated to an α WT strain (IH3186). α strains were WT (IH2350), fus2Δ (JP257), and fus1 fus2 (IH2351); they were mated to an a WT strain (IH3196). Values are the means of three experiments ± SD.
Percentage of prezygotes represents the number of prezygotes/(zygotes + prezygotes).
Additional evidence that the cell fusion defect of fps1Δ mutants is not due to an absolute increase in glycerol but rather to a difference between intracellular and extracellular glycerol comes from comparing the behavior of wild-type cells overexpressing GPD1 and fps1Δ mutants. Both of these strains contained approximately twofold more intracellular glycerol than wild-type strains containing vector alone (Table III, sixth and seventh lines), yet the fps1Δ mutant exhibited a more severe cell fusion defect (Table IV, sixth and seventh lines). One explanation for this difference is that the wild-type cells, which contain a higher level of glycerol as a result of overexpression of GPD1, can efficiently release this glycerol, so that extracellular glycerol levels also increase. The fps1Δ mutant, which releases glycerol inefficiently, has higher intracellular glycerol and decreased extracellular glycerol (Luyten et al., 1995). These data suggest that osmotic imbalance, rather than the absolute level of intracellular glycerol, accounts for the cell fusion defect of fps1Δ mutants.
Activated Alleles of PKC1 Inhibit Cell Fusion
The protein kinase C pathway is induced by conditions in which intracellular solute is higher than extracellular solute (Davenport et al., 1995). Because fps1Δ mutants have higher intracellular glycerol concentrations than wild-type cells, we wondered whether the cell fusion defect of fps1Δ mutants was influenced or mediated by the PKC1 pathway. If activation of the PKC1 pathway is responsible for the defect in cell fusion of fps1Δ mutants, then activation of Pkc1p in an otherwise wild-type background should give a similar defect. We therefore examined the effect of expressing an activated allele of PKC1 on mating and cell fusion.
A CEN-ARS plasmid containing such an allele of PKC1 (PKC1-R398P) was introduced into a wild-type strain (IH3196) to generate strain JP300. PKC1-R398P alters the pseudosubstrate binding site of Pkc1p, creating a dominant, activated allele (Nonaka et al., 1995). Expression of this allele under control of its own promoter had no detectable effect on cell viability (data not shown). The activated allele did, however, result in a mating defect similar to that of the fps1Δ mutant (Fig. 5 a). Assays of cell cycle arrest and shmoo formation indicated that JP300 responded normally to pheromone (data not shown). Furthermore, JP300 produced pheromone normally, as assayed by halo formation. When JP300 was mated to a wild-type strain, prezygotes accumulated. JP300 exhibited normal partnership, indicating that its defect did not result from inability to respond or adhere to its partner, but rather to a defect in cell fusion (Table VI, first and second lines; Fig. 1 e). We conclude that activation of the PKC1 pathway is sufficient to cause a defect in cell fusion. We were able to activate the PKC1 pathway at the time of mating by expressing an activated allele of PKC1 (PKC1-R398A) under control of the GAL1 promoter (Watanabe et al., 1994). Expression of this activated allele when cells were mating resulted in a mating defect, whereas no significant mating defect was seen when cells expressed wild-type PKC1 or PKC1-K853R, an allele mutated in the kinase domain (Fig. 5 b). Hence, a constitutively active allele of Pkc1p expressed during mating causes a mating defect.
Figure 5.
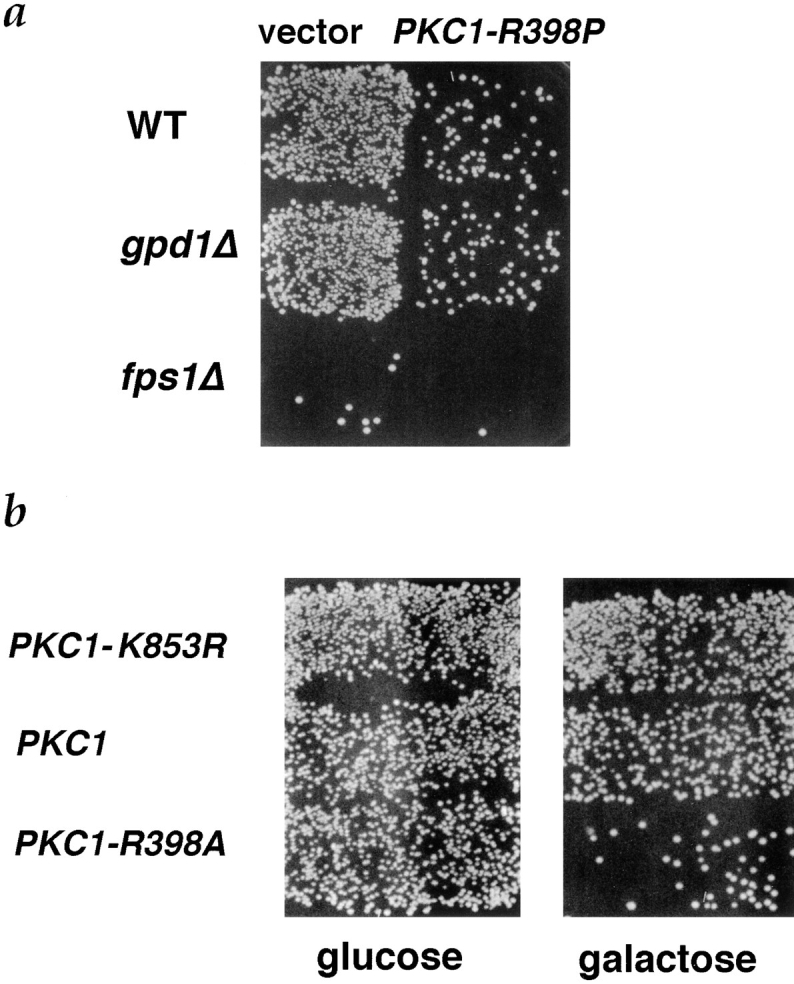
Inhibition of mating by activated PKC1 alleles. (a) Patches of MATa strains, WT (IH3196), gpd1Δ (JP168), and fps1Δ (JP163), containing YCp50 (left) or YCp50- PKC1-R398P (pJP67) (right), were mated to a MATα fus1 fus2 strain (IH2351). (b) A wild-type strain (IH3196) containing plasmids pDL295 (pGAL1[PKC1-K853R::HA]), pDL242 (pGAL1[PKC1- R398A]), or pDL293 (pGAL1 [PKC1::HA]), was mated to a MATα fus1 fus2 strain (IH2351) on either YEPD (left) or YEPGalactose (right) as described in Materials and Methods.
Table VI.
The PKC1-R398P Allele Causes a Defect in Cell Fusion
| Genotype | Plasmid | Percentage of prezygotes* | Percentage of partnership | |||
|---|---|---|---|---|---|---|
| a WT | vector | 2.0 ± 0.7 | 41.6 ± 1.6 | |||
| a WT | PKC1-R398P | 16.3 ± 1.1 | 46.3 ± 3.9 | |||
| a fps1Δ | vector | 26.3 ± 6.4 | 40.0 ±2.5 | |||
| a fps1Δ | PKC1-R398P | 82.6 ± 4.9 | 44.0 ± 0.7 | |||
| a gpd1Δ | vector | 2.0 ± 0.7 | 40.7 ± 7.9 | |||
| a gpd1Δ | PKC1-R398P | 18.3 ± 1.5 | 41.0 ± 1.9 |
Percentage of partnership represents the number of (zygotes + prezygotes)/total cells. a strains were WT (IH3196), fps1Δ (JP163), and gpd1Δ (JP168). The strains were transformed with either YCp50 containing PKC1-R398P (pJP67) or YCp50 alone and were mated to a wild-type α strain (IH3186). More than 100 partnered cells were counted per experiment to determine the percentage of prezygotes. More than 200 total cells were counted per experiment to determined the percentage of partnership. Values are the mean of three experiments ± SD.
Percentage of prezygotes represents the number of prezygotes/(zygotes + prezygotes).
Pkc1p Does Not Act through Fps1p to Inhibit Cell Fusion
Activated alleles of Pkc1p and loss of function of FPS1 caused quantitatively very similar cell fusion defects (Table VI), raising the possibility that Pkc1p negatively regulates cell fusion by inhibiting Fps1p. If the only target of Pkc1p for inhibiting cell fusion were Fps1p, then the activated allele of Pkc1p should not exacerbate the defect in fusion of an fps1Δ strain. However, we found that the PKC1-R398P allele increased the defect in cell fusion of the fps1Δ strain (Table VI, third and fourth lines). In addition, if Pkc1p negatively regulates Fps1p, then deletion of GPD1 should suppress the PKC1-R398P allele, as it partially suppressed an fps1Δ mutant (Fig. 4 a; Table IV). Deletion of GPD1 did not, however, suppress the PKC1-R398P allele, suggesting that Pkc1p acts downstream or parallel to glycerol accumulation (Table VI, second and sixth lines; Figs. 5 and 7).
Figure 7.

Regulation of cell fusion by osmosensing pathways. (a) Increased intracellular glycerol levels due to deletion of FPS1 and exacerbated by overexpression of GPD1 activate the protein kinase C pathway, which responds to hypoosmotic shock. Pkc1p is part of a checkpoint that inhibits cell fusion if cells are hypoosmotically shocked. (b) Pkc1p is a negative regulator of cell fusion that is turned off or overridden in order for cells to fuse. When cells are osmotically stressed, because of deletion of fps1Δ and exacerbated by increased expression of GPD1, Pkc1p is activated, preventing cell fusion.
fps1Δ mpk1Δ Double Mutants Require Osmotic Support for Viability
One explanation for the defect in fps1Δ mutants is that increased activity of the PKC1 pathway accounts, at least in part, for the defect in cell fusion. Additional evidence that this pathway is active came from analysis of a mutant defective in both FPS1 and MPK1, which codes for the MAP kinase regulated by Pkc1p (Lee et al., 1993; Torres et al., 1991). Although both fps1Δ and mpk1Δ mutants grow normally at 25°C (Van Aelst et al., 1994; Lee et al., 1993), we were unable to obtain double mutant segregants when spores were germinated at 25°C on YEPD in eight tetrads analyzed. The fps1Δ mpk1Δ double mutant was obtained, however, when spores were germinated in the presence of 1 M sorbitol: 14 double mutants were obtained from 18 tetrads; all were unable to grow on YEPD lacking sorbitol. The remaining 58 spores, none of which were double mutants, grew normally on unsupplemented YEPD at 25°C (Fig. 6). The inviability of the fps1Δ mpk1Δ strain demonstrates that the fps1Δ mutant, in the absence of osmotic stabilizing agents, requires MPK1 for viability, and thus that Mpk1p is active in fps1Δ mutants.
Figure 6.

Synthetic lethality of fps1Δ mpk1Δ. (a) mpk1Δ (JP326), fps1Δ (JP325), mpk1Δ fps1Δ (JP327), and wild-type strains were grown on YEPD + 1 M sorbitol at 25°C for 2 d. (b) mpk1Δ (JP326), fps1Δ (JP325), mpk1Δ fps1Δ (JP327), and wild-type strains were grown on YEPD 25°C for 2 d.
Mutations in FUS1 and FUS2 Are Not Suppressed by Altering Osmotic Conditions
We have carried out a variety of analyses to determine whether the cell fusion defect of fus1 and fus2 mutants has a similar basis to that of fps1 mutants. First, we determined whether deletion of GPD1 restored mating and cell fusion to fus1 or fus2 mutants. Although deletion of GPD1 improved mating in fps1Δ mutants, it did not improve matings of fus1 and fus2 mutants (Fig. 4, a and b). Similarly, microscopic examination of mating mixes did not reveal a significant change in prezygote accumulation in fus1 gpd1 or fus2 gpd1 double mutants compared with fus1 GPD1 or fus2 GPD1 strains. Moreover, overexpression of GPD1 did not exacerbate the cell fusion defect of fus1 mutants (data not shown). We conclude that the cell fusion defect of fus1 and fus2 strains is unlikely to be due to an accumulation of intracellular glycerol. Additional evidence that the defect in cell fusion in fus1 and fus2 mutants differs from that in fps1Δ mutants came from analyzing the ability of mutants to mate in the presence of 1 M sorbitol. Accumulation of prezygotes was not detectably altered for fus1Δ mutants by mating in the presence of 1 M sorbitol, whereas fus2Δ and fus1 fus2 strains showed an exacerbation of the fusion defect, a result opposite to that seen with fps1Δ mutants (Table VI). Thus, fus1 and fus2 mutants differ from fps1Δ mutants in their genetic interactions with GPD1 and in their response to 1 M sorbitol. We conclude that it is unlikely that the cell fusion defect associated with mutations in FUS1 and FUS2 is due to an inability to maintain osmotic stability.
Discussion
During conjugation, haploid cells of opposite mating type reorganize their cell walls to allow cell fusion. The signals that control cell wall degradation and membrane fusion and the machinery that mediates these processes are not known. We have found that the FPS1 gene, which codes for a glycerol transporter, is essential for efficient cell fusion. We identified fps1 mutants based on their defect in mating to an enfeebled (fus1 fus2) mating partner and showed that they are specifically defective in cell fusion: they accumulated prezygotes during mating but were normal for pheromone signaling. The fusion defect of fps1 mutants correlates with their increased level of intracellular glycerol relative to wild-type cells: the defect was partially suppressed by reducing intracellular glycerol and exacerbated by further increasing intracellular glycerol levels. The defect appears to result from osmotic imbalance rather than a high level of glycerol per se since extracellular 1 M sorbitol partially relieved the fusion defect. We propose that, during mating, cells monitor their osmotic state before committing to breaking down and remodeling their cell wall. In particular, under conditions of osmotic imbalance, such as in strains lacking Fps1p, cells interrupt cell wall breakdown. The fps1 mutant thus reveals a checkpoint for cell wall breakdown during mating.
Because fps1 mutants accumulate a high level of intracellular glycerol, we reasoned that they experience a situation analogous to that of wild-type cells exposed to hypotonic conditions, which would lead to activation of the PKC1 pathway (Davenport et al., 1995; Kamada et al., 1995). We therefore anticipated that constitutive activation of the PKC1 pathway, e.g., due to alteration of Pkc1p itself, would cause a fusion defect similar to that of fps1 mutants. This prediction was borne out. Additional analyses of the constitutively activated PKC1 mutant indicate that Pkc1p functions downstream of Fps1p and glycerol accumulation. We suggest that activation of Pkc1p couples sensation of hypoosmotic conditions to inhibition of cell fusion. Other genes required for cell fusion, in particular FUS1 and FUS2, do not appear to participate in this osmolarity checkpoint, as mutants defective in these genes are not influenced by osmolarity.
Osmotic Balance Governs Cell Fusion
During vegetative growth, fps1 mutants accumulate approximately twofold more intracellular glycerol than do wild-type cells, which does not cause any apparent growth defect (Van Aelst et al., 1991; Philips, J., unpublished observations). This increase in intracellular glycerol causes a defect in cell fusion during mating. In principle, Fps1p, which contains six putative membrane-spanning domains (Van Aelst et al., 1991), might play a role in mating that is distinct from its role in transport. Our analyses, however, indicate that its role in mating is a consequence of its function in glycerol transport and that, in its absence, the process of cell wall breakdown is inhibited. Our observations indicate that glycerol functions as an osmolyte during the mating process rather than being directly involved, e.g., in signaling between the mating partners. This conclusion comes from our observations that glycerol per se is not required during mating since the defect of fps1 mutants can be suppressed by extracellular sorbitol or by deleting GPD1, which reduces intracellular glycerol levels. We thus favor the hypothesis that mating cells of yeast are exceptionally sensitive to osmotic disequilibrium before undergoing the potentially lethal morphogenetic changes required for cell fusion. Cells lacking FPS1 sense that they are not osmotically balanced and respond by blocking cell wall breakdown. In other words, cells possess a checkpoint that ensures that they do not degrade their cell wall under hypoosmotic conditions.
It is unclear whether the osmotic imbalance perceived by the fps1 mutant results from a difference between the mutant and its environment or a disparity between mating partners. If the osmolarity between mating partners were monitored, we might expect that an a fps1 and α fps1 mutant mated to each other would form zygotes at normal frequency. Although the number of prezygotes does not return to wild-type levels when fps1Δ mutants are mated with fps1Δ mutants, the reduction in the number of prezygotes compared with fps1Δ mutants mated to wild type may be significant (Table II), indicating that a difference in osmolarity between mating partners may be monitored.
The PKC1 Pathway May Mediate the Cell Fusion Checkpoint
Support for the hypothesis that osmosensing pathways regulate cell fusion was obtained by analyses of the PKC1 pathway. This pathway is required for maintenance of cell integrity and cell wall construction (for review see Errede and Levin, 1993) and is induced when cells are subjected to conditions, such as hypoosmotic shock, that threaten cell integrity (Davenport et al., 1995; Kamada et al., 1995). We found that activation of Pkc1p using a constitutively activated allele of PKC1 led to a fusion defect essentially identical to that of fps1 mutants: PKC1-R398P and fps1 strains accumulated prezygotes that were morphologically indistinguishable from each other.
One possibility is that Pkc1p blocks cell fusion by inhibiting FPS1. Two experiments suggest that this is not the case. First, the PKC1-R398P mutation has a more severe phenotype in an fps1Δ mutant than in a wild-type strain. Additionally, cell fusion is not restored to the PKC1-R398P mutant by reducing intracellular glycerol levels due to deletion of GPD1. These data, in conjunction with the previously demonstrated role of Pkc1p in responding to hypoosmotic shock, lead us to propose that Pkc1p lies downstream of Fps1p and Gpd1p and functions as a part of a checkpoint to monitor osmotic balance during mating (Fig. 7).
If activation of the PKC1 pathway is responsible for the defect in cell fusion of fps1Δ mutants, then Pkc1p should be active in fps1Δ mutants and deletion of pkc1 should suppress the mating defect. Because 1 M sorbitol suppresses the mating defect of fps1Δ mutants, and pkc1Δ mutants require 1 M sorbitol for viability (Levin and Bartlett-Heubusch, 1992; Paravicini et al., 1992), it is impossible to ask whether pkc1Δ suppresses the mating defect. However, evidence that the PKC1 pathway is active in fps1 mutants comes from the analysis of mutants defective in both Fps1p and Mpk1p, the MAP kinase in the PKC1 pathway (Lee et al., 1993; Torres et al., 1991). We have observed that fps1Δ mpk1Δ mutants are inviable unless supported osmotically, indicating that Mpk1p is active in the fps1 mutants. The FPS1 mpk1 strains are inviable only at high temperatures (37°C) (Lee et al., 1993), whereas the fps1 mpk1 strains are also inviable at low temperatures (25°C). One explanation for the inviability of the fps1 mpk1 double mutant is that fps1 mutants depend upon Mpk1 activity to respond to the osmotic imbalance caused by their inefficient release of glycerol.
At least two different models could explain the role of PKC1 in cell fusion. According to one view, Pkc1p negatively regulates cell fusion as part of a checkpoint to ensure that cells are not osmotically vulnerable before fusion (Fig. 7 a). In this case, Pkc1p is used only under conditions, such as hypoosmotic shock, that make cell fusion particularly dangerous to cell integrity. According to a second view, Pkc1p constitutively inhibits cell fusion, so that cell fusion occurs only when Pkc1p is antagonized (Fig. 7 b). According to this model, Pkc1p is a key regulator of cell fusion, inhibiting fusion until a signal turns it off. In this case, the cell fusion defect of fps1 mutants results from activation of the osmotic sensing pathway, which prevents the Pkc1p-dependent inhibition from being relieved during cell fusion.
Previous work has shown that Mpk1p is activated in response to mating pheromone (Errede et al., 1995; Zarzov et al., 1996), presumably because of activation of Pkc1p, and is necessary for viability of yeast cells under these conditions (Errede et al., 1995). Our observations indicate, in contrast, that activation of Pkc1p inhibits a late step in mating. To explain the apparently paradoxical actions of the PKC1 pathway in mating, we suggest, as in the second model above, that Pkc1p is a negative regulator of cell fusion (Fig. 7 b) that, during normal mating, is activated and then subsequently inhibited. We propose that Pkc1p is first activated in response to pheromone and thereby inhibits cell wall degradation during initial stages of pheromone response and projection formation. When mating partners come in contact with each other, they generate a mechanical force as the cell walls become irreversibly adhered to each other, which generates a signal to turn off the PKC1 pathway. We suggest that the signal to turn off the PKC1 pathway is a mechanical signal. It has been previously suggested that the PKC1 pathway responds to membrane stretch due to various stimuli, such as low external osmolarity, high temperature, and drug-induced membrane stretch (Kamada et al., 1995).
Proteins Required for Cell Fusion: Regulators and Machinery
Genes required for cell fusion have been identified by a number of different strategies (Trueheart et al., 1987; McCaffrey et al., 1987; Elion et al., 1990, 1995; Kurihara et al., 1994; Elia and Marsh, 1996). The studies presented here allow us to make a distinction between proteins that regulate the process of fusion (such as Pkc1p or, more indirectly, Fps1p) and potential participants in fusion itself (such as Fus1p and Fus2p). This distinction comes from the following observations. Fps1p is not directly required for cell fusion: its requirement can be relieved by deleting GPD1 or by mating in the presence of high osmolarity. Rather, Fps1p is required to maintain osmotic balance and thereby avoid tripping a checkpoint that inhibits cell fusion. We have shown here that Pkc1p also governs fusion: cell expressing activated forms are defective in fusion presumably because they activate the cell fusion checkpoint. It is not clear whether Fus1p and Fus2p are regulators of fusion or play more direct roles in this process, or whether they are targets for the inhibition mediated by the PKC1 pathway. The fusion defect of fus1 and fus2 mutants is not influenced by deletion of GPD1 or osmotic stabilizers, and thus it is unlikely that these mutants exhibit a fusion defect by evoking the osmotic checkpoint.
Our findings on fusion during yeast mating may have implications for other examples of cell fusion. Maintenance of cell integrity is essential for all cell fusion processes. We therefore expect that processes such as sperm–egg and myoblast fusion may also be regulated by osmolarity. It would be striking if protein kinase C were involved in monitoring osmotic balance during sperm–egg or myoblast fusion. Eyster and McFarland (1995) have, in fact, reported that endogenous modulators of protein kinase C can regulate myogenesis. Perhaps activated versions of protein kinase C could block sperm–egg or myoblast fusion as they block fusion between mating partners in yeast. The targets of the inhibition triggered by the osmolarity-induced checkpoint in yeast remain to be determined. We are seeking to identify such targets by genetic strategies. These proteins might be directly involved in cell fusion during mating and illuminate mechanisms relevant to other cell fusion events.
Acknowledgments
We thank members of our laboratory and S. Doberstein for valuable discussion, S. Hohmann, D. Levin, Y. Takai, J. Chenevert, and J. Whistler for providing plasmids; and S. Hohmann for communicating results before publication. We also thank R. Tabtiang, L. Huang, and F. Banuett for helpful comments on the manuscript, as well as C. Boone for suggesting the RAS2-GFP plasmid.
This work was supported by Research and Program Project Grants from the National Institutes of Health (NIH) to I. Herskowitz. J. Philips was supported by a Medical Scientist Training Program grant from the NIH, supplemented by the Sussman Fund, the Markey Program in Biological Sciences, and the Herbert W. Boyer Fund.
Abbreviations used in this paper
- DAPI
4′,6-diamidino-2-phenylindole
- GFP
green fluorescent protein
- MAP
mitogen-activated protein
- ORF
open reading frame
- YEPD
yeast extract/peptone/dextrose
Footnotes
Please address all correspondence to Ira Herskowitz, Department of Biochemistry and Biophysics, Programs in Genetics and Cell Biology, University of California, San Francisco, San Francisco, CA 94143-0448. Tel.: (415) 476-4985. Fax: (415) 502-5145. e-mail: philips@socrates.ucsf.edu
References
- Adames N, Blundell K, Ashby MN, Boone C. Role of yeast insulin-degrading enzyme homologs in propheromone processing and bud site selection. Science (Wash DC) 1995;270:464–467. doi: 10.1126/science.270.5235.464. [DOI] [PubMed] [Google Scholar]
- Adams AEM, Pringle JR. Relationship of actin and tubulin distribution to bud growth in wild-type and morphogenetic-mutant Saccharomyces cerevisiae. . J Cell Biol. 1984;98:934–945. doi: 10.1083/jcb.98.3.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertyn J, Hohmann S, Prior BA. Characterization of the osmotic-stress response in Saccharomyces cerevisiae: osmotic stress and glucose repression regulate glycerol-3-phosphate dehydrogenase independently. Curr Genet. 1994a;25:12–18. doi: 10.1007/BF00712960. [DOI] [PubMed] [Google Scholar]
- Albertyn J, Hohmann S, Thevelein JM, Prior BA. b. GPD1, which encodes glycerol-3-phosphate dehydrogenase, is essential for growth under osmotic stress in Saccharomyces cerevisiae, and its expression is regulated by the high-osmolarity glycerol response pathway. Mol Cell Biol. 1994;14:4135–4144. doi: 10.1128/mcb.14.6.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Baba N, Ohsumi Y, Kanaya K, Osumi M. Three-dimensional analysis of morphogenesis induced by mating pheromone α-factor in Saccharomyces cerevisiae. . J Cell Sci. 1989;94:207–216. doi: 10.1242/jcs.94.2.207. [DOI] [PubMed] [Google Scholar]
- Bardwell L, Cook JG, Inouye CJ, Thorner J. Signal propagation and regulation in the mating pheromone response pathway of the yeast Saccharomyces cerevisiae. . Dev Biol. 1994;166:363–379. doi: 10.1006/dbio.1994.1323. [DOI] [PubMed] [Google Scholar]
- Brizzio V, Gammie AE, Nijbroek G, Michaelis S, Rose MD. Cell fusion during yeast mating requires high levels of a-factor mating pheromone. J Cell Biol. 1996;135:1727–1740. doi: 10.1083/jcb.135.6.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson M, Botstein D. Two differentially regulated mRNAs with different 5′ ends encode secreted intracellular forms of yeast invertase. Cell. 1982;28:145–154. doi: 10.1016/0092-8674(82)90384-1. [DOI] [PubMed] [Google Scholar]
- Chenevert J, Valtz N, Herskowitz I. Identification of genes required for pheromone-induced cell polarization in Saccharomyces cerevisiae. . Genetics. 1994;136:1287–1297. doi: 10.1093/genetics/136.4.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport KR, Sohaskey M, Kamada Y, Levin DE, Gustin MC. A second osmosensing signal transduction pathway in yeast. Hypotonic shock activates the PKC1 protein kinase-regulated cell integrity pathway. J Biol Chem. 1995;270:30157–30161. doi: 10.1074/jbc.270.50.30157. [DOI] [PubMed] [Google Scholar]
- Elia L, Marsh L. Role of the ABC transporter Ste6 in cell fusion during yeast conjugation. J Cell Biol. 1996;135:741–752. doi: 10.1083/jcb.135.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elion EA, Grisafi PL, Fink GR. FUS3encodes a cdc2/CDC28 related kinase required for the transition from mitosis into conjugation. Cell. 1990;60:649–664. doi: 10.1016/0092-8674(90)90668-5. [DOI] [PubMed] [Google Scholar]
- Elion EA, Trueheart J, Fink GR. Fus2 localizes near the site of cell fusion and is required for both cell fusion and nuclear alignment during zygote formation. J Cell Biol. 1995;130:1283–1296. doi: 10.1083/jcb.130.6.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errede B, Levin D E. A conserved kinase cascade for MAP kinase activation in yeast. Curr Opin Cell Biol. 1993;5:254–260. doi: 10.1016/0955-0674(93)90112-4. [DOI] [PubMed] [Google Scholar]
- Errede B, Cade RM, Yashar BM, Kamada Y, Levin DE, Irie K, Matsumoto K. Dynamics and organization of MAP kinase signal pathways. Mol Reprod Dev. 1995;42:477–485. doi: 10.1002/mrd.1080420416. [DOI] [PubMed] [Google Scholar]
- Eyster K, McFarland D. Production of an endogenous inhibitor of protein kinase C by embryonic myoblasts undergoing differentiation. Comp Biochem Physiol. 1995;112B:549–555. doi: 10.1016/0305-0491(95)00107-7. [DOI] [PubMed] [Google Scholar]
- Field C, Schekman R. Localized secretion of acid phosphatase reflects the pattern of cell surface growth in Saccharomyces cerevisiae. . J Cell Biol. 1980;86:123–128. doi: 10.1083/jcb.86.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gancedo C, Gancedo JM, Sols A. Glycerol metabolism in yeasts: pathways of utilization and production. Eur J Biochem. 1968;5:165–172. doi: 10.1111/j.1432-1033.1968.tb00353.x. [DOI] [PubMed] [Google Scholar]
- Guthrie, C., and G. Fink. 1991. Guide to yeast genetics and molecular biology. In Methods in Enzymology. Volume 194. Academic Press, Inc., San Diego, CA. 933. [PubMed]
- Hasek J, Rupes I, Svobodova J, Streiblova E. Tubulin and actin topology during zygote formation of Saccharomyces cerevisiae. . J Gen Microbiol. 1987;133:3355–3363. doi: 10.1099/00221287-133-12-3355. [DOI] [PubMed] [Google Scholar]
- Herskowitz I. MAP kinase pathways in yeast: for mating and more. Cell. 1995;80:187–197. doi: 10.1016/0092-8674(95)90402-6. [DOI] [PubMed] [Google Scholar]
- Hirayama T, Maeda T, Saito H, Shinozaki K. Cloning and characterization of seven cDNAs for hyperosmolarity-responsive (HOR) genes of Saccharomyces cerevisiae. . Mol Gen Genet. 1995;249:127–138. doi: 10.1007/BF00290358. [DOI] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CL, Hartwell LH. Courtship in S. cerevisiae: both cell types choose mating partners by responding to the strongest pheromone signal. Cell. 1990;63:1039–1051. doi: 10.1016/0092-8674(90)90507-b. [DOI] [PubMed] [Google Scholar]
- Kamada Y, Jung US, Piotrowski J, Levin DE. The protein kinase C-activated MAP kinase pathway of Saccharomyces cerevisiaemediates a novel aspect of the heat shock response. Genes & Dev. 1995;9:1559–1571. doi: 10.1101/gad.9.13.1559. [DOI] [PubMed] [Google Scholar]
- Kuchler K, Sterne RE, Thorner J. Saccharomyces cerevisiae STE6gene product: a novel pathway for protein export in eukaryotic cells. EMBO (Eur Mol Biol Organ) J. 1989;8:3973–3984. doi: 10.1002/j.1460-2075.1989.tb08580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara LJ, Beh CT, Latterich M, Schekman R, Rose MD. Nuclear congression and membrane fusion: two distinct events in the yeast karyogamy pathway. J Cell Biol. 1994;126:911–923. doi: 10.1083/jcb.126.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurjan J. Pheromone response in yeast. Annu Rev Biochem. 1992;61:1097–1129. doi: 10.1146/annurev.bi.61.070192.005313. [DOI] [PubMed] [Google Scholar]
- Lee KS, Irie K, Gotoh Y, Watanabe Y, Araki H, Nishida E, Matsumoto K, Levin DE. A yeast mitogen-activated protein kinase homolog (Mpk1p) mediates signalling by protein kinase C. Mol Cell Biol. 1993;13:3067–3075. doi: 10.1128/mcb.13.5.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin D, Bartlett-Heubusch E. Mutants in the S. cerevisiae PKC1gene display a cell cycle-specific osmotic stability defect. J Cell Biol. 1992;116:1221–1229. doi: 10.1083/jcb.116.5.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipke PN, Kurjan J. Sexual agglutination in budding yeasts: structure, function, and regulation of adhesion glycoproteins. Microbiol Rev. 1992;56:180–194. doi: 10.1128/mr.56.1.180-194.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipke PN, Taylor A, Ballou CE. Morphogenic effects of α-factor on Saccharomyces cerevisiaecells. J Bacteriol. 1976;127:610–618. doi: 10.1128/jb.127.1.610-618.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luyten K, Albertyn J, Skibbe WF, Prior BA, Ramos J, Thevelein JM, Hohmann S. Fps1, a yeast member of the MIP family of channel proteins, is a facilitator for glycerol uptake and efflux and is inactive under osmotic stress. EMBO (Eur Mol Biol Organ) J. 1995;14:1360–1371. doi: 10.1002/j.1460-2075.1995.tb07122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden K, Snyder M. Specification of sites of polarized growth in Saccharomyces cerevisiaeand the influence of external factors on site selection. Mol Biol Cell. 1992;3:1025–1035. doi: 10.1091/mbc.3.9.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey G, Clay FJ, Kelsay K, Sprague GF. Identification and regulation of a gene required for cell fusion during mating of the yeast Saccharomyces cerevisiae. . Mol Cell Biol. 1987;7:2680–2690. doi: 10.1128/mcb.7.8.2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka H, Tanaka K, Hirano H, Fujiwara T, Kohno H, Umikawa M, Mino A, Takai Y. A downstream target of RHO1 small GTP-binding protein is PKC1, a homolog of protein kinase C, which leads to activation of the MAP kinase cascade in Saccharomyces cerevisiae. . EMBO (Eur Mol Biol Organ) J. 1995;14:5931–5938. doi: 10.1002/j.1460-2075.1995.tb00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P, Botstein D. Phenotypic analysis of temperature-sensitive yeast actin mutants. Cell. 1985;40:405–416. doi: 10.1016/0092-8674(85)90154-0. [DOI] [PubMed] [Google Scholar]
- Osumi M, Shimoda C, Yanagishima N. Mating reaction in Saccharomyces cerevisiae.V. Changes in fine structure during the mating reaction. Arch Microbiol. 1974;97:27–38. [PubMed] [Google Scholar]
- Paravicini G, Cooper M, Friedli L, Smith D, Carpentier J, Klig L, Payton M. The osmotic integrity of the yeast cell requires a functional PKC1gene product. Mol Cell Biol. 1992;12:4896–4905. doi: 10.1128/mcb.12.11.4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers S, Michaelis S, Broek D, Santa S, Anna, Field J, Herskowitz I, Wigler M. RAM, a gene of yeast required for a functional modification of RAS proteins and for production of mating pheromone a-factor. Cell. 1986;47:413–422. doi: 10.1016/0092-8674(86)90598-2. [DOI] [PubMed] [Google Scholar]
- Read EB, Okamura HH, Drubin DG. Actin- and tubulin-dependent functions during Saccharomyces cerevisiaemating projection formation. Mol Biol Cell. 1992;3:429–444. doi: 10.1091/mbc.3.4.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reizer J, Reizer A, Saier M H., Jr The MIP family of integral membrane channel proteins: sequence comparisons, evolutionary relationships, reconstructed pathway of evolution, and proposed functional differentiation of the two repeated halves of the proteins. Crit Rev Biochem Mol Biol. 1993;28:235–257. doi: 10.3109/10409239309086796. [DOI] [PubMed] [Google Scholar]
- Rose, M.D., F. Winston, and P. Hieter. 1990. Methods in Yeast Genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 545 pp.
- Schekman R, Brawley V. Localized deposition of chitin on the yeast cell surface in response to mating pheromone. Proc Natl Acad Sci USA. 1979;76:645–649. doi: 10.1073/pnas.76.2.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segall JE. Polarization of yeast cells in spatial gradients of α-mating factor. Proc Natl Acad Sci USA. 1993;90:8332–8336. doi: 10.1073/pnas.90.18.8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague, G.F., and J.W. Thorner. 1992. Pheromone response and signal transduction during the mating process of Saccharomyces cerevisiae. In The Molecular and Cellular Biology of the Yeast Saccharomyces. E.W. Jones, J.R. Pringle, and J.R. Broach, editors. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 657–744.
- Tkacz JS, MacKay VL. Sexual conjugation in yeast. J Cell Biol. 1979;80:326–333. doi: 10.1083/jcb.80.2.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres L, Martin H, Garcia-Saez MI, Arroyo J, Molina M, Sanchez M, Nombela C. A protein kinase gene complements the lytic phenotype of Saccharomyces cerevisiae lyt2mutants. Mol Microbiol. 1991;5:2845–2854. doi: 10.1111/j.1365-2958.1991.tb01993.x. [DOI] [PubMed] [Google Scholar]
- Trueheart J, Fink GR. The yeast cell fusion protein FUS1 is O-glycosylated and spans the plasma membrane. Proc Natl Acad Sci USA. 1989;86:9916–9920. doi: 10.1073/pnas.86.24.9916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trueheart J, Boeke JD, Fink GR. Two genes required for cell fusion during yeast conjugation: evidence for a pheromone-induced surface protein. Mol Cell Biol. 1987;7:2316–2328. doi: 10.1128/mcb.7.7.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtz N, Herskowitz I. Pea2 protein of yeast is localized to sites of polarized growth and is required for efficient mating and bipolar budding. J Cell Biol. 1996;135:725–739. doi: 10.1083/jcb.135.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Aelst L, Hohmann S, Zimmermann FK, Jans AW, Thevelein JM. A yeast homologue of the bovine lens fibre MIP gene family complements the growth defect of a Saccharomyces cerevisiaemutant on fermentable sugars but not its defect in glucose-induced RAS-mediated cAMP signalling. EMBO (Eur Mol Biol Organ) J. 1991;10:2095–2104. doi: 10.1002/j.1460-2075.1991.tb07742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Chen CY, Levin DE. Saccharomyces cerevisiaePKC1 encodes a protein kinase C (PKC) homolog with a substrate specificity similar to that of mammalian PKC. J Biol Chem. 1994;269:16829–16836. [PubMed] [Google Scholar]
- Zarzov P, Mazzoni C, Mann C. The SLT2(MPK1) MAP kinase is activated during periods of polarized cell growth in yeast. EMBO (Eur Mol Biol Organ) J. 1996;15:83–91. [PMC free article] [PubMed] [Google Scholar]