

Abstract
Mouse strains AcB55 and AcB61 are resistant to malaria by virtue of a mutation in erythrocyte pyruvate kinase (PklrI90N). Linkage analysis in [AcB55 × A/J] F2 mice detected a second locus (Char9; logarithm of odds = 4.74) that regulates the blood-stage replication of Plasmodium chabaudi AS independently of Pklr. We characterized the 77 genes of the Char9 locus for tissue-specific expression, strain-specific alterations in gene expression, and polymorphic variants that are possibly associated with differential susceptibility. We identified Vnn1/Vnn3 as the likely candidates responsible for Char9. Vnn3/Vnn1 map within a conserved haplotype block and show expression levels that are strictly cis-regulated by this haplotype. The absence of Vnn messenger RNA expression and lack of pantetheinase protein activity in tissues are associated with susceptibility to malaria and are linked to a complex rearrangement in the Vnn3 promoter region. The A/J strain also carries a unique nonsense mutation that leads to a truncated protein. Vanin genes code for a pantetheinase involved in the production of cysteamine, a key regulator of host responses to inflammatory stimuli. Administration of cystamine in vivo partially corrects susceptibility to malaria in A/J mice, as measured by reduced blood parasitemia and decreased mortality. These studies suggest that pantetheinase is critical for the host response to malaria.
Malaria causes severe morbidity and mortality with an estimated 300–500 million cases worldwide and >1 million deaths annually in sub-Saharan Africa alone (www.who.int). Disease control has been hampered by the spread of drug resistance in both the Plasmodium parasites and the Anopheles gambiae insect vector and by the lack of an efficacious vaccine (1). A better understanding of malaria pathogenesis, including the identification of innate or adaptive host defense mechanisms against the blood-stage Plasmodium parasite, may provide new targets for intervention in this disease. Such mechanisms may manifest themselves as genetic determinants of susceptibility in areas of endemic diseases and during epidemics and as strain variations in mouse models of experimental infections (for review see reference 2).
In humans, malaria provides a clear example of host genetic factors influencing the onset, progression, type of disease developed, and ultimate outcome of infection (3). Epidemiological data together with linkage and association studies have shown that selection pressure from the parasite has caused retention of disease- associated but malaria-protective alleles in the human population, suggesting coevolution of the host and parasite. Such otherwise deleterious alleles include those causing sickle cell anemia (4, 5), thalassemias (6), and glucose-6-phosphate dehydrogenase deficiency (7). Polymorphisms in other erythroid proteins, including common variants of the Duffy antigen (8), the erythrocyte band 3 (anion exchanger; reference 9), and glycophorin C (10), as well as variants in the TNFα cytokine (11) and the CD36 scavenger receptor (12) are also associated with protection against malaria. Additional linkage studies in Burkina Faso have suggested a complex genetic component of susceptibility showing blood parasitemia levels linked to the 5q31-q33 region (13). Overall, the genetic component of malaria susceptibility is acknowledged to be very complex and heterogeneous in humans and is further modified by environmental factors (14).
Among the mouse malarial parasites, Plasmodium chabaudi AS provides a unique experimental model to study the erythroid stage of the disease (15). P. chabaudi AS produces an infection in mice that shares many similarities with Plasmodium falciparum malaria in humans, including anemia, splenomegaly, hepatomegaly, renal alterations, hypoglycemia, and parasite sequestration (16, 17). Inbred mouse strains vary considerably in their degree of susceptibility to infection with P. chabaudi AS, as measured by blood parasitemia at the peak of infection (peak parasitemia) and overall survival, with strains such as A/J, BALB/c, and C3H/HeJ being highly susceptible and strains such as C57BL/6J, CBA, and DBA/2 being resistant (18). We and others have shown that the genetic control of susceptibility to P. chabaudi AS infection in mice is complex and multigenic. Quantitative trait locus (QTL) mapping studies by whole genome scan in informative backcross and F2 mice have located several significant QTLs (logarithm of odds [LOD] > 4.0) on chromosomes 9 (Char1), 8 (Char2), and 3 (Char4) controlling peak parasitemia and an MHC-linked locus (Char3) regulating parasite clearance after the peak of infection (19–22). Recently, four additional suggestive linkages (LOD < 2.0) have been detected in F11 advanced intercross lines that have a modest effect on blood-stage replication of P. chabaudi and that map to chromosomes 5 (Char5 and Char6), 17 (Char7 related to Char3), and 11 (Char8 syntenic with human 5q31-q33; references 23, 24). Overall, the various Char loci individually account only for a small fraction of the phenotypic variance (∼10–20%), and they are each defined by a relatively large genetic interval (∼20–30 cM). These factors have severely complicated the positional cloning of the genes involved.
As an alternate approach, we have used the AcB/BcA set of recombinant congenic strains (RCSs) derived from C57BL/6J (B) and A/J (A) progenitors to identify novel gene effects affecting the blood-stage replication of P. chabaudi AS (25). The AcB/BcA set was produced by the systematic inbreeding of pairs of A × C57BL/6 (B6)/B6 × A double backcross (N3) mice, resulting in individual RCSs showing ∼12.5% of donor genome fixed as small congenic fragments on the background of the other strain (87.5%). This strain set is particularly useful to study multigenic traits because (a) individual resistance/susceptibility loci may have segregated in individual RCSs, enabling their study in isolation; (b) the small size of the congenic segments fixed in individual strains should facilitate positional cloning; and (c) the reassortment of parental haplotypes or the appearance of novel mutations may generate hyperphenotypes that segregate as simple traits (26–28). Phenotyping of 18 AcB/BcA strains for susceptibility to blood-stage malaria identified strains AcB55 and AcB61 as discordant, being highly resistant to P. chabaudi AS infection (low peak parasitemia; 100% survival) despite a susceptible A/J-derived genetic background, including the presence of susceptibility alleles at Char1 and Char2 (29). Linkage studies in informative [AcB55 × A] F2 mice showed that resistance was inherited as a recessive trait that was controlled by a QTL (LOD = 6.57) on the distal portion of chromosome 3, which was designated Char4 (22). Additional studies in AcB55 and AcB61, including transcript profiling in the spleen, suggested the presence of anemia in these mice characterized by expansion of the erythroid compartment, extramedullary erythropoiesis in the liver, reduced RBC counts, and constitutive reticulocytosis (30). Genetic linkage studies showed that alterations in erythroid parameters behaved as a monogenic trait that mapped to the Char4 region on chromosome 3 (28). Subsequently, we showed that the Char4 effect was caused by a loss of function mutation in Pklr (liver erythrocyte-specific pyruvate kinase [PK]) that occurred during the breeding of the AcB/BcA set and that was fixed in both AcB55 and AcB61. The PklrI90N mutation causes hemolytic anemia, which, in turn, protects against blood-stage replication of the parasite in AcB55/AcB61, including reduced peak parasitemia, rapid clearance of the parasite, and dramatic increase in survival (28).
In addition to Char4, linkage studies in [AcB55 × A] F2 mice identified a second suggestive QTL on chromosome 10 (LOD score = 2.53; D10Mit189) that maps to a 14-Mb B6-derived congenic segment fixed in AcB55. B6 alleles at this locus are protective (reduced peak parasitemia), inherited in a codominant fashion, and show an additive effect with Char4 (22). This locus has been given the temporary designation Char9. In this study, we have characterized the 77 genes contained within the 14-Mb Char9 interval with respect to (a) tissue-specific expression, (b) the presence of strain-specific alterations in the level of gene expression, and (c) strain-specific polymorphic variants in coding and regulatory regions of positional candidates. We report that alterations in the pantetheinase-encoding genes Vnn3/Vnn1 are likely responsible for the Char9 effect.
RESULTS
QTL mapping in F2 mice
Genetic linkage studies in F2 mice derived from malaria-resistant AcB55 and susceptible A/J showed that parasitemia at the peak of infection was determined, in part, by Char4 on chromosome 3, with the contribution of a second QTL on chromosome 10 (maximum linkage at D10Mit189; LOD = 2.83; P = 0.001; Table I) that had an additive effect on peak parasitemia (22). The protective effect of Char4 in AcB55 is caused by a loss of function at Pklr (PklrI90N; reference 28), which controls ∼30% of the phenotypic variance. As the marker initially used to detect the Char4 effect was located ∼10 cM distal to Pklr, we reevaluated the effect of the chromosome 10 QTL (allelic combinations at D10Mit189) on peak parasitemia in [AcB55 × A] F2 mice, taking into account their genotypes at Pklr itself (Fig. 1). This analysis shows that the chromosome 10 effect on peak parasitemia is noticeable for all Pklr allelic combinations, with B6 alleles at D10Mit189 being protective (lower parasitemia) and inherited in a codominant fashion and with a more prominent effect detected in males (Fig. 1 A) than females (Fig. 1 B). In addition, this second QTL appears to have no statistically significant effect on reticulocytosis, the phenotypic trait determined by Pklr alleles (Fig. 1, C and D). This suggests that the effect of Char9 on peak parasitemia is unrelated to PK deficiency–induced hemolytic anemia and is likely to involve a distinct mechanism. We performed regression analysis and interval mapping in 200 [AcB55 × A] F2 mice while controlling for Pklr genotypes to remove the major effect of this locus and obtained revised LOD scores of 4.74 (D10Mit167; P < 0.00002) for the chromosome 10 region (Table I), with all informative markers from the B6-derived congenic fragment showing significant linkage (LOD > 3.5) and controlling ∼8% of the phenotypic variance. Together, these results indicate that the chromosome 10 locus contributes substantially to the control of blood-stage replication of the P. chabaudi parasite. This locus was given the temporary designation Char9.
Table I.
Linkage analysis of Char9 in [AcB55 × A/J] F2 mice using peak parasitemia as a quantitative trait
| STS marker | Position | Free regression model | Control for Pklr | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr | cM | Megabasea | χ2 | LOD | Percent variance |
P-value | χ2 | LOD | Percent variance |
P-value | |
| D10Mit246 | 10 | 5 | 12.6 | 10.5 | 2.28 | 4 | 0.00527 | 18.5 | 4.02 | 7 | 9.90E-05 |
| D10Mit167 | 10 | 4 | 19.1 | 12.3 | 2.67 | 5 | 0.00219 | 21.8 | 4.74 | 8 | 1.90E-05 |
| D10Mit189 | 10 | 7 | 19.1 | 13 | 2.83 | 5 | 0.00151 | 21 | 4.57 | 8 | 2.70E-05 |
| D10Mit51 | 10 | 9 | 19.4 | 13.1 | 2.85 | 5 | 0.00141 | 21.1 | 4.59 | 8 | 2.70E-05 |
| D10Mit213 | 10 | 11 | 21.2 | 9.9 | 2.15 | 4 | 0.00717 | 16.1 | 3.50 | 6 | 3.20E-04 |
| D10Mit106 | 10 | 17 | 25.3 | 12.1 | 2.63 | 5 | 0.0023 | 18.6 | 4.04 | 7 | 9.00E-05 |
| D10Mit214 | 10 | 19 | 26.4 | 12.9 | 2.80 | 5 | 0.00158 | 17.7 | 3.85 | 7 | 1.40E-04 |
| Pklr | 3 | 44 | 91.3 | 61.6 | 13.39 | 27 | 4.10E-14 | NA | NA | NA | NA |
| D3Mit109 | 3 | 61.8 | 131.1 | 20.3 | 4.41 | 9 | 4.00E-05 | 7.3 | 1.59 | 3 | 2.36E-02 |
Chr, chromosome; cM, centimorgans; NA, not applicable; STS, sequence-tagged site.
Megabase position is based on Mouse Genome Assembly v33 (Ensembl).
Figure 1.

The effect of Char9 alleles on peak parasitemia and reticulocytosis. The effect of chromosome 10 (Char9) alleles on peak parasitemia (percentage of infected RBCs; A and B) and reticulocytosis (C and D) is shown in the context of WT or mutant (I90N) alleles at pklr (Char4) in [AcB55 × A/J] F2 mice. Each dot represents a mouse. Alleles at D10Mit189 are represented by the following: a, A/J; b, AcB55; h, heterozygote. Genotypes at Pklr are represented by the following: +/+, WT; +/−, heterozygote; −/−, mutant. Males (A and C) and females (B and D) are shown separately. Horizontal bars indicate the mean peak parasitemia value for each group.
Tissue-specific expression of candidates at the Char9 locus
The 14-Mb B6-derived segment underlying the Char9 effect in AcB55 was scrutinized for the presence of positional candidates. From public assemblies of the mouse genome (National Center for Biotechnology Information [NCBI]; Ensembl/v33), the Char9 segment is predicted to contain 77 annotated transcripts. This list includes 16 copies of the trace amine-associated receptor (Taar1, 2, 3, etc.) and 61 other genes identified in the RefSeq database (http://www.ncbi.nih.gov) that form the transcriptome of the region. These were prioritized for study by messenger RNA (mRNA) expression profiling in different tissues and in different mouse strains by haplotype mapping and nucleotide sequencing.
As a first step, we analyzed all genes from the region for RNA expression in the spleen and liver, which are two organs known to play a key role in both the erythroid and early immune response to P. chabaudi infection. Tissue expression data were obtained from two sources: first, by in silico analysis of the Genomics Institute of the Novartis Research Foundation (GNF) expression database (http://symatlas.gnf.org) and, second, by direct qualitative end point RT-PCR analysis using kidney RNA as an additional control (Table II). Using both methods, we obtained tissue expression data for 59/61 genes, whereas we were unable to generate data for two (by either RT-PCR and/or in silico data), including the Taar gene copies. Of those, 33 genes showed at least detectable mRNA expression in the spleen, whereas 28 displayed fairly robust expression in this organ (Table II).
Table II.
Target tissue expression of Char9 candidate genes
| Position | Gene | Spleen | Liver | Kidney |
|---|---|---|---|---|
| 12.7 | Sf3b5a | Low ubiquitous | Low ubiquitous | Low ubiquitous |
| 12.8 | Plagl1 | * | * | ** |
| 12.8 | 4930519B02Rik | N | * | N |
| 12.9 | Ltv1 | ** | ** | *** |
| 12.9 | Phactr2 | * | N | * |
| 13.2 | Fuca2a | Low ubiquitous | Low ubiquitous | Low ubiquitous |
| 13.2 | Pex3 | ** | *** | *** |
| 13.3 | Deadc1 | ** | ** | *** |
| 13.4 | Aig1 | ** | ** | *** |
| 13.7 | Hivep2 | N | N | N |
| 14.1 | Gpr126a | Placenta specific | Placenta specific | Placenta specific |
| 14.4 | 1110059P08Rik | * | * | * |
| 14.5 | Nmbr | N | N | N |
| 17.4 | Cited2 | N | N | * |
| 17.5 | Txlnb | Muscle specific | Muscle specific | Muscle specific |
| 17.6 | Heca | No data | No data | No data |
| 17.6 | 3110003A17Rik | ** | * | ** |
| 17.8 | Reps1 | ** | ** | ** |
| 17.9 | Ccdc28a | ** | ** | ** |
| 18.2 | Nhsl1a | Low ubiquitous | Low ubiquitous | Low ubiquitous |
| 18.3 | Hebp2 | N | * | * |
| 18.3 | D10Bwg1379ea | Brain | Brain | Brain |
| 18.6 | Perp | N | *** | *** |
| 18.7 | Tnfaip3 | *** | N | * |
| 19.1 | Olig3a | Very low ubiquitous | Very low ubiquitous | Very low ubiquitous |
| 19.3 | Ifngr1 | *** | ** | ** |
| 19.3 | Il22ra2 | * | N | N |
| 19.4 | 9230106D20Rik | N | N | * |
| 19.4 | Il20ra | * | N | N |
| 19.6 | Slc35d3 | *** | * | ** |
| 19.6 | Pex7 | * | ** | ** |
| 19.7 | Map3k5 | *** | ** | *** |
| 19.9 | Mtap7 | ** | * | *** |
| 20.0 | 4933406P04Rika | Testes specific | Testes specific | Testes specific |
| 20.0 | Bclaf1 | *** | *** | *** |
| 20.1 | 2610016C23Rika | Oocyte specific | Oocyte specific | Oocyte specific |
| 20.1 | Pde7b | * | * | * |
| 20.7 | Ahi1 | ** | * | ** |
| 20.9 | Myb | *** | * | * |
| 21.0 | Hbs1l | *** | *** | ** |
| 21.1 | Aldh8a1 | N | *** | *** |
| 21.2 | 1700021A07Rik | * | * | * |
| 21.3 | 1700020N01Rik | N | N | N |
| 21.6 | Sgk | ** | ** | *** |
| 21.9 | Raet1c | N | ** | N |
| 22.4 | Slc2a12 | N | N | ** |
| 22.4 | Tbpl1 | * | * | * |
| 22.6 | Tcf21 | ** | N | * |
| 22.9 | Eya4 | N | N | * |
| 23.5 | Rps12a | Low ubiquitous | Low ubiquitous | Low ubiquitous |
| 23.6 | Vnn3 | * | ** | N |
| 23.6 | Vnn1 | N | ** | *** |
| 23.7 | Taar1-9 | No data | No data | No data |
| 23.9 | Stx7 | ** | * | ** |
| 24.0 | Moxd1 | N | * | N |
| 24.3 | Ctgf | ** | * | ** |
| 24.4 | Enpp1 | * | * | ** |
| 24.6 | Crsp3 | *** | *** | *** |
| 24.7 | Arg1 | N | *** | N |
| 24.9 | Akap7a | Oocyte/intestine | Oocyte/intestine | Oocyte/intestine |
| 25.1 | Epb4.1l2 | ** | * | ** |
Position is given in megabases. N, not expressed. *, **, and *** represent low, intermediate, and high levels of expression, respectively.
Tissue expression was obtained from the GNF Expression database (http://symatlas.gnf.org).
This latter group of 28 genes was then investigated for possible differences in expression levels associated with resistance or susceptibility to P. chabaudi AS infection as determined by Char9. For this, we compared expression levels in spleens from susceptible A/J (PklrWT) and resistant AcB55 (PklrI90N; B6-derived Char9 region on chromosome 10) mice. Because the PklrI90N mutation has pleiotropic effects on the cellular composition of the spleen, including expansion of the erythroid compartment and reduction of the lymphoid compartment (and associated changes in cell-specific transcript profiles; references 28, 30), spleen RNA from the AcB61 strain was used as an additional control. Although AcB61 has the PklrI90N mutation and displays the associated cellular and transcriptional changes in the spleen (28, 30), it lacks the B6-congenic Char9 segment of AcB55 and instead harbors an intact A/J-derived chromosome 10 (25). Thus, mRNA expression was measured and compared in spleens of A/J, B6, AcB55, and AcB61 mice using real-time RT-PCR (see Materials and methods). Three housekeeping genes, β-actin, Gapdh, and Hprt, were amplified in parallel and used as internal controls for comparison among experimental groups. Fig. 2 shows relative expression levels of the 28 positional candidates (three to six replicates) in spleens of A/J, AcB55, and AcB61 mice. These are expressed as ratios using expression levels in B6 mice as an internal control for normalization (B6 level = 1.0 for all genes). As predicted, the PklrI90N mutation has a strong effect on cell populations and gene expression in the spleen (30), and the majority of transcripts showing differential expression between A/J (susceptible) and AcB55 (resistant) were also differentially expressed between AcB61 and A/J. Representative examples of such expression patterns include Map3k5, Pde7b, and Enpp1 (Fig. 2). These effects are most likely unrelated to Char9.
Figure 2.

mRNA expression of positional candidates in the Char9 interval. 28 genes from the Char9 region showing expression in the spleen were examined for strain-specific differences in RNA expression and possible association with malaria susceptibility or resistance. Real-time RT-PCR was performed using a LightCycler (see Materials and methods). The histograms represent the ratio of mRNA expression of each transcript measured in A/J (blue), AcB55 (purple), and AcB61 (yellow) relative to the levels measured in B6 (red). The expression level has been normalized for various control transcripts, including Gapd, actin, and Hprt (representative results with Hprt are shown). Error bars represent the SEM of normalized expression (2−ΔCt) for three to six replicates. The unique ratios detected for Vnn3 and indicative of a Char9 effect are outlined with a box, and the highly significant difference in expression levels between A/J and AcB55 for Vnn3 is indicated.
Of the 28 positional candidates examined by real-time RT-PCR, a single transcript Vnn3 (GenBank/EMBL/DDBJ accession no. NM_011979) showed a highly significant (P < 0.0001) 12-fold difference in mRNA expression level between A/J and AcB55 (AcB55 > A/J; Fig. 2, boxed area). This difference was attributable to a Char9 effect and not to PklrI90N-associated changes in the spleen because the undetectable level of Vnn3 mRNA expression in A/J was identical to that found in AcB61 and was clearly distinct from AcB55. These findings establish Vnn3 as the only gene of the 28 spleen-expressed positional candidates for which expression is differentially regulated by Char9.
Haplotype structure of the Char9 locus
A complementary approach was taken to further prioritize positional candidates. For this, the haplotype structure of the B6-derived Char9 congenic segment was determined in different inbred strains of mice and was analyzed with respect to previously reported resistance and susceptibility of these strains to infection with P. chabaudi AS (blood parasitemia and survival; reference 29). The haplotype map of 10 inbred mouse strains was determined for a 23-Mb chromosome 10 segment using available single-nucleotide polymorphic (SNP) marker information (Wellcome Trust Complex Trait Consortium [WTCTC] database; Fig. 3 A). This analysis highlighted a conserved haplotype block of ∼4 Mb that correlates most considerably with susceptibility or resistance to P. chabaudi, with five (SJL/J, C3H/HeJ, BALB/cJ, AKR/J, and A/J) of six known susceptible strains harboring the same haplotype. Closer inspection of this haplotype block revealed six transcripts showing detectable levels of expression in the spleen, including the Vnn3 gene (Fig. 3 A). To determine whether the two distinct haplotypes defining this region were associated with differential transcript levels of the six genes mapping within the block, we performed additional quantitative RT-PCR on spleen RNA. We chose five strains carrying the B6-like haplotype in the 4-Mb region and five strains carrying the A/J-like haplotype and examined the expression level of each gene relative to B6 (Fig. 3 B). Of the six genes mapping to the conserved haplotype block, only Vnn3 shows 100% correlation between the genotype and transcript expression level. This indicates a strong cis-regulation of the gene by the haplotype, thus providing additional support for this Char9 candidate.
Figure 3.

Haplotype structure of the Char9 congenic segment of AcB55. (A) The position of the B6-derived chromosome 10 segment (gray) of AcB55 is shown on the A/J chromosome 10 background (white). Microsatellite markers used in Char9 linkage mapping studies are identified along with their positions on the physical map (in megabases). SNP-based genotypes (spacing of ∼100 kB) for 11 inbred strains (identified on top) were obtained from the WTCTC SNP database. B6 alleles are gray, and the minor alleles are white. Inbred strains were segregated into P. chabaudi–susceptible (S) and –resistant (R) strains (*, DBA/1J is susceptible), and blocks of conserved haplotypes that segregated with susceptibility were noted. The region of the haplotype covering the Char9 congenic segment is outlined, and the minimal interval is indicated. The 11 genes mapping to this minimal interval are shown, with genes expressed in the spleen highlighted in black. (B) The level of mRNA expression of positional candidates Vnn3, Stx7, Ctgf, Enpp1, Crsp3, and Epb4.1l2 was determined in the spleen of 10 inbred mouse strains by quantitative RT-PCR. Strains carrying the A/J-like haplotype in this region are shown in white, and strains with the B6-like haplotype are shown in gray. Expression level is shown as a mean of three replicates and relative to B6 (B6 = 1) after normalization to Hprt. Error bars represent SEM. (C) Inbred strains of mice display two major haplotypes defined by SNPs in the Vnn3 gene that are associated with susceptibility (white) or resistance (gray) to infection with P. chabaudi (P.c.) susceptibility. Int, intronic; sil, silent/synonymous; MS, missense/nonsynonymous; NS, nonsense/premature STOP.
We sequenced all coding exons as well as the intron/exon boundaries of Vnn3 in the 10 aforementioned informative mouse strains as well as in the AcB55 RCS (Table S2, available at http://www.jem.org/cgi/content/full/jem.20061252/DC1). This information was used to construct a detailed haplotype for Vnn3 in these strains (Fig. 3 C). As expected from the haplotype structure of the region (Fig. 3 A), the analysis of 15 informative SNPs within or near Vnn3 shows the segregation of inbred mouse strains into two haplotype groups that display good correlation with susceptibility/resistance to P. chabaudi infection and 100% correlation with the expression level of the transcript. In the coding region of Vnn3, the two haplotypes are defined by a number of silent substitutions but also show three distinguishing missense mutations (M137V, I209V, and E345C; Table S2). In addition, the A/J Vnn3 transcript carries a single nucleotide substitution at position 1,592 (C1592T) that introduces a premature termination codon (Q494Stop) and causes a seven–amino acid truncation of the pantetheinase enzyme encoded by Vnn3. These results provide additional evidence identifying Vnn3 as the positional candidate for Char9.
Absence of Vanin encoded pantetheinase activity in the A/J liver
There are two Vanin genes in the mouse genome, Vnn1 and Vnn3, that encode highly similar proteins (63% identity; BLOSUM score = 624) with identical pantetheinase enzymatic activity. The Vnn1 and Vnn3 genes are adjacent to each other, are separated by 30 kb, and are contained within the minimal haplotype interval. They are expressed in different tissues and cell types: Vnn1 mRNA is found primarily in epithelial cells and is abundant in the intestine, liver, and kidney (31), whereas Vnn3 is preferentially expressed in myeloid cells with additional lower ubiquitous expression in many tissues (32). RT-PCR experiments in Table II showed that (a) as opposed to Vnn3, Vnn1 is not expressed in the spleen and that (b) Vnn1 and Vnn3 are both expressed in the liver. We investigated the effect of the Char9-associated haplotype block on the expression of Vnn1 and Vnn3 by real-time RT-PCR (not depicted) and by Northern blotting (Fig. 4 A) in the liver. Vnn1 (2.3 kb) and Vnn3 (1.8 kb) mRNAs were found abundantly expressed in the liver of B6 and AcB55 mice (B6 haplotype) but were both undetectable in the liver of A/J and AcB61 mice (A/J haplotype). These results suggest that both genes are regulated in cis and that Vanin expression in multiple tissues (liver and spleen) is affected in the malaria susceptibility haplotype of A/J strains.
Figure 4.

Effect of the Char9 genomic haplotype on Vanin mRNA expression and pantetheinase activity. Levels of Vanin mRNA and pantetheinase enzyme activity in mouse strains carrying the A/J or B6 haplotype. (A) Northern blot analysis of total liver RNA from A/J, B6, AcB55, and AcB61 using isoform-specific hybridization probes corresponding to the Vnn1 and Vnn3 genes. Ethidium-bromide staining of the agarose gel is shown to demonstrate similar RNA loading on each lane. (B) Levels of pantetheinase activity in the liver (n = 4) of A/J, B6, AcB55, and AcB61 mice as measured by spectrophotometric enzyme assay. Activity is measured in milliunits/milligram of tissue. The limit of detection was 0.1 mU/mg (dashed line). Horizontal bars indicate the mean peak parasitemia value for each group.
To determine whether the absence of transcript in the A/J strain is associated with an absence or decrease of functional protein levels, we measured pantetheinase activity in the liver from A/J, B6, AcB55, and AcB61 mice using a spectrophotometric quantitative assay. Fig. 4 B shows a complete absence of pantetheinase activity in A/J compared with B6 and AcB55, with zero out of four mice showing levels above the detection limit of the assay (<0.1 mU/mg of tissue). On the other hand, B6 and AcB55 show detectable levels of activity in all mice tested. The AcB61 strain displayed results similar to A/J, with two out of four mice having undetectable activity and two mice showing barely detectable levels of activity (0.2–0.3 mU/mg). These results demonstrate that the lack of Vnn3/Vnn1 mRNA expression in A/J mice results in the loss of functional pantetheinase activity.
Analysis of the Vnn3 putative proximal promoter region
The molecular basis for differential Vnn1/Vnn3 expression in the A and B6 haplotypes was investigated. Vnn3 and Vnn1 are in the same transcriptional orientation: the Vnn3 gene is located upstream of Vnn1, with ∼30 kb separating the most 3′ exon of Vnn3 (exon 7) from the first exon of Vnn1 (32). Thus, we first investigated the 5′ region of Vnn3 as a possible site for genetic alteration affecting transcription. A 2-kb segment directly upstream of the Vnn3 gene transcription start site was isolated as four overlapping PCR fragments (Fig. 5 A), and its nucleotide sequence was determined in different mouse strains bearing the B6-expressing haplotype (C57BL/6J, AcB55, and DBA/2J) or the A/J transcriptionally inactive haplotype (A/J, AcB61, and BALB/cJ). The sequences were aligned with the reference B6 genomic sequence (Ensembl; build v35), resulting in the identification of a number of SNPs that distinguish the two haplotypes (Fig. 5 B), including a substantially different dinucleotide repeat at position −660. More importantly, PCR amplification of the −1,000 to −500-bp region revealed an insertion of ∼340 bp of additional sequence in A/J just upstream of the variable repeat region (Fig. 5 A). The presence of additional sequence in this region of the A/J haplotype was validated by Southern blotting analysis of total genomic DNA digested with BamHI and probed with a hybridization fragment overlapping exons 1–2 of Vnn3 (Fig. 5, A and C). Sequence analysis of the −1,000 to −500-bp PCR fragment shows a complex genomic rearrangement in the A/J haplotype, including two insertions of 301 and 31 bp upstream the di-nucleotide repeat (Fig. 5 A). Together, these results show that the loss of transcriptional activity of the Vnn locus is associated with genomic rearrangement in the promotor region of the Vnn3 gene.
Figure 5.

Analysis of the Vnn3 proximal promoter region. Examination of the genomic sequence found upstream of the Vnn3 transcription start site. (A) A schematic representation of the genomic region in the putative proximal promoter of Vnn3. The 2,000 bp that were analyzed for nucleotide sequence integrity are displayed with PCR primers indicated by arrows and SNPs indicated by triangles. BamH1 restriction sites and the localization of the specific probe for Southern blotting are labeled. The major genomic rearrangement distinguishing the B6-like strains from the A/J-like strains is shown in greater detail with repeat regions and deleted regions indicated. (B) All identified polymorphisms in the promoter region are listed with respect to the reference (B6) genomic sequence. The double asterisks represent a deletion at this position. (C) The Southern blot from a BamH1 digest of genomic DNA displaying the size of the genomic rearrangement; A/J-like strains carry ∼340 bp of extra sequence. The arrows indicate the two distinct bands produced by the genomic rearrangement: the smaller band is representative of the B6-like promoters, and the larger band is representative of the A/J-like promoters.
Reduction in early parasite burden through cystamine treatment in vivo
Pantetheinases produce the low molecular weight thiol cysteamine, and the loss of pantetheinase activity can be reversed in vivo by passive treatment with cystamine (the oxidized form of cysteamine; reference 33). To further investigate the role of pantetheinase activity in Char9-associated susceptibility to malaria, we tested the effect of cystamine treatment on blood-stage replication of P. chabaudi in susceptible A/J mice. A/J and control C57BL/6J mice were treated with cystamine given daily from 2 d before infection with P. chabaudi and continued for 15 d alongside PBS-injected untreated controls, and the extent of blood-stage replication of the parasite was investigated. Cystamine treatment was found to substantially reduce P. chabaudi replication (Fig. 6 A). At day 7 after infection (near the peak of parasitemia in control mice), A/J males treated with cystamine showed a reduction in parasitemia from ∼60% (in controls) to only ∼20% parasitized RBCs (pRBCs; P < 0.0001; two-tailed Student's t test), whereas in females, the reduction was from ∼40 to ∼10% parasitemia (P = 0.017; two-tailed Student's t test). Cystamine treatment also reduced parasitemia in B6 mice, albeit less so than in A/J mice, with an effect visible in B6 males only (from ∼35 to ∼10%; P = 0.0003; two-tailed Student's t test). Finally, cystamine treatment increased overall survival to infection in A/J females by 30% (P = 0.06; log-rank test), whereas it delayed the onset of mortality in A/J males (Fig. 6 B). Thus, the passive administration of cystamine can partially correct malaria susceptibility in A/J mice, as witnessed by reduced parasite replication before the peak of infection and increased survival. These results provide further evidence for a functional link between Vnn genes and susceptibility to malaria.
Figure 6.

Administration of cystamine in vivo and the effect on P. chabaudi replication in A/J and B6 mice. The effect of cystamine treatment on P. chabaudi AS infection in A/J and B6 mice. (A) Male and female mice were treated with cystamine (120 mg/kg of body weight) or with PBS for 2 d before infection with 106 parasitized erythrocytes (pRBCs) and continuing daily for 15 d. Boxes and whisker plot shows parasitemia (percent pRBC) levels for 8–10 mice per group at day 7 after infection for control (gray bars) and treated mice (white bars). The median parasitemia is indicated by a horizontal line, whereas the box represents the quartiles, and the whiskers represent the end points of the parasitemia distribution. Error bars represent SEM. Statistical differences between groups are indicated by asterisks: **, P < 0.001; *, P < 0.02. (B) Effect of cystamine treatment on the survival of A/J mice after P. chabaudi AS infection. Dashed lines represent treated mice, whereas solid lines represent control animals. Cont, control; treat, treated; pRBC, parasitized RBC.
DISCUSSION
The malarial parasite has a complex life cycle in its mammalian host that involves sequential replication in the erythrocyte and sequestration in different tissues such as liver and brain microvasculature. Protective host defenses against Plasmodium species are not fully understood but involve different cell types and physiological and biochemical pathways at each stage of the infection. In humans and mice (for review see references 34, 35), the genetic component of susceptibility to malaria is very complex and may reflect the plurality of sites at which genetic variations can exert an influence on pathophysiology and disease outcome. However, isolating single gene effects and identifying their molecular basis may provide valuable new insight into protective host mechanisms. RCSs of mice (25) represent a valuable tool to break down multigenic traits into single gene effects.
The AcB55 strain is very resistant to P. chabaudi AS and shows low parasitemia at the peak of infection, early clearance of the parasite, and uniform survival to infection (29). We have previously shown that resistance in AcB55 is caused by two genetic effects (22). The first (Char4), on chromosome 3, is caused by a loss of function in the erythrocyte form of PK (28). This is a mutation that arose on the A/J background of the AcB set early during their derivation and is also found in AcB61. In AcB55 and AcB61, deficiency in pklr causes hemolytic anemia (30). The malaria-protective effect in PK-deficient erythrocytes is linked to the increased expression of senescence markers, increased phagocytosis of intact and Plasmodium-infected cells, and increased turnover of erythrocytes (unpublished data). The second genetic effect (Char9; LOD score = 4.74) is specific to AcB55 (and is not seen in AcB61) and maps to a 14-Mb B6-derived segment of chromosome 10 fixed in AcB55 (22). In informative [AcB55 × A] F2 mice, the Char9 effect is additive to Char4, but Char9 alleles have no consequence on anemia-associated hematological parameters (e.g., reticulocyte counts), suggesting that the effect of Char9 on peak parasitemia is unrelated to PK deficiency–induced hemolytic anemia and likely involves another pathway.
In this study, we have undertaken the positional cloning of the gene underlying the Char9 QTL. Through a combination of systematic RNA expression analysis of 77 transcripts in the region and SNP-based haplotype data, we have converged on the Vnn genes as underlying the Char9 effect and contributing to P. chabaudi resistance in the AcB55 strain. The evidence pointing to Vnn3 as the strongest candidate in the region includes the following: (a) Vnn3 expression in the spleen, which is a key target organ for erythroid response to P. chabaudi–induced anemia and for general mononuclear phagocyte-mediated early immune response; (b) differential expression of Vnn3 between AcB55, AcB61, and A/J strains that is independent of the Pklr alleles; (c) location within a conserved haplotype showing good cosegregation with P. chabaudi susceptibility; (d) a unique cis-regulation of transcript levels based on this discriminating haplotype; and (e) a complex rearrangement in the promoter region associated with the A/J haplotype and lack of Vnn1/Vnn3 mRNA expression in the spleen and liver. In addition, we have shown that in the malaria susceptibility haplotype, the complete absence of Vnn1/Vnn3 expression at the mRNA level results in a lack of Vanin-encoded pantetheinase enzyme in the susceptible A/J strain (compared with AcB55 and B6). In addition, we have shown that passive administration in vivo of the pantetheinase reaction product cysteamine can partially correct susceptibility to P. chabaudi infection in A/J mice. This provides a functional link between Char9, pantetheinase activity, and host response to malaria.
There are three genes in humans (VNN1, VNN2, and VNN3) clustered on 6q22, whereas there are only two in mice (Vnn1 and Vnn3; reference 32). The genes show a very similar structure consisting of seven exons and encode proteins with a high degree of similarity (∼65% sequence identity; reference 36). In the mouse, Vnn1 mRNA is abundantly expressed in epithelial cells of the kidney, liver, and intestine; Vnn3 mRNA is expressed in myeloid cells, including circulating granulocytes and monocytes (but not lymphoid cells) and is abundant in the spleen and liver (32). No Vnn1 mRNA was detected in hematopoietic tissues (32). Vnn1 and Vnn3 code for pantetheinases, a group of ectoenzymes in the catabolic pathway of coenzyme A that hydrolyze d-pantetheine to pantothenic acid (vitamine B5) and cysteamine (37, 38). Although pantothenic acid is critical to many metabolic processes as a key component of CoA (39), cysteamine has potent antioxidant properties and acts as an inhibitor of γ-glutamyl/cysteine synthetase, the rate-limiting step in the biosynthesis of glutathione (GSH; reference 40). In addition to distinct tissue and cell expression, Vnn1 and Vnn3 act at different subcellular sites by virtue of distinct C-terminal domains (32). Although Vnn1 is attached to the membrane via a glycosyl-phosphatidylinositol anchor and a putative transmembrane domain, Vnn3 lacks these features and appears to be cytoplasmic and/or secreted (32). Vanins were originally described as cell adhesion proteins that are expressed by thymus stromal cells and play a role in the entry of hematopoietic precursors in this organ (41). They are also expressed on neutrophils and are required for the transendothelial migration of these cells in tissues (42).
Importantly, recent studies in Vnn1-deficient mice (Vnn1 −/−) have established that through the production of cysteamine, pantetheinases play a critical role in host response to oxidative and inflammatory stimuli (31, 33, 43). Pantetheinase-produced cysteamine acts to up-regulate the inflammatory response locally in the liver and intestine (membrane-bound Vnn1) but also at distant sites through Vnn3 expressed by myeloid cells. This model is based on the observations that Vnn1 −/− mice show reduced inflammatory response (infiltration of myeloperoxidase+ cells and MIP2 chemokine production), are protected from tissue damage, and show increased survival in acute (high-dose indomethacin) and chronic (Schistosoma mansoni infection) intestinal models of oxidative and inflammatory stress (43). In addition, Vnn1 and Vnn3 mRNA expression is induced by γ irradiation, and Vnn1 −/− mice are resistant to radiation-induced oxidative damage (33). Protection is associated with the reduced recruitment of myeloid cells at the site of inflammation, reduced production of proinflammatory cytokines (IL-1β, IL-6, and MIP2), and increased survival (33). In both models, the protective effect is associated with increased GSH production and can be abrogated by passively administered cysteamine.
Our analysis of the Vnn1/Vnn3 locus identified several sequence polymorphisms, including three missense mutations in the Vnn3 protein. These variations do not constitute obvious loss of function, as the substitutions are either very conservative (M137V and I209V) or affect a residue (E347C) that is not conserved in distant fly and fish relatives (ClustalW; http://www.ebi.ac.uk/clustalw/). In addition, an A/J-specific nonsense mutation was detected that introduces a termination codon and causes a seven-residue C-terminal truncation of Vnn3. Although the truncated segment is highly conserved in rat and human Vnn3 homologues, it is not conserved in the more distant frog Vnn3 relative and is absent from the glycosyl-phosphatidylinositol–linked pantetheinases (VNN1 and VNN2; ClustalW; reference 32). Therefore, the consequences of this A/J-specific alteration on the pantetheinase activity of Vnn3 await formal biochemical characterization of the mutant enzyme. However, we observed that the A/J haplotype at Vnn1/Vnn3 results in a loss of pantetheinase activity in the liver. This loss of expression is associated with a complex rearrangement in the promotor region of Vnn3 involving either the loss of key regulatory elements or the insertion of silencing sequences. Whether the loss in activity is a direct result of transcriptional silencing, is a consequence of the truncated A/J protein, or is a combination of the two remains to be formally tested. The lack of pantetheinase activity in the A/J strain may consequently affect cysteamine levels and GSH metabolism in vivo more profoundly than the loss of Vnn1 activity in the corresponding mouse mutant, in which the deletion of Vnn1 is compensated for by increased Vnn3 mRNA expression (in the liver; reference 33), which is not seen in A/J mice (Fig. 4 B).
How would the loss of pantetheinase activity in the spleen, liver, and myeloid cells affect susceptibility to malaria? Although a formal answer to this question awaits the creation and characterization of mutant mice bearing loss of function mutations at both Vnn1 and Vnn3, we may speculate on a possible mechanism. During blood-stage replication of the Plasmodium parasite, an early inflammatory response is required to control the initial burst of intraerythrocytic parasite multiplication (for review see reference 44). This is dependent on sensing the presence and phagocytosis of parasitized erythrocytes and parasite products by spleen cells of the myeloid lineage, including macrophages, DCs, and NK cells that quickly respond by producing proinflammatory cytokines such as IL-12, TNFα, IFNγ, and IL-6 (for review see 45). Interfering in this response with specific antibodies or with gene mutations results in increased parasitemia and decreased survival (46–49). The normal inflammatory response is progressively dampened by the counteraction of regulatory cytokines such as TGF-β and IL-10, and this response is essential for the expression of adaptive immune mechanisms and for resolving the infection (for review see references 50, 51). The balance between the activating (IL-12/IFNγ) and suppressing signals (TGF-β/IL-10) is critical and carefully controlled, and aberrations in either response have pathological consequences (52, 53). Given the documented role of pantetheinases and their cysteamine reaction product in response to inflammatory stimuli such as S. mansoni infection and γ irradiation, it is tempting to hypothesize that a reduction in cysteamine levels in the spleen, liver, and peripheral myeloid cells of animals bearing the Vnn1/Vnn3 nonexpressing susceptibility haplotype may cause a decreased inflammatory response to rising parasitemia in such P. chabaudi–infected animals. We have shown that the administration of cystamine in vivo diminishes parasitemia levels before the peak of infection and increases survival to the infection in the otherwise highly susceptible A/J strain. This model is exciting, as it points to cysteamine as a major regulator of host inflammatory response during Plasmodium infection.
Cysteamine could additionally affect blood-stage replication of Plasmodium by modulating the intracellular redox environment of the erythrocyte itself. Intracellular Plasmodium parasites are known to be sensitive to oxidant stress and O2 − radicals, and observable intraerythrocytic death of blood-stage parasites through the oxidative stress response has been proposed to contribute substantially to the control of infection by the host (for reviews see references 54, 55). GSH is a key mediator of cellular redox, and, when cysteamine levels are low, there is an increase in the stores of GSH, and cell redox status is altered (33). Thus, increased GSH levels in erythrocytes and/or the spleen of pantetheinase-compromised animals may further dampen inflammatory response and facilitate parasite replication. A related mechanism has been proposed to explain the protective nature of G6PD deficiency in which the lack of G6PD uncouples the pentose phosphate shunt and impairs the reduction of reactive oxygen species by GSH (56, 57).
Our findings suggest a role for pantetheinase and associated inflammatory response modulation in malaria susceptibility. These findings in the mouse provide possible new targets for parallel intervention in the human disease.
MATERIALS AND METHODS
Mice and parasites.
A/J and C57BL/6J mice were purchased from the Jackson ImmunoResearch Laboratories. The AcB/BcA panel of RCSs was generated according to a breeding scheme described previously (58) and has been genotyped for >600 informative markers (25). AcB55 and AcB61 strains were obtained as breeding pairs from Emerillon Therapeutics, Inc., and were subsequently maintained as breeding colonies at McGill University. All mice were maintained and handled according to guidelines of the Canadian Council on Animal Care.
P. chabaudi AS infection.
A lactate dehydrogenase virus-free isolate of P. chabaudi AS, which was originally obtained from D. Walliker (University of Edinburgh, Edinburgh, Scotland, UK), was maintained by weekly passage in B6 mice by i.p. infection with 106 pRBCs suspended in 1 ml pyrogen-free saline. After infection, the percentage of pRBCs was determined daily on duplicate thin blood smears stained with Dif-Quick (American Scientific Products) on days 4–28 after infection as described previously (22).
Cystamine (Sigma-Aldrich) was resuspended in PBS and dosed at 120 mg/kg of body weight. Injections were performed i.p. on mice starting 2 d before infection with P. chabaudi (as described in the previous paragraph) and continued until day 13 after infection. Untreated control animals were injected with PBS alone. A group of 8–10 mice were tested per condition (treated and control), per strain, and per sex.
RNA isolation and cDNA synthesis.
Total cellular RNA was extracted from the spleen and liver using a commercial reagent and according to the manufacturer's recommended instructions (TRIzol; Invitrogen). Tissues were snap frozen in liquid nitrogen, and 100 mg of tissue was homogenized by mechanical disruption using a homogenizer (Polytron; Brinkmann Instruments) in a final 2-ml volume of TRIzol reagent. The samples were incubated for 5 min at 20°C followed by chloroform extraction. The aqueous phase was removed, and nucleic acids were precipitated with isopropanol. Pellets were washed with 75% ethanol and dissolved in ribonuclease-free water treated with 0.1% diethlypyrocarbamate. The quality of RNA preparations was verified by electrophoresis on 1% formaldehyde-containing agarose gels before use. To ensure high quality RNA, further purification was performed using RNeasy columns (QIAGEN) on 100 μg of total RNA according to the recommended protocol from QIAGEN.
For cDNA synthesis, 2 μg of total RNA pooled from three male mice was converted to cDNA in a 20-μl reaction containing 1 U of moloney mouse leukemia virus reverse transcriptase (Invitrogen), dinucleotide triphosphates (dNTPs; 500 μM each), 5 μM oligonucleotide d(T) primers, First Strand Buffer, 0.1 M dithiothreitol, and 1 μL RNAGuard (GE Healthcare), and the reaction was allowed to proceed for 50 min at 37°C. The reaction was inactivated by 15 min of incubation at 70°C. A minimum of two replicate RT reactions along with a negative control lacking reverse transcriptase (−RT) were performed for each sample.
Tissue-specific expression by RT-PCR.
The expression of individual genes in select tissues was tested by standard PCR amplification of RT products (RT-PCR) using total oligonucleotide d(T)–primed cDNA from the spleen, liver, or kidney (as described in the previous section) from mouse strains A/J, B6, and AcB55. The reaction mixture contained 10 pmol of gene-specific oligonucleotides (Table S1, available at http://www.jem.org/cgi/content/full/jem.20061252/DC1), 200 μM dNTPs, 2 mM MgCl2, 2 μL cDNA (1:10 dilution), and Taq polymerase (Invitrogen). Conditions for the PCR amplification of each cDNA were kept standard to allow for the qualitative comparison of expression level and were as follows: 28 cycles of 95°C for 30 s, 57°C for 30 s, and 72°C for 30 s followed by a 10-min final extension. PCR products were separated by agarose gel electrophoresis, stained with ethidium bromide (0.1% final), and photographed under UV. Oligonucleotide pairs spanning intron/exon boundaries were designed for each gene (one to three sets) from published sequences and using Primer3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). Amplicons were situated near the 3′ end of the transcript when possible. The sequences of primer pairs used for the amplification of each transcript are listed in Table S1.
Real-time RT-PCR.
cDNA samples generated with spleen, liver, and kidney RNA from mouse strains A/J, B6, AcB55, and AcB61 were used for real-time RT-PCR analysis. PCR amplification was performed using the SYBR Green I PCR kit (QIAGEN). A typical reaction included 2 μL cDNA template (1:10 dilution of initial RT reaction; see previous section), 3 mM of final MgCl2 concentration, 10 pmol of gene-specific oligonucleotide primer pairs (Table S1), and Quantitect Mix (includes SYBR green, dNTPs, Taq polymerase, and buffer; QIAGEN). Oligonucleotides were designed as in the previous section with high melting temperatures and amplicon sizes of 150–200 bp to ensure high specificity and PCR efficiency appropriate for real-time monitoring. Standard curves for PCR amplification were generated for each set of oligonucleotide primer pairs using a pool of cDNAs from A/J, B6, and AcB55 serially diluted at 1:10, 1:100, 1:1,000, 1:10,000, and 1:100,000. This was done to ensure optimal PCR amplification (90% or above) and similar reaction efficiencies for all sample sets. This allowed for use of the 2−ΔΔCt method of relative quantification, which was described previously (59). The data was normalized against three housekeeping genes (Hprt, Gapdh, and β-actin) that were used as internal controls and assayed under the same conditions. Results normalized to Hprt were chosen as representative, as relative expression levels were similar when calculated using any of the tested housekeeping genes. The calibrator sample, which allows for comparison between runs, was chosen as B6. Three to six replicate PCR reactions were performed for each gene and were used to calculate averages.
Real-time PCR was performed on a standard 32-capillary LightCycler instrument (Roche). Cycle threshold values representing the cycle crossing point were calculated by the LightCycler software (Roche) after setting a baseline threshold value to exclude fluorescence noise. For the calculation of normalized expression, the mean cycle threshold value of the reference gene (i.e., Hprt) was subtracted from the target gene for individual runs and was converted to expression by 2−ΔCt. The mean expression values from separate runs were calculated and normalized to the value for B6 (i.e., B6 value set at 1.0). SEM normalized expression was calculated from the following formula (60):
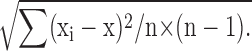 |
Sequencing.
Individual exons from the Ifngr1, Tnfaip3, Il22ra2, Sgk, Tcf21, Vnn3, and Vnn1 genes were PCR amplified from genomic DNA of A/J, C57BL/6J, AcB61, and AcB55. The PCR products were visualized by agarose gel electrophoresis to monitor purity followed by purification using Exo-SapIT (USB Corporation). Purified PCR products were subjected to cycle sequencing in the presence of fluorescently labeled BigDye Terminator (Applied Biosystems), and the products were analyzed using an automated instrument (ABI 3100; Applied Biosystems). Sequences from all strains were aligned with the reference sequence (GenBank/EMBL/DDBJ accession no. NM_011979) in BioEdit (http://www.mbio.ncsu.edu/BioEdit/bioedit.html) using a ClustalW base algorithm. The Vnn3 coding exons from AKR/J, CBA/J, 129/Sv, C3H/HeJ, DBA/2J, and BALB/cJ were sequenced using the same procedure. The 5′ upstream region of the Vnn3 gene from different inbred strains was amplified by PCR as four overlapping segments tiling 2,000 bp upstream from the transcription start site. PCR products were either sequenced directly or after subcloning into TOPO TA vector (Invitrogen). A total of eight independent plasmid subclones were sequenced for each fragment and for each inbred strain, and sequences were aligned to the Ensembl build v34 reference upstream genomic sequence (gene ID: ENSMUSG00000020010) using the BioEdit software. Oligonucleotide primers used for sequencing the Vnn3 coding region and promoter are listed in Table S1.
Northern blotting.
Aliquots of 20 μg of total spleen RNA were resolved by electrophoresis in a 1% agarose denaturing gel containing 8% formaldehyde. The RNA was transferred onto a Hybond-N (GE Healthcare) nylon membrane according to the manufacturer's instructions and fixed to the membrane by UV cross-linking. The membranes were prehybridized for 16 h at 65°C in buffer containing 1% sodium dodecyl sulfate, 1 M NaCl, 10% dextran sulfate, and 100 ng/ml of heat-denatured salmon sperm DNA. Membranes were hybridized for 16 h at 65°C in the same buffer containing radiolabeled probes (1 × 106 cpm/ml). Blots were washed in a series of buffer conditions of increasing stringency up to 0.1× SSC (20× SSC is 3 M sodium chloride and 0.3 M sodium citrate, pH 7.0) and 0.5% SDS (1 h at 65°C) and exposed to XAR films (−80°C; Kodak) with intensifying screens. Hybridization probes included cDNAs for Vnn3 and Vnn1. The 651-bp Vnn3 probe was PCR amplified as cDNA from B6 liver (generated by RT with moloney mouse leukemia virus as described in the RNA isolation and cDNA synthesis section) using the primers 5′-AGTTCCATTCCATTCGGTGTG-3′ and 5′-GACGTCCATCTCTTGAGACTTC-3′. The 487-bp Vnn1 probe was amplified from B6 liver cDNA using the primers 5′-TTCCCAGGGTAAACTGGTTGC-3′ and 5′-GGTTTTGGTTGGGGTTGATTC-3′.
Pantetheinase activity assay.
1 g of mouse livers were homogenized in 50 mM phosphate buffer, pH 7.0 (2.0 ml), and 1 mM dithiothreitol, were ultracentrifuged for 1 h at 100,000 g, and supernatants were used for the soluble enzymatic assay. Pellets were suspended in 50 mM phosphate buffer, pH 7.0 (1.0 mL), 1 mM dithiothreitol, and 0.1% DOC, were ultracentrifuged for 1 h at 100,000 g after 30 min at 37°C, and supernatants were used for membrane-bound activity. Pantetheinase activity was measured spectrophotometrically at 25°C; 100–300 μl of tissue extracts was added to 10 mM K-phosphate buffer, pH 8.0, containing 300 μM pantothenate-p-nitroanilide, and the increase in absorbance at 387 nm (extinction coefficient = 12,144 M−1 cm−1) was recorded over time. 1 U pantetheine hydrolase is defined as the amount of enzyme that hydrolyzes 1 μmol pantothenate-p-nitroanilide per minute at 25°C.
Southern blotting.
For Southern blotting, 15 μg of genomic DNA was digested to completion with a 10× excess (10 U/μg of DNA) of restriction enzymes BamH I or SacI followed by ethanol precipitation. Digested genomic DNA was separated by electrophoresis on 1% agarose gel in Tris-acetate-EDTA buffer (0.04 M Tris, 0.03 M acetic acid, 0.001 M EDTA, and 0.02 M sodium acetate, pH 7.6) and transferred to a nylon hybrization membrane (GeneScreen; NEN Life Science Products) by capillary blotting in 10× SSC (sodium chloride–sodium citrate buffer). The membranes were prehybridized for 16 h and were then hybridized with 32P-radiolabeled DNA probes (specific activity of 5 × 108 cpm/μg of DNA) for 16 h at 42°C in 50% formamide, 5× SSC, 1% SDS, 10% dextran sulfate, 20 mM Tris, pH 7.5, 1× Denhardt's solution, and 200 μg/ml of sonicated denatured salmon sperm DNA. The membrane was washed to a final stringency of 0.1× SSC and 0.5% SDS at 65°C for 30 min and was exposed to XAR film (Kodak) at −80°C with an intensifying screen. The hybridization probe consisted of a 447-bp PCR fragment corresponding to nucleotide positions 25–472 downstream of the transcription start site comprising the first two exons of Vnn3.
Haplotype construction.
Haplotypes for the 16 Mb overlapping the Char9 region were obtained from M. Daly (Massachussetts Institute of Technology, Cambridge, MA) as well as from the WTCTC Mouse Strain SNP database (http://www.well.ox.ac.uk/mouse/INBREDS) and were examined using Excel software (Microsoft). Additional single-nucleotide polymorphisms were derived for the Vnn3 locus from sequencing (see the Sequencing section) and were subsequently genotyped in various inbred strains by PCR amplification from genomic DNA using oligonucleotide primer pairs listed in Table S1 followed by DNA sequencing. The two alleles at each polymorphism were color coded to aid in the visualization of shared haplotypes, with the B6 allele represented in gray and the other allele shown as white (Fig. 3 A).
Statistical analysis.
The effect of Char9 on the quantitative trait peak parasitemia while controlling for the dominating effect of pklr alleles was analyzed using Map Manager QT (61), generating chi-square values using interval regression in 200 [AcB55 × A] F2 mice. Student's t tests were performed using Prism software (GraphPad).
Online supplemental material.
Table S1 provides information about the oligonucleotides used for quantitative RT-PCR and nucleotide sequencing, and Table S2 provides results of the nucleotide sequencing of candidate genes. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20061252/DC1.
Acknowledgments
We thank Melissa Mathieu for help with nucleotide sequencing of candidate genes.
This work was supported by a Canadian Institute of Health Research (CIHR) Team Grant in Malaria (MT-13721) and by grants to P. Gros and M.M. Stevenson from CIHR and the Canadian Genetic Diseases Network. G. Min-Oo is supported by a CIHR Canada Graduate Scholarship, and P. Gros is supported by a James McGill Professorship.
The authors have no conflicting financial interests.
Abbreviations used: B6, C57BL/6; dNTP, dinucleotide triphosphate; GSH, glutathione; LOD, logarithm of odds; mRNA, messenger RNA; PK, pyruvate kinase; pRBC, parasitized RBC; QTL, quantitative trait locus; RCS, recombinant congenic strain; SNP, single-nucleotide polymorphic.
References
- 1.Moorthy, V.S., M.F. Good, and A.V. Hill. 2004. Malaria vaccine developments. Lancet. 363:150–156. [DOI] [PubMed] [Google Scholar]
- 2.Kwiatkowski, D.P. 2005. How malaria has affected the human genome and what human genetics can teach us about malaria. Am. J. Hum. Genet. 77:171–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hill, A.V. 1998. The immunogenetics of human infectious diseases. Annu. Rev. Immunol. 16:593–617. [DOI] [PubMed] [Google Scholar]
- 4.Allison, A.C. 1954. Protection afforded by sickle-cell trait against subtertian malarial infection. Br. Med. J. 1:290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willcox, M., A. Bjorkman, J. Brohult, P.O. Pehrson, L. Rombo, and E. Bengtsson. 1983. A case-control study in northern Liberia of Plasmodium falciparum malaria in haemoglobin S and beta-thalassaemia traits. Ann. Trop. Med. Parasitol. 77:239–246. [DOI] [PubMed] [Google Scholar]
- 6.Weatherall, D.J. 2001. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat. Rev. Genet. 2:245–255. [DOI] [PubMed] [Google Scholar]
- 7.Ruwende, C., S.C. Khoo, R.W. Snow, S.N. Yates, D. Kwiatkowski, S. Gupta, P. Warn, C.E. Allsopp, S.C. Gilbert, N. Peschu, et al. 1995. Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature. 376:246–249. [DOI] [PubMed] [Google Scholar]
- 8.Miller, L.H., S.J. Mason, D.F. Clyde, and M.H. McGinniss. 1976. The resistance factor to Plasmodium vivax in blacks. The Duffy-blood-group genotype, FyFy. N. Engl. J. Med. 295:302–304. [DOI] [PubMed] [Google Scholar]
- 9.Allen, S.J., A. O'Donnell, N.D. Alexander, C.S. Mgone, T.E. Peto, J.B. Clegg, M.P. Alpers, and D.J. Weatherall. 1999. Prevention of cerebral malaria in children in Papua New Guinea by southeast Asian ovalocytosis band 3. Am. J. Trop. Med. Hyg. 60:1056–1060. [DOI] [PubMed] [Google Scholar]
- 10.Patel, S.S., R.K. Mehlotra, W. Kastens, C.S. Mgone, J.W. Kazura, and P.A. Zimmerman. 2001. The association of the glycophorin C exon 3 deletion with ovalocytosis and malaria susceptibility in the Wosera, Papua New Guinea. Blood. 98:3489–3491. [DOI] [PubMed] [Google Scholar]
- 11.McGuire, W., A.V. Hill, C.E. Allsopp, B.M. Greenwood, and D. Kwiatkowski. 1994. Variation in the TNF-alpha promoter region associated with susceptibility to cerebral malaria. Nature. 371:508–510. [DOI] [PubMed] [Google Scholar]
- 12.Aitman, T.J., L.D. Cooper, P.J. Norsworthy, F.N. Wahid, J.K. Gray, B.R. Curtis, P.M. McKeigue, D. Kwiatkowski, B.M. Greenwood, R.W. Snow, et al. 2000. Malaria susceptibility and CD36 mutation. Nature. 405:1015–1016. [DOI] [PubMed] [Google Scholar]
- 13.Rihet, P., Y. Traore, L. Abel, C. Aucan, T. Traore-Leroux, and F. Fumoux. 1998. Malaria in humans: Plasmodium falciparum blood infection levels are linked to chromosome 5q31-q33. Am. J. Hum. Genet. 63:498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwiatkowski, D. 2000. Genetic susceptibility to malaria getting complex. Curr. Opin. Genet. Dev. 10:320–324. [DOI] [PubMed] [Google Scholar]
- 15.Li, C., E. Seixas, and J. Langhorne. 2001. Rodent malarias: the mouse as a model for understanding immune responses and pathology induced by the erythrocytic stages of the parasite. Med. Microbiol. Immunol. (Berl.). 189:115–126. [DOI] [PubMed] [Google Scholar]
- 16.Cox, J., S. Semoff, and M. Hommel. 1987. Plasmodium chabaudi: a rodent malaria model for in-vivo and in-vitro cytoadherence of malaria parasites in the absence of knobs. Parasite Immunol. 9:543–561. [DOI] [PubMed] [Google Scholar]
- 17.Landau, I., and P. Gautret. 1998. Animal models: rodents. In Malaria: Parasite Biology, Pathogenesis, and Protection. I.W. Sherman, editor. ASM Press, Washington, DC. 401–417.
- 18.Stevenson, M.M., J.J. Lyanga, and E. Skamene. 1982. Murine malaria: genetic control of resistance to Plasmodium chabaudi. Infect. Immun. 38:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foote, S.J., R.A. Burt, T.M. Baldwin, A. Presente, A.W. Roberts, Y.L. Laural, A.M. Lew, and V.M. Marshall. 1997. Mouse loci for malaria-induced mortality and the control of parasitaemia. Nat. Genet. 17:380–381. [DOI] [PubMed] [Google Scholar]
- 20.Fortin, A., A. Belouchi, M.F. Tam, L. Cardon, E. Skamene, M.M. Stevenson, and P. Gros. 1997. Genetic control of blood parasitaemia in mouse malaria maps to chromosome 8. Nat. Genet. 17:382–383. [DOI] [PubMed] [Google Scholar]
- 21.Burt, R.A., T.M. Baldwin, V.M. Marshall, and S.J. Foote. 1999. Temporal expression of an H2-linked locus in host response to mouse malaria. Immunogenetics. 50:278–285. [DOI] [PubMed] [Google Scholar]
- 22.Fortin, A., L.R. Cardon, M. Tam, E. Skamene, M.M. Stevenson, and P. Gros. 2001. Identification of a new malaria susceptibility locus (Char4) in recombinant congenic strains of mice. Proc. Natl. Acad. Sci. USA. 98:10793–10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hernandez-Valladares, M., J. Naessens, J.P. Gibson, A.J. Musoke, S. Nagda, P. Rihet, O.K. Ole-MoiYoi, and F.A. Iraqi. 2004. Confirmation and dissection of QTL controlling resistance to malaria in mice. Mamm. Genome. 15:390–398. [DOI] [PubMed] [Google Scholar]
- 24.Hernandez-Valladares, M., P. Rihet, O.K. Ole-MoiYoi, and F.A. Iraqi. 2004. Mapping of a new quantitative trait locus for resistance to malaria in mice by a comparative mapping approach with human Chromosome 5q31-q33. Immunogenetics. 56:115–117. [DOI] [PubMed] [Google Scholar]
- 25.Fortin, A., E. Diez, D. Rochefort, L. Laroche, D. Malo, G.A. Rouleau, P. Gros, and E. Skamene. 2001. Recombinant congenic strains derived from A/J and C57BL/6J: a tool for genetic dissection of complex traits. Genomics. 74:21–35. [DOI] [PubMed] [Google Scholar]
- 26.Bodnar, J.S., A. Chatterjee, L.W. Castellani, D.A. Ross, J. Ohmen, J. Cavalcoli, C. Wu, K.M. Dains, J. Catanese, M. Chu, et al. 2002. Positional cloning of the combined hyperlipidemia gene Hyplip1. Nat. Genet. 30:110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Wezel, T., A.P. Stassen, C.J. Moen, A.A. Hart, M.A. van der Valk, and P. Demant. 1996. Gene interaction and single gene effects in colon tumour susceptibility in mice. Nat. Genet. 14:468–470. [DOI] [PubMed] [Google Scholar]
- 28.Min-Oo, G., A. Fortin, M.F. Tam, A. Nantel, M.M. Stevenson, and P. Gros. 2003. Pyruvate kinase deficiency in mice protects against malaria. Nat. Genet. 35:357–362. [DOI] [PubMed] [Google Scholar]
- 29.Fortin, A., M.M. Stevenson, and P. Gros. 2002. Complex genetic control of susceptibility to malaria in mice. Genes Immun. 3:177–186. [DOI] [PubMed] [Google Scholar]
- 30.Min-Oo, G., A. Fortin, M.F. Tam, P. Gros, and M.M. Stevenson. 2004. Phenotypic expression of pyruvate kinase deficiency and protection against malaria in a mouse model. Genes Immun. 5:168–175. [DOI] [PubMed] [Google Scholar]
- 31.Pitari, G., F. Malergue, F. Martin, J.M. Philippe, M.T. Massucci, C. Chabret, B. Maras, S. Dupre, P. Naquet, and F. Galland. 2000. Pantetheinase activity of membrane-bound Vanin-1: lack of free cysteamine in tissues of Vanin-1 deficient mice. FEBS Lett. 483:149–154. [DOI] [PubMed] [Google Scholar]
- 32.Martin, F., F. Malergue, G. Pitari, J.M. Philippe, S. Philips, C. Chabret, S. Granjeaud, M.G. Mattei, A.J. Mungall, P. Naquet, and F. Galland. 2001. Vanin genes are clustered (human 6q22-24 and mouse 10A2B1) and encode isoforms of pantetheinase ectoenzymes. Immunogenetics. 53:296–306. [DOI] [PubMed] [Google Scholar]
- 33.Berruyer, C., F.M. Martin, R. Castellano, A. Macone, F. Malergue, S. Garrido-Urbani, V. Millet, J. Imbert, S. Dupre, G. Pitari, et al. 2004. Vanin-1−/− mice exhibit a glutathione-mediated tissue resistance to oxidative stress. Mol. Cell. Biol. 24:7214–7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fortin, A., M.M. Stevenson, and P. Gros. 2002. Susceptibility to malaria as a complex trait: big pressure from a tiny creature. Hum. Mol. Genet. 11:2469–2478. [DOI] [PubMed] [Google Scholar]
- 35.Hill, A.V. 1996. Genetics of infectious disease resistance. Curr. Opin. Genet. Dev. 6:348–353. [DOI] [PubMed] [Google Scholar]
- 36.Granjeaud, S., P. Naquet, and F. Galland. 1999. An ESTs description of the new Vanin gene family conserved from fly to human. Immunogenetics. 49:964–972. [DOI] [PubMed] [Google Scholar]
- 37.Maras, B., D. Barra, S. Dupre, and G. Pitari. 1999. Is pantetheinase the actual identity of mouse and human vanin-1 proteins? FEBS Lett. 461:149–152. [DOI] [PubMed] [Google Scholar]
- 38.Dupre, S., M. Graziani, M. Rosei, A. Fabi, and E. Del Grosso. 1970. The enzymatic breakdown of pantethine to pantothenic acid and cystamine. Eur. J. Biochem. 16:571–578. [DOI] [PubMed] [Google Scholar]
- 39.Robishaw, J.D., and J.R. Neely. 1985. Coenzyme A metabolism. Am. J. Physiol. 248:E1–E9. [DOI] [PubMed] [Google Scholar]
- 40.Lebo, R.V., and N.M. Kredich. 1978. Inactivation of human gamma-glutamylcysteine synthetase by cystamine. Demonstration and quantification of enzyme-ligand complexes. J. Biol. Chem. 253:2615–2623. [PubMed] [Google Scholar]
- 41.Aurrand-Lions, M., F. Galland, H. Bazin, V.M. Zakharyev, B.A. Imhof, and P. Naquet. 1996. Vanin-1, a novel GPI-linked perivascular molecule involved in thymus homing. Immunity. 5:391–405. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki, K., T. Watanabe, S. Sakurai, K. Ohtake, T. Kinoshita, A. Araki, T. Fujita, H. Takei, Y. Takeda, Y. Sato, et al. 1999. A novel glycosylphosphatidyl inositol-anchored protein on human leukocytes: a possible role for regulation of neutrophil adherence and migration. J. Immunol. 162:4277–4284. [PubMed] [Google Scholar]
- 43.Martin, F., M.F. Penet, F. Malergue, H. Lepidi, A. Dessein, F. Galland, M. de Reggi, P. Naquet, and B. Gharib. 2004. Vanin-1(−/−) mice show decreased NSAID- and Schistosoma-induced intestinal inflammation associated with higher glutathione stores. J. Clin. Invest. 113:591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stevenson, M.M., and E.M. Riley. 2004. Innate immunity to malaria. Nat. Rev. Immunol. 4:169–180. [DOI] [PubMed] [Google Scholar]
- 45.Malaguarnera, L., and S. Musumeci. 2002. The immune response to Plasmodium falciparum malaria. Lancet Infect. Dis. 2:472–478. [DOI] [PubMed] [Google Scholar]
- 46.van der Heyde, H.C., B. Pepper, J. Batchelder, F. Cigel, and W.P. Weidanz. 1997. The time course of selected malarial infections in cytokine- deficient mice. Exp. Parasitol. 85:206–213. [DOI] [PubMed] [Google Scholar]
- 47.Su, Z., and M.M. Stevenson. 2000. Central role of endogenous gamma interferon in protective immunity against blood-stage Plasmodium chabaudi AS infection. Infect. Immun. 68:4399–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su, Z., and M.M. Stevenson. 2002. IL-12 is required for antibody-mediated protective immunity against blood-stage Plasmodium chabaudi AS malaria infection in mice. J. Immunol. 168:1348–1355. [DOI] [PubMed] [Google Scholar]
- 49.Riopel, J., M. Tam, K. Mohan, M.W. Marino, and M.M. Stevenson. 2001. Granulocyte-macrophage colony-stimulating factor-deficient mice have impaired resistance to blood-stage malaria. Infect. Immun. 69:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langhorne, J., F.R. Albano, M. Hensmann, L. Sanni, E. Cadman, C. Voisine, and A.M. Sponaas. 2004. Dendritic cells, pro-inflammatory responses, and antigen presentation in a rodent malaria infection. Immunol. Rev. 201:35–47. [DOI] [PubMed] [Google Scholar]
- 51.Stevenson, M.M., and B.C. Urban. 2006. Antigen presentation and dendritic cell biology in malaria. Parasite Immunol. 28:5–14. [DOI] [PubMed] [Google Scholar]
- 52.Omer, F.M., and E.M. Riley. 1998. Transforming growth factor β production is inversely correlated with severity of murine malaria infection. J. Exp. Med. 188:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li, C., L.A. Sanni, F. Omer, E. Riley, and J. Langhorne. 2003. Pathology of Plasmodium chabaudi chabaudi infection and mortality in interleukin-10-deficient mice are ameliorated by anti-tumor necrosis factor alpha and exacerbated by anti-transforming growth factor beta antibodies. Infect. Immun. 71:4850–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Becker, K., L. Tilley, J.L. Vennerstrom, D. Roberts, S. Rogerson, and H. Ginsburg. 2004. Oxidative stress in malaria parasite-infected erythrocytes: host-parasite interactions. Int. J. Parasitol. 34:163–189. [DOI] [PubMed] [Google Scholar]
- 55.Hunt, N.H., and R. Stocker. 1990. Oxidative stress and the redox status of malaria-infected erythrocytes. Blood Cells. 16:499–526 (discussion 527–530). [PubMed]
- 56.Eckman, J.R., and J.W. Eaton. 1979. Dependence of plasmodial glutathione metabolism on the host cell. Nature. 278:754–756. [DOI] [PubMed] [Google Scholar]
- 57.Eaton, J.W., J.R. Eckman, E. Berger, and H.S. Jacob. 1976. Suppression of malaria infection by oxidant-sensitive host erythrocytes. Nature. 264:758–760. [DOI] [PubMed] [Google Scholar]
- 58.Demant, P., and A.A. Hart. 1986. Recombinant congenic strains–a new tool for analyzing genetic traits determined by more than one gene. Immunogenetics. 24:416–422. [DOI] [PubMed] [Google Scholar]
- 59.Pfaffl, M.W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muller, P.Y., H. Janovjak, A.R. Miserez, and Z. Dobbie. 2002. Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques. 32:1372–1374, 1376, 1378–1379. [PubMed] [Google Scholar]
- 61.Manly, K.F., and J.M. Olson. 1999. Overview of QTL mapping software and introduction to map manager QT. Mamm. Genome. 10:327–334. [DOI] [PubMed] [Google Scholar]