

Abstract
The ATM protein kinase is a primary activator of the cellular response to DNA double-strand breaks (DSBs). In response to DSBs, ATM is activated and phosphorylates key players in various branches of the DNA damage response network. ATM deficiency causes the genetic disorder ataxia-telangiectasia (A-T), characterized by cerebellar degeneration, immunodeficiency, radiation sensitivity, chromosomal instability and cancer predisposition. The MRN complex, whose core contains the Mre11, Rad50 and Nbs1 proteins, is involved in the initial processing of DSBs. Hypomorphic mutations in the NBS1 and MRE11 genes lead to two other genomic instability disorders: the Nijmegen breakage syndrome (NBS) and A-T like disease (A-TLD), respectively. The order in which ATM and MRN act in the early phase of the DSB response is unclear. Here we show that functional MRN is required for ATM activation, and consequently for timely activation of ATM-mediated pathways. Collectively, these and previous results assign to components of the MRN complex roles upstream and downstream of ATM in the DNA damage response pathway and explain the clinical resemblance between A-T and A-TLD.
Keywords: ATM/ataxia-telangiectasia/DNA damage response/double-strand breaks/MRN complex
Introduction
Double strand breaks (DSBs) are extremely cytotoxic DNA lesions that activate an extensive array of responses that lead to damage repair and allow continuation of cellular life (Hopfner et al., 2002; Jackson, 2002; Bradbury and Jackson, 2003). The nuclear protein kinase ATM is regarded as the primary activator of this network (Shiloh, 2003). ATM is missing or inactivated in patients with the genetic disorder ataxia-telangiectasia (A-T), characterized by cerebellar degeneration, immunodeficiency, radiation sensitivity, chromosomal instability and cancer predisposition (Crawford, 1998; Gatti et al., 2001). Following the induction of DSBs, ATM’s kinase activity is enhanced and it phosphorylates key proteins in numerous signaling pathways (Shiloh and Kastan, 2001; Shiloh, 2003). ATM’s activation was recently shown to involve autophosphorylation, which is essential for turning inactive ATM into a very potent protein kinase (Bakkenist and Kastan, 2003). Concomitantly with ATM activation, a fraction of the nuclear content of ATM adheres to the DSB sites (Andegeko et al., 2001). The mode by which the DSB signal is conveyed to ATM is not clear, nor are the proteins that function in the DSB response between damage induction and ATM activation.
DSBs are repaired in eukaryotes by the concerted action of mechanisms based on homologous recombination (HR) or non-homologous end joining (NHEJ) (Hopfner et al., 2002; Jackson, 2002). The highly conserved MRN complex, whose core contains the proteins Mre11, Rad50 and Nbs1, plays a role in both modes of DSB repair, particularly in the HR pathway (D’Amours and Jackson, 2002; Tauchi et al., 2002). Prior to these highly structured processes, the random broken ends need to be detected and processed. The MRN complex is involved in the initial processing of DSBs due to its nuclease activity and DNA binding capability (Petrini et al., 2001; D’Amours and Jackson, 2002). These activities reside in the Mre11 protein, and partially depend on Mre11’s interaction with the Rad50 ATPase. The Nbs1 protein is thought to be involved in the nuclear localization and proper assembly of the complex at DSB ends, probably via its ability to interact directly with the histone protein H2AX, which is rapidly phosphorylated following DSB induction (Kobayashi et al., 2002; Tauchi et al., 2002). The MRN complex adheres to the sites of DSBs immediately following their induction, and this process is independent of ATM (Mirzoeva and Petrini, 2001; Petrini et al., 2001).
The order in which ATM and the MRN complex act in the early phase of the DSB response is unclear. Initial clues to the close relationships between ATM and the MRN complex in this pathway are provided by the phenotypes associated with deficiencies of each of the proteins. Hypomorphic mutations in the NBS1 locus lead to the Nijmegen breakage syndrome (NBS), expressed as a combination of microcephaly, mental deficiency, immunodeficiency, radiation sensitivity, chromosomal instability and cancer predisposition (Digweed et al., 1999; Tauchi et al., 2002). A variant of this disease is caused by RAD50 mutations (R.Bendix et al., presented at The International Meeting on the Nijmegen Breakage Syndrome, Prague, 2002). MRE11 mutations, on the other hand, cause the A-T like disease (A-TLD), a late-onset variant of A-T (Stewart et al., 1999). Importantly, while A-T and A-TLD are highly similar, NBS does not share the cerebellar degeneration typical of these diseases. These differences may point to a certain specificity in the functions of Nbs1 and Rad50 on the one hand, and the Mre11 protein on the other hand. The cellular phenotype of the three disorders is, however, quite similar and is characterized by various degrees of radiosensitivity and impairment of the cellular response to DSBs.
Direct functional links between ATM and components of the MRN complex were evidenced by ATM-mediated phosphorylation of Nbs1 and Mre11 in response to DSBs (Gatei et al., 2000; Lim et al., 2000; Wu et al., 2000; Zhao et al., 2000; Stewart et al., 2001; Yuan et al., 2002a,b). Phosphorylated Nbs1 plays an as yet undefined role in major ATM-mediated pathways, the intra-S and G2/M cell cycle checkpoints (Lim et al., 2000; Zhao et al., 2000; Buscemi et al., 2001). Furthermore, at low damage levels, Nbs1 appears to facilitate ATM-mediated phosphorylation of several other ATM substrates, such as the checkpoint kinase Chk2 (Buscemi et al., 2001; Girard et al., 2002; Lee et al., 2003), the chromatin remodeling protein SMC1 (Kim et al., 2002b; Yazdi et al., 2002), Chk1 (Gatei et al., 2003), and Mre11 itself (Dong et al., 1999). These observations assign to Nbs1 and Mre11 roles downstream of ATM in the DNA damage response pathway. On the other hand, the activities of the MRN complex in the very early stage of the DSB response, and the A-TLD phenotype caused by Mre11 deficiency suggest functional links between ATM and the MRN complex also at the very early phase of the DNA damage response, between damage induction and ATM activation.
We hypothesized that the clinical resemblance between A-T and A-TLD might reflect a dependence of ATM activation and the subsequent ATM-mediated damage response on functional MRN complex, particularly on proper action of the Mre11 protein. We present here evidence that supports this hypothesis.
Results
ATM activation is defective in cells with MRN deficiencies
We compared the response to treatment with the radiomimetic chemical neocarzinostatin (NCS) of cell lines from healthy donors and patients with A-T, NBS and A-TLD (Figure 1). A-TLD is represented by patients with two variants of this disorder. The moderate variant, A-TLD(M), results from compound heterozygosity for a nonsense and a missense MRE11 mutation, and exhibits reduced levels of partially active Mre11. The severe form of A-TLD, A-TLD(S), is caused by homozygosity for a nonsense MRE11 mutation that leaves extremely low levels of truncated protein (Stewart et al., 1999; Pitts et al., 2001). The NBS cell line is homozygous for a hypomorphic NBS1 mutation, which is common for the majority of NBS patients (Digweed et al., 1999; Tauchi et al., 2002). It produces two truncated versions of the Nbs1 protein, one of which is contained in the Mre11 complex and probably has residual activity (Maser et al., 2001).
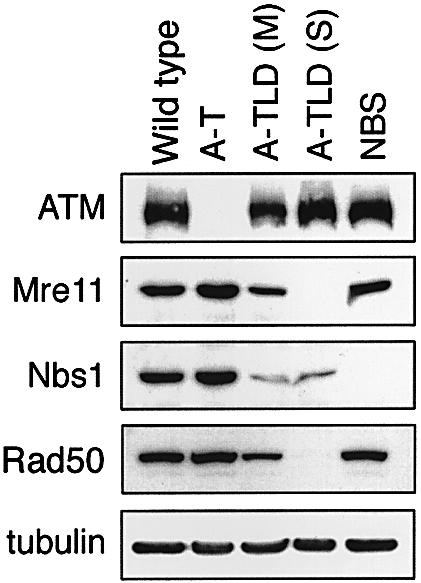
Fig. 1. Immunoblotting analysis of lymphoblastoid cell lines from patients with various genome instability syndromes. A-TLD is represented by patients with the two variants of this disorder, A-TLD(M) and A-TLD(S) (see text). Note the reduced levels of the Nbs1 and Rad50 in A-TLD patients (Stewart et al., 1999).
An initial response of the ATM protein to DSBs is its activation, which is reflected in vitro as rapid enhancement of its kinase activity (Banin et al., 1998; Canman et al., 1998). We noticed that the elevation in ATM’s catalytic activity following NCS treatment was moderately reduced in A-TLD(M) cells and completely abolished in A-TLD(S) cells, while NBS cells exhibited variable extents of reduction in ATM activation, which on the average did not differ significantly from those of wild-type cells (data not shown). Bakkenist and Kastan (2003) have recently shown, however, that the damage-induced increase in ATM’s kinase activity in vitro is just a modest reflection of the actual activation of ATM in cells, and demonstrated that this activation involves intermolecular autophosphorylation of ATM on Ser1981. Thus, a considerably more sensitive measure of ATM activation in cells can be obtained by monitoring ATM autophosphorylation on Ser1981 using a phospho-specific antibody (Bakkenist and Kastan, 2003). Dose response and time course experiments (Figure 2) indicated that ATM activation was indeed retarded in NBS cells, was more pronouncedly moderate in A-TLD(M), and most severely affected in A-TLD(S).

Fig. 2. ATM activation, reflected by its autophosphorylation, in lymphoblastoid lines from genomic instability syndromes. Following NCS treatments, cellular extracts were subjected to immunoblotting analysis using an antibody directed against phosphorylated Ser1981 of ATM. An anti-ATM antibody was used to control for ATM amounts. (A) ATM activation in response to increasing NCS doses (treatment time 15 min). (B) Time course of ATM autophosphorylation following treatment with 5 ng/ml of NCS.
Nuclear retention of ATM is defective in cells with MRN deficiencies
Recently, we found another early and visible hallmark of ATM’s response to DSBs: nuclear relocalization and enhanced binding to the damaged sites of a fraction of the nuclear ATM content. This phenomenon is demonstrated as damage-induced increase in the resistance of a fraction of ATM to detergent extraction, termed ‘nuclear retention’ (Andegeko et al., 2001). Nuclear retention of ATM following radiomimetic damage was increasingly attenuated in NBS, A-TLD(M) and A-TLD(S) (Figure 3). Interestingly, the order in which the different genotypes increasingly affected ATM activation and retention was similar for both phenomena, suggesting that they reflect two parallel facets of ATM’s response to DNA damage.

Fig. 3. Nuclear retention of ATM following radiomimetic treatment in cell lines with different genotypes. (A) Biochemical demonstration of damage-induced nuclear retention of ATM, reflected as increased resistance of this protein to detergent extraction. Lymphoblastoid cells were treated with the indicated doses of NCS, and protein extracts underwent successive fractionations with detergents. ATM was visualized in the resultant fractions using immunoblotting analysis. The extraction-resistant fraction of ATM (‘retained ATM’) represents a portion of ATM that adheres to DSB sites in a dose-dependent manner (see top panel representing a normal control). ‘Non-retained ATM’ represents a clarified cell extract from the first fractionation that contains unbound ATM in each sample. Dose-dependent nuclear retention of ATM is abrogated in A-TLD cells in correlation with the extent of Mre11 deficiency. Note intermediate impairment of ATM retention in the NBS cell line. (B) Demonstration of nuclear retention of ATM in lymphoblast cells using in situ detergent extraction followed by immunostaining (Andegeko et al., 2001). In untreated cells most of the nuclear content of ATM is extracted and washed out, while prior NCS treatment (50 ng/ml) renders a significant portion of nuclear ATM extraction-resistant. This process is impaired to different extents in A-TLD and NBS cells, most seriously in the severe A-TLD variant. (C) Quantitation of the amount of ATM fluorescence in lymphoblast cells after treatment with 50 ng/ml of NCS and subsequent detergent extraction in situ. The fluorescent signal was quantitated in at least 16 cells for each point.
The importance of the DSB-bound fraction of ATM was recently underscored by Lukas et al. (2003), who showed that ATM-mediated phosphorylation of Nbs1 and the checkpoint kinase Chk2 occurs exclusively at the DSB sites. Phosphorylated Chk2 is then released into the soluble nucleoplasmic fraction, and in turn phosphorylates other downstream targets, while Nbs1 remains at the DSB sites. Indeed, we noticed that both of the retained and soluble ATM fractions contained activated ATM molecules (Supplementary figure 1, available at The EMBO Journal Online).
Defective phosphorylation of ATM targets in MRN-deficient cells
The attenuated response of ATM to DNA damage in A-TLD cells was reflected in the phosphorylation of ATM’s downstream targets. Assessment of the specific effect of Mre11 deficiency on the phosphorylation of ATM substrates is complicated by the fact that some of these substrates require functional Nbs1 protein for optimal phosphorylation (Buscemi et al., 2001; Girard et al., 2002; Kim et al., 2002b; Yazdi et al., 2002; Gatei et al., 2003; Lee et al., 2003). Hence, the low levels of Nbs1 in A-TLD cells (Figure 1) may obscure the net effect of Mre11 deficiency. We therefore examined three ATM-dependent phosphorylation events that depend on the presence of Nbs1 to different extents: phosphorylation of the Chk2 kinase (Matsuoka et al., 2000), which at low treatment doses is dependent on the presence of Nbs1 (Buscemi et al., 2001; Girard et al., 2002) (Figure 4, top panel); phosphorylation of p53 on Ser15 (Banin et al., 1998; Canman et al., 1998), which we found to be moderately and variably dependent on Nbs1 (middle panel); and phosphorylation of Hdm2, the human ortholog of murine Mdm2, on Ser395 (Khosravi et al., 1999; Maya et al., 2001), which we found to be Nbs1-independent (bottom panel). Significantly, all three phosphorylations were increasingly reduced in A-TLD cells in correlation with the degree of Mre11 deficiency, regardless of their dependence on Nbs1 (Figure 4). Significantly, the response of A-TLD(S) cells was almost indistinguishable from that of A-T cells.

Fig. 4. Phosphorylation of ATM effectors in lymphoblastoid cells following treatment with 10 ng/ml of NCS. Top panel: phosphorylation of the Chk2 kinase reflected as retarded electrophorectic migration of the phosphorylated protein. Note Nbs1 dependence of this phosphorylation at the treatment dose used in this experiment, the delayed and reduced phosphorylation of Chk2 in A-TLD(M) cells, and the lack of phosphorylation in A-TLD(S) cells. Middle panel: phosphorylation of p53 on Ser15 detected by a phospho-specific antibody. A moderate retardation of this phosphorylation is noticed in NBS cells, and the response is attenuated in the A-TLD variants. Bottom panel: phosphorylation of Hdm2 on Ser395. This phosphorylation abolishes the immunoreactivity of Hdm2 with the monoclonal antibody 2A10 and is therefore expressed as loss of the Hdm2 band on immunoblots (Khosravi et al., 1999). The band below the Hdm2 band represents a cross-reacting protein and conveniently serves as a loading control. Hdm2 phosphorylation occurs at a normal rate in NBS cells, is moderately impaired in A-TLD(M) cells, and is abolished in A-TLD(S) cells.
In order to obtain a more general view of the consequences of ATM activation in cells, we performed immunostaining experiments with an antibody raised against a group of potential ATM phosphorylation sites. ATM phosphorylates serine or threonine residues followed by glutamine (‘SQ’ or ‘TQ’ motifs) (Kim et al., 1999; O’Neill et al., 2000). This antibody was raised against a collection of peptides containing these motifs with the pertinent serine or threonine phosphorylated. It detects the phosphorylated forms of some known ATM substrates, such as p53 and Chk2, and several other substrates currently under investigation in our laboratory. Upon NCS treatment, this antibody reveals a strong, ATM-dependent nuclear response in a variety of cell lines (Figure 5A). Quantitation of this response indicated that it is significantly impaired in A-TLD(M) cells and abolished in A-TLD(S) cells, while being variably low in NBS cells (Figure 5B).

Fig. 5. Integrative visualization of phosphorylation of ATM substrates in human fibroblast lines in response to DSBs, visualized by immunostaining with an antibody raised against phosphorylated ATM target sequence (‘anti-phospho-[SQ/TQ]’). (A) Thirty minutes following NCS treatment, a vigorous response is observed in wild-type cells while the diffuse background staining in A-T cells is barely changed. Phase contrast images of the cells indicate that this response is confined to the nucleus. (B) Phosphorylation of ATM substrates in fibroblast cell lines with various genotypes following treatment with 50 ng/ml of NCS. Fluorescence intensity after staining with the anti-phospho-(SQ/TQ) antibody was averaged over ∼20 cells for each genotype and treatment. The numbers below the line represent the ratios between fluorescence values after NCS treatment and without treatment. Note the similar lack of response in A-T and A-TLD(S) cells and the variably low responses of all other genotypes. In some cell lines initial high level of phosphorylation is noticed. This constitutive damage response is occasionally observed in cells with defective damage response and represents elevated basal levels of DNA damage (Gatei et al., 2001; Kamsler et al., 2001).
Reconstitution of the MRN complex restores ATM activation
In order to further establish the link between functional MRN complex and ATM activation, we reconstituted the endogenous MRN complex in A-TLD(S) cells. Since the life span of primary A-TLD fibroblasts is considerably limited, primary A-TLD(S) fibroblasts were first immortalized by ectopic expression of the catalytic subunit of human telomerase (hTERT) (Wood et al., 2001). It was previously shown that hTERT immortalization does not alter the phenotypic characteristics associated with the defective DNA damage response of A-T cells (Wood et al., 2001) and A-TLD(M) cells (Stracker et al., 2002). The immortalized cells were then transduced with a retroviral vector expressing recombinant Mre11. Mre11 expression reconstituted the normal levels and nuclear co-localization of Rad50 and Nbs1 (Figure 6A), suggesting reassembly of nuclear MRN complex. Importantly, ATM activation (Figure 6B) and phosphorylation of downstream substrates (not shown) were subsequently reconstituted. An analogous system of NBS cells stably reconstituted with recombinant Nbs1 was previously established by Tauchi et al. (2001). In these cells too, ectopic expression of Nbs1 leads to reassembly of the MRN complex in the nucleus (Tauchi et al., 2001) and restores ATM-mediated Chk2 phosphorylation (Buscemi et al., 2001). Indeed, the attenuation in ATM activation typical of the parental NBS cells disappeared in the reconstituted cells (Figure 6C).

Fig. 6. Reconstitution of the MRN complex and ATM activation in A-TLD(S) and NBS cells by ectopic expression of Mre11 and Nbs1, respectively. (A) Immunofluorescence images of the three components of the MRN complex in hTERT-immortalized A-TLD(S) fibroblasts following transduction with a control vector expressing GFP and a vector expressing wild-type Mre11 protein. In each panel the same field is shown in all photographs. (B) Immunoblotting analysis showing ATM activation in A-TLD(S) cells transduced with the control and Mre11 vectors. Note reconstitution of endogenous Rad50 and Nbs1 levels following ectopic expression of Mre11 and reconstitution of damage-induced ATM activation. (C) ATM activation in NBS cells reconstituted by ectopic expression of Nbs1 (Tauchi et al., 2001).
We concluded from these results that the presence of functional MRN complex in the nucleus is required for proper ATM activation. One of the functions of the MRN complex at the early stage of the DSB response is thought to involve initial processing of the DSB prior to its repair (Bradbury and Jackson, 2003; D’Amours and Jackson, 2002). We asked whether a nuclease-defective Mre11 protein would complement the defective ATM activation in A-TLD(S) cells. The mutant Mre11-3 contains two amino acid substitutions (H129L/D130V) that alter its phosphodiesterase domain, thereby reducing its endonuclease activity (Stracker et al., 2002). Interestingly, expression of the the Mre11-3 mutant protein in A-TLD(S) cells seemed to reconstitute nuclear MRN complex similarly to wild-type Mre11(Figure 7A), but failed to fully restore damage-induced ATM activation (Figure 7B). This result suggested to us that the mere assembly of the MRN complex is not sufficient for proper ATM activation, and its nuclease activity, which resides in the Mre11 protein, is specifically required for this process.

Fig. 7. Inability of nuclease-defective Mre11 to fully reconstitute ATM activation in A-TLD(S) cells. (A) Expression of recombinant, mutant Mre11 and endogenous Rad50 and Nbs1 in A-TLD(S) fibroblasts transduced with GFP and Mre11-3 vectors. Note reconstitution of endogenous Rad50 and Nbs1 levels and nuclear localization. (B) Visualization of ATM activation in the same cells by immunostaining with the antibody against phosphorylated Ser1981. Note the defective ATM activation in cells transduced with the Mre11-3 mutant.
Discussion
It is becoming evident that the DNA damage response, which is essential to the organism’s ability to cope with environmental hazards, is based on a multi-branched, finely tuned network of signaling pathways that involves many aspects of cellular metabolism. Less is known about the very early events that follow infliction of DNA damage but that precede the spread of the damage signal throughout the cell. Our understanding of the DSB response is a case in point: while much is known about the DSB-activated pathways such as the cell cycle checkpoints, less is known about the order of early events that follow DSB formation. The response to this deadly DNA lesion must be swift, hence immediate detection of the lesion and timely recruitment of damage processing and signaling proteins are critical. Current models of the DSB response describe a linear progression beginning with sensors that convey the initial damage signal to transducers, which in turn convey it to numerous effectors. In this model ATM seems to play the role of the transducer, with its plethora of downstream effectors. What, then, are the sensors that act upstream of ATM, possibly recruiting this powerful transducer and leading to its activation?
D’Amours and Jackson (2002) recently proposed a model in which the initial DNA lesions, whose structural and chemical characteristics are usually random, are first turned by ‘signal modifiers’ into more conventional structures (e.g. single-stranded ends) that are amenable to repair and recruit the signal transducers. In the case of DSBs, the MRN complex is thought to carry out initial resection of the broken ends that creates such structures. The notion that single-stranded DNA is a common intermediate in the recruitment of response proteins following DNA damage is indeed gaining support (Zou and Elledge, 2003). Petrini and Stracker (2003) have also suggested that the MRN complex fulfills a set of criteria that defines a damage sensor. MRN is a good candidate for a sensor/signal modifier also in view of its ATM-independent, rapid recruitment to DSB sites immediately following DSB formation (Mirzoeva and Petrini, 2001; Mirzoeva and Petrini, 2003), and Nbs1’s ATM-independent interaction with the chromatin (Zhao et al., 2002), particularly with phosphorylated histone H2AX (Kobayashi et al., 2002). Indeed, in the budding yeast, damage-induced activation of ATM’s ortholog, Tel1p, subsequent phosphorylation of its downstream targets, and activation of pertinent pathways such as the cell cycle checkpoints are dependent on functional Mre11/Rad50/Xrs2 complex (the yeast ortholog of the mammalian MRN complex)—specifically on the nuclease activity of Mre11 (D’Amours and Jackson, 2001; Grenon et al., 2001; Usui et al., 2001). The importance of the enzymatic activity of Mre11 for proper activation of ATM (Figure 7) may indeed reflect the role of this activity in a process that turns initial DSBs into a structural intermediate that signals to transducer molecules. Worthy of note, the chromatin remodeling protein SMC1, itself an ATM substrate, is also recruited to damaged sites in an MRN-dependent, ATM-independent manner (Kim et al., 2002a), further indicating the requirement for MRN activity for the recruitment of damage response players to the damaged sites.
These notions and the similar phenotypes of ATM and Mre11 deficiencies in humans prompted us to test this model in human cells. Our data suggest that proper ATM activation and nuclear retention and subsequent ATM substrate phosphorylations are dependent on the presence of functional MRN complex. Importantly, we were able to distinguish between ATM-dependent substrate phosphorylations that specifically require the mediating function of Nbs1 downstream of ATM, and those that do not require this function, thereby controlling for this potential confounder of the specific role of the Mre11 protein upstream of ATM. While the binding of the MRN complex to DSB sites is ATM-independent (Mirzoeva and Petrini, 2001), we find that ATM’s binding to these sites is dependent on functional Mre11, further pointing to an order of recruitment of the MRN complex and ATM to the damaged site. The demonstration of ATM-mediated phosphorylation of Chk2 specifically and exclusively at the damaged sites (Lukas et al., 2003) attests to the importance of the binding of ATM to these sites, which is reflected in the retention phenomenon (Andegeko et al., 2001). We noticed that, indeed, bound ATM is autophosphorylated, i.e., activated. Experiments are in progress to examine whether ATM activation is required for its binding to DSB sites.
We conclude that functional MRN complex is necessary for proper ATM-mediated response to DSBs (Figure 8). Due to the physical association and functional cooperation between the three core components of the MRN complex, even partial loss of one of them leads to abrogation of the complex’s functions. This makes it difficult to assess the relative contribution of individual components of the complex to the damage response. The different phenotypes associated with genetic Nbs1 and Mre11 deficiencies may reflect specific roles of each of them in the damage response, as well as the extent to which the function of the entire MRN complex is impaired by the corresponding hypomorphic mutations. Notably, the extent of the the defect in ATM-mediated responses to DSBs was reported to be variable in NBS cell lines (Girard et al., 2002), and appears to be more pronounced in A-TLD cells (this study).

Fig. 8. A model depicting early events in the cellular response to moderate DSB levels. The MRN complex is essential for the initial damage processing. Processed DNA lesions lead to the recruitment and activation of ATM, which in turn phosphorylates its substrates, among them Nbs1 and Mre11. Phosphorylated Nbs1 facilitates the phosphorylation of certain ATM substrates and plays a role in the activation of cell cycle checkpoint. It is not clear whether Nbs1 acts alone in this capacity or in the context of the MRN complex.
The sequence of events at the early stage of the DNA damage response may not necessarily be a simple hierarchy in which each protein has a single position in the damage signaling cascade (Bradbury and Jackson, 2003; Petrini and Stracker, 2003). There is evidence to the contrary: both the Nbs1 and Mre11 proteins were shown to be phosphorylated in an ATM-dependent manner in response to DSBs (Gatei et al., 2000; Lim et al., 2000; Wu et al., 2000; Zhao et al., 2000; Stewart et al., 2001; Yuan et al., 2002a), placing them also downstream of ATM in the damage response pathway. Moreover, phosphorylated Nbs1 plays a separate role in the activation of specific cell cycle checkpoints (Buscemi et al., 2001; Lim et al., 2000). However, Nbs1 phosphorylation is not required for the recruitment of MRN to the DSB sites (Lukas et al., 2003), emphasizing the distinction between the sensor role of MRN and the downstream roles of the phosphorylated Nbs1. Interestingly, replication protein A (RPA), itself an ATM downstream substrate, was recently shown to play an essential role in the initial recruitment of ATR to the damaged site and its subsequent activation (Zou and Elledge, 2003), providing another example of a protein that plays roles upstream of the transducers, and then, following its transducer-mediated phosphorylation, separate roles in downstream steps of the damage response.
Nbs1 also has a role in facilitating the phosphorylation of other ATM substrates following low damage levels (Buscemi et al., 2001; Yazdi et al., 2002; Lee et al., 2003). It may carry out these activities alone, possibly while being detached from the MRN complex. Petrini and Stracker (2003) suggested that following the initial action of the MRN complex as a sensor, the subsequent phosphorylation of Nbs1 turns it into a mediator in further stages of the damage response. Indeed, in response to DSBs, the MRN complex forms two types of nuclear foci: the first is rapid and ATM-independent and probably reflects its sensor mode of action, while the second is ATM-dependent and prolonged and probably corresponds to MRN’s downstream roles (Mirzoeva and Petrini, 2001; Mirzoeva and Petrini, 2003). We showed here, however, that not every substrate of ATM requires Nbs1 for its phosphorylation. Foray et al. (2003) recently showed that a subset of ATM- and ATR-dependent phosphorylations require the presence of the Brca1 protein, further attesting to the importance of such mediators in the phosphorylation of some but not all the substrates of these protein kinases. The list of such mediators is growing. Two other proteins, 53BP1 and MDC1(NFBD1), recently emerged as possible adaptors that facilitate the phosphorylation of specific ATM substrates at low damage levels. Similar to Nbs1 and Brca1, they contain a BRCT domain, they are recruited to nuclear foci together with MRN complex early in the damage response, and are themselves ATM substrates (Abraham, 2002; DiTullio et al., 2002; Fernandez-Capetillo et al., 2002; Wang et al., 2002; Goldberg et al., 2003; Lou et al., 2003a,b; Stewart et al., 2003). Thus, it appears that the transfer of ATM-mediated signal to certain subsets of substrates requires the involvement of specific mediators/adaptors, pointing once again to a complex, fine-tuned process.
The importance of the treatment dose applied to animals or cultured cells in laboratory studies on the relationships between various proteins acting in the DNA damage response is important. Because of the complex nature of this response and the redundancy of certain pathways, the dependence of one pathway on the other may differ considerably at different levels of damage (Fernandez-Capetillo et al., 2002; Girard et al., 2002; Wang et al., 2002). A well-documented example is ATM-dependent pathways. ATM’s activity is required primarily for the initial phase of the DNA damage response, immediately after damage infliction. In A-T cells that completely lack ATM activity, the damage response pathways eventually get activated because of the action of other kinases, such as ATR, which belongs to the same protein family as ATM (Abraham, 2001; Shiloh, 2001; Shiloh and Kastan, 2001). This redundancy is particularly evident at high doses of damaging agents, and may even mask the effect of ATM loss at such doses or at later time points (T.Uziel, unpublished data). A recent example of this notion concerns the dependence of the phosphorylation of certain ATM substrates on Nbs1, which is clearly observed at low radiation doses but less so at high doses (Buscemi et al., 2001; Fernandez-Capetillo et al., 2002; Girard et al., 2002; T.Uziel, unpublished observations). Our experiments were therefore carried out with relatively low treatment doses—a dose of 50 ng/ml of NCS is roughly equivalent to 2 Gy of ionizing radiation—and early time points that clearly demonstrate the subtle effect of abrogated activation of otherwise intact ATM protein. Thus, different treatment protocols may account for the discrepancy between the A-T-like phenotype of A-TLD(S) cells observed by us and the moderate cellular phenotype reported for these cells by Stewart et al. (1999) and Falck et al. (2002). Presumably, however, the low treatment doses reflect more realistic physiological situations and better explain human phenotypes. Conceivably, the evolution of damage response mechanisms happened under natural stresses and these mechanisms are probably structured to respond to real-life genotoxic stress.
The A-TLD phenotype and our results indicate that the clinical presentation of A-T can be caused merely by defective activation of ATM, and not necessarily by its absence or complete inactivation as in classical A-T patients. They also suggest that the progressive cerebellar degeneration in A-T, which is the prominent and most devastating symptom of the disease, may result primarily from the defect in the DNA damage response. The etiology of this degenerative process has long been debated and has sometimes been attributed to possible functions of ATM that may not be associated with the DNA damage response. We believe our results swing the pendulum of this debate back to the DNA damage response. This conclusion has important implications for future attempts to slow down the progression of the neurodegenerative process in A-T.
Materials and methods
Antibodies
MAT3-4G10/8, a monoclonal antibody directed against the ATM protein, was raised in our laboratory (Andegeko et al., 2001). Visualization of ATM by immunofluorescence was performed using the mouse monoclonal antibody 5C2 (from E.Lee). The phospho-specific antibody directed against phosphorylated Ser1981 of human ATM was a generous gift from M.Kastan. Rabbit polyclonal antibody for hMre11 (Ab-1, Oncogene Research Products, Cambridge, MA) and mouse monoclonal antibodies for Mre11 (Ab12D7) and Rad50 (Ab 13B3) (Genetex, San Antonio, TX) were used for immunoblotting and immunofluorescence analyses. Immunoblotting of other proteins was carried out using polyclonal antibodies against the Nbs1 protein (Ab-1, Oncogene Research Products), tubulin (clone B-5-1-2, Sigma Israel Chemicals, Rehovot, Israel), the Chk2 kinase (from S.Elledge), anti-phospho-p53(Ser15), anti-phospho-(SQ/TQ) ATM/ATR substrate (Cell Signaling Technology, Beverley, MA) and the monoclonal antibody 2A10 against Hdm2 (from A.Levine).
Cell culture
The following lymphobalstoid cell lines were used: C3ABR (from M.Lavin) and L40 derived from healthy donors; L6, AT24RM and AT59RM (the latter two from L.Chessa) derived from A-T patients; D5136 and D5143 from two related A-TLD(M) patients with identical MRE11 genotype, and D5722 derived from an A-TLD(S) patient (Stewart et al., 1999) (A-TLD cell lines are from A.M.Taylor), and GM07078A was derived from an NBS patient (obtained from W.Kleijer). Primary human fibroblasts from a healthy donor (F2054) and an A-T patient (F2071) were established in our lab. A-TLD(M) (D6857, D6852), A-TLD(S) (D6807) fibroblasts (Stewart et al., 1999) were obtained from A.M.Taylor, and NBS fibroblasts (93RD204 and 93RD469, 89RD202) are fromW.Kleijer. NBS cells (GM7166VA7) and the same cells reconstituted by stable expression of Nbs1 (Tauchi et al., 2001) are originally from the laboratory of K.Komatsu and were obtained from D.Delia.
The A-TLD(S) D6087 fibroblast line was immortalized by transducing the cells with a retroviral vector expressing the catalytic subunit of human telomerase (hTERT) (obtained from T.Pandita), as described previously (Wood et al., 2001). Recombinant Mre11 proteins were ectopically expressed in these cells by infecting them with retroviral vectors pCNCX-Mre11 (wild type) and pCNCX-Mre11-3 (H129L/D130V mutant) (Naviaux et al., 1996; Stracker et al., 2002), as described previously (Stracker et al., 2002). pCNCX-GFP vector was used as control (vectors obtained from M.Weitzman).
Experimental protocols
Immunoblotting analysis was performed according to standard techniques.
Immunofluorescent visualization of cellular proteins was carried out after fixation of the cells with either cold absolute methanol for 5 min at –20°C or 4% paraformaldehyde for 10 min at room temperature. Permeabilization was carried out by incubation of the slides in 0.5% Triton X-100 for 10 min, and blocking was performed with 1% BSA, 10% NDS in phosphate-buffered saline for 45 min. Incubation with the primary antibody was for 1.5 h to overnight.
Nuclear retention of ATM was demonstrated using biochemical fractionation and in situ extraction followed by immunofluorescence analysis (Andegeko et al., 2001). The fluorescent readings in the nuclear retention experiments were calculated using the Percentage of Positive Area (PPA) algorithm (Andegeko et al., 2001). The reading of the amount of immunostaining with the anti-phospho-(SQ/TQ) antibody represents the average fluorescence intensity of all the pixels in the nuclear area of each cell, obtained using the Histogram function contained in the Photoshop package (version 6.0).
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank A.Malcolm Taylor for A-TLD cell lines, Michael B.Kastan for a gift of the anti-phospho-Ser1981 antibody, Luciana Chessa, Kenshi Komatsu, Domenico Delia, Martin Lavin and Wim Kleijer for patient cell lines, Tej Pandita for hTERT vector, Matthew Weitzman for Mre11 cDNA clones, Eva Lee for antibody 5C2, Stephen Elledge for anti-Chk2 antibody, and Arnold Levine for the 2A10 antibody. We gratefully acknowledge the contribution of Nechama Smorodinsky, Ella Harness, Margalit Yaakobowicz and Liat Ben-Senior for the establishment of our anti-ATM antibodies. We are indebted to Yael Ziv, Dganit Shkedy, Mali Weisz and Yuval Landau for critical reading of the manuscript, and to Ruth Shiloh for artwork. This study was supported by research grants from the A-T Medical Research Foundation, the A-T Children’s Project, and the National Institutes of Health (RO1 NS 31763). This work was carried out in partial fulfillment of the requirements for the Ph.D. degree of T.Uziel.
References
- Abraham R.T. (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev., 15, 2177–2196. [DOI] [PubMed] [Google Scholar]
- Abraham R.T. (2002) Checkpoint signalling: focusing on 53BP1. Nat. Cell Biol., 4, E277–E279. [DOI] [PubMed] [Google Scholar]
- Andegeko Y., Moyal,L., Mittelman,L., Tsarfaty,I., Shiloh,Y. and Rotman,G. (2001) Nuclear retention of ATM at sites of DNA double strand breaks. J. Biol. Chem., 276, 38224–38230. [DOI] [PubMed] [Google Scholar]
- Bakkenist C.J. and Kastan,M.B. (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature, 421, 499–506. [DOI] [PubMed] [Google Scholar]
- Banin S. et al. (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science, 281, 1674–1677. [DOI] [PubMed] [Google Scholar]
- Bradbury J.M. and Jackson,S.P. (2003) The complex matter of DNA double-strand break detection. Biochem. Soc. Trans, 31, 40–44. [DOI] [PubMed] [Google Scholar]
- Buscemi G. et al. (2001) Chk2 activation dependence on Nbs1 after DNA damage. Mol. Cell Biol., 21, 5214–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman C.E. et al. (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science, 281, 1677–1679. [DOI] [PubMed] [Google Scholar]
- Crawford T.O. (1998) Ataxia telangiectasia. Semin. Pediatr. Neurol., 5, 287–294. [DOI] [PubMed] [Google Scholar]
- D’Amours D. and Jackson,S.P. (2001) The yeast Xrs2 complex functions in S phase checkpoint regulation. Genes Dev., 15, 2238–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amours D. and Jackson,S.P. (2002) The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol., 3, 317–327. [DOI] [PubMed] [Google Scholar]
- Digweed M., Reis,A. and Sperling,K. (1999) Nijmegen breakage syndrome: consequences of defective DNA double strand break repair. BioEssays, 21, 649–656. [DOI] [PubMed] [Google Scholar]
- DiTullio R.A., Mochan,T.A., Venere,M., Bartkova,J., Sehested,M., Bartek,J. and Halazonetis,T.D. (2002) 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat. Cell Biol., 4, 998–1002. [DOI] [PubMed] [Google Scholar]
- Dong Z., Zhong,Q. and Chen,P.L. (1999) The Nijmegen breakage syndrome protein is essential for Mre11 phosphorylation upon DNA damage. J. Biol. Chem., 274, 19513–19516. [DOI] [PubMed] [Google Scholar]
- Falck J., Petrini,J.H., Williams,B.R., Lukas,J. and Bartek,J. (2002) The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat. Genet., 30, 290–294. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O. et al. (2002) DNA damage-induced G2–M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol., 4, 993–997. [DOI] [PubMed] [Google Scholar]
- Foray N., Marot,D., Gabriel,A., Randrianarison,V., Carr,A.M., Perricaudet,M., Ashworth,A. and Jeggo,P. (2003) A subset of ATM- and ATR-dependent phosphorylation events requires the BRCA1 protein. EMBO J., 22, 2860–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatei M., Shkedy,D., Khanna,K.K., Uziel,T., Shiloh,Y., Pandita,T.K., Lavin,M.F. and Rotman,G. (2001) Ataxia-telangiectasia: chronic activation of damage-responsive functions is reduced by alpha-lipoic acid. Oncogene, 20, 289–294. [DOI] [PubMed] [Google Scholar]
- Gatei M. et al. (2003) Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J. Biol. Chem., 278, 14806–14811. [DOI] [PubMed] [Google Scholar]
- Gatei M. et al. (2000) ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat. Genet., 25, 115–119. [DOI] [PubMed] [Google Scholar]
- Gatti R.A. et al. (2001) The pathogenesis of ataxia-telangiectasia. Learning from a Rosetta Stone. Clin. Rev. Allergy Immunol., 20, 87–108. [DOI] [PubMed] [Google Scholar]
- Girard P.M., Riballo,E., Begg,A.C., Waugh,A. and Jeggo,P.A. (2002) Nbs1 promotes ATM dependent phosphorylation events including those required for G1/S arrest. Oncogene, 21, 4191–4199. [DOI] [PubMed] [Google Scholar]
- Goldberg M., Stucki,M., Falck,J., D’Amours,D., Rahman,D., Pappin,D., Bartek,J. and Jackson,S.P. (2003) MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature, 421, 952–956. [DOI] [PubMed] [Google Scholar]
- Grenon M., Gilbert,C. and Lowndes,N.F. (2001) Checkpoint activation in response to double-strand breaks requires the Mre11/Rad50/Xrs2 complex. Nat. Cell Biol., 3, 844–847. [DOI] [PubMed] [Google Scholar]
- Hopfner K.P., Putnam,C.D. and Tainer,J.A. (2002) DNA double-strand break repair from head to tail. Curr. Opin. Struct. Biol., 12, 115–122. [DOI] [PubMed] [Google Scholar]
- Jackson S.P. (2002) Sensing and repairing DNA double-strand breaks. Carcinogenesis, 23, 687–696. [DOI] [PubMed] [Google Scholar]
- Kamsler A., Daily,D., Hochman,A., Stern,N., Shiloh,Y., Rotman,G. and Barzilai,A. (2001) Increased oxidative stress in ataxia telangiectasia evidenced by alterations in redox state of brains from Atm-deficient mice. Cancer Res., 61, 1849–1854. [PubMed] [Google Scholar]
- Khosravi R., Maya,R., Gottlieb,T., Oren,M., Shiloh,Y. and Shkedy,D. (1999) Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc. Natl Acad. Sci. USA, 96, 14973–14977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.S., Krasieva,T.B., LaMorte,V., Taylor,A.M. and Yokomori,K. (2002a) Specific recruitment of human cohesin to laser-induced DNA damage. J. Biol. Chem., 277, 45149–45153. [DOI] [PubMed] [Google Scholar]
- Kim S.T., Lim,D.S., Canman,C.E. and Kastan,M.B. (1999) Substrate specificities and identification of putative substrates of ATM kinase family members. J. Biol. Chem., 274, 37538–37543. [DOI] [PubMed] [Google Scholar]
- Kim S.T., Xu,B. and Kastan,M.B. (2002b) Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev., 16, 560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi J. et al. (2002) NBS1 Localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr. Biol., 12, 1846–1851. [DOI] [PubMed] [Google Scholar]
- Lee J.H. et al. (2003) Distinct functions of nijmegen breakage syndrome in ataxia telangiectasia mutated-dependent responses to DNA damage. Mol. Cancer Res., 1, 674–681. [PubMed] [Google Scholar]
- Lim D.S., Kim,S.T., Xu,B., Maser,R.S., Lin,J., Petrini,J.H. and Kastan,M.B. (2000) ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature, 404, 613–617. [DOI] [PubMed] [Google Scholar]
- Lou Z., Chini,C.C., Minter-Dykhouse,K. and Chen,J. (2003a) Mediator of DNA damage checkpoint protein 1 regulates BRCA1 localization and phosphorylation in DNA damage checkpoint control. J. Biol. Chem., 278, 13599–13602. [DOI] [PubMed] [Google Scholar]
- Lou Z., Minter-Dykhouse,K., Wu,X. and Chen,J. (2003b) MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature, 421, 957–961. [DOI] [PubMed] [Google Scholar]
- Lukas C., Falck,J., Bartkova,J., Bartek,J. and Lukas,J. (2003) Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat. Cell Biol., 5, 255–260. [DOI] [PubMed] [Google Scholar]
- Maser R.S., Zinkel,R. and Petrini,J.H. (2001) An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele. Nat. Genet., 27, 417–421. [DOI] [PubMed] [Google Scholar]
- Matsuoka S., Rotman,G., Ogawa,A., Shiloh,Y., Tamai,K. and Elledge,S.J. (2000) Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl Acad. Sci. USA, 97, 10389–10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya R. et al. (2001) ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev., 15, 1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva O.K. and Petrini,J.H. (2001) DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol. Cell Biol., 21, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva O.K. and Petrini,J.H. (2003) DNA replication-dependent nuclear dynamics of the mre11 complex. Mol. Cancer Res., 1, 207–218. [PubMed] [Google Scholar]
- Naviaux R.K., Costanzi,E., Haas,M. and Verma,I.M. (1996) The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol., 70, 5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill T. et al. (2000) Utilization of oriented peptide libraries to identify substrate motifs selected by ATM. J. Biol. Chem., 275, 22719–22727. [DOI] [PubMed] [Google Scholar]
- Petrini H.J. and Stracker,T.H. (2003) The cellular response to DNA double strand breaks: defining the sensors and mediators. Trends Cell Biol., in press. [DOI] [PubMed] [Google Scholar]
- Petrini J.H., Maser,R.S. and Bressan,D.A. (2001) The Mre11-Rad50 complex. In Hoekstra,M.F. (ed.), DNA damage and repair Vol. 3. Humana Press, Totowa, NJ, pp. 147–172. [Google Scholar]
- Pitts S.A. et al. (2001) hMRE11: genomic structure and a null mutation identified in a transcript protected from nonsense-mediated mRNA decay. Hum. Mol. Genet., 10, 1155–1162. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. (2001) ATM and ATR: networking cellular responses to DNA damage. Curr. Opin. Genet. Dev., 11, 71–77. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. (2003) ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer, 3, 155–168. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. and Kastan,M.B. (2001) ATM: genome stability, neuronal development and cancer cross paths. Adv. Cancer Res., 83, 209–254. [DOI] [PubMed] [Google Scholar]
- Stewart G.S., Last,J.I., Stankovic,T., Haites,N., Kidd,A.M., Byrd,P.J. and Taylor,A.M. (2001) Residual ataxia telangiectasia mutated protein function in cells from ataxia telangiectasia patients, with 5762ins137 and 7271T→G mutations, showing a less severe phenotype. J. Biol. Chem., 276, 30133–30141. [DOI] [PubMed] [Google Scholar]
- Stewart G.S. et al. (1999) The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell, 99, 577–587. [DOI] [PubMed] [Google Scholar]
- Stewart G.S., Wang,B., Bignell,C.R., Taylor,A.M. and Elledge,S.J. (2003) MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature, 421, 961–966. [DOI] [PubMed] [Google Scholar]
- Stracker T.H., Carson,C.T. and Weitzman,M.D. (2002) Adenovirus oncoproteins inactivate the Mre11–Rad50–NBS1 DNA repair complex. Nature, 418, 348–352. [DOI] [PubMed] [Google Scholar]
- Tauchi H. et al. (2001) The forkhead-associated domain of NBS1 is essential for nuclear foci formation after irradiation but not essential for hRAD50·hMRE11·NBS1 complex DNA repair activity. J. Biol. Chem., 276, 12–15. [DOI] [PubMed] [Google Scholar]
- Tauchi H., Matsuura,S., Kobayashi,J., Sakamoto,S. and Komatsu,K. (2002) Nijmegen breakage syndrome gene, NBS1 and molecular links to factors for genome stability. Oncogene, 21, 8967–8980. [DOI] [PubMed] [Google Scholar]
- Usui T., Ogawa,H. and Petrini,J.H. (2001) A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol. Cell, 7, 1255–1266. [DOI] [PubMed] [Google Scholar]
- Wang B., Matsuoka,S., Carpenter,P.B. and Elledge,S.J. (2002) 53BP1, a mediator of the DNA damage checkpoint. Science, 298, 1435–1438. [DOI] [PubMed] [Google Scholar]
- Wood L.D. et al. (2001) Characterization of ataxia telangiectasia fibroblasts with extended life-span through telomerase expression. Oncogene, 20, 278–288. [DOI] [PubMed] [Google Scholar]
- Wu X. et al. (2000) ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature, 405, 477–482. [DOI] [PubMed] [Google Scholar]
- Yazdi P.T., Wang,Y., Zhao,S., Patel,N., Lee,E.Y. and Qin,J. (2002) SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev., 16, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S.S., Chang,H.L., Hou,M.F., Chan,T.F., Kao,Y.H., Wu,Y.C. and Su,J.H. (2002a) Neocarzinostatin induces Mre11 phosphorylation and focus formation through an ATM- and NBS1-dependent mechanism. Toxicology, 177, 123–130. [DOI] [PubMed] [Google Scholar]
- Yuan S.S., Su,J.H., Hou,M.F., Yang,F.W., Zhao,S. and Lee,E.Y. (2002b) Arsenic-induced Mre11 phosphorylation is cell cycle-dependent and defective in NBS cells. DNA Repair (Amst), 1, 137–142. [DOI] [PubMed] [Google Scholar]
- Zhao S., Renthal,W. and Lee,E.Y. (2002) Functional analysis of FHA and BRCT domains of NBS1 in chromatin association and DNA damage responses. Nucleic Acids Res., 30, 4815–4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S. et al. (2000) Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature, 405, 473–477. [DOI] [PubMed] [Google Scholar]
- Zou L. and Elledge,S.J. (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science, 300, 1542–1548. [DOI] [PubMed] [Google Scholar]