

Abstract
Lumican, a prototypic leucine-rich proteoglycan with keratan sulfate side chains, is a major component of the cornea, dermal, and muscle connective tissues. Mice homozygous for a null mutation in lumican display skin laxity and fragility resembling certain types of Ehlers-Danlos syndrome. In addition, the mutant mice develop bilateral corneal opacification. The underlying connective tissue defect in the homozygous mutants is deregulated growth of collagen fibrils with a significant proportion of abnormally thick collagen fibrils in the skin and cornea as indicated by transmission electron microscopy. A highly organized and regularly spaced collagen fibril matrix typical of the normal cornea is also missing in these mutant mice. This study establishes a crucial role for lumican in the regulation of collagen assembly into fibrils in various connective tissues. Most importantly, these results provide a definitive link between a necessity for lumican in the development of a highly organized collagenous matrix and corneal transparency.
Collagen architecture plays an important role in optimal functioning of various extracellular matrix–rich tissues such as skin, tendon, muscle connective tissue, and cornea of the eye. Lumican is a keratan sulfate (KS)1-carrying member of the leucine-rich proteoglycan family (LRP) that colocalizes with fibrillar collagens in these connective tissues (2, 7). Other LRP members are decorin, biglycan, fibromodulin, epiphycan, osteoglycin, and keratocan (10, 17). In in vitro collagen fibril–forming assays, some of these proteoglycans inhibit the lateral growth of spontaneously forming fibrils, and are thus implicated in regulating certain aspects of collagen fibril structure as extracellular matrix is deposited and assembled in vivo (31, 34, 37, 45). Thus in the eye, corneal transparency is proposed to arise from minimal scattering of incident light by a lattice arrangement of uniformly thick collagen fibrils maintained by LRP members (24, 31, 44).
The lumican core protein, without the keratan sulfate side chains, is equally efficient in inhibiting in vitro collagen fibrillogenesis, suggesting this function to be entirely core protein mediated (31). Recombinant decorin core protein fragments have also been shown to bind collagen and affect collagen fibril formation in vitro (34). The leucine-rich repeat motifs have been historically implied in interactions with collagens. A recent study has identified the sixth leucine motif of decorin as the major collagen I–binding site in vitro (21). For lumican, specific sites of interactions with collagens have yet to be identified. The three- dimensional structures of the LRP members have been modeled based on the crystal structure of porcine ribonuclease inhibitor (20), which also contains these leucine motifs and shares 18% amino acid identity with the LRPs. The LRP core proteins are envisioned as having an arch or horse shoe shape with the glycosaminoglycan (GAG) chains protruding from the convex surface, whereas the concave surface is free to bind parallel collagen microfibrils and regulate their lateral association (38, 46). However, each LRP may bind to distinct sites along a collagen fibril as suggested by immunoelectron microscopic localization and biochemical studies (15, 37). Thus in vivo they are likely to control different aspects of collagen morphology.
The GAG side chains of the proteoglycans retain water and maintain normal tissue hydration. In the cornea the KS–GAGs play a significant role. KS levels show a strong correlation with acquisition of corneal transparency during development and healing of stromal wounds (8, 9).
However, functions of each of these proteoglycans in vivo remain to be elucidated. Subtle differences in their interactions with collagens, GAG content and expression patterns in tissues would imply that, in addition to functions that overlap, they may have evolved to serve certain unique functions in different tissues.
To understand functions of lumican in vivo, we delineated the effect of lumican deficiency on connective tissues of mice homozygous for a lumican null mutation. The mutant mice develop bilateral corneal opacification, display Ehlers-Danlos syndrome (EDS)-like skin laxity, and some of the same collagen fibril anomalies that have been described in EDS patients (40). Our results emphasize the importance of these proteoglycan genes as “primary defect” candidates for certain types of EDS and corneal dystrophies.
Materials and Methods
In Situ Hybridization
Embryos were fixed in 4% paraformaldehyde for 12–24 h, dehydrated, embedded in paraffin, and then sectioned at 6 μm. For in situ hybridization, a 324-bp mouse coding sequence PCR amplified from exon 2 of a mouse lumican genomic clone was cloned into pBluescript and used to synthesize an antisense riboprobe (33). Washes were carried out at 65°C.
Null Homozygous Mice
A replacement vector was constructed from an 11-kb mouse lumican genomic clone in λ Fix II (premade mouse 129SVJ genomic DNA library; Stratagene, La Jolla, CA) using the following steps. A Not–BamHI lumican genomic insert, 5.5 kb in length, was subcloned into pBluescript from the original λ Fix II genomic clone. An EcoRI–EcoRI fragment of 1.6 kb, containing exon 2 was deleted from this clone. A EcoRI–BamHI 1.33-kb PGK-neo cassette in which the BamHI site was repaired by blunt-ending, ligated to EcoRI linkers, and then digested with EcoRI, to obtain a 1.33-kb insert with EcoRI ends. This 1.33-kb PGK-neo cassette, missing the 500-bp poly(A) signal sequence at its 3′ end, was inserted into the EcoRI site of the 5.5-kb Not–BamHI lumican genomic clone. Clones containing the PGK-neo cassette in the 5′ to 3′ orientation downstream of the 2.6-kb Not–EcoRI lumican genomic DNA segment were then selected (see Fig. 2 a). This Not–BamHI lumican clone derivative has a lumican genomic DNA segment of 2.6 kb to serve as the 5′ arm of homology in the final targeting construct, a PGK neoA insert in the correct orientation and a 3′ arm of homology of 1.4 kb. The latter 1.4-kb BamHI–XhoI genomic DNA segment was replaced with a 5-kb BamHI–XhoI fragment from the λ Fix II genomic clone to obtain a longer 3′ arm of homology. Correct targeting in the embryonic stem (ES) cell line CT 120#25 was detected by Southern (DNA) hybridization of ES cell DNA digested with HindIII, BamHI (see Fig. 2), SacI and NcoI (data not shown) and probed with an internal 2-kb probe. The targeting frequency was 1/200. Blastocyst (CD-1 background) injection and development of chimeric animals were performed according to published methods (16).
Figure 2.

Gene targeting at the lum locus. Exons are marked as E1, E2, and E3. (a) Diagram of the wild-type lum + allele, targeting vector, and targeted lumtm1Sc allele. Correct targeting should replace the 3-kb EcoRI–BamHI segment containing exon 2 (E2) with the 1.33-kb pGKneo cassette for positive selection. (b) Diagnostic Southern hybridization on ES clone DNA. Presence of a 4.5-kb BamHI and an 11-kb HindIII fragment indicates one correctly targeted allele. (c) PCR analysis of offsprings from heterozygous matings. (d) Western blot analysis on total protein extracted from whole eyes of lumtm1Sc/ lumtm1Sc (−/−) and lum +/lumtm1Sc (+/−) mice. Undigested extract from heterozygous mice show a broad band at 65–90 kD, indicating the presence of lumican. Lack of a sharp band is due to heterogenous KS modification of lumican. After keratanase treatment and removal of most of the KS a sharper and faster migrating band results, a characteristic feature of KS proteoglycans. The lumtm1Sc/lumtm1Sc tissue lacks any immunoreactive material confirming the absence of any lumican gene product.
A mixture of two primer pairs, one pair internally from E2 (forward primer, 5′-CCTGAGGAATAACCAAATCGACC-3′ and reverse primer, 5′-AGGCAGCTTGCTCATCTGAATTG-3′), the second pair from the pGKneo cassette (forward primer 5′-CATTCGACCACCAAGCGAAAC-3′ and reverse primer 5′-AGCTCTTCAGCAATATCACGGG-3′) were used to amplify a 380-bp and a 300-bp product from the wild type and mutant allele, respectively. All mice were housed in the animal care facility of Case Western Reserve University according to IACUC approved protocols.
Immunoblotting
Total proteoglycan was extracted from 8 to 10 whole adult eyes in 4 M guanidine HCl, 100 mM sodium acetate buffer, pH 6.0, containing the following protease inhibitors: 1 mM PMSF, 5 mM benzamidine HCl, 5 mM iodoacetamide, 5 μg/ml leupeptin, and 5 μg/ml pepstatin (12). An aliquot of the extracted and dialyzed sample was digested with keratanase (Sigma Chemical Co., St. Louis, MO) for KSPG detection according to published methods (36). Digested and undigested samples were resolved by SDS-PAGE and transferred to nitrocellulose membrane. Lumican was identified using a rabbit antiserum generated against a synthetic peptide from the NH2-terminal end spanning residues 20–37 of mouse lumican (Research Genetics Inc., Huntsville, AL). Presence of decorin in the lumican null mice was confirmed using LF113, a rabbit anti–mouse decorin antiserum generated against a synthetic peptide spanning amino acid residues 36–49 of mouse decorin (13). Both antisera were used at a dilution of 1:2,000 and a goat anti–rabbit secondary antibody (1:10,000) conjugated to HRP for substrate detection with the SuperSignal Substrate Western blotting kit (Pierce Chemical Co., Rockford, IL).
Skin Tension Test
Two tensile samples were prepared from the dorsal skin of 9-mo male control (n = 5) and mutant mice (n = 5). The skin was harvested and tested within 1 h of sacrifice. Thickness measurements were taken on a small shaved area of the skin using a micrometer. Test samples were produced using a template with a gauge region of 10-mm wide by 20-mm long. The long axis of the sample coincided with the superior–inferior direction of the mouse. The samples were placed in a custom alignment jig and the ends were clamped between two aluminium plates with a piece of sandpaper glued to the surfaces. The tension tests were performed on an Instron (Canton, MA) servohydraulic materials test machine and loaded to failure in tension at a constant rate of 1 mm/s. Stress was determined by normalizing the change in actuator displacement for initial sample length. Data was collected at a sampling frequency of 50 Hz using LabView software (National Instruments, Austin, TX). Differences in skin tensile properties between the control and mutant mice were analyzed by Student's t test.
Slit-Lamp Examination of Eyes
Eyes of anesthetized mice were examined and photographed using a vertical slit-lamp biomicroscope (Marco, Jacksonville, FL). For the external photographs, additional external light was oriented towards the mice. For slit views, the lens was adjusted to 50° angle. To photograph whole eyes and slit-lamp views, Ektachrome 160T and 1600 (Eastman Kodak, Rochester, NY) films were used, respectively.
Transmission Electron Microscopy
Small portions of tail and whole cornea from eight homozygous mutants, two heterozygous, and four homozygous wild-type mice were fixed in 2.5% glutaraldehyde, 25 mM sodium acetate buffer, pH 5.7, containing 0.3 M MgCl2, and 0.05% cuprolinic blue (39). Thin sections, with or without further uranyl acetate staining were examined in an electron microscope (100CX; JEOL USA Inc., Peabody, MA). Diameter of 100 to 250 fibrils were measured manually.
Results
Expression by In Situ Hybridization
During mouse embryonic development, lumican transcript is detectable by in situ hybridization as early as E9.5 in all mesenchymal tissues (Fig. 1 a). By E13.5 stage lumican mRNA is abundant throughout the corneal stroma, extrinsic ocular muscles, and to a lesser extent in the sclera of the eye (Fig. 1 b), as well as the subepithelial dermis (Fig. 1 c). It is also expressed in the perichondrium of developing rib primordia, developing circular and longitudinal muscles of the gut (data not shown). In the developing heart, a strong signal for lumican message is apparent in the leaflets of the pulmonary and aortic valves (Fig. 1 d). Expression of lumican in early stages of development implies a role in the migration/proliferation of mesenchymal cells and in the formation of an embryonic collagenous connective tissue matrix.
Figure 1.
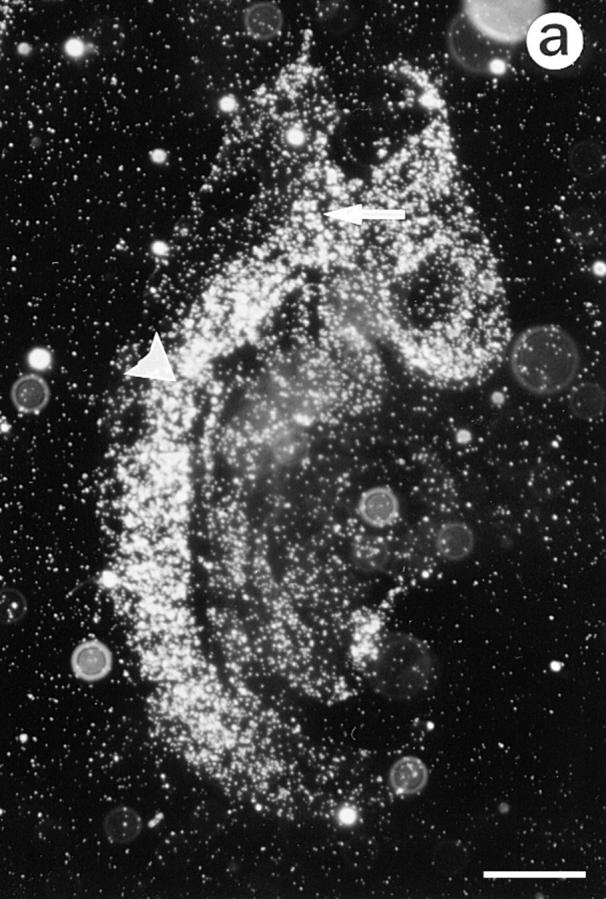
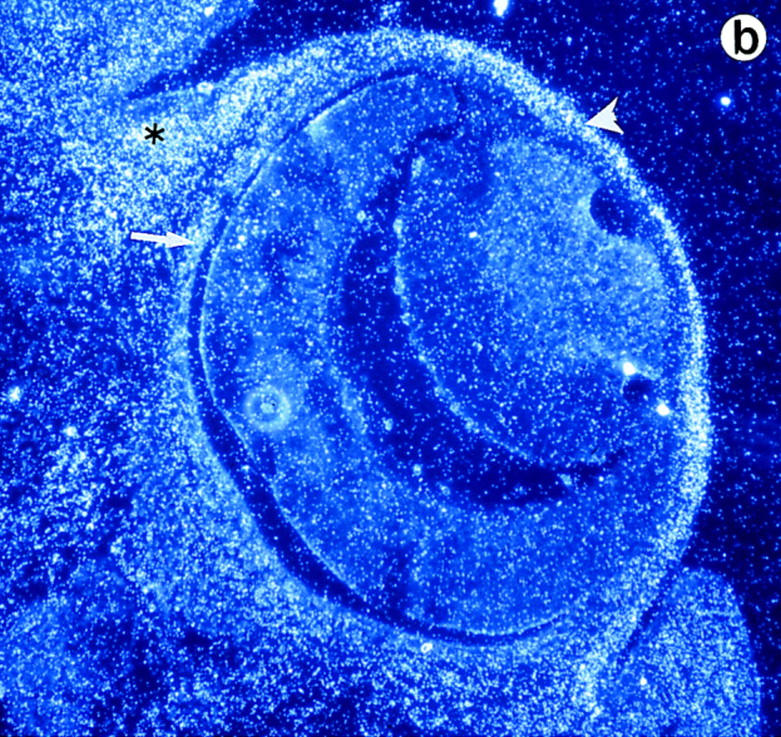
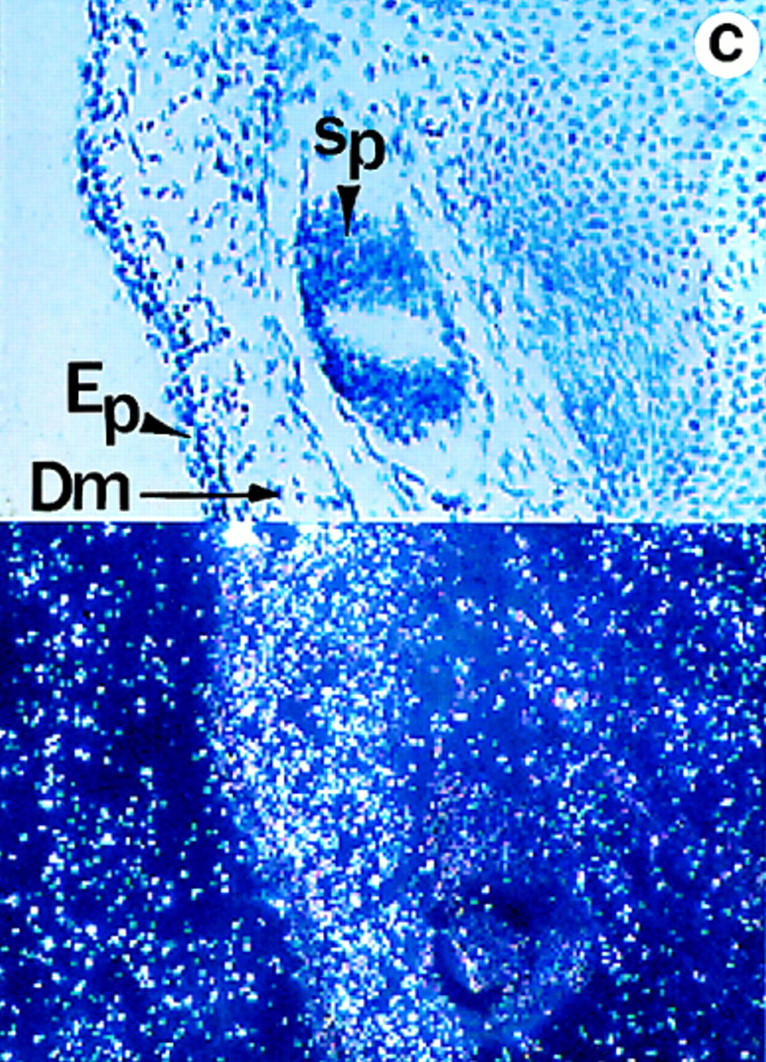
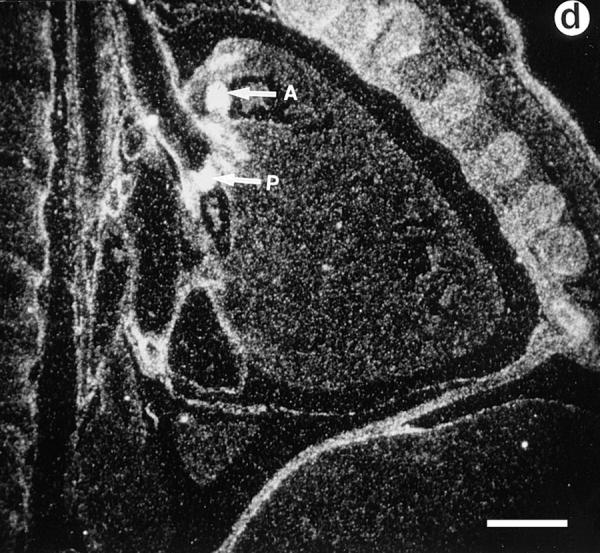
Lumican expression during mouse embryonic development. In situ hybridization analysis. (a) Sagittal section of E.9.5. Lumican is expressed at high levels in head (arrow) and lateral mesenchyme (arrowhead). (b) Transverse section of E13.5 embryo. At E13.5 lumican expression is focused in the subepithelial dermis of eyelid area (asterisk), cornea (arrowhead), with lesser amounts in the sclera (arrow). The lens and retina do not express lumican. (c) Section through E15.5 stage dorsal skin. Top panel is a bright field view of a toluidine blue–stained section, and bottom panel is a dark field view of same section showing silver grain deposits. Note that darkly stained epidermal layer (Ep) does not contain lumican mRNA. Dermal layer (Dm) displays a strong signal for lumican transcript. No lumican mRNA in central canal in sacro-coccygeal region of spinal cord (Sp). (d) Parasagittal section through E15.5 embryo showing heart pulmonary (P) and aortic valve (A) leaflets with strong signal for lumican transcript. Bars, 100 μm.
Gene Targeting at the Lumican Locus
A replacement gene targeting vector (Fig. 2 a) was designed such that a 3-kb EcoRI–BamHI segment containing all of exon 2, the largest of the three exons was replaced with a 1.33-kb PGKneo expression cassette for positive selection of ES cells. The 500-bp poly(A) signal sequence was deleted from the original 1.8-kb PGKneo cassette so that correct transcription of the cassette would be contingent upon the presence of a chromosomal poly(A) signal downstream of the construct. Correctly targeted clones were identified by diagnostic Southern hybridization using an internal probe and several restriction enzymes with sites outside the vector DNA (Fig. 2 b). Germline transmission of the mutated lumican allele, targeted mutation 1 Sc (lumtm1Sc), was achieved by mating founder chimeras generated by injecting heterozygous ES cells into eight-cell CD1 host embryos. Heterozygous lum +/lumtm1Sc CD-1 mice were intercrossed to develop homozygous lumtm1Sc/lumtm1Sc mice. The mice were genotyped by PCR analysis (Fig. 2 c).
Homozygous Lumican Null Mutants Are Viable
The homozygous mutant mice were born alive, in the expected Mendelian ratio (n = 122; lum +/lum + = 26%, lum +/lumtm1/Sc= 50%, lumtm1Sc/lumtm1Sc= 24%), and were fertile. Approximately 10–15% of the lumtm1Sc/lumtm1Sc mice were significantly smaller at birth, weighing 70–80% of the body weight of heterozygous or wild-type littermates. Western blot analysis of total protein from extracts of whole eyes indicated no expression of lumican in lumtm1Sc homozygous mice indicating that lumtm1Sc is a null allele (Fig. 2 d).
The lumican gene localizes to the same general region as decorin, a related LRP member, on distal mouse chromosome 10 (6, 17). Therefore, a targeted deletion at the lumican locus could conceivably disrupt decorin gene expression. To exclude this possibility we determined the presence of the decorin core protein in the lumican null mutants. Using LF113, an anti-decorin (murine) antibody (13), in Western blots on total proteoglycans extracted from whole eyes, we confirmed that lumican-deficient mice have normal levels of decorin (data not shown).
Routine histological examinations of null mice failed to detect major cardiac valvular anomalies (data not shown) although lumican is expressed fairly early in these tissues (Fig. 1 d). Lumican is also expressed in the developing gastrointestinal tract, however, intestinal sections of adult mice revealed normal tissue organization (data not shown). Additionally, development of the corneal stroma (between E13.5 and E16.5 stages, data not shown) appeared largely unaffected although lumican is expressed in the early stages of corneal development (Fig. 1 b).
Reduced Tensile Strength of Skin from lumtm1Sc/lumtm1Sc Mice
The null mutants had extremely loose and fragile skin apparent during routine handling of the mice. Decorin-deficient mice developed recently also exhibited similar skin anomalies and reduced load to failure in tension tests (12). We measured the tensile strength of freshly isolated skin samples from 10 9-mo-old animals, 5 wild type, and 5 lumican-deficient mice. Mutant mice without any visible lesions were chosen for skin testing. Test samples were produced in duplicate using a template with a gauge region 10-mm wide by 20-mm long. Stress, in Newtons/mm2 (N/ mm2) was determined by normalizing the load for initial cross sectional area, and strain was determined by normalizing the change in actuator displacement for initial sample length. The stress versus strain curves revealed significant differences in skin properties between mutant and wild type (Fig. 3 a). First, the modulus or the slope of the linear portion of the stress-strain curve of mutant skin (4.2 ± 1.6 N/mm2) was 83% (P < 0.0004) lower than modulus of wild-type mouse skin (25.4 ± 7.9 N/mm2). This indicates that the lumican-null mutation causes a dramatic increase in skin compliance. Second, the strength of the lumican-deficient skin was compromised greatly (Fig. 3 b): skin from mutant mice failed at 1.15 ± 0.39 N/mm2, whereas the wild-type skin samples failed at 8.28 ± 1.04 N/mm2. This 86% (P < 0.0001) reduction in tensile strength of the lumican-deficient skin is consistent with the increased incidence of skin lesions in the mutants.
Figure 3.


Skin tensile stress-strain measurements. (a) Representative tensile stress-strain curves for skin samples from wild type (+/+) and lumtm1Sc homozygous null mutants (−/−), n = 5 for each genotype. Tension tests were performed in duplicate on freshly isolated skin samples with a 10 × 20-mm gauge region from the dorsal surface of each animal. Samples were loaded to failure in tension at a constant rate of 1 mm/s using an Instron servohydraulic materials test machine. Stress in N/mm2 was plotted versus the strain. Note the significant reduction in both the modulus and the stress to failure in the −/− animals. (b) Tensile strength in wild type (+/+) and lumtm1Sc homozygous null mutants (−/−). The average tensile strength or maximum stress in the (−/−) skin was 86% lower than the wild-type (+/+) skin.
Trichrome-stained sections of skin samples taken from dorsal and ventral areas of adult wild-type and null mutant mice (non-lesioned areas sampled in the mutants) revealed disorganized and a more loosely arranged dermal connective tissue (stained blue) in the mutants (Fig. 4, b and d, arrowhead). In wild-type animals dermal fibroblasts are aligned more in parallel to the epithelial layer (Fig. 4, a and c). In the mutants, the fibroblasts appear poorly aligned (Fig. 4, b and d, arrows). Heterozygous mice do not harbor any of these phenotypic abnormalities.
Figure 4.



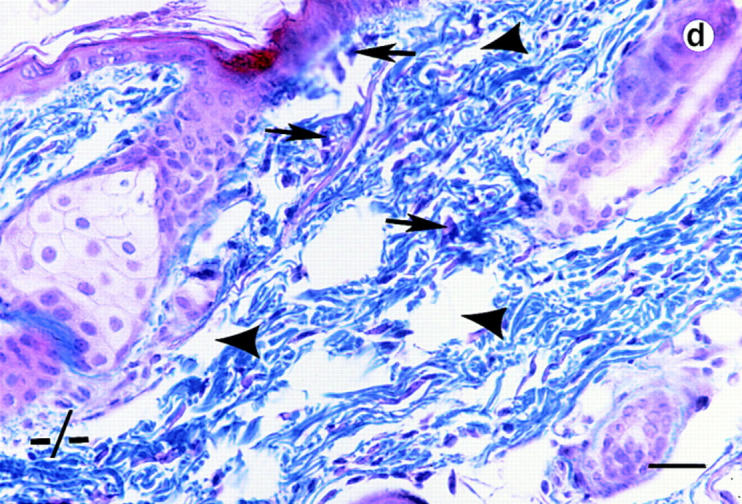
Histological analyses on skin alterations. Sections through skin from dorsal (a and b) and ventral (c and d) surfaces. Wild-type animals (+/+) shown in a and c and lumican null mutants (−/−) shown in b and d. The sections were trichrome stained in which the connective tissue, collagens primarily stain blue. Note disoriented fibroblasts (arrows) and open spaces (arrowheads) in the dermis of b and d. Bar, 50 μm.
Corneal Opacification in the Absence of Lumican
Lumtm1Sc homozygous mice were examined for loss of corneal clarity by slit lamp biomicroscopy to assess lumican's role in regulating corneal transparency. Out of 51 lumtm1Sc/ lumtm1Sc mice, between the ages of 5 to 34 wk, 44 displayed bilateral corneal clouding, with a peripheral ring-like clear zone, a characteristic feature of many human corneal stromal dystrophies (5). We scored corneal clouding as 1+ (clouding barely detectable) to 4+ (total opacification until no iris details visible). The maximum opacification associated with the lumtm1Sc null phenotype was 3.5+ as shown in a full view of the eye (Fig. 5 b). The cornea of wild-type and heterozygous mice (not shown) appeared clear with normal stromal granularity (Fig. 5, a and c; arrow in c), whereas the stroma of lumtm1Sc/lumtm1Sc mice appeared uniformly hazy (Fig. 5 b) and opalescent in slit view resulting from increased scattering of light (Fig. 5 d, arrowhead).
Figure 5.




Eye examination for corneal clarity. Full view photobiomicroscopy of wild type (a) and lumtm1Sc homozygous mutants (b) eyes. Wild-type mice show clear corneas through which iris details can be seen (a). A continuous cloudiness with a clear ringlike peripheral zone is noticeable in the null mutant (b). Slit-lamp photobiomicrography of wild type (c) and lumtm1Sc homozygous mutant (d). Note clear beam with normal granularity in c and enhanced brightness because of increased light scattering and extensive granular material in d.
The corneal clouding phenotype appeared to be clearly age dependent; the 7 out of the 51 that had clear corneas during our initial examination were between the ages of 5 and 6 wk, by 8–12 wk most of the mice had cloudy corneas. Follow-up eye examinations over a period of 4 mo were performed on 14 additional mutants and age-matched wild type. Over time the corneal opacification showed an increase; in 11 out of 14, opacity increased from a level of 0 or 1 to 3.5 (Fig. 6). However, all of the older mutants examined did not display maximal clouding of corneas. The variable expressivity of the corneal clouding phenotype may be due to the effect of other modifier genes. Whether this variable expressivity extends to other collagenous tissues is currently being investigated. Limited analysis of lumtm1Sc homozygosity on an inbred 129/Sv+Tyr+p genetic background also showed corneal clouding in adult mice. Further analyses of the lumtm1Sc mutation in 129/Sv+Tyr+p mice are underway to address the impact of genetic background on age of onset.
Figure 6.

Changes in corneal opacification over time. Out of 14 lumtm1Sc homozygous mutants that were given follow-up eye exams over 7–8 mo, progression in opacity is shown for four mutants here. All 14 animals showed increase in opacity over time.
Abnormal Collagen Fibril Morphology
To determine whether abnormal collagen architecture underlies the skin and corneal manifestations, corneal and tail sections (skin and tendon areas) of age matched mice were examined by transmission electron microscopy. In wild-type (Fig. 7, a and e) and heterozygous mice (not shown), the corneal collagen fibrils were uniformly thin in cross section (diameter distribution = 20–40 nm) and closely packed. In the null mutant mouse corneas (Fig. 7, b and f) three differences were obvious. First, in addition to fibrils of normal diameter there were thicker fibrils. Mean fibril diameter was 47 ± 1.4 nm in mutants and 30 ± 1.7 nm in wild type (Fig. 7, f and e, respectively). Second, a cross sectional view revealed many irregular shaped fibrils, which were most likely due to abnormal lateral fusion of fibrils in the null mutants (Fig. 7 b, arrow). Third, there was an increase in the interfibrillar spacing. Another feature of the mutant corneas was a dramatic disorganization in the arrangement of collagen fibrils and keratocytes evident under low magnification electron microscopy (not shown). In lumtm1Sc/lumtm1Sc tail preparations, dermal areas also displayed increased interfibrillar spacing and a wide range of fibril diameter in cross section (50–150 nm in wild type, and 33–250 nm in mutants; Fig. 7, g and h). Compared with corneal collagen however, tail skin collagen fibrils were less affected by the lumican null mutation (mean fibril diameter was 90 ± 1 in wild type and 107 ± 7 in null mutants). Larger and abnormally shaped collagen fibrils in cross section were also evident in electron micrographs of dorsal skin preparations (not shown). Interestingly, although lateral fusion of collagen fibrils were frequent in dermis and tendon of decorin-deficient mice their corneal collagen were normal (12).
Figure 7.
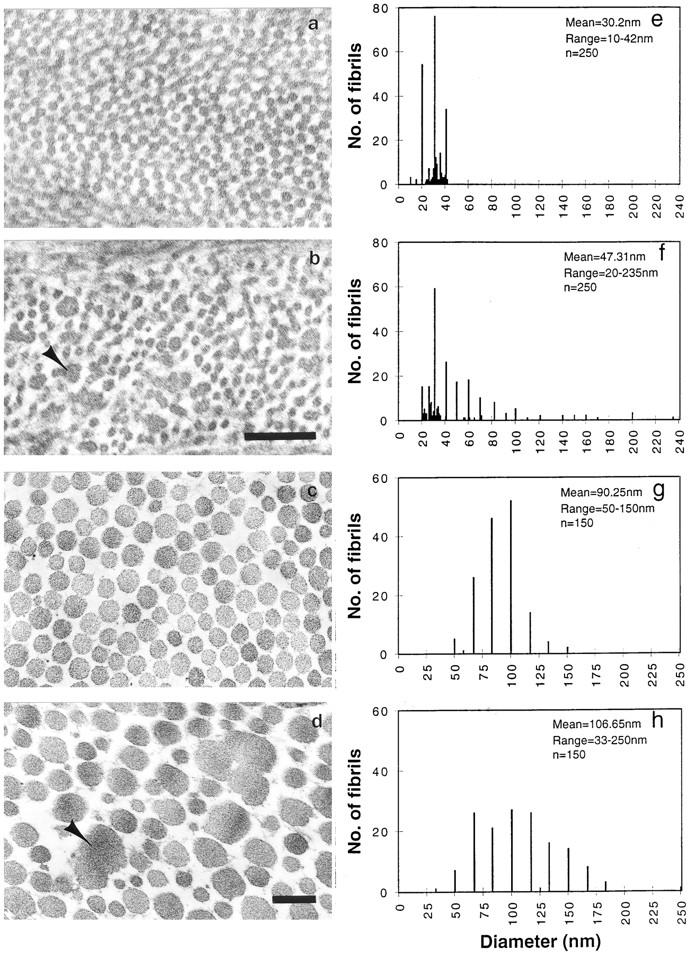
Transmission electron micrography of cornea (a and b) and tail dermis (c and d) collagen preparations (17). (a and c) Homozygous wild type, and (b and d) Lumtm1Sc homozygous. Note larger abnormal shaped fibrils in cross section (arrowhead) in b and d. Fibril diameter frequency (e–h). Homozygous wild-type cornea and tail collagen (e and g) and lumtm1Sc homozygous cornea and tail collagen (f and h). Bar, 0.2 μm.
Discussion
Mice homozygous for a null mutation of the lumican gene are viable, with major effects on two distinct connective tissues: the skin and cornea. The mutant mice display skin laxity and fragility with easy bruising of the skin. Corneal development in the mutant mice appear normal but the cornea loses transparency between 4 and 5 wk of age. To elucidate the underlying defect that leads to this phenotype we focused on understanding the changes in the architecture of the lumican-deficient collagenous extracellular matrix of these connective tissues.
Fibrillar collagens constitute the bulk of all connective tissues and provide the biomechanical support for optimal functioning of these tissues. A large number of common and rare connective tissue disorders have been linked to altered collagen morphology. Not surprisingly much of these studies have concentrated on identifying collagen gene defects (22, 25). However, assembly and final properties of the fibrillar collagen matrix is also influenced by other collagen types such as collagen type V (1) and associated glycoproteins and proteoglycans. In vitro collagen-binding and fibrillogenesis assays (31, 34, 35, 45) suggest strongly that in vivo molecular interactions of lumican and other LRP members with collagen fibrils are likely to influence collagen architecture and overall tissue biomechanics. The phenotypes of the lumican-deficient mice presented in this study and the decorin-deficient mice by Danielson and coworkers (12) clearly bring into focus the LRPs as major regulators of collagen fibril assembly and as likely primary causes for many of these connective tissue disorders.
Lumican Regulates Collagen Fibril Assembly
Lumican deficiency has a profound effect on the structure of collagen fibrils as evidenced by transmission electron microscopy on corneal and tail skin sections. The main features of the altered collagen morphology are the presence of thicker fibrils in addition to fibrils of normal diameter, with frequent lateral fusions of fibrils, increased and non-uniform interfibrillar spacing. The emerging notion is that the LRP core proteins bind to the surface of collagen fibrils as they form by lateral association of trimeric collagen molecules. The LRPs maintain narrow diameter and discourage lateral fusion of fibrils. Laterally fused fibrils may be somewhat displaced in space, thus locally increasing interfibrillar space in the mutants. Absence of lumican may also disturb normal GAG levels, or lead to overexpression of other collagens or proteoglycans that contribute to increased interfibrillar spacing. However, by immunoblot analyses, we found that the lumican null mice make normal levels of the decorin core protein.
Skin Fragility in Lumtm1Sc Homozygous Mice
There is a remarkable association of skin laxity and fragility with the targeted lumican null mutation seen in this study, and in the targeted decorin and more recently developed thrombospondin 2 null mutations (12, 23). We found a major disruption in the biomechanical properties of skin in the lumican null mutants. Compared with lumican wild type, the lumican-deficient mice exhibited an eightfold reduction in the load to failure in tension tests. Decorin-deficient mice, on the other hand, developed a fourfold reduction in the load to failure compared with their wild-type background (12). Our lumican-deficient skin tensile test results were independent of any differences in skin thickness between the mutant and wild-type animals and, therefore, represent a direct biomechanical measure of tissue-level alterations associated with the mutation. At the histological level, lumican-deficient skin tissue displayed disorganized and loosely packed collagen fibers. In particular, in the subepithelial areas there seemed to be a lack of orientation in the collagen fibers and fibroblasts. These histological anomalies correlate well with the electron microscopically observed poorly organized collagen matrix, increased interfibrillar spacing and altered fibril morphology. Both decorin and thrombospondin-deficient mice displayed similar structural anomalies of the skin as well (12, 23). From a biomechanical standpoint, the reduced tensile strength of the lumican-deficient skin is due to a disruption in the normal transmission of load from the matrix to the collagen fibrils and, thus a direct consequence of the altered collagen morphology (11, 27, 29).
EDS, a highly heterogenous group of connective tissue disorders is also characterized by skin laxity and fragility, and displays some of the same collagen fibril anomalies seen in the lumican null mice (30, 40). Mutations in the fibrillar collagen genes are leading causes for several types of EDS. Additionally, mutations in enzymes involved in processing of nascent collagens, and those that may aid fibril assembly, are beginning to be recognized as causes of EDS. Mutations in lumican and other LRPs will fall in this last category, which includes recent detection of mutations in COL5A1 gene (42) and Tenascin-X (3).
Lumican Required for Postnatal Corneal Development and Maturation
The effect of lumican deficiency on the cornea is quite dramatic: initially clear corneas become cloudy postnatally at a certain age in these mice. This has important implications for lumican's role in acquiring and maintaining corneal transparency. Our results show that although lumican is not necessary for early embryonic stages of corneal development, it is essential for postnatal maturation of the cornea. An understanding of how collagen fibrils are assembled during corneal development will answer the question of why, as in many human corneal dystrophies, corneal clouding in the lumican null mutants display an age dependence. Keeping pace with the rest of the organism, the cornea continues to increase in diameter for sometime after birth. This process requires increased synthesis of collagen, proteoglycans, and degradative remodeling of the extracellular matrix (32). Lumican appears to play a critical role during this post natal assembly and organization of collagen fibrils in the cornea.
Lumican deficiency in the cornea poses two problems: loss of core protein as well as its KS functions. Whether lumican deficiency affects overall corneal KS levels and whether some of the other lumican-like core proteins are abnormally modified with KS chains have to be carefully evaluated. Indeed a preliminary investigation of GAGs in the lumican-deficient mice suggests a reduction in GAG levels (data not shown). The corneal clouding may thus be the result of multiple interacting factors: abnormal fibril assembly, lateral fusion of fibrils resulting from lumican core protein deficiency, and altered interfibrillar spacing because of an absence of lumican-associated KS chains. In decorin-deficient mice, which reportedly have normal corneal collagen architecture (12), it is possible that other LRP members such as lumican complement decorin core protein functions in the eye. Furthermore, chondroitin-4-sulfate, the GAG side chain of decorin is believed to play a minor role in corneal transparency (8, 9). Therefore, any reduction in its level in the decorin null mice is less likely to affect corneal transparency.
Lumtm1Sc Homozygous Corneal Phenotype and Human Corneal Stromal Dystrophies
The lumtm1Sc corneal phenotype may be compared with two broad categories of corneal stromal dystrophies. Perturbed GAG levels in lumtm1Sc homozygosity is a feature that biochemically resembles type I and type II macular corneal dystrophies (MCD I and II), which also show progessive bilateral corneal clouding. Antigenic KS is absent from serum and cornea in MCD I whereas other GAGs may be affected in MCD II (19, 26). Additionally, in MCD non-glycanated core proteins of the proteoglycans precipitate and contribute to the pathogenesis. Both MCD types localize to human chromosome 16q22 (43). Whereas lumican localizes to human chromosome 12q21.3-q22 (7), four other stromal dystrophies with amyloid type deposits that somewhat resemble the lumican null corneal phenotype are all because of mutations in the gene encoding kerato-epithelin on chromosome 5q31.1 (28). The second category includes stromal dystrophies with altered collagen fibril manifestations (see online Mendelian Inheritance in Man database on the World Wide Web at http://www3.ncbi.nlm.nih.gov/omim/) (4, 25). Lack of lumican–collagen interactions and resulting disruption of collagen fibril assembly mimics the second category. The recessive eye bleb mutation in mouse, with clear blistering and eventual corneal opacity also maps to distal 10 and could be due to mutations in lumican, or keratocan this region, or one of the other related LRP that map to this region (12a, 14). A class of corneal dystrophies that map on human chromosome 12q, same chromosome as lumican, are dominant and recessive forms of Cornea Plana Congenita, involving corneal maldevelopment. However, these are phenotypically quite different from the lumican null corneas, and may be due to keratin gene defects (41). Meesmann's corneal dystrophy, a defect of the corneal epithelium, has recently been shown to be resulting from mutations in the keratin K3 and k12 genes on chromosome 12q and 17q, respectively (18).
Conclusions
In conclusion we have generated lumican null homozygous mutants by gene targeting in the mouse. Lumican deficiency causes skin laxity and fragility resembling EDS and will lead to mouse models for specific types of EDS. By comparing the targeted null mutation phenotypes of lumican, decorin (12), and thrombospondin-2 (23) we will gain further insights into how these proteoglycans and glycoproteins regulate fibril assembly in vivo and affect functions of different organs and tissues. Although tissue ultrastructure and skin tensile strength is affected in all three null mutants, the lumican null mutation also disrupts corneal transparency. Corneal proteoglycans have been historically implicated in maintaining a collagen architecture that is conducive to corneal transparency. Our study provides the first in vivo evidence that loss of lumican, one such proteoglycan from the cornea, causes deregulation of collagen fibril assembly and corneal opacification. The lumtm1Sc/lumtm1Sc mice will serve as an in vivo system for understanding the basis of corneal transparency.
Acknowledgments
We are indebted to J. Polak and R.Z. Wang for the electron microscopy work; E. Diaconu for help in the slit-lamp examinations; K.D. Lute, G. Terasaki, L. Yuzon, and P. Gyalzen for technical assistance. We thank J.R. Hassell (Shriners Hospital for Children, Tampa, FL) and H.F. Willard (Genetics, Case Western Reserve University) for helpful discussions and critical reading of the manuscript; S.H. Yuspa (National Cancer Institute, National Institutes of Health [NIH], Bethesda, MD) for interpretation of skin histology.
This work was supported by funds from the Department of Genetics and Medicine, AHA102BGA, NIH EY11654 (to S. Chakravarti); HD 26722 (to T. Magnuson); NIH core grant EY11373, Research to Prevent Blindness (to J.H. Lass).
Abbreviations used in this paper
- EDS
Ehlers-Danos syndrome
- ES
embryonic stem
- GAG
glycosaminoglycan
- KS
keratan sulfate
- LRP
leucine-rich proteoglycan family
Footnotes
Address all correspondence to Shukti Chakravarti, Ph.D., Departments of Medicine and Genetics, School of Medicine, Case Western Reserve University, 10900 Euclid Avenue, Office: 423 BRB, Cleveland, OH 44106-4952. Tel.: (216) 368-1828. Fax: (216) 368-1674. E-mail: sxc76@po.cwru.edu
References
- 1.Birk DE, Fitch JM, Babiarz JP, Doane KJ, Linsenmayer TE. Collagen fibrillogenesis in vitro: interactions of types I and V collagen regulates fibril diameter. J Cell Sci. 1990;95:649–657. doi: 10.1242/jcs.95.4.649. [DOI] [PubMed] [Google Scholar]
- 2.Blochberger TC, Vergnes J-P, Hempel J, Hassell JR. cDNA to chick lumican (corneal keratan sulfate proteoglycan) reveals homology to the small interstitial proteoglycan gene family and expression in muscle and intestine. J Biol Chem. 1992;267:347–352. [PubMed] [Google Scholar]
- 3.Burch GH, Gong Y, Liu W, Dettman RW, Curry CJ, Smith L, Miller WL, Bristow J. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat Genet. 1997;17:104–108. doi: 10.1038/ng0997-104. [DOI] [PubMed] [Google Scholar]
- 4.Byers PH, Holbrook KA, Hall JG, Bornstein P, Chandler JW. A new variety of spondyoepiphyseal dysplasia characterized by punctate corneal dystrophy and abnormal collagen fibrils. Hum Genet. 1978;40:157–169. doi: 10.1007/BF00272296. [DOI] [PubMed] [Google Scholar]
- 5.Casey, T.A., and K.W. Sharif. 1991. A Color Atlas of Corneal Dystrophies and Degenerations. Wolfe Publishing Limited, London. 21–51.
- 6.Chakravarti S, Magnuson T. Localization of mouse lumican (keratan sulfate proteoglycan) to distal chromosome 10. Mamm Genome. 1995;6:367–368. doi: 10.1007/BF00364803. [DOI] [PubMed] [Google Scholar]
- 7.Chakravarti S, Stalling R, Fite D, SundarRaj N, Hassell JR. Primary structure of human lumican (keratan sulfate proteoglycan) and localization of the gene (LUM) to chromosome 12q21.3-q22. Genomics. 1995;27:481–488. doi: 10.1006/geno.1995.1080. [DOI] [PubMed] [Google Scholar]
- 8.Cintron C, Gregory JD, Damle SP, Kublin CL. Biochemical analyses of proteoglycans in rabbit corneal scars. Investig Ophthalmol Vis Sci. 1990;31:1975–1981. [PubMed] [Google Scholar]
- 9.Cornuet PK, Blochberger TC, Hassell JR. Molecular polymorphisms of lumican during corneal development. Investig Ophthalmol Vis Sci. 1994;35:870–876. [PubMed] [Google Scholar]
- 10.Corpuz LM, Funderburgh JL, Funderburgh ML, Bottomley GS, Prakash S, Conrad GW. Molecular cloning and tissue distribution of keratocan. J Biol Chem. 1996;271:9759–9763. doi: 10.1074/jbc.271.16.9759. [DOI] [PubMed] [Google Scholar]
- 11.Daly CH. Biomechanical properties of dermis. J Invest Dermatol. 1982;79:17S–20S. doi: 10.1111/1523-1747.ep12544620. [DOI] [PubMed] [Google Scholar]
- 12.Danielson KG, Baribault H, Holmes DF, Graham H, Kadler KE, Iozzo RV. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol. 1997;136:729–743. doi: 10.1083/jcb.136.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12a.Dunlevy JR, Chakravarti S, Gyalzen P, Vergnes J-P, Hassell JR. Cloning and chromosomal localization of mouse keratocan, a corneal keratan sulfate proteoglycan. Mamm Genome. 1998;9:316–319. doi: 10.1007/s003359900757. [DOI] [PubMed] [Google Scholar]
- 13.Fisher LW, Stubbs JT, Young MF. Antisera and cDNA robes to human and certain animal bone matrix noncollagenous proteins. Acta Orthop Scand Suppl. 1995;266:61–65. [PubMed] [Google Scholar]
- 14.Green, E.L. 1975. Biology of the Laboratory Mouse. E.L. Green, editor. Dover Publications, New York. 94–95.
- 15.Hedbom E, Heinegard D. Binding of fibromodulin and decorin to separate sites on fibrillar collagens. J Biol Chem. 1993;268:27307–27312. [PubMed] [Google Scholar]
- 16.Hogan, B., R. Bedington, F. Constantini, and E. Lacy. 1994. Manipulating the Mouse Embryo. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 219–323.
- 17.Iozzo RV, Murdoch AD. Proteoglycans of the extracellular environment: clues from the gene and protein side offer novel perspectives in molecular diversity and function. FASEB (Fed Am Soc Exp Biol) J. 1996;10:598–614. [PubMed] [Google Scholar]
- 18.Irvine AD, Corden LD, Swensson O, Swensson B, Moore JE, Frazer DG, Smith FJD, Christophers RGK, Rochels R, Uitto J, McLean WHI. Mutations in cornea-specific keratin K3 or K12 genes cause Meesmann's corneal dystrophy. Nat Genet. 1997;16:184–187. doi: 10.1038/ng0697-184. [DOI] [PubMed] [Google Scholar]
- 19.Klintworth GK, Meyer R, Dennis R. Macular corneal dystrophy. Lack of keratan sulfate in serum and cornea. Ophthalmic Paediatr Genet. 1986;7:139–143. doi: 10.3109/13816818609004130. [DOI] [PubMed] [Google Scholar]
- 20.Kobe B, Deisenhofer J. Crystal structure of porcine ribonuclease inhibitor, a protein with leucine-rich repeats. Nature. 1993;366:751–756. doi: 10.1038/366751a0. [DOI] [PubMed] [Google Scholar]
- 21.Kresse H, Liszio C, Schonherr E, Fisher LW. Critical role of glutamate in a central leucine-rich repeat of decorin for interaction with type I collagen. J Biol Chem. 1997;272:18404–18410. doi: 10.1074/jbc.272.29.18404. [DOI] [PubMed] [Google Scholar]
- 22.Kuivaniemi H, Tromp G, Prockop D. Mutations in collagen genes: causes of rare and some common diseases in humans. FASEB (Fed Am Soc Exp Biol) J. 1991;5:2052–2060. doi: 10.1096/fasebj.5.7.2010058. [DOI] [PubMed] [Google Scholar]
- 23.Kyriakides TR, Zhu Y-H, Smith LT, Bain SD, Yang Z, Lin MT, Danielson KG, Iozzo RV, LaMarca M, McKinney CF, Ginns EI. Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density, and a bleeding diathesis. J Cell Biol. 1998;140:419–430. doi: 10.1083/jcb.140.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maurice DM. The structure and transparency of the cornea. J Physiol. 1957;136:263. doi: 10.1113/jphysiol.1957.sp005758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKusick, V.A. 1992. Mendelian Inheritance in Man. Vol. 1 and 2. The Johns Hopkins University Press, Baltimore.
- 26.Midura RJ, Hascall V, MacCallum DK, Meyer RF, Thonar EJ, Hassell JR, Smith CF, Klintworth GK. Proteoglycan biosynthesis by human corneas from patients with types 1 and 2 macular corneal dystrophy. J Biol Chem. 1990;265:15947–15955. [PubMed] [Google Scholar]
- 27.Mohanaradhakrishnan V, Muthiah PL, Hadhanyi A. A few factors contributing to the mechanical strength of collagen fibres. Arzneimittelforschung. 1975;25:726–735. [PubMed] [Google Scholar]
- 28.Munier FL, Korvatska E, Djemai A, Le Paslier D, Zografos L, Pescia G, Schorderet DF. Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet. 1997;15:247–251. doi: 10.1038/ng0397-247. [DOI] [PubMed] [Google Scholar]
- 29.Parkinson J, Brass A, Canova G, Brechet Y. The mechanical properties of simulated collagen fibrils. J Biomech. 1997;30:549–554. doi: 10.1016/s0021-9290(96)00151-0. [DOI] [PubMed] [Google Scholar]
- 30.Pope FM, Burrows NP. Ehlers-Danlos syndrome has varied molecular mechanisms. J Med Genet. 1997;34:400–410. doi: 10.1136/jmg.34.5.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rada JA, Cornuet PK, Hassell JH. Regulation of corneal collagen fibrillogenesis in vitro by corneal keratan sulfate proteoglycan (lumican) and decorin core proteins. Exp Eye Res. 1993;56:635–648. doi: 10.1006/exer.1993.1081. [DOI] [PubMed] [Google Scholar]
- 32.Rada JA, Fini ME, Hassell JR. Regionalized growth patterns of young chicken corneas. Investigative Ophthalmol Vis Sci. 1996;37:2060–2067. [PubMed] [Google Scholar]
- 33.Sassoon, D., and N. Rosenthal. 1993. Detection of messenger RNA by in situ hybridization. In Guide to Techniques in Mouse Development. P.M. Wasserman, and M.L. DePamphilis, editors. Academic Press, New York. 384–404. [DOI] [PubMed]
- 34.Schonherr E, Hausser H, Beavan L, Kresse H. Decorin-type I collagen interaction. J Biol Chem. 1995;270:8877–8883. doi: 10.1074/jbc.270.15.8877. [DOI] [PubMed] [Google Scholar]
- 35.Schonherr E, Witsch-Prehm P, Harach B, Robenek H, Raurerberg J, Kresse H. Interactions of biglycan with Type I collagen. J Biol Chem. 1995;270:2776–2783. doi: 10.1074/jbc.270.6.2776. [DOI] [PubMed] [Google Scholar]
- 36.Schrecengost PK, Blochberger TC, Hassell JR. Identification of chick corneal keratan sulfate proteoglycan precursor protein in whole corneas and in cultured corneal fibroblasts. Arch Biochem Biophys. 1992;292:54–61. doi: 10.1016/0003-9861(92)90050-7. [DOI] [PubMed] [Google Scholar]
- 37.Scott JE. Proteoglycan-fibrillar collagen interactions. Review article. Biochem J. 1988;252:313–323. doi: 10.1042/bj2520313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott JE. Proteodermatan and proteokeratan sulfate (decorin, lumican/fibromodulin)proteins are horseshoe shaped. Implications for their interactions with collagen. Biochemistry. 1996;35:8795–8799. doi: 10.1021/bi960773t. [DOI] [PubMed] [Google Scholar]
- 39.Scott JE, Orford CR. Dermatan sulfate rich proteoglycan associates with rat tail tendon collagen at the d band in the gap region. Biochem J. 1981;197:213–216. doi: 10.1042/bj1970213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steinmann, B., P.M. Royce, and A. Superti-Furga. 1993. The Ehlers-Danlos Syndrome. In Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects. P.M. Royce, and B. Steinmann, editors. Wiley-Liss Inc., New York. 351–408.
- 41.Tahvanainen E, Villnueva AS, Forsius H, Salo P, de la Chapelle A. Dominantly and recessively inherited Cornea Plana Congenita map to the same small region of chromosome 12. Genome Res. 1996;6:249–253. doi: 10.1101/gr.6.4.249. [DOI] [PubMed] [Google Scholar]
- 42.Toriello HV, Glover TW, Takahara K, Byers PH, Miller DE, Higgins JV, Greenspan DS. A translocation interrupts the COL5A1 gene in a patient with Ehlers-Danlos syndrome and hypomelanosis of Ito. Nat Genet. 1996;13:361–365. doi: 10.1038/ng0796-361. [DOI] [PubMed] [Google Scholar]
- 43.Vance JM, Jonasson F, Lennon F, Sarrica J, Damji KF, Stauffer J, Pericak-Vance MA, Klintworth GK. Linkage of a gene for macular corneal dystrophy to chromosome 16. Am J Hum Genet. 1996;58:757–762. [PMC free article] [PubMed] [Google Scholar]
- 44.Vogel KG, Trotter JA. The effect of proteoglycans on the morphology of collagen fibrils formed in vitro. Collagen Relat Res. 1987;7:105–114. doi: 10.1016/s0174-173x(87)80002-x. [DOI] [PubMed] [Google Scholar]
- 45.Vogel KG, Paulsson M, Heinegard D. Specific inhibition of type I and type II collagen fibrillogenesis by the small proteoglycans of tendon. Biochem J. 1984;223:587–597. doi: 10.1042/bj2230587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weber IT, Harrison RW, Iozzo RV. Model structure of decorin and implications for collagen fibrillogenesis. J Biol Chem. 1996;271:31767–31770. doi: 10.1074/jbc.271.50.31767. [DOI] [PubMed] [Google Scholar]