

Abstract
E1B 19K, the adenovirus Bcl-2 homologue, is a potent inhibitor of apoptosis induced by various stimuli including Fas and tumor necrosis factor-α. Fas and TNFR-1 belong to a family of cytokine-activated receptors that share key components in their signaling pathways, Fas-associating protein with death domain (FADD) and FADD-like interleukin-1β–converting enzyme (FLICE), to induce an apoptotic response. We demonstrate here that E1B 19K and Bcl-xL are able to inhibit apoptosis induced by FADD, but not FLICE. Surprisingly, apoptosis was abrogated by E1B 19K and Bcl-xL when FADD and FLICE were coexpressed. Immunofluorescence studies demonstrated that FADD expression produced large insoluble death effector filaments that may represent oligomerized FADD. E1B 19K expression disrupted FADD filament formation causing FADD and FLICE to relocalize to membrane and cytoskeletal structures where E1B 19K is normally localized. E1B 19K, however, does not detectably bind to FADD, nor does it inhibit FADD and FLICE from being recruited to the death-inducing signaling complex (DISC) when Fas is stimulated. Thus, E1B 19K may inhibit Fas-mediated cell death downstream of FADD recruitment of FLICE but upstream of FLICE activation by disrupting FADD oligomerization and sequestering an essential component of the DISC.
Fas/Apo-1/CD95 (Fas) and tumor necrosis factor receptor-1 (TNFR-1)1 are related receptor molecules that play a critical role in inducing cells to commit apoptosis. Fas plays an integral role in the maintenance of a homeostatic balance within the immune system through the removal of activated and self-reactive T and B cells (for review see Nagata, 1997). Cells undergoing Fas- and TNF-α–mediated apoptosis display classic apoptotic signatures marked by membrane blebbing, chromatin condensation, nuclear degradation, and the formation of apoptotic bodies that are engulfed by neighboring cells (Laster et al., 1988; Itoh et al., 1991).
Fas or Fas ligand deficiency in mice causes lymphoproliferative and autoimmune diseases that resemble human disorders (for review see Nagata, 1997), such as systemic lupus erythematosus (for review see Thompson, 1995). TNF-α has also been implicated in the pathogenesis of several inflammatory, infectious, and autoimmune diseases. It may play a central role in the development of a wide range of diseases including diabetes, rheumatoid arthritis, bowel disease, and multiple sclerosis (Klinkert et al., 1997; Probert et al., 1997).
The cytokines Fas ligand or TNF-α are the critical components necessary for triggering receptor-mediated cell death in vivo. For example, in the immune privilege sites of the eye and testes, constitutive expression of Fas ligand on its cell surface allows these organs to induce apoptosis in activated T cells that express Fas before an inflammatory response can be mounted (Bellgrau et al., 1995; Griffith et al., 1995). This mechanism for evading an immune system attack has also been used advantageously by tumor cells. Fas ligand upregulation in melanoma cells induces Fas-bearing cytotoxic T lymphocytes and natural killer cells to undergo apoptosis (Hahne et al., 1996). Thus, cancer cells have subverted the Fas pathway to defend themselves from immune destruction. As a cellular defense mechanism, cytotoxic T lymphocytes and natural killer cells also express Fas ligand, which contributes to immune surveillance of virally infected cells that are stimulated to undergo apoptosis and eliminated through the Fas pathway (for review see Nagata, 1997). Thus, the Fas and TNF-α apoptosis signaling pathways are required for maintaining homeostasis in the immune system and for defense against viral infection.
As premature death of an infected host cell can compromise virus replication, viruses have adapted mechanisms for escaping apoptosis by expression of antiapoptotic genes. Examples of viral gene products that interfere with Fas and TNFR-1 death signaling include: BHRF1 from Epstein-Barr virus (Henderson et al., 1993; Foghsgaard and Jäättelä, 1997); p35 from Autographa californica nuclear polyhedrosis virus (Clem et al., 1991; Beidler et al., 1995); MC159 and MC160 from Molluscum contagiosum virus (Hu et al., 1997; Thome et al., 1997); E8 from equine herpesvirus 2 (Hu et al. 1997; Thome et al. 1997); and CrmA from the cowpox virus (Ray et al., 1992; Enari et al., 1995; Miura et al., 1995; Tewari and Dixit, 1995). Adenovirus encodes numerous genes whose products block Fas- and TNF-α–induced apoptosis. These genes include E3 14.7, 10.4, and 14.5 gene products (Gooding et al., 1988, 1991a ,b; Shisler et al., 1997), and the E1B 19K gene product (Gooding et al., 1991a ; Hashimoto et al., 1991; White et al., 1992).
In addition to effectively inhibiting Fas- and TNF-α–mediated apoptosis, E1B 19K blocks apoptosis induced by apparently unrelated stimuli. During productive adenovirus infection of human cells, the early gene product E1A stimulates host cell DNA synthesis, thereby causing cells to aberrantly go through the cell cycle (Moran, 1993). In response to cell cycle deregulation, the host cell undergoes apoptosis (White et al., 1991). As a defense mechanism, the E1B 19K protein inhibits this E1A-induced apoptosis and allows assembly of viral progeny to be completed before the cell commits suicide (White et al., 1991; Rao et al., 1992). Bcl-2 is able to functionally complement E1B 19K during a permissive infection of human cells (Chiou et al., 1994b ). In primary baby rat kidney (BRK) cells, E1A also deregulates cell cycle control and induces apoptosis; however, inhibition of apoptosis in this setting by E1B 19K or Bcl-2 results in transformation (Rao et al. 1992; White et al., 1992). The induction of apoptosis by E1A in BRK cells is exclusively dependent on wild-type p53 (Debbas and White, 1993). E1B 19K, Bcl-2, and Bcl-xL share the ability to block this p53-mediated apoptosis (Debbas and White, 1993; Chiou et al., 1994a ; Schott et al., 1995). Although Bcl-2, Bcl-xL, and E1B 19K are all capable of conferring a protective effect against apoptosis, the effectiveness of each varies and may be dependent upon the specific cell type and the death stimuli administered.
In BRK cells, E1A induces accumulation of p53 that transcriptionally upregulates the death-promoting protein Bax, leading to activation of downstream caspases and the final executionary steps in apoptosis (Han et al., 1996a ; Sabbatini et al., 1997). The biochemical mechanism by which E1B 19K and Bcl-2 disable p53-mediated apoptosis is partly a consequence of direct binding to Bax and inhibition of caspase activation (Han et al. 1996a ; Sabbatini et al. 1997). Despite these insights into the mechanism by which E1B 19K inhibits p53-dependent apoptosis, nothing is known about how it functions to block Fas- and TNF-α–mediated apoptosis.
Over the past several years, the mechanism through which Fas mediates cell death is beginning to be elucidated. Upon receptor trimerization with Fas ligand or an anti–APO-1 antibody, Fas is activated and recruits the adaptor molecule Fas-associating protein with death domain (FAAD)/mediator of receptor-induced toxicity (MORT1) (FADD) to its cytoplasmic region via their corresponding conserved death domains (DD) (Boldin et al., 1995; Chinnaiyan et al., 1995; Kischkel et al., 1995). FADD also contains a death effector domain (DED) at its NH2 terminus responsible for the recruitment of FADD-like ICE (interleukin-1β–convertase enzyme) (FLICE)/MORT1-associated Ced-3 homologue (MACH)/caspase-8 (FLICE) via its two DEDs present on its NH2-terminal prodomain (Boldin et al., 1996; Muzio et al., 1996). These protein interactions are required for the formation of the death-inducing signaling complex (DISC) that leads to downstream apoptotic events (Kischkel et al., 1995). At its COOH terminus, FLICE is homologous to a cysteine protease that is activated upon cleavage and is responsible for activation of downstream caspases leading to the amplification of the cell death signal and the final executionary steps in apoptosis. FADD and FLICE are also activated in the TNFR-1 pathway upon interaction with TNF-α (for review see Nagata, 1997). Receptor stimulation allows FADD, and thereby FLICE, to interact with the adaptor molecule TRADD that is recruited to the cytoplasmic region of TNFR-1 causing FLICE activation and apoptosis (Hsu et al., 1996).
Here we investigate the ability of E1B 19K to block apoptosis downstream of Fas. The results implicate FADD multimerization through its DED as a novel mechanism for induction of apoptosis. E1B 19K inhibits FADD-induced cell death coincident with FADD filament disruption and relocalization to membranes and cytoskeletal structures. Recruitment of FLICE by FADD allows E1B 19K to block FLICE-induced apoptosis only in the presence of FADD. Since E1B 19K does not directly associate with FADD or FLICE, we conclude another essential DISC component is mediating this protein interaction. Therefore, we propose E1B 19K abrogates Fas-mediated apoptosis by disrupting FADD multimerization and possibly sequestering an integral protein from the DISC.
Materials and Methods
Antibodies
The following antibodies were used for indirect immunofluorescence, immunoprecipitation, and Western blotting analyses: E1B 19K was visualized with the 2F3 monoclonal antibody and a rabbit polyclonal antibody directed against the E1B 19K protein (Han et al., 1996a ; White and Cipriani, 1990). The monoclonal AU1 antibody directed against an AU1 epitope tag was purchased from Berkeley Antibody Co. (Richmond, CA). The monoclonal HA.11 and the polyclonal HA.11 antibodies directed against the hemagglutinin (HA) epitope tag were also purchased from Berkeley Antibody Co. The monoclonal Myc antibody-1 directed against a Myc epitope tag was purchased from Oncogene Science (Cambridge, MA). The monoclonal antibody directed against actin was purchased from Amersham Corp. (Arlington, IL). The anti-Flag M5 monoclonal antibody was purchased from Scientific Imaging Systems (New Haven, CT). The monoclonal antibody directed against Apo-1/Fas was purchased from Alexis Biochemicals Corp. (San Diego, CA). A polyclonal antibody directed against FADD was a gift of V. Dixit (University of Michigan, Ann Arbor, MI).
Plasmids
pCMV-E1B 19K (White and Cipriani, 1989, 1990) expresses the wild-type adenovirus 2 E1B 19K protein from the cytomegalovirus (CMV) promoter and has been previously characterized. pcDNA3-E1B 19K (Han et al., 1996a ) expresses E1B 19K from a CMV promoter and has been previously described. pCMV-7dl (White et al., 1992) is identical to pCMV-E1B 19K except that it does not express a functional protein due to a frameshift mutation at codon 7 within the coding region of the E1B 19K gene. pCMV-7dl was used as a negative control for E1B 19K function. pcDNA3-AU1-FADD and pcDNA3-AU1-FADD-DN (Muzio et al., 1996) are CMV expression vectors for an NH2-terminal AU1-tagged human FADD and an NH2-terminal AU1-tagged truncated form of FADD lacking 18 NH2-terminal amino acids, respectively. Both expression vectors were generously provided by V. Dixit. pcDNA3-HA-FLICE (Muzio et al., 1997) expresses the human FLICE with an influenza virus hemagglutinin (HA) epitope tag at its COOH terminus and driven by a CMV promoter, and was a gift of V. Dixit. pcDNA3-CrmA (Miura et al., 1993) expresses the cowpox virus ICE inhibitor CrmA from a CMV promoter and was a gift of J. Yuan (Massachusetts General Hospital, Boston, MA). pcDNA3-Flag-Bcl-xL, (Merino et al., 1995) a CMV-driven expression plasmid expressing the human Bcl-xL protein with a Flag-epitope tag at its NH2 terminus was kindly provided by G. Nunez (University of Michigan, Ann Arbor, MI). pcDNA3-Myc-hNbk expresses a human Nbk/Bik protein, with a Myc-epitope tag at its NH2 terminus, from a T7 promoter for in vitro transcription/translation as previously described (Han et al., 1996b ). The pcDNA3-Myc-Bcl-2 plasmid expresses human Bcl-2 under the control of the CMV promoter and was cloned from pGEM2-Bcl-2 (Han et al. 1996a ). A specific 5′ primer was used to engineer a KpnI site in front of the Myc tag and ATG start site. The PCR products were digested with KpnI and XhoI sites and then ligated into pcDNA3. The pCMV-β-galactosidase (β-gal) expression vector expresses β-gal from the CMV promoter and was provided by C. Abate-Shen (Center for Advanced Biotechnology and Medicine and the University of Medicine and Dentistry of New Jersey, Piscataway, NJ).
Indirect Immunofluorescence
HeLa cells were transfected with 30 μg of the pCMV-7dl control vector, 8 μg of pcDNA3-HA-FLICE, 10 μg of pcDNA3-AU1-FADD, 10 μg of pcDNA3-FADD-DN, 12 μg of pCMV-E1B 19K, 12 μg of pcDNA3-Myc-Bcl-2, and 12 μg of pcDNA3-Flag-Bcl-xL expression plasmids. Cells were also transfected with 12 μg of pcDNA3-CrmA expression plasmid to retain cell viability. Cells were fixed at 48 h posttransfection in a 2% paraformaldehyde solution and then permeabilized with PBS containing 0.5% Triton X-100 (Sigma Chemical Co., St. Louis, MO). Cells were double labeled with the indicated antibodies and visualized with goat anti– mouse rhodamine-conjugated and goat anti–rabbit fluorescein-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Photography was performed using a microscope equipped with epifluorescence optics (model FXA; Nikon Inc., Garden City, NY).
Western Blotting
Whole cell extracts were prepared for Western blot analysis 48 h posttransfection. 20 μg of protein from each sample were analyzed by SDS-PAGE and semidry-blotted onto nitrocellulose membranes. Proteins were detected by enhanced chemiluminescence and used according to the manufacturer's specifications (Amersham Corp.).
In Vitro Protein Interaction Assay
Expression vectors encoding E1B 19K (pcDNA3-E1B 19K), FADD (pcDNA3-AU1-FADD), FADD-DN (pcDNA3-AU1-FADD-DN), FLICE (pcDNA3-HA-FLICE), and Nbk/Bik (pcDNA3-Myc-hNbk), were in vitro transcribed and translated according to the manufacturer's specifications (Promega Corp., Madison, WI). The in vitro translated 35S-labeled proteins were titrated to equal concentrations and incubated with an anti-FADD polyclonal antibody or an anti-E1B 19K polyclonal antibody (see figure legend) in the presence of 0.5 ml NETN lysis buffer (20 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.2% NP-40) (Han et al., 1996a ). Protein complexes were collected with protein A–Sepharose (Pharmacia Biotech Inc., Piscataway, NJ) and washed three times with NETN buffer. Proteins were resolved on a 17% SDS-PAGE gel and then subjected to autoradiography.
Immunoprecipitation
HeLa cells were transiently transfected with 3 μg of pcDNA3-HA-FLICE, 3 of μg pcDNA3-AU1-FADD, 12 μg of pCMV-E1B 19K, and 12 μg of pcDNA3-CrmA expression plasmids as indicated in Fig. 8, and then subjected to immunoprecipitation analysis at 48 h posttransfection (Kischkel et al. 1995). Briefly, a 10-cm plate of cells was trypsinized, centrifuged at 1,000 g, and then resuspended in 37°C DME. Fas receptor was stimulated by preincubation with 2 μg/ml of anti–APO-1 antibody for 15 min and then immediately placed on ice. Cells were washed in PBS and lysed in 1 ml of lysis buffer (as stated above) containing 1% Triton X-100 and 10% glycerol for 30 min at 4°C. Lysates were centrifuged and protein A–Sepharose (Pharmacia Biotech Inc.) was added to the supernatant. As a negative control, 2 μg/ml of anti–APO-1 antibody and protein A–Sepharose were added to the lysates of unstimulated cells. Lysates were incubated at 4°C on a rotator (Labquake Shaker model 400110; Barnstead/Thermolyne Inc., Dubuque, IA) for 3 h. The beads were washed five times in lysis buffer, boiled for five minutes in Laemmli buffer (Laemmli, 1970), and then resolved by SDS-PAGE analysis. Immunoprecipitated FADD and FLICE were detected using Western blot analysis by probing with anti-FADD polyclonal and anti-HA polyclonal antibodies, respectively.
Figure 8.
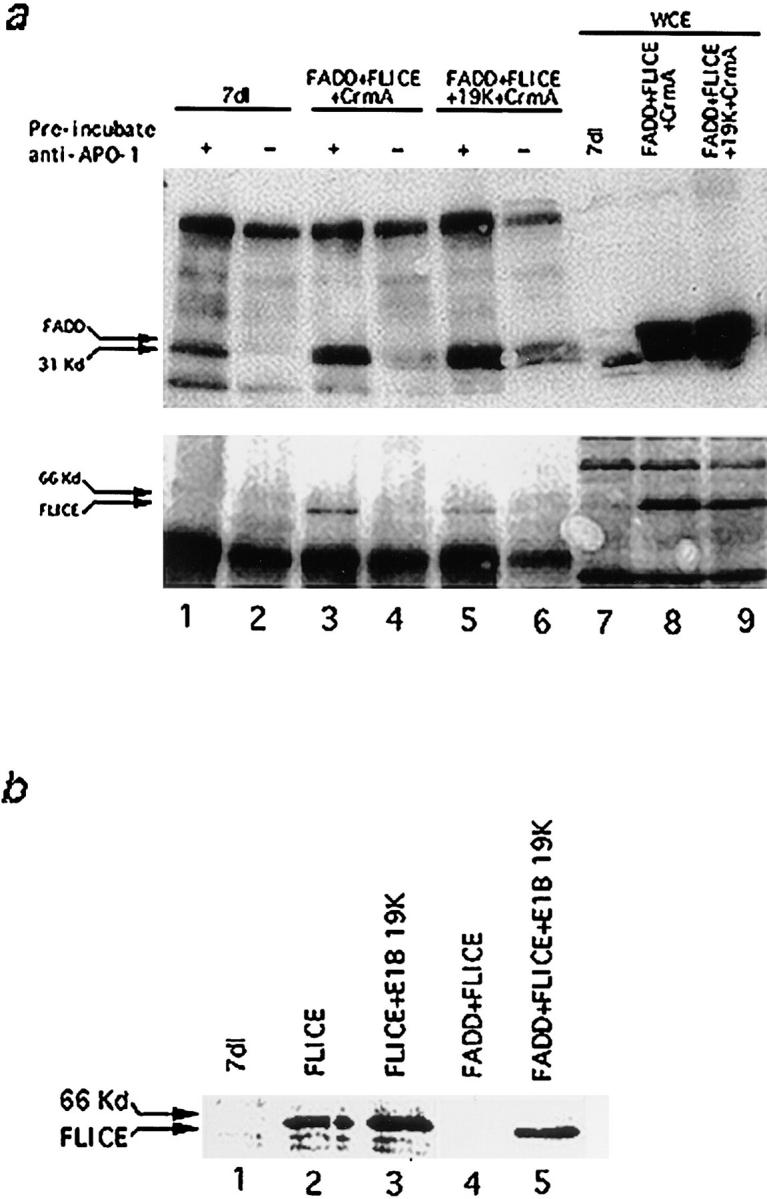
(a) FADD and FLICE coimmunoprecipitate with Fas in the presence of E1B 19K. HeLa cells were transfected with 7dl vector control (lanes 1 and 2), FADD, FLICE, and CrmA (lanes 3 and 4), or FADD, FLICE, E1B 19K, and CrmA (lanes 5 and 6) and then immunoprecipitated with an anti–APO-1 antibody 48 h posttransfection. In lanes 1, 3, and 5, cells were first stimulated with anti–APO-1 antibody, lysed, and then immunoprecipitated with the addition of protein A–Sepharose beads. In lanes 2, 4, and 6, cells were lysed first and then immunoprecipitated with anti–APO-1 antibody. Lanes 7–9 correspond to whole cell extracts used to verify equal protein expression. Samples were resolved on a 15% SDS-PAGE and subjected to Western analysis with an anti-FADD polyclonal antibody (top panel) and an anti-HA polyclonal antibody (bottom panel). (b) E1B 19K blocks upstream of FLICE activation. HeLa cells were transfected with 7dl vector control, FLICE, or FLICE plus FADD alone or in the presence of E1B 19K at concentrations indicated in the viability assay (refer to Materials and Methods). Whole cell extracts were made 48 h posttransfection, resolved by SDS-PAGE, and then immunoblotted with an anti-HA polyclonal antibody.
Viability Assays
Induction of cell death by FADD and its inhibition by Bcl-2 family members was assessed by a β-gal viability assay (Han et al., 1996b ). HeLa cells (6-cm plates) were electroporated, as previously described (Chiou et al., 1994a ), with 18 μg of the pCMV-7dl control vector, 6 μg of pcDNA3-AU1-FADD, 12 μg of pCMV-E1B 19K, 12 μg of pcDNA3-Myc-Bcl-2, and 12 μg of pcDNA3-Flag-Bcl-xL expression plasmids, as indicated in the text. All samples were also transfected with 6 μg of pCMV-β-gal DNA. Total DNA transfected was kept constant at 18 μg. 48 h posttransfection, cells were fixed and the percentage of viable blue cells assessed by 5-bromo-4-chloro-3-indoxyl-β-d-galactopyranoside staining and examined by phase-contrast microscopy as previously described (Han et al., 1996b ).
FLICE functional assays were performed by transfecting HeLa cells with 30 μg of the pCMV-7dl control vector, 8 μg of pcDNA3-HA-FLICE, 10 of μg pcDNA3-AU1-FADD, 12 μg of pCMV-E1B 19K, 12 μg of pcDNA3-Myc-Bcl-2, and 12 μg of pcDNA3-Flag-Bcl-xL expression constructs as indicated in Fig. 1 b. 10 μg of pCMV-β-gal was added to all the samples and the total DNA concentration was kept constant at 30 μg. Viability was assessed as stated above.
Figure 1.
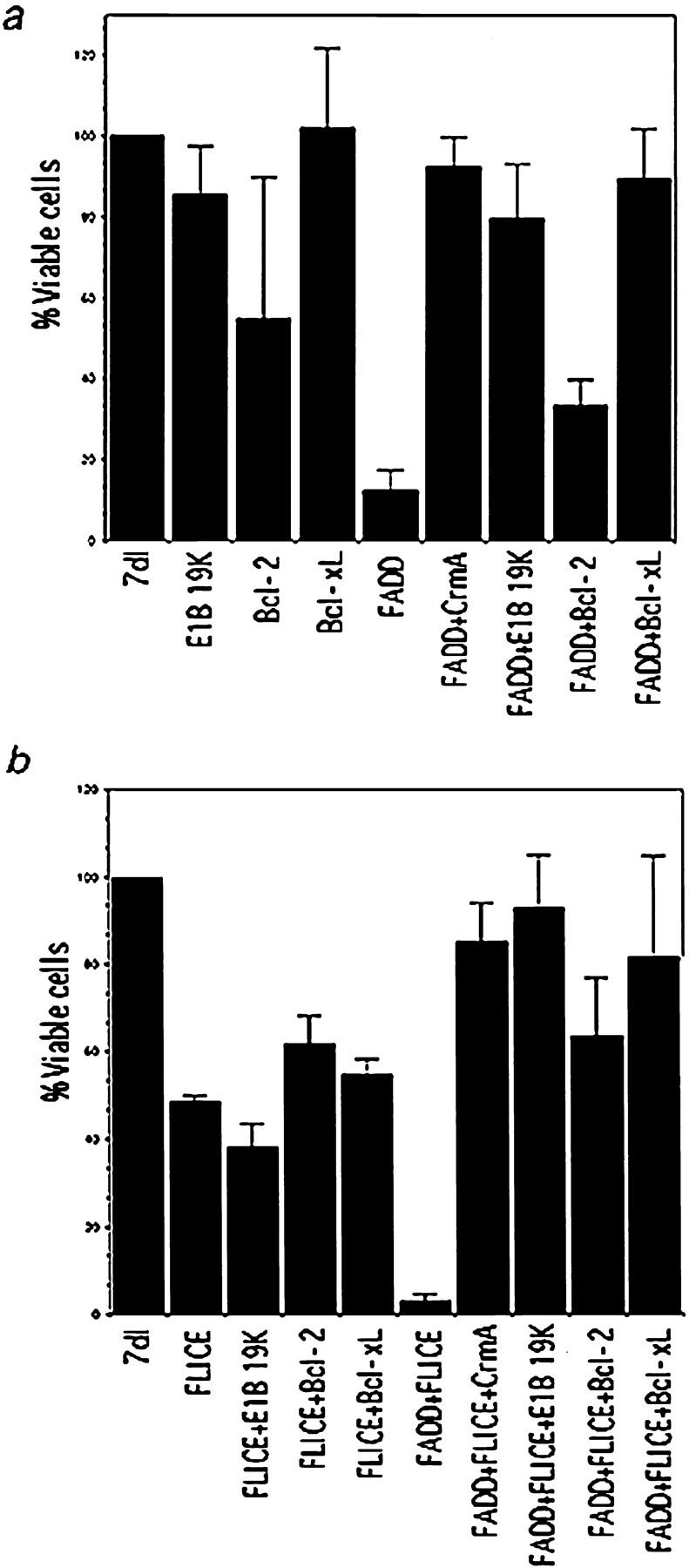
(a) E1B 19K and Bcl-xL abrogate FADD-mediated apoptosis. HeLa cells were transfected with FADD alone or cotransfected with Bcl-2, Bcl-xL, or E1B 19K. Bcl-2, Bcl-xL, and E1B 19K were also each expressed individually as controls. A β-gal viability assay was performed 48 h posttransfection. The percentage of viable cells was compared with 7dl transfected vector control cells. The values represent data from a single experiment that was reproducible in three independent experiments. (b) E1B 19K and Bcl-xL block FLICE-mediated cell death only in the presence of the FADD adaptor molecule. HeLa cells were transfected with FLICE alone or coexpressed with Bcl-2, Bcl-xL, E1B 19K, or FADD. Cells were transfected with FADD and FLICE in the presence of Bcl-2, Bcl-xL, or E1B 19K. Bcl-2, Bcl-xL, and E1B 19K were also each expressed alone as a control. Viability was assessed 48 h posttransfection and compared with the 7dl vector control.
Results
E1B 19K and Bcl-xL Inhibit FADD but Not FLICE-induced Apoptosis
The adenoviral E1B 19K protein is a potent inhibitor of Fas- and TNF-α–induced apoptosis (Gooding et al. 1991a ; Hashimoto et al. 1991; White et al. 1992). Other Bcl-2 family members, however, vary in their ability to block death receptor–mediated apoptosis (Chiou et al. 1994b ; Armstrong et al., 1996; Chinnaiyan et al., 1996a ; Nagata 1997). We sought to identify at which point in the signaling cascade E1B 19K, Bcl-2, and Bcl-xL interfere with apoptosis induction by Fas and TNF-α, and whether they act by a similar mode to inhibit cell death.
E1B 19K, and to a lesser extent Bcl-2, have been shown to abrogate Fas- and TNF-α–induced apoptosis in HeLa cells (Gooding et al., 1991a ; White et al., 1992; Chiou et al., 1994b ; Armstrong et al., 1996). Since E1B 19K does not affect the presence of the TNF receptors at the cell surface of adenovirus infected cells, we have postulated E1B 19K must be acting at, or downstream of, the receptor to block cell death (White et al., 1992). To determine at which point downstream of death receptors the survival signal is exerted, we examined the ability of E1B 19K, Bcl-2, and Bcl-xL to inhibit FADD- and FLICE-induced death.
Transient expression of FADD in HeLa cells caused only 10% of the transfected cells to remain viable compared with transfection with a plasmid control pm7dl (7dl), that does not express a functional protein (Fig. 1 a). CrmA acts as a competitive inhibitor for the ICE-related proteases, and has been shown to abrogate both Fas- and TNF-α–mediated apoptosis (Enari et al., 1995; Miura et al., 1995; Tewari and Dixit, 1995). As a positive control for apoptosis inhibition, FADD was coexpressed with the ICE inhibitor CrmA, which restored cell viability, as expected (Fig. 1 a). FADD was then coexpressed with E1B 19K, to determine if apoptosis could be abrogated (Fig. 1 a). Approximately 90% of the cells survived when E1B 19K protein was coexpressed with FADD. Thus, E1B 19K potently inhibits both Fas- and FADD-induced apoptosis.
Bcl-2 and Bcl-xL were similarly examined for suppression of FADD-induced death. Hela cells were cotransfected with FADD, and either Bcl-2 or Bcl-xL. Although both Bcl-2 and Bcl-xL were highly expressed, Bcl-2 was not able to block FADD-induced death. In fact, overexpression of Bcl-2 in HeLa cells appears to be partially toxic as indicated by the 50% reduction in viability (Fig. 1 a). In contrast, Bcl-xL restored viability to FADD-transfected cells to a similar extent as E1B 19K expression (Fig. 1 a). These results correlate with the ability of E1B 19K, and not Bcl-2, to nearly completely abrogate Fas-induced cell death (Gooding et al., 1991a ; White et al., 1992; Chiou et al., 1994b ). Therefore, we conclude Bcl-xL and E1B 19K, but not Bcl-2, can ablate the apoptotic function of FADD downstream of Fas.
FLICE activation is induced upon its recruitment to the DISC that is mediated through DEDs in the prodomain of FLICE interacting with FADD (Boldin et al., 1996; Muzio et al., 1996). We addressed whether inhibition of cell death was acting at FADD or further downstream at FLICE. FLICE was expressed alone or in the presence of E1B 19K, Bcl-2, and Bcl-xL (Fig. 1 b). FLICE overexpression caused a 50% reduction in cell viability compared with the plasmid control 7dl. Coexpression of E1B 19K, Bcl-2, or Bcl-xL did not significantly increase cell survival upon FLICE overexpression. In conclusion, E1B 19K and Bcl-xL, but not Bcl-2, can inhibit FADD-induced cell death, but neither E1B 19K or Bcl-xL can block cell death upon FLICE overexpression.
E1B 19K and Bcl-xL Inhibit FLICE-induced Apoptosis in the Presence of FADD
As implicated in the FADD and FLICE functional assays, inhibition of apoptosis was upstream of FLICE and thus possibly acting at FADD. To further address where the Bcl-2 family of inhibitors exert their protective effects, FADD and FLICE were coexpressed alone or in the presence of E1B 19K, Bcl-xL, or Bcl-2. When FADD was coexpressed with FLICE, apoptosis was increased significantly compared with FLICE overexpression (Fig. 1 b). E1B 19K expression nearly completely abrogated cell death when FADD and FLICE were coexpressed (Fig. 1 b). This inhibition in cell death by E1B 19K was just as efficient as that of the CrmA control. Thus, E1B 19K, despite being unable to block cell death upon FLICE activation, was able to block FLICE-induced apoptosis in the presence of overexpressed FADD.
Similarly, Bcl-xL and Bcl-2 were coexpressed with FADD and FLICE to determine if they possessed a similar activity to that of the E1B 19K protein. A dramatic increase in viable cell number was observed when Bcl-xL, and less profoundly when Bcl-2, were coexpressed with FADD and FLICE (Fig. 1 b). Taken together, these results suggest that Bcl-2 is not very proficient at blocking Fas-mediated apoptosis, or may be acting differently from E1B 19K and Bcl-xL.
E1B 19K Disrupts FADD Filament Formation
E1B 19K is found to associate with the nuclear lamina as well as the cytoplasmic and nuclear membranes (White et al., 1984; White and Cipriani, 1990). Previous experiments have shown E1B 19K partially colocalizes with Bax in cytoplasmic membranes and inhibits cell death through direct interaction with Bax during p53-mediated apoptosis (Han et al., 1996). To gain insight into the mechanism through which E1B 19K abrogated FADD-induced, as well as FLICE-induced apoptosis in the presence of FADD, indirect immunofluorescence was performed. HeLa cells were transfected with either an AU1-tagged FADD, or an HA-tagged FLICE, alone or in the presence of E1B 19K. Double-label indirect immunofluorescence at 48 h posttransfection was performed to ascertain whether the subcellular localization of FADD or FLICE could be altered in the presence of E1B 19K.
HeLa cells were transfected with HA-tagged FLICE and protein expression was visualized with an anti-HA polyclonal antibody. Cells transiently transfected with FLICE demonstrated a diffuse cytoplasmic and nuclear staining that was not observed in vector control transfected cells (Fig. 2, A and B). Although diffuse staining patterns as we have observed for FLICE are less obvious by indirect immunofluorescence than proteins displaying a discrete localization pattern, FLICE-positive cells were clearly visible. E1B 19K localized to the cytoplasmic and nuclear membranes and the cytoskeletal structures as previously reported (Fig. 2 D) (White and Cipriani, 1989, 1990) Co-expression of E1B 19K with FLICE did not alter the subcellular localization of FLICE, nor was any colocalization of the two proteins observed (Fig. 2, C and D).
Figure 2.


FLICE has a diffuse cytosolic and nuclear staining pattern and E1B 19K does not alter its subcellular localization. HeLa cells were transfected with the expression plasmids indicated and processed for indirect immunofluorescence 48 h posttransfection. HeLa cells were transfected with the 7dl vector control and stained with an anti-HA polyclonal antibody (A). HeLa cells were transfected with HA-tagged FLICE and stained with an anti-HA polyclonal antibody (B). In C and D, cells were transfected with HA-tagged FLICE plus E1B 19K and stained with an anti-HA polyclonal antibody (C) and an antibody directed against E1B 19K (D). Cells were also transfected with CrmA to retain cell viability.
HeLa cells transiently expressing AU1-tagged FADD were stained with an anti-FADD polyclonal or anti-AU1 monoclonal antibodies. The staining pattern of FADD was strikingly different from FLICE, with FADD overexpression causing the formation of large filamentous structures in the nucleus and cytoplasm that react with anti-FADD or anti-FADD epitope tag antibodies (Fig. 3 B). These structures were not present in the 7dl vector control–transfected cells (Fig. 3 A). Yeast two-hybrid experiments have shown FADD self-associates and this oligomerization process may be essential for the induction of cell death (Boldin et al., 1995). This is similar to the manner in which Fas receptor trimerization is required for transduction of the death signal (Kischkel et al., 1995). Thus, filament formation due to FADD overexpression may result from FADD multimerization.
Figure 3.

Overexpression of FADD and not a FADD-DN mutant forms filamentous structures that are disrupted by E1B 19K expression. HeLa cells were transfected with the 7dl vector control and stained with an anti-FADD antibody (A). In B, cells were transfected with AU1-tagged FADD and protein expression detected with an anti-FADD polyclonal antibody. In C and D, HeLa cells coexpressed AU1-tagged FADD and E1B 19K and were stained with an anti-FADD polyclonal antibody (C) and an anti-E1B 19K monoclonal antibody (D). HeLa cells were transfected with 7dl vector control and stained with an anti-AU1 monoclonal antibody (E). Cells were transfected with AU1-tagged FADD-DN alone (F) or in the presence of E1B 19K (G and H). FADD-DN protein expression was visualized with an anti-AU1 monoclonal antibody (F and G) and E1B 19K detected with a polyclonal antibody directed against it (H). All cells were also transfected with CrmA to retain cell viability.
To address whether E1B 19K can alter the subcellular localization of FADD, immunofluorescence studies with E1B 19K coexpression were performed. As shown in Fig. 3 C, coexpression of FADD with E1B 19K dramatically altered the localization pattern of FADD, with complete disruption of the filaments into large aggregates in the cytoplasm. However, FADD filaments within the nucleus did not appear affected as indicated by phase-contrast and electron microscopy (data not shown). E1B 19K does not localize within the nucleus and, therefore, may not be able to disrupt nuclear FADD filaments. Double labeling with an anti-E1B 19K monoclonal antibody and an anti-FADD polyclonal antibody revealed complete colocalization of the two proteins, with FADD displaying the typical localization of the E1B 19K protein (cytoplasmic and nuclear membranes as well as the cytoskeletal structures) (Fig. 3, C and D).
An AU1-tagged FADD-DN, a death effector domain deletion mutant that does not self-associate or induce death, was visualized with an anti-AU1 monoclonal antibody (Boldin et al., 1995; Chinnaiyan et al., 1996b ). Its expression pattern was diffuse within the cytoplasm and nucleus similar to that of FLICE (Fig. 3 F). This result suggests that FADD filament formation, and thus oligomerization, may play an integral role in mediating apoptosis. As seen in Fig. 3, G and H, addition of E1B 19K did not change FADD-DN subcellular localization, suggesting that colocalization of E1B 19K with FADD requires either full-length or the DED of FADD.
FADD Recruits FLICE into Filaments That Are Disrupted by E1B 19K
To ascertain whether FADD could alter the localization of FLICE, Hela cells transiently coexpressing AU1-tagged FADD and HA-tagged FLICE were examined by indirect immunofluorescence with anti-FADD and anti-HA antibodies, respectively. The intracellular staining pattern of FADD remained as large filaments, however, FLICE no longer had a diffuse cytosolic pattern (Fig. 4 A) but was recruited by FADD into the large filamentous structures as evidenced by the colocalization of FADD and FLICE (Fig. 4 B). Thus, FADD was able to alter the localization pattern of FLICE most likely through a direct protein– protein interaction with FLICE. We were, however, not able to detect endogenous FADD or FLICE filaments because antibodies sufficient to detect the endogenous proteins by indirect immunofluorescence were not available.
Figure 4.


E1B 19K disrupts FADD filament formation and colocalizes with FLICE in the presence of the FADD adaptor molecule. HeLa cells were transfected with HA-tagged FLICE or coexpressed with HA-tagged FLICE plus AU1-tagged FADD (A and B, respectively). FLICE was visualized with an anti-HA antibody (A and B). In C–E HeLa cells transiently coexpressed AU1-tagged FADD, HA-tagged FLICE, and E1B 19K. A polyclonal antibody directed against FADD (C), an anti-HA polyclonal antibody directed against the epitope tag on FLICE (D), and monoclonal antibody directed against E1B 19K (E) was used to visualize protein expression. All cells were also transfected with CrmA to retain cell viability.
To test if E1B 19K could alter the localization of both FADD and FLICE, E1B 19K was coexpressed with both, and the transfected cells were examined by indirect immunofluorescence for E1B 19K, FADD, and FLICE. Fig. 4, C–E demonstrates that coexpression of FLICE and FADD in the presence of E1B 19K significantly altered the diffuse cytosolic distribution of FLICE such that FLICE colocalized with both FADD and E1B 19K (Fig. 4 D). FADD directly binds to FLICE and by E1B 19K altering the localization of FADD, E1B 19K apparently alters the localization of FLICE (Fig. 4, C–E). The alteration in localization of FADD and FLICE by E1B 19K suggests that the ability of E1B 19K to protect the cells from apoptosis may be through functional sequestration of both FADD and FLICE.
Bcl-xL Expression Attenuates FADD Filament Formation
To address whether other Bcl-2 homologues may also implement a similar mechanism for inhibition of FADD- induced apoptosis, immunofluorescence studies were executed with Bcl-2 and Bcl-xL. Bcl-2 and Bcl-xL localize at the mitochondrial, endoplasmic reticulum, and nuclear membranes (Figs. 5 B and 6 B) (for review see White, 1996). In HeLa cells, transient FLICE expression remained diffuse throughout the cytoplasm and nucleus even when FLICE was coexpressed with Bcl-2 or Bcl-xL, (Figs. 5 A and 6 A, respectively).
Figure 5.

Bcl-2 does not substantially disrupt FADD filaments. HeLa cells were transfected with the expression plasmids indicated and indirect immunofluorescence was performed 48 h posttransfection. HeLa cells were cotransfected with HA-tagged FLICE and Myc-tagged Bcl-2 and then stained with an anti-HA polyclonal antibody (A) or an anti-Myc monoclonal antibody (B). In C, cells were transfected with AU1-tagged FADD alone and visualized with an anti-FADD polyclonal antibody. In D and E, cells were transfected with AU1-tagged FADD and Myc-tagged Bcl-2 and protein expression detected with an anti-FADD polyclonal antibody (D) and an anti-Myc monoclonal antibody (E). In panels F–H, cells were transfected with AU1-tagged FADD, HA-tagged FLICE, and Myc-tagged Bcl-2. Proteins were visualized with anti-FADD polyclonal antibody (F), anti-HA monoclonal antibody (G), and anti-Myc monoclonal antibody (H). All cells were also transfected with CrmA to retain cell viability.
Figure 6.

Bcl-xL attenuates the formation of FADD filaments. HeLa cells were transfected with the expression plasmids indicated and indirect immunofluorescence was performed 48 h posttransfection. HeLa cells were cotransfected with HA-tagged FLICE and Flag-tagged Bcl-xL and then stained with an anti-HA polyclonal antibody (A) or an anti-Flag monoclonal antibody (B). Cells were transfected with AU1-tagged FADD alone and visualized with an anti-FADD polyclonal antibody (C). In D and E, cells were transfected with AU1-tagged FADD and Flag-tagged Bcl-xL and protein expression was detected with an anti-FADD polyclonal antibody (D) and an anti-Flag monoclonal antibody (E). In panels F–H, cells were transfected with AU1-tagged FADD, HA-tagged FLICE, and Flag-tagged Bcl-xL. Proteins were visualized with anti-FADD polyclonal antibody (F), anti-HA monoclonal antibody (G), and anti-Flag monoclonal antibody (H). All cells were also transfected with CrmA to retain cell viability.
When FADD was coexpressed with Bcl-2, FADD filaments were still apparent but reduced in length or in number (Fig. 5 D) compared with that of FADD overexpression alone (Fig. 5 C). In contrast, overexpression of Bcl-xL substantially attenuated the degree of FADD filament formation. Bcl-xL expression caused FADD filaments to be highly disrupted (Fig. 6 D). Occasionally, FADD-expressing cells displayed filament formation in the presence of Bcl-xL. These cells, however, routinely expressed unusually high levels of Bcl-xL, suggesting a concentration dependence of the ability of Bcl-xL to disrupt FADD filaments. However, in contrast to E1B 19K, Bcl-xL does not colocalize with FADD. These results suggest that Bcl-2 and Bcl-xL have a differential effect on FADD filament formation and that Bcl-xL inhibits Fas-mediated apoptosis differently than E1B 19K.
When HeLa cells were transiently transfected with FADD, FLICE, and either Bcl-2 or Bcl-xL, FLICE remained diffuse throughout the cell (Figs. 5 G and 6 G, respectively). When FADD, FLICE, and Bcl-2 were coexpressed, the appearance of FADD filaments remained consistent with that of FADD and Bcl-2 coexpression alone. Together, these results suggest that FADD failed to recruit FLICE into these filaments in the presence of Bcl-2 (Fig. 5, G and F). Coexpression of Bcl-xL with FADD and FLICE profoundly disrupted FADD filaments in almost all transfected cells (Fig. 6 F). In contrast to the results in E1B 19K–expressing cells, there was no colocalization of FLICE and FADD when Bcl-2 or Bcl-xL was coexpressed (Figs. 5 and 6). Therefore, we conclude, Bcl-xL alters the ability of FADD to polymerize and recruit FLICE, whereas E1B 19K apparently disrupts FADD organization without affecting FLICE recruitment.
E1B 19K Does Not Interact with FADD or FLICE In Vitro
The E1B 19K protein is predominantly insoluble and bound to the membranes and cytoskeleton in adenovirus infected and transformed cells (White et al., 1984; White and Cipriani, 1989, 1990). Alteration in FADD and FLICE subcellular localization by E1B 19K and colocalization with E1B 19K suggested that E1B 19K, FADD, and FLICE may directly associate to form an insoluble protein complex. To examine the ability of E1B 19K to associate with FADD or FLICE in vitro, E1B 19K, FADD, and FLICE were in vitro transcribed/translated and analyzed by coimmunoprecipitation. As expected, an anti-FADD antibody was able to coimmunoprecipitate FADD and FLICE but there was no detectable interaction between FADD-DN and FLICE (Fig. 7, lanes 1 and 2). Interestingly, coincubation of FADD and FLICE caused the appearance of a 21-kD band that was not detectable in individually in vitro–translated FADD or FLICE. Whether this band represents a FADD or FLICE cleavage product remains to be determined. An E1B 19K-specific antibody was not able to coimmunoprecipitate E1B 19K with either FADD or FLICE individually or when cotranslated (Fig. 8, lanes 3–5). The E1B 19K interacting protein Nbk/Bik served as a positive control for the presence of a direct protein–protein interaction with E1B 19K (Fig. 7, lane 6).
Figure 7.

E1B 19K does not associate with FADD or FLICE in vitro. In vitro translated [35S]methionine FADD, FLICE, FADD-DN, E1B 19K, and Nbk/Bik were incubated as indicated and immunoprecipitated with an anti-FADD antibody (lanes 1 and 2) and an anti-E1B 19K antibody (lanes 3–6). Arrows, in vitro translated products.
Yeast two-hybrid analysis has also confirmed no detectable direct association between FADD or the prodomain of FLICE with E1B 19K (data not shown). Thus, we can conclude E1B 19K does not directly interact with FADD or FLICE, or that FADD and/or FLICE require modification that does not take place in vitro or in yeast to interact with E1B 19K.
E1B 19K Does Not Inhibit FADD and FLICE from Being Recruited to the DISC
Coimmunoprecipitation experiments revealed that engagement of Fas receptor with anti–APO-1 antibody recruits FADD and FLICE to the cytoplasmic domain of Fas, and this protein complex collectively forms the DISC (Kischkel et al., 1995). This multiprotein interaction may allow FLICE to undergo autocatalytic activation and to induce apoptosis. To address whether E1B 19K was able to abrogate DISC formation, HeLa cells overexpressing FADD, FLICE, and CrmA, with and without E1B 19K, were subjected to Fas activation using an anti–APO-1 monoclonal antibody (Fig. 8 a). HeLa cells were first preincubated with an anti–APO-1 antibody for 15 min before cell lysis (Fig. 8 a, lanes 1, 3, and 5). Immune complexes were collected with protein A and subjected to immunoblot analysis using an anti-FADD polyclonal antibody. As a negative control for Fas-interacting proteins, the cell lysates were generated without receptor engagement with the addition of an anti–APO-1 antibody after cell lysis (Fig. 8 a, lanes 2, 4, and 6). FADD and FLICE were able to coimmunoprecipitate with Fas even in the presence of E1B 19K (Fig. 8 a, lanes 3 and 5). This data suggests the mechanism for protection by E1B 19K is not solely the sequestration of FADD and FLICE to an insoluble intracellular compartment, since both proteins were able to interact with Fas at the plasma membrane.
E1B 19K Inhibits FLICE Activation
To address whether overexpression of FADD and FLICE autocatalyzes FLICE activation in the presence of E1B 19K, HeLa cells were transiently transfected with FLICE or FADD plus FLICE alone or in the presence of E1B 19K (Fig. 8 b). 7dl vector control was overexpressed as a negative control for FLICE expression. Whole cell lysates were produced 20 (data not shown) and 48 h posttransfection and then probed with an anti-HA polyclonal antibody. Full-length proFLICE appeared as a 66-kD band in cells that overexpressed FLICE or FLICE plus E1B 19K (Fig. 8 b, lanes 2 and 3). Due to ∼40% of the transfected cells remaining viable, the inactive zymogen form of FLICE was still detectable. Unprocessed full-length proFLICE, however, disappeared when FADD and FLICE were coexpressed, indicating that increased processing and activation of FLICE had taken place (Fig. 8 b, lane 4). We were not able to detect the 10-kD COOH-terminal cleavage product, which suggested that the activated forms of cytosolic FLICE may have a short half-life. Coexpression of E1B 19K, FADD, and FLICE inhibited FLICE activation as indicated by the abundance of unprocessed proFLICE (Fig. 8 b, lane 5). These results suggest E1B 19K blocks FLICE-mediated apoptosis, in a FADD-dependent fashion, upstream of FLICE activation.
Discussion
We have established E1B 19K is highly efficient at inhibiting FADD- but not FLICE-induced apoptosis. Coexpression of FADD and FLICE profoundly diminishes cell viability and E1B 19K dramatically rescues cells from this apoptosis. This mechanism for inhibition of cell death may be functionally homologous to the C. elegans model for abrogation of apoptosis. C. elegans encodes three integral genes necessary for the regulation of programmed cell death (Hengartner, 1996). The ced-3 gene encodes an effector protein that is homologous to the interleukin-1β–converting enzyme ICE-like family members (Yuan et al., 1993). Its activation is inhibited by the Ced-9 gene product that is able to sequester Ced-3 through protein–protein interactions (Chinnaiyan et al., 1997a ,b; Seshagiri and Miller, 1997; Spector et al., 1997; Wu et al., 1997). The Ced-3–Ced-9 interaction is mediated by a third bridging protein encoded by the ced-4 gene. This model for protection of cell death can be applied to mammalian cells through protein substitution. Several labs have shown Bcl-xL can inhibit FLICE activation and, subsequently, cell death when in the presence of the Ced-4 gene product (Chinnaiyan et al., 1997b ; Seshagiri and Miller, 1997; Wu et al., 1997).
The adenoviral E1B 19K protein is able to abrogate Fas-mediated apoptosis likely through protein–protein interactions with key DISC components. The FADD adaptor molecule is required for E1B 19K to block FLICE-induced cell death. However, another protein(s), posttranslational modification and/or a conformational change of FADD appears to be required for E1B 19K to interact with FADD and for disruption of FADD filament formation.
Immunofluorescence studies reveal overexpression of FADD causes FADD to form filaments within the cell (refer to Fig. 3 C) (Siegel et al., 1998). Electron microscopy indicates FADD forms higher order filamentous structures arranged in bundles (data not shown). These filaments are localized in the cytosol as well as in the nucleus. Furthermore, they do not associate with cytoskeletal structures such as vimentin (data not shown) or tubulin (Siegel et al., 1998). Full-length FADD is known to self-associate (Boldin et al., 1995), suggesting the filaments result from FADD multimerization.
A FADD-DN expression vector containing a deletion within the death effector domain does not self-associate nor does it induce cell death (Boldin et al., 1995; Chinnaiyan et al., 1996b ). Overexpression of FADD-DN does not cause filament formation but is rather localized diffusely throughout the cell. This result implies that FADD oligomerization may be required for mediating cell death. This is similar to other death domain–containing molecules such as Fas, whereby trimerization is necessary for transduction of the death signal (Kischkel et al., 1995). The DED, but not the DD, of FADD is able to directly bind to full length FADD (Boldin et al., 1995). This suggests the DED is essential for both multimerization and for inducing cell death.
E1B 19K does not alter the subcellular localization of FLICE but is able to strikingly disrupt FADD filament formation and relocalize FADD into the membranous and cytoskeletal elements of the cell where the E1B 19K protein is normally found. The disruption of FADD filaments is concurrent with the ability of E1B 19K to block FADD-induced apoptosis. E1B 19K also blocks FLICE-induced cell death in the presence of FADD but apparently does not block FLICE from associating with FADD in the cytoplasm or the DISC.
Indirect immunofluorescence (refer to Fig. 4) and subcellular fractionation (data not shown) studies demonstrate that E1B 19K is also able to sequester FLICE to an insoluble intracellular compartment only in the presence of FADD. These results further indicate FADD is acting as an adaptor molecule bridging FLICE with E1B 19K. Other viruses such as Molluscum contagiosum and equine herpes-virus 2 are capable of blocking FADD-mediated cell death, but not FLICE-induced apoptosis, through expression of viral inhibitory proteins MC159 and E8, respectively (Hu et al., 1997; Thome et al., 1997). The mechanism used by these viral proteins is by direct interaction with the inducers of cell death, FADD and FLICE, which abrogates formation of a complete DISC and results in inhibition of apoptosis. Therefore, we propose E1B 19K may act analogously and form a probably indirect physical interaction with FADD, thereby causing an alteration in its subcellular localization and ability to oligomerize. Since we have been unable to detect binding of E1B 19K to FADD, another protein, posttranslational modification, and/or a conformational change of FADD may be required for E1B 19K to interact with the FADD–FLICE complex and disrupt its function.
The current model of Fas activation proposes that heterooligomerization of FADD and FLICE at the DISC results in FLICE activation. Coimmunoprecipitation of FADD and FLICE with Fas demonstrates FADD and FLICE are recruited to the DISC upon Fas activation by anti–APO-1 antibody (Kischkel et al., 1995; Muzio et al., 1996). Thus, sequestration of FADD/FLICE into an insoluble intracellular compartment by E1B 19K may abrogate activation of FLICE by preventing recruitment to the receptor complex upon Fas receptor stimulation. However, FADD and FLICE recruitment occurs even in the presence of E1B 19K. Thus, we can conclude FADD and FLICE are present at the DISC, but E1B 19K is still able to abrogate cell death. Recent data has shown that FLICE needs to be clustered for full activation and induction of apoptosis (Muzio et al., 1998). Thus, we propose a mechanism in which FADD needs to be multimerized at the DISC, similar to Fas, to activate FLICE. E1B 19K may sequester a portion of the FADD molecules, thereby depleting the number of FADD molecules recruited to the DISC and subsequently inhibiting its oligomerization and activation of FLICE.
Conflicting data has been reported by many labs as to whether other Bcl-2 family members, particularly Bcl-2 and Bcl-xL, are able to block Fas-mediated apoptosis. Even within the same cell line, Bcl-2 has been shown to be either nonfunctional or act as a potent inhibitor of Fas- mediated death (Armstrong et al., 1996; Chinnaiyan et al., 1996a ). These discrepancies may be due to differences in cell type specificity, strength of the death signal, and levels of expression of the inhibitory proteins. Our results demonstrate Bcl-xL and E1B 19K efficiently block Fas-induced cell death, whereas Bcl-2 functions poorly in the same assay. Bcl-xL inhibits FADD- but not FLICE-induced death and is also able to block FLICE-mediated death in the presence of FADD. These results are coincident with Bcl-xL disrupting the formation of FADD filaments. Bcl-2, however, appears to be only marginally effective in inhibiting Fas-mediated cell death and does not substantially alter FADD filament formation. Recent evidence has shown Bcl-2 is cleaved subsequent to anti-Fas antibody treatment, generating a cleavage product that has proapoptotic activity similar to that of Bax and Bak (Cheng et al., 1997). E1B 19K, however, is not cleaved during the apoptotic process to generate a proapoptotic derivative (Han et al., 1996). The difference between the potent inhibitory activity of E1B 19K, in contrast to the weak effect of Bcl-2, in Fas-mediated apoptosis may reside within this intrinsic difference between E1B 19K and Bcl-2.
Coexpression of FADD and FLICE without E1B 19K caused increased FLICE activation as indicated by the disappearance of full-length FLICE. In contrast, the presence of FLICE in the unprocessed pro form when coexpressed with FADD and E1B 19K, indicates that E1B 19K expression inhibits FLICE activation. This result, however, does not rule out the possibility that undetectable levels of processed FLICE are present.
Bcl-xL appears to affect FADD and FLICE differently than E1B 19K. Rather than diverting FADD and FLICE into large aggregates, Bcl-xL appears to prevent FADD filament formation and FLICE recruitment into the filaments. This may be a mechanism for inhibiting FADD from promoting FLICE activation. However, Bcl-xL has been reported to act downstream of FLICE activation (Boise and Thompson, 1997). Whether dispersed FLICE, in FADD- and Bcl-xL-expressing cells, is activated, has yet to be determined. Nonetheless, E1B 19K appears to function differently than Bcl-xL to inhibit the Fas and TNFR-1 death-signaling pathway by blocking FLICE activation subsequent to FADD recruitment.
Acknowledgments
We wish to thank V. Dixit, G. Nunez, and J. Yuan for expression plasmids and antibodies. We would also like to thank K. Degenhart and G. Kasoff (both from Center for Advanced Biotechnology and Medicine, Piscataway, NJ) for advice and critical reading of the manuscript.
This work has been supported by a grant from the National Institutes of Health (CA53370) to E. White.
Abbreviations used in this paper
- β-gal
β-galactosidase
- BRK
baby rat kidney
- CMV
cytomegalovirus
- DD
death domain
- DED
death effector domain
- DISC
death-inducing signaling complex
- HA
hemagglutinin
- ICE
interleukin-1β–convertase enzyme
- TNFR-1
tumor necrosis factor receptor-1
Footnotes
Address all correspondence to Eileen White, Center for Advanced Biotechnology and Medicine, Rutgers University, 679 Hoes Lane, Piscataway, NJ 08854. Tel.: (732) 235-5329. Fax: (732) 235-5795. E-mail: ewhite@mbcl.rutgers.edu
References
- Armstrong RC, Aja T, Xioang J, Gaur S, Krebs JF, Hoang K, Bai X, Korsmeyer SJ, Karanewsky DS, Fritz LC, Tomaselli KJ. Fas- induced activation of the cell death-related protease CPP32 is inhibited by Bcl-2 and by ICE family protease inhibitors. J Biol Chem. 1996;27128:16850–16855. doi: 10.1074/jbc.271.28.16850. [DOI] [PubMed] [Google Scholar]
- Beidler DR, Tewari M, Friesen PD, Poirier G, Dixit VM. The baculovirus p35 protein inhibits Fas- and tumor necrosis factor-induced apoptosis. J Biol Chem. 1995;270:16526–16528. doi: 10.1074/jbc.270.28.16526. [DOI] [PubMed] [Google Scholar]
- Bellgrau D, Gold D, Selawry H, Moore J, Franzusoff A, Duke RC. A role for CD95 ligand in preventing graft rejection. Nature. 1995;377:630–632. doi: 10.1038/377630a0. [DOI] [PubMed] [Google Scholar]
- Boise LH, Thompson CB. Bcl-xLcan inhibit apoptosis in cells that have undergone Fas-induced protease activation. Proc Natl Acad Sci USA. 1997;94:3759–3764. doi: 10.1073/pnas.94.8.3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldin MP, Varfolomeev EE, Pancer Z, Mett IL, Camonis JH, Wallach D. A novel protein that interacts with the death domain of Fas/ APO1 contains a sequence motif related to the death domain. J Biol Chem. 1995;270:7795–7798. doi: 10.1074/jbc.270.14.7795. [DOI] [PubMed] [Google Scholar]
- Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- Cheng EH-Y, Kirsch DG, Clem RJ, Ravi F, Kastan MB, Bedi A, Ueno K, Hardwick JM. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997;278:1966–1968. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-constraining protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, Orth K, O'Rourke K, Duan H, Poirier GG, Dixit VM. Molecular ordering of the cell death pathway. J Biol Chem. 1996a;271:4573–4576. doi: 10.1074/jbc.271.9.4573. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, Tepper CG, Seldin MF, O'Rourke K, Kischkel FC, Hellbardt S, Krammer PH, Peter ME, Dixit VM. FADD/ MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J Biol Chem. 1996b;271:4961–4965. doi: 10.1074/jbc.271.9.4961. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, Chaudhary D, O'Rourke K, Koonin EV, Dixit VM. Role of CED-4 in the activation of CED-3. Nature. 1997a;388:728–729. doi: 10.1038/41913. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, O'Rourke K, Lane BR, Dixit VM. Interaction of CED-4 with CED-3 and CED-9: a molecular framework for cell death. Science. 1997b;275:1122–1126. doi: 10.1126/science.275.5303.1122. [DOI] [PubMed] [Google Scholar]
- Chiou S-K, Rao L, White E. Bcl-2 blocks p53-dependent apoptosis. Mol Cell Biol. 1994a;14:2556–2563. doi: 10.1128/mcb.14.4.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou S-K, Tseng CC, Rao L, White E. Functional complementation of the adenovirus E1B 19K protein with Bcl-2 in the inhibition of apoptosis in infected cells. J Virol. 1994b;68:6553–6566. doi: 10.1128/jvi.68.10.6553-6566.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RJ, Fechheimer M, Miller LK. Prevention of apoptosis by a Baculovirus gene during infection of insect cells. Science. 1991;254:1388–1390. doi: 10.1126/science.1962198. [DOI] [PubMed] [Google Scholar]
- Debbas M, White E. Wild-type p53 mediates apoptosis by E1A which is inhibited by E1B. Genes Dev. 1993;7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- Enari M, Hug H, Nagata S. Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature. 1995;375:78–81. doi: 10.1038/375078a0. [DOI] [PubMed] [Google Scholar]
- Foghsgaard L, Jäättelä M. The ability of BHRF1 to inhibit apoptosis is dependent on stimulus and cell type. J Virol. 1997;171:7509–7571. doi: 10.1128/jvi.71.10.7509-7517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooding LR, Elmore LW, Tollefson AE, Brady HA, Wold WSM. A 14,700 MW protein from the E3 region of adenovirus inhibits cytolysis by tumor necrosis factor. Cell. 1988;53:341–346. doi: 10.1016/0092-8674(88)90154-7. [DOI] [PubMed] [Google Scholar]
- Gooding LR, Aquino L, Duerksen-Hughes PJ, Day D, Horton TM, Yei S, Wold WSM. The E1B-19K protein of group C adenoviruses prevents cytolysis by tumor necrosis factor of human cells but not mouse cells. J Virol. 1991a;65:3083–3094. doi: 10.1128/jvi.65.6.3083-3094.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooding LR, Ranheim R, Tollefson AE, Aquino L, Duerksen-Hughes P, Horton TM, Wold WSM. The 10,400- and 14,500-dalton proteins encoded by region E3 of adenovirus function together to protect many but not all mouse cell lines against lysis by tumor necrosis factor. J Virol. 1991b;65:4114–4123. doi: 10.1128/jvi.65.8.4114-4123.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270:1189–1192. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- Hahne M, Rimoldi D, Schröter M, Romero P, Schreier M, French LE, Schneider P, Bornand T, Fontana A, Lienard D, et al. Melanoma cell expression of Fas(Apo-1/CD95) ligand: implications for tumor immune escape. Science. 1996;274:1363–1366. doi: 10.1126/science.274.5291.1363. [DOI] [PubMed] [Google Scholar]
- Han J, Sabbatini P, Perez D, Rao L, Mohda D, White E. The E1B 19K protein blocks apoptosis by interacting with and inhibiting the p53-inducible and death-promoting Bax protein. Genes Dev. 1996a;10:461–477. doi: 10.1101/gad.10.4.461. [DOI] [PubMed] [Google Scholar]
- Han J, Sabbatini P, White E. Induction of apoptosis by human Nbk/Bik, a BH3 containing E1B 19K interacting protein. Mol Cell Biol. 1996b;16:5857–5864. doi: 10.1128/mcb.16.10.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto S, Ishii A, Yonehara S. The E1B oncogene of adenovirus confers cellular resistance to cytotoxicity of tumor necrosis factor and monoclonal anti-Fas antibody. Int Immunol. 1991;3:343–351. doi: 10.1093/intimm/3.4.343. [DOI] [PubMed] [Google Scholar]
- Henderson S, Huen D, Rowe M, Dawson C, Johnson G, Rickinson A. Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc Natl Acad Sci USA. 1993;90:8479–8483. doi: 10.1073/pnas.90.18.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengartner MO. Programmed cell death in invertebrates. Curr Opin Genet Dev. 1996;6:34–38. doi: 10.1016/s0959-437x(96)90007-6. [DOI] [PubMed] [Google Scholar]
- Hsu H, Shu H-B, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interaction define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- Hu S, Vincenz C, Buller M, Dixit VM. A novel family of viral death effector domain-containing molecules that inhibit both CD-95- and tumor necrosis factor receptor-1-induced apoptosis. J Biol Chem. 1997;272:9621–9624. doi: 10.1074/jbc.272.15.9621. [DOI] [PubMed] [Google Scholar]
- Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S. The polypeptides encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 1991;66:233–243. doi: 10.1016/0092-8674(91)90614-5. [DOI] [PubMed] [Google Scholar]
- Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO (Eur Mol Biol Organ) J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinkert WE, Kojima K, Lesslauer W, Rinner W, Lassmann H, Wekerle H. TNF-α receptor fusion protein prevents experimental auto-immune encephalomyelitis and demyelination in Lewis rats: an overview. J Neuroimmunol. 1997;72:163–168. doi: 10.1016/s0165-5728(96)00183-x. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Laster SM, Good JG, Gooding LR. Tumor necrosis factor can induced both apoptic and necrotic forms of cell lysis. J Immunol. 1988;141:2629–2634. [PubMed] [Google Scholar]
- Merino R, Grillot DA, Simonian PL, Muthukkumar S, Fanslow WC, Bondada S, Nunez G. Modulation of anti-IgM-induced B cell apoptosis by Bcl-xL and CD40 in WEHI-231 cells. Dissociation from cell cycle arrest and dependence on the avidity of the antibody-IgM receptor interaction. J Immunol. 1995;155:3830–3838. [PubMed] [Google Scholar]
- Miura M, Zhu H, Rotello R, Hartwieg EA, Yuan J. Induction of apoptosis in fibroblasts by IL-1β-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. . Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- Miura M, Friedlander RM, Yuan J. Tumor necrosis factor-induced apoptosis is mediated by a CrmA-sensitive cell death pathway. Proc Natl Acad Sci USA. 1995;92:8318–8322. doi: 10.1073/pnas.92.18.8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran E. DNA tumor virus transforming proteins and the cell cycle. Cur Opin Gen Dev. 1993;3:63–70. doi: 10.1016/s0959-437x(05)80342-9. [DOI] [PubMed] [Google Scholar]
- Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, Mann M, Krammer PH, Peter ME, Dixit VM. FLICE, a novel FADD-homologous ICE/ CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Muzio M, Salvesen GS, Dixit VM. FLICE-induced apoptosis in a cell-free system. J Biol Chem. 1997;272:2952–2956. doi: 10.1074/jbc.272.5.2952. [DOI] [PubMed] [Google Scholar]
- Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM. An induced proximity model for caspase-8 activation. J Biol Chem. 1998;273:2926–2930. doi: 10.1074/jbc.273.5.2926. [DOI] [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Probert L, Akassoglou K, Kassiotis G, Pasparakis M, Alexopoulou L, Kollias G. TNF-a transgenic and knockout models of CNS inflammation and degeneration. J Neuroimmunol. 1997;72:137–141. doi: 10.1016/s0165-5728(96)00184-1. [DOI] [PubMed] [Google Scholar]
- Rao L, Debbas M, Sabbatini P, Hockenberry D, Korsmeyer S, White E. The adenovirus E1A proteins induce apoptosis which is inhibited by the E1B 19K and Bcl-2 proteins. Proc Natl Acad Sci USA. 1992;89:7742–7746. doi: 10.1073/pnas.89.16.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray CA, Black RA, Kronheim SR, Greenstreet TA, Sleath PR, Salvesen GS, Pickup DJ. Viral inhibition of inflammation: cowpox virus encodes an inhibitor of the interleukin-1β converting enzyme. Cell. 1992;69:597–604. doi: 10.1016/0092-8674(92)90223-y. [DOI] [PubMed] [Google Scholar]
- Sabbatini P, Han JH, Chiou S-K, Nicholson D, White E. Interleukin 1β converting enzyme-like proteases are essential for p53-mediated transcriptionally dependent apoptosis. Cell Growth Diff. 1997;8:643–653. [PubMed] [Google Scholar]
- Schott AF, Apel IJ, Nuñez G, Clarke MF. Bcl-XLprotects cancer cells from p53-mediated apoptosis. Oncogene. 1995;11:1389–1394. [PubMed] [Google Scholar]
- Seshagiri S, Miller LK. Caenorhabditis elegansCED-4 stimulates CED-3 processing and CED-3-induced apoptosis. Curr Biol. 1997;7:455–460. doi: 10.1016/s0960-9822(06)00216-8. [DOI] [PubMed] [Google Scholar]
- Shisler J, Yang C, Walter B, Ware CF, Gooding LR. The adenovirus E3-10.4K/14.5K complex mediates loss of cell surface Fas (CD95) and resistance to Fas-induced apoptosis. J Virol. 1997;71:8299–8306. doi: 10.1128/jvi.71.11.8299-8306.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector MS, Desnoyers S, Hoeppner DJ, Hengartner MO. Interaction between the C. eleganscell-death regulators CED-9 and CED-4. Nature. 1997;385:653–656. doi: 10.1038/385653a0. [DOI] [PubMed] [Google Scholar]
- Tewari M, Dixit VM. Fas- and TNF-induced apoptosis is inhibited by the poxvirus crmAgene product. J Biol Chem. 1995;270:3255–3260. doi: 10.1074/jbc.270.7.3255. [DOI] [PubMed] [Google Scholar]
- Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer J-L, Schröter M, Scaffidl C, Krammer PH, Peter ME, Tschopp J. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- White E. Life, death, and the pursuit of apoptosis. Genes Dev. 1996;10:1–15. doi: 10.1101/gad.10.1.1. [DOI] [PubMed] [Google Scholar]
- White E, Cipriani R. Specific disruption of intermediate filaments and the nuclear lamina by the 19-kDa product of the adenovirus E1B oncogene. Proc Natl Acad Sci USA. 1989;86:9886–9890. doi: 10.1073/pnas.86.24.9886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E, Cipriani R. Role of adenovirus E1B proteins in transformation: altered organization of intermediate filaments in transformed cells that express the 19-kilodalton protein. Mol Cell Biol. 1990;10:120–130. doi: 10.1128/mcb.10.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E, Blose SH, Stillman B. Nuclear envelope localization of an adenovirus tumor antigen maintains the integrity of cellular DNA. Mol Cell Biol. 1984;4:2865–2875. doi: 10.1128/mcb.4.12.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E, Cipriani R, Sabbatini P, Denton A. The adenovirus E1B 19-kilodalton protein overcomes the cytotoxicity of E1A proteins. J Virol. 1991;65:2968–2978. doi: 10.1128/jvi.65.6.2968-2978.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E, Sabbatini P, Debbas M, Wold WSM, Kusher DI, Gooding L. The 19-kilodalton adenovirus E1B transforming protein inhibits programmed cell death and prevents cytolysis by tumor necrosis factor a. Mol Cell Biol. 1992;12:2570–2580. doi: 10.1128/mcb.12.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Wallen HD, Nuñez G. Interaction and regulation of subcellular localization of CED-4 by CED-9. Science. 1997;275:1126–1128. doi: 10.1126/science.275.5303.1126. [DOI] [PubMed] [Google Scholar]
- Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. eleganscell death gene ced-3 encodes a protein similar to mammalian interleukin-1β-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]