

Abstract
In the biflagellated alga Chlamydomonas, adhesion and fusion of the plasma membranes of gametes during fertilization occurs via an actin-filled, microvillus-like cell protrusion. Formation of this ∼3-μm-long fusion organelle, the Chlamydomonas fertilization tubule, is induced in mating type plus (mt+) gametes during flagellar adhesion with mating type minus (mt−) gametes. Subsequent adhesion between the tip of the mt+ fertilization tubule and the apex of a mating structure on mt− gametes is followed rapidly by fusion of the plasma membranes and zygote formation. In this report, we describe the isolation and characterization of fertilization tubules from mt+ gametes activated for cell fusion. Fertilization tubules were detached by homogenization of activated mt+ gametes in an EGTA-containing buffer and purified by differential centrifugation followed by fractionation on sucrose and Percoll gradients. As determined by fluorescence microscopy of samples stained with a fluorescent probe for filamentous actin, the method yielded 2–3 × 106 fertilization tubules/μg protein, representing up to a 360-fold enrichment of these organelles. Examination by negative stain electron microscopy demonstrated that the purified fertilization tubules were morphologically indistinguishable from fertilization tubules on intact, activated mt+ gametes, retaining both the extracellular fringe and the internal array of actin filaments. Several proteins, including actin as well as two surface proteins identified by biotinylation studies, copurified with the fertilization tubules. Most importantly, the isolated mt+ fertilization tubules bound to the apical ends of activated mt− gametes between the two flagella, the site of the mt− mating structure; a single fertilization tubule bound per cell, binding was specific for gametes, and fertilization tubules isolated from trypsin-treated, activated mt+ gametes did not bind to activated mt− gametes.
The defining moment of fertilization is the adhesion and fusion of the plasma membranes of two interacting gametes. Although gametes are nonfusogenic when they first encounter their partners, their initial adhesive interactions generate a signal transduction cascade that renders the gametes capable of cell fusion (Snell and White, 1996). In multicellular organisms, adhesion-induced signaling prepares sperm for adhesion and fusion of the plasma membranes by inducing the acrosome reaction, an exocytic event that exposes previously cryptic membrane domains that contain adhesion/fusion molecules (Primakoff et al., 1980; Ward and Kopf, 1993). Moreover, hydrolytic enzymes released from the sperm during the acrosome reaction digest the extracellular matrix surrounding the egg, thereby exposing the adhesion/fusion molecules on the egg plasma membrane (Primakoff et al., 1980).
Although the molecules that mediate adhesion and fusion vary widely among species (Glabe, 1985; Hong and Vacquier, 1986; Blobel et al., 1992; Snell and White, 1996), evolution appears to have favored a common cellular structure, an actin-filled cell protrusion, that provides the scaffolding for presentation of adhesion/fusion proteins (for example see Monroy, 1985; Yanagimachi, 1988). Microscopic analysis of sperm–egg interactions in mouse and many other vertebrate and invertebrate species has shown that sperm selectively bind to the microvillus-rich portion of the egg plasma membrane (Tegner and Epel, 1976; Hylander and Summers, 1977; Yanagimachi, 1988; Foltz and Lennarz, 1992; Hart et al., 1992; Ohlendieck et al., 1994). In a manner analogous to eggs, the adhesion/fusion proteins on sperm also appear to be restricted to specialized regions of the plasma membrane. Although eutherian sperm bind and fuse via their apparently microvillus-free equatorial region, the male gametes in most other organisms studied form an apically localized, actin-filled cell protrusion or fusion organelle similar to a microvillus (Jessen et al., 1973; Tilney et al., 1973; Tilney, 1975; Yanagimachi, 1988).
The nearly universal use of actin-filled fusion organelles in fertilization suggests that the regulated fusion of cell membranes requires a complex interplay of adhesion/fusion molecules and other, as yet unidentified features of these microvillus-like structures. Therefore, and by analogy to studies of fusion of intracellular membranes (Rothman, 1994), a complete understanding of the cellular and molecular events that underlie gamete fusion will require both the development of methods for isolating these domains of the cell surface in a structurally and functionally intact form as well as assays for in vitro reconstitution of adhesion and fusion. To this end, we have begun to study a fusion organelle, the mating type plus (mt+)1 fertilization tubule, in the genetically tractable organism, Chlamydomonas. The initial stage of fertilization in this unicellular, biflagellated alga occurs when gametes of opposite mating types undergo a signal-transduction cascade, induced by flagellar adhesion, that generates fusion-competent gametes (Goodenough, 1991; Snell, 1993). Cyclic AMP generated during signaling induces activation of the mating structures, specialized regions of the apical plasma membrane where gametic adhesion and fusion will occur (Pasquale and Goodenough, 1987). Activation of the mt+ mating structure results in both the rapid recruitment of membrane and the explosive polymerization of actin to form the ∼3-μm-long, microvillus-like fertilization tubule (Friedmann et al., 1968; Goodenough et al., 1982; Detmers et al., 1985). Ultrastructural analysis has shown that the mt+ mating structure contains an overlying extracellular coat, called fringe, as well as a densely staining submembranous structure, the doublet zone, proposed to nucleate actin filament polymerization during activation. In contrast, while the activated, dome-shaped mating type minus (mt−) mating structure displays fringe, it does not contain filamentous actin as determined by electron microscopy and staining with antiactin antibodies and NBD-phallacidin (Friedmann et al., 1968; Goodenough et al., 1982; Detmers et al., 1985).
Both thin section and scanning electron microscopic analyses have indicated that binding and subsequent fusion occur between the tip of the mt+ fertilization tubule and the apex of the activated mt− mating structure (Friedmann et al., 1968; Goodenough et al., 1982; Detmers et al., 1983; Forest, 1983). Consistent with this observation, freeze-fracture electron microscopic studies have revealed that, in contrast to membrane on other portions of the fertilization tubule, the membrane at the tip of this structure contains a cluster of intramembranous particles (Weiss et al., 1977). These results suggest that molecules involved in the adhesion and fusion of gametes not only are sequestered in the fertilization tubule but may be further restricted to the tips of these fusion organelles.
Although much is known about their morphology, little biochemical information about the mating structures is available. For example, ultrastructural analysis has demonstrated that actin is present within the fertilization tubule, but the overall protein composition of this organelle is unknown. Similarly, identifying adhesion/fusion molecules, ascertaining whether their expression is restricted to mating structures, and determining if adhesion/fusion molecules are expressed on unactivated mating structures have remained elusive because of the lack of in vitro methods for studying these organelles. To learn more about the mechanism of cell fusion in Chlamydomonas, we have used cell biological and biochemical methods to characterize the mt+ mating structure, the fertilization tubule. Here, we describe a method for isolating fertilization tubules from Chlamydomonas mt+ gametes. We identify several proteins associated with this structure and show that two are surface proteins. Finally, by use of an in vitro assay, we demonstrate that: (a) isolated fertilization tubules bind to activated mt− mating structures; (b) there is little if any binding to unactivated mt− mating structures; and (c) adhesion molecules on fertilization tubules are protease sensitive.
Materials and Methods
Materials
Fluoromount-G was from Fisher Scientific (Pittsburgh, PA). Formvar, EM-grade paraformaldehyde, EM-grade glutaraldehyde, lead citrate, uranyl acetate, Spurr's resin, osmium tetroxide, and grids were from Electron Microscopy Sciences (Ft. Washington, PA). Phalloidin and [4-(2-aminoethyl)benzenesulfonylfluoride, HCl] were obtained from Calbiochem (La Jolla, CA). Saponin was obtained from ICN Biomedicals, Inc. (Costa Mesa, CA). KCl, sucrose, NaCl, Na2HPO4, NaH2PO4, CaCl2, K2HPO4, and KH2PO4 were from Mallinckrodt Inc. (Paris, KY). DMSO (catalog No. D2650), n-amyl alcohol, cytochrome c, trypsin, and soybean trypsin inhibitor were from Sigma Chemical Co. (St. Louis, MO). All other chemicals were of reagent grade.
Cells and Cell Culture
Chlamydomonas reinhardtii strains 21gr (mt+) [cc-1690] and 6145c (mt−) [cc-1691] (available from the Chlamydomonas Genetics Center, Duke University, Chapel Hill, NC) were cultured with aeration at 22°C in Medium I or Medium II of Sager and Granick (1954) on a 13:11-h light/dark cycle. Gametogenesis was induced by transferring vegetatively growing cells to nitrogen free medium (N-free medium) as previously described (Snell, 1976a ). To harvest gametes in 6-liter cultures in N-free medium, cells were allowed to accumulate in the bottom of the culture bottles by negative phototaxis for 30 min under illumination by a bank of fluorescent lights. The supernatant was removed by aspiration with a water pump (model 1P579E; Teel Water Systems; Dayton Electric Mfg., Co., Chicago, IL), and the sedimented cells were resuspended in fresh N-free medium. This procedure was repeated once or twice to ensure that only highly motile cells were used for experiments.
Bodipy Phallacidin Staining
Fertilization tubules on cells and in cell fractions were visualized by fluorescent staining with bodipy phallacidin (Molecular Probes, Eugene, OR), a fluorescein-containing molecule that binds filamentous actin (Nothnagel et al., 1981; Detmers et al., 1985). To do this, wells of an eight-well glass slide (Cel-Line Associates, Inc., Newfield, NJ) were precoated with 10 μl of an aqueous solution of 0.1% polyethylenimine (Sanders and Salisbury, 1994). Excess polyethylenimine was removed by blotting from the side of the well, and the slide was allowed to air-dry. Samples of cells and cell fractions were fixed by mixing with an equal volume of freshly prepared 4% paraformaldehyde in 10 mM Hepes, pH 7.2, and 5-μl portions were applied to wells of the precoated glass slide. After the samples had dried, the slides were immersed for 6 min in 80% acetone, 30 mM NaCl, and 2 mM sodium phosphate buffer, pH 7.0, at −20°C followed by immersion in 100% acetone at −20°C for 6 min. Slides were air-dried and incubated with 20 μl/well of bodipy phallacidin staining solution for 25 min at 37°C in the dark. To prepare the staining solution, an aliquot of a 200 U/ml stock solution of bodipy phallacidin in 100% HPLC-grade methyl alcohol was allowed to dry in a microfuge tube in the dark and the dried sample was resuspended in PBS (150 mM NaCl, 10 mM sodium phosphate buffer, pH 7.0) to yield a 50 U/ml solution. After the incubation with the bodipy phallacidin, slides were washed by immersion in PBS for 6 min, and No. 0 coverslips (Thomas Scientific, Swedesboro, NJ) were applied to the slides with Fluoromount-G mounting medium containing 2.5% 1,4-diazabicyclo- [2.2.2]octane (Sigma Chemical Co.), an antiquenching agent. Samples were examined with a 100× plan apo, 1.3 N.A. objective (Carl Zeiss, Inc., Thornwood, NY) fitted on an epifluorescent microscope (model IM35; Carl Zeiss, Inc.). Images were acquired either by a 25-s exposure on Kodak TMAX 400 film (Rochester, NY) or 1600 daylight Provia Fujichrome color slide film (Tokyo, Japan), or by using a chilled CCD camera and an Argus 20 image processor (Hamamatsu Phototonics, Hamamatsu City, Japan). Final composite images were constructed using the image processing program Adobe Photoshop (San Jose, CA) on a Power Macintosh 8500/120 computer (Apple Computer Co., Cupertino, CA) and printed with a Kodak XLS 8600 printer.
Cell Wall Loss Assay
Cell wall loss was determined as previously described (Snell, 1982). Cell suspensions (1–3 × 106 cells) to be tested for wall loss were added to 1 ml of 0.075% Triton X-100 in 0.5 mM EDTA in a microfuge tube, vortexed briefly, and centrifuged for 20 s at 14,000 rpm in a Hermle microfuge (Labnet, Woodbridge, NJ) at room temperature. The absence of any chlorophyll in the sedimented material when compared to a non–detergent-treated control indicated that ∼100% of the cells had lost their walls.
Isolation of Fertilization Tubules
Fertilization tubules were isolated from activated, homogenized mt+ gametes by differential centrifugation and fractionation on sucrose and Percoll gradients. Flagellated, highly motile cells from 12 liters of mt+ gametes were obtained by negative phototaxis as described above and collected by centrifugation in 1-liter polycarbonate centrifuge bottles (Nalgene, Rochester, NY) at 4,600 g for 20 min (model H6000A rotor, RC-3B centrifuge; Dupont Sorvall, Newtown, CT). Sedimented cells were concentrated ∼300-fold over their starting concentration by resuspension in ∼40 ml of N-free medium (final cell concentration was ∼3 × 109 cells/ ml). To activate mt+ gametes, a concentrated dibutyryl cAMP solution (0.3 M made up in N-free medium) was added to cells to obtain a final concentration of 15 mM, and a 100× stock solution (freshly prepared in 100% DMSO) of the phosphodiesterase inhibitor, papaverine, also was added to yield a final concentration of 0.15 mM. (It was important to use a freshly opened vial of DMSO to prepare the 15 mM papaverine stock.) During this incubation, cells were agitated by vigorous aeration under illumination at room temperature for 30–40 min; activation of mt+ gametes was confirmed both by the cell wall loss assay described above and by the appearance of fertilization tubules on cells as determined by bodipy phallacidin staining.
Activated mt+ gametes were washed several times with ice-cold fertilization tubule stabilization buffer (FTSB, 40 mM KCl, 50 mM EGTA, 0.5 mM EDTA, 1 mM [4-(2-aminoethyl)benzenesulfonylfluoride, HCl], 1 μM leupeptin [100× stock made up in water], 1 μM pepstatin A [100× stock made up in 100% methyl alcohol], 20 μM chymostatin [100× stock made up in 100% DMSO], and 10 mM imidazole, pH 7.2), resuspended in ∼80 ml of FTSB, and homogenized on ice (model 5000 homogenizer, 32 mm generator probe; Omni International, Gainesville, VA) until 80–90% of the cells appeared to have been disrupted upon examination by phase contrast microscopy. All subsequent steps were carried out at 4°C or on ice. To remove unbroken cells, cell fragments, and larger cell organelles, the homogenate was centrifuged at 2,600 g for 4 min in 50-ml conical polycarbonate centrifuge tubes (Nalgene), followed by centrifugation of the supernatant in 50-ml round-bottomed polycarbonate centrifuge tubes (Dupont Sorvall) at 6,000 g for 6 min (model SA-600 rotor, RC-5C centrifuge; Dupont Sorvall). Fertilization tubules and other particulate material in the supernatant were harvested by centrifugation at 37,000 g for 20 min in 50-ml polycarbonate centrifuge tubes and resuspended in ∼4.5 ml of 0.5× FTSB containing 60% sucrose (wt/vol) to yield a suspension whose density was equivalent to ∼30% sucrose. Aliquots (0.6 ml) of this suspension were loaded onto sucrose gradients consisting of 1 ml steps of 40, 50, and 60% sucrose in 6-ml polycarbonate centrifuge tubes (Dupont Sorvall), and the samples were overlaid with 1-ml steps of 20 and 15% sucrose. (The sucrose solutions were prepared by dilution of a 60% sucrose solution with FTSB.) The gradients were centrifuged at 14,000 g (model HB-4 rotor, RC-5C centrifuge; Dupont Sorvall) for 25 min. Fractions (0.5 ml) were collected from the top of each gradient, pooled with equivalent fractions from the other gradients, and examined for the presence of fertilization tubules by staining with bodipy phallacidin. Fertilization tubule– enriched fractions were diluted with FTSB, transferred to 3-ml polycarbonate centrifuge tubes (Beckman Instruments, Inc., Houston, TX) and centrifuged at 68,000 g for 30 min (model TLA100.3 rotor, Optima TLX Ultracentrifuge; Beckman Instruments). Sedimented fertilization tubules were resuspended in 1.2 ml of 150 mM NaCl, 8% sucrose, FTSB, sheared by seven passages through a 25-gauge needle, and mixed with 100% Percoll and 60% sucrose that was further diluted with FTSB to generate a 30% Percoll, 8% sucrose solution. This solution was mixed by inversion, and a Percoll gradient was formed in situ by centrifugation in 6-ml polycarbonate tubes (Dupont Sorvall) at 37,000 g for 22 min. Fractions were collected from above, pooled, and assayed for the presence of fertilization tubules by staining with bodipy phallacidin. Fertilization tubule–containing fractions were diluted with FTSB and harvested by centrifugation in 3-ml polycarbonate centrifuge tubes (Beckman Instruments) for 1 h at 100,000 g (model TLA100.3 rotor, Optima TLX Ultracentrifuge; Beckman Instruments). The fertilization tubules, which formed a fluffy, white sediment on top of the clear Percoll pellet, were resuspended in a small volume of FTSB, flash-frozen, and stored in liquid nitrogen until used. In some cases, small amounts of remaining Percoll were removed by centrifugation in 1-ml polycarbonate tubes (Dupont Sorvall) at 128,000 g.
For isolation of fertilization tubules from protease-treated mt+ gametes, cells activated as described above were diluted 10-fold with ice-cold N-free medium, and the suspension was divided into two equal portions and placed on ice. A freshly prepared stock solution of trypsin (36 mg/ml in 0.1 N HCl) was added to one portion to a final concentration of 1 mg/ml. After 16 min on ice, at which time cells had lost their ability to undergo flagellar agglutination with tester mt− gametes, a freshly prepared stock of soybean trypsin inhibitor (200 mg/ml in N-free medium) was added to a final concentration of 2 mg/ml, and fertilization tubules were isolated from both the trypsin-treated cells and the control cells after surface labeling with biotin as described below. Fertilization tubules isolated from the trypsin-treated and control mt+ gametes were indistinguishable in bodipy phallacidin-stained samples.
Surface Biotinylation
Surface proteins were biotin-labeled following the method of Reinhart and Bloodgood (1988). mt+ gametes were activated with dibutyryl cAMP and papaverine as described above, washed two to four times in FTSB (free of protease inhibitors) by centrifugation at 2,600 g for 4 min at 4°C, and resuspended in ∼40 ml of buffer to which Sulfo-NHS-Biotin (Pierce, Rockford, IL) was added to a final concentration of 1 mg/ml. After incubation on ice for 10–20 min, the reaction was terminated by the addition of 3–4 vol of FTSB containing 1% glycine and centrifuged at 2,600 g for 4 min at 4°C, followed by three additional washes by centrifugation with FTSB containing 1% glycine. After the final wash, mt+ gametes were resuspended in glycine-free FTSB containing protease inhibitors, and fertilization tubules were isolated as described above.
Determination of Protein Concentration
The protein concentration of samples was determined using the BCA Protein Assay kit (Pierce) according to the manufacturer's instructions. BSA supplied in the kit was used as a standard.
Quantification of Fertilization Tubule Enrichment
The number of fertilization tubules per milliliter in samples from various steps of purification was determined by counting bodipy phallacidin– stained fertilization tubules in a fluorescent microscope. Fertilization tubules within the area defined by the photographic field in the microscope were counted. 20 random fields per sample and two to three different dilutions per sample were counted in duplicate. The average number of fertilization tubules per field was used to calculate the total number of fertilization tubules in the original sample based on the dilution and the calculated number of photographic fields per well.
Determination of Chlorophyll Content
Chlorophyll content was determined by a modification of the Wintermans and De Mots (1965) method described in Harris (1989). To do this, samples that had been flash-frozen in liquid nitrogen were quickly thawed and 20–50-μl portions were transferred to microfuge tubes containing 1.45 ml of 95% ethyl alcohol, vortexed briefly, and allowed to incubate at room temperature for 30–60 min. After vortexing again and centrifugation at 14,000 rpm for 2 min, the green, chlorophyll-containing supernatants were removed and the OD665 and OD649 were determined in a spectrophotometer (model DU-68; Beckman Instruments).
Electron Microscopy
For visualization of fertilization tubules on intact mt+ gametes, the cells were activated by mixing with an equal number of mt− gametes in N-free medium for ∼1 min and fixed on ice by addition of 50% EM-grade glutaraldehyde to a final concentration of 1%. After 10 min, the cells were concentrated by centrifugation (14,000 rpm in a Hermle microfuge for 3 s) to 2 × 108 cells/ml, and 10 μl of the sample was placed onto Formvar- and carbon-coated grids that had been treated by glow-discharge in a Med 010 Mini deposition system (Balzers Union Ltd., Balzers, Liechtenstein). Grids were rinsed with distilled water and stained with two drops of 1% aqueous uranyl acetate. Excess stain was removed by blotting from the side with Whatman No. 1 filter paper; grids were allowed to air-dry and examined on an electron microscope (model 1200EX; JEOL Ltd., Tokyo, Japan).
Isolated fertilization tubules were analyzed by negative stain electron microscopy using a modification of the method of Berryman et al. (1995). Fertilization tubules were collected by centrifugation in 1-ml polycarbonate centrifuge tubes at 128,000 g for 30 min. The sedimented fertilization tubules were resuspended in 5 mM MgCl2, 50 mM KCl in 0.1 M sodium phosphate buffer, pH 7.2, and mixed with an equal volume of 4% EM-grade glutaraldehyde, 4% EM-grade paraformaldehyde, 2% tannic acid, 20 μM phalloidin, 0.5 mg/ml saponin, 5 mM MgCl2, 50 mM KCl, in 0.1 M sodium phosphate buffer, pH 7.2. After 1–5 min, 2 μl of the fixed sample was placed onto Formvar- and carbon-coated grids that had been treated by glow-discharge. Grids were rinsed with three drops of distilled water, two drops of 1% n-amyl alcohol containing 0.02% cytochrome c, followed by three drops of 1% aqueous uranyl acetate (Snell, 1983). Excess stain was removed by blotting from the side with Whatman No. 1 filter paper, and grids were allowed to air-dry and were examined on an electron microscope (model 1200EX; JEOL Ltd.).
SDS-PAGE
Samples (diluted to ∼0.5 mg/ml) were boiled for 4 min in SDS-PAGE sample buffer (10% glycerol, 10% SDS, 0.1% bromphenol blue, 0.1 M DTT, and 0.0625 M Tris, pH 6.8) and subjected to electrophoresis on Laemmli (1970) mini-slab gels made as linear 4–20% polyacrylamide gradient gels at 30 mA (Jarvik and Rosenbaum, 1980). Typically, each well was loaded with 20–30 μl of sample containing 12 μg of protein. After electrophoresis, the gels either were fixed and stained for protein by Coomassie blue (De St. Groth et al., 1963; Meyer and Lamberts, 1965) or silver (Merril et al., 1980) or prepared for transfer for antibody or streptavidin blot analysis.
Antibody and Streptavidin Blotting
For blots to be probed with streptavidin or with antiactin antibody, proteins were transferred to Immobilon-P membranes (Millipore, Bedford, MA) in buffer containing 192 mM glycine, 20% methyl alcohol, 25 mM Tris, pH 8.3, at 100 V for 1 h at room temperature or 20 V overnight at 4°C (Towbin et al., 1979). All subsequent steps were carried out at room temperature on an orbital shaker (Bellco Biotechnology, Vineland, NJ). For streptavidin blots, membranes were fixed by incubation in TBS (137 mM NaCl, 20 mM Tris, pH 7.6) containing 0.2% glutaraldehyde (Hulen et al., 1991) for 45 min and then washed twice for 10 min each with TBS. Membranes were blocked in TBS containing 5% Carnation Dry Milk and 0.5% Triton X-100 for 1 h. Blocked membranes were rinsed and incubated for 15 min with wash buffer (0.2% Carnation Dry Milk, 0.2% Triton X-100 in TBS). For detection of biotinylated proteins, membranes were incubated in peroxidase-streptavidin (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) diluted 1:7,500 in antibody dilution buffer (1% Carnation Dry Milk, 0.2% Triton X-100 in TBS) for 1 h, rinsed twice in wash buffer, and then washed once for 15 min followed by four changes of wash buffer every 5 min. Membranes were developed with enhanced chemiluminescence as described by the manufacturer (Amersham Life Science, Arlington Heights, IL), exposed to Hyperfilm (Amersham Life Science), and the film was developed in an x-ray developer (model Konica QX-60A; Konishiroku Photo Ind. Co., Ltd., Japan).
For antiactin blots, membranes were blocked and washed as described above. Membranes were incubated with an anti-Volvox actin polyclonal antibody (Ehler et al., 1995) at a 1:1,000 dilution for 2 h or overnight at 4°C, rinsed twice in wash buffer, and then incubated once in wash buffer for 15 min followed by two changes of wash buffer every 5 min. Membranes were incubated with a 1:30,000 dilution of peroxidase-labeled goat anti–rabbit antibody (Organon Teknika Corp., West Chester, PA) in antibody dilution buffer for 1 h, washed, and developed as described above.
For anticentrin blots, proteins were transferred to Immobilon-P membranes in 25 mM potassium phosphate buffer, pH 7.0, at 20 V overnight at 4°C (Hulen et al., 1991). All subsequent steps were carried out at room temperature on an orbital shaker (Bellco Biotechnology). After transfer, membranes were fixed for 45 min in 0.2% glutaraldehyde, 25 mM potassium phosphate buffer, pH 7.0, washed twice for 10 min in TBS, and incubated with primary and secondary antibodies as described above. The anticentrin monoclonal antibody, 17E10 (Salisbury and Sanders, 1988), was used at a 1:500 dilution and peroxidase-labeled goat anti–mouse antibody (Organon Teknika Corp.) was used at a 1:30,000 dilution. Washing and detection of signal were as described above.
Binding of Fertilization Tubules to mt− Gametes
Isolated fertilization tubules were incubated with activated mt− gametes, and fertilization tubules that remained bound after washing were detected by staining with bodipy phallacidin followed by examination in a fluorescence microscope. To do this, mt− gametes were activated either with dibutyryl cAMP and papaverine as described above or by incubation with isolated mt+ gametic flagella at a ratio of 10 flagella/cell; activation was assessed by loss of cell walls (Witman et al., 1972; Weiss et al., 1977; Kaska and Gibor, 1982; Snell, 1982). Bodipy phallacidin staining could not be used as an assay for activation of mt− gametes because the mt− mating structure does not contain filamentous actin (Goodenough et al., 1982; Detmers et al., 1985). To assay binding, 1–5 μl of freshly isolated fertilization tubules or fertilization tubules that had been flash-frozen were incubated with activated mt− cells (2 × 107 cells/ml, 100 μl final volume, ∼10 fertilization tubules/cell) in N-free medium supplemented with 2.5 mM CaCl2. After 12–15 min of agitation at room temperature on an orbital shaker (Bellco Biotechnology), the samples were diluted with 1.4 ml of N-free medium supplemented with 2.5 mM CaCl2, and cells were separated from unbound fertilization tubules by centrifugation at 14,000 rpm for 9 s. After removal of the supernatant, the sedimented cells were resuspended in 50–100 μl of 3.7% formaldehyde (Fisher Scientific) freshly diluted in 2 mM MgSO4, 0.7 mM CaCl2, 20 mM KCl, and 40 mM sodium phosphate buffer, pH 6.4 (Detmers et al., 1985), and bound fertilization tubules were detected by staining with bodipy phallacidin as described above. In some experiments, activated mt− gametes were fixed before incubation with isolated fertilization tubules. To do this, activated cells were mixed with an equal volume of freshly prepared 4% paraformaldehyde, 10 mM Hepes, pH 7.2. After 10 min on ice, the fixed cells were washed several times in 1% glycine in N-free medium by centrifugation at room temperature, resuspended in N-free medium, and incubated with isolated fertilization tubules followed by washing with N-free medium as described above. The sedimented samples were resuspended in N-free medium, mixed with an equal volume of 4% paraformaldehyde, 10 mM Hepes, pH 7.2, and bound fertilization tubules were detected as described above. In control experiments with mt− vegetative cells, cell walls were removed by incubation with g-lysin (Buchanan et al., 1989) before the binding assay. To quantify binding, the number of cells that had bound a fertilization tubule was determined by examining bodipy phallacidin-stained samples in the fluorescence microscope. For each cell population tested in the binding assay with fixed cells, 100–200 cells/well and 2–4 wells/sample were scored for binding of fertilization tubules. Data in Table II are averages from two experiments and are presented ± SEM. In the assays with live cells, a more qualitative assessment was made, and binding was scored as no binding (<5% of the cells had bound fertilization tubules), low binding (∼10–20% of the cells had bound fertilization tubules), or extensive binding (30–60% of cells had bound fertilization tubules). Data from several independent isolations of fertilization tubules are included.
Table II.
Binding of Fertilization Tubules to mt− Gametes
| mt− gametes | Fertilization tubules | Percentage of cells with bound fertilization tubules | ||
|---|---|---|---|---|
| Unactivated* | Control | 4 ± 3 | ||
| Activated* | Control | 27 ± 8 | ||
| Activated‡ | Control | 30–50 | ||
| Activated‡ | Trypsin-treated | <5 |
mt− gametes were fixed with 2% paraformaldehyde before use in the binding assay.
Live mt− gametes were used in the binding assay.
Results
Isolation of Fertilization Tubules
Negative stain electron microscopy shows a fertilization tubule emerging from the apical end of an activated mt+ gamete between the two flagella (Fig. 1 A). Fig. 1, B and C, shows higher magnification views of fertilization tubules on activated mt+ gametes and reveals the characteristic actin filaments within these microvillus-like structures. Fig. 2 shows a fluorescence micrograph of activated mt+ gametes stained with bodipy phallacidin. To learn about the biochemical and cell biological properties of fertilization tubules, it was necessary to develop a method for obtaining fractions highly enriched in these fusion organelles, which represent only ∼1/500 of the cell volume. For detection of fertilization tubules, which are not visible by phase contrast microscopy (Cavalier-Smith, 1975; Pasquale and Goodenough, 1987), we took advantage of the fact that the fertilization tubule is the only cellular constituent in activated mt+ gametes that contains filamentous actin (Detmers et al., 1985). To assay rapidly for the presence of fertilization tubules on cells and in cell fractions, samples were adsorbed onto the wells of eight-well glass microscope slides, stained with bodipy phallacidin, a fluorescein-containing phallotoxin that binds to filamentous actin (Nothnagel et al., 1981; Detmers et al., 1985), and examined in a fluorescence microscope. We also refined previously published methods for activation of mt+ gametes (Pasquale and Goodenough, 1987) so that we were able to consistently induce formation of long fertilization tubules in large numbers of highly concentrated cells. Finally, the buffer used for the purification of fertilization tubules was a modification of previously described buffers used for the isolation of microvilli (Bretscher and Weber, 1978; Bretscher, 1986) and other actin-containing structures (Rosenberg et al., 1981). A critical modification was the use of increased EGTA to chelate Ca2+; this divalent cation is known to destabilize actin filaments (Glenney et al., 1980; Walsh et al., 1984; Neely and Gesemann, 1994) and is reported to be present in high levels in Chlamydomonas gametes (Goodenough et al., 1993).
Figure 1.

Negative stain electron microscopy of fertilization tubules on activated mt+ gametes. mt+ gametes activated by mixing with an equal number of mt− gametes were negatively stained and examined by electron microscopy. (A) Low magnification view of an activated mt+ gamete with a fertilization tubule extending from the apical region of the cell between the two flagella. (B and C) Higher magnification views of fertilization tubules on intact mt+ gametes. Bars: (A) 1 μm; (B and C) 0.05 μm.
Figure 2.

Bodipy phallacidin staining of activated mt+ gametes. mt+ gametes were activated with dibutyryl cAMP and papaverine, and fertilization tubules were visualized by fluorescent microscopy after staining with bodipy phallacidin. Bar, 5 μm.
The method for isolating fertilization tubules is summarized in Fig. 3. mt+ gametes were induced to undergo sexual signaling by incubation with 15 mM dibutyryl cAMP and 0.15 mM papaverine for 30 min. Activated mt+ gametes were separated by centrifugation from cell walls that had been released during signaling and resuspended in FTSB. Examination by phase contrast microscopy indicated that, in the majority of cells, homogenization detached the apical region of the cell from the rest of the cell body. Cell bodies, flagella, and other large cellular components were separated from the fertilization tubules and smaller particulate material by differential centrifugation. As evidenced by the dark green color, partially disrupted chloroplasts remained a major contaminant of the crude preparation of fertilization tubules harvested after differential centrifugation.
Figure 3.

Diagrammatic representation of the method for isolation of fertilization tubules.
The next step in the isolation, the sucrose gradient, generated a colorful set of fractions, including more rapidly sedimenting dark green fractions (Fig. 4 A, fractions 7–10) that contained fragments of chloroplasts and small numbers of flagella as indicated by microscopic inspection, and more slowly sedimenting orange and light-green fractions (Fig. 4 A, fractions 4 and 5) that contained the majority of the fertilization tubules. Sucrose gradient fractions enriched in fertilization tubules were collected by centrifugation and further fractionated on Percoll gradients. The Percoll step separated the orange and the remaining green material from the denser, milky-white fraction that contained the majority of the fertilization tubules (Fig. 4 B). Fractions enriched in fertilization tubules were collected by centrifugation and stained with bodipy phallacidin. Fig. 5 A illustrates the large number of fertilization tubules observed upon examination of these samples in the fluorescence microscope. No fluorescence was visible when the same field was examined in the rhodamine channel, indicating that contamination of the purified fertilization tubules with chloroplast components (which autofluoresce red) was below the level of detection in the fluorescence microscope (Fig. 5 B).
Figure 4.

Enrichment of fertilization tubules by fractionation on sucrose and Percoll gradients. (A) Crude preparations of fertilization tubules from differential centrifugation were fractionated on 15–60% sucrose gradients as described in Materials and Methods. After centrifugation, 0.5-ml fractions were collected from above, and the numbers of fertilization tubules/μg protein in each fraction were determined (gray line, protein; black line, fertilization tubules). (B) Fractions 4 and 5 from the sucrose gradients in A were pooled, collected by centrifugation, and further fractionated on 30% Percoll gradients as described in Materials and Methods. Fractions of the indicated volumes were collected from above, and the numbers of fertilization tubules/μg protein in each fraction were determined (gray bars, protein; black bars, fertilization tubules).
Figure 5.

Bodipy phallacidin staining of purified fertilization tubules. Isolated fertilization tubules were harvested from the Percoll gradient fraction enriched in fertilization tubules, fixed, and diluted 1:80 with FTSB before staining with bodipy phallacidin. Large numbers of fertilization tubules were visible in the fluorescein channel (A), while no obvious chloroplast contamination was observed in the rhodamine channel (B). Bar, 5 μm.
Table I shows the fold-enrichment of fertilization tubules from key steps of the isolation; homogenized cells, pooled fractions from the sucrose gradient, and purified fertilization tubules from the Percoll gradient were stained with bodipy phallacidin, and the numbers of fertilization tubules were determined. Routinely, 2–3 × 106 fertilization tubules/μg protein were obtained in the final fraction of purified fertilization tubules with a ∼0.1% recovery of protein and 15–60% recovery of fertilization tubules. With this procedure, it was possible to obtain up to ∼360-fold enrichment of fertilization tubules, indicating a substantial purification of this important organelle.
Table I.
Enrichment of Fertilization Tubules
| Step | Number tubules/μg (× 106) | Percentage of protein recovery | Percentage of tubule recovery | Fold enrichment | ||||
|---|---|---|---|---|---|---|---|---|
| Homogenized cells | .014 | 100 | 100 | 1 | ||||
| Sucrose gradient* | .29 | 1 | 20 | 21 | ||||
| Percoll gradient* | 2.35 | 0.1 | 15 | 168 | ||||
| (2.2–3.1)‡ | (76–360)‡ |
Fractions from the gradient enriched in fertilization tubules that were pooled and collected by centrifugation.
Ranges of the number of fertilization tubules/μg protein and fold enrichment over starting material are shown in parentheses.
Morphology of Isolated Fertilization Tubules
Examination of purified fertilization tubules by negative stain electron microscopy indicated that the isolation procedure preserved their key ultrastructural features. The organelles ranged from 1 to 3 μm in length with diameters of ∼0.05–0.08 μm (Fig. 6). Although a significant number of fertilization tubules retained a small portion of the basal region of the mating structure and some of the surrounding plasma membrane, most appeared as simple tubes (Figs. 6 and 7). Both actin filaments as well as the extracellular coat of fringe, two of the defining ultrastructural characteristics of fertilization tubules, are visible in many of the fertilization tubules shown in Fig. 6 and are more evident in the higher magnification views shown in Fig. 7, A and B.
Figure 6.
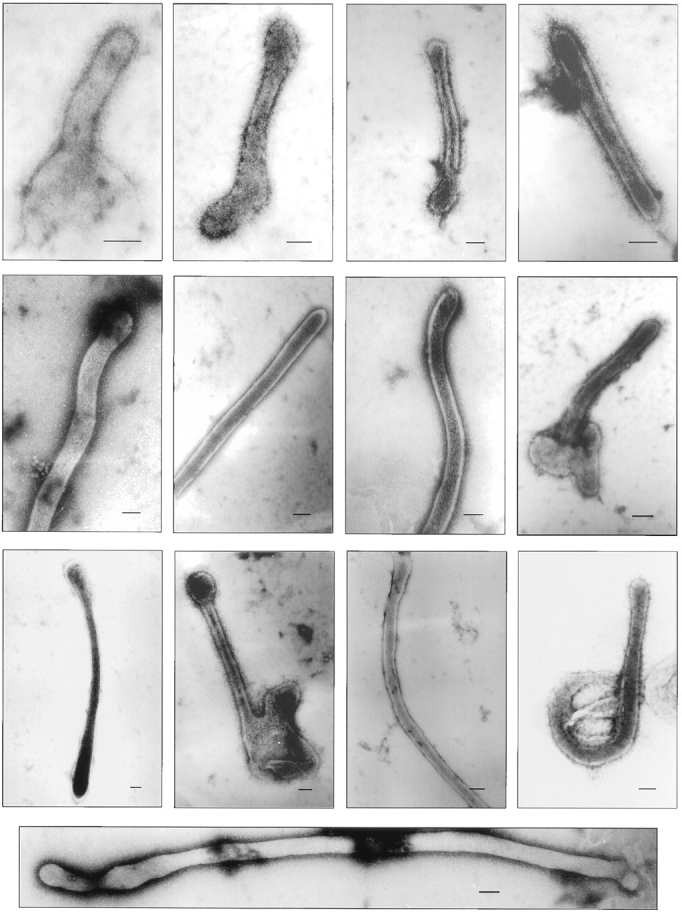
Negative stain electron microscopic analysis at low magnification of isolated fertilization tubules. Isolated fertilization tubules were harvested from the Percoll gradient fraction enriched in fertilization tubules, fixed, stained as described in Materials and Methods, and examined by electron microscopy. Actin filaments and fringe are visible in these images of isolated fertilization tubules. Bars, 0.05 μm.
Figure 7.

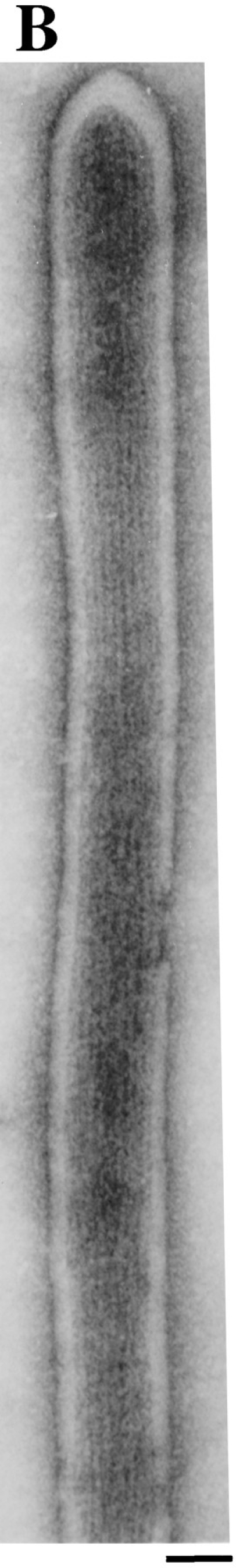
Negative stain electron microscopic analysis at high magnification of isolated fertilization tubules. (A) High magnification view of an isolated fertilization tubule showing the extracellular fringe on the distal portion of the fertilization tubule. (B) Enlargement of an isolated fertilization tubule from the panel in Fig. 6, showing the large number of actin filaments in this structure. Bars, 0.05 μm.
De-enrichment of Other Cellular Organelles
In addition to monitoring numbers of fertilization tubules during the isolation procedure, the levels of other key cellular constituents were assessed. Examination by phase contrast microscopy of the final purified fraction of fertilization tubules indicated that, as expected, it contained few if any flagella and no cell walls, and it was composed almost exclusively of fine, particulate material (data not shown).
Spectrophotometric analysis indicated that the amount of chlorophyll in the starting material was consistent with levels previously reported for Chlamydomonas (Harris, 1989) (Fig. 8 A, homogenized cells [HC]). As expected from their much lighter green appearance, the fertilization tubule–enriched fractions from the sucrose gradient contained significantly less chlorophyll (Fig. 8 A, sucrose pellet [SP]), and the milky-white preparation of purified fertilization tubules contained little if any chlorophyll (Fig. 8 A, Percoll pellet [PP]). These results were consistent with the absence of chlorophyll autofluorescence observed in the rhodamine channel of the fluorescence microscope (Fig. 5 B) and indicated that the purified fertilization tubules were essentially free of chloroplasts.
Figure 8.
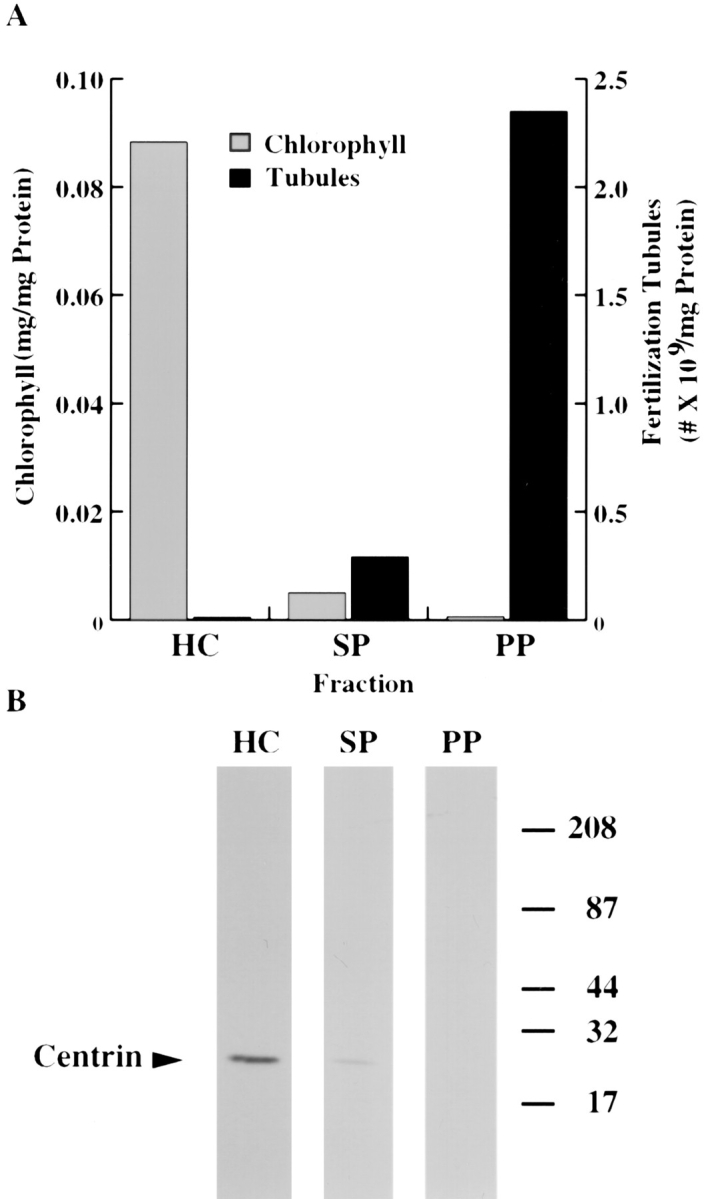
De-enrichment of chlorophyll and centrin. (A) The amounts of chlorophyll present at various steps in the isolation of fertilization tubules (homogenized cells [HC], sucrose pellets [SP], and Percoll pellets [PP]) were determined as described in Materials and Methods (gray bars, chlorophyll; black bars, fertilization tubules). (B) Immunoblot analysis with anticentrin monoclonal antibody, 17E10, of various steps in the isolation of fertilization tubules. Samples (12 μg) of homogenized cells (lane HC), sucrose pellets (lane SP), and Percoll pellets (lane PP) were separated by SDS-PAGE and analyzed by immunoblotting as described. The location of centrin is indicated by the arrowhead on the left. The migration of prestained molecular weight markers is shown on the right.
We also assessed the levels of centrin, a component of the apically localized, nucleus–basal body connector that ultrastructural studies have shown to be associated with basal portions of the mt+ mating structure (Goodenough and Weiss, 1978; Salisbury and Sanders, 1988). Immunoblot analysis demonstrated that although centrin was detectable in homogenized cells (Fig. 8 B, lane HC), the fertilization tubule–enriched fraction from the sucrose gradient (Fig. 8 B, lane SP) contained much less centrin, and centrin was not detectable in purified fertilization tubules (Fig. 8 B, lane PP).
Identification of Proteins that Enriched during Isolation of Fertilization Tubules
SDS-PAGE and silver stain analysis demonstrated that a number of proteins (indicated by asterisks in Fig. 9 A) in the starting material were de-enriched as fertilization tubules were purified (Fig. 9 A, compare homogenized cells, lane HC, to Percoll pellet, lane PP). On the other hand, although some proteins neither enriched nor de-enriched, examination of profiles from several isolations indicated that a number of proteins consistently enriched during purification of fertilization tubules including proteins with molecular masses of ∼500, 350, 200, 134, 56, 55, 51, 45, 37, 34, 30, and 27 kD. The more prominent of these, including proteins with molecular masses of 350, 200, 56, 55, 51, 45, and 30 kD, are indicated by arrows in Fig. 9 A.
Figure 9.

SDS-PAGE analysis of fertilization tubule proteins. (A) Samples (12 μg) of homogenized cells (lane HC), sucrose pellets (lane SP), and Percoll pellets (lane PP) were separated by SDS-PAGE and stained with silver. Asterisks indicate proteins that de-enriched, and arrows indicate proteins that enriched during isolation of fertilization tubules. The migration of unstained molecular weight markers is indicated on the left. (B) Migration of ft51 above flagellar tubulin on SDS-PAGE. Samples of purified fertilization tubules (12 μg, lane PP) and isolated mt+ gametic flagella (∼3 μg, lane FLG) were analyzed by SDS-PAGE and silver staining as described.
Among these more prominent fertilization tubule (ft) proteins, an ∼45-kD protein (ft45; Fig. 9 A, lane PP) was identified as actin based on its comigration on SDS-PAGE with purified rabbit actin and its reactivity with the monoclonal antibody N350 (Amersham Life Science) (Detmers et al., 1985; Harper et al., 1992), a broad-specificity antiactin antibody (data not shown). In addition, ft45 reacted with a polyclonal antiactin antibody (raised against a COOH-terminal, eight–amino acid peptide from Volvox actin) that has been shown to cross-react with Chlamydomonas actin (Cresnar et al., 1990; Ehler et al., 1995). Fig. 10, showing the relative amounts of the ft45 antigen in homogenized cells (lane HC), sucrose pellet (lane SP), and purified fertilization tubules (lane PP), documents the enrichment of actin during purification of fertilization tubules. An ∼51-kD protein (ft51; Fig. 9 A) was the most prominent and as yet unidentified protein of purified fertilization tubules; ft51 migrated slightly above tubulin as shown in Fig. 9 B, which compares flagellar tubulin (lane FLG) with fertilization tubule proteins (lane PP). In addition to ft45 and ft51, an ∼350-kD protein (ft350; shown in Fig. 9 A, lane PP) was another prominent protein of purified fertilization tubules. Based on its comigration with an ∼350-kD protein in flagella (not shown), it is likely that ft350 is a previously described, ubiquitous cell surface protein found on the plasma membranes both of flagella (Witman et al., 1972; Bergman et al., 1975; Bloodgood and May, 1982) and of cell bodies (Hunnicutt et al., 1990).
Figure 10.
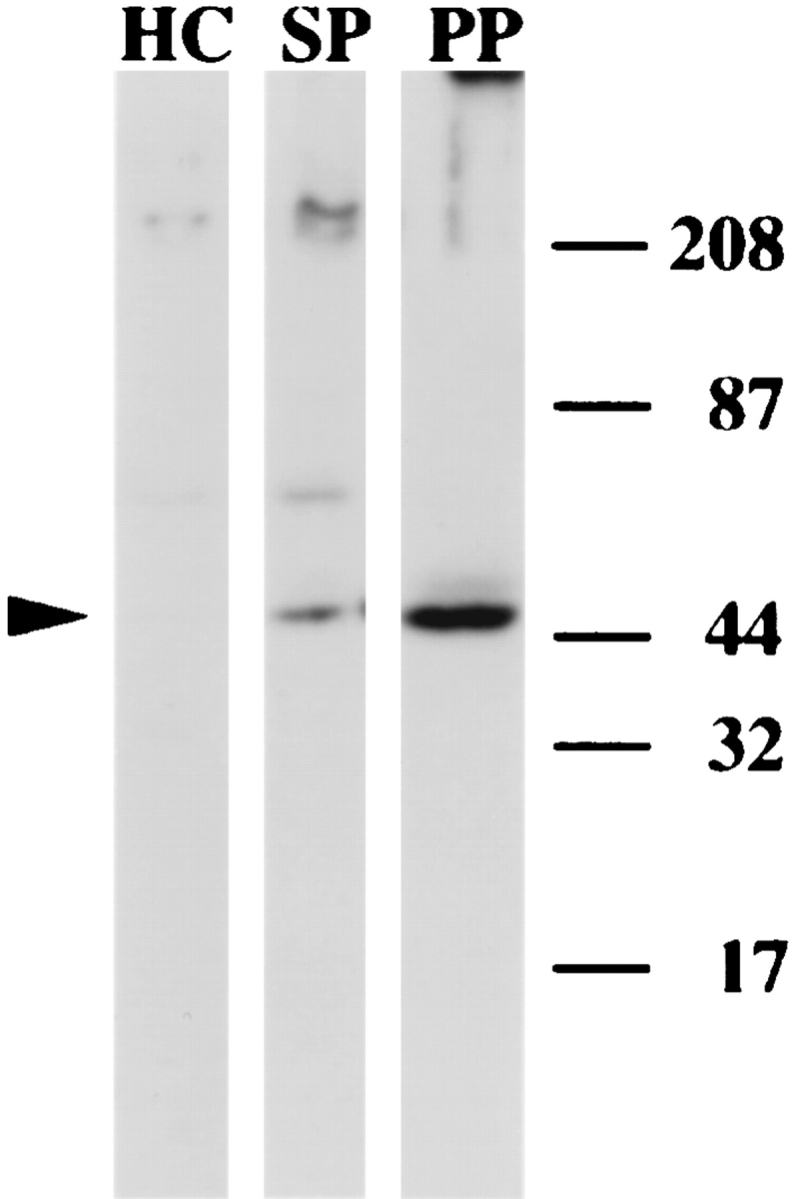
Enrichment of actin during purification of fertilization tubules. Samples (12 μg) of homogenized cells (lane HC), sucrose pellets (lane SP), and Percoll pellets (lane PP) were separated by SDS-PAGE and analyzed by immunoblotting with an anti-Volvox actin polyclonal antibody as described. The location of actin is indicated by the arrowhead on the left. Prestained molecular weight markers are shown on the right.
Surface Biotinylation of Fertilization Tubule Proteins
Surface-labeling studies also were consistent with the identification of ft350 as a previously identified cell surface protein. Fertilization tubules were isolated from activated mt+ gametes that had been surface labeled with biotin, and biotinylated proteins were detected by SDS-PAGE and streptavidin blotting (see Materials and Methods). As shown in Fig. 11 A, ft350 enriched with fertilization tubules. With equal amounts of protein loaded in each lane, ft350 was not detectable in the streptavidin blots of homogenized cells (Fig. 11 A, lane HC), appeared in the sucrose pellet (lane SP), and was a prominently labeled protein in isolated fertilization tubules (lane PP). A second surface-biotinylated protein, ft500, also copurified with fertilization tubules as shown in Fig. 11 A. ft500, which stained poorly with silver, was most evident after Coomassie blue staining; Fig. 11 B shows Coomassie blue staining of the biotinylated samples shown in Fig. 11 A and also documents the enrichment of ft500 in purified fertilization tubules. These results suggest that ft350 and ft500 are surface proteins that contain a domain accessible to labeling with biotin.
Figure 11.

Identification of surface proteins by vectorial labeling with biotin. (A) Activated mt+ gametes were labeled with Sulfo-NHS-Biotin, fertilization tubules were isolated, and samples (12 μg) of homogenized cells (lane HC), sucrose pellets (lane SP), and Percoll pellets (lane PP) were separated by SDS-PAGE and analyzed by streptavidin blotting. Arrows indicate the locations of surface-biotinylated proteins of 500 (ft500) and 350 kD (ft350). (B) Staining with Coomassie blue of the same samples as in A. Arrows indicate the locations of ft500 and ft350. (C) To identify proteins biotinylated in disrupted cells, activated mt+ gametes were homogenized before biotinylation (lane H → B) and 1.5 μg of protein was analyzed by SDS-PAGE and streptavidin blotting. In addition, potential streptavidin-binding proteins were identified by analyzing both nonbiotinylated homogenized cells (12 μg, HC, −B) and fertilization tubules purified from nonbiotinylated cells (12 μg, PP, −B).
Several controls for the biotinylation experiment are presented in Fig. 11 C; the biotinylation pattern of the homogenized cell fraction of cells biotinylated after homogenization was significantly different from the pattern obtained when cells were biotinylated before homogenization (compare lane HC, H → B in Fig. 11 C with lane HC in Fig. 11 A), indicating that the cells remained intact during biotinylation. In addition, although streptavidin bound two proteins in the homogenized cell fraction from nonbiotinylated cells (Fig. 11 C, lane HC, −B), no binding of streptavidin was observed in purified fertilization tubules from the nonbiotinylated cells (compare lane PP, −B in Fig. 11 C with lane PP of Fig. 11 A), indicating the specificity of streptavidin binding.
Isolated Fertilization Tubules Contain Functional Adhesion Molecules
Having developed methods for isolating fertilization tubules that were morphologically intact, we wanted to determine if the isolated organelles retained functional adhesion molecules. To do this, fertilization tubules were incubated with activated and unactivated mt− gametes, and binding was assessed as described in Materials and Methods. As shown in Table II, nearly 30% of fixed, activated mt− gametes bound a fertilization tubule, whereas 4% of fixed, unactivated mt− gametes bound fertilization tubules. In similar experiments, 30–50% of live, activated mt− gametes bound a fertilization tubule (Table II). Fig. 12 shows images of control mt− cells and activated mt− gametes incubated with fertilization tubules. Fig. 12 A, showing fixed, activated mt− gametes incubated with FTSB alone, documents the absence of filamentous actin in mt− gametes. Fluorescent images of fixed, activated mt− gametes that had bound fertilization tubules (Fig. 12, C and D) show that cells bound just a single fertilization tubule. Moreover, examination of the cells with bound fertilization tubules in both the fluorescein and rhodamine channels (the latter was used to locate chloroplasts) indicated that the fertilization tubules bound to the apical ends of the cells.
Figure 12.
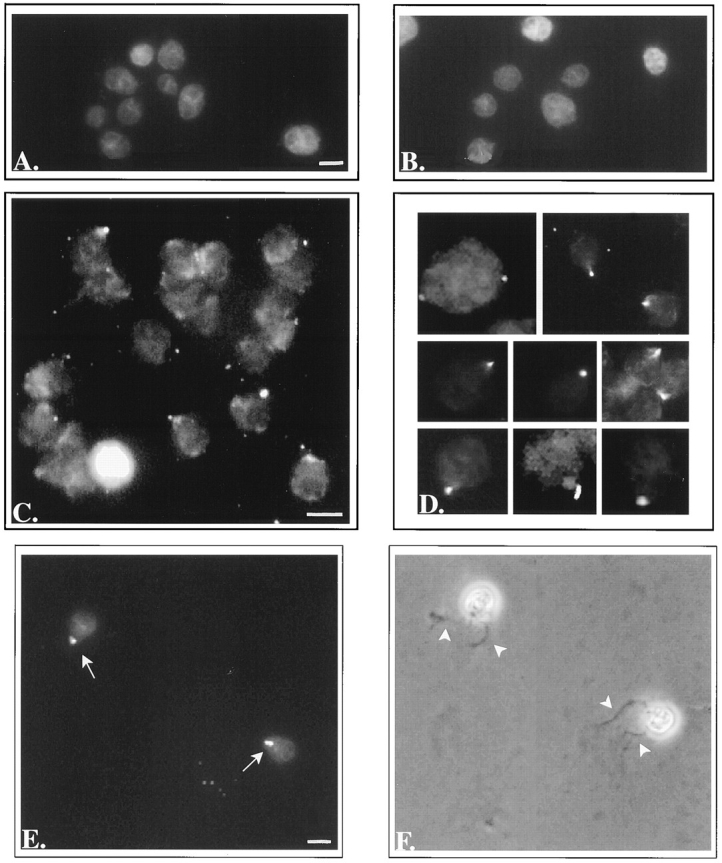
Binding of fertilization tubules to activated mt− gametes. (A) Fluorescence microscopy of bodipy phallacidin stained mt− gametes activated and fixed as described in Materials and Methods and incubated with FTSB alone. (B) De-walled, fixed, mt− vegetative cells incubated with isolated fertilization tubules. (C and D) Activated, fixed mt− gametes incubated with isolated fertilization tubules. (E) Fluorescence and (F) phase contrast micrographs of activated, live mt− gametes incubated with isolated fertilization tubules. E and F are images of the same cells. Arrows point to the fertilization tubules visible in the fluorescent image shown in E, and arrowheads in F indicate the apically located flagella on the same cells. Bars (for pairs A and B, C and D, and E and F), 5 μm.
Cell morphology was better preserved in experiments in which live, activated mt− gametes were incubated with fertilization tubules. As shown in fluorescence (Fig. 12 E) and phase contrast images (Fig. 12 F) of the same cells, fertilization tubules (Fig. 12 E, arrows) bound at the apical ends of the cells, between the two flagella (Fig. 12 F, arrowheads), the site of the mt− mating structure. Fertilization tubules were not observed to bind to flagella on mt− gametes (Fig. 12, E and F) or to mt− vegetative cells (Fig. 12 B). Moreover, in mt− gametes to which a long fertilization tubule was bound, the fertilization tubule appeared to have bound by one end only, with the other end extending away from the mt− cell body (Fig. 12 D). This observation, which was consistent with ultrastructural studies (Goodenough et al., 1982; Forest, 1983) showing interactions in vivo between the tip of the fertilization tubule and the apex of the mt− mating structure, suggested that adhesion molecules were restricted to the tips of the fertilization tubules. The binding of fertilization tubules to mt− gametes shown here demonstrated that isolated fertilization tubules retained functional adhesion molecules.
Characterization of Adhesive Interactions Between Isolated Fertilization Tubules and mt− Mating Structures
Development of this in vitro adhesion assay allowed us to bypass the requirement for flagellar adhesion and begin to examine directly the adhesive properties of isolated fertilization tubules and their interaction with mating structures on mt− gametes. Thus, it became possible to determine if the fertilization tubule molecules responsible for adhesion to mt− mating structures were protease sensitive. To do this, fertilization tubules isolated from mt+ gametes that had been treated with trypsin after activation were assayed for their ability to bind to live, activated mt− gametes. As shown in Table II, protease-treated fertilization tubules were inactive in the binding assay. While 30–50% of mt− gametes bound fertilization tubules isolated from non–trypsin-treated mt+ gametes, only 5% of mt− gametes bound the protease-treated fertilization tubules, further documenting the specificity of binding and indicating that fertilization tubule adhesion is protease sensitive.
We also used the binding assay to study adhesion molecules on mating structures of mt− gametes. Although the data in Table II indicate that fertilization tubules did not bind to unactivated cells, the cells used in those experiments were enclosed by their cell walls. To determine whether mating structures on unactivated mt− gametes displayed functional adhesion molecules, cell walls were removed from otherwise untreated mt− gametes by incubation with g-lysin, a cell wall–degrading enzyme (Buchanan et al., 1989). Results varied from experiments in which 15% of the cells in samples of de-walled, unactivated mt− gametes bound fertilization tubules to experiments in which there was no binding of fertilization tubules; in those same experiments, 25–50% of the activated mt− gametes bound fertilization tubules (data not shown). These results suggested that in the experiments where there was some binding by cells in the unactivated cell samples, either a small percentage of unactivated gametes expressed functional adhesion molecules, or more likely, a small percentage of untreated mt− gametes had undergone spontaneous activation.
Discussion
Actin, Ca2+, and the Isolation of Fertilization Tubules
This report describes a method for isolation of fertilization tubules from activated mt+ gametes as well as the first cell biological and biochemical analysis of a plasma membrane fraction specialized for cell fusion. One of the key discoveries in the development of the isolation method was that preservation of fertilization tubules during isolation required a buffer with a high concentration of the Ca2+ chelator, EGTA. This requirement for chelation of Ca2+ during isolation of actin-containing structures has been observed in a number of systems (Bretscher and Weber, 1978; Rosenberg et al., 1981; Bretscher, 1986) and presumably derives from the actin filament destabilizing activity of several cytoplasmic Ca2+-binding proteins, including proteases (Phillips and Jakabova, 1977; Rosenberg et al., 1981), actin-severing proteins, and actin-nucleating proteins (Vandekerckhove, 1990; Hartwig and Kwiatkowski, 1991). The higher level of EGTA required for isolation of fertilization tubules compared to that required for isolation of actin-containing structures in other systems is likely due to the high concentrations of Ca2+ in Chlamydomonas gametes. It has been shown that 2 × 107 gametes/ml release enough Ca2+ in 30 min to increase the concentration of Ca2+ in their medium by 20–40 μM (Goodenough et al., 1993), and the starting cell concentration for isolation of fertilization tubules was ∼150-fold higher than in the experiments by Goodenough et al. (1993).
Using the methods described here a highly enriched fraction of isolated fertilization tubules was obtained. Routinely, this method yielded 15–60% recovery, up to a 360-fold enrichment of fertilization tubules, and 2–3 × 106 fertilization tubules/μg protein (Table I). As expected for a microvillus-like structure, actin was a major constituent of the purified fertilization tubules (ft45 in Figs. 9 A and 10, lane PP). In addition, a number of other proteins copurified with fertilization tubules. Although many of these remain unidentified, two are surface proteins (see below). In contrast to the enrichment of actin and several other proteins, purification of fertilization tubules was accompanied by de-enrichment of two key cytoplasmic components, chlorophyll and centrin. The absence of chlorophyll, a marker for the chloroplast (which occupies ∼40% of the cell volume) (Schotz et al., 1972; Harris, 1989), in purified fertilization tubules (Fig. 8 A) was consistent with the absence of rhodamine autofluorescence upon examination of the isolated fertilization tubules in a fluorescence microscope (Fig. 5 B). The levels of centrin, a component of the nucleus–basal body connector, also were followed during purification of fertilization tubules. The nucleus–basal body connector comprises a series of contractile fibers that extend from the apically localized basal bodies to form a basket around the nucleus. Although ultrastructural studies have shown that this structure interacts with the basal-most aspect of the mating structure through a connective lattice (Goodenough and Weiss, 1978), immunoblot analysis revealed that the isolation method described here disrupted this interaction, as illustrated by the absence of centrin in purified fertilization tubules (Fig. 8 B).
Negative Stain Electron Microscopic Analysis of Isolated Fertilization Tubules
Examination of the isolated fertilization tubules by negative stain electron microscopy revealed a morphology similar to that seen by electron microscopy of fertilization tubules on activated mt+ gametes (compare Fig. 1, B and C, with Figs. 6 and 7). As is apparent in the panel of isolated fertilization tubules (Fig. 6), some of the fertilization tubules appeared as simple tubes, while others maintained their association with the basal aspect of the mating structure, including the cell body plasma membrane in this region. Many of the fertilization tubules retained their extracellular coat of fringe, indicating that the integrity of the membrane was preserved during isolation (Figs. 6 and 7 A). In addition, closely packed actin filaments were readily observed (Figs. 6 and 7 B), consistent with the presence of filamentous actin visible by staining with bodipy phallacidin (Fig. 5 A). The preservation of ultrastructural features suggests that future biochemical, genetic, and molecular genetic investigations with purified fertilization tubules should provide new information on the assembly and disassembly of these microvillus-like structures.
Surface Proteins of Fertilization Tubules
Biotinylation studies identified ft350 and ft500 as two major surface proteins that copurified with fertilization tubules. Other minor surface proteins were detected using this method, but ft350 and ft500 were consistently the most prominently labeled proteins. Although it is likely that ft350 and ft500 are not the only surface proteins of the fertilization tubules, they may be much more abundant than other surface proteins, or the two proteins may be more accessible to biotinylation on live cells. This relatively simple profile of surface proteins on fertilization tubules is consistent with studies of membrane proteins of microvilli isolated from other cell types. While membranes of microvilli isolated from some cells have been shown to contain a complex protein profile detected by surface iodination (Truman and Ford, 1986), protein profiles of membrane fractions from brush border microvilli (Bretscher and Weber, 1978) and rat adenocarcinoma cells are dominated by a small number of proteins (Carraway et al., 1982).
The ft350 protein (Fig. 11 A, lane PP), a previously identified, abundant cell surface molecule (Witman et al., 1972; Bergman et al., 1975; Bloodgood and May, 1982; Hunnicutt et al., 1990), may be distributed over the entire surface of the fertilization tubule, or it could be restricted to the basal region of this organelle, whose membrane is continuous with the cell body plasma membrane. The ubiquitous distribution of ft350 suggests that it may play more of a structural, rather than a unique functional role in adhesion/fusion. Although the second surface protein, ft500 (Fig. 11 A, lane PP), remains unidentified, several roles for it can be proposed: (a) It could protect the surface of this transient fusion organelle; (b) it may anchor proteins to the underlying actin cytoskeleton; or (c) it might establish membrane domains within the fertilization tubule (e.g., at the tip). In addition, ft500 could be an adhesion/fusion molecule or a component of fringe, the extracellular coat overlying both mt+ and mt− mating structures, that has been proposed to function in an adhesive capacity during mating (Goodenough et al., 1982). Recently, Ferris et al. (1996) reported on the cloning and sequencing of fus1, a gene that was found to restore expression of fringe and fusion competence to impotent mt+ mutants. These workers proposed that mt+ fringe represents a single glycoprotein encoded by fus1 (Ferris et al., 1996). Although these results are promising and have the potential to offer new insights into molecules involved in adhesion/fusion, the native fus1 protein has not been identified yet. Thus, the questions of whether the fus1 protein indeed is a fertilization tubule protein and whether it is on the cell surface remain to be elucidated. When more molecular information is gained about ft500 and when specific probes for ft500 and the fus1 protein become available, it should be possible to learn more about the location of these molecules, their relationship to each other, and their roles, if any, in gametic adhesion and fusion.
Adhesion Molecules on Mating Structures
The observation that isolated fertilization tubules bound to activated mt− gametes indicated that the molecules required for adhesion between activated mt+ and mt− mating structures were retained during the isolation method (Fig. 12, C–E). Binding of the fertilization tubules was specific to gametic cells, a single fertilization tubule bound per cell, and fertilization tubules did not bind to flagella or randomly on the cell surface. Instead, binding occurred only at the apical ends of the mt− gametes between the two flagella, the site of the mt− mating structure. Thus, adhesion molecules on fertilization tubules indeed are distinct from flagellar agglutinins (Adair et al., 1982; Pasquale and Goodenough, 1987); moreover, mt− adhesion molecules are not distributed over the entire cell surface but are exquisitely localized to a specific cell surface domain. These results are consistent with previous studies on gametes of other species, showing that molecules involved in the initial adhesive interactions between unactivated gametes are distinct from the molecules involved in the final adhesion/fusion of the fusogenic regions of the plasma membranes of the two gametes (Snell and White, 1996).
The development of the in vitro binding assay was particularly exciting because it allowed us to bypass the requirement for flagellar adhesion and permitted direct examination of the adhesive properties of isolated fertilization tubules and their interaction with mating structures on mt− gametes. Studying the effects of proteases, for example, on the adhesion/fusion properties of activated mating structures has been difficult since the flagellar adhesion and sexual signaling required for mating structure activation also are affected by incubation of whole cells with proteases (Wiese and Hayward, 1972; Snell, 1976b ; Forest, 1983). Experiments to address these issues are theoretically possible with flagella-less and agglutinin-defective mutants, which can be induced to form mating structures by addition of dibutyryl cAMP, but the rates of cell fusion are low (Pasquale and Goodenough, 1987). Furthermore, although microscopic studies of fusion-defective mutants have suggested that adhesion and fusion of mating structures are distinct events (Goodenough et al., 1982; Forest, 1983, 1987; Ferris et al., 1996), the in vitro assay described here makes it possible to study adhesion separately from fusion in wild-type and mutant cells. Our studies showing that fertilization tubules isolated from activated, protease-treated mt+ gametes did not bind to mt− gametes (Table II) indicated that, as expected, protease-sensitive molecules are critical in this first step in cell fusion. In future studies, it will be interesting to determine if, in addition to their ability to bind to mt− gametes, the isolated fertilization tubules retain the ability to fuse with mt− gametes, thereby making it possible to study eukaryotic cell–cell fusion using methods similar to those used in studies of viral fusion with their target cells (White, 1996).
We also used the assay to begin to study expression of adhesion molecules on mt− gametes. The observation that very low numbers of cells in samples of unactivated mt− gametes bound isolated fertilization tubules, even if their cell walls had been removed, suggested that gametic activation is required for the expression of functional adhesion molecules. Although it is possible that unactivated mt− gametes display a low number of functional adhesion molecules on their mating structures, it is more likely that the small numbers of cells that bound fertilization tubules in the unactivated samples in some experiments were a subset of the mt− gametic population that had undergone spontaneous activation. This interpretation is consistent with previous studies on fusion of agglutinin-defective mutants. Pasquale and Goodenough (1987) showed that agglutinin-defective mutant gametes did not undergo flagellar adhesion, and therefore did not activate their mating structures after mixing with mt− gametes. If, however, the mt+ and mt− gametes were activated by addition of dibutyryl cAMP before they were mixed, 75% of them fused. On the other hand, only 5% of de-walled, unactivated cells underwent fusion with activated mt− gametes. Taken together, these data suggest that unactivated mt− mating structures contain few, if any, functional adhesion/fusion molecules and that expression of active molecules requires either modification of preexisting, inactive molecules or insertion of new molecules into the membrane overlying the mating structures.
This reconstitution in vitro of interactions between mt+ fertilization tubules and activated mt− gametes should make it possible to learn more about the properties inherent in microvillus-like structures that have resulted in their ubiquitous use as scaffolds for display of adhesion/fusion molecules (Poste and Allison, 1971; Monroy, 1985; Carpen et al., 1992; Berlin et al., 1995; von Andrian et al., 1995) and may be one of the first systems available for studying a fusogenic cell surface membrane domain that contains the elements required for the final steps in fertilization.
Acknowledgments
We would like to thank Dr. Judith M. White (University of Virginia, Charlottesville, VA) for helpful discussions and for encouraging us to assay for binding of purified fertilization tubules to activated mt− gametes, Dave Kirk (Washington University, St. Louis, MO) for the anti-Volvox actin polyclonal antibody, and Dr. Jeff Salisbury (Mayo Clinic, Rochester, MN) for the anticentrin monoclonal antibody. We also thank our colleagues, Drs. George Bloom and Helen Yin, for suggestions on preserving the integrity of actin filaments and Dr. Fred Grinnell for critically reading the manuscript.
This work was supported by National Science Foundation Grant IBN-9318708 to W.J. Snell. N.F. Wilson was supported in part by a Minority Supplement to National Institutes of Health Grant GM 25661 to W.J. Snell.
Abbreviations used in this paper
- ft
fertilization tubule
- FTSB
fertilization tubule stabilization buffer
- mt
mating type
- N-free medium
nitrogen-free medium
Footnotes
Address all correspondence to William J. Snell, Department of Cell Biology and Neuroscience, The University of Texas Southwestern Medical Center, Dallas, TX 75235. Tel.: (214) 648-2332. Fax: (214) 648-8694.
This work was submitted in partial fulfillment of the requirements for the Ph.D. degree for Nedra F. Wilson, University of Texas Southwestern Graduate School of Biomedical Sciences, Dallas, TX.
References
- Adair WS, Monk BC, Cohen R, Hwang C, Goodenough UW. Sexual agglutinins from the Chlamydomonasflagellar membrane: partial purification and characterization. J Biol Chem. 1982;257:4593–4602. [PubMed] [Google Scholar]
- Bergman K, Goodenough UW, Goodenough DA, Jawitz J, Martin H. Gametic differentiation in Chlamydomonas reinhardtii.II. Flagellar membranes and the agglutination reaction. J Cell Biol. 1975;67:606–622. doi: 10.1083/jcb.67.3.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin C, Bargatze RF, Campbell JJ, von Andrian UH, Szabo MC, Hasslen SR, Nelson RD, Berg EL, Erlandsen SL, Butcher EC. α4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell. 1995;80:413–422. doi: 10.1016/0092-8674(95)90491-3. [DOI] [PubMed] [Google Scholar]
- Berryman M, Gary R, Bretscher A. Ezrin oligomers are major cytoskeletal components of placental microvilli: a proposal for their involvement in cortical morphogenesis. J Cell Biol. 1995;131:1231–1242. doi: 10.1083/jcb.131.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobel CP, Wolfsberg TG, Turck CW, Myles DG, Primakoff P, White JM. A potential fusion peptide and an integrin ligand domain in a protein active in sperm-egg fusion. Nature (Lond) 1992;356:248–252. doi: 10.1038/356248a0. [DOI] [PubMed] [Google Scholar]
- Bloodgood RA, May GS. Functional modification of the Chlamydomonasflagellar surface. J Cell Biol. 1982;93:88–96. doi: 10.1083/jcb.93.1.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher A. Purification of the intestinal microvillus cytoskeletal proteins villin, fimbrin, and ezrin. Methods Enzymol. 1986;134:24–37. doi: 10.1016/0076-6879(86)34072-2. [DOI] [PubMed] [Google Scholar]
- Bretscher A, Weber K. Purification of microvilli and an analysis of the protein components of the microfilament core bundle. Exp Cell Res. 1978;116:397–407. doi: 10.1016/0014-4827(78)90463-9. [DOI] [PubMed] [Google Scholar]
- Buchanan MJ, Imam SH, Eskue WA, Snell WJ. Activation of the cell wall degrading protease, lysin, during sexual signalling in Chlamydomonas: the enzyme is stored as an inactive, higher relative molecular mass precursor in the periplasm. J Cell Biol. 1989;108:199–207. doi: 10.1083/jcb.108.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpen O, Pallai P, Staunton DE, Springer TA. Association of intercellular adhesion molecule-1 (ICAM-1) with actin-containing cytoskeleton and α-actinin. J Cell Biol. 1992;118:1223–1234. doi: 10.1083/jcb.118.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraway KL, Cerra RF, Jung G, Carraway CAC. Membrane-associated actin from the microvillar membranes of ascites tumor cells. J Cell Biol. 1982;94:624–630. doi: 10.1083/jcb.94.3.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier-Smith T. Electron and light microscopy of gametogenesis and gamete fusion in Chlamydomonas reinhardii. . Protoplasma. 1975;86:1–18. doi: 10.1007/BF01275619. [DOI] [PubMed] [Google Scholar]
- Cresnar B, Mages W, Muller K, Salbaum JM, Schmitt R. Structure and expression of a single actin gene in Volvox carteri. . Curr Genet. 1990;18:337–346. doi: 10.1007/BF00318215. [DOI] [PubMed] [Google Scholar]
- De St. Groth SF, Webster RG, Datyner A. Two new staining procedures for quantitative estimation of proteins on electrophoretic strips. Biochim Biophys Acta. 1963;71:377–391. doi: 10.1016/0006-3002(63)91092-8. [DOI] [PubMed] [Google Scholar]
- Detmers PA, Goodenough UW, Condeelis J. Elongation of the fertilization tubule in Chlamydomonas: new observations on the core microfilaments and the effect of transient intracellular signals on their structural integrity. J Cell Biol. 1983;97:522–532. doi: 10.1083/jcb.97.2.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmers PA, Carboni JM, Condeelis J. Localization of actin in Chlamydomonasusing anti-actin and NBD-phallacidin. Cell Motil. 1985;5:415–430. doi: 10.1002/cm.970050505. [DOI] [PubMed] [Google Scholar]
- Ehler LL, Holmes JA, Dutcher SK. Loss of spatial control of the mitotic spindle apparatus in a Chlamydomonas reinhardtiimutant strain lacking basal bodies. Genetics. 1995;141:945–960. doi: 10.1093/genetics/141.3.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris PJ, Woessner JP, Goodenough UW. A sex recognition glycoprotein is encoded by the plus mating-type gene fus1 of Chlamydomonas reinhardtii. . Mol Biol Cell. 1996;7:1235–1248. doi: 10.1091/mbc.7.8.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz KR, Lennarz WJ. Identification of the sea urchin egg receptor for sperm using an antiserum raised against a fragment of its extracellular domain. J Cell Biol. 1992;116:647–658. doi: 10.1083/jcb.116.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forest CL. Specific contact between mating structure membranes observed in conditional fusion defective Chlamydomonasmutants. Exp Cell Res. 1983;148:143–154. doi: 10.1016/0014-4827(83)90194-5. [DOI] [PubMed] [Google Scholar]
- Forest CL. Genetic control of plasma membrane adhesion and fusion of Chlamydomonasgametes. J Cell Sci. 1987;88:613–621. doi: 10.1242/jcs.88.5.613. [DOI] [PubMed] [Google Scholar]
- Friedmann I, Colwin AL, Colwin LH. Fine-structural aspects of fertilization in Chlamydomonas reinhardi. . J Cell Sci. 1968;3:115–128. doi: 10.1242/jcs.3.1.115. [DOI] [PubMed] [Google Scholar]
- Glabe CG. Interaction of the sperm adhesive protein, bindin, with phospholipid vesicles. II. Bindin induces the fusion of mixed-phase vesicles that contain phosphatidylcholine and phosphatidylserine in vitro. J Cell Biol. 1985;100:800–806. doi: 10.1083/jcb.100.3.800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenney JR, Jr, Bretscher A, Weber K. Calcium control of the intestinal microvillus cytoskeleton: its implications for the regulation of microfilament organizations. Proc Natl Acad Sci USA. 1980;77:6458–6462. doi: 10.1073/pnas.77.11.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough, U.W. 1991. Chlamydomonas mating interactions. In Microbial Cell-Cell Interactions. M. Dworkin, editor. American Society of Microbiologists, New York. 71–112.
- Goodenough UW, Weiss RL. Interrelationships between microtubules, a striated fiber, and the gametic mating structures of Chlamydomonas reinhardi. . J Cell Biol. 1978;76:430–438. doi: 10.1083/jcb.76.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough UW, Detmers PA, Hwang C. Activation for cell fusion in Chlamydomonas: analysis of wild-type gametes and nonfusing mutants. J Cell Biol. 1982;92:378–386. doi: 10.1083/jcb.92.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough UW, Shames B, Small L, Saito T, Crain RC, Sanders MA, Salisbury JL. The role of calcium in the Chlamydomonas reinhardtiimating reaction. J Cell Biol. 1993;121:365–374. doi: 10.1083/jcb.121.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JDI, McCurdy DW, Sanders MA, Salisbury JL, John PCL. Actin dynamics during the cell cycle in Chlamydomonas reinhardtii. . Cell Motil Cytoskel. 1992;22:117–126. doi: 10.1002/cm.970220205. [DOI] [PubMed] [Google Scholar]
- Harris, E.H. 1989. The Chlamydomonas Sourcebook: A Comprehensive Guide to Biology and Laboratory Use. Academic Press, San Diego. 780 pp. [DOI] [PubMed]
- Hart NH, Becker KA, Wolenski JS. The sperm entry site during fertilization of the zebrafish egg: localization of actin. Mol Reprod Dev. 1992;32:217–228. doi: 10.1002/mrd.1080320306. [DOI] [PubMed] [Google Scholar]
- Hartwig JH, Kwiatkowski DJ. Actin-binding proteins. Curr Opin Cell Biol. 1991;3:87–97. doi: 10.1016/0955-0674(91)90170-4. [DOI] [PubMed] [Google Scholar]
- Hong K, Vacquier VD. Fusion of liposomes induced by a cationic protein from the acrosome granule of abalone spermatozoa. Biochemistry. 1986;25:543–549. doi: 10.1021/bi00351a004. [DOI] [PubMed] [Google Scholar]
- Hulen D, Baron A, Salisbury J, Clarke M. Production and specificity of monoclonal antibodies against calmodulin from Dictyostelium discoideum. . Cell Motil Cytoskel. 1991;18:113–122. doi: 10.1002/cm.970180206. [DOI] [PubMed] [Google Scholar]
- Hunnicutt GR, Kosfiszer MG, Snell WJ. Cell body and flagellar agglutinins in Chlamydomonas reinhardtii: the cell body plasma membrane is a reservoir for agglutinins whose migration to the flagella is regulated by a functional barrier. J Cell Biol. 1990;111:1605–1616. doi: 10.1083/jcb.111.4.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylander BL, Summers RG. An ultrastructural analysis of the gametes and early fertilization in two bivalve molluscs, Chama macerophylla and Spisula solidissimawith special reference to gamete binding. Cell Tiss Res. 1977;182:469–489. doi: 10.1007/BF00219830. [DOI] [PubMed] [Google Scholar]
- Jarvik JW, Rosenbaum JL. Oversized flagellar membrane protein in paralyzed mutants of Chlamydomonas reinhardtii. . J Cell Biol. 1980;85:258–272. doi: 10.1083/jcb.85.2.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen H, Behnke O, Wingstrand KG, Rostgaard J. Actin–like filaments in the acrosomal apparatus of spermatozoa of a sea urchin. Exp Cell Res. 1973;80:47–54. doi: 10.1016/0014-4827(73)90273-5. [DOI] [PubMed] [Google Scholar]
- Kaska DD, Gibor A. Initiation of cell wall lysis in gametes of Chlamydomonas reinhardiby isolated flagella of the complementary mating type. Exp Cell Res. 1982;138:121–125. doi: 10.1016/0014-4827(82)90097-0. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (Lond) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Merril CR, Goldman D, Sedman SA, Eber MH. Ultrasensitive stain for proteins in polyacrylamide gels shows a regional variation in cerebrospinal fluid proteins. Science (Wash DC) 1980;211:1437–1438. doi: 10.1126/science.6162199. [DOI] [PubMed] [Google Scholar]
- Meyer TS, Lamberts BL. Use of Coomassie brilliant blue R250 for the electrophoresis of microgram quantities of parotid saliva proteins on acrylamide-gel strips. Biochim Biophys Acta. 1965;107:144–145. doi: 10.1016/0304-4165(65)90403-4. [DOI] [PubMed] [Google Scholar]
- Monroy A. Processes controlling sperm-egg fusion. Eur J Biochem. 1985;152:51–56. doi: 10.1111/j.1432-1033.1985.tb09162.x. [DOI] [PubMed] [Google Scholar]
- Neely MD, Gesemann M. Disruption of microfilaments in growth cones following depolarization and calcium influx. J Neurosci. 1994;14:7511–7520. doi: 10.1523/JNEUROSCI.14-12-07511.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothnagel EA, Barak LS, Sanger JW, Webb WW. Fluorescence studies on modes of cytochalasin B and phallotoxin action on cytoplasmic streaming in Chara. . J Cell Biol. 1981;88:364–372. doi: 10.1083/jcb.88.2.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlendieck K, Partin JS, Stears RL, Lennarz WJ. Developmental expression of the sea urchin egg receptor for sperm. Dev Biol. 1994;165:53–62. doi: 10.1006/dbio.1994.1233. [DOI] [PubMed] [Google Scholar]
- Pasquale SM, Goodenough UW. Cyclic AMP functions as a primary sexual signal in gametes of Chlamydomonas reinhardtii. . J Cell Biol. 1987;105:2279–2292. doi: 10.1083/jcb.105.5.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips DR, Jakabova M. Ca2+-dependent protease from human platelets. J Biol Chem. 1977;252:5602–5605. [PubMed] [Google Scholar]
- Poste G, Allison AC. Membrane fusion reaction: a theory. J Theor Biol. 1971;32:165–184. doi: 10.1016/0022-5193(71)90144-5. [DOI] [PubMed] [Google Scholar]
- Primakoff P, Myles DG, Bellve AR. Biochemical analysis of the released products of the mammalian acrosome reaction. Dev Biol. 1980;80:324–331. doi: 10.1016/0012-1606(80)90408-x. [DOI] [PubMed] [Google Scholar]
- Reinhart FD, Bloodgood RA. Membrane-cytoskeleton interactions in the flagellum: a 240,000 Mr surface-exposed glycoprotein is tightly associated with the axoneme in Chlamydomonas moewusii. . J Cell Sci. 1988;89:521–531. doi: 10.1242/jcs.89.4.521. [DOI] [PubMed] [Google Scholar]
- Rosenberg S, Stracher A, Lucas RC. Isolation and characterization of actin and actin-binding protein from human platelets. J Cell Biol. 1981;91:201–211. doi: 10.1083/jcb.91.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JE. Mechanisms of intracellular protein transport. Nature (Lond) 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Sager R, Granick S. Nutritional control of sexuality in Chlamydomonas reinhardi. . J Gen Physiol. 1954;37:729–742. doi: 10.1085/jgp.37.6.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salisbury JL, Sanders MA. The centrin-based cytoskeleton of Chlamydomonas reinhardtii:distribution in interphase and mitotic cells. J Cell Biol. 1988;107:635–641. doi: 10.1083/jcb.107.2.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders MA, Salisbury JL. Centrin plays an essential role in microtubule severing during flagellar excision in Chlamydomonas reinhardtii. . J Cell Biol. 1994;124:795–805. doi: 10.1083/jcb.124.5.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotz F, Bathelt H, Arnold CG, Schimmer O. The architecture and organization of the Chlamydomonascell. Results of serial-section electron microscopy and a three-dimensional reconstruction. Protoplasma. 1972;75:229–254. doi: 10.1007/BF01279818. [DOI] [PubMed] [Google Scholar]
- Snell WJ. Mating in Chlamydomonas: a system for the study of specific cell adhesion. I. Ultrastructural and electrophoretic analysis of flagellar surface components involved in adhesion. J Cell Biol. 1976a;68:48–69. doi: 10.1083/jcb.68.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell WJ. Mating in Chlamydomonas: a system for the study of specific cell adhesion. II. A radioactive flagella-binding assay for quantitation of adhesion. J Cell Biol. 1976b;68:70–79. doi: 10.1083/jcb.68.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell WJ. Study of the release of cell wall degrading enzymes during adhesion of Chlamydomonasgametes. Exp Cell Res. 1982;138:109–119. doi: 10.1016/0014-4827(82)90096-9. [DOI] [PubMed] [Google Scholar]
- Snell WJ. Characterization of the Chlamydomonasflagellar collar. Cell Motil. 1983;3:273–280. [Google Scholar]
- Snell, W.J. 1993. Signal transduction during fertilization in Chlamydomonas. In Signal Transduction: Procaryotic and Simple Eucaryotic Systems. J. Kurjan and B.L. Taylor, editors. Academic Press, New York. 255–277.
- Snell WJ, White JM. The molecules of mammalian fertilization. Cell. 1996;85:629–637. doi: 10.1016/s0092-8674(00)81230-1. [DOI] [PubMed] [Google Scholar]
- Tegner MJ, Epel D. Scanning electron microscope studies of sea urchin fertilization. J Exp Zool. 1976;197:31–58. doi: 10.1002/jez.1401970105. [DOI] [PubMed] [Google Scholar]
- Tilney LG. Actin filaments in the acrosomal reaction of Limulussperm. J Cell Biol. 1975;64:289–310. doi: 10.1083/jcb.64.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney LG, Hatano S, Ishikawa H, Mooseker MS. The polymerization of actin: its role in the generation of the acrosomal process of certain echinoderm sperm. J Cell Biol. 1973;59:109–126. doi: 10.1083/jcb.59.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman P, Ford HC. Proteins of human placental microvilli. II. Identification and topology of the membrane proteins. Placenta. 1986;7:111–120. doi: 10.1016/s0143-4004(86)80002-9. [DOI] [PubMed] [Google Scholar]
- Vandekerckhove J. Actin-binding proteins. Curr Opin Cell Biol. 1990;2:41–50. doi: 10.1016/s0955-0674(05)80029-8. [DOI] [PubMed] [Google Scholar]
- von Andrian UH, Hasslen SR, Nelson RD, Erlandsen SL, Butcher EC. A central role for microvillous receptor presentation in leukocyte adhesion under flow. Cell. 1995;82:989–999. doi: 10.1016/0092-8674(95)90278-3. [DOI] [PubMed] [Google Scholar]
- Walsh TP, Weber A, Davis K, Bonder E, Mooseker M. Calcium dependence of villin-induced actin depolymerization. Biochemistry. 1984;23:6099–6102. doi: 10.1021/bi00320a030. [DOI] [PubMed] [Google Scholar]
- Ward CR, Kopf GS. Molecular events mediating sperm activation. Dev Biol. 1993;158:9–34. doi: 10.1006/dbio.1993.1165. [DOI] [PubMed] [Google Scholar]
- Weiss RL, Goodenough DA, Goodenough UW. Membrane differentiations at sites specialized for cell fusion. J Cell Biol. 1977;72:144–160. doi: 10.1083/jcb.72.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JM. Membrane fusion: the influenza paradigm. Cold Spring Harbor Symp Quant Biol. 1996;60:581–588. doi: 10.1101/sqb.1995.060.01.062. [DOI] [PubMed] [Google Scholar]
- Wiese L, Hayward PC. On sexual agglutination and mating-type substances in isogamous dioecious Chlamydomonads. III. The sensitivity of sex cell contact to various enzymes. Am J Bot. 1972;59:530–536. [Google Scholar]
- Wintermans JFGM, De Mots A. Spectrophotometric characteristics of chlorophylls a and band their pheophytins in ethanol. Biochim Biophys Acta. 1965;109:448–453. doi: 10.1016/0926-6585(65)90170-6. [DOI] [PubMed] [Google Scholar]
- Witman GB, Carlson K, Berliner J, Rosenbaum JL. Chlamydomonasflagella. I. Isolation and electrophoretic analysis of microtubules, matrix, membranes, and mastigonemes. J Cell Biol. 1972;54:507–539. doi: 10.1083/jcb.54.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagimachi R. Sperm-egg fusion. Curr Top Membr Transp. 1988;32:3–43. [Google Scholar]