

Abstract
Sphingosine kinase 1 is an agonist-activated signalling enzyme that catalyses the formation of sphingosine 1-phosphate, a lipid second messenger that has been implicated in a number of agonist-driven cellular responses, including stimulation of cell proliferation, inhibition of apoptosis and expression of inflammatory molecules. Although agonist-induced stimulation of sphingosine kinase activity is critical in a number of signalling pathways, nothing has been known of the molecular mechanism of this activation. Here we show that this activation results directly from phosphorylation of sphingosine kinase 1 at Ser225, and present several lines of evidence to show compellingly that the activating kinase is ERK1/2 or a close relative. Furthermore, we show that phosphorylation of sphingosine kinase 1 at Ser225 results not only in an increase in enzyme activity, but is also necessary for translocation of the enzyme from the cytosol to the plasma membrane. Thus, these studies have elucidated the mechanism of agonist-mediated sphingosine kinase activation, and represent a key finding in understanding the regulation of sphingosine kinase/sphingosine 1-phosphate-controlled signalling pathways.
Keywords: ERK/phosphorylation/sphingosine kinase/sphingosine 1-phosphate/translocation
Introduction
Sphingosine kinase has emerged as an important signalling enzyme through its role in the formation of the bioactive lipid sphingosine 1-phosphate (S1P). Among the remarkably wide spectrum of biological processes regulated by S1P are calcium mobilization, cell growth, survival, differentiation, motility and cytoskeletal organization (Pyne and Pyne, 2000; Spiegel and Milstien, 2002). In recent years the reasons for these diverse actions of S1P have begun to be elucidated, with the discovery that this lipid mediator appears to function as both an intracellular second messenger and a ligand for cell-surface receptors (Hla et al., 2001; Spiegel and Milstien, 2002).
The cellular levels of S1P are controlled by its formation from sphingosine through the activity of sphingosine kinase, and by its degradation by S1P lyase (Van Veldhoven et al., 2000) and S1P phosphatases (Mandala, 2001). In the basal state, this balance between S1P generation and degradation results in low cellular levels of S1P (Pyne and Pyne, 2000). However, when cells are exposed to specific growth factors and other agonists like tumour necrosis factor-α (TNFα) or phorbol esters the cellular levels of S1P can increase rapidly and transiently (Pitson et al., 2000b). This results in the triggering of various signalling pathways through as yet unidentified intracellular S1P targets, as well as through the engagement of cell surface S1P receptors following its release from cells (Hobson et al., 2001). This agonist-induced increase in cellular S1P is a direct consequence of a rapid increase in sphingosine kinase activity in the cell. Use of a dominant-negative form of this enzyme, developed in this laboratory, has demonstrated that this activation of sphingosine kinase, above a basal catalytic level, is required for its function in signal transduction cascades (Pitson et al., 2000b). This places sphingosine kinase, and in particular its activation, in a central and obligatory role in controlling many of the observed cellular effects attributed to S1P.
The activation pathway leading from agonist binding to sphingosine kinase activation has yet to be elucidated. We have recently described the essential involvement of TNFα receptor-associated factor 2 (TRAF2) in TNFα-induced human sphingosine kinase 1 (hSK1) activation (Xia et al., 2002), but the details of this involvement remain unclear. In this study, we reveal that the activation of sphingosine kinase is directly elicited by its phosphorylation, and that this phosphorylation is mediated by a mitogen-activated serine/threonine protein (MAP) kinase. Furthermore we find that phosphorylation not only results in an increase in enzyme activity, but is also necessary for translocation of the enzyme from the cytosol to the plasma membrane.
Results
Phosphorylation of hSK1
To elucidate the mechanism of hSK1 activation we examined the phosphorylation of this enzyme in HEK293T cells in response to TNFα or PMA (phorbol 12-myristate 13-acetate), both known activators of hSK1 (Pitson et al., 2000b). Consistent with an earlier study (Johnson et al., 2002), metabolic labelling with 32Pi of cultured cells overexpressing human sphingosine kinase 1 (hSK1) revealed a low level of phosphorylation of this protein, even in the absence of stimulatory agonists (Figure 1). However, the presence of TNFα or PMA induced marked increases in the phosphorylation of hSK1 that closely paralleled the increases in sphingosine kinase activity in the cells in response to these agonists (Figure 1). Thus, we speculated that phosphorylation of hSK1 may lead to the activation of this enzyme, and consequently initiated a search for activating phosphorylation sites.
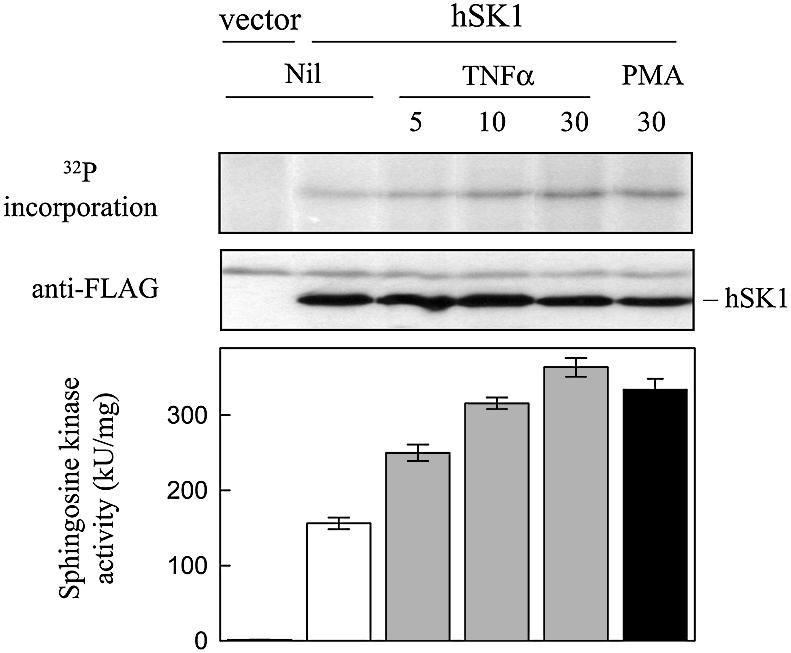
Fig. 1. Agonist-induced phosphorylation of hSK1. Time course of in vivo hSK1 phosphorylation and activity following TNFα (1 ng/ml) and PMA (10 ng/ml) stimulation of cells. HEK293T cells over expressing hSK1 were metabolically labelled with 32P prior to treatment with TNFα and PMA for the indicated times (min). Immunoprecipitated hSK1 from these cells was then subjected to SDS–PAGE and the incorporation of 32P into hSK1 determined. Loading controls for hSK1 in the immunoprecipitates were visualized by western blot via their FLAG epitope. Quantitation of 32P incorporation into hSK1 following 5, 10 and 30 min of TNFα treatments showed fold increases of 1.7 ± 0.3, 2.3 ± 0.3 and 3.4 ± 0.4, respectively, over that seen in the untreated cell extracts. Similarly, PMA treatment for 30 min resulted in a 2.9 ± 0.3-fold increase in 32P incorporation into hSK1 compared with the untreated cell extracts. Sphingosine kinase activities in the extracts were determined prior to immunoprecipitation. All data are represented as means (± SD) from more than three experiments.
The observations that the protein kinase C (PKC) activator, PMA, activates hSK1, and that the hSK1 polypeptide has four putative PKC phosphorylation sites (Pitson et al., 2000a) has lead to suggestions that PKC may have a direct role in the phosphorylation of sphingosine kinase (Hannun et al., 2001; Shu et al., 2002). However, a direct effect of PKC in activating sphingosine kinase in vivo is unlikely since we have been unable to show significant phosphorylation of hSK1 by PKC in vitro (data not shown). Previous studies have also failed to establish a substantial direct effect of PKC on sphingosine kinase activity (Buehrer et al., 1996; Shu et al., 2002).
Analysis of the hSK1 polypeptide sequence using the NetPhos phosphorylation site prediction algorithm (Blom et al., 1999) revealed five possible phosphorylation sites in hSK1 that were conserved in the mouse, rat and macaque hSK1 homologues. To examine the possible phosphorylation of hSK1 at these sites, we performed alanine mutagenesis at each of these residues, followed by transfection of the mutants into HEK293T cells for metabolic labelling studies. Four of the mutations had no effect on hSK1 phosphorylation (Figure 2A). In contrast, however, the Ser225→Ala mutation completely ablated both basal and agonist-induced phosphorylation of hSK1 (Figure 2A). This, combined with the lack of effect on hSK1 phosphorylation of alanine mutations at serine or threonine residues close to Ser225 (Ser220 and Thr222; Figure 2A) indicated that Ser225 was the sole site of hSK1 phosphorylation. Mutation of the analogous residue in mouse sphingosine kinase 1 (Ser224; Figure 2B) similarly eliminated basal and PMA-stimulated phosphorylation of this enzyme (Figure 2C), supporting the notion that this is a conserved phosphorylation site.

Fig. 2. Site-directed mutagenesis reveals that hSK1 is phosphorylated Ser225. (A) In vivo phosphorylation of hSK1 mutants, determined by 32P incorporation, prior to and following treatment of transiently transfected HEK293T cells with PMA (10 ng/ml) for 30 min. Loading controls for hSK1 in the immunoprecipitates were visualized by western blot via their FLAG epitope. (B) Sequence alignment of sphingosine kinases from higher organisms in the region of Ser225 of hSK1. (C) In vivo phosphorylation of wild-type mouse sphingosine kinase 1 (mSK1) and mSK1S224A, determined by 32P incorporation, prior to and following treatment of transiently transfected HEK293T cells with PMA (10 ng/ml) for 30 min. Loading controls for mSK1 in the immunoprecipitates were visualized by western blot via their FLAG epitope. Quantitation of 32P incorporation into wild-type mSK1 following PMA treatment showed a 4.7 ± 0.6-fold increase compared with that seen in untreated cell extracts. (D) Mutation of Ser225 ablates activation of hSK1. Sphingosine kinase activities shown are from HEK293T cells transiently transfected with wild-type hSK1 (hSK1WT) and hSK1S225A, prior to and following treatment with TNFα (1 ng/ml) and PMA (10 ng/ml). All data are represented as means (± SD) from more than three experiments.
Activation of hSK1 by phosphorylation
To demonstrate that phosphorylation of Ser225 is required for agonist-dependent activation, we examined the effect of ablating this phosphorylation on the catalytic activity and activation of hSK1. Sphingosine kinase activity in HEK293T cells expressing hSK1S225A (the hSK1 mutant with the Ser225→Ala mutation) was measured after TNFα or PMA treatment. Remarkably, this non-phosphorylatable hSK1 mutant could not be activated by these agonists (Figure 2D). In contrast, however, this mutation had only a minor effect on the basal catalytic activity of the enzyme when expressed in HEK293T cells (Figure 2D). Similarly, wild-type hSK1 and hSK1S225A possessed identical catalytic activity when expressed as recombinant proteins in Escherichia coli (69 ± 5 U/ng and 73 ± 6 U/ng, respectively). This is consistent with our earlier findings that hSK1 has considerable intrinsic catalytic activity that is not dependent on post-translational modifications (Pitson et al., 2000a), and supports our previous model that hSK1 has both a basal ‘housekeeping’ function and an activated signalling function. These data provide compelling evidence that phosphorylation of hSK1 is an obligatory step in the mechanism of activation of this enzyme.
Examination of the amino acid sequence surrounding Ser225 (SKTPAS225PVVVQ) suggested that it is a likely phosphorylation site for proline-directed protein kinases (e.g. the MAP and cyclin-dependent kinases) considering the presence of a proline immediately C-terminal to Ser225 (Lu et al., 2002). In particular, the PASP sequence of this region is reminiscent of the extracellular signal-regulated kinase (ERK) 1 and 2 substrate recognition motif, PXS/TP (where X represents a neutral or basic amino acid) (Clark-Lewis et al., 1991; Songyang et al., 1996), suggesting a direct role for ERK1/2 in hSK1 phosphorylation. Furthermore, the hSK1 amino acid sequence contains a region (144RRLLSPMNLLSL155) that has similarity to a known ERK1/2 docking site motif, which has been shown to confer specificity and efficiency of phosphorylation in other ERK1/2 substrates (Sharrocks et al., 2000). Therefore, we examined the ability of ERK1 and ERK2 to phosphorylate purified recombinant hSK1 in vitro. Indeed, we found that both enzymes phosphorylated hSK1, although surprisingly, considering the similarity of these two enzymes (Pearson et al., 2001), ERK2 showed much higher efficiency for this substrate than ERK1 (Figure 3A). Cyclin-dependent kinase 2 (CDK2) was found to phosphorylate hSK1 also, but with much lower efficiency than either ERK2 or -1 (Figure 3A). Importantly, in vitro phosphorylation of hSK1 by ERK2 occurred specifically at Ser225 since no phosphorylation was seen with the recombinant hSK1S225A mutant. This result was mirrored in the equivalent mouse SKS224A mutant (Figure 3A). Furthermore, alanine mutations at the two other possible ERK1/2 phosphorylation sites in hSK1 had no effect on in vitro phosphorylation of the recombinant hSK1 mutants by ERK2 (Figure 3A). In contrast, in vitro phosphorylation of hSK1 by ERK1 or CDK2 was only partially ablated by the Ser225→Ala mutation, indicating some phosphorylation by these enzymes at other sites.

Fig. 3. In vitro phosphorylation and activation of hSK1 by ERK2. (A) In vitro phosphorylation of recombinant wild-type and mutant hSK1 and mSK1proteins with ERK2, ERK1 and CDK2. Phosphorylation was measured by 32P incorporation from [γ-32P]ATP, while protein levels of hSK1 were determined using Coomassie Brilliant Blue staining. Quantitation of 32P incorporation, corrected for hSK1 protein levels, showed that ERK2 incorporated similar levels of 32P into the hSK1S148A and hSK1T222A mutants (94 ± 5% and 108 ± 9%, respectively), compared with wild-type hSK1. In contrast, ERK2 was unable to mediate any detectable 32P incorporation into hSK1S225A, but did show a small level of 32P incorporation into the corresponding mSK1S224A mutant (8 ± 4% compared with wild-type mSK1). ERK1 incorporated 32P into hSK1S225A, hSK1S148A and hSK1T222A, at 26 ± 7%, 87 ± 11% and 112 ± 15%, respectively, of that seen with wild-type hSK1. Similarly, CDK2 incorporated 32P into hSK1S225A, hSK1S148A and hSK1T222A, at 19 ± 6%, 101 ± 8% and 120 ± 14%, respectively, compared with wild-type hSK1. (B) Sphingosine kinase activity of hSK1 prior to and following in vitro phosphorylation of hSK1 at Ser225 by ERK2. Values are corrected for the proportion of hSK1 phosphorylated in the assay mix. All data are represented as means (± SD) from three experiments.
While establishing that phosphorylation of hSK1 at Ser225 is necessary for activation of this enzyme, the studies described above in intact cells did not establish whether phosphorylation activates hSK1 by a direct or an indirect mechanism. The ability to specifically phosphorylate recombinant hSK1 at Ser225 in vitro with purified ERK2, however, afforded us the opportunity to examine the direct effect of this phosphorylation on the catalytic activity of hSK1. We found that phosphorylation of purified hSK1 in vitro with ERK2 resulted in a dramatic increase in its sphingosine kinase activity. Utilizing the known specific activity of the [32P]ATP added to this reaction, and knowing that only a single phosphorylation site is used, we determined that 42% of the hSK1 protein became phosphorylated. Given the 5.8-fold increase in activity in this substoichiometric phosphorylation reaction, we calculated that phosphorylation of hSK1 at Ser225 results in an ∼14-fold increase in its catalytic activity (Figure 3B). Substrate kinetic analysis demonstrated that the phosphorylation-dependent activation is primarily due to an increased turnover number of the enzyme, with phosphorylation raising the kcat of hSK1 13.6-fold from 93 s–1 to 1265 s–1. In contrast, phosphorylation of hSK1 only resulted in a slightly lower KM value for ATP (56 ± 8 µM compared with 81 ± 12 µM for non-phosphorylated hSK1), and an unaltered KM value for sphingosine (15 ± 4 µM and 13 ± 3 µM for non-phosphorylated and phosphorylated hSK1, respectively).
This increase in catalytic activity of in vitro-activated hSK1 parallels that seen with sphingosine kinase activation in intact cells, which is also the result of an increase in Vmax, with no change in KM for either substrate (Rius et al., 1997; Xia et al., 1999). Thus, although we have previously shown that hSK1 possesses considerable intrinsic catalytic activity in the absence of post-translational modifications (Pitson et al., 2000a), phosphorylation of this enzyme at Ser225 directly results in a marked, 14-fold increase in its catalytic efficiency.
Acidic residues are known to occasionally mimic phosphorylated amino acids in inducing conformational changes in protein structure to generate constitutively activated enzyme mutants. Thus, we attempted to generate a constitutively activated hSK1 mutant by substitution of Ser225 in hSK1 with glutamate and aspartate, however these mutants were without increased activity (data not shown).
Phospho-hSK1-specific antibodies
To follow phosphorylation of both endogenous and overexpressed hSK1 in HEK293T cells in a more facile manner, we generated and verified phospho-hSK1-specific polyclonal antibodies raised in rabbits against a phosphopeptide centred around phospho-Ser225 of hSK1. In cells overexpressing hSK1 this antibody recognized in vivo phosphorylated wild-type hSK1, but not the hSK1S225A mutant (Figure 4). Consistent with our metabolic labelling studies, the phospho-hSK1 antibody showed basal hSK1 phosphorylation that was increased following exposure of the cells to stimulatory agonists (Figure 4). To confirm that there was an agonist-stimulated phosphorylation of Ser225 in the endogenous sphingosine kinase we immunoprecipitated this enzyme from cell cultures using an anti-hSK1 antibody that recognizes the protein regardless of phosphorylation status and probed the immunoprecipitates with the anti-phospho-hSK1 antibody (Figure 4). In these untransfected cells, basal phosphorylation of the endogenous enzyme could not be detected. However, following treatment with either TNFα or PMA, strong phosphorylation of endogenous hSK1 at Ser225 was observed. Therefore, it is clear that phosphorylation of Ser225 is a relevant modification in the activation of hSK1 and not an artefact of overexpression. Notably, basal phosphorylation of hSK1 in the absence of stimulatory agonists was only clear in cells overexpressing hSK1. The mechanism of this low-level activation in unstimulated hSK1-overexpressing cells is not currently understood. However, it does not depend on a feed-forward loop of hSK1 activity in overexpressing cells since we see a similar basal phosphorylation of the catalytically inactive hSK1G82D form of the enzyme (Figure 4).

Fig. 4. Phospho-hSK1-specific polyclonal antibodies detect phosphorylation of hSK1. The phospho-hSK1 specific polyclonal antibodies show high specificity in western blots for the phosphorylated hSK1 in cell extracts, with no reactivity to non-phosphorylated hSK1S225A. The endogenous hSK1 cannot be directly detected in cell lysates with either the anti-phospho-hSK1 or rabbit anti-hSK1 antibodies (left lanes). However, following immunoprecipitation of cell lysates with a chicken anti-hSK1 antibody, phosphorylation of endogenous hSK1 can be seen following stimulation of cells with TNFα (1 ng/ml for 10 min) or PMA (10 ng/ml for 30 min).
ERK1/2 phosphorylates hSK1 in vivo
Our findings that ERK2 efficiently phosphorylates hSK1 in vitro, and does so at the correct site, suggested that this enzyme may be the physiologically relevant protein kinase that phosphorylates hSK1 in vivo. To test the involvement of ERK1/2 in intact cells, we examined the effect of the well characterized ERK1/2 pathway inhibitors, PD98059 and U0126, on hSK1 phosphorylation and activation. These both act specifically on MEK1/2 to block ERK1/2 activation (Davies et al., 2000). Both chemical inhibitors potently blocked TNFα and PMA-induced increases in hSK1 phosphorylation and catalytic activity (Figure 5A) in cells overexpressing hSK1. Consistent with this, the U0126 inhibitor also blocked increases in phosphorylation and catalytic activity of the endogenous enzyme in non-overexpressing cells (Figure 5B). This is strong evidence for an in vivo role for ERK1/2 in the activation of hSK1. To establish that there is a direct interaction between ERK1/2 and hSK1 in vivo, as suggested by the in vitro experiments, we examined co-immunoprecipitation of the two proteins from cell extracts (Figure 5C). We detected an interaction between hSK1 and ERK1/2 using antibodies against either component for immunoprecipitation. Importantly, we could detect an interaction between endogenous ERK1/2 and endogenous hSK1, confirming that this was not an interaction that was forced by overexpression. If ERK1/2 is the directly activating kinase for hSK1, the time-course of activation of these kinases should parallel the phosphorylation status of hSK1 and the resultant changes in sphingosine kinase activity. We found this to be the case for both TNFα or PMA activation (Figure 5D). Notably, compared with empty vector transfected cells, the phosphorylation and activation of hSK1 by TNFα is considerably more sustained in cells overexpressing hSK1. This is only partially explained by the more sustained activation of ERK1/2 in the cells, which is consistent with previous studies (Lacaná et al., 2002), and may also indicate that overexpression of hSK1 saturates the hSK1 dephosphorylation mechanism. The combined results of the in vitro and in vivo experiments demonstrate activation and association of hSK1 with ERK1/2, and are a strong indication that hSK1 is a bona fide direct effector of ERK1/2.
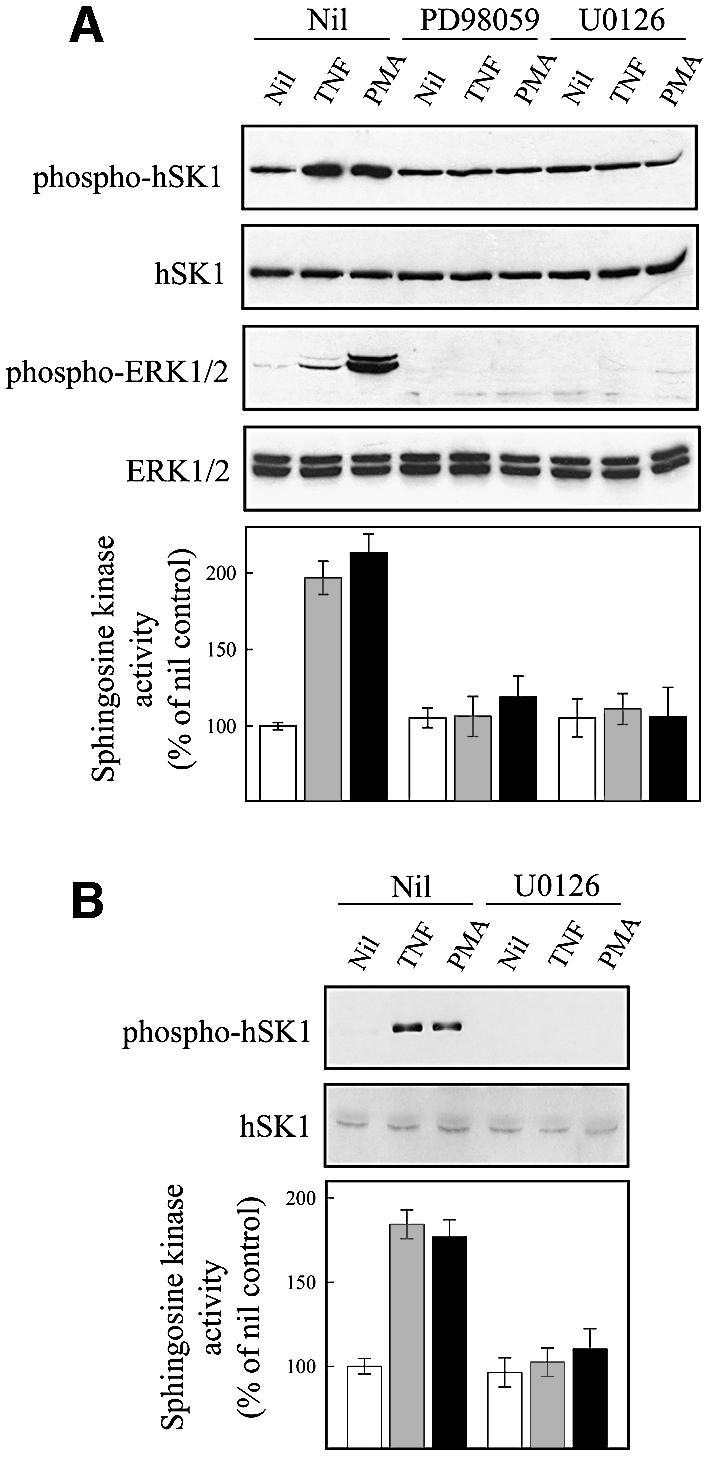

Fig. 5. ERK1 and -2 phosphorylate and activate hSK1 in vivo. (A and B) Agonist-induced phosphorylation and activation of hSK1 in HEK293T cells is blocked by ERK1/2 pathway inhibitors. Phosphorylation and activation of hSK1 in transiently transfected HEK293T cells (A) was followed by western blot using the phospho-hSK1 specific polyclonal antibodies (phospho-hSK1) and sphingosine kinase enzyme assays. HEK293T cells overexpressing wild-type hSK1 were treated with TNFα (1 ng/ml) or PMA (10 ng/ml) for 30 min in the presence or absence of the ERK1/2 pathway inhibitors PD98059 (10 µM) and U0126 (2 µM), added 30 min prior to TNFα or PMA. Total hSK1 levels were determined via the FLAG epitope, while ERK1/2 activation was followed by phospho-ERK1/2 and ERK1/2 antibodies. Phosphorylation of endogenous hSK1 in untransfected HEK293T cells showed similar results following treatment with TNFα (1 ng/ml for 10 min) and PMA (10 ng/ml for 30 min) in the presence or absence of U0126 (B). (C) Immunoprecipitation of ERK1/2 and hSK1. HEK293T cells overexpressing either wild-type hSK1 or hSK1TB2 were treated with TNFα or PMA for 30 min. hSK1 (via the FLAG epitope) or ERK1/2 from clarified cell lysates were immunoprecipitated, subjected to SDS–PAGE, and the protein complexes probed by western blot for ERK1/2, FLAG or directly for hSK1. (D) The time course of sphingosine kinase and ERK1/2 activation by TNFα (closed circles) and PMA (open circles) correlate in both empty vector and hSK1-transfected cells.
Interestingly, we have previously established, through the use of a dominant-negative sphingosine kinase (hSK1G82D), that ERK1/2 activation by TNFα is dependent on sphingosine kinase activation (Pitson et al., 2000b). Similar results have also been reported for vascular endothelial growth factor (VEGF)-mediated ERK1/2 activation (Shu et al., 2002). These studies, combined with our current work, indicate that sphingosine kinase resides both upstream and downstream of ERK1/2 in the signalling pathways leading from TNFα and VEGF receptor engagement. Consistent with this, we observed that TNFα treatment of cells expressing hSK1G82D did not enhance the phosphorylation of this protein as it does with wild-type hSK1 (Figure 4). In contrast, hSK1G82D was phosphorylated in response to cell exposure to PMA (Figure 4) where ERK1/2 activation occurs in a sphingosine kinase-independent manner (Pitson et al., 2000b).
Interaction of hSK1 with TRAF2 is essential for TNFα-induced hSK1 activation (Xia et al., 2002). Consistent with this, we have found that TNFα-induced hSK1 phosphorylation is also dependent on the interaction of hSK1 with TRAF2 since a previously described hSK1 mutant (hSK1TB2) deficient in its ability to bind TRAF2 (Xia et al., 2002) was not phosphorylated in response to cell treatment with TNFα (Figure 4). One possibility is that TRAF2 may mediate the interaction between hSK1 and ERK1/2 to facilitate hSK1 phosphorylation. This appears unlikely, however, since hSK1TB2 also formed a complex with ERK1/2 (Figure 5C). Instead, our data indicates TRAF2 interacts with the pre-formed hSK1–ERK1/2 complex. This may facilitate the recruitment of upstream components of the MAP kinase pathway to this complex following engagement of TNFα, allowing for localized activation of ERK1/2 and subsequent phosphorylation of the associated hSK1. Indeed, TRAF2 is known to be involved in the recruitment and subsequent activation of MEKK1 at the TNF receptor (see Kyriakis and Avruch, 2001). Such an adaptor molecule role for TRAF2 would allow for enhanced hSK1 activation and explain how TNFα, a relatively weak ERK1/2 activator, results in similar hSK1 activation as PMA.
Translocation of hSK1 to the plasma membrane depends on hSK1 phosphorylation
Recent reports suggest that activation of hSK1 by PMA, platelet-derived growth factor (PDGF) or IgE-antigen is accompanied by translocation of the enzyme from the cytosol to the plasma membrane (Rosenfeldt et al., 2001; Johnson et al., 2002; Melendez and Khaw, 2002). To test whether this translocation depends on agonist-dependent phosphorylation of hSK1 we measured the levels of both total and phosphorylated hSK1 in cytosolic and membrane fractions with and without stimulation by TNFα or PMA of HEK293T cells overexpressing either wild-type hSK1 or the phosphorylation-deficient hSK1S225A mutant (Figure 6A). Stimulation with PMA, as previously reported (Johnson et al., 2002), or TNFα consistently induced an increase in membrane-associated wild-type hSK1. In contrast the Ser225→Ala mutation completely blocked any increase in membrane-associated hSK1, confirming that phosphorylation at that site is required for the translocation event. Further confocal microscopy of HEK293T cells expressing hSK1–green fluorescent protein (GFP) fusion proteins also confirmed phosphorylation-dependent translocation of hSK1 since PMA induced cytosol to plasma membrane translocation of wild-type hSK1, but not the phosphorylation-deficient hSK1S225A mutant (Figure 6B). This PMA-induced effect on wild-type hSK1 was blocked by the U0126 inhibitor, further demonstrating the requirement of ERK1/2-mediated phosphorylation in the translocation of hSK1 to the plasma membrane. These findings suggest that phosphorylation of hSK1 at Ser225 not only results in an increase in enzyme activity, but may also facilitate interaction of hSK1 with another protein that results in translocation of the enzyme from the cytosol to the plasma membrane.

Fig. 6. Phosphorylation of hSK1 leads to its translocation to the plasma membrane. (A) Lysates from HEK293T cells transfected with wild-type hSK1 or hSK1S225A were fractionated into cytosol to membranes following treatment for 30 min with TNFα (1 ng/ml) and PMA (10 ng/ml). Cell fractions were then probed via western blot for total hSK1 (with anti-FLAG) and phospho-hSK1 to show phosphorylation-dependent translocation to the membrane fraction. Immunoblots for E-cadherin, an integral plasma membrane protein, were used to show equal membrane loading. Data are representative of three independent experiments. (B) Confocal microscopy of HEK293T cells transfected with either wild-type hSK1-GFP or hSK1S225A-GFP, with or without PMA (10 ng/ml), for 30 min showed phosphorylation-dependent translocation of hSK1 from the cytosol to the plasma membrane. This PMA-induced translocation of wild-type hSK1 was blocked by 30 min preincubation of the cells with U0126 (2 µM). Confocal images are representative of >50% of cells observed in three independent experiments.
Agonist-induced increases in intracellular and extracellular sphingosine 1-phosphate levels depend on hSK1 phosphorylation
Phosphorylation is not only required for agonist-stimulated increases in enzyme activity, as demonstrated above, but is also essential for the activation-dependent elevation in S1P, the second messenger produced by hSK1 (Figure 7). Overexpression of wild-type hSK1 resulted in large increases in both intracellular and extracellular S1P over that of vector-transfected cells, even in the absence of stimulus. Stimulation of these cells with either TNFα or PMA, however, resulted in further increases in both intracellular and extracellular S1P. This mirrored well the changes in S1P, at lower overall levels, in empty vector-transfected cells. In contrast, mutation of the Ser225 phosphorylation site completely eliminated any agonist-dependent changes in either intracellular or extracellular S1P. In addition, compared with wild-type hSK1 transfected cells, reduced basal S1P levels were observed in cells overexpressing the hSK1S225A mutant. This was most marked for extracellular S1P, and was despite the wild-type hSK1 and hSK1S225A-overexpressing cells possessing similar basal sphingosine kinase activities. Consistent with ERK1/2 being the activator(s) of endogenous as well as overexpressed hSK1, inhibition with the ERK1/2 pathway inhibitor U0126 blocked TNFα- and PMA-induced increases in intracellular and extracellular S1P in both vector-transfected and wild-type hSK1-transfected cells. The inability of this inhibitor to reduce S1P levels in wild-type hSK1-transfected cells to that of the hSK1S225A mutant is explained by an incomplete block of basal hSK1 phosphorylation (Figure 5A).

Fig. 7. Phosphorylation of hSK1 leads to increased intracellular and extracellular sphingosine 1-phosphate levels. Intracellular and extracellular S1P levels were determined in HEK293T cells transiently transfected with empty vector, or plasmids encoding for wild-type hSK1 and hSK1S225A following 30 min treatment with TNFα (1 ng/ml) and PMA (10 ng/ml), and in the presence or absence of the ERK1/2 pathway inhibitor U0126 (2 µM, added 30 min prior to TNFα or PMA). Data are represented as means (± SD) from three independent experiments.
Discussion
Here we have shown that activation of sphingosine kinase by at least two agonists is directly mediated by phosphorylation of the enzyme at Ser225 and, furthermore, that this phosphorylation is required for translocation of hSK1 from the cytosol to the plasma membrane. Thus, remarkably, phosphorylation of hSK1 at a single site results in two distinct effects on the enzyme.
In comparison with other signalling enzymes, it is likely that phosphorylation of hSK1 at Ser225 induces a conformational change in the enzyme to facilitate the observed increase in its catalytic efficiency. Unfortunately, sequence analysis has failed to identify in hSK1 any recognizable features of many other signalling kinases, such as an ‘activation loop’ (Adams, 2003), which may be altered by phosphorylation at Ser225. Thus, in the current absence of a crystal structure for hSK1, or any related protein, the details of these phosphorylation-induced structural changes that lead to increased catalytic activity are unknown.
Interestingly, the 14-fold increase in catalytic activity that results from phosphorylation of hSK1 is considerably greater than the 2- to 3-fold increase in sphingosine kinase activity observed in cells following treatment with various agonists. This indicates only partial phosphorylation of hSK1 in cells, even with maximal agonist stimulation. This may suggest that hSK1 exists in discrete pools in the cell, differing in the association with specific protein complexes and/or subcellular location, and that these pools are differentially activatable. Such a signalling-specific compartmentalization is consistent with the observations that strong activators of ERK1/2, such as phorbol esters, result in similar fold activation of sphingosine kinase in cells as comparatively weak ERK1/2-activating agonists like TNFα and PDGF.
In addition to the quantitative change in the catalytic activity of hSK1, phosphorylation at Ser225 is also necessary for its translocation to the plasma membrane. It is tempting to speculate that this is most likely achieved via a protein interaction that is dependent on the phosphorylation status of hSK1. Although this most commonly occurs with phosphotyrosine residues, several protein domains are now known to interact specifically with phosphoserine and phosphothreonine residues (Pawson and Scott, 1997; Yaffe and Cantley, 1999; Sudol and Hunter, 2000). Studies are currently being undertaken to identify such phosphorylation-dependent hSK1-interacting proteins. Interestingly, Young et al. (2003) have recently reported that translocation of hSK1 to the plasma membrane in response to calcium-mobilizing stimuli occurs in a calmodulin-dependent manner since it is blocked by the calmodulin inhibitor W-7. Since hSK1 interacts with calmodulin in vitro (Pitson et al., 2000a), Young et al. (2003) have suggested a direct role for calmodulin in hSK1 translocation to the plasma membrane. Notably, however, calmodulin predominantly translocates from the cytosol to the nucleus, rather than the plasma membrane, upon increases in free calcium (Chin and Means, 2000). Furthermore, it is also noteworthy that the hSK1–calmodulin interaction is not phosphorylation dependent, since the non-phosphorylated recombinant hSK1 generated in bacteria still binds to calmodulin in vitro (Pitson et al., 2000a).
Using a combination of approaches, including chemical inhibitors, co-immunoprecipitation and in vitro phosphorylation, we have generated compelling evidence that ERK1 and -2 are the relevant kinases responsible for phosphorylating and activating hSK1. We cannot rule out the possibility, however, that other closely related kinase(s) might also play this role in certain cell types, or in response to some other agonists. Notably, a recent study has suggested a direct role for PKC in phosphorylating and activating hSK1 based on a very small increase in catalytic activity of recombinant murine sphingosine kinase 1 following its incubation in vitro with PKC and ATP (Shu et al., 2002). However, in light of our current findings and other previous work (Buehrer et al., 1996), a direct role for PKC in hSK1 activation appears unlikely, with its reported minor effect on sphingosine kinase activity in vitro (Shu et al., 2002) probably due to forced, non-specific phosphorylation at Ser225.
Given the widespread involvement of ERK1/2-mediated pathways in cellular proliferation, it is perhaps not surprising that sphingosine kinase, which has proliferative effects (Olivera et al., 1999; Xia et al., 2000), is an ERK1/2 effector. Importantly, since many of the agonist-mediated signalling cascades that result in ERK1/2 activation have been elucidated, this information allows, for the first time, some clarity to be revealed in the signal transduction pathways leading from receptor engagement to sphingosine kinase activation. The identification of hSK1 as an ERK1/2 effector is also interesting since hSK1 activation is an essential element in ERK1/2 activation by TNFα (Pitson et al., 2000b) and VEGF (Shu et al., 2002). Clearly some level of hSK1-independent, agonist-mediated ERK1/2 activation must be possible in order to initiate these pathways. Our data indicate that this would lead to activation of hSK1, resulting in further activation of ERK1/2, possibly via an autocrine mechanism involving S1P receptor engagement following S1P release (Hobson et al., 2001; Pyne and Pyne, 2002). Thus, in such a system hSK1 would form an important part of a positive feedback loop for ERK1/2, similar to other such ERK1/2 amplification mechanisms (Xu et al., 1999; Ma et al., 2001; Garcia et al., 2002). Since we (Figure 5) and others (Lacaná et al., 2002) have observed prolonged ERK1/2 activation in the presence of high sphingosine kinase activity, it is tempting to speculate that such a positive feedback loop involving hSK1 may modulate the amplitude and duration of ERK1/2 activation, which now appears vital in regulating the diverse signalling pathways ERK1/2 control (Bhalla et al., 2002; Murphy et al., 2002; Werlen et al., 2003).
Taken together, our findings suggest that phosphorylation of hSK1 leads to a quantitative change in the catalytic activity of the enzyme, but of equal or greater importance, a translocation of hSK1 to plasma membrane sites. This translocation may have the dual function of localizing hSK1 to available pools of substrate and the generation of localized ‘signalling’ pools of S1P, or its secretion to engage cell-surface S1P receptors (Rosenfeldt et al., 2001; Johnson et al., 2002). We found that in the absence of agonist, the non-phosphorylatable (hSK1S225A) mutant produced less S1P in overexpressing cells than the wild-type enzyme, despite similar overall levels of sphingosine kinase activity in cell extracts. This is especially notable in measurements of extracellular S1P. This indicates that the small amount of wild-type enzyme that is phosphorylated under basal conditions is disproportionately responsible for the production and secretion of S1P. This may be explained by a phosphorylation-dependent targeting mechanism to sites specialized for substrate utilization and product secretion. Such a mechanism would amplify the relatively modest agonist-stimulated increase in hSK1 activity. Furthermore, this compartmentalization would separate the signalling and ‘housekeeping’ functions that have been proposed for hSK1 in controlling the levels of sphingolipid metabolites in the cell (Pitson et al., 2000a,b).
Material and methods
Cell culture, transfection and cell fractionation
Human embryonic kidney cells (HEK293T, ATCC CRL-1573) were cultured, transiently transfected and harvested as described previously (Pitson et al., 2000b). For cell fractionation, HEK293T cells were scraped into lysis buffer lacking Triton X-100, sonicated, and centrifuged at 1000 g for 10 min. The supernatants were then centrifuged at 100 000 g for 60 min at 4°C to yield membrane and cytosolic fractions. The membrane fraction was then resuspended in lysis buffer containing 0.8% Triton X-100, sonicated, left on ice for 30 min, and centrifuged at 10 000 g for 10 min at 4°C to remove Triton-insoluble material. Protein concentrations in cell homogenates were determined with Coomassie Brilliant Blue (Sigma) reagent using bovine serum albumin (BSA) as standard. Immunoblots of membrane fractions with anti-E-cadherin monoclonal antibodies (Transduction Laboratories) were used as a membrane loading control.
Sphingosine kinase assays
Sphingosine kinase activity was routinely determined using d-erythro-sphingosine (Biomol) and [γ-32P]ATP (Geneworks) as substrates, as described previously (Pitson et al., 2000a). A unit of sphingosine kinase activity is defined as the amount of enzyme required to produce 1 pmol S1P/min. Substrate kinetics were analysed using Michaelis–Menten kinetics with the non-linear regression program, Hyper 1.1s.
Site-directed mutagenesis
Wild-type hSK1 cDNA (DDBJ/EMBL/GenBank accession No. AF200328) was FLAG epitope tagged at the 3′ end and subcloned into the pALTER site-directed mutagenesis vector (Promega Corp.), as described previously (Pitson et al., 2000b). Single-stranded DNA was prepared and used as template for oligonucleotide-directed mutagenesis, as detailed in the manufacturer’s protocol. The mutagenic oligonucleotides used to generate the point mutant constructs were as follows: for hSK1S148A, 5′-CGGCTGCTGGCGCCCATGAAC-3′; hSK1S181A, 5′-TG TGGACCTCGAGGCTGAGAAGTA-3′; hSK1Y184A, 5′-AGTGAGAA GGCTCGGCGCCTGGGGGAG-3′; hSK1S225A, 5′-AAGACACCTGC GGCGCCCGTTGTG-3′; hSK1T250A, 5′-TCTCACTGGGCAGTGGT GC-3′; hSK1S220A, 5′-AAGAGTGGGCGCCAAGACAC-3′; hSK1T222A, 5′-AAGAGTGGGATCCAAGGCGCCTGCCTCC-3′; hSK1S225E, 5′-AC ACCTGCCGAACCGGTTGTGGTC-3′; and hSK1S225D, 5′-GACACC TGCGGATCCCGTTGTG-3′. The mutants were sequenced to verify incorporation of the desired modification and the cDNA subsequently subcloned into pcDNA3 (Invitrogen) for transient transfection into HEK293T cells.
In vivo labelling of hSK1
HEK293T cells overexpressing hSK1 were incubated for 4 h in phosphate-free DMEM, then labelled with fresh phosphate-free DMEM containing [32P]orthophosphate (0.2 mCi/ml) and incubated for 4 h at 37°C. The cells were treated with 1 ng/ml TNFα or 100 ng/ml PMA, harvested into cold PBS and then lysed in 50 mM Tris–HCl buffer (pH 7.4) containing 1% Triton X-100, 1% deoxycholate, 0.1% SDS, 150 mM NaCl, 10 mM NaF, 10 mM Na3VO4 and protease inhibitors. Immuno precipitated hSK1 from the soluble fraction, achieved via its FLAG epitope with M2 anti-FLAG antibody (Sigma), was then subjected to SDS–PAGE and the incorporation of 32P into hSK1 in the dried gel determined by phosphorimager analysis.
In vitro phosphorylation of hSK1
Recombinant hSK1 proteins were generated in either E.coli as glutathione S-transferase fusion proteins and purified using glutathione–Sepharose as detailed previously (Pitson et al., 2000a), or insect cells as His-tagged proteins, purified with Ni-NTA beads as detailed previously (Pitson et al., 2002). In vitro phosphorylation of 1 µg of these proteins was performed using 0.1 U of purified recombinant protein kinases (Upstate biotechnology) and 2 mM [γ-32P]ATP (0.2 mCi/ml). The activities of the recombinant protein kinases were quantitated using myelin basic protein according to the manufacturer’s protocol. Incorporation of 32P into hSK1 was then determined by phosphorimaging following SDS–PAGE.
Generation of phospho-hSK1 antibodies
Phospho-hSK1 specific polyclonal antibodies were raised in rabbits against a phosphopeptide (CGSKTPApSPVVVQQ) centred around phospho-Ser225 of hSK1. Prior to injection into rabbits, the phosphopeptide was conjugated to maleimide-activated keyhole limpet hemocyanin (Pierce) via its N-terminal cysteine. Antibodies active against the corresponding non-phosphorylated peptide were removed from the antiserum using SulfoLink beads (Pierce) to which the peptide was conjugated according to the manufacturer’s instructions.
Immunoprecipitations and western blotting
Cell lysates were clarified by centrifugation at 13 000 g for 15 min at 4°C, equalized for protein concentration, and complexed with M2 anti-FLAG (Sigma) or anti-ERK1/2 (Promega) antibodies at 4°C for 2 h with constant agitation. The anti-FLAG and anti-ERK1/2 immune complexes were precipitated by incubation with protein A/G PLUS-agarose beads (Santa Cruz) for 1 h at 4°C, and washed three times in lysis buffer. Endogenous hSK1 was immunoprecipitated with chicken anti-hSK1 antibodies raised against recombinant hSK1 generated in E. coli (Pitson et al., 2000a), with the immune complexes collected by incubation with biotinylated sheep anti-chicken IgG and streptavidin sepharose (Amersham) for 1 h at 4°C, followed by three washes in lysis buffer. The samples were then subjected to SDS–PAGE and western blotting. hSK1 expression levels in cell lysates were quantitated with either the monoclonal M2 anti-FLAG antibody, or a polyclonal anti-hSK1 antibody raised against recombinant hSK1 generated in E.coli (Pitson et al., 2000a). Expression of H-Ras was determined using anti-H-Ras polyclonal antibodies (Santa Cruz Biotech nology). ERK1/2 activation was followed by phospho-ERK1/2 and ERK1/2 polyclonal antibodies (Promega). The immunocomplexes were detected with HRP anti-mouse or anti-rabbit IgG (Pierce) using an enhanced chemiluminescence kit (ECL; Amersham Pharmacia Biotech).
Confocal microscopy
For fluorescence microscopy, wild-type hSK1 and hSK1S225A were tagged at the N-terminus with GFP. Briefly, EGFP was PCR amplified from pEGFP-1 (Clontech) with the following primers: 5′-TAAAGCTTG CCACCATGGTGAGCAAG-3′ and 5′-ATGGATCCATCTTGTACAG CTCGTCCATG-3′, and the resultant product was digested with HindIII and BamHI and cloned into pcDNA3-hSK1-FLAG (Pitson et al., 2000a) and pcDNA3-hSK1S225A-FLAG. HEK293T cells were transfected with these plasmids, as described above, cultured for 24 h, and then plated onto eight-well glass chamber slides at 1 × 104 cells/well. After 24 h, cells were treated for 30 min with 1 µg/ml PMA or DMSO control, fixed with 4% paraformaldehyde, and viewed using a 60× oil-immersion objective on an Olympus IX70 inverted microscope linked to a BioRad Radiance 2100 confocal microscope after excitation at 488 nm.
S1P levels
To determine both intracellular and extracellular S1P levels, HEK293T cells were metabolically labelled with [32P]orthophosphate as described above. Cells were then transferred to fresh phosphate-free DMEM and treated with 1 ng/ml TNFα or 100 ng/ml PMA for 30 min. To determine extracellular S1P release following stimulation, the media was then removed, centrifuged at 1000 g, and 2.5 ml of the supernatant added to 2.5 ml chloroform, 2.5 ml methanol and 20 µl concentrated HCl. The organic phase was then dried under vacuum, resuspended in chloroform, and S1P resolved by thin-layer chromatography (TLC) on silica gel 60 with l-butanol/ethanol/acetic acid/water (8:2:1:2 v/v). Intracellular S1P levels were determined by harvesting the cells into 400 µl methanol containing 25 µl concentrated HCl. Lipids were then extracted under alkaline conditions by the addition of 400 µl chloroform, 400 µl KCl and 40 µl 3 M NaCl. The aqueous phase, containing S1P under these conditions, was then acidified through the addition of 50 µl concentrated HCl and re-extracted with 400 µl chloroform. The organic phase was then dried under vacuum and resuspended in chloroform, and was S1P resolved by TLC as described above.
Acknowledgments
Acknowledgements
We thank Bruce Kemp for assistance during identification of the phosphorylation site, and Christopher Bagley, Vidya Limaye and Jennifer Gamble for their contributions towards generating antibodies. This work was supported by a Georgina Dowling Medical Research Fellowship from the University of Adelaide and an R.Douglas Wright Biomedical Research Fellowship from the National Health and Medical Research Council to S.M.P., and grants from the National Health and Medical Research Council.
References
- Adams J.A. (2003) Activation loop phosphorylation and catalysis in protein kinases: is there functional evidence for the autoinhibitor model? Biochemistry, 42, 601–607. [DOI] [PubMed] [Google Scholar]
- Bhalla U.S., Ram,P.T. and Iyengar,R. (2002) MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science, 297, 1018–1023. [DOI] [PubMed] [Google Scholar]
- Blom N., Gammeltoft,S. and Brunak,S. (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol., 294, 1351–1362. [DOI] [PubMed] [Google Scholar]
- Buehrer B.M., Bardes,E.S. and Bell,R.M. (1996) Protein kinase C-dependent regulation of human erythroleukemia (HEL) cell sphingosine kinase activity. Biochim. Biophys Acta, 1303, 233–242. [DOI] [PubMed] [Google Scholar]
- Chin D. and Means,A.R. (2000) Calmodulin: a prototypical calcium sensor. Trends Cell Biol., 10, 322–328. [DOI] [PubMed] [Google Scholar]
- Clark-Lewis I., Sanghera,J.S. and Pelech,S.L. (1991) Definition of a consensus sequence for peptide substrate recognition by p44mpk, the meiosis-activated myelin basic protein kinase. J. Biol. Chem., 266, 15180–15184. [PubMed] [Google Scholar]
- Davies S.P., Reddy,H., Caivano,M. and Cohen,P. (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J., 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia J., Ye,Y., Arranz,V., Letourneux,C., Pezeron,G. and Porteu,F. (2002) IEX-1: a new substrate involved in both ERK survival activity and ERK activation. EMBO J., 21, 5151–5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannun Y.A., Luberto,C. and Argraves,K.M. (2001) Enzymes of sphingolipid metabolism: from modular to integrative signaling. Biochemistry, 40, 4893–4903. [DOI] [PubMed] [Google Scholar]
- Hla T., Lee,M.J., Ancellin,N., Paik,J.H. and Kluk,M.J. (2001) Lysophospholipids—receptor revelations. Science, 294, 1875–1878. [DOI] [PubMed] [Google Scholar]
- Hobson J.P., Rosenfeldt,H.M., Barak,L.S., Olivera,A., Poulton,S., Caron,M.G., Milstien,S. and Spiegel,S. (2001) Role of the sphingosine-1-phosphate receptor EDG-1 in PDGF-induced cell motility. Science, 291, 1800–1803. [DOI] [PubMed] [Google Scholar]
- Johnson K.R., Becker,K.P., Facchinetti,M.M., Hannun,Y.A. and Obeid,L.M. (2002) PKC-dependent activation of sphingosine kinase 1 and translocation to the plasma membrane. Extracellular release of sphingosine-1-phosphate induced by phorbol 12-myristate 13-acetate (PMA). J. Biol. Chem., 277, 35257–35262. [DOI] [PubMed] [Google Scholar]
- Kyriakis J.M. and Avruch,J. (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev., 81, 807–869. [DOI] [PubMed] [Google Scholar]
- Lacaná E., Maceyka,M., Milstien,S. and Spiegel,S. (2002) Cloning and characterization of a protein kinase A achoring protein (AKAP)-related protein that interacts with and regulates sphingosine kinase 1 activity. J. Biol. Chem., 277, 32947–32953. [DOI] [PubMed] [Google Scholar]
- Lu K.P., Liou,Y.C. and Zhou,X.Z. (2002) Pinning down proline-directed phosphorylation signaling. Trends Cell Biol., 12, 164–172. [DOI] [PubMed] [Google Scholar]
- Ma Z., Webb,D.J., Jo,M. and Gonias,S.L. (2001) Endogenously produced urokinase-type plasminogen activator is a major determinant of the basal level of activated ERK/MAP kinase and prevents apoptosis in MDA-MB-231 breast cancer cells. J. Cell Sci., 114, 3387–3396. [DOI] [PubMed] [Google Scholar]
- Mandala S.M. (2001) Sphingosine-1-phosphate phosphatases. Prostaglandins, 64, 143–156. [DOI] [PubMed] [Google Scholar]
- Melendez A.J. and Khaw,A.K. (2002) Dichotomy of Ca2+ signals triggered by different phospholipid pathways in antigen stimulation of human mast cells. J. Biol. Chem., 277, 17255–17262. [DOI] [PubMed] [Google Scholar]
- Murphy L.O., Smith,S., Chen,R., Fingar,D.C. and Blenis,J. (2002) Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol., 4, 556–564. [DOI] [PubMed] [Google Scholar]
- Olivera A., Kohama,T., Edsall,L., Nava,V., Cuvillier,O., Poulton,S. and Spiegel,S. (1999) Sphingosine kinase expression increases intracellular sphingosine-1-phosphate and promotes cell growth and survival. J. Biol. Chem., 147, 545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T. and Scott,J.D. (1997) Signaling through scaffold, anchoring and adaptor proteins. Science, 278, 2075–2080. [DOI] [PubMed] [Google Scholar]
- Pearson G., Robinson,F., Gibson,T.B., Xu,B.E., Karandikar,M., Berman,K. and Cobb,M.H. (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev., 22, 153–183. [DOI] [PubMed] [Google Scholar]
- Pitson S.M., D’Andrea,R.J., Vandeleur,L., Moretti,P.A.B., Xia,P., Gamble,J.R., Vadas,M.A. and Wattenberg,B.W. (2000a) Human sphingosine kinase: purification, molecular cloning and characterisation of the native and recombinant enzymes. Biochem. J., 350, 429–441. [PMC free article] [PubMed] [Google Scholar]
- Pitson S.M., Moretti,P.A.B., Zebol,J.R., Xia,P., Gamble,J.R., Vadas,M.A., D’Andrea,R.J. and Wattenberg,B.W. (2000b) Expression of a catalytically inactive sphingosine kinase mutant blocks agonist-induced sphingosine kinase activation: a dominant-negative sphingosine kinase. J. Biol. Chem., 275, 33945–33950. [DOI] [PubMed] [Google Scholar]
- Pitson S.M. et al. (2002) The nucleotide-binding site of human sphingosine kinase 1. J. Biol. Chem., 277, 49545–49553. [DOI] [PubMed] [Google Scholar]
- Pyne S. and Pyne,N.J. (2000) Sphingosine 1-phosphate signalling in mammalian cells. Biochem. J., 349, 385–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne S. and Pyne,N.J. (2002) Sphingosine 1-phosphate signalling and termination at lipid phosphate receptors. Biochim. Biophys. Acta, 1582, 121–131. [DOI] [PubMed] [Google Scholar]
- Rius R.A., Edsall,L.C. and Spiegel,S. (1997) Activation of sphingosine kinase in pheochromocytoma PC12 neuronal cells in response to trophic factors. FEBS Lett., 417, 173–176. [DOI] [PubMed] [Google Scholar]
- Rosenfeldt H.M., Hobson,J.P., Maceyka,M., Olivera,A., Nava,V.E., Milstien,S. and Spiegel,S. (2001) EDG-1 links the PDGF receptor to Src and focal adhesion kinase activation leading to lamellipodia formation and cell migration. FASEB J., 15, 2649–2659. [DOI] [PubMed] [Google Scholar]
- Sharrocks A.D., Yang,S.H. and Galanis,A. (2000) Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem. Sci., 25, 448–453. [DOI] [PubMed] [Google Scholar]
- Shu X., Wu,W., Mosteller,R.D. and Broek,D. (2002) Sphingosine kinase mediates vascular endothelial growth factor-induced activation of Ras and mitogen-activated protein kinases. Mol. Cell. Biol., 22, 7758–7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z. et al. (1996) A structural basis for substrate specificities of protein Ser/Thr kinases: primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5 and Erk1. Mol. Cell. Biol., 16, 6486–6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S. and Milstien,S. (2002) Sphingosine 1-phosphate, a key cell signaling molecule. J. Biol. Chem., 277, 25851–25854. [DOI] [PubMed] [Google Scholar]
- Sudol M. and Hunter,T. (2000) New wrinkles for an old domain. Cell, 103, 1001–1004. [DOI] [PubMed] [Google Scholar]
- Van Veldhoven P.P., Gijsbers,S., Mannaerts,G.P., Vermeesch,J.R. and Brys,V. (2000) Human sphingosine-1-phosphate lyase: cDNA cloning, functional expression studies and mapping to chromosome 10q22(1). Biochim. Biophys. Acta, 1487, 128–134. [DOI] [PubMed] [Google Scholar]
- Werlen G., Hausmann,B., Naeher,D. and Palmer,E. (2003) Signaling life and death in the thymus: timing is everything. Science, 299, 1859–1863. [DOI] [PubMed] [Google Scholar]
- Xia P., Vadas,M.A., Rye,K.-A., Barter,P.J. and Gamble,J.R. (1999) High density lipoproteins (HDL) interrupt the sphingosine kinase signaling pathway. J. Biol. Chem., 274, 33143–33147. [DOI] [PubMed] [Google Scholar]
- Xia P., Gamble,J.R., Wang,L., Pitson,S.M., Moretti,P.A.B., D’Andrea,R.J., Wattenberg,B.W. and Vadas,M.A. (2000) An oncogenic role of sphingosine kinase. Curr. Biol., 10, 1527–1530. [DOI] [PubMed] [Google Scholar]
- Xia P., Wang,L., Moretti,P.A.B., Albanese,N., Chai,F., Pitson,S.M., D’Andrea,R.J., Gamble,J.R. and Vadas,M.A. (2002) Sphingosine kinase interacts with TRAF2 and dissects TNF signalling. J. Biol. Chem., 277, 7996–8003. [DOI] [PubMed] [Google Scholar]
- Xu R., Seger,R. and Pecht,I. (1999) Extracellular signal-regulated kinase activates syk: a new potential feedback regulation of Fc epsilon receptor signaling. J. Immunol., 163, 1110–1114. [PubMed] [Google Scholar]
- Yaffe M.B. and Cantley,L.C. (1999) Grabbing phosphoproteins. Nature, 402, 30–31. [DOI] [PubMed] [Google Scholar]
- Young K.W., Willets,J.M., Parkinson,M.J., Bartlett,P., Spiegel,S., Nahorski,S.R. and Challiss,R.A.J. (2003) Ca2+/calmodulin-dependent translocation of sphingosine kinase: role in plasma membrane translocation but not activation. Cell Calcium, 33, 119–128. [DOI] [PubMed] [Google Scholar]