

Abstract
The Chediak-Higashi syndrome (CHS) is a human recessive autosomal disease caused by mutations in a single gene encoding a protein of unknown function, called lysosomal-trafficking regulator. All cells in CHS patients bear enlarged lysosomes. In addition, T- and natural killer cell cytotoxicity is defective in these patients, causing severe immunodeficiencies. We have analyzed major histocompatibility complex class II functions and intracellular transport in Epstein Barr Virus–transformed B cells from CHS patients. Peptide loading onto major histocompatibility complex class II molecules and antigen presentation are strongly delayed these cells. A detailed electron microscopy analysis of endocytic compartments revealed that only lysosomal multilaminar compartments are enlarged (reaching 1–2 μm), whereas late multivesicular endosomes have normal size and morphology. In contrast to giant multilaminar compartments that bear most of the usual lysosomal markers in these cells (HLA-DR, HLA-DM, Lamp-1, CD63, etc.), multivesicular late endosomes displayed reduced levels of all these molecules, suggesting a defect in transport from the trans-Golgi network and/or early endosomes into late multivesicular endosomes. Further insight into a possible mechanism of this transport defect came from immunolocalizing the lysosomal trafficking regulator protein, as antibodies directed to a peptide from its COOH terminal domain decorated punctated structures partially aligned along microtubules. These results suggest that the product of the Lyst gene is required for sorting endosomal resident proteins into late multivesicular endosomes by a mechanism involving microtubules.
Major histocompatibility complex (MHC)1 class II molecules are composed of an αβ dimer that associates in the ER with a third membrane molecule, the invariant chain (Ii; 33, 24). The αβ−Ii chain complexes are transported via the Golgi apparatus to the endocytic pathway, directed by a signal localized in the cytoplasmic tail of Ii chain (7, 41). Ii chain is then degraded (12), and upon complete removal of the remaining Ii fragments (60), antigenic peptides are loaded onto class II molecules under the control of HLA-DM (65, 22).
Ii chain cleavage and antigen processing to fitting peptides occurs in endosomal and/or lysosomal compartments (24). Depending on the species origin of the cell, cell types, or even on the maturation status in the case of dendritic cells, accumulation of MHC class II molecules may occur in different endocytic compartments (43, 51). In human Epstein Barr virus–transformed B (EBV-B) cells, HLA-DR molecules accumulate in lysosomal compartments named MHC class II compartments (MIICs; 49). In murine splenic lipopolysaccharide-activated B cells (18) as well as in macrophages and human melanoma cells (30, 52), MHC class II is found all along the endocytic pathway, from early endosomes to lysosomes. In contrast, A20 murine B lymphoma cells accumulate MHC class II molecules in endosomal compartments, the class II vesicles (2, 4), whereas few class II molecules are found in conventional endosomes and lysosomes. However, upon inhibition of Ii chain degradation, class II molecules redistribute into lysosomal compartments (14).
Recent results from the laboratory of H. Geuze (50, 35) showed that the distribution of MHC class II molecules in EBV-B cells is not as restricted as initially envisioned. Indeed, HLA-DR accumulates in two types of compartments: (a) in endosomes containing multiple internal vesicles that are reached by fluid phase markers after 20–30 min of internalization and contain some Ii chain (multivesicular late endosomes); and (b) in vesicles containing internal membranes organized in onion-like structures that accumulate fluid phase markers only after 60 min and contain no Ii chain (multilaminar lysosomal compartments). Both types of compartments also contain Lamp1/2, CD63, and HLA-DM.
The functional relevance of this heterogeneity of endocytic MHC class II–containing compartments is still unclear, and the precise role of multivesicular and multilaminar endosomes in MHC class II transport and Ii chain degradation is not known. Moreover, it has recently been shown that the antigenic peptides generated in endosomal and lysosomal compartments might not be the same (30). In addition, we have recently shown that antigen internalization through different membrane receptors that may deliver antigens to particular endocytic compartments results in presentation of different antigenic peptides (3).
To evaluate the role of this heterogeneity of endocytic compartments in MHC class II transport and function, we examined EBV-B cells of patients suffering from a rare genetic immunodeficiency disease, the Chediak-Higashi Syndrome (CHS), which affects the morphology and function of endocytic compartments. CHS results from mutations in a gene encoding a large cytosolic protein called lysosomal trafficking regulator (LYST), which displays limited sequence homology to a regulatory subunit of the yeast phosphatidyl-inositol-3 kinase (PI3K), VPS15 (9, 45). LYST also includes several WD40 and HEAT/ARM domains, a domain of limited homology to stathmin, as well as a unique domain that has been called BEACH (9, 8, 10, 45).
Despite having identified several subdomains in the CHS protein, the precise function of the protein is not known. We do know, however, that mutations in this gene result in immunological disorders and susceptibility to multiple childhood infections. The lysosomal compartments in all cell types of CHS patients are enlarged, reaching over 1 μm/vesicle (70). In hematopoietic cells, including T lymphocytes, NK cells, and granulocytes, cytotoxicity is defective, most likely because of a defect in regulated secretion (61, 29, 6). In nonhematopoietic cells such as melanocytes and kidney cells, enlarged lysosomal morphology and defects in lysosomal enzyme secretion have been reported (15). It is yet unclear whether the defect in the secretory function of lysosomes in hematopoietic cells is a consequence or a cause of the abnormal lysosomal morphology. It is also possible that both phenotypes arise from a unique upstream defect in the endocytic pathway.
Here we show that antigen presentation and MHC class II intracellular transport are affected in EBV-B cells from CHS patients. Surprisingly, only lysosomal multilaminar MHC class II–containing compartments are enlarged, while multivesicular late endosomes displayed normal size and morphology. However, a severe reduction in the staining of multivesicular endosomes for MHC class II, Lamp 1/2, CD63, CD82, and β-hexosaminidase was observed, suggesting that transport of these markers from the TGN and/or early endosomes into late endosomes is affected. Missorting of resident lysosomal proteins to the plasma membrane and early endosomes was also observed, as well as a striking redistribution of the cation-dependent mannose-6-phosphate receptor (CD-MPR) into giant multilaminar lysosomes. In addition, we showed that LYST partially colocalizes with microtubules, which have previously been shown to play a critical role in transport from early to late endosomes (19). Together, these results show severe missorting of membrane proteins along the endocytic pathway of CHS cells, and suggest that LYST may be directly involved in microtubule-dependent transport into late endocytic compartments.
Materials and Methods
Antibodies and Cell Lines
All cell lines were maintained in RPMI 1640, 10% FCS (Sigma Chemical Co., St. Louis, MO), 5 mM heat-inactivated glutamine, and β-mercaptoethanol. Two CHS EBV-B cell lines were used: HLA DRB1* 0314 (JH) and DRB1* 0113 (EA). JH cells were stably transfected with the β-DR1 encoding cDNA to reconstitute the DR1 expression. Three human EBV B cell lines—Hom-2 (homozygous DR1; 11), COX (homozygous DR3, DRB1*0301), and PALA (homozygous DR3; 42)—were used as normal controls in different sets of experiments. Anti-H3 specific T cells (TH1.7) were Jurkat cells transfected with the α and β chains from a DR1/HA 306–319–specific T cell receptor (31).
The anti-MHC class II antibodies used in this study are: Tü36 (64), L243 (63), Tal.1B5 (1), and DA6.147 (28). MHC class I molecules were detected using HC10 (67). The antibodies against lysosomal markers were Lamp1, a gift of Dr. Sven Carlsson (University of Umeå, Sweden), and CD63 (37), obtained from B. Hoflack (Institut Pasteur Lille, France). The CD-MPR antibody was a gift from Dr. A. Hille-Rehfeld (University of Göttingen, Germany; 68), and the anti-HLA-DM antibody was from J. Trowsdale (Imperial Cancer Research Fund, London, United Kingdom). The anti-DNP antibody was from Molecular Probes, Inc. (Eugene, OR). The anti LYST peptide-antiserum was raised against peptide 3426–3445 (KLNIEGELPAAVGLLVQFAF coupled to KLH). Specific antibodies were isolated by affinity purification on the same peptide. The anti-α-tubulin monoclonal antibody was purchased from Amersham Corp. (Arlington Heights, IL). Secondary antibodies were: FITC-conjugated donkey anti– mouse F(ab′)2 fragments, FITC-conjugated donkey anti–rabbit F(ab′)2 fragments, and TR-conjugated donkey anti–mouse F(ab′)2 fragments from Jackson Laboratories (Bar Harbor, ME).
FACS Analysis
Cells were washed twice in PBS and incubated for 20 min. with the primary antibodies and 20 min. with the secondary antibody. Acquisition and analysis were performed on a FACscan (Becton Dickinson).
Intracellular Immunofluorescence and Confocal Analysis
Paraformaldehyde fixation and immunofluorescent staining were performed as previously described (13). In brief, the cells were fixed for 10 min at RT with 3% paraformaldehyde, and were washed with 5 mM PBS/ glycine to quench PFA excess. Permeabilization was performed with 0.05% saponin, 0.2% BSA in PBS for 30 min.
For anti-CHS and tubulin stainings, fibroblasts were fixed in ice-cold methanol (−20°C) for 5 min, followed by a Saponin/BSA incubation as described above. The cells were then analyzed by confocal microscopy (TCS microscope; Leica AG, Heerbrugg, Switzerland).
Immunoelectron Microscopy
The B lymphoblastoid cells lines PALA and Hom-2, and the B-CHS cell lines JH and EA, were fixed with 2% paraformaldehyde in 0.1 M phosphate buffer (PB), pH 7.4, for 1 h at room temperature. After blocking with 50 mM glycine in PB, the cells were embedded in 7.5% gelatin. Small gelatin blocks were infused in 2.3 M sucrose and frozen in liquid nitrogen (57). Ultrathin cryosections were prepared with an ultracryomicrotome (FCS; Leica AG) and a diamond knife (Drukker International, Cuijk, The Netherlands), and were collected using a mixture of 2.3 M sucrose and methylcellulose (vol/vol; 40). Ultrathin frozen/thawed sections were single or double immunogold–labeled with different antibodies and protein A gold conjugates (PAG 5, 10, 15; reference 57). Internalization of colloidal gold-coupled bovine serum albumin (BSA-G) was performed by incubating 107 cells for 10 min at 37°C with BSA-G in RPMI (OD = 5). After washing with ice-cold RPMI supplemented with 5% FCS, the endocytic tracer was chased for 20, 50, or 120 min at 37°C. To analyze the acidity of intracellular compartments, 107 cells were washed with RPMI, and were incubated with N-3-(2,4-dinitroanilino)-3-amino-N-methyldipropylamine (DAMP; Molecular Probes, Inc.) at a final concentration of 30 μg/ml for 30 min with ice-cold RPMI. After incubation with the endocytic tracer or with DAMP, at the indicated times cells were fixed and processed as described above.
Pulse Chase and Immunoprecipitations
Pulse chase, surface biotinylation, and immunoprecipitations were performed as previously described (14). In brief, EBV-B cells were pulse- labeled with 250 μCi/ml [35S]Trans-Label™ (Amersham Corp.). The cells were then chased in nonradioactive 5% RPMI1640 FCS, and were surface-biotinylated with NHSS-biotin (Pierce Chemical Co., Rockford, IL) at a final concentration of 2 mg/ml. The cells were lysed in 1 ml lysis buffer (0.5% NP-40, 300 mM NaCl, 50 mM Tris pH 7.4, 20 mM NEM with an addition of protease inhibitors at a concentration of 10 μg/ml mix of leupeptin, chemostatin, aprotinin and pepstatin). MHC class II molecules were immunoprecipitated using specific antibodies, and biotinylated class II molecules were reprecipitated with streptavidin-agarose beads (Pierce Chemical Co.). SDS-PAGE was carried out using 12% acrylamide gels (39). Gels were fixed before drying, and were exposed for autoradiography.
Antigen Presentation Assay
UV-inactivated influenza virus and the hemagglutinin (HA) peptide 307– 318 were added to the plates at the indicated concentrations. 5 × 104 TH 1.7 cells and 5 × 104 EBV-B cells were added per well. After 24 h of incubation at 37°C, IL-2 production was measured using the CTLL-2–dependent T cell line (3). The B cells were pulsed with UV-inactivated influenza virus or the M1 peptide for 3 or 24 h, and M1 presentation was assayed with M1-specific cytotoxic T cell clone D9.1 in a 51Cr release assay (53, 54).
Mass Spectrometry
DR-associated peptides were analyzed by mass spectrometry as previously described (38). In brief, 5 × 108 EBV-transformed B-LCL were used for isolation in a micropreparative scale. About 5–10 μg of purified HLA-DR molecules were extensively washed with aqua bidest in an ultrafree ultrafiltration tube with a 30-kD cutoff (Millipore Corp., Bedford, MA). Peptides were eluted by incubation in 0.1% trifluoro acetic acid for 0.5 h at 37°C. After separating the protein by ultrafiltration, peptides were lyophilized and prepared for mass spectrometry by dissolving them in 0.5 ml 1.4-diydroxybenzoic acid/acetonitrile (2:1). Spectra were recorded on a Lasermat™ (Finnigan MAT GmbH, Bremen, Germany), and were collected by averaging the ion signals from 20–50 individual laser shots.
Results
Antigen Presentation in B-EBV Cells from CHS Patients
First, the ability of B-EBV cells from CHS patients to present antigens in an MHC class II-restricted manner was evaluated. We used T lymphocytes specific for two DR1-restricted epitopes from the influenza virus: the H3 epitope, corresponding to the 307–318 peptide from hemagglutinin (HA), and the M1 epitope, corresponding to the 18–29 peptide from matrix protein. Control DR1+ B-EBV cells (HOM-2) or B-EBV cells from two different patients (JH and EA) were used as antigen-presenting cells (APCs). These cells expressed similar levels of HLA-DR at their surface, as assessed by FACscan analysis using L243 and Tü36 mAbs (not shown). The JH cells did not express HLA-DR1 endogenously, and were therefore transfected with the cDNAs encoding the HLA-DR1 β chain (JH-DR1). Although difficult to evaluate directly, it is most likely that the levels of HLA-DR1 in these cells are reduced as compared with HOM-2 or EA cells.
In the case of the HA epitope, APCs were incubated with complete antigen (i.e., UV-inactivated influenza virus) for different times (from 1 to 24 h). After glutaraldehyde fixation of the cells, anti-H3 TH 1.7 T cells were added for 24 h, and IL-2 production was measured. As shown in Fig. 1 A, the presentation of the H3 epitope was delayed by 4–6 h in the two CHS EBV-B cells, as compared with normal HOM-2 cells. All the cells presented the H3 peptide with similar efficiencies and kinetics (not shown).
Figure 1.
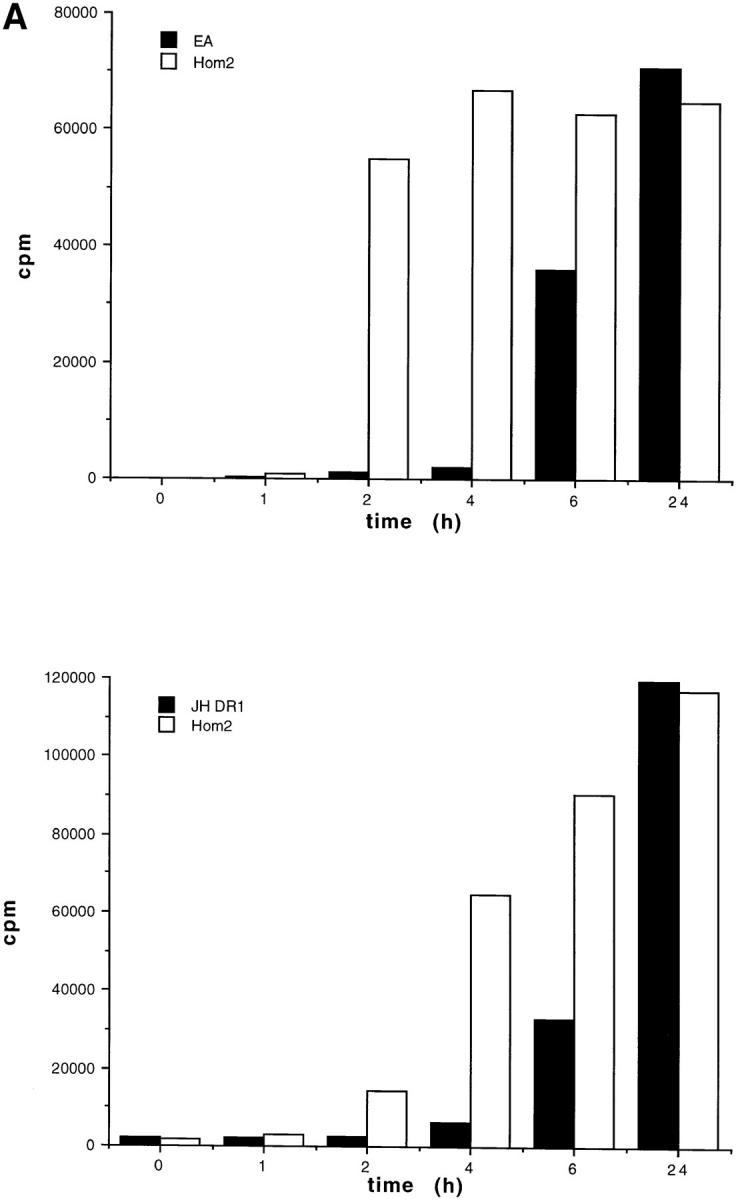

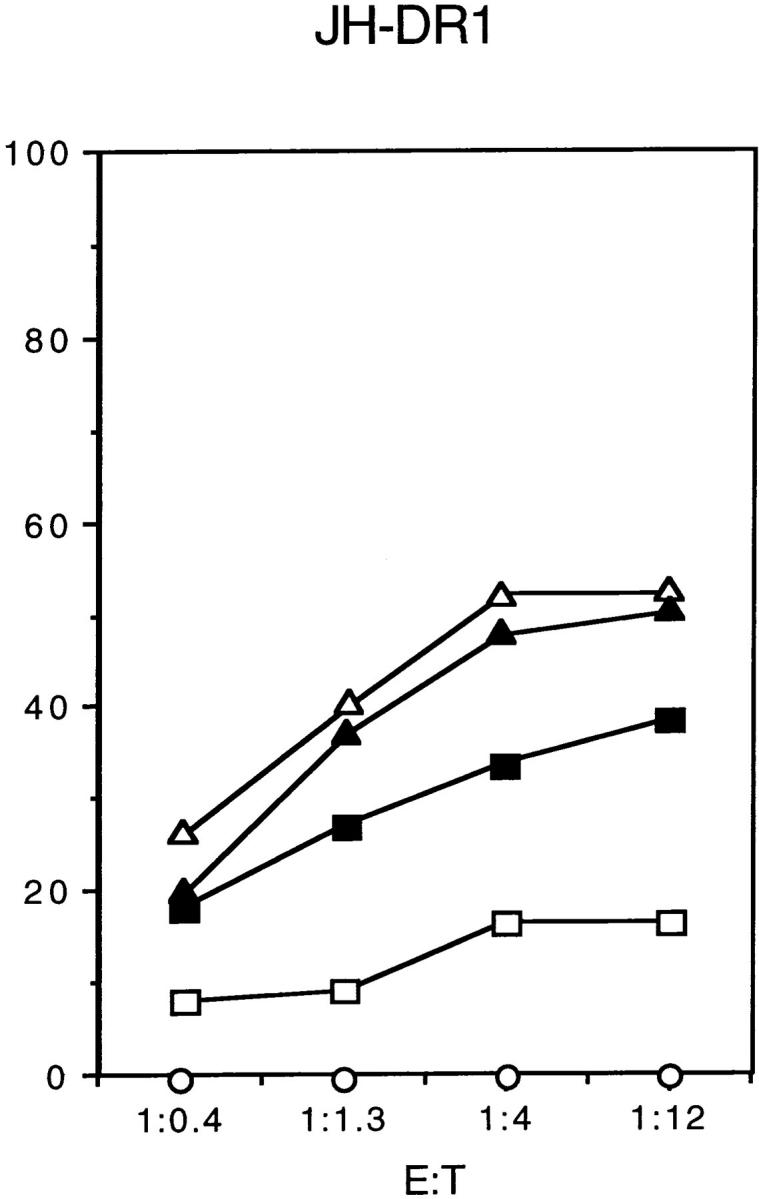
(A) Presentation of the influenza hemagglutinin epitope H3 to a class-II–restricted T cell hybridoma was delayed in two CHS B cell lines compared with control B cells. HOM-2 control cells or two EBV-B cell lines from CHS patients (EA and JH-DR1) were incubated with UV-inactivated influenza virus for the indicated time periods, and were fixed before incubation with an anti-H3 T cell hybridoma. Antigen presentation was measured by IL-2 secretion. (B) MHC class II–restricted presentation of the influenza matrix epitope M1 was delayed in CHS B cells as compared with controls cells. The cells were incubated without (circles), or with either the UV-inactivated influenza virus (squares) or with the M1 peptide (triangles; 18–29) for 3 (open symbols) or 24 h (closed symbols). Control HOM2 cells and the CHS cell lines EA or JH-DR1 were assayed for cytotoxicity by the DR1 restricted T cell line D1.9. The effector target ratios are indicated at the x axis.
To measure presentation of the M1 epitope, control EBV-B cells (HOM-2) or CHS EBV-B cells (JH-DR1 and EA) were cultured for either 3 or 24 h in the presence of inactivated influenza virus, or of the corresponding 18–29 peptide from the M1 matrix protein. The cells were then used as targets for cytotoxicity by a specific CD4+ DR1- restricted cytotoxic T cell clone. As shown in Fig. 1 B, incubation of antigen-presenting cells with the peptides for 3 or 24 h induced efficient antigen presentation and lysis of the target cells. In contrast, when intact antigen was used, presentation by the two CHS EBV-B cells was strongly delayed, as compared with control HOM-2 cells (Fig. 1 B). In the case of the EA cells, the overall efficiency of presentation was reduced after both 3 and 24 h of incubation, whereas in the case of the JH-DR1 cells, presentation was delayed (presentation after 24 h of incubation was similar to that found in HOM2 cells).
Therefore, the overall process of antigen presentation is affected in CHS. This problem with antigen presentation may result from different mechanisms, including defective loading of the peptides onto MHC class II molecules and/ or an inefficient transport of the peptide/class II complexes to the cell surface. We next tested these two possibilities.
Peptide Loading Onto MHC Class II Molecules in CHS
The binding of certain peptides to MHC class II molecules results in production of SDS-stable αβ-dimers (25). The kinetics of SDS-stable MHC class II αβ-dimers were analyzed in control Hom-2 and CHS JH cells. After pulse-chase labeling of control and CHS cells and surface biotinylation, total and surface MHC class II molecules were sequentially precipitated with the monoclonal antibody Tü36 and streptavidin beads, respectively. The precipitates were then either incubated at room temperature or boiled before analysis by SDS-PAGE in order to visualize peptide-associated SDS-stable MHC class II molecules.
As shown in Fig. 2 A, SDS-stable dimers appeared after 2 h of chase in control cells as a band of 55 kD that was absent when the samples were boiled (upper left). In CHS cells, SDS-stable forms were barely detected, even after 6 h of chase (Fig. 2 A, lower left), indicating that peptide loading is strongly delayed in these cells. This delay was even more clear when the surface MHC class II molecules were analyzed (compare the upper and lower right panels). However, SDS-stable dimers were detected in similar amounts in CHS and control cells after 24 h of chase in both total and surface MHC class II (Fig. 2 B). Therefore, peptide loading is strongly delayed, but not absent in CHS cells.
Figure 2.



(A) Delayed formation of SDS-stable dimers in CHS B-cells. Immunoprecipitation of total (Tü36 mAb) and surface (streptavidin-agarose) MHC class II molecules was performed after pulse-chase and cell surface biotinylation. The precipitates were boiled or not before the samples were separated on SDS-PAGE. (B) Similar amounts of SDS-stable dimers in CHS and control cells were found after a 24-h chase. (C ) Mass spectometry profiles of DR-associated self-peptides from JH CHS cells and control B-LCL COX cells. In CHS cells the mass range varied between 1200 and 2600 D with a mode of 1700 D, whereas in control cells the masses ranged from 1500 to 2900 D with a mode of 1900 D. Assuming an average amino acid mass of 115 D, peptides from CHS cells are two amino acids shorter than those in other B cells. Typical CLIP variants are marked by number: CLIP (83–102; m/z 5 2221; peak 1); CLIP (82–102; m/z 5 2334; peak 2); CLIP (82–104; m/z 5 2562; peak 3); and CLIP (81–104; m/z 2675; peak 4; 62).
To evaluate if the peptides found on MHC class II molecules in normal and CHS cells are qualitatively different from each other, mass spectography of peptides eluted from purified HLA-DR in CHS and control cells was performed. As shown in Fig. 2 C, peptides eluted from both CHS and control cells exhibited similar degrees of heterogeneity. Strikingly, however, the peptides eluted from CHS cells were overall shorter by two amino acids than were the peptides eluted from control cells. Likewise, only the shorter forms of the class II–associated Ii chain peptide (83–102 and 82–102) were found in CHS, whereas normal cells also contained the 81–104 and 82–104 forms. These results demonstrate that the peptides found associated to HLA-DR in CHS are overall shorter than those in normal cells.
MHC Class II Transport to the Cell Surface in CHS
To investigate the kinetics of HLA-DR maturation and transport to the cell surface, pulse-chase experiments were performed on cells that were first surface-biotinylated. After lysis, MHC class II molecules were precipitated with the DA6.147 monoclonal antibody that recognizes all MHC class II molecules, or with the L243 antibody that recognizes only mature, Ii chain-free, αβ dimers. The proportion of mature HLA-DR molecules delivered to the cell surface was evaluated by reprecipitating biotinylated molecules with strepavidin-agarose.
As shown in Fig. 3 A (top), the total amounts of newly synthesized MHC class II and Ii chain, as detected with the monoclonal antibody DA6.147, was similar in control and CHS cells. This antibody efficiently precipitated αβIi complexes, and thus allowed us to evaluate the rate of Ii degradation. No significant difference was observed in Ii degradation between control and CHS EBV-B cells. Using direct precipitation of Ii chain with the mAb PIN1, no difference in the rates of degradation of Ii were observed (not shown).
Figure 3.



(A) Delayed maturation of MHC class II molecules. CHS and control Hom-2 cells were labeled for 30 min with [35S]cysteine-methionine and chased for the indicated time points. Split parts of the lysates were immunoprecipitated with the monoclonal antibodies DA6.147 (nonconformational mAb) and L243 (which only binds to mature Ii chain–free MHC class II molecules). Total amounts of class II molecules precipitated from the two cell lines were similar (top). Generation of the L243 epitope was delayed in the CHS cell line (top). Its surface appearance was further delayed (bottom). (B) Densitometric quantification of the autoradiographs in Fig. 3 A (top and bottom). Ratios of total to surface L243 MHC class II molecules were calculated and plotted against time. (C) No difference in rate of cell surface arrival of MHC class I molecules (as detected by HC10 mAb) was observed between control and CHS B-cells. Pulse-chase experiments and immunoprecipitations of class I molecules were performed in JH CHS B cells and control PALA cells.
In contrast, appearance of mature L243-positive HLA-DR molecules was delayed by 1 h, and the maximum levels were slightly lower in JH CHS cells as compared with control cells (Fig. 3 A, middle). In contrast, a strong delay was observed in the delivery of mature L243-positive HLA-DR to the cell surface (Fig. 3 A, bottom). Quantification of these results and calculation of the ratios of total-to-surface molecules showed that the delay in the maturation of HLA-DR molecules does not account for the delay in cell surface appearance. Indeed, the rate of arrival of mature HLA-DR to the plasma membrane, as measured by the ratio of surface/total L243-positive HLA-DR molecules at different times of chase, was decreased in CHS cells (Fig. 3 B). In contrast, no difference in the rates of cell surface transport of MHC class I molecules, which do not transit through the endocytic pathway, was observed between normal and CHS cells (Fig. 3 C). Therefore, at least two different steps in MHC class II biogenesis are effected in CHS: (a) formation of mature, peptide loaded, αβ dimers; and (b) transport of mature MHC class II molecules to the plasma membrane.
MHC Class II–containing Compartments in CHS Cells
To analyze the basis of the functional disorders observed in CHS EBV-B cells, we next analyzed the morphology of MHC class II–containing compartments. It has previously been shown that all cells from CHS patients bear large cytoplasmic inclusions with characteristics of late endosomes and lysosomes, which have been termed macrolysosomes. In NK cells and cytotoxic T lymphocytes too, secretory lysosomes are enlarged. To determine if MHC class II– containing compartments in EBV-B lymphocytes from CHS patients were enlarged, we first examined these cells by immunoflurescence confocal microscopy using different specific markers.
In normal EBV-B cells, MHC class II antibodies strongly stained the cell surface, as well as numerous internal vesicles in the perinuclear area and the cell periphery (Fig. 4, C and D). The internal vesicles were also labeled by anti-HLA-DM and anti-Lamp1 antibodies, two markers of late endosomes and lysosomes. In CHS cells, the morphology of the internal vesicles labeled by these antibodies was strikingly different (Fig. 4, A and B). The vesicles were larger and, although they costained for MHC class II and either HLA-DM or Lamp1, the distribution of the markers was different within each of the macrovesicles: MHC class II staining was homogenous throughout the compartments, whereas HLA-DM and Lamp1 antibodies decorated the periphery of the vesicles (Fig. 4, A and B).
Figure 4.

MHC class II molecules colocalize with Lamp1 and HLA-DM in macrolysosomes of JH CHS cells. JH (A and B) and Pala (C and D) cells were fixed, permeabilized, and stained with the different antibodies. The cells were incubated with either an anti-Lamp1 (A and C) or an anti-DM (B and D) rabbit serum (Texas Red) and L243 anti-MHC class II mAb (A and D; FITC).
Multivesicular and Multilaminar MHC class II–containing Compartments in CHS EBV-B Cells
In human EBV-B cell lines, the bulk of the intracellular MHC class II molecules localize to late endosomal and lysosomal compartments (49). A recent semiquantitative analysis at the ultrastructural level of the localization of MHC class II molecules in human and murine B cells indicated that MHC class II distribution is broader than previously envisioned (35). The compartments where MHC class II molecules were found can be distinguished on the basis of their morphology, accessibility to endocytic tracers (fluid phase markers or ligands internalized by receptor mediated-endocytosis), specific markers, and acidity. Low amounts of MHC class II molecules are detected in tubulovesicular, transferrin receptor–positive early endosomes. MHC class II molecules are progressively enriched in a set of later endocytic compartments that contain in their lumen increasing numbers of internal 60–80 nm vesicles (multivesicular compartments). Finally, one of the major sites of accumulation of MHC class II molecules are compartments displaying both internal vesicles and concentrically arranged membrane sheets (intermediate type), or only membrane sheets (multilaminar compartments). Examples of these compartments in human EBV-B cells are shown in Figs. 5 A (multilaminar MIICs) and 7 C (multivesicular MIICs). Multivesicular and multilaminar MIICs were similarly enriched in Lamp 1/2 (Figs. 5 A and 7 C), HLA-DM, and CD63 (not shown).
Figure 5.
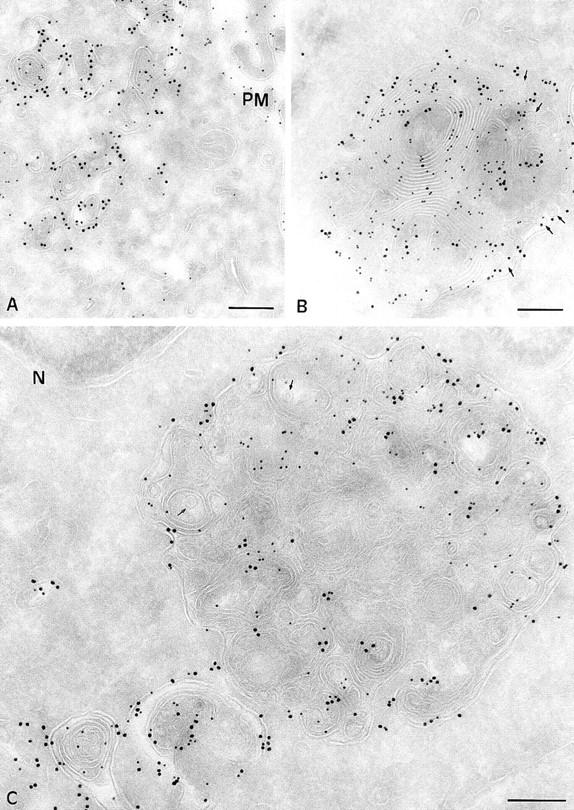
Immunogold localization of MHC class II molecules and Lamp1 in control Pala B cells and JH CHS EBV-B cells. Ultrathin cryosections were double immunogold labeled with anti-MHC class II (PAG 10) and anti-lamp1 antibodies (PAG 15). (A) In control B cells, MHC class II molecules were found in compartments showing concentrically arranged membrane sheets (multilaminar MIICs), and had a mean diameter of 200 nm. (B) At the same magnification, the multilaminar compartments containing MHC class II molecules and Lamp1 in B cells from CHS patients (JH) have a mean diameter of 1 μm. (C) An example of an enlarged multilaminar compartment in JH cells double immunogold–labeled with anti-class II (PAG 10) and anti-Lamp 1 (PAG 15) antibodies at a higher magnification. Small membrane vesicles are eventually detected inbetween membrane sheets (arrows in B and C). PM, plasma membrane. Bars, 200 nm.
Figure 7.
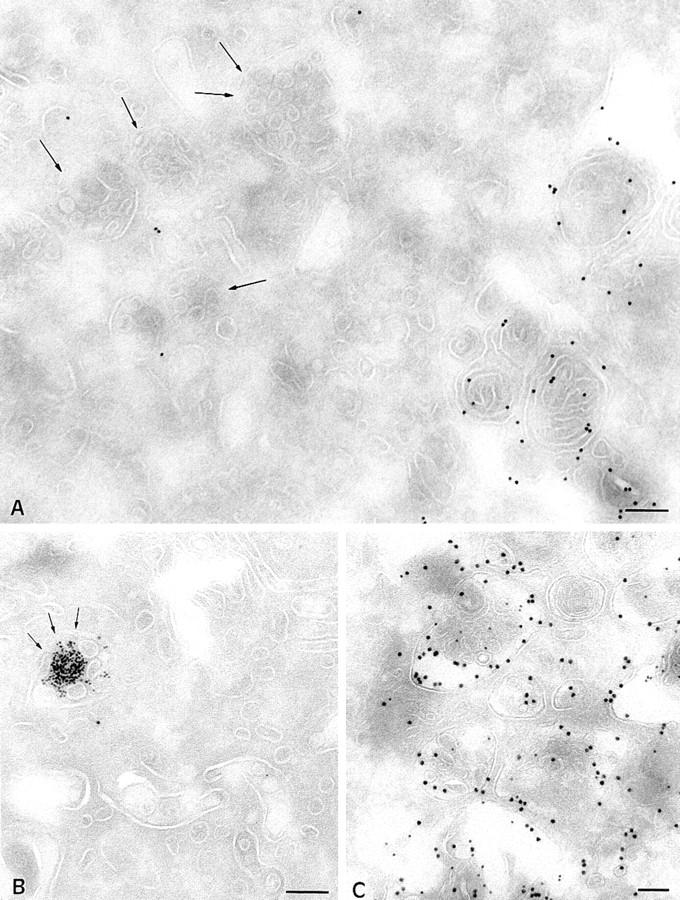
Multivesicular MIICs B cells from CHS patients (JH) and control Pala B cells. (A) In JH cells, compartments displaying intralumenal vesicles have normal morphology and size (mean diameter of 200–300 nm). In these cells the majority of multivescular compartments do not label with anti-class II antibodies, while strong labeling is observed in multilaminar compartments. (B) MHC class II–negative multivesicular compartments contain BSA-G after 30 min of internalization. (C) In control B cells, the majority of the multivesicular compartments are double immunogold–labeled with anti-class II (PAG 10) and anti-Lamp 1 antibodies (PAG 15). Bars, 200 nm.
In JH (see Fig. 9) and EA (not shown) cells, MHC class II molecules were detected at the plasma membrane at levels similar to those found in other B cell lines. Intracellularly, most class II molecules were found in large (generally >1 μm diameter) multilaminar compartments (Fig. 5, B and C). Occasionally, these compartments also contained a small number of internal vesicles (arrows in Fig. 5 C). In addition to MHC class II molecules, all the protein markers described to be enriched in multilaminar MIICs, namely Lamp 1/2 (Fig. 5, B and C), HLA-DM, CD63, and β-hexosaminidase (not shown), also accumulated in the giant CHS compartments. Macrolysosomes also accumulated the weak base DAMP, showing that they acidified normally (Fig. 6 B). Since giant compartments displaying only internal vesicles were not found, we concluded that only the multilaminar MIICs are enlarged in EBV-B lymphocytes from CHS patients.
Figure 9.
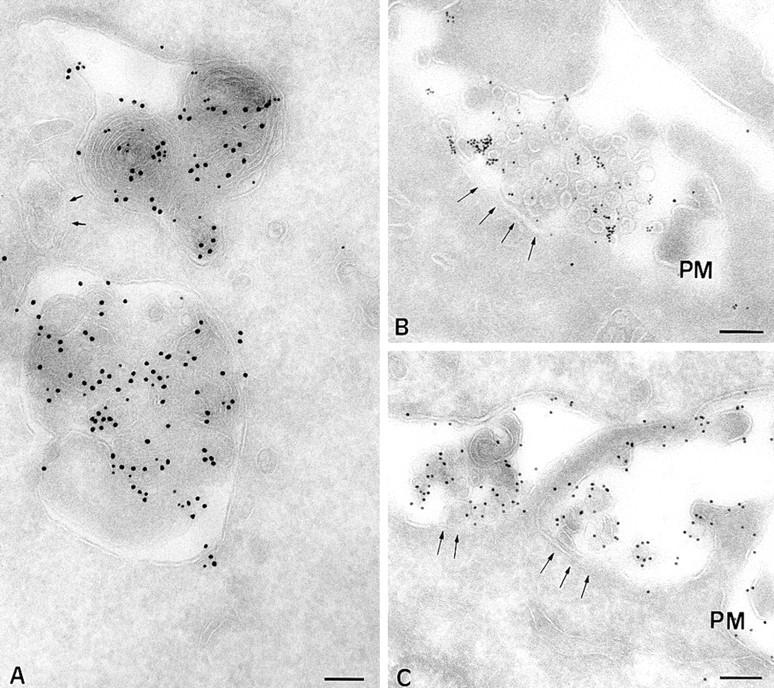
(A) The CD-MPR accumulates in macrolysosomes in B cells from CHS patients (JH cells). Immunogold localization of MHC class II molecules (PAG 10) and CD-MPR (PAG 15) in B-CHS cells. (B) Multivesicular compartments in JH cells fuse with the cell surface. Previously internalized BSAG is reexternalized together with internal vesicles. MHC class II molecules are not detected in the externalized vesicles. (C) In control B cells (PALA), the internal vesicles of multivesicular compartments are strongly labeled with anti-class II antibodies. PM, plasma membrane. Bars, 200 nm.
Figure 6.
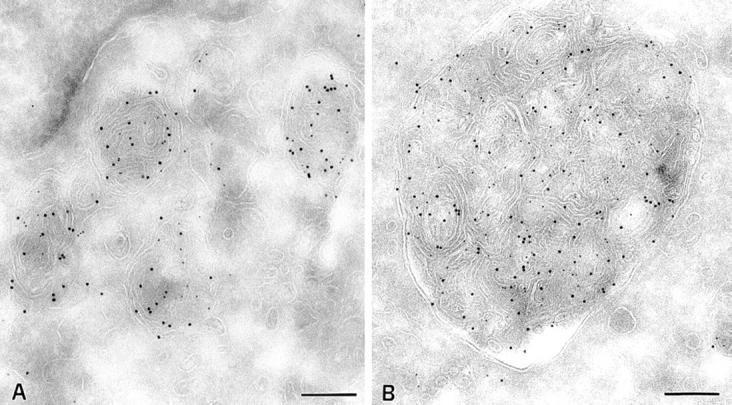
Acidity of multilaminar MHC class II–containing compartments in control Pala B cells and CHS B cells. Cells were incubated with DAMP for 30 min before fixation and processing for cryosectioning. DAMP is visualized on ultrathin cryosections with anti-DNP antibodies, and PAG 5. MHC class II molecules were detected with an anti-class II antibody (PAG 10). The weak base DAMP accumulates in multilaminar MIICs of control cells (A) as well as in the enlarged MIIC in B cells from CHS patients (JH; B). Bars, 200 nm.
In normal cells, multivesicular late endosomal compartments have been proposed to play a critical role in MHC class II transport and Ii chain degradation (50, 26, 58), and to be involved in peptide loading onto MHC class II molecules (69, 44). As shown in Fig. 7 C, multivesicular MIICs in EBV-B cells contained MHC class II molecules and Lamp 1/2 as well as CD63 and β-hexosaminidase (not shown; see reference 36 for review). These vesicles were also weakly stained by anti-Ii chain antibodies (50). Compartments displaying intralumenal vesicles were present in the cytoplasm of CHS B cells. They were abundant, normally sized (mean diameter of 200 nm), and contained normal numbers of internal vesicles in their lumen (Fig. 7 A). However, in contrast to normal EBV-B cells, very low levels of MHC class II molecules and other protein markers (Lamp 1/2 as well as CD63 and β-hexosaminidase, not shown) were detected in multivesicular compartments of JH cells (Fig. 7 A). A semiquantitative analysis of the immunolabeling for MHC class II in multivesicular compartments of normal EBV-B cells and JH CHS cells revealed that only 8% of the compartments in CHS-B cells were labeled, and that the labeling never exceeded three gold particles. In normal EBV-B cells, 87% of the compartments were labeled, and showed a mean of six gold particles per compartment.
To further characterize multivesicular compartments and the multilaminar macrolysosome in JH cells, we tested the kinetics of accessibility to the endocytic tracer BSA-G. Cells were incubated for 10 min with the tracer, and were chased for 20 or 150 min as described in Materials and Methods. Multivesicular compartments of JH cells, despite the fact that they do not label with anti–class II antibodies, were reached by the tracer after 20 min of chase, confirming their late endosomal/prelysosomal origin (Fig. 7 B). BSA-G was found in enlarged multilaminar compartments only after 150 min of chase (not shown). Therefore, BSA-G reached late endosomes and lysosomes with normal kinetics and efficiencies in CHS-B cells.
We also examined transport of membrane immunoglobulins (mIg) to multivesicular endosomes after cross-linking at the cell surface with rabbit anti–human IgG antibodies. After a continuous internalization (30 min at 37°C), analysis of cryosections with anti-human IgG revealed that mIgs were abundant in MHC class II/CD63 negative multivesicular compartments in JH cells (not shown). In contrast to the late endosomal/lysosomal compartment of CHS B cells, early endosomes had normal morphology and function. Tubulovesicular structures enriched in transferrin receptor were present beneath the plasma membrane and in proximity to the Golgi complex (not shown). In addition, uptake and recycling of radioactive Tf exhibited similar kinetics in normal EBV-B and JH CHS B cells (results not shown).
Taken together, these observations show that MHC class II–containing multilaminar compartments are selectively enlarged in B-CHS cells. The morphology of multivesicular late endosomes is not altered, but expression of resident membrane proteins is strongly reduced in these compartments. Therefore, transport of endosomal/lysosomal resident proteins in multivesicular late endosomes, but not fluid phase markers or cross-linked mIg, is affected in CHS EBV-B lymphocytes.
Missorting of Lysosomal Resident Membrane Proteins in CHS
Late endosomes receive membrane proteins mainly from two different compartments: early endosomes and the TGN. We reasoned that if transport of resident membrane proteins into multivesicular late endosomes is defective in CHS, an accumulation of molecules normally transported to late endosomes and lysosomes should occur in the compartments from where these proteins reach late endosomes, i.e. early endosomes, plasma membrane, and/or TGN.
No evidence of accumulation of MHC class II or any other lysosomal marker in the Golgi/TGN was observed by electron or confocal microscopy (not shown). In contrast, increased expression of Lamp1 and CD63 at the cell surface was observed in JH (Fig. 8) and EA (not shown) by FACscan analysis. Similar results were obtained with HLA-DM (not shown). In addition, Lamp1, CD63, and HLA-DM were also found to accumulate in transferrin receptor– containing early endosomes, and the plasma membrane by immunoelectron microscopy (not shown). These results are consistent with a defective sorting of membrane molecules from early endosomes and/or the TGN to late endosomes in CHS, causing accumulation of lysosomal resident proteins in early endosomes and the plasma membrane.
Figure 8.

Cell surface accumulation of lysosomal markers in B cells from CHS patients. FACS analysis of MHC class II molecules (A and B), Lamp1 (C and D), and CD63 (E and F) in control PALA cells (A, C, and E) and JH CHS B cells (B, D, and F).
Striking differences were also found in the case of CD-MPR. In normal EBV-transformed B lymphocytes, MPR accumulated in the TGN, and was barely detectable in multivesicular compartments and totally absent from multilaminar compartments (27 and not shown). Surprisingly, in CHS cells, the CD-MPR strongly accumulated in multilaminar macrolysosomes (Fig. 9 A), but was not detected in multivesicular late endosomes, supporting an endosomal sorting defect in CHS.
Fusion of MHC class II-containing Compartments with the Plasma Membrane
The results shown thus far may account for the delay in MHC class II maturation and peptide loading observed in CHS. However, our biochemical analysis showed that transport of mature MHC class II to the cell surface is also delayed in CHS. The pathways of MHC class II transport to the cell surface are still unclear. Direct fusion of multivesicular MHC class II–containing compartments and the plasma membrane results in both delivery of the proteins of the external membrane of the endosome to the cell surface and secretion of the internal vesicles into the extracellular medium. In addition, it has recently been proposed that a defect in fusion of cytolytic granules with the plasma membrane may occur in cytotoxic T lymphocytes. Therefore, we next tested the possibility that fusion of multivesicular endosomes with the cell surface is deficient in CHS.
In normal EBV-transformed B cells, fusion of multivesicular MHC class II–containing compartments and the cell surface were often observed (Fig. 9 C). The fusion profiles contained previously internalized BSA gold, and stained abundantly for MHC class II and different lysosomal markers (including CD63 and CD82, not shown). In CHS cells, fusion of multivesicular BSA-G–containing compartments with the cell surface was also observed with a frequency similar to that of normal cells (Fig. 9 B and not shown). In contrast to normal cells, the vesicles secreted in the extracellular medium in CHS cells did not label for MHC class II (Fig. 9 B) or CD63 (not shown), consistent with the observation that little MHC class II and CD63 was found in multivesicular late endosomes in these cells. Occasionally, we also observed fusion of enlarged MHC class II–positive multilaminar lysosomes with the plasma membrane (not shown). In normal EBV-transformed B lymphocytes, fusion of multilaminar compartments with the plasma membrane was not observed. These results indicate that direct fusion of endosomal multivesicular compartments with the plasma membrane is not deficient in CHS EBV-transformed B cells.
Subcellular Distribution of LYST
The defect in transport of membrane proteins into multivesicular late endosomes and the resulting missorting of endosomal resident proteins that we have observed in CHS may represent a direct or an indirect consequence of the mutation in the Lyst gene. To analyze the intracellular distribution of LYST, we raised a rabbit antiserum against a peptide from the COOH-terminal region of LYST. Specific antibodies were affinity-purified. These antibodies precipitated a 400-kD protein, consistent with the expected molecular weight of LYST (not shown).
The intracellular distribution of LYST was analyzed in HeLa cells by immunofluorescence and confocal microscopy. The affinity-purified antibody decorated small punctated structures in the cytoplasm of HeLa cells (Fig. 10 A, left). A subset (∼50%) of these structures was aligned along tubular processes that labeled for α-tubulin with specific antibodies (Fig. 10 A, right). At higher magnifications in regions where microtubules were less concentrated, alignment of the LYST-positive structures along individual microtubules were observed (Fig. 10 A, bottom, arrows). The staining with the anti-LYST antibodies was abolished by the presence of 10 μM of the LYST peptide (not shown). In addition, antibodies directed against a peptide from the NH2 terminus of LYST also decorated structures aligned along microtubules (F. Barrat and G. De Saint-Basile, not shown), indicating that this staining is not due to a cross-reaction of our antipeptide antibodies with an unrelated protein.
Figure 10.
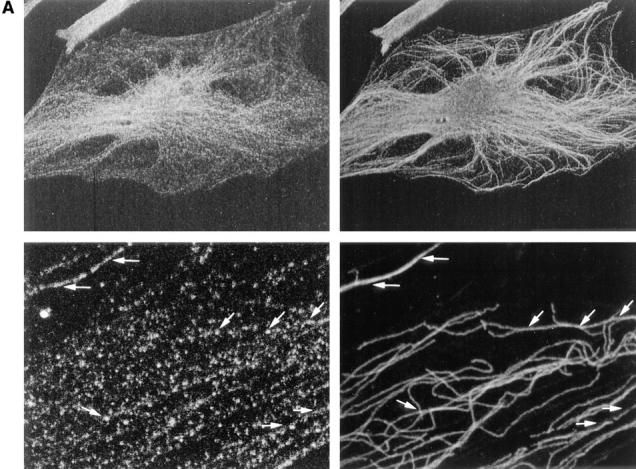
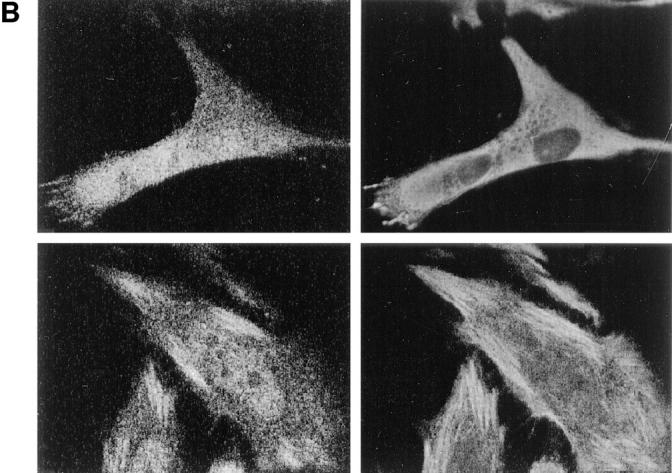
The rabbit anti-LYST antibodies decorate structures aligned along microtubules in fibroblasts (Hela cells). (A) Cells were double-labeled with rabbit anti-LYST affinity-purified antibodies (left) and monoclonal anti-α tubulin antibodies (right). Higher magnification in lower panels shows alignment of the LYST and microtubules (arrows). (B) Cells were treated with either nocodazole (2 h at final concentrations of 10 mM; top) or taxol (0.1 mM for 5 h; bottom) and stained for either LYST (left) or microtubules (right). Nocodazole treatement disorganizes the structures labeled by both antimicrotubule antibodies and LYST. LYST is reorganized together with microtubules upon taxol treatment.
LYST interaction with microtubules was confirmed by treatment of the cells with nocodazole, which resulted in microtubule dispersion (Fig. 10 B, top right), and of the LYST-containing structures (Fig. 10 B, top left). Similar results were found in EBV-B lymphocytes (not shown). In addition, treatment of the cells with taxol, which stimulates microtubule polymerization, resulted in a strong redistribution of both the anti-tubulin and anti-LYST stainings (Fig. 10 B, bottom). These results indicate that the LYST protein is found in punctated structures that interact with microtubules.
Discussion
Although mutations in the lyst gene (in cells from CHS patients) results in dramatic and selective alterations of lysosomal morphology and function, the precise function of LYST is still unclear. Importantly, other membrane compartments such as early endosomes, the ER, or the Golgi apparatus, are not affected in CHS, suggesting that LYST function is specifically related to late endosomes and lysosomes. The only endocytic function that was known to be deficient in CHS hematopoietic cells thus far was cell- mediated cytotoxicity. We now document that another important endocytic function in hematopoietic cells, i.e., antigen presentation, is also affected. Our results on endosomal sorting in late endosomes and the localization of LYST to microtubule-associated structures, suggest a novel role for the lyst gene product.
Endosomal Sorting Defect in CHS
The functional organization of the endocytic pathway in EBV-B cells has been extensively analyzed (35, 49, 69, 18, 2). The first compartment where internalized membrane proteins and fluid are delivered is a heterogenous population of tubovesicular structures present in the cell periphery, most often called early or sorting endosomes. Most of the molecular sorting between the molecules that recycle back to the plasma membrane and those that are delivered to late endosomes and lysosomes occurs within early endosomes. Late endosomes most likely form by fission of a large portion of early endosomes (endocytic carrier vesicles), which then mature into and/or fuse with multivesicular late endosomes. Multivesicular endosomes mature by extensive exchange of membrane material with the TGN. Mature multivesicular late endosomes eventually fuse with preexisting multilaminar lysosomes, forming mixed compartments that may then again mature to become multilaminar lysosomes.
It has been previously suggested that LYST may play a role in endosomal sorting (70, 17). However, our results first characterize a transport defect outside CHS macrolysosomes in late endosomal compartments. The absence of resident endosomal proteins in late multivesicular endosomes, as well as mislocalization of several of these proteins to either early endosomes/plasma membrane or lysosomes, suggests a defect in sorting of membrane proteins in late endocytic compartments. We would like to propose that the CHS defects in lysosomal shape and functions are an indirect consequence of downstream sorting defects of integral membrane proteins in late endosomes. Although the molecules controlling lysosomal homeostasis (shape, size, function, dynamics, etc.) are largely unknown, most likely some of them are integral membrane proteins, whose missorting would result in the morphological and functional lysosomal defects characteristic of CHS.
This defect, however, does not affect delivery of internalized fluid phase markers to late endosomes. Indeed, CHS cells internalize and degrade α2-macroglobulin normally (17), which is consistent with our findings with BSA-G, since α2-macroglobulin dissociates from its receptor in early endosomes and reaches late endosomes and lysosomes by fluid phase. These results are not surprising, since fluid phase marker delivery to late endosomes and lysosomes is thought to occur by default in the absence of active sorting. The observation that cross-linked mIg also reaches multivesicular endosomes (not shown) indicates that transport from early to late endosomes is not completely abolished in CHS. One important difference between mIg and the endosomal resident proteins that we have shown to be absent from late endosomes in CHS is that only cross-linked mIg must transit via early endosomes and not via the TGN. Thus, the transport step defective in CHS might be from TGN to late endosomes, and not from early to late endosomes. However, mlg are only transported to lysosomes upon cross-linking, which activates protein tyrosine kinases (PTKs). Sorting of mlg into late endosomes may therefore require a different cytosolic sorting machinery than other constitutive resident endosomal proteins (which do not require cross-linking or activate PTKs). Therefore, our results do not us allow to distinguish between an early-to-late endosome or a TGN-to-late endosome defect in CHS.
Accumulation of lysosomal markers in the macrolysosomes of CHS B cells, despite the apparent defect in transport to late endosomes, may be accounted for in different ways. It is possible that reduced levels of protein transport through late endosomes are sufficient to allow accumulation in lysosomes, which represent a terminal compartment from where recycling back to the cell surface is slow and inefficient. Alternatively, other transport pathways from the TGN to lysosomes may also exist as described in yeast, which may not involve late endosomes and could be intact in CHS (55). The presence of the CD-MPR in macrolysosomes may also be accounted for by missorting at the TGN, and direct transport through a putative direct TGN–lysosome pathway. The normal rates of Ii chain degradation, too, may result from direct transport from the TGN to lysosomes. However, it is also possible that the sorting defects in CHS result in accumulation of cathepsins (maybe cathepsin S, which is thought to be involved in Ii degradation in early endosomes (59) and Ii degradation therein.
The Lyst Gene Product
The recent cloning of the lyst gene has revealed several interesting features of LYST protein structure. The lyst gene includes six exons, and encodes a large cytosolic protein. Two major mRNA species of ∼6 and 13 kb were found by Northern blot. The expected sizes of the Lyst gene products are ∼175 and 425 kD (8, 9). A functional domain of 345 amino acid residues called BEACH, highly conserved between the human and murine putative proteins, is found in the COOH-terminal region of LYST. The LYST protein also displays several HEAT and ARM repeats that are present in various cytosolic proteins involved in membrane transport (5). In the COOH-terminal region of LYST, there is a cluster of seven WD40 motifs that is involved in protein–protein interactions. This series of HEAT/ARM and WD40 repeats in the COOH-terminal domain of LYST is very similar to the structural organization of VPS15, a regulator of the yeast PI3K (VPS 34; 66). The human homologue of VPS15, a p150, has recently been cloned and also presents homology to LYST COOH-terminal domain (47). However, in contrast to VPS15 and p150, LYST sequence does not suggest the presence of a kinase domain.
The Vps15 mutant presents a defect in sorting to the vacuole, causing secretion of vacuolar hydrolases. In yeast and mammalian cells, PI3K is involved in endosomal transport and sorting into late endosomes and lysosomes (66, 21). This fact has been clearly demonstrated for the PDGFR whose lysosomal transport is inhibited by mutations that prevent PI3K activation (34). Furthermore, wortmannin, a PI3K inhibitor, inhibits transport of membrane proteins from endosomes to lysosomes (16). In addition, reminiscent of the phenotype of CHS cells, wortmannin caused accumulation of MPRs in swollen lysosomal compartments in normal fibroblasts (20). The effect of PI3K inhibitors on lysosomal transport in mammalian cells and mutations in yeast, as well as the structural homology of the lyst gene product and a regulatory subunit of the PI3K, and the sorting defects in CHS cells suggest a functional analogy in LYST and the PI3K membrane transport involvement. The role of the PI3K in sorting endosomal proteins is consistent with our findings and the hypothesis that LYST is involved in protein sorting into late endosomes.
How is LYST Involved in Intracellular Transport?
A variety of different mechanisms have been proposed to account for the CHS defect, including defects in lysosomal fusion and fission, microtubules, and cytoskeleton. Although a defect in the cytolytic granule fusion with the cell surface was thought to occur in T-lymphocytes (6), the evidence for it was indirect. Arguing against a defect in fusion of endocytic compartments with the cell surface is the observation that mast cells from beige mice do degranulate (32). In addition, Ca++-dependent secretion of lysosomal enzymes occurs in fibroblasts (A. Rodriguez, personal communication) from CHS patients. Our observation of normal frequencies of fusion profiles of multivesicular late endosomes and plasma membrane in CHS EBV-B cells also supports the idea that the actual fusion of endocytic compartments and the plasma membrane is not deficient in CHS. This observation is also consistent with our proposal that CHS cells present a membrane- sorting defect in endosomes rather than a fusion defect, as previously envisioned. The formation of enlarged macrolysosomes would then represent an indirect consequence of endosomal mis-sorting of membrane proteins involved in the control of lysosomal morphology.
How then could the lyst gene product be involved in membrane traffic? Our observations that antibodies to the LYST protein decorate structures that partially codistribute with microtubules, and that are sensitive to nocodazole, strongly suggest a microtubule-related function for this protein. The presence of a domain homologous to stathmin in the NH2-terminal domain of LYST could be relevant from this point of view (45, 9). However, this homology is low, and its significance is still unclear. In addition, it was previously shown that CHS fibroblasts and macrophages have normal microtubules (23), and that lysosomal movement along microtubules is also normal in CHS (48). However, a difference in microtubule growth after stimulation was found in neutrophils from at least certain CHS patients (56). Microtubules are directly involved in different membrane transport steps, including transport from early to late endosomes (19). Since we have shown that transport into late endosomes is affected in CHS, it is tempting to speculate that LYST is involved in microtubule-dependent membrane traffic between early and late endosomes. However, a microtubule-dependent transport step in the TGN has also been described (19), and a defect at this level could also account for the transport defect in CHS cells.
Antigen Presentation and MHC Class II Transport in CHS
Our results identify two steps in MHC class II intracellular traffic that are affected in CHS: first, a delay in maturation and peptide loading, and second, a delay in transport from the endocytic pathway to the cell surface. The overall delay in antigen presentation to helper T lymphocytes might be related to this modification of MHC class II traffic. It is also possible that other defects related to antigen internalization and/or processing are also deficient in CHS, and may contribute to the antigen presentation defect that we describe here. However, antigen presentation was only delayed, and not abolished in CHS. Normal levels of SDS-stable dimers were consistently detected at steady state. For these reasons, it is unlikely that this defect in antigen presentation participates significantly in the immunodeficiency in CHS patients, which is more readily accounted for by defects in cell-mediated cytotoxicity.
It was striking, however, to observe that the repertoire of peptides bound to MHC class II are overall shorter by one to two amino acids (the repertoire of peptides eluted may not be compared, since JH cells are DR3/DR11 and COX cells DR3/DR3). Given that MHC class II intracellular transport is significantly slower than that of normal cells, peptides loaded onto MHC class II peptides might be exposed to putative exopeptidases for longer periods of time, which may result in more severe trimming of the NH2-and/or COOH-terminal residues. The reduced levels of MHC class II found in multivesicular late endosomes in CHS, however, suggest that most peptide loading occurs in enlarged macrolysosomes in CHS EBV-transformed B cells. If this was indeed the case, the shorter mean length of MHC class II-associated peptides in CHS cells may reflect a difference in the peptides generated in multivesicular late endosomes vs. multilaminar lysosomes. In any case, this difference in the length of the loaded peptides in CHS could influence the repertoire of T cell receptors selected during thymic T cell maturation.
Whatever the molecular mechanism of the CHS defect will turn out to be, it is striking and informative that defects in late endosomal and lysosomal compartments in all cells of the organism primarily affect important biological functions in hematopoietic cells. The resulting immunodeficiency in CHS stresses the critical role of late endocytic compartments in the systems of immune surveillance and defense against infectious pathogens.
Acknowledgments
We would like to thank Roy Goldsteyn for critically reading the manuscript and Evelyne Coudrier, Michel Bornens, and Manfred Schliwa for helpful advice.
Abbreviations used in this paper
- BSA-G
colloidal gold–coupled bovine serum albumin
- CD-MPR
cation-dependent mannose-6-phosphate receptor
- CHS
Chediak-Higashi syndrome
- DAMP
N-3-(2,4-dinitroanilino)- 3-amino-N-methyldipropylamine
- EBV-B cell
Epstein Barr virus–transformed B cell
- HA
hemagglutinin
- Ii
invariant chain
- LYST
lysosomal trafficking regulator
- MHC
major histocompatibility complex
- MIIC
MHC class II compartment
- mIg
membrane immunoglobulins
- PI3K
phosphatidylinositol-3 kinase
Footnotes
W. Faigle is supported by the INSERM. This work was supported by grants from the Curie Institute, the Institut National de la Santé et Recherche Medicale, the Centre National de la Recherche Scientifique, Vaincre les Maladies Lysosomales, Ligue Nationale Contre le Cancer, and Association pour la Recherche Contre le Cancer.
W. Faigle and G. Raposo contributed equally to this work.
Address all correspondence to S. Amigorena, CJF 95-01 INSERM, Institut CURIE, Section Recherche, 12 Rue Lhomond, 75005, Paris, France. Tel.: 33-1-4234-6389; Fax: 33-1-4234-6382; E-mail: s.amigorena@curie.fr
References
- 1.Adams TE, Bodmer JG, Bodmer WF. Production and characterization of monoclonal antibodies recognizing the alpha-chain subunits of human ia alloantigens. Immunology. 1983;50:613–624. [PMC free article] [PubMed] [Google Scholar]
- 2.Amigorena S, Drake JR, Webster P, Mellman I. Transient accumulation of new class II MHC molecules in a novel endocytic compartment in B lymphocytes (see comments) Nature. 1994;369:113–120. doi: 10.1038/369113a0. [DOI] [PubMed] [Google Scholar]
- 3.Amigorena S, Lankar D, Briken V, Laurent G, Viguier M, Bonnerot C. Type II and III receptors for immunoglobulin G (IgG) control the presentation of different T cell epitopes from single IgG-complexed antigens. J Exp Med. 1998;187:505–515. doi: 10.1084/jem.187.4.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amigorena S, Webster P, Drake J, Newcomb J, Cresswell P, Mellman I. Invariant chain cleavage and peptide loading in major histocompatibility complex class II vesicles. J Exp Med. 1995;181:1729–1741. doi: 10.1084/jem.181.5.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andrade MA, Bork P. HEAT repeats in the Huntington's disease protein. Nat Genet. 1995;11:115–116. doi: 10.1038/ng1095-115. [DOI] [PubMed] [Google Scholar]
- 6.Baetz K, Isaaz S, Griffiths GM. Loss of cytotoxic T lymphocyte function in Chediak-Higashi syndrome arises from a secretory defect that prevents lytic granule exocytosis. J Immunol. 1995;154:6122–6131. [PubMed] [Google Scholar]
- 7.Bakke O, Dobberstein B. MHC class II-associated invariant chain contains a sorting signal for endosomal compartments. Cell. 1990;63:707–716. doi: 10.1016/0092-8674(90)90137-4. [DOI] [PubMed] [Google Scholar]
- 8.Barbosa MD, Barrat FJ, Tchernev VT, Nguyen QA, Mishra VS, Colman SD, Pastural E, Dufourcq-Lagelouse R, Fischer A, Holcombe RF, et al. Identification of mutations in two major mRNA isoforms of the Chediak-Higashi syndrome gene in human and mouse. Hum Mol Genet. 1997;6:1091–1098. doi: 10.1093/hmg/6.7.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbosa MD, Nguyen QA, Tchernev VT, Ashley JA, Detter JC, Blaydes SM, Brandt SJ, Chotai D, Hodgman C, Solari RC, et al. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature. 1996;382:262–265. doi: 10.1038/382262a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barrat FJ, Auloge L, Pastural E, Lagelouse RD, Vilmer E, Cant AJ, Weissenbach J, Le Paslier D, Fischer A, de Saint G, Basile Genetic and physical mapping of the Chediak-Higashi syndrome on chromosome 1q42-43. Am J Hum Genet. 1996;59:625–632. [PMC free article] [PubMed] [Google Scholar]
- 11.Benaroch P, Yilla M, Raposo G, Ito K, Miwa K, Geuze HJ, Ploegh HL. How MHC class II molecules reach the endocytic pathway. EMBO (Eur Mol Biol Organ) J. 1995;14:37–49. doi: 10.1002/j.1460-2075.1995.tb06973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blum JS, Cresswell P. Role for intracellular proteases in the processing and transport of class II HLA antigens. Proc Natl Acad Sci USA. 1988;85:3975–3979. doi: 10.1073/pnas.85.11.3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonnerot C, Lankar D, Hanau D, Spehner D, Davoust J, Salamero J, Fridman WD. Role of B cell receptor Igα and Igβ subunits in MHC class II restricted antigen presentation. Immunity. 1995;3:335–347. doi: 10.1016/1074-7613(95)90118-3. [DOI] [PubMed] [Google Scholar]
- 14.Brachet V, Raposo G, Amigorena S, Mellman I. Ii chain controls the transport of major histocompatibility complex class II molecules to and from lysosomes. J Cell Biol. 1997;137:51–65. doi: 10.1083/jcb.137.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brandt EJ, Elliott RW, Swank RT. Defective lysosomal enzyme secretion in kidneys of Chediak-Higashi (Beige) mice. JCell Biol. 1975;67:774–788. doi: 10.1083/jcb.67.3.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown WJ, DeWald DB, Emr SD, Plutner H, Balch WE. Role for phophatidylinositol 3-kinase in the sorting and transport of newly synthesized lysosomal enzymes in mammalian cells. JCell Biol. 1995;130:781–796. doi: 10.1083/jcb.130.4.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burkhardt JK, Wiebel FA, Hester S, Argon Y. The giant organelles in Beige and Chediak-Higashi fibroblasts are derived from late endosomes and mature lysosomes. J Exp Med. 1993;178:1845–1856. doi: 10.1084/jem.178.6.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castellino F, Germain RN. Extensive trafficking of MHC class II-invariant chain complexes in the endocytic pathway and appearance of peptide-loaded class II in multiple compartments. Immunity. 1995;2:73–88. doi: 10.1016/1074-7613(95)90080-2. [DOI] [PubMed] [Google Scholar]
- 19.Cole NB, Lippincott-Schwartz J. Organization of organelles and membrane traffic by microtubules. Curr Opin Cell Biol. 1995;7:55–64. doi: 10.1016/0955-0674(95)80045-x. [DOI] [PubMed] [Google Scholar]
- 20.Davidson HW. Wortmannin causes mistargeting of procathepsin D. Evidence for the involvement of a phosphatidylinositol 3-kinase in vesicular transport to lysosomes. J Cell Biol. 1995;130:797–805. doi: 10.1083/jcb.130.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Camilli P, Emr SD, McPherson PS, Novick P. Phosphoinositides as regulators in membrane traffic. Science. 1996;271:1533–1538. doi: 10.1126/science.271.5255.1533. [DOI] [PubMed] [Google Scholar]
- 22.Denzin LK, Cresswell P. HLA-DM induces CLIP dissociation from MHC class II alpha beta dimers and facilitates peptide loading. Cell. 1995;82:155–165. doi: 10.1016/0092-8674(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 23.Frankel FR, Tucker RW, Bruce J, Stenberg R. Fibroblasts and nacrophages of mice with the Chediak-Higashi-like syndrome have microtubules and actin cables. J Cell Biol. 1978;79:401–408. doi: 10.1083/jcb.79.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76:287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 25.Germain RN, Hendrix LR. MHC class II structure, occupancy and surface expression determined by post-endoplasmic reticulum antigen binding. Nature. 1991;353:134–139. doi: 10.1038/353134a0. [DOI] [PubMed] [Google Scholar]
- 26.Glickman JN, Morton PA, Slot JW, Kornfeld S, Geuze HJ. The biogenesis of the MHC class II compartment in human I-cell disease B lymphoblasts. J Cell Biol. 1996;132:769–785. doi: 10.1083/jcb.132.5.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griffiths G, Hoflack B, Simons K, Mellman I, Kornfeld S. The mannose6 phosphate receptor and the biogenesis of lysosomes. Cell. 1988;52:329–341. doi: 10.1016/s0092-8674(88)80026-6. [DOI] [PubMed] [Google Scholar]
- 28.Guy K, Van Heyningen V, Cohen BB, Deane DL, Steel CM. Differential expression and serologically distinct subpopulations of human Ia antigens detected with monoclonal antibodies to Ia alpha and beta chains. Eur J Immunol. 1982;12:942–948. doi: 10.1002/eji.1830121109. [DOI] [PubMed] [Google Scholar]
- 29.Haliotis T, Roder J, Klein J, Ortaldo J, Fauci AS, Herbermann RB. Chediak-Higashi gene in humans. I. Impairment of Natural-Killer function. J Exp Med. 1980;151:1039–1048. doi: 10.1084/jem.151.5.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harding CV, Geuze HJ. Antigen processing and intracellular traffic of antigens and MHC molecules. Curr Opin Cell Biol. 1993;5:596–605. doi: 10.1016/0955-0674(93)90128-d. [DOI] [PubMed] [Google Scholar]
- 31.Hewitt CRA, Lamb JR, Haydall J, Hill M, Owen MJ, O'Hehir RE. Major histocompatibility complex independent clonal T cell anergy by direct interaction of Staphylococcus aureus Enterotoxin B with the T cell antigen receptor. J Exp Med. 1992;175:1493–1499. doi: 10.1084/jem.175.6.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jippo-Kanemoto T, Kasugai T, Yamatodani A, Ushio H, Mochizuki T, Tohya K, Kimura M, Nishimura M, Kitamura Y. Supernormal histamine release and normal cytotoxic activity of beige (Chediak-Higashi syndrome) rat mast cells with giant granules (see comments) Int Arch Allergy Immunol. 1993;100:99–106. doi: 10.1159/000236395. [DOI] [PubMed] [Google Scholar]
- 33.Jones PP, Murphy DB, Hewgill D, McDewitt HO. Detection of a common polypeptide chain in I-A and I-E subregion immunoprecipitates. Immunochemistry. 1979;16:51–60. doi: 10.1016/0161-5890(79)90027-0. [DOI] [PubMed] [Google Scholar]
- 34.Kapeller R, Chakrabarti R, Cantley L, Fay F, Corvea S. Internalization of activated platelet-derived growth factor receptor-phosphatidylinositol-3′ kinase complexes: potential interactions with the microtubule cytoskeleton. Mol Cell Biol. 1993;13:6052–6063. doi: 10.1128/mcb.13.10.6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kleijmeer MJ, Morkowski S, Griffith JM, Rudensky AY, Geuze HJ. Major histocompatibility complex class II compartments in human and mouse B Lymphoblasts represent conventional endocytic compartments. J Cell Biol. 1997;139:639–649. doi: 10.1083/jcb.139.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kleijmeer MJ, Ossevoort MA, van Veen CJ, van Hellemond JJ, Neefjes JJ, Kast WM, Melief CJ, Geuze HJ. MHC class II compartments and the kinetics of antigen presentation in activated mouse spleen dendritic cells. J Immunol. 1995;154:5715–5724. [PubMed] [Google Scholar]
- 37.Knol EF, Mul FP, Jansen H, Calafat J, Roos D. Monitoring human basophil activation via CD63 monoclonal antibody 435. J Allergy Clin Immunol. 1991;88:328–338. doi: 10.1016/0091-6749(91)90094-5. [DOI] [PubMed] [Google Scholar]
- 38.Kropshofer H, Vogt AB, Stern LJ, Hammerling GJ. Self- release of CLIP in peptide loading of HLA-DR molecules. Science. 1995;270:1357–1359. doi: 10.1126/science.270.5240.1357. [DOI] [PubMed] [Google Scholar]
- 39.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 40.Liou W, Geuze HJ, Slot JW. Improving structural integrity of cryosections for immunogold labeling. Histochem Cell Biol. 1996;106:41–58. doi: 10.1007/BF02473201. [DOI] [PubMed] [Google Scholar]
- 41.Lotteau V, Teyton L, Peleraux A, Nilsson T, Karlsson L, Schmid S, Quaranta V, Peterson P. Intracellular transport of class II MHC molecules directed by invariant chain. Nature. 1990;348:600–605. doi: 10.1038/348600a0. [DOI] [PubMed] [Google Scholar]
- 42.Markert ML, Cresswell P. Polymorphism of human B-cell alloantigens: evidence for three loci within the HLA system. Proc Natl Acad Sci USA. 1980;77:6101–6104. doi: 10.1073/pnas.77.10.6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mellman I, Pierre P, Amigorena S. Lonely MHC molecules seeking immunogenic peptides for meaningful relationships. Curr Opin Cell Biol. 1996;7:564–572. doi: 10.1016/0955-0674(95)80014-x. [DOI] [PubMed] [Google Scholar]
- 44.Morkowski S, Raposo G, Kleijimeer M, Geuze HJ, Rudensky AY. Assembly of an abundant endogenous major histocompatibility complex class II/peptide complex in class II compartments. Eur J Immunol. 1997;27:609–617. doi: 10.1002/eji.1830270306. [DOI] [PubMed] [Google Scholar]
- 45.Nagle DL, Karim MA, Woolf EA, Holmgren L, Bork P, Misumi DJ, McGrail SH, Dussault BJ, Jr, Perou CM, Boissy RE, et al. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome (see comments) Nat Genet. 1996;14:307–311. doi: 10.1038/ng1196-307. [DOI] [PubMed] [Google Scholar]
- 46.Neer EJ, Schmidt CJ, Nambudripad R, Smith TF. The ancient regulatory-protein family of WD-repeat proteins. Nature. 1994;371:297–300. doi: 10.1038/371297a0. [DOI] [PubMed] [Google Scholar]
- 47.Panaretou C, Domin J, Cockcroft S, Waterfield MD. Characterization of p150, an adaptor protein for the human phosphatidylinositol (PtdIns) 3-kinase. J Biochem Chem. 1997;272:2477–2485. doi: 10.1074/jbc.272.4.2477. [DOI] [PubMed] [Google Scholar]
- 48.Perou CM, Kaplan J. Chediak-Higashi syndrome is not due to a defect in microtubule-based lysosomal mobility. J Cell Sci. 1993;106:99–107. doi: 10.1242/jcs.106.1.99. [DOI] [PubMed] [Google Scholar]
- 49.Peters PJ, Neefjes JJ, Oorschot V, Ploegh HL, Geuze HJ. Segregation of MHC class II molecules from MHC class I molecules in the Golgi complex for transport to lysosomal compartments (see comments) Nature. 1991;349:669–676. doi: 10.1038/349669a0. [DOI] [PubMed] [Google Scholar]
- 50.Peters PJ, Raposo G, Neefjes JJ, Oorschot V, Leijendekker RL, Geuze HJ, Ploegh HL. Major histocompatibility complex class II compartments in human B lymphoblastoid cells are distinct from early endosomes. J Exp Med. 1995;182:325–334. doi: 10.1084/jem.182.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pierre P, Turley SJ, Gatti E, Hull M, Meltzer J, Mirza A, Inaba K, Steinmann RM, Mellmann I. Developmental regulation of MHC II transport on mouse dendritic cells. Nature. 1997;388:787–792. doi: 10.1038/42039. [DOI] [PubMed] [Google Scholar]
- 52.Pieters J, Horstmann H, Bakke O, Griffiths G, Lipp J. Intracellular transport and localization of major histocompatibility complex class II molecules and associated invariant chain. Eur Mol Biol Lab. 1991;115:1213–1223. doi: 10.1083/jcb.115.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pinet V, Malnati MS, Long EO. Two processing pathways for the MHC class II-restricted presentation of exogenous influenza virus antigen. J Immunol. 1994;152:4852–4860. [PubMed] [Google Scholar]
- 54.Pinet V, Vergelli M, Martin R, Bakke O, Long EO. Antigen presentation mediated by recycling of surface HLA-DR molecules. Nature. 1995;375:603–606. doi: 10.1038/375603a0. [DOI] [PubMed] [Google Scholar]
- 55.Piper RC, Bryant NJ, Stevens TH. The membrane protein alkaline phosphatase is delivered to the vacuole by a route that is distinct from the VPS-dependent pathway. J Cell Biol. 1997;138:531–545. doi: 10.1083/jcb.138.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pryzwansky KB, Schliwa M, Boxer LA. Microtubule organization of unstimulated and stimulated adherent human neutrophils in Chediak-Higashi syndrome. Blood. 1985;66:1398–1403. [PubMed] [Google Scholar]
- 57.Raposo, G., M.J. Kleijmeer, G. Posthuma, J.W. Slot, and H.J. Geuze. 1997b. Immunogold labeling of ultrathin cryosections: application in immunology. In Handbook of Experimental Immunology. 5th ed. I. Blackwell Science, editor. Elsevier Trends Journals, Cambridge, MA. 1–11.
- 58.Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, Geuze HJ. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996;183:1161–1172. doi: 10.1084/jem.183.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Riese RJ, Wolf PR, Bromme D, Natkin LR, Villadangos JA, Ploegh HL, Chapman HA. Essential role for cathepsin S in MHC class II-restricted invariant chain processing and peptide loading. Immunity. 1996;4:357–366. doi: 10.1016/s1074-7613(00)80249-6. [DOI] [PubMed] [Google Scholar]
- 60.Roche PA, Cresswell P. Proteolysis of the class II-associated invariant chain generates a peptide binding site in intracellular HLA-DR molecules. Proc Natl Acad Sci USA. 1991;88:3150–3154. doi: 10.1073/pnas.88.8.3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roder JC, Haliotis T, Klein M, Korec S, Jett JR, Ortaldo J, Hebermann RB, Katz P, Fauci AS. A new immunodeficiency disorder in humans involving NK cells. Nature. 1980;284:553–555. doi: 10.1038/284553a0. [DOI] [PubMed] [Google Scholar]
- 62.Sette A, Ceman S, Kubo RT, Sakaguchi K, Appella E, Hunt DF, Davis TA, Michel H, Shabanowitz J, Rudersdorf R, et al. Invariant chain peptides in most HLA-DR molecules of an antigen-processing mutant. Science. 1992;258:1801–1804. doi: 10.1126/science.1465617. [DOI] [PubMed] [Google Scholar]
- 63.Shackelford DA, Lampson LA, Strominger JL. Analysis of HLA-DR antigens by using monoclonal antibodies: recognition of conformational difference in biosynthetic intermediates. J Immunol. 1981;127:1403–1410. [PubMed] [Google Scholar]
- 64.Shaw S, Ziegler A, DeMars R. Specificity of monoclonal antibodies directed against human and murine class II histocompatibility antigens as analyzed by binding to HLA-deletion mutant cell lines. Hum Immunol. 1985;12:191–211. doi: 10.1016/0198-8859(85)90336-2. [DOI] [PubMed] [Google Scholar]
- 65.Sloan VS, Cameron P, Porter G, Gammon M, Amaya M, Mellins E, Zaller DM. Mediation by HLA-DM of dissociation of peptides from HLA-DR. Nature. 1995;375:802–806. doi: 10.1038/375802a0. [DOI] [PubMed] [Google Scholar]
- 66.Stack JH, Herman PK, Schu PV, Emr SD. A membrane- associated complex containing the Vps15 protein kinase and the Vps34 PI 3-kinase is essential for protein sorting to the yeast lysosome like vacuole. EMBO (Eur Mol Biol Organ) J. 1993;12:2195–2204. doi: 10.1002/j.1460-2075.1993.tb05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stam NJ, Spits H, Ploegh HL. Monoclonal antibodies raised against denatured HLA-B locus heavy chains permit biochemical characterization of certain HLA-C locus products. J Immunol. 1986;137:2299–2306. [PubMed] [Google Scholar]
- 68.von Figura K, Gieselmann V, Hasilik A. Antibody to mannose 6-phosphate specific receptor induces receptor deficiency in human fibroblasts. EMBO (Eur Mol Biol Organ) J. 1984;3:1281–1286. doi: 10.1002/j.1460-2075.1984.tb01963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.West MA, Lucocq JM, Watts C. Antigen processing and class II MHC peptide-loading compartments in human B-lymphoblastoid cells [see comments] Nature. 1994;369:147–151. doi: 10.1038/369147a0. [DOI] [PubMed] [Google Scholar]
- 70.White JG. The Chediak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143–156. [PubMed] [Google Scholar]