

Abstract
By incubating at 30°C in the presence of an energy source, p34cdc2/cyclin B was activated in the extract prepared from a temperature-sensitive mutant, tsBN2, which prematurely enters mitosis at 40°C, the nonpermissive temperature (Nishimoto, T., E. Eilen, and C. Basilico. 1978. Cell. 15:475–483), and wild-type cells of the hamster BHK21 cell line arrested in S phase, without protein synthesis. Such an in vitro activation of p34cdc2/cyclin B, however, did not occur in the extract prepared from cells pretreated with protein synthesis inhibitor cycloheximide, although this extract still retained the ability to inhibit p34cdc2/cyclin B activation. When tsBN2 cells arrested in S phase were incubated at 40°C in the presence of cycloheximide, Cdc25B, but not Cdc25A and C, among a family of dual-specificity phosphatases, Cdc25, was lost coincidentally with the lack of the activation of p34cdc2/cyclin B. Consistently, the immunodepletion of Cdc25B from the extract inhibited the activation of p34cdc2/cyclin B. Cdc25B was found to be unstable (half-life < 30 min). Cdc25B, but not Cdc25C, immunoprecipitated from the extract directly activated the p34cdc2/cyclin B of cycloheximide-treated cells as well as that of nontreated cells, although Cdc25C immunoprecipitated from the extract of mitotic cells activated the p34cdc2/cyclin B within the extract of cycloheximide-treated cells. Our data suggest that Cdc25B made an initial activation of p34cdc2/cyclin B, which initiates mitosis through the activation of Cdc25C.
The activation of the cdks is a central issue of cell cycle regulation. From S phase, cyclin B accumulates and binds to p34cdc2. Upon binding to cyclin B, p34cdc2 is phosphorylated on Thr-161, a site essential for the catalytic activity of p34cdc2. Simultaneously, p34cdc2 is phosphorylated on Thr-14 and Tyr-15 to prevent the premature activation of p34cdc2/cyclin B (for review see by Dunphy, 1994). The phosphorylation of Thr-161 is carried out by cyclin-dependent kinase (CDK)1-activating kinase (CAK; Shuttleworth et al., 1990; Solomon et al., 1992, 1993; Fesquet et al., 1993; Poon et al., 1993), and the phosphorylation at Thr-14 and Tyr-15 is performed by both Wee1/Mik1 (Featherstone and Russell, 1991; Parker and Piwnica-Worms, 1992; Lee et al., 1994b ; Mueller et al., 1995a ) and Myt1 kinases (Mueller et al., 1995b ). Xenopus Cdc25, a dual-specificity phosphatase, dephosphorylates both Thr-14 and Tyr-15 (Izumi et al., 1992; Kumagai and Dunphy, 1992). During S phase, the activity of Thr-14 and Tyr-15 kinase is held high, and the activity of Cdc25 is low. When DNA replication is completed, the balance between Thr-14 and Tyr-15 kinase and Cdc25 phosphatase is switched to activate p34cdc2/cyclin B. In mammalian cells, Cdc25 is comprised of Cdc25A, Cdc25B, and Cdc25C (Sadhu et al., 1990; Galaktionov and Beach, 1991; Nagata et al., 1991; Millar et al., 1991). All three Cdc25 species are functional homologues of the Schizosaccharomyces pombe cdc25 + gene. Cdc25A is expressed in late G1 and has been reported to be required for initiation of S phase (Jinno et al., 1994), whereas the role of Cdc25B in the cell cycle remains vague. It is thought to be required for mitosis (Galaktionov and Beach, 1991; Nagata et al., 1991; Honda et al., 1993) and for development of human cancer (Galaktionov et al., 1995). On the other hand, Cdc25C, which is phosphorylated and thereby activated by p34cdc2/cyclin B, induces the full activation of p34cdc2 by forming a positive feedback mutual activation loop (Kumagai and Dunphy, 1992; Hoffmann et al., 1993; Izumi and Maller, 1993). A trigger for a full activation of p34cdc2/cyclin B that initiates mitosis is unknown. It is supposed to be carried out by the balance between cyclin and PP2A (Solomon et al., 1990; Lee et al., 1991, 1994a ). PP2A inhibits Thr-161 phosphorylation (Lee et al., 1994a ), and the increment of cyclin accumulates the p34cdc2/cyclin B complexes, which titrates out the stoichiometric inhibitors of the p34cdc2/cyclin B complex (Kumagai and Dunphy, 1995; Lee and Kirschner, 1996). Recently, Cdc25C has been found to be phosphorylated and thereby activated by Plx1 POLO-like kinase (Kumagai and Dunphy, 1996). Wee1 tyrosine kinase is inactivated by Nim1 serine/threonine kinase in S. pombe (Wu and Russell, 1993). Nim1p is localized within the cytoplasm and Wee1p within the nucleus (Wu et al., 1996). Similarly, in BHK21 cells, Cdc25C is localized within the cytoplasm and Wee1p within the nucleus (Heald et al., 1993). In HeLa cells, cyclin B has been reported to be localized within the cytoplasm (Pines and Hunter, 1991, 1994). To initiate mitosis, therefore, Nim1p, Cdc25C, and cyclin B are supposed to enter the nucleus. Thus, in addition to protein phosphorylation, the cellular compartment is thought to play a role in the cell cycle regulation.
In hamster BHK21 cells arrested in S phase, p34cdc2/cyclin B is prematurely activated, and cells enter mitosis either by temperature-sensitive (ts) BN2 mutation of BHK21 cell line or by the administration of caffeine, resulting in premature chromatin condensation (PCC; Nishimoto et al., 1978; Schlegel and Pardee, 1986). In both cases, protein synthesis inhibitors block PCC induction (Nishimoto et al., 1981; Schlegel and Pardee, 1986; Nishitani et al., 1991). These findings have been considered to indicate that a new synthesis of proteins is required for the activation of p34cdc2/cyclin B. Wasserman and Masui (1975) originally found that protein synthesis inhibitors block the Xenopus oocyte maturation induced by progesterone. As a protein that is newly synthesized by progesterone treatment, cyclin B was plausible, since it is synthesized from S phase onwards and is essential for p34cdc2/cyclin B activation (Evans et al., 1983). However, cyclin B already exists in Xenopus oocytes (Minshull et al., 1991) and also in BHK21 cells arrested in S phase (Nishitani et al., 1991). Another candidate for a newly synthesized activator is a c-mos proto-oncogene product (MOS)-like protein. In Xenopus oocyte, MOS that is synthesized just before meiosis I is essential for the activation of p34cdc2/cyclin B (Sagata et al., 1988). Microinjected MOS protein can induce meiosis I, but not meiosis II, without protein synthesis (Yew et al., 1992; Furuno et al., 1994). Thus, the synthesis of protein(s) other than MOS appears to be required to complete Xenopus oocyte maturation. Alternatively, if Xenopus oocytes and BHK21 cells arrested in S phase contain all the molecules required for p34cdc2/cyclin B activation, the inhibition of p34cdc2/cyclin B activation caused by blocking protein synthesis may reflect the loss of endogenous activators of p34cdc2/cyclin B that are rapidly turning over.
tsBN2 mutation causes the loss of RCC1 function, resulting in either G1 arrest or premature activation of p34cdc2/cyclin B depending on the cell cycle stage (Nishimoto et al., 1978; Nishitani et al., 1991). RCC1 is localized on chromatin (Frasch, 1991) and functions as a GDP/GTP exchanging factor on Ran, a Ras-like nuclear G protein (Bischoff and Ponstingle, 1991). GTP hydrolysis of Ran is essential for the nuclear import of karyophilic proteins (Melchior et al., 1993; Moore and Blobel, 1993). It can be argued, therefore, that all of the phenotypes of rcc1− are indirect consequences of the role of Ran in nuclear transport. However, PCC induced by the loss of RCC1 can be inhibited by microinjection of both GTP- and GTPγS-bound Ran (Ohba et al., 1996), indicating that GTP-Ran plays some role in the nucleus. This finding is consistent with the fact that Yrb2p that possesses a Ran-binding domain similar to RanBP1/Yrb1p (Dingwall et al., 1995), thereby specifically binding to the GTP-bound Gsp1p S. cerevisiae Ran homologue, is localized in the nucleus (Noguchi et al., 1997). The disruptant of the YRB2 gene, which is cold sensitive, has no defect in the nucleus/cytosol exchange of macromolecules (Noguchi et al., 1997).
When tsBN2 cells are incubated in S phase at 40°C, the nonpermissive temperature, Cdc25C enters the nucleus parallel with PCC induction (Seki et al., 1992). This is inconsistent with the findings that the loss of RCC1 function retards nuclear protein import (Tachibana et al., 1994; Dickmanns et al., 1996). We noticed the fact that the inhibition of protein synthesis blocks the nuclear import of Cdc25C as well as the activation of p34cdc2/cyclin B (Seki et al., 1992), which suggests that loss of RCC1 function induces a new protein synthesis required either for nuclear entrance of Cdc25C or for the activation of p34cdc2/cyclin B. To clarify the relationship between the loss of RCC1 and the p34cdc2/cyclin B activation, we began to identify the protein(s) required for p34cdc2/cyclin B activation induced by loss of RCC1 (Nishitani et al., 1991). To do this, we have first established a method to activate the p34cdc2/ cyclin B complexes in vitro by using the extract prepared from BHK21 cells arrested in S phase (Seki et al., 1992). In this paper, we confirmed our previous results by using tsBN2 cells. We found that the in vitro activation of p34cdc2/cyclin B occurred without new protein synthesis and that p34cdc2/cyclin B was not activated in the extract of cells pretreated with protein synthesis inhibitor. Instead of a new synthesis of p34cdc2/cyclin B activators, therefore, an endogenous activator(s) of p34cdc2/cyclin B was suggested to be lost by blocking protein synthesis. Cdc25B, but not Cdc25C or Cdc25A, was identified as such an unstable activator of p34cdc2/cyclin B.
Materials and Methods
Cells and Cell Culture
The BHK21 cell line derived from golden hamsters and its ts mutant tsBN2 cell line (Nishimoto et al., 1978) and human HeLa cell line were grown in DME containing 10% calf or FCS in a humidified atmosphere containing 10% CO2. HeLa cells were cultured at 37.5°C, and BHK21 and tsBN2 cells were cultured at 33.5°C. As the nonpermissive temperature, 40°C was used for tsBN2 cells.
Synchronization of Cells
Wild-type BHK21 and tsBN2 cells were cultured in isoleucine-free medium for 24–30 h (G0/G1 arrest) and then in DME containing 10% FCS and 2.5 mM hydroxyurea (HU) for 15.5 h (S phase arrest) as described (Nishitani et al., 1991). To synchronize in mitosis, after the release from HU block, cells were incubated in DME medium containing 10% FCS and 0.4 μg/ml Nocodazole for 8 h.
HeLa cells were cultured in medium containing thymidine (2 mM) for 14 h, in a normal growth medium for 13 h, and finally in medium containing 2.5 mM HU for 14 h (S phase arrest).
Preparation of Cell Extracts
Cells were collected with a scraper and then sequentially washed, first with TD buffer containing 136.8 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4, and 25 mM Tris-HCl (pH 7.4), with the EB buffer, and finally with the 0.5 × EB buffer as described (Seki et al., 1992). The EB buffer contained 20 mM Hepes-KOH (pH 7.4), 50 mM KCl, 50 mM 2-glycerophosphate, 15 mM EGTA, and 10 mM MgCl2. Washed cells were lysed by pipetting and vortexing in the buffer (0.5 × EB-1), 0.5 × EB buffer supplemented with 5 μg/ml cytochalasin B, 5 mM DTT, a mixture of protease inhibitors, 1 mM ATP-γ-S, and 0.5% NP-40. After being kept on ice for 30 min, the extract was centrifuged at 2,000 g for 5 min. The supernatant was further centrifuged at 80,000 g for 30 min. The resulting supernatant was used as the cytosolic extracts. The total cell extract was prepared with the same procedure, except that the cells were lysed directly in the 0.5 × EB-1 buffer supplemented with 500 mM NaCl. The mixture of protease inhibitors contained 2 μg/ml of aprotinin, antipain dihydrochloride, leupeptin, and pepstatin A, and 100 μM p-amidinophenyl methansulfonyl fluoride (pAPMSF).
The cell extracts were desalted, concentrated by centricon (Amicon Corp., Danvers, MA), and then subjected to use as the cell extracts for the in vitro activation assay of p34cdc2 protein kinase.
Preparation of E. coli-Produced Hamster Cdc25C
E. coli BL21 (DE3)LysS/pGEX-KG-hamster Cdc25C (a gift of K. Yamashita, Kyushu University, Fukuoka, Japan) strains were cultured at 30°C up to an OD 600 of 0.4, treated with IPTG (final concentration, 1.0 mM) for 3 h, and then collected by centrifugation. Cell-pellet was lysed and glutathione-S-transferase (GST)-Cdc25C was purified using glutathione Sepharose 4B beads (Pharmacia Fine Chemicals, Piscataway, NJ) as described previously (Noguchi et al., 1997). Eluted GST-Cdc25C was further purified by gel filtration using Sephacryl S-100 HR column (Pharmacia Fine Chemicals).
Immunodepletion of cyclin B, cdc25B, and cdc25C from Cytoplasmic Extract
The antibody was mixed with protein G Sepharose (Pharmacia Fine Chemicals), rotated at 4°C for 2 h. Beads were pulled down by centrifugation, washed with 0.5 × EB-1 buffer, and then suspended in the cytosolic extracts. After rotation for 2 h at 4°C, the beads were pulled down by centrifugation. The resulting supernatant was used as immunodepleted extracts.
In Vitro Activation of p34cdc2 Histone H1 Kinase and Assay of Histone H1 Kinase Activity
The extract of 5–15 μl (protein concentration: 3 mg/ml) was incubated at 30°C in the presence of 1 mM ATP, 10 mM creatine phosphate, and 2.5 μg/ml creatine kinase as an energy source (Kumagai and Dunphy, 1992). The reaction was stopped by a buffer containing 40 mM Hepes-NaOH (pH 7.5), 60 mM 2-glycerophosphate, 20 mM p-nitrophenyl phosphate, 0.5 mM Na3VO4, 250 mM NaCl, 15 mM MgCl2, 1% Triton X-100, 1 mM DTT, and a mixture of protease inhibitors. p34cdc2/cyclin B complexes were precipitated with the anti-cyclin B1 antibody or the p13suc1 beads (Nishitani et al., 1991), as indicated. The histone H1 kinase activity of precipitates was assayed by using the histone H1 as the substrate, as described (Nishitani et al., 1991).
The reaction was carried out more than twice by using different extracts.
Immunoblotting
Antibodies used were the anti-cdc2 antibodies against PSTAIR peptide (Nishitani et al., 1991), the anti-phosphotyrosine antibody, pTyr (a gift of Dr. J.Y.J. Wang, University of California, San Diego, CA; Morla and Wang, 1986), and the antibodies against cdc25A, cdc25B and cdc25C antibodies (Seki et al., 1992), cyclin B, and cdk2 (all from Santa Cruz Biotechnology, Santa Cruz, CA). The procedure for immunoblotting was carried out as described elsewhere (Nishitani et al., 1990), except that the detection was achieved using an ECL detection system (Amersham).
Pulse–Chase of Cdc25B
Cells were preincubated in methionine-free medium for 1 h and then labeled with 100 μCi/ml of [35S]l-methionine (ICN Biochemicals Inc., Irvine, CA) for 3 h. After washing out the labeled medium, cells were given the medium containing ten times the concentration (2 mM) of methionine. Cdc25B was immunoprecipitated using the anti-Cdc25B antibody, electrophoresed in SDS–10% polyacrylamide gel, and analyzed by autoradiography.
Transient Over-Expression of Cdc25B
Wild-type BHK21 cells were seeded, and after 18 to 23 h of incubation at 33.5°C, cells were transfected with plasmid pcD2-human Cdc25B (a gift from H. Okayama, Tokyo University, Tokyo, Japan) using lipofection (LipofectaminTM; GIBCO BRL, Gaithersburg, MD) as described previously (Makishima et al., 1997).
Results
In Vitro Activation of p34cdc2/cyclin B
A series of cultures of tsBN2 cells arrested in S phase (throughout this paper, cells were arrested in S phase except for the case mentioned) was incubated at 40°C, the nonpermissive temperature. Every hour, cells were harvested and lysed. From the total cell extracts prepared, p34cdc2/cyclin B complexes were precipitated with p13suc1 beads and assayed for histone H1 kinase activity as described in Materials and Methods. As previously reported (Nishitani et al., 1991), the histone H1 kinase activity of p34cdc2/cyclin B increased by incubating tsBN2 cells at 40°C (Fig. 1 A). When the same extracts were incubated in vitro at 30°C, the histone H1 kinase activity of p34cdc2/cyclin B was also increased (Fig. 1 B). Such an in vitro activation of p34cdc2/cyclin B occurred in the extract of tsBN2 cells prepared before increasing the temperature (Fig. 1 A, 0 h), as well as in the extract of tsBN2 cells that had been incubated at 40°C for 1 h and had no histone H1 kinase activity yet (Fig. 1 A, 1 h). The finding that p34cdc2/cyclin B was activated in the cell extracts prepared from tsBN2 cells that had been harvested before increasing the temperature indicated that the disruption of cells, rather than the mutation of tsBN2 cells, induced an in vitro activation of p34cdc2/cyclin B. To address this issue, the extract was prepared from wild-type BHK21 cells and incubated at 30°C for 1 h. To specifically detect the activity of p34cdc2/cyclin B complex, we used, henceforth, the anti-cyclin B antibody instead of p13suc1 beads. The p34cdc2/cyclin B was activated in the extract of wild-type BHK21 cells (Fig. 2, columns 3 and 4) as reported (Seki et al., 1992).
Figure 1.

In vivo and in vitro activation of p34cdc2/cyclin B in tsBN2 cells. (A) Cultures of tsBN2 cells synchronized in S phase with HU were incubated at 40°C in the presence of HU. Every hour, cells were harvested and lysed to prepare the total cell extract. From 10 μl (protein concentration: 3 mg/ml) of the total cell extract, the p34cdc2/cyclin B complexes were precipitated using p13suc1 beads and assayed for histone H1 kinase activities. (B) The total cell extracts prepared from tsBN2 cells that had been incubated at 40°C for 0 (–□–), 1 ( –♦–), 2 (–▪–), and 3 (–⋄–) h were incubated at 30°C. Every hour, 10 μl of the extracts (protein concentration: 3 mg/ml) were sampled and mixed with the reaction-stopping buffer. The p34cdc2/cyclin B complexes were precipitated using p13suc1 beads and assayed for histone H1 kinase activities. Histone H1 kinase activities shown in A correspond to the value at the beginning of incubation (time: 0) in B as shown by arrows.
Figure 2.

In vitro activation of p34cdc2/cyclin B in wild-type BHK21 cells. Cultures of wild-type BHK21 cells synchronized in S phase with HU were incubated in the presence (CHX-treated, columns 1 and 2) or absence (non-treated, columns 3–6) of cycloheximide (10 μg/ml) for 1 h and then lysed to prepare the cytosolic extracts, as described in Materials and Methods. The cytosolic extracts were incubated at 30°C in the absence (columns 1 to 4) or presence (columns 5 and 6) of cycloheximide (10 μg/ml). Before incubation (columns 1, 3, and 5) or after incubation for 1 h (columns 2, 4, and 6), 8 μl of the extracts (protein concentration: 3 mg/ml) were mixed with the reaction-stopping buffer. p34cdc2/ cyclin B complexes were immunoprecipitated using the anti- cyclin B antibodies and assayed for histone H1 kinase activities.
In parallel with the activation of p34cdc2/cyclin B, the molecular mass of p34cdc2 was reduced, as reported (Atherton-Fessler et al., 1994), consistent with the dephosphorylation of Tyr-15 of p34cdc2, which was confirmed by using the anti-pTyr antibody that specifically recognizes phosphorylated tyrosine (Morla and Wang, 1986) (Fig. 3).
Figure 3.

Dephosphorylation of p34cdc2 occurs in parallel with the activation of p34cdc2/cyclin B. Wild-type BHK21 cells synchronized in S phase with HU were incubated in the presence (lanes 1 and 2) or absence (lanes 3 and 4) of cycloheximide (10 μg/ml) for 1 h and then lysed to prepare the total cell extracts. The total cell extracts were incubated at 30°C. Before incubation (lanes 1 and 3), and after incubation for 1 h (lanes 2 and 4), the extracts were mixed with SDS-sample buffer and boiled. Total proteins of the mixtures were electrophoresed in SDS-10% polyacrylamide gel and analyzed by immunoblotting using as a probe, anti-pTyr (top) and the anti-PSTAIR (bottom), respectively.
Protein Synthesis Required for Activation of p34cdc2/cyclin B
In the presence of protein synthesis inhibitor, while RCC1 disappears due to the mutation, the activation of p34cdc2/ cyclin B does not occur in tsBN2 cells arrested in S phase, at 40°C, the nonpermissive temperature (Nishimoto et al., 1981; Nishitani et al., 1991). To investigate the effect of protein synthesis inhibitor on the activation of p34cdc2/cyclin B, we prepared the extracts from S phase-arrested wild-type BHK21 cells that had been incubated in the presence of protein synthesis inhibitor cycloheximide (10 μg/ml) for 1 h before cell harvest (henceforth designated cycloheximide treated). When the extract of cycloheximide-treated cells was incubated at 30°C for 1 h, there was no activation of p34cdc2/cyclin B (Fig. 2, columns 1 and 2), consistent with the finding that p34cdc2 was not dephosphorylated (Fig. 3, lanes 1 and 2). Therefore, cycloheximide-treated cells had no ability to activate the p34cdc2/cyclin B complexes, both in vivo and in vitro.
To exclude the possibility that cycloheximide carried over from cycloheximide-treated cells inhibited the activation of p34cdc2/cyclin B, the extract was prepared from wild-type BHK21 cells that had not been treated with cycloheximide, and then incubated in vitro either in the presence or absence of 10 μg/ml of cycloheximide. In both cases, p34cdc2/cyclin B was activated (Fig. 2, compare columns 4 and 6), indicating that BHK21 cells that were arrested in S phase possessed all molecules required for the observed in vitro activation of p34cdc2/cyclin B. The lack of activation of p34cdc2/cyclin B in the extract prepared from cycloheximide-treated cells, therefore, suggested that some molecules or protein modifications required for the activation of p34cdc2/cyclin B were lost during the blocking of protein synthesis.
Activator(s), but Not Inhibitor(s) of p34cdc2/cyclin B Was Lost by Blocking Protein Synthesis
To investigate the possibility that the activator(s) of p34cdc2/cyclin B was lost by blocking protein synthesis, the extract was prepared from S phase-arrested BHK21 cells that had been either treated with cycloheximide or not treated with cycloheximide (henceforce designated nontreated) as described in Materials and Methods. To exclude the possibility that p34cdc2/cyclin B is involved in the activation step, the p34cdc2/cyclin B complexes were depleted using the anti-cyclin B antibodies, from these extracts. The cyclin B-depleted extracts were then mixed and incubated at 30°C, with the extract prepared either from cycloheximide-treated or from nontreated cells, respectively.
The cyclin B-depleted extract prepared from nontreated cells, while it had no activity of p34cdc2/cyclin B (Fig. 4 C), possessed the ability to activate the p34cdc2/cyclin B within the extract prepared from cycloheximide-treated cells (Fig. 4 A). On the other hand, the cyclin B-depleted extract prepared from cycloheximide-treated cells had no ability to activate the p34cdc2/cyclin B within the extract prepared from cycloheximide-treated cells. To the contrary, the cyclin B-depleted extract of cycloheximide-treated cells was found to have the ability to inhibit the in vitro activation of p34cdc2/cyclin B (Fig. 4 B). These findings indicated that wild-type BHK21 cells that were arrested in S phase possessed both the activator(s) and the inhibitor(s) of p34cdc2/cyclin B. By blocking protein synthesis, the activator(s), which was not the endogenous p34cdc2/cyclin B complex, was lost, but the inhibitor(s) remained. The inhibitory activity for p34cdc2/cyclin B of the cycloheximide-treated cell extracts may reflect either the activation of p34cdc2/cyclin B inhibitor(s) or the loss of balance between the inhibitor and the activator of p34cdc2/cyclin B.
Figure 4.



Both activator(s) and inhibitor(s) of p34cdc2/cyclin B are present in HU-arrested BHK21 cells. Cultures of wild-type BHK21 cells synchronized in S phase with HU were incubated in the absence (nontreated) or presence (CHX-treated) of cycloheximide (10 μg/ ml) for 1 h and then lysed to prepare the cytosolic extracts. When indicated as cyclin B-depleted, the p34cdc2/cyclin B complexes were immunoprecipitated using the anti-cyclin B antibody and the supernatant was used as the cyclin B-depleted extracts as described in Materials and Methods. 8 μl of the total cytosolic extracts prepared from CHX-treated cells (A), from nontreated cells (B), and as a control of the buffer (0.5× EB-1; C) were mixed with 0, 4, 8, 16, and 32 μl of the cyclin B-depleted extracts prepared from nontreated (–•–) or CHX-treated cells (–○–). In each case, the final volume was adjusted to 40 μl with the buffer (0.5 × EB-1). After incubation at 30°C for 1 h, the p34cdc2/cyclin B complexes were immunoprecipitated using the anti-cyclin B antibodies and assayed for histone H1 kinase activities.
Cdc25B Was Lost by Blocking Protein Synthesis
To identify proteins that were required for p34cdc2/cyclin B activation and disappeared during treatment of cells with protein synthesis inhibitors, cultures of tsBN2 cells were synchronized in S phase at 33.5°C and then incubated at 40°C, either in the absence or presence of cycloheximide (10 μg/ml). Every hour, cells were harvested and assayed for the presence of p34cdc2/cyclin B activators, such as Cdc25A, B, and C, and as controls, p34cdk2, p34cdc2, and cyclin B, by immunoblotting analysis (Fig. 5).
Figure 5.


Cdc25B was lost by treatment with cycloheximide. A series of cultures of tsBN2 cells synchronized in S phase with HU was incubated at 40°C in the absence (nontreated) or presence (CHX-treated) of cycloheximide (10 μg/ml). Every hour, cells were harvested to prepare the total cell extracts. (A) p34cdc2/cyclin B complexes of total cell extracts prepared from nontreated (–•–) or cycloheximide-treated cells (–○–) were immunoprecipitated using the anti-cyclin B antibody and assayed for histone H1 kinase activities. (B) Proteins of total cell extracts prepared from nontreated (−CHX) and CHX-treated (+CHX) cells were electrophoresed in SDS-10% polyacrylamide gel and analyzed by immunoblotting using as a probe the antibodies against the proteins indicated.
While p34cdc2/cyclin B was activated in tsBN2 cells without cycloheximide, no activation of p34cdc2/cyclin B occurred in the presence of cycloheximide as previously reported (Nishitani et al., 1991; Fig. 5 A). Parallel with the activation of p34cdc2/cyclin B, the molecular mass of p34cdc2 was shifted down to the faster-migration form, while that of Cdc25C was shifted up to the retarded-migration form (Fig. 5 B), as previously reported (Izumi et al., 1992; Kumagai and Dunphy, 1992; Seki et al., 1992; Hoffmann et al., 1993). In tsBN2 cells treated with cycloheximide, the molecular mass of both Cdc25C and p34cdc2 did not change, consistent with the fact that there was no activation of p34cdc2/cyclin B. The amount of Cdc25A and C and p34cdc2 seemed to be constant during treatment with cycloheximide. However, Cdc25B became undetectable after inhibiting protein synthesis for 1 h. Loss of Cdc25B was also observed in cycloheximide-treated wild-type BHK21 cells (Fig. 6 A), indicating that Cdc25B was an unstable protein in BHK21 cells.
Figure 6.



Immunodepletion of Cdc25B blocks the in vitro activation of p34cdc2/cyclin B. Cytosolic extracts of wild-type BHK21 cells treated (CHX-treated) or not treated (non-treated) with cycloheximide (10 μg/ml) were prepared as described in Fig. 4. From the extract of nontreated cells, Cdc25B or Cdc25C was immunoprecipitated using the antibodies against Cdc25B and Cdc25C, respectively, and the supernatants were used as the Cdc25B- or Cdc25C-immunodepleted extracts. (A) Proteins of the total and immunodepleted cytosolic extracts were electrophoresed in SDS-10% polyacrylamide gel and analyzed by immunoblotting using the antibodies against indicated proteins. Lanes 1 and 2, total cytosolic extract of cycloheximide-treated (CHX) and nontreated (non) cells. Lanes 3 and 4, Cdc25B- and Cdc25C-depleted extract, respectively. (B) Total cytosolic extracts of cycloheximide-treated cells (CHX-treated) and nontreated (non-treated) cells and the Cdc25B- or Cdc25C-depleted extracts of nontreated cells (designated as −cdc25B and −cdc25C, respectively) were incubated at 30°C. Before incubation (open column) and after incubation for 1 h (shaded column), the p34cdc2/cyclin B complexes were immunoprecipitated using the anti-cyclin B antibody and assayed for histone H1 kinase activities. (C) From the cytosolic extract of nontreated cells, cyclin B and then either Cdc25B or Cdc25C were immunodepleted using the antibodies against cyclin B, Cdc25B or Cdc25C, respectively: cyclin B-depleted (−cyclin B), cyclin B- and Cdc25B-depleted (−cyclinB and −cdc25B), and cyclin B- and Cdc25C-depleted (−cyclin B and −cdc25C) extracts. Total cytosolic extracts of cycloheximide-treated cells (CHX-treated) and the immunodepleted extracts from nontreated cells (non-treated) were incubated alone (open and shaded column) or after mixing with the total cytosolic extract of CHX-treated cells (filled column) at 30°C. Before incubation (open column) and after incubation for 1 h (shaded and filled column), the p34cdc2/cyclin B complexes were immunoprecipitated using the anti-cyclin B antibody and assayed for histone H1 kinase activities.
Interestingly, even in the absence of cycloheximide, the amount of Cdc25B was gradually reduced and finally lost in parallel with the reduction in histone H1 kinase activity of p34cdc2/cyclin B, similar to cyclin B (Fig. 5, compare A and B).
Cdc25B Is an In Vitro Activator of p34cdc2/cyclin B
To confirm that Cdc25B is an activator required for the in vitro activation of p34cdc2/cyclin B, Cdc25B, and as a control, Cdc25C, which is essential for p34cdc2/cyclin B activation induced by loss of RCC1 (Seki et al., 1992), were depleted from the cytosolic extract prepared from nontreated, wild-type BHK21 cells, using the anti-Cdc25B and Cdc25C antibodies, respectively. Depletion of Cdc25B and Cdc25C was confirmed by immunoblotting analysis (Fig. 6 A). In both cases, Cdc25A remained in the extracts.
Cdc25B- and Cdc25C-depleted extracts were then incubated at 30°C for 1 h. The p34cdc2/cyclin B complex was activated in the Cdc25C-depleted extract, but not in the Cdc25B-depleted extract (Fig. 6 B, cdc25B and cdc25C). The effect of Cdc25B depletion was further confirmed using the extracts from which p34cdc2/cyclin B had been depleted, in addition to Cdc25B or CdC25C. These extracts alone possessed no activity of p34cdc2/cyclin B. However, as shown in Fig. 6 C, the cyclin B- and Cdc25C-depleted extract contained the ability to activate the p34cdc2/cyclin B complex of the extract prepared from cycloheximide-treated cells, whereas the cyclin B- and Cdc25B-depleted extract did not.
To further confirm the involvement of Cdc25B in the in vitro activation of p34cdc2/cyclin B, proteins were immunoprecipitated from the extracts of cycloheximide-treated and nontreated cells, using the anti-Cdc25B antibody and assayed for their abilities to activate the p34cdc2/cyclin B complex within the extract of cycloheximide-treated cells. Proteins that were immunoprecipitated from the extracts of nontreated cells activated p34cdc2/cyclin B, but those precipitated from cycloheximide-treated cells did not (Fig. 7 A). In both cases, immunoprecipitated proteins alone had no activity of histone H1 kinase. As a control, Cdc25C was immunoprecipitated from the extract of BHK21 cells arrested either in S phase or in mitosis. While Cdc25C derived from mitotic cells had an ability to activate the p34cdc2/cyclin B complex within the extract of cycloheximide-treated cells, Cdc25C derived from S phase-arrested cells did not (Fig. 7 B). These findings indicated that Cdc25B was an activator of the p34cdc2/cyclin B complex, which was lost by cycloheximide treatment.
Figure 7.
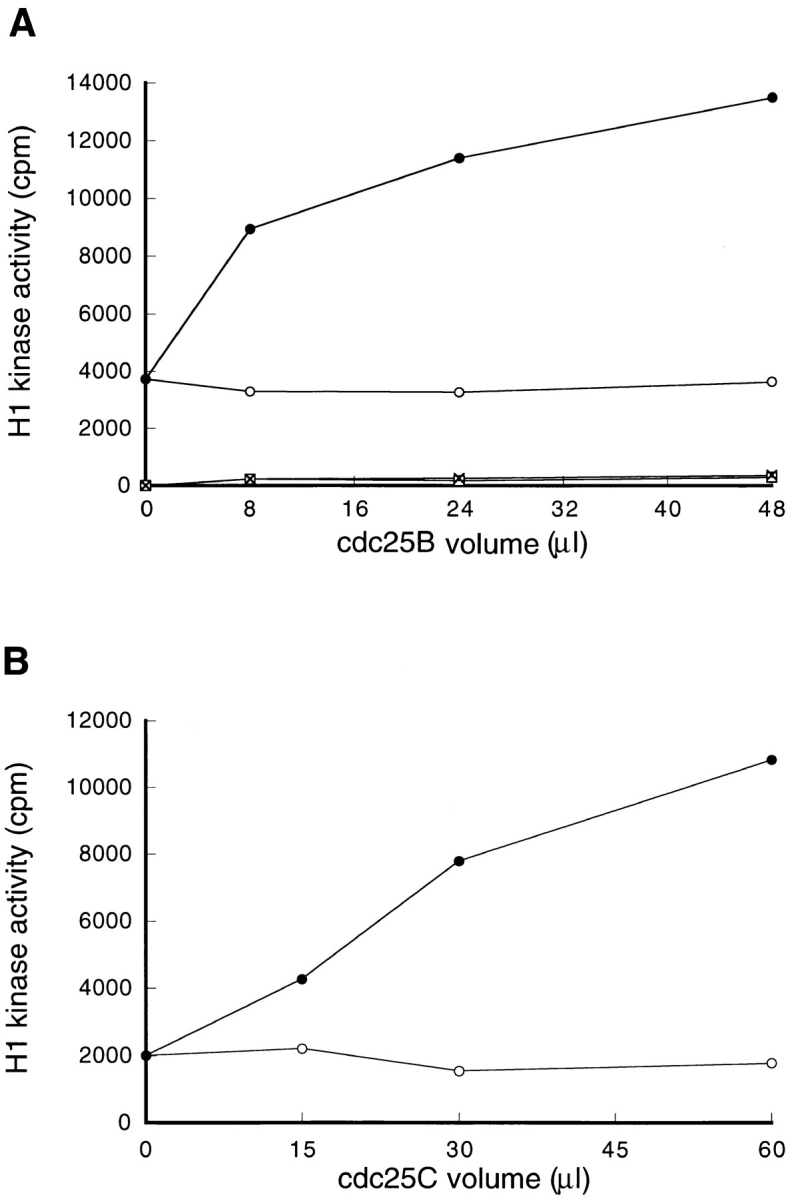
Cdc25B, but not Cdc25C, activates the p34cdc2/cyclin B complexes of cycloheximide-treated cells. (A) Cdc25B proteins were immunoprecipitated using the anti-Cdc25B antibody from 8, 24, and 48 μl of the cytosolic extracts derived from nontreated (–•–) and, as a control, CHX-treated (–○–) wild-type BHK21 cells that had been arrested in S phase. The immunoprecipitated Cdc25B prepared from the indicated volume of the cytosolic extract was then mixed with 8 μl of total cytosolic extract prepared from cycloheximide-treated cells and as a control, with 8 μl of the buffer (0.5 × EB-1; nontreated: –□–; CHX-treated: -X-). A final volume was adjusted to 30 μl. After incubation at 30°C for 1 h, p34cdc2/cyclin B was immunoprecipitated using the anti-cyclin B antibody and assayed for histone H1 kinase activities. (B) A series of cultures of BHK21 cells was synchronized with HU in S phase, and then half of the cultures was released from HU block and arrested in M phase, as described in Materials and Methods. The cytosolic extracts were prepared from the S phase-arrested (–○–) and the M phase-arrested cells (–•–), respectively, and then Cdc25C proteins were immunoprecipitated using the anti-Cdc25C antibody from 15, 30, and 60 μl of the S and M phase extract. The immunoprecipitated Cdc25C prepared from the indicated volume of the cytosolic extract was then mixed with 8 μl of total cytosolic extract prepared from cycloheximide-treated cells. A final volume was adjusted to 30 μl. After incubation at 30°C for 1 h, p34cdc2/cyclin B was immunoprecipitated using the anti-cyclin B antibody and assayed for histone H1 kinase activities.
Cdc25B Directly Activated p34cdc2/cyclin B
To address the question of whether Cdc25B directly activate p34cdc2/cyclin B, Cdc25B was immunoprecipitated from the extract of nontreated cells, and as a control, from the extract of cycloheximide-treated cells. On the other hand, the p34cdc2/cyclin B complexes were immunoprecipitated using the anti-cyclin B antibody, from the extracts of cycloheximide-treated and nontreated cells. Proteins immunoprecipitated with the anti-Cdc25B antibody alone had no activity of histone H1 kinase (Fig. 8, base line). By mixing with the p34cdc2/cyclin B, which had no histone H1 kinase activity by itself (Fig. 8, zero point), Cdc25B of nontreated cells activated p34cdc2/cyclin B complexes isolated from both the extracts of nontreated and cycloheximide-treated cells, with a similar kinetics (Fig. 8, A and B). As a control, mock-precipitated Cdc25B from cycloheximide-treated cells had no ability to activate p34cdc2/cyclin B.
Figure 8.
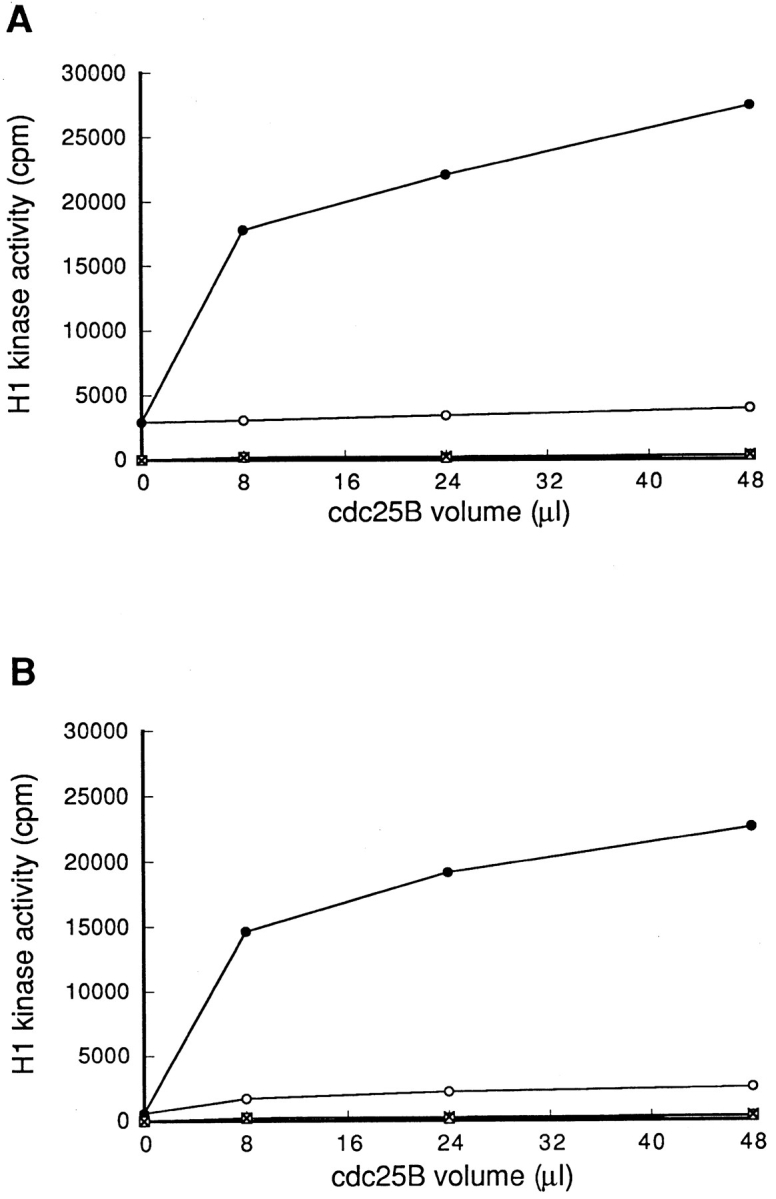
Immunoprecipitated Cdc25B equally activated p34cdc2/ cyclin B complexes of cycloheximide-treated and nontreated cells. From 8 μl of cytosolic extracts of nontreated (A) or cycloheximide-treated (B) wild-type BHK21 cells, p34cdc2/cyclin B complexes were immunoprecipitated using the anti-cyclin B antibody and then mixed with proteins immunoprecipitated with the anti-Cdc25B antibodies from 8, 24, and 48 μl of cytosolic extracts derived from cycloheximide-treated (–○–) and non-treated (–•–) wild-type BHK21 cells. As a control, proteins immunoprecipitated with the anti-Cdc25B antibodies were incubated by themselves in the buffer (0.5 × EB-1; nontreated: –□–; CHX-treated: -X-). A final volume was adjusted to 30 μl. After incubation at 30°C for 1 h, p34cdc2/cyclin B was immunoprecipitated using the anti-cyclin B antibody and assayed for histone H1 kinase activities.
Compared to the p34cdc2/cyclin B retained within the extract, the isolated p34cdc2/cyclin B complexes were highly activated by Cdc25B (compare Fig. 7 and 8). This finding may suggest that the inhibitors for p34cdc2/cyclin B activation retained within the extracts were removed from the p34cdc2/cyclin B complexes by immunoprecipitation. With these results taken together, we concluded that Cdc25B directly activated the p34cdc2/cyclin B complex, while we could not exclude the possibility that some proteins that were co-precipitated with p34cdc2/cyclin B or with Cdc25B, activated Cdc25B before the activation of p34cdc2/cyclin B (Galaktionov and Beach, 1991). These findings also indicated that the protein modification required for the activation of p34cdc2/cyclin B was not lost by blocking protein synthesis.
Cdc25B Is a Short-Life Protein in Mammalian Cells
Our present results suggest that Cdc25B was lost in tsBN2 and wild-type BHK21 cells treated with protein synthesis inhibitor cycloheximide. Since the accumulation of truncated, abnormal proteins may enhance ubiquitin-dependent proteolysis (Hershko, 1983), it is possible that Cdc25B, which is stable in a normal growth condition, becomes unstable by blocking protein synthesis. To address this question, Cdc25B was pulse labeled with [35S]methionine and chased in the presence of an excess amount of cold methionine in wild-type BHK21 cells that had been arrested in S phase. As shown in Fig. 9 A, Cdc25B was rapidly lost even in the absence of cycloheximide. To further confirm the instability of Cdc25B, human Cdc25B (a gift of H. Okayama) was transfected into wild-type BHK21 cells, and the expressed human Cdc25B was pulse–chased in randomly growing wild-type BHK21 cells. Exogenously expressed human Cdc25B was rapidly lost with a half-life of <30 min, similar to the endogenous Cdc25B (Fig. 9, A and B). In the same cultures of wild-type BHK21 cells expressing human Cdc25B, the endogenous hamster Cdc25B was lost with the kinetics similar to that of human Cdc25B (data not shown). Thus, Cdc25B was unstable in BHK21 cells, and the stability of Cdc25B was not changed by arresting cells in S phase with hydroxyurea. Even in HeLa cells, Cdc25B, but not Cdc25A and Cdc25C, was lost during treatment with cycloheximide (Fig. 9 C). Taking these results together, we concluded that Cdc25B was rapidly turning over in mammalian cells.
Figure 9.



Cdc25B is an unstable protein. (A) Endogenous hamster Cdc25B. Wild-type BHK21 cells were seeded at 1 × 106/100-mm dish, synchronized in S phase, incubated in Met-free medium containing HU, labeled with [35S]l-methionine, and chased as described in Materials and Methods. At the indicated times (min), cells were lysed and Cdc25B was immunoprecipitated and analyzed by autoradiography. Human Cdc25B expressed in hamster BHK21 cells. Wild-type BHK21 cells were seeded at 2 × 106/100-mm dish. After 18 h of incubation at 33.5°C, cells were transfected with plasmid pcD2-human Cdc25B (16 μg/100-mm dish) using lipofection. After incubation in a normal medium for an additional 48 h at 33.5°C, transfected cells were incubated in Met-free medium, labeled with [35S]l-methionine, and chased as described in Materials and Methods. At the indicated times (min), cells were lysed and Cdc25B was immunoprecipitated and analyzed by autoradiography. (B) Half-life of Cdc25B. The radioactivity of immunoprecipitated, labeled endogenous hamster (–○–) and expressed human (–•–) Cdc25B was counted by an analyzer (BAS2000; Bio Image, Ann Arbor, MI). The vertical value indicates the relative count of radioactivity (Ct/C0). (C) Cultures of HeLa cells synchronized in S phase as described in Materials and Methods were incubated for 1 h in the presence (CHX) or absence (non) of cycloheximide (10 μg/ml) and then lysed. Total proteins of the extracts were electrophoresed in SDS-10% polyacrylamide gel and analyzed by immunoblotting using the antibodies against Cdc25A, Cdc25B, and Cdc25C.
All of p34cdc2 in the Extract Was Not Activated by Cdc25B
To address the question whether the observed in vitro activation of p34cdc2/cyclin B complex reflects a full activation of p34cdc2 within the extract, we added the E. coli-produced Cdc25C to the extract of BHK21 cells. By the addition of E. coli-produced Cdc25C, the histone H1 kinase activity was increased compared to the extract alone (Fig. 10). This finding indicated that all of p34cdc2 in the extract was not activated by Cdc25B. Probably, in our reaction condition, the activity of p34cdc2/cyclin B complexes obtained by the in vitro incubation was not enough to overcome the threshold for mitotic entrance, which is supposed to be carried out by forming a positive feedback mutual activation loop between Cdc25C and p34cdc2/cyclin B complexes (Kumagai and Dunphy, 1992; Hoffmann et al., 1993; Izumi and Maller, 1993). To address this issue, we added an increasing amount of p34cdc2/cyclin B complexes that had been activated by Cdc25B, to the extract of S phase-arrested BHK21 cells. However, the high activation of the p34cdc2 that was obtained by the addition of E. coli-produced Cdc25C, did not occur by the addition of extra p34cdc2/cyclin B complexes (data not shown). The similar result has been reported by Lee et al. (1994a) using Xenopus egg extract.
Figure 10.

Activation of p34cdc2 within the cytosolic extract of cycloheximide-treated and nontreated cells by E. coli-produced hamster Cdc25B. Cultures of BHK21 cells were synchronized with HU as described in Materials and Methods, and half of the cultures were treated with cycloheximide (10 μg/ml) for 1 h before cell disruption. 8 μl of cytosolic extracts of nontreated (–•–, –▪–) or cycloheximide-treated (–○–, –□–) cultures were incubated at 30°C for indicated time (min), in the presence (–▪–, –□–) or absence (–•–, –○–) of E. coli-produced hamster Cdc25C (final concentration 100 μg/ml). A final volume was adjusted to 30 μl. After incubation, p34cdc2/cyclin B was immunoprecipitated using the anti-cyclin B antibody and assayed for histone H1 kinase activities.
Over-Expression of Cdc25B Enhanced Chromatin Condensation
To address whether Cdc25B is rate limiting for entry into mitosis, increasing doses of Cdc25B cDNA were introduced into wild-type BHK21 cells using lipofection. Transfected cells were synchronized at G1 by isoleucine deprivation and then at S phase with HU. After incubation for 15.5 h in the presence of HU, cells were stained with Hoechst 33342, and cells showing PCC were scanned with photomicroscope. There were no cells showing PCC (<0.1%). Next we examined whether over-expressed Cdc25B can enhance chromatin condensation induced by addition of caffeine. HU-arrested cells were given 5 mM of caffeine along with 0.2 μg/ml of nocodazole and every hour, cells were scanned for cells showing PCC by photomicroscope. As shown in Fig. 11, the frequency of cells showing PCC was increased in a dose-dependent manner of transfected Cdc25B cDNA. When > 6 μg/35-mm dish of Cdc25B cDNA was introduced into cells, cells rapidly died as previously reported (Makishima et al., 1997). Thus, the question whether the amount of Cdc25B is rate limiting for entry into mitosis still remains to be answered. But our present finding indicates that Cdc25B enhanced chromatin condensation. Alternatively, it is possible that over-expressed Cdc25B could not stimulate cells to enter mitosis due to the tight cellular regulation of Cdc25B activity, since no cells showing PCC appeared until 2 h after caffeine addition, even in the case of a higher dose of Cdc25B cDNA.
Figure 11.

Over-expression of Cdc25B enhances chromatin condensation induced by caffeine. Wild-type BHK21 cells were seeded at 2 × 105/35-mm dish. After 23 h of incubation at 33.5°C, cells were transfected with the indicated doses of pcD2-Cdc25B DNA. After transfection, cells were synchronized at S phase by isoleucine deprivation and HU as described in Materials and Methods. After S phase arrest, cells were given caffeine (final concentration 5 mM) and nocodazole (final concentration 0.2 μg/ ml) in the presence of HU and then incubated at 37.5°C. Every 2 h cells were fixed with cold methanol and stained with Hoechst 33342. More than 1,000 cells were scored for PCC. The frequency of cells showing PCC is shown on the vertical axis. Amount of transfected DNA: –○–, 0 μg/dish ; –•–, 1 μg/dish; –□–, 2 μg/dish; –▪–, 4 μg/dish.
Discussion
Cdc25B Is a Short-Life Protein
The activation of p34cdc2/cyclin B occurs in the extract prepared from S phase-arrested hamster BHK21 cells without protein synthesis, but not in the extract of cells pretreated with protein synthesis inhibitor cycloheximide, which inhibits the in vivo activation of p34cdc2/cyclin B as well. Taking these results together, it can be deduced that S phase- arrested hamster BHK21 cells possess all the molecules required for the observed activation of p34cdc2/cyclin B and that either some molecules or protein modifications that are required for the activation of p34cdc2/cyclin B were lost by blocking protein synthesis. Since the p34cdc2/cyclin B complexes of cycloheximide-treated cells can be directly activated by immunoprecipitated Cdc25B, the modification of p34cdc2/cyclin B may not be lost. It is, therefore, rational to consider that some molecule(s) essential for the activation of p34cdc2/cyclin B is lost by blocking protein synthesis.
Cyclins are most important proteins for the mitotic activation of p34cdc2/cyclin B, and their cellular contents change depending on the cell cycle (Norbury and Nurse, 1992). In hamster BHK21 cells, cyclin B is present in S phase-arrested cells as reported (Nishitani et al., 1991). The amount of cyclin B, however, is not reduced by cycloheximide treatment. The other protein that is essential for the activation of p34cdc2/cyclin B is a family of dual-specificity phosphatase Cdc25 that perform dephosphorylation of Thr-14 and Tyr-15 (Dunphy, 1994). Among the family of Cdc25 phosphatases, we have found that Cdc25B, while Cdc25A and Cdc25C are stable, is lost by blocking protein synthesis. Indeed, the half-life of Cdc25B is <30 min in hamster BHK21, and also in HeLa cells. In this context, Cdc25B is unique among the Cdc25 family. The finding that human Cdc25B expressed in hamster wild-type BHK21 has a short life similar to hamster Cdc25B suggests that mammalian Cdc25B possesses a character of rapid turn over. Consistent with the loss of Cdc25B by cycloheximide treatment, the immunodepletion of Cdc25B from the extract abrogates the in vitro activation of p34cdc2/cyclin B.
Cdc25B Functions as a Trigger for Mitosis
Previously we have found that Cdc25C is essential for p34cdc2/cyclin B activation induced by the loss of RCC1 (Seki et al., 1992). However, Cdc25C is not required for the observed in vitro activation of p34cdc2/cyclin B. Cdc25C from mitotic cells activates p34cdc2/cyclin B within the extract of cycloheximide-treated cells, but Cdc25C of S phase-arrested cells does not. This finding is consistent with the previous finding that during G2/M transition, Cdc25C is phosphorylated and thereby activated, parallel with the activation of p34cdc2/cyclin B (Kumagai and Dunphy, 1992; Izumi and Maller, 1993; Hoffmann et al., 1993). In fact, the molecular mass of Cdc25C in S phase-arrested cells is not high like in mitotic cells. It has been reported that Cdc25C has a low activity of tyrosine phosphatase even in the interphase (Kumagai and Dunphy, 1992; Izumi and Maller, 1993; Hoffmann et al., 1994). We think that the discrepancy between our results and the previous ones may be due to the difference in materials. We stringently synchronized BHK21 cells into S phase by using the stepwise method; that is, first, cells were arrested in G0/G1 by isoleucine deprivation and then in S phase with HU.
In tsBN2 cells arrested in S phase, the activation of p34cdc2/cyclin B can be induced at 40°C, the nonpermissive temperature, and in parallel with the activation of p34cdc2/ cyclin B, p34cdc2 is dephosphorylated and Cdc25C is phosphorylated (Nishitani et al., 1991; Seki et al., 1992; current study), as found in Xenopus extract (Kumagai and Dunphy, 1992; Izumi and Maller, 1993; Hoffmann et al., 1993). Based on these observations, we assume that the premature activation of p34cdc2/cyclin B induced by tsBN2 mutation mimics a normal entrance of mitosis. In this context, we notice that loss of Cdc25B is coincident with loss of p34cdc2/cyclin B activation, both in vivo and in vitro. Interestingly, the content of Cdc25B was reduced in parallel with the activation of p34cdc2/cyclin B and the molecular shift of Cdc25C. These findings may suggest that Cdc25B initially activates the p34cdc2/cyclin B complexes, which, in turn, phosphorylate Cdc25C, forming a positive feedback, mutual activation loop between Cdc25C and p34cdc2/cyclin B. Consistent with the notion that Cdc25B functions as a trigger for the initiation of mitosis, the in vitro activation of p34cdc2/cyclin B carried out by Cdc25B is lower compared to the activation of p34cdc2/cyclin B that is carried out by constitutively active E. coli-produced Cdc25C.
To prove that Cdc25B is a “starter” for the mitotic activation of p34cdc2, it will be essential to show that the antibody to Cdc25B that neutralizes the enzyme activity of Cdc25B blocks the initiation of mitosis in mammalian cells.
Cellular Regulation of Cdc25B Function
While Cdc25C is activated by phosphorylation (Izumi et al., 1992; Kumagai and Dunphy, 1992, 1996), it is not yet clear how Cdc25B is activated. The finding that Cdc25B immunoprecipitated before the in vitro incubation activates the p34cdc2/cyclin B complexes that were isolated from cycloheximide-treated cells indicates that the Cdc25B of S phase-arrested BHK21 cells is active. If Cdc25B is already active in cells, there must be some regulatory system to prevent the p34cdc2/cyclin B from the premature activation. In this context, the in vitro spontaneous activation of p34cdc2/cyclin B in the cell extract suggests that the cell disruption that may mimic loss of the cellular compartment causes an activation of p34cdc2/cyclin B. In the interphase, therefore, we assume that the compartment mechanism that restrains an interaction between an enzyme and its substrate may play a role in the regulation of p34cdc2/cyclin B activation. In human HeLa cells, cyclin B1 is anchored in the cytoplasm during the interphase and enters the nucleus at the beginning of mitosis (Pines and Hunter, 1991, 1994). Recently, Nim1, which phosphorylates and thereby inhibits Wee1, is found to be localized in the cytoplasm, whereas Wee1 is localized in the nucleus (Parker and Piwnica-Worms, 1992; McGowan and Russell, 1993; Heald et al., 1993; Wu et al., 1996). In both mammalian and yeast, thus, the cellular compartment of proteins seems to play a role in regulating the initiation of mitosis. In this regard, we notice that loss of RCC1, which performs GDP/GTP exchange of Ran GTPase (Bischoff and Ponstingl, 1991), induces the activation of p34cdc2/cyclin B (Nishitani et al., 1991). The Ran GTPase is essential for nucleus/cytosol exchange of macromolecules (Melchior et al., 1993; Moore and Blobel, 1993) and for nuclear processing of mRNA (Sazer, 1996; Seki et al., 1996). Loss of RCC1 may accumulate the GDP-bound form of Ran GTPase, thereby, disturbing the Ran GTPase cycle. We are tempted to speculate, therefore, that the cellular compartment required for the cell cycle progression is regulated by the Ran GTPase.
Unstable Triggers and Stable Inhibitors for the Initiation of Mitosis
Although we could not exclude the possibility that the other unstable proteins like Cdc25B are also required for the initiation of mitosis, our findings indicate that the activator(s) is rapidly turning over, but the inhibitor(s) is stable. In S phase-arrested BHK21 cells, the activator(s) and the inhibitor(s) for p34cdc2/cyclin B seem to be balanced, and thereby, cells do not enter mitosis. The instability of the p34cdc2/cyclin B activator(s) may reflect an adaptation of cells to stresses, including the inhibition of protein synthesis. In such circumstances, it is better for cells to remain in a less active state of p34cdc2/cyclin B, or otherwise cells will suffer further damage from proceeding through the cell cycle. In this context, it seems to be reasonable that the ‘starter' for the mitotic entrance is labile.
By loss of the activator(s), the presence of p34cdc2/cyclin B inhibitor(s) became obvious. It is also possible that the inhibitor(s) was activated by cycloheximide treatment. In mammalian cells, the inhibitory kinases for p34cdc2/cyclin B like Wee1 and Myt1 (Parker and Piwnica-Worms, 1992; Mueller et al., 1995a ,b), the phosphatase INH/PP2A (Cyert and Kirschner, 1988; Lee et al., 1991, 1994a ) and the stoichiometric inhibitors (Kumagai and Dunphy, 1995; Lee and Kirschner, 1996) have been reported to prevent the activation of p34cdc2/cyclin B in the interphase. The finding that the addition of p34cdc2/cyclin B complexes pre-activated by Cdc25B could not induce a full activation of p34cdc2/cyclin B in the extract, suggests that our extracts contain the inhibitory kinase of p34cdc2/cyclin B or the phosphatase INH/PP2A.
In summary, our results indicate that Cdc25B functions as a trigger for mitosis. Our finding that Cdc25B is an unstable trigger for the entrance of mitosis will provide a new aspect regarding the check point control. In S. pombe, the level of Cdc25 is regulated by ubiquitin ligase Pub1 (Nefsky and Beach, 1996). Probably it is also true in mammalian cells. The stabilization of Cdc25B, therefore, may disturb the cell cycle control at the G2/M transition.
Acknowledgments
We thank J.Y.J. Wang for the anti-phosphotyrosine antibody, H. Okayama for human Cdc25B, K. Yamashita for pGEX-KG-hamster Cdc25C, and Miss K. Miller (Royal English Language Centre, Fukuoka, Japan) for proofreading the English used here.
This work was supported by Grants in Aid for Specially Promoted Research and for Cancer Research from the Ministry of Education, Science and Culture and from the Science and Technology Agency and by Human Frontier Science Program.
Abbreviations used in this paper
- CAK
CDK-activating kinase
- CDK
cyclin-dependent kinase
- HU
hydroxyurea
- PCC
premature chromatin condensation
- ts
temperature sensitive
Footnotes
Please address all correspondence to Dr. Takeharu Nishimoto, Department of Molecular Biology, Graduate School of Medical Science, Kyushu University, Higashi-ku, 3-1-1, Maidashi, Fukuoka 812-82, Japan. Tel.: (81) 92-642-6175; Fax: (81) 92-642-61831; E-mail: tnishi@mailserver.med.kyushu-u.ac.jp
Hideo Nishitani's present address is Imperial Cancer Research Fund, Lincoln's Inn Fields, London WC2A 3PX, UK.
Takashi Seki's present address is Imperial Cancer Research Fund, Clare Hall Laboratories, South Mimms, Potters Bar, Hers EN6 3LD, UK.
References
- Atherton-Fessler S, Liu F, Gabrielli B, Lee MS, Peng C-Y, Piwnica-Worms H. Cell cycle regulation of the p34cdc2inhibitory kinases. Mol Biol Cell. 1994;5:989–1001. doi: 10.1091/mbc.5.9.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff FR, Ponstingle H. Catalysis of guanine nucleotide exchange on Ran by the mitotic regulator RCC1. Nature (Lond) 1991;354:80–82. doi: 10.1038/354080a0. [DOI] [PubMed] [Google Scholar]
- Cyert MS, Kirschner MW. Regulation of MPF activity in vitro. Cell. 1988;53:185–195. doi: 10.1016/0092-8674(88)90380-7. [DOI] [PubMed] [Google Scholar]
- Dickmanns A, Bischoff FR, Marshallsay C, Luhrmann R, Ponstingle H, Fanning E. The thermolability of nuclear protein import in tsBN2 cells is suppressed by microinjected Ran-GTP or Ran-GDP, but not by RanQ69L or RanT24N. J Cell Science. 1996;109:1449–1457. doi: 10.1242/jcs.109.6.1449. [DOI] [PubMed] [Google Scholar]
- Dingwall C, Kandels-Lewis S, Seraphin B. A family of Ran binding proteins that includes nucleoporins. Proc Natl Acad Sci USA. 1995;92:7525–7529. doi: 10.1073/pnas.92.16.7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy WG. The decision to enter mitosis. Trends Cell Biol. 1994;4:202–207. doi: 10.1016/0962-8924(94)90142-2. [DOI] [PubMed] [Google Scholar]
- Evans T, Rosenthal ET, Youngblom J, Distel D, Hunt T. Cyclin: a protein specified by maternal mRNA in Sea Urchineggs that is destroyed at each cleavage division. Cell. 1983;33:389–396. doi: 10.1016/0092-8674(83)90420-8. [DOI] [PubMed] [Google Scholar]
- Fesquet D, Labbe JC, Derancourt J, Capony JP, Galas S, Girard F, Lorca T, Shuttleworth J, Doree M, Cavadore JC. The MO15 gene encodes the catalytic subunit of a protein kinase that activates cdc2 and other cyclin-dependent kinases (CDKs) through phosphorylation of Thr 161 and its homologues. EMBO (Eur Mol Biol Organ) J. 1993;12:3111–3121. doi: 10.1002/j.1460-2075.1993.tb05980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone C, Russell P. Fission yeast p107wee1 mitotic inhibitor is a tyrosine/serine kinase. Nature (Lond) 1991;349:808–811. doi: 10.1038/349808a0. [DOI] [PubMed] [Google Scholar]
- Frasch, M. 1991. The maternally expressed Drosophila gene encoding the chromatin-binding protein BJ1 is a homolog of the vertebrate gene Regulator of Chromatin Condensation, RCC1. EMBO (Eur. Mol. Biol. Organ.) J. 10: 1225–1236. [DOI] [PMC free article] [PubMed]
- Furuno N, Nishizawa M, Okazaki K, Tanaka H, Iwashita J, Nakajo N, Ogawa Y, Sagata N. Suppression of DNA replication via Mos function during meiotic divisions in Xenopusoocytes. EMBO (Eur Mol Biol Organ) J. 1994;13:2399–2410. doi: 10.1002/j.1460-2075.1994.tb06524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaktionov K, Beach D. Specific activation of cdc25 tyrosine phosphatases by B-type cyclins: evidence for multiple roles of mitotic cyclins. Cell. 1991;67:1181–1194. doi: 10.1016/0092-8674(91)90294-9. [DOI] [PubMed] [Google Scholar]
- Galaktionov K, Lee AK, Eckstein J, Draetta G, Meckler J, Loda M, Beach D. Cdc25 phosphatases as potential human oncogenes. Science (Wash DC) 1995;269:1575–1577. doi: 10.1126/science.7667636. [DOI] [PubMed] [Google Scholar]
- Heald R, McLoughlin M, McKeon F. Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated cdc2 kinase. Cell. 1993;74:463–474. doi: 10.1016/0092-8674(93)80048-j. [DOI] [PubMed] [Google Scholar]
- Hershko A. Ubiquitin; roles in protein modification and breakdown. Cell. 1983;34:11–12. doi: 10.1016/0092-8674(83)90131-9. [DOI] [PubMed] [Google Scholar]
- Hoffmann I, Clarke PR, Marcote MJ, Karsenti E, Draetta G. Phosphorylation and activation of human cdc25-C by cdc2/cyclin B and its involvement in the self-amplification of MPF at mitosis. EMBO (Eur Mol Biol Organ) J. 1993;12:53–63. doi: 10.1002/j.1460-2075.1993.tb05631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann I, Draetta G, Karsenti E. Activation of the phosphatase activity of human cdc25A by a cdk2-cyclin E dependent phosphorylation at the G1/S transition. EMBO (Eur Mol Biol Organ) J. 1994;13:4302–4310. doi: 10.1002/j.1460-2075.1994.tb06750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R, Ohba Y, Nagata A, Okayama H, Yasuda H. Dephosphorylation of human p34cdc2 kinase on both Thr-14 and Tyr-15 by human cdc25B phosphatase. FEBS (Fed Eur Biochem Soc) Lett. 1993;318:331–334. doi: 10.1016/0014-5793(93)80540-b. [DOI] [PubMed] [Google Scholar]
- Izumi T, Maller JL. Elimination of cdc2 phosphorylation sites in the cdc25 phosphatase blocks initiation of M-phase. Mol Biol Cell. 1993;4:1337–1350. doi: 10.1091/mbc.4.12.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi T, Walker DH, Maller JL. Periodic changes in phosphorylation of the Xenopuscdc25 phosphatase regulate its activity. Mol Biol Cell. 1992;3:927–939. doi: 10.1091/mbc.3.8.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinno S, Suto K, Nagata A, Igarashi M, Kanaoka Y, Nijima H, Okayama H. Cdc25A is a novel phosphatase functioning early in the cell cycle. EMBO (Eur Mol Biol Organ) J. 1994;13:1549–1556. doi: 10.1002/j.1460-2075.1994.tb06417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Dunphy WG. Regulation of the cdc25 protein during the cell cycle in Xenopusextracts. Cell. 1992;70:139–151. doi: 10.1016/0092-8674(92)90540-s. [DOI] [PubMed] [Google Scholar]
- Kumagai A, Dunphy WG. Control of the Cdc2/cyclin B complex in Xenopus egg extracts arrested at a G2/M checkpoint with DNA synthesis inhibitors. Mol Biol Cell. 1995;6:199–213. doi: 10.1091/mbc.6.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Dunphy WG. Purification and molecular cloning of Plx1, a Cdc25-regulatory kinase from Xenopusegg extracts. Science (Wash DC) 1996;273:1377–1380. doi: 10.1126/science.273.5280.1377. [DOI] [PubMed] [Google Scholar]
- Lee TH, Kirschner MW. An inhibitor of p34cdc2/cyclin B that regulates the G2/M transition in Xenopusextracts. Proc Natl Acad Sci USA. 1996;93:352–356. doi: 10.1073/pnas.93.1.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TH, Solomon MJ, Mumby MC, Kirschner MW. INH, a negative regulator of MPF, is a form of protein phosphatase 2A. Cell. 1991;64:415–423. doi: 10.1016/0092-8674(91)90649-j. [DOI] [PubMed] [Google Scholar]
- Lee TH, Turck C, Kirschner MW. Inhibition of cdc2 activation by INH/PP2A. Mol Biol Cell. 1994a;5:323–338. doi: 10.1091/mbc.5.3.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Enoch T, Piwnica-Worms H. mik1 encodes a tyrosine kinase that phosphorylates p34cdc2on tyrosine-15. J Biol Chem. 1994b;269:30530–30537. [PubMed] [Google Scholar]
- McGowan CH, Russell P. Human Wee1 kinase inhibits cell division by phosphorylating p34cd c2exclusively on Tyr 15. EMBO (Eur Mol Biol Organ) J. 1993;12:75–85. doi: 10.1002/j.1460-2075.1993.tb05633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima T, Nakashima T, Nagata-Kuno K, Fukushima K, Iida H, Sakaguchi M, Ikehara Y, Komiyama S, Nishimoto T. The highly conserved DAD1 protein involved in apoptosis is required for N-linked glycosylation. Genes Cells. 1997;2:129–141. doi: 10.1046/j.1365-2443.1997.1070303.x. [DOI] [PubMed] [Google Scholar]
- Melchior F, Paschal B, Evans E, Gerace L. Inhibition of nuclear protein import by nonhydrolyzable analogues of GTP and identification of the small GTPase Ran/TC4 as an essential transport factor. J Cell Biol. 1993;123:1649–1659. doi: 10.1083/jcb.123.6.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JBA, Blevitt J, Gerace L, Sadhu K, Featherstone C, Russell P. p55CD C25is a nuclear protein required for the initiation of mitosis in human cells. Proc Natl Acad Sci USA. 1991;88:10500–10504. doi: 10.1073/pnas.88.23.10500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshull J, Murray A, Colman A, Hunt T. Xenopusoocyte maturation does not require new cyclin synthesis. J Cell Biol. 1991;114:767–772. doi: 10.1083/jcb.114.4.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morla A, Wang JYJ. Protein tyrosine phosphorylation in the cell cycle of GALB/c 3T3 fibroblasts. Proc Natl Acad Sci USA. 1986;83:8191–8195. doi: 10.1073/pnas.83.21.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MS, Blobel G. The GTP-binding protein Ran/TC4 is required for protein import into the nucleus. Nature (Lond) 1993;365:661–663. doi: 10.1038/365661a0. [DOI] [PubMed] [Google Scholar]
- Mueller PR, Coleman TR, Dunphy WG. Cell cycle regulation of a Xenopuswee1-like kinase. Mol Biol Cell. 1995a;6:119–134. doi: 10.1091/mbc.6.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller PR, Coleman TR, Kumagai A, Dunphy WG. Myt1: a membrane-associated inhibitory kinase that phosphorylates cdc2 on both threonine-14 and tyrosine-15. Science (Wash DC) 1995b;270:86–90. doi: 10.1126/science.270.5233.86. [DOI] [PubMed] [Google Scholar]
- Nagata A, Igarashi M, Jinno S, Suto K, Okayama H. An additional homolog of the fission yeast cdc25+gene occurs in humans and is highly expressed in some cancer cells. New Biol. 1991;3:959–968. [PubMed] [Google Scholar]
- Nefsky B, Beach D. Pub1 acts as an E6-AP-like protein ubiquitin ligase in the degradation of cdc25. EMBO (Eur Mol Biol Organ) J. 1996;15:1301–1312. [PMC free article] [PubMed] [Google Scholar]
- Nishimoto T, Eilen E, Basilico C. Premature chromosome condensation in a ts DNA−Mutant of BHK cells. Cell. 1978;15:475–483. doi: 10.1016/0092-8674(78)90017-x. [DOI] [PubMed] [Google Scholar]
- Nishimoto T, Ishida R, Ajiro K, Yamamoto S, Takahashi T. The synthesis of protein(s) for chromosome condensation may be regulated by a post-transcriptional mechanism. J Cell Physiol. 1981;109:299–308. doi: 10.1002/jcp.1041090213. [DOI] [PubMed] [Google Scholar]
- Nishitani H, Ohtsubo M, Yamashita K, Iida H, Pines J, Yasuda H, Shibata Y, Hunter T, Nishimoto T. Loss of RCC1, a nuclear DNA-binding protein, uncouples the completion of DNA replication from the activation of cdc2 protein kinase and mitosis. EMBO (Eur Mol Biol Organ) J. 1991;10:1555–1564. doi: 10.1002/j.1460-2075.1991.tb07675.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi E, Hayashi N, Nakashima N, Nishimoto T. Yrb2p, Nup2p-related yeast protein has functional overlap with Rna1p, yeast RanGAP protein. Mol Cell Biol. 1997;17:2235–2246. doi: 10.1128/mcb.17.4.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbury C, Nurse P. Animal cell cycles and their control. Annu Rev Biochem. 1992;61:441–470. doi: 10.1146/annurev.bi.61.070192.002301. [DOI] [PubMed] [Google Scholar]
- Ohba T, Seki T, Azuma Y, Nishimoto T. Premature chromatin condensation induced by loss of RCC1 is inhibited by GTP- and GTPgS-Ran, but not GDP-Ran. J Biol Chem. 1996;27:14665–14667. doi: 10.1074/jbc.271.25.14665. [DOI] [PubMed] [Google Scholar]
- Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science (Wash DC) 1992;257:1955–1957. doi: 10.1126/science.1384126. [DOI] [PubMed] [Google Scholar]
- Pines J, Hunter T. Human cyclins A and B1 are differentially located in the cell and undergo cell cycle-dependent nuclear transport. J Cell Biol. 1991;115:1–17. doi: 10.1083/jcb.115.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines J, Hunter T. The differential localization of human cyclins A and B is due to a cytoplasmic retention signal in cyclin B. EMBO (Eur Mol Biol Organ) J. 1994;13:3772–3781. doi: 10.1002/j.1460-2075.1994.tb06688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon RYC, Yamashita K, Adamczewski JP, Hunt T, Shuttleworth J. The cdc2-related protein p40MO15 is the catalytic subunit of a protein kinase that can activate p33cdk2 and p34cdc2 . EMBO (Eur Mol Biol Organ) J. 1993;12:3123–3132. doi: 10.1002/j.1460-2075.1993.tb05981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadhu K, Reed SI, Richardson H, Russell P. Human homolog of fission yeast cdc25 mitotic inducer is predominantly expressed in G2 . Proc Natl Acad Sci USA. 1990;87:5139–5143. doi: 10.1073/pnas.87.13.5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagata N, Oskarsson M, Copeland T, Brumbaugh J, Vande GF, Woud Function of c-mos proto-oncogene product in meiotic maturation in Xenopusoocytes. Nature (Lond) 1988;335:519–525. doi: 10.1038/335519a0. [DOI] [PubMed] [Google Scholar]
- Sazer S. The search for the primary function of the Ran GTPase continues. Trends Cell Biol. 1996;6:81–85. doi: 10.1016/0962-8924(96)80992-5. [DOI] [PubMed] [Google Scholar]
- Seki T, Yamashita K, Nishitani H, Takagi T, Russell P, Nishimoto T. Chromosome condensation caused by loss of RCC1 function requires the cdc25C protein that is located in the cytoplasm. Mol Biol Cell. 1992;3:1373–1388. doi: 10.1091/mbc.3.12.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki T, Hayashi N, Nishimoto T. RCC1 in the Ran pathway. J Biochem. 1996;120:207–214. doi: 10.1093/oxfordjournals.jbchem.a021400. [DOI] [PubMed] [Google Scholar]
- Schlegel R, Pardee AB. Caffeine-induced uncoupling of mitosis from the completion of DNA replication in mammalian cells. Science (Wash DC) 1986;232:1264–1266. doi: 10.1126/science.2422760. [DOI] [PubMed] [Google Scholar]
- Shuttleworth J, Godfrey R, Colman A. p40MO15, a cdc2-related protein kinase involved in negative regulation of meiotic maturation of Xenopusoocytes. EMBO (Eur Mol Biol Organ) J. 1990;9:3233–3240. doi: 10.1002/j.1460-2075.1990.tb07522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon MJ, Glotzer M, Lee TH, Philippe M, Kirschner MW. Cyclin activation of p34 cdc2. Cell. 1990;63:1013–1024. doi: 10.1016/0092-8674(90)90504-8. [DOI] [PubMed] [Google Scholar]
- Solomon MJ, Lee T, Kirschner MW. Role of phosphorylation in p34cdc2activation: identification of an activating kinase. Mol Biol Cell. 1992;3:13–27. doi: 10.1091/mbc.3.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon MJ, Harper JW, Shuttleworth J. CAK, the p34cdc2 activating kinase, contains a protein identical or closely related to p40MO15 . EMBO (Eur Mol Biol Organ) J. 1993;12:3133–3142. doi: 10.1002/j.1460-2075.1993.tb05982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana T, Imamoto N, Seino H, Nishimoto T, Yoneda Y. Loss of RCC1 leads to suppression of nuclear protein import in living cells. J Biol Chem. 1994;269:24542–24545. [PubMed] [Google Scholar]
- Wasserman WJ, Masui Y. Effects of cycloheximide on a cytoplasmic factor initiating meiotic maturation in Xenopus laevisoocytes. Exp Cell Res. 1975;91:381–388. doi: 10.1016/0014-4827(75)90118-4. [DOI] [PubMed] [Google Scholar]
- Wu L, Shiozaki K, Aligue R, Russell P. Spatial organization of the Nim1-Wee1-Cdc2 mitotic control network in Schizosaccharomyces pombe. . Mol Biol Cell. 1996;7:1749–1758. doi: 10.1091/mbc.7.11.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Russell P. Nim1 kinase promotes mitosis by inactivating Wee1 tyrosine kinase. Nature (Lond) 1993;363:738–741. doi: 10.1038/363738a0. [DOI] [PubMed] [Google Scholar]
- Yew N, Mellini ML, Vande GF, Woude Meiotic initiation by the mos protein in Xenopus. . Nature (Lond) 1992;355:649–652. doi: 10.1038/355649a0. [DOI] [PubMed] [Google Scholar]