

Abstract
Cell–matrix and cell–cell adhesion are recognized physiological determinants of cell growth and survival. In epithelial and endothelial cell systems, oncogenic transformation has in several cases been shown to confer resistance to apoptosis upon depriving cells of substrate adhesion. We examined the effects of oncogenic transformation in adherent versus adhesion- deprived primary embryonic fibroblasts. Whereas untransformed early passage fibroblasts undergo cell cycle arrest, their Myc/Ras- or E1A/Ras-transformed counterparts rapidly enter apoptosis when placed into suspension. This phenomenon also occurs upon incubation with a soluble, RGD-containing integrin ligand and is blocked by a peptide antagonist to ICE family proteases or by aggregation of cells plated at high density. Loss of wild-type p53 modulates the kinetics but does not abrogate this death pathway. Transformation with activated Src rather than Ras rendered fibroblasts selectively resistant to adhesion-dependent apoptosis, an effect likely related to Src's role in integrin signaling, while simultaneously sensitizing the cells to radiation-induced apoptosis. Thus cell adhesion events regulate transformation-selective apoptosis in fibroblasts and provide potentially important targets for understanding and interfering with tumor cell viability.
Adhesion to the extracellular matrix (ECM)1 is a process largely mediated by the integrin family of cell surface receptors, which modulates a number of pivotal cellular decisions ranging from embryonic development and differentiation to tumor cell growth and metastasis (Yamada and Miyamoto, 1995; Ruoslahti, 1996). In addition to inducing tissue-specific gene expression, adhesion of cells to the ECM has recently been shown to serve as a survival mechanism in several anchorage-dependent cell types. Primary cells of epithelial and endothelial origin rapidly undergo apoptosis when denied proper substrate adhesion in a process called anoikis (Frisch and Francis, 1994; Ruoslahti and Reed, 1994). Human umbilical vein endothelial cells (HUVEC) plated on agarose or MDCK cells incubated in saturating amounts of either soluble vitronectin or synthetic RGD-containing peptides are prevented from adhering and rapidly undergo anoikis (Meredith et al., 1993; Frisch and Francis, 1994). These studies are consistent with others showing that ECM degradation during normal development results in programmed cell death of matrix-dependent cells (Re et al., 1994). During cavitation of the vertebrate embryo, inner endodermal cells that do not contact the basement membrane are triggered into apoptosis, while those retaining direct contact are rescued (Coucouvanis and Martin, 1995).
The mechanism by which integrin-mediated adhesion suppresses anoikis in these cell types, however, remains unclear. Focal adhesion kinase (FAK), which is known to bind and mediate integrin-dependent signals (Schaller et al., 1992), has been shown to be required for the survival of both primary (Hungerford et al., 1996) as well as tumor cells (Xu et al., 1996) in the process of adhesion. Up-regulation of Bcl-2 expression via the α5β1 integrin in CHO cells attached to fibronectin has been suggested as a potential mechanism for the ECM's ability to prevent apoptosis (Zhang et al., 1995). Activation of the Jun-NH2-terminal kinase (JNK) in adhesion-deprived MDCK cells was also recently suggested as a downstream signaling event during anoikis (Frisch et al., 1996a ). The p53 tumor suppressor gene has also been suggested to modulate anoikis of fibroblasts in an experimental metastasis system, in which p53 loss was associated with enhanced survival of transformed fibroblasts in suspension (Nikiforov et al., 1996). Finally, inhibition of apoptosis by oncogenic transformation has been suggested as a mechanism for achieving anchorage-independent growth potential of epithelial and endothelial cells that have lost substrate attachment (Ruoslahti and Reed, 1994).
Apoptosis, now recognized as a major mode of action of chemotherapy and radiotherapy, is a process whereby individual cells are triggered to self destroy in a manner that will neither harm neighboring cells nor induce an inflammatory response (Martin, 1993; Williams and Smith, 1993; Fisher, 1994). During the normal development of most if not all multicellular organisms, apoptosis is an efficient homeostatic mechanism whereby unwanted cells are eliminated (Raff, 1992). The interleukin-1β converting enzyme (ICE) family of cysteine proteases, conserved from nematodes to mammals, has been implicated in mediating downstream events of apoptosis in a variety of systems (Martin and Green, 1995; Nicholson et al., 1995; Tewari et al., 1995; Chinnaiyan and Dixit, 1996). A number of specific substrates for ICE family proteases have been described, such as polyADP ribose polymerase (PARP), which is rapidly degraded within hours after ionizing radiation and other apoptotic stimuli (Lazebnik et al., 1994). Tumor cells harboring genetic mutations that block apoptosis may gain a growth advantage under various stressful growth conditions (Lowe et al., 1994; Graeber et al., 1996).
While sharing certain anchorage-dependent growth properties with the stationary cells that make up epithelial and endothelial layers, fibroblasts are motile and do not extensively adhere to one another (Stroker, 1968; Ben-Ze'ev et al., 1980). Although these three cell types require matrix adhesion and spreading to proliferate, nonadherent primary fibroblasts differ in that they arrest in the G0/G1 phase of the cell cycle when deprived of adhesion and reduce their rate of macromolecular synthesis instead of undergoing rapid apoptosis (Otsuka and Moscovitz, 1975; Campisi and Medrano, 1983). The mechanisms that underlie this basic difference are not well understood. While oncogenic transformation has been suggested to rescue nonadherent epithelial and endothelial cells from apoptosis, its effect on nonadherent fibroblasts is less well understood. A recent study by Nikiforov et al. (1996) reports that transformation with E1A and ras oncogenes sensitizes cells to p53-dependent anoikis. Anchorage-independent growth of fibroblasts is a well established characteristic of oncogenic transformation and is routinely assessed by growth in a semi-solid medium. However such conditions may not mimick true substrate detachment, thereby obscuring a potential role for matrix adhesion in regulating survival.
In this study we show that transformed fibroblasts are triggered into apoptosis as a result of detachment from the ECM, in contrast with primary fibroblasts, which arrest in G0/G1. Both Myc/Ras and E1A/Ras oncogene combinations transfected into rat embryo fibroblasts (REF) trigger apoptosis after substrate detachment, as determined by cellular morphology, DNA fragmentation, PARP cleavage, and the ability of an ICE family peptide inhibitor to block this effect. A role for integrin-mediated signal transduction is suggested through the ability of an RGD-containing peptide to similarly trigger apoptosis in these cells. While p53 null-transformed mouse embryo fibroblasts were also sensitive to the loss of adhesion, p53 wild-type cells displayed more rapid kinetics of death, suggesting a p53-modulated pathway. In striking contrast, REF transformed with either E1A/Src or Myc/Src were profoundly resistant to apoptosis triggered by the loss of ECM, suggesting that Src can mimick ECM survival signals normally found in adherent cells. In addition, cells grown at high densities became resistant to this apoptosis through the formation of cell–cell contacts, which could be selectively inhibited by the calcium chelator EDTA. These observations suggest an important role for both cell–matrix and cell–cell signaling pathways in regulating the survival of transformed fibroblasts.
Materials and Methods
Cell Culture Media and Reagents
Early passage primary REF (P1-P5; Rat Embryo Fibroblasts; BioWhitaker, Walkersville, MD) were grown in minimal essential medium (MEM; GIBCO BRL, Gaithersburg, MD), supplemented with 20% FBS (GIBCO BRL). REF were transfected by calcium phosphate coprecipitation or lipofectamine reagent (GIBCO BRL) with long terminal repeat-driven human c-myc or E1A and activated ras to generate the Myc/Ras- and E1A/Ras-REF lines, respectively, or plasmid pKVS (containing v-src) and E1A, or pMKVS plasmid (containing both v-myc and v-src, gift of B. Mathey-Prevot, Dana Farber Cancer Institute, Boston, MA; D'Emilia et al., 1991) to generate E1A/Src- and Myc/Src-REF lines. Transformed foci were picked 10–12 d after transfections. pB-SK carrier plasmid was used for control transfection. All REF lines were grown in MEM with 20% FBS. The Myc/Rat1 cell line (gift of Andrew Wagner and Nissim Hay, Ben May Institute, Chicago, IL) was maintained in 10% FCS containing DME with G418 sulfate (Geneticin/GIBCO BRL) and Hygromycin B (Sigma Chemical Co., St. Louis, MO) at final concentrations of 500 and 200 μg/ml, respectively.
The murine fibroblast line C8 (E1A/Ras transfected mouse embryo fibroblasts [MEF] p53+/+) and A8 (E1A/Ras transfected MEF p53−/− [both gifts of Scott Lowe, David Housman, and Tyler Jacks, MIT, Cambridge, MA]) were grown in DME with 20% FBS. Cells were grown without antibiotics (unless otherwise noted) and maintained in a humidified atmosphere at 37°C in 5% CO2 in media supplemented with l-glutamine.
Suspension Dishes
100-mm tissue culture dishes (Falcon; Becton Dickinson Labware, Franklin Lakes, NJ) were used to maintain all cell lines. Suspension dishes were made by coating 100-mm tissue culture dishes with 2 ml of a 12% solution of polyhydroxyethylmethacrylate polymer (poly-HEMA; Aldrich Chemical Co., Milwaukee, WI) in 190 proof ethanol (Folkman and Moscona, 1978) and allowed to evaporate at 37°C for 40–48 h. Plates were washed with PBS before use.
DAPI Staining
105 REF and E1A/Ras-REF were plated on tissue culture plastic (TCP) or polyHEMA-coated TCP 100-mm dishes for 24 h before harvesting by centrifugation. Cells were diluted in PBS and cytospun onto glass slides for 5 min. Tissue culture cells were then fixed in a 3:1 solution of methanol/acetic acid for 20 min. Slides, plates, or coverslips were subsequently incubated in the dark in a 0.01 mg/ml aqueous solution of DAPI (Sigma Chemical Co.) at room temperature for 15 min and destained in 100% methanol for 3 h before fluorescence microscopy.
DNA Fragmentation Assay
Cells from either adherent or suspension cultures were pooled for DNA fragmentation analysis (Wagner et al., 1994). 5 × 106 cells were washed once with PBS and subsequently trypsinized with trypsin-EDTA (Sigma Chemical Co.). Cells were pelleted and resuspended in 1 ml lysis solution (10 mM Tris, pH 8.0, 100 mM NaCl, 25 mM EDTA, pH 8.0, 0.5% SDS, 0.5 mg/ml proteinase K), incubated ∼15 h at 37°C, phenol/chloroform extracted twice, ethanol precipitated, and resuspended in 10 mM Tris, 1 mM EDTA buffer, pH 7.5 (TE). Samples were then RNase A treated for 1 h at 37°C (final concentration of 100 μg/ml in TE), phenol/chloroform extracted twice, ethanol precipitated, resuspended in TE, and resolved on a 1.5% TAE-agarose gel, stained with ethidium bromide, visualized, and photographed under short-wave UV.
Flow Cytometry and Viability Assays
Cell cycle analysis of REF or E1A/Ras-REF was performed 24 h after plating 105 cells on either TCP or polyHEMA-coated TCP. Cells were pelleted, washed, resuspended into 1 ml PBS on ice, fixed by adding 1 ml of ice-cold 80% ethanol dropwise while vortexing, and incubated for 30 min on ice. After washing again with 1 ml PBS, 5 μl of propidium iodide solution (0.5 mg/ml in PBS with 50 U/mg RNase A) was added and the cells subsequently incubated in the dark at 37°C for 30 min. Cells remained on ice until flow cytometric analysis was performed at the Dana Farber Cancer Institute Cell Sorting Facility on a FACScan® Flow Cytometer (Becton Dickinson Immunocytometry Systems, Bedford, MA). Typically, 10,000 cells were collected per sample and analyzed on LYSYS II and CELLFIT software.
For quantitation of apoptotic cells by flow cytometry and terminal deoxynucleotidyl transferase-mediated dUTP-Biotin Nick-end Labeling (TUNEL; Gavrieli et al., 1992), 105 cells were grown on normal TCP or polyHEMA-coated dishes and harvested 24, 48, 72, and 96 h after plating. Cells plated on both types of dishes were trypsinized and washed with PBS before fixation in 1% paraformaldehyde, pH 7.4, for 10 min on ice. Labeling of DNA strand breaks with terminal transferase was subsequently performed using the ApopTag Fluorescein kit (Oncor, Gaithersburg, MD). After incubation with anti–digoxigenin-fluorescein antibody, cells were counterstained with a 5 μg/ml propidium iodide and 50 μg/ml RNase A solution for 30 min in the dark. The green (FITC) and red (PI) fluorescence intensities of cells were measured using a FACScan® Flow Cytometer (Becton Dickinson Immunocytometry Systems). Cells were collected and data stored using CELLFIT software and analyzed using LYSYS II software. High and low fluorescence regions were determined using adherent control samples and subsequently used to gate experimental samples to assess the number of apoptotic cells. Percent apoptosis was calculated by dividing the number of cells displaying high fluorescence levels with the total number of collected cells.
Trypan blue exclusion was also used to assess viability. To score as nonviable, cells were counted only if 50% the diameter of viable cells, thereby excluding over representation of apoptotic bodies (though perhaps slightly underestimating the full magnitude of cell death). Data are presented as means ± SD from three to five independent experiments.
Inhibition of Apoptosis Using an ICE Peptide Inhibitor
105 E1A/Ras-REF and primary REF were plated on polyHEMA-coated TCP either in the presence or absence of 100 μM Z-FA-fmk control peptide or 100 μM Z-VAD-fmk inhibitor peptide for 12 h. 5 mg of both peptides (Enzyme Systems Products, Dublin, CA) were dissolved in cell culture grade DMSO (Sigma Chemical Co.) to yield 20 mM stock concentrations, and diluted into growth medium at 100 μM final concentration by adding 40 μl of stock to 8 ml of medium just before plating cells. Cells plated on polyHEMA with and without DMSO alone or on TCP were used as controls.
Inhibiting Cellular Attachment with RGD Peptide
5 × 106 A8 (p53−/−) cells were plated onto normal tissue culture plastic in the presence of 1.5 mg/ml of either Gly-D-Arg-Gly-Asp-Ser-Pro (GdRGDSP) synthetic peptide (fibronectin/vitronectin ligand competitor; GIBCO BRL) or Gly-Arg-Gly-Glu-Ser-Pro (GRGESP) control peptide (GIBCO BRL). Peptide stocks were prepared in DME supplemented with 10% FCS with 100 U/ml penicillin and 100 μg/ml streptomycin and were added at the time of plating. DNA fragmentation analysis was carried out after 12 h incubation with either peptide.
In Vitro Cleavage of PARP by Cell Extracts
Cytosolic extracts were isolated from E1A/Ras-transformed REF 6 h after plating onto polyHEMA or TCP and prepared essentially as previously described (Nicholson et al., 1995). All adherent and floating cells from each culture were pooled and 2 × 106 cells lysed in 50 μl 10 mM Hepes/ KOH (pH 7.4), 2 mM EDTA, 0.1% CHAPS, 5 mM dithiothreitol, 1 mM phenylmethylsulfonylfluoride, 10 μg/ml pepstatin A, 20 μg/ml leupeptin, 10 μg/ml aprotinin on ice for 10 min, followed by centrifugation at 10,000 g. An NH2-terminal PARP fragment that spans the specific cysteine protease cleavage site was expressed as a His-tagged chimera (plasmid generously provided by Dr. John Collier, Harvard Medical School, Boston, MA), and the protein was purified by nickel chelate chromatography in 6 M guanidine HCl per manufacturers recommendations (Qiagen, Chatsworth, CA). 800 ng of recombinant PARP was added to 40 μl of 10,000 g supernatant and incubated at 37°C for 1 h. Samples were resolved on SDS-PAGE minigels, transferred to nitrocellulose for 40 min at 50 V, blocked overnight at 4°C in 150 mM NaCl, 50 mM Tris (pH 8.0), and 5% powdered milk, washed with Tris-buffered saline and Tween 20 (TBST), incubated with C2-10 αPARP monoclonal antibody (gift of Guy Poirier Université Lavat, Quebec, Canada) 1:10,000 in TBST for 2 h at room temperature, washed with TBST, and finally incubated with Peroxidase-conjugated goat anti–mouse antibody 1:10,000 in TBST (Cappel Laboratories, Cochranville, PA). ECL (Renaissance; Dupont NEN, Boston, MA) was then performed for 1 min at room temperature according to the manufacturer's recommendations.
Results
Loss of ECM Adhesion Triggers Transformation-specific Apoptosis in Fibroblasts
We have studied the response of primary untransformed and transformed REF to the loss of matrix adhesion. Myc/ Ras- or E1A/Ras-transformed lines were generated from parental REF cells by lipofection and plated on tissue culture plastic (TCP) or polyHEMA-coated TCP. Agarose gel electrophoresis of genomic DNA isolated from Myc/ Ras-REF (Fig. 1 A, lane 7) but not primary REF (lane 4) plated onto polyHEMA displayed oligonucleosomal laddering of ∼180–200 bp in length, a characteristic feature of apoptosis. This behavior was also observed in Myc-transformed Rat1 fibroblasts (Fig. 1 B), an oncogene/apoptosis system that has been extensively studied for apoptosis upon growth factor withdrawal (Askew et al., 1991; Evan et al., 1991; Wagner et al., 1994). While previous studies have demonstrated polyHEMA's ability to prevent matrix deposition on wettable tissue culture polystyrene and to be nontoxic to cells (Folkman and Moscona, 1978; Ben-Ze'ev et al., 1980; O'Neill et al., 1986; Schwartz et al., 1989; Frisch and Francis, 1994; Re et al., 1994), we found that bacterial Petri dishes also prevented cellular adhesion, thereby controlling for the effect of this polymer coating. Myc/Ras-REF on bacterial Petri dishes behaved similarly to those on polyHEMA-coated TCP and displayed the same DNA ladder pattern after 24 h (Fig. 1 A, lanes 6 and 7), suggesting that the loss of anchorage (and not the polyHEMA treatment itself) was responsible for apoptosis. Nuclear staining of cells with DAPI revealed shrinkage and nuclear condensation, characteristic of apoptotic cell death (Fig. 1 C). In contrast, untransformed, primary REF lacked these morphological changes. As early as 12 h after plating onto polyHEMA, viability of E1A/Ras- and Myc/ Ras-REF was greatly reduced as compared to the parental primary REF (Fig. 1 D) using trypan blue exclusion. This effect was reproducible in multiple, independently derived foci for each oncogene combination.
Figure 1.

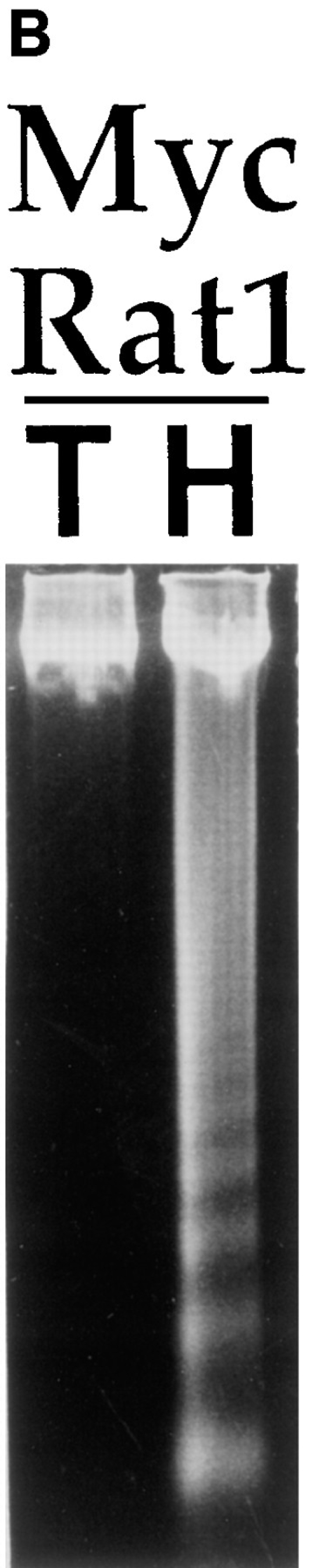


Rat embryo fibroblasts transformed with Myc/Ras or E1A/Ras are triggered into apoptosis when deprived of substrate adhesion. (A) Oligonucleosomal genomic DNA fragmentation. Early passage primary REF and Myc/Ras-REF were plated on either tissue culture plastic (lanes 2 and 5), bacterial petri (3 and 6), or polyHEMA-coated TCP dishes (4 and 7), and genomic DNA was isolated from 5 × 106 cells as described in the Materials and Methods. (B) Myc/Rat1 cells were plated on TCP (T) or polyHEMA (H) as above, and genomic DNA was isolated from 5 × 106 cells as described in Materials and Methods. (C) E1A/Ras-REF and parental REF were plated on TCP or polyHEMA-coated TCP for 12 h before nuclear staining with DAPI as described in Materials and Methods. (D) REF clones transformed with oncogenes Myc/Ras or E1A/Ras or early passage primary REF were plated at 105 cells/100-mm dish on TCP (open bars) or polyHEMA-coated TCP (closed bars). Viability was measured 12 h after plating by calculating the ratio of trypan blue-positive cells to live cells (see Materials and Methods).
A predominant sub-G1 population characteristic of apoptosis was also observed in nonadherent, transformed cells by propidium iodide staining and flow cytometric analysis (Fig. 2 D). In agreement with earlier studies (Campisi and Medrano, 1983), primary REF were found to arrest when grown in suspension (Fig. 2 B). Cell cycle quantitation was derived from histograms shown in Fig. 2 (see Materials and Methods) and revealed both a 13% increase in the G1 population (from 56 to 69%) and a 13% decrease in S phase (from 29 to 16%); G2/M remained constant at 15%. Suspended REF remained >90% viable over at least 72 h (data not shown). Controls on regular TCP for primary and transformed REF showed DNA distribution of log-phase cells (Fig. 2, A and C).
Figure 2.

Flow cytometric analysis of REF and E1A/Ras-REF plated on TCP or polyHEMA-coated TCP dishes. 24 h after plating, cells were washed, fixed, and stained with propidium iodide as described in Materials and Methods. DNA content was plotted on a logarithmic scale to expand the sub-G1 apoptotic fraction. Cell cycle fractions are given in the text. 10,000 events were counted in all, but the E1A/Ras-REF in suspension represented fewer events, as indicated by the y axis amplitude due to cell loss by apoptosis. (D, Note that fewer events were counted in the apoptotic versus nonapoptotic samples as indicated by the abscissa.)
Cysteine Protease Activity Accompanies Apoptosis Triggered by Loss of ECM Adhesion
We asked whether ICE-family cysteine protease activity is required for apoptosis to occur in transformed fibroblasts denied ECM (Fig. 3). To test this, we incubated 105 E1A/ Ras-REF cells on polyHEMA with either the ICE protease inhibitor Z-VAD-fmk, or as control, the cathepsin B protease inhibitor Z-FA-fmk both at 100 μM final concentration in medium as previously described (Pronk et al., 1996). Whereas >80% of the suspended transformed cells died by 12 h in the absence of peptide, we observed a marked decrease in death (down to ∼15%) for cells incubated with Z-VAD-fmk (Fig. 3 A). The viability of cells in the presence of Z-FA-fmk remained unchanged at ∼85%. Furthermore, addition of recombinant PARP to S-10 extract of suspension E1A/Ras-REF (see Materials and Methods) revealed the specific cleavage characteristic of ICE family/CPP32 activation in apoptotic cells (Fig. 3 B).
Figure 3.

(A) Apoptosis triggered by the loss of ECM adhesion is blocked by inhibition of an ICE-like protease. 105 E1A/Ras-REF per 100-mm dish were plated on TCP or on polyHEMA-coated TCP in the presence or absence of 100 μM Z-FA-fmk control peptide or 100 μM Z-VAD-fmk inhibitor peptide (the same amount of DMSO carrier used to dissolve the peptides was added to cells as a control). Viability was determined by trypan blue staining after 12 h. Bars represent the average of three experiments (±SD). (B) Loss of ECM adhesion in transformed fibroblasts leads to activation of the Yama/CPP32 cysteine protease and results in PARP cleavage.
Apoptosis Triggered by the Loss of Adhesion Can Occur in the Absence of p53
Oncogene-driven apoptosis induced by a variety of triggers is commonly modulated by wild-type p53 protein (Yonish-Rouach et al., 1991; Debbas and White, 1993). E1A/Ras-transformed p53 wild-type– or null MEF have served as a model of genetically related cell lines to examine the capacity of p53 to regulate apoptosis after treatment with chemotherapeutic agents, ionizing radiation, or hypoxia (Lowe et al., 1993a , 1994; Graeber et al., 1996). We investigated whether suspension-induced apoptosis was modulated by the presence or absence of p53. Quantitation of cell death after 72 h on polyHEMA showed extensive death of p53 wild-type–transformed fibroblasts and significant, but reproducibly lower levels of death in the p53 null cells (Fig. 4 A). Oligonucleosomal DNA degradation was observed for both C8 and A8 lines on polyHEMA, confirming that the loss of viability observed by trypan blue is due to apoptosis (Fig. 4 B). Using DNA fragmentation TUNEL staining and flow cytometry, the kinetics of apoptotic death was delayed by ∼2 d in E1A/ Ras-transformed p53−/− cells as compared to p53+/+ (data not shown). Thus this death pathway may be potentiated by the presence of p53 but is not abrogated by loss of p53.
Figure 4.


E1A/Ras-transformed MEF lacking p53 are sensitive to apoptosis triggered by the loss of substrate adhesion. (A) Viability of E1A/Ras-transformed MEF+/+ and MEF−/− plate on polyHEMA-coated TCP or regular TCP dishes after 72 h (determined as described in Fig. 1 A). (B) Oligonucleosomal genomic DNA fragmentation. E1A/ Ras-transformed MEF+/+ and MEF−/− were plated on either TCP (lanes 2 and 4) or polyHEMA-coated TCP dishes (3 and 5) for 48 h. Genomic DNA was isolated from 5 × 106 cells as previously described.
Inhibition of Integrin Signaling Triggers Apoptosis in Transformed Fibroblasts
We investigated the role of integrins in mediating the ECM's ability to rescue cells from apoptosis. An RGD-containing peptide that competes for the binding of the fibronectin and vitronectin receptors to their respective ECM substrates was used to test this hypothesis. E1A/ Ras-MEF (p53−/−) were plated onto TCP in the presence of 1.5 mg/ml of either RGD- or RGE-soluble peptides (in culture medium). A dose titration of RGD peptide added at the time of plating revealed this concentration to completely block adhesion to TCP (data not shown). Genomic DNA fragmentation analysis performed 12 h after plating cells demonstrated an oligonucleosomal DNA ladder for RGD-treated cells, whereas the same cells incubated with control RGE peptide were morphologically healthy and displayed minimal DNA degradation (Fig. 5).
Figure 5.

Inhibition of fibronectin/ vitronectin-mediated adhesion of p53−/− transformed fibroblasts triggers apoptosis. E1A/Ras-MEF (p53−/−) were plated onto regular TCP dishes in the presence of saturating amounts (1.5 mg/ml) of either GRGDSP (lane 3) or GRGESP control peptide (lane 2). 12 h after plating, cells were harvested and genomic DNA was isolated as previously described.
v-Src Transformation Sensitizes to Radiation but Blocks Adhesion-dependent Apoptosis
Recognizing a potential role for integrin-mediated signal transduction in triggering apoptosis of transformed cells denied ECM contact, we sought to examine this cascade further downstream. Clustering of ECM-bound integrins and formation of focal contacts results in the localization of FAK to these sites and subsequent recruitment and activation of Src tyrosine kinase (Guan and Shaloway, 1992; Kornberg et al., 1992; Schaller et al., 1992; Schlaepfer et al., 1994). Transfection of Src has previously been shown to confer anchorage-independent growth to epithelial, endothelial, and fibroblast cells (MacCauley and Pawson, 1988; Frisch and Francis, 1994; Re et al., 1994). We therefore asked whether Src-transformed fibroblasts display the same sensitivity to apoptosis by traditional triggers, such as ionizing radiation, as well as loss of substrate adhesion. Multiple independent E1A/Src- and Myc/Src-REF–transformed clones were generated from primary REF cells and tested for radiation sensitivity and survival in suspension (Fig. 6). Src transformation profoundly sensitized cells to γ radiation, an oncogene sensitizing effect similar to that seen in E1A/Ras- and Myc/Ras-transformed rat fibroblasts (Fig. 6) and E1A/Ras-transformed mouse fibroblasts, relative to primary fibroblast cells (Lowe et al., 1994). In contrast, however, all Src-transformed cell lines were resistant to apoptosis triggered by loss of adhesion. In fact a number of these cell clones were able to proliferate (and be passaged) in suspension. Thus Ras-dependent transformation differed profoundly from Src in sensitizing fibroblasts to adhesion-dependent apoptosis.
Figure 6.

Src rescues transformed fibroblasts from apoptosis triggered by the loss of ECM adhesion. Three separately grown clones were isolated from REF cultures transfected with either E1A and Src or Myc and Src (filled bars). 105 cells from each clone were plated onto polyHEMA-coated TCP (HEMA), regular TCP dishes (TCP), or regular TCP dishes and irradiated after 24 h with 500 cGy ionizing radiation (5Gy XRT). Viability was determined by trypan blue 24 h after plating onto TCP or polyHEMA and 24 h after radiation for XRT controls. Bars represent the average of three experiments (±SD).
Cell–Cell Aggregation Can Rescue Ras-transformed Fibroblasts from Apoptosis in Suspension
In comparing suspension growth characteristics of primary versus Ras-transformed REF cells, we observed a correlation between plating density, cellular aggregation, and cell survival. While primary REF plated at high density on polyHEMA remain mostly single cells after plating (Fig. 7 A), Myc/Ras-REF (or E1A/Ras-REF, data not shown) are largely aggregated and predominantly viable. We therefore considered the possibility that cell–cell contacts could be protective in the absence of ECM adhesion. E1A/Ras-REF were plated at varying cell densities both on TCP- and polyHEMA-coated, 100-mm dishes in an attempt to vary the propensity for cell–cell aggregation and uncouple this phenomenon from that of cell–matrix adhesion. At densities of 5 × 104 or 105 per plate, cells remained single and unaggregated (data not shown) and displayed maximum levels of death, whereas at densities of 106 or 5 × 106 per plate, cellular aggregation was observed together with markedly reduced death (Fig. 7 B, closed circles). The nonlinear shape of the viability curve suggested the presence of a critical cell density to achieve rescue from apoptosis. The same E1A/Ras-REF plated on regular TCP showed insignificant levels of cell death at all densities (Fig. 7 B, open squares).
Figure 7.
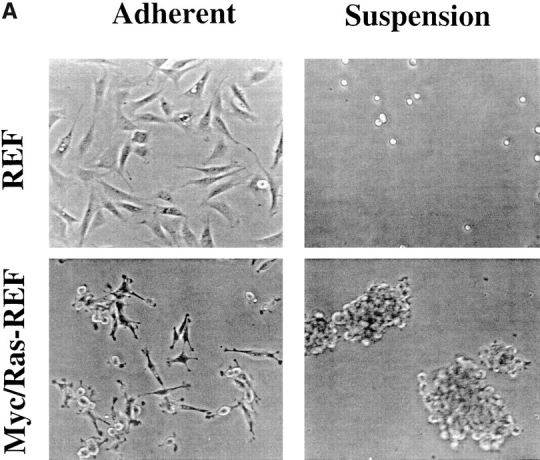


Cell–cell clustering rescues transformed fibroblasts in the absence of cell–matrix contact. (A) Early passage, primary REF or Myc/Ras-REF were plated onto TCP or polyHEMA-coated TCP at high density (2 × 106 cells per 100-mm dish) and photographed under phase contrast microscopy after 24 h. For transformed cells in suspension, representative aggregates are shown. (B) E1A/Ras-REF cells were plated on polyHEMA-coated TCP or regular TCP 100-mm dishes at indicated cell densities. Viability was determined by trypan blue staining 24 h after plating. Bars represent the average of three experiments (±SD). (C) 1 mM of the calcium chelator EDTA restores apoptosis of E1A/Ras-transformed fibroblasts plated at high density (106), while having no effect on adherent cells or REF under the same conditions.
The precise molecular determinants of this cell–cell aggregation and rescue are unknown. However, since numerous cell adhesion molecules require Ca2+, we tested the effect of the calcium chelator EDTA on the aggregation and viability of E1A/Ras-REF plated at high density on polyHEMA. Parental REF cells and adherent transformed cells served as controls to assess nonspecific toxicity of EDTA. At a dose of 1 mM EDTA added to the usual growth media, untransformed REF displayed no significant change in viability. Similarly, the viability and adhesion properties of E1A/Ras-transformed REF on normal TCP at densities of 105 or 106 was essentially unaffected by EDTA. In contrast, 1 mM EDTA resulted in a ∼60% increase in cell death of transformed cells plated at 106 cells/ 100-mm dish, a density that typically favors cell–cell contacts and rescues cells from apoptosis (Fig. 7, A and C). A substantial decrease in the number and size of cell aggregates was observed, although many dead cells also appeared to remain in clusters (data not shown). While the identity of the specific molecules affected by this low dose EDTA is not yet known, it may be interesting that under these conditions, minimal loss of matrix adhesion occurred for the adherent controls, suggesting that integrin attachment was spared despite its divalent cation requirements. Thus, low dose EDTA produces a transformation-dependent and suspension-specific restoration of apoptotic death.
Discussion
Preventing ECM adhesion of fibroblasts transformed by Myc and Ras or E1A and Ras leads to rapid cell death by apoptosis. This phenomenon was observed in a number of independently derived foci and displays the classical features of apoptotic cell death (DNA fragmentation, morphology, ICE-family cysteine protease activity). This response contrasts with that of untransformed fibroblasts that remain >95% viable and lack morphological or biochemical evidence of apoptosis. In agreement with a number of previous studies examining fibroblast adhesion (Stroker, 1968; Folkman and Moscona, 1978; Ben-Ze'ev et al., 1980; Campisi and Medrano, 1983), cell cycle analysis of primary rat fibroblasts revealed an increase in G0/G1- arrested cells. Interestingly, recent analysis of primary chick embryo fibroblasts has revealed a greater propensity for anoikis, particularly in the absence of serum (Hungerford et al., 1996). In transformed rat fibroblasts, a soluble RGD-containing peptide that blocks attachment to fibronectin and vitronectin (Pierschbacher and Ruoslahti, 1987) also triggered apoptosis, suggesting that integrin-mediated events (e.g., direct signaling and/or regulation of cell shape) can regulate transformation-specific death decisions in fibroblasts. The effect of this peptide on cell adhesion and viability suggests that integrin engagement without clustering is insufficient to protect from apoptosis and also implies that protection in these cells may be specifically mediated via either the α5β1 or αvβ1 receptors. The α5β1 integrin has recently been shown to support the survival of CHO cells on fibronectin in a manner that correlates with increased levels of Bcl-2 mRNA and protein (Zhang et al., 1995). Whether other combinations of integrin heterodimers also transmit protective signals is uncertain, especially in light of studies suggesting that the response to the loss of ECM adhesion may be cell-type specific (i.e., growth arrest in primary fibroblasts versus apoptosis in epithelial or endothelial cells).
The behavior of fibroblasts denied attachment appears to differ from epithelial and endothelial cell suspension systems in which primary cells appeared sensitive to apoptosis and oncogene transformation produced resistance (see below). It is unclear why primary rodent fibroblasts survive adhesion-free conditions, while epithelial and endothelial cells do not, but this behavior may also underlie the differing effects of transformation, particularly with activated Ras, which sensitizes, rather than protects fibroblasts from adhesion-dependent apoptosis. Transformation of fibroblasts with either Myc or E1A has previously been shown to sensitize them to apoptosis after growth factor deprivation (Askew et al., 1991; Evan et al., 1991; Debbas and White, 1993). In fact the behavior after deprivation of substrate attachment mimicks growth factor deprivation. Of note, for Ras-transformed cells the sensitivity to anoikis was greatest at low rather than high cell density, arguing against the possibility that growth factor deprivation itself was actually triggering death in these studies. E1A/Ras transformation has been found to sensitize fibroblasts to apoptosis from ionizing radiation and a variety of chemotherapeutic agents (Lowe et al., 1993a ,b). Oncogene-sensitized apoptosis often appears to be mediated by p53, whose loss renders transformed fibroblasts substantially resistant to radiation and many chemotherapeutic apoptosis triggers in vitro, in murine solid tumors (Lowe et al., 1993b , 1994), and in human cancers where p53 status is prognostically meaningful (Hollstein et al., 1991; Harris and Hollstein, 1993; Fisher, 1994; Fung and Fisher, 1995; Shimamura and Fisher, 1996). The same genotoxic treatments that trigger p53-mediated apoptosis in transformed fibroblasts typically induce cell-cycle arrest (without death) in untransformed cells, a behavior observed here with loss of substrate attachment as well.
Using genetically related E1A/Ras-transformed mouse fibroblast lines differing in the presence or absence of p53, we observed significant apoptosis in both the presence and absence of p53, although the kinetics of this death was slower in the latter case. This delay could provide a selective advantage to tumor cells lacking functional p53 and manifest itself in altered propensity for tumor cell metastasis. In fact, our observations support the recent report of p53-modulated anoikis (Nikiforov et al., 1996), in which p53 loss was associated with enhanced survival of transformed fibroblasts in suspension. Importantly, however, the p53 null transformed fibroblasts studied here did undergo massive (albeit delayed) adhesion-dependent apoptosis, suggesting the possibility that drugs that interfere with tumor cell-ECM signaling might hold therapeutic promise in p53 deficient tumors.
Our observations using Myc- (or E1A) plus Ras-transformed fibroblasts confirm the notion that the ECM may act as a cell survival factor and that the loss of ECM attachment is cell-type specific. Studies examining anchorage-dependent apoptosis in epithelial or endothelial cells have suggested that this phenomenon is likely to play a role in cell positioning (Ruoslahti and Reed, 1994). Unlike primary fibroblasts, untransformed epithelial and endothelial cells undergo apoptosis rather than growth arrest when placed in suspension (Meredith et al., 1993; Frisch and Francis, 1994; Re et al., 1994; Ruoslahti and Reed, 1994; Boudreau et al., 1995). HUVEC plated on agarose-coated tissue culture dishes and prevented from adhesion rapidly die by apoptosis, while plating them on dishes coated with the β1 integrin (but not HLA class I or VCAM-1) rescues them (Meredith et al., 1993). Consistent with these results are studies by Re et al. (1994) and Frisch and Francis (1994) that show that blocking integrin receptors of HUVEC or MDCK cells with either saturating amounts of soluble vitronectin or soluble RGD peptide prevents substrate attachment and leads to apoptosis. Boudreau et al. (1995) demonstrate that, in vitro, attachment of mammary epithelial cells (MEC) to a basement membrane via the β1 integrin reduces ICE mRNA levels. Their results also suggest that active ECM degradation during normal mammary gland involution results in apoptosis of cells that depend on it. Ectopic expression of the ECM-degrading metalloproteinase stromelysin-1 in MEC leads to apoptosis, which is inhibited by the specific protease inhibitor GM6001. In addition, transgenic mice expressing stromelysin-1 under the control of a promoter active during mid- to late pregnancy results in apoptosis of MEC during this period (Boudreau et al., 1995). Unlike stationary epithelial and endothelial cells that form tight sheets as a result of intercellular junctions, one might expect that migratory cells of mesenchymal origin like primary fibroblasts would not possess the same apoptosis program in response to the loss of ECM contact.
While Myc/Ras or E1A/Ras transformation of fibroblasts sensitizes them to apoptosis triggered by the loss of ECM as well as to genotoxic stress, this response is not paralleled in endothelial and epithelial cell types, where oncogenic transformation has been found to confer resistance to anchorage-dependent apoptosis (Frisch and Francis, 1994; Re et al., 1994; Rak et al., 1995). Endothelial cells transformed with either SV40 large-T antigen or v-Ras and grown on polyHEMA are less susceptible to apoptosis than their untransformed counterparts (Re et al., 1994). Immortalized intestinal epithelial cells IEC-18 conditionally expressing mutant c-H-Ras protein are rescued from apoptosis induced by three-dimensional growth conditions (Rak et al., 1995). In addition, in a reverse-transformation model of the HT1080 human fibrosarcoma line (Frisch, 1991), E1A sensitized these cells to apoptosis upon loss of anchorage (Frisch and Francis, 1994). HT1080 cells are also triggered into apoptosis when incubated with the laminin-mimetic peptide YIGSR (Kim et al., 1994), suggesting that reverse transformation is not absolutely required to sensitize these cells to apoptosis in suspension. One possible connection between Ras behavior in fibroblasts and its protection against anoikis in epithelial or endothelial cells could relate to intercellular adhesion. Prior studies (Jamal et al., 1994) have suggested that Ras activity may stimulate expression of CD44, a modulator of intracellular adhesion. The propensity of Ras-transformed cells to aggregate (shown here to correlate with rescue from anoikis) may thereby confer survival via a specific cell–cell adhesion pathway in both fibroblasts and epithelial cell types.
Given Src's involvement in integrin signaling, we hypothesized that v-Src transformation might be protective to fibroblasts grown in suspension. In striking contrast to Ras-transformed REF, both Myc/Src-REF and E1A/Src-REF were profoundly resistant to the modulation of apoptosis when plated onto polyHEMA-coated tissue culture plates. However, the same Src-transformed cell lines were simultaneously sensitized (relative to parental REF) to ionizing radiation, a trigger of p53-modulated apoptosis (Lowe et al., 1993), suggesting that apoptosis is not generally disabled in these cells. These results agree with studies showing that Src transformation protects MDCK cells and HUVECs from anoikis (Frisch and Francis, 1994; Re et al., 1994). Although a variety of mechanisms could potentially account for this behavior, Src activation may mimick ECM survival signals required by transformed cells (irrespective of cell type) to survive in suspension, thereby conferring anchorage-independent growth potential. Another point raised by this observation is the possibility that Src-transformed cells could display a different survival behavior within animals, possibly relating to metastatic potential for example.
Recent studies have shown that permanent membrane localization of FAK in MDCK cells prevents apoptosis in suspension, supporting the notion that activation of signaling intermediates in this adhesion pathway (like Src) may protect cells from anoikis (Frisch et al., 1996b ). Moreover, preventing integrin binding to FAK through competition with microinjected synthetic peptides triggers rapid apoptosis in primary chick embryo fibroblasts grown in the absence of serum (Hungerford et al., 1996). The apoptosis resistance we observed with primary REF may reflect integrin-independent activation of such signaling intermediates or alternatively distinct downstream nuclear events that signal arrest versus apoptose. For the current studies, we have attempted to uncouple growth factor influences from anoikis by use of 20% serum in all experiments. Given the specific role of Ras in growth factor signaling as well as in the transformation-specific anoikis reported here, these apoptotic pathways may share certain mechanistic components that should be amenable to study in these genetically related cells.
Whereas integrins may mediate cell–matrix signals to prevent growth arrest or apoptosis, cell–cell adhesion via integrin-dependent or independent mechanisms may also generate signals that block apoptosis. Establishment and maintenance of cell–cell contacts in epithelial and endothelial cell types plays a central role in determining cellular polarity and ensuring correct tissue function (Birchmeier et al., 1993; Birchmeier and Birchmeier, 1994). In addition, studies have demonstrated that cell–cell contacts can modulate a drug-induced cell death pathway (Kobayashi et al., 1993; Kerbel et al., 1995). Inhibition of intercellular contact has been shown to trigger rapid apoptosis in the colon carcinoma line LIM 1863, which normally grows in aggregates termed “organoids” (Bates et al., 1994). In this study, we observed a striking difference in the behavior of primary and transformed fibroblasts in suspension. Unlike primary REF which remain mostly as single cells in suspension, E1A/Ras- and Myc/Ras-transformed REF aggregate into clusters. In these aggregates, transformed cells likely require specific signals generated from cell–cell contacts to survive. The suspension-dependent effect of low dose EDTA on these cells further suggests that cell–cell survival signals are mediated by a Ca2+-requiring molecule(s) like integrins and/or the cadherin family of intercellular receptors (Grunwald, 1993; Takeichi, 1993; Gumbiner, 1996). Other potential candidates include CD44, a molecule associated with metastasis propensity (Günthert et al., 1991) and shown to be overexpressed upon both Ras and Src transformation (Hofmann et al., 1993; Jamal et al., 1994). Interestingly, Src-transformed fibroblast lines showed a much greater propensity to grow as single cells in suspension (data not shown), consistent with the likelihood that Src's survival signal does not require cell–cell contact.
The observations reported here strongly suggest that survival in suspension is not synonymous with “anchorage independence,” a term traditionally employed to describe colony formation in semi-solid medium. Growth in semi-solid medium distinguishes transformed from untransformed REF, and oncogene-transformed cells used in these studies readily form colonies in soft agar (data not shown) while undergoing rapid anoikis in suspension. Moreover, the conferral of anoikis sensitivity in fibroblasts upon oncogene transfection is a relatively stringent assay of oncogene function given that it would produce a negative selection bias during the establishment of transformed lines.
The in vivo relevance of cell–cell signaling in modulating apoptosis of tumor cells in suspension is implicit in studies demonstrating the importance of intercellular junctions during colon carcinogenesis. The identification of the product of the adenomatous polyposis coli (APC) tumor suppressor gene as a protein that binds to β-catenin (Rubinfeld et al., 1993) as well as its anti-apoptotic activity (Morin et al., 1996) suggest that its tumor suppressive role could be related to a role in cell–cell adhesion and survival. Further analysis of signaling events both from integrin/Src and cell–cell adhesion receptors may reveal tumor-specific survival pathways and potential drug targets in malignancies such as sarcomas.
Acknowledgments
The authors thank Scott Lowe, David Chang, Martin Hemler, Tim Hemesath, John Abraham, Patti Harrigan, and Amy C. Vollmer for helpful discussions at various stages of this work; Clifford Takemoto for E1A/Ras-REF cells; Scott Lowe, Tyler Jacks, and David Housman for the C8 and A8 lines; Andrew Wagner and Nissim Hay for the Myc/Rat1 line; Guy Poirier for the αPARP C2-10 antibody; John Collier for the His-tagged PARP plasmid; and Bernard Mathey-Prevot for plasmids pKVS and pMKVS.
This work was supported by National Institutes of Health grant CA69531 (D.E. Fisher) and research funds from the Swarthmore College Department of Biology (G. McGill). R.C. Bates is a CJ Martin Fellow supported by the National Health and Medical Research Council (Australia), and D.E. Fisher is a fellow of the Pew Foundation and the James S. McDonnell Foundation.
Abbreviations used in this paper
- ECM
extracellular matrix
- FAK
focal adhesion kinase
- HUVEC
human umbilical vein endothelial cells
- ICE
interleukin-1β converting enzyme
- MEC
mammary epithelial cells
- MEF
mouse embryo fibroblasts
- PARP
polyADP ribose polymerase
- REF
rat embryo fibroblasts
- polyHEMA
polyhydroxyethylmethacrylate
Footnotes
Please address all correspondence to David E. Fisher, Children's Hospital, Harvard Medical School, 44 Binney Street, Boston, MA 02115. Tel.: (617) 632-4916; Fax: (617) 632-2085.
References
- Askew D, Ashmun R, Simmons B, Cleveland J. Constitutive c-mycexpression in IL-3-dependent myeloid cell line suppresses cycle arrest and accelerates apoptosis. Oncogene. 1991;6:1915–1922. [PubMed] [Google Scholar]
- Bates RC, Buret A, van Helden DF, Horton MA, Burns GF. Apoptosis induced by inhibition of intercellular contact. J Cell Biol. 1994;125:403–415. doi: 10.1083/jcb.125.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ze'ev A, Farmer SR, Penman S. Protein synthesis requires cell-surface contact while nuclear events respond to cell shape in anchorage-dependant fibroblasts. Cell. 1980;21:365–372. doi: 10.1016/0092-8674(80)90473-0. [DOI] [PubMed] [Google Scholar]
- Birchmeier W, Birchmeier C. Mesenchymal-epithelial transitions. Bioessays. 1994;16:305–307. doi: 10.1002/bies.950160503. [DOI] [PubMed] [Google Scholar]
- Birchmeier W, Weidner KM, Behrens J. Molecular mechanisms leading to loss of differentiation and gain of invasiveness in epithelial cells. J Cell Sci. 1993;17:159–164. doi: 10.1242/jcs.1993.supplement_17.23. [DOI] [PubMed] [Google Scholar]
- Boudreau N, Sympson CJ, Werb Z, Bissell MJ. Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science (Wash DC) 1995;267:891–893. doi: 10.1126/science.7531366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, Medrano EE. Cell cycle perturbations in normal and transformed fibroblasts caused by detachment from the substratum. J Cell Physiol. 1983;114:53–60. doi: 10.1002/jcp.1041140109. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, Dixit VM. The cell-death machine. Curr Biol. 1996;6:555–562. doi: 10.1016/s0960-9822(02)00541-9. [DOI] [PubMed] [Google Scholar]
- Coucouvanis E, Martin GR. Signals for death and survival: a two-step mechanism for cavitation in the vertebrate embryo. Cell. 1995;83:279–287. doi: 10.1016/0092-8674(95)90169-8. [DOI] [PubMed] [Google Scholar]
- D'Emilia JC, Mathey-Prevot B, Jaros K, Wolf B, Steele GJ, Summerhayes IC. Preneoplastic lesions induced by myc and src oncogenes in a heterotopic rat colon. Oncogene. 1991;6:303–310. [PubMed] [Google Scholar]
- Debbas D, White E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 1993;7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1991;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Fisher DE. Apoptosis in cancer therapy: crossing the threshold. Cell. 1994;78:539–542. doi: 10.1016/0092-8674(94)90518-5. [DOI] [PubMed] [Google Scholar]
- Folkman J, Moscona A. Role of cell shape in growth control. Nature (Lond) 1978;273:345–349. doi: 10.1038/273345a0. [DOI] [PubMed] [Google Scholar]
- Frisch SM. Antioncogenic effect of adenovirus E1A in human tumor cells. Proc Natl Acad Sci USA. 1991;88:9077–9081. doi: 10.1073/pnas.88.20.9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;4:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Vuori K, Kelaita D, Sicks S. A role for Jun-N-Terminal kinase in anoikis; suppression by bcl-2 and crmA. J Cell Biol. 1996a;135:1377–1382. doi: 10.1083/jcb.135.5.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Vuori K, Ruoslahti E, Chan-Hui P-Y. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996b;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung CY, Fisher DE. p53: From molecular mechanisms to prognosis in cancer. J Clin Oncol. 1995;13:808–811. doi: 10.1200/JCO.1995.13.4.808. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumors. Nature (Lond) 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- Grunwald GB. The structural and functional analysis of cadherin calcium-dependent cell adhesion molecules. Curr Opin Cell Biol. 1993;5:797–805. doi: 10.1016/0955-0674(93)90028-o. [DOI] [PubMed] [Google Scholar]
- Guan J-L, Shaloway D. Regulation of focal adhesion-associated protein tyrosine kinase by both cellular adhesion and oncogenic transformation. Nature (Lond) 1992;358:690–692. doi: 10.1038/358690a0. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- Günthert U, Hofmann M, Rudy W, Reber S, Zöller M, Haubmann I, Matzku S, Wenzel A, Ponta H, Herrlich P. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell. 1991;65:13–24. doi: 10.1016/0092-8674(91)90403-l. [DOI] [PubMed] [Google Scholar]
- Harris CC, Hollstein M. Clinical implications of the p53 tumor suppressor gene. New Engl J Med. 1993;329:1318–1327. doi: 10.1056/NEJM199310283291807. [DOI] [PubMed] [Google Scholar]
- Hofmann M, Rudy W, Günthert U, Zimmer SG, Zawadzki V, Zöller M, Lichter RB, Herrlich P, Ponta H. A link between ras and metastatic behavior of tumor cells: ras induces CD44 promoter activity and leads to low-level of metastasis-specific variants of CD44 in CREF cells. Cancer Res. 1993;53:1516–1521. [PubMed] [Google Scholar]
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science (Wash DC) 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- Hungerford JE, Compton MT, Matter ML, Hoffstrom BG, Otey CA. Inhibition of pp125FAKin cultured fibroblasts results in apoptosis. J Cell Biol. 1996;135:1383–1390. doi: 10.1083/jcb.135.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamal HH, Cano-Gauci DF, Buick RN, Filmus J. Activated ras and src induce CD44 overexpression in rat intestinal epithelial cells. Oncogene. 1994;9:417–423. [PubMed] [Google Scholar]
- Kerbel RS, Brauer MJ, Powers VC, Wu JJ, Umansky SR, Tomei LD, Bar PJ. Multicellular resistance: a new paradigm to explain aspects of acquired drug resistance of solid tumors. Cold Spring Harbor Symp Quant Biol Mol Gen Cancer. 1995;59:661–672. doi: 10.1101/sqb.1994.059.01.076. [DOI] [PubMed] [Google Scholar]
- Kim WH, Schnaper W, Nomizu M, Yamada Y, Kleinman HK. Apoptosis in human fibrosarcoma cells is induced by a multimeric synthetic Tyr-Ile-Gly-Ser-Arg (YIGSR)-containing polypeptide from laminin. Cancer Res. 1994;54:5005–5010. [PubMed] [Google Scholar]
- Kobayashi H, Man S, Kapitain SJ, Teicher BA, Kerbel RS. Acquired multicellular-mediated resistance to alkylating agents in cancer. Proc Natl Acad Sci USA. 1993;90:3294–3298. doi: 10.1073/pnas.90.8.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg L, Earp HS, Parsons JT, Schaller M, Juliano RL. Cell adhesion or integrin clustering increases phosphorylation of a focal adhesion-associated tyrosine kinase. J Biol Chem. 1992;267:23439–23442. [PubMed] [Google Scholar]
- Lazebnik YA, Kaufmann SH, Dersnoyers S, Poirier GG, Earnshaw WC. Cleavage of the poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature (Lond) 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE, Jacks T, Houseman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993a;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature (Lond) 1993b;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, Housman DE, Jacks T. p53Status and the efficacy of cancer therapy in vivo. Science (Wash DC) 1994;266:807–810. doi: 10.1126/science.7973635. [DOI] [PubMed] [Google Scholar]
- MacCauley A, Pawson T. Cooperative transforming activities of ras, myc, and srcviral oncogenes in nonestablished rat adrenocortical cells. J Virol. 1988;62:4712–4721. doi: 10.1128/jvi.62.12.4712-4721.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ. Apoptosis: suicide, execution, or murder? . Trends Cell Biol. 1993;3:141–144. doi: 10.1016/0962-8924(93)90128-n. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Green DR. Protease activation during apoptosis: death by a thousand cuts. Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- Meredith JE, Fazeli B, Schwartz MA. The extracellular matrix as a cell survival factor. Mol Biol Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin PJ, Vogelstein B, Kinsler KW. Apoptosis and APC in colorectal tumorigenesis. Proc Natl Acad Sci USA. 1996;93:7950–7954. doi: 10.1073/pnas.93.15.7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson DW, All A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Mazebnik YA, Munday NA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature (Lond) 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Nikiforov MA, Hagen K, Ossovskaya VS, Connor TM, Lowe SW, Deichman GI, Gudkov AV. p53 modulation of anchorage independent growth and experimental metastasis. Oncogene. 1996;13:1709–1719. [PubMed] [Google Scholar]
- O'Neill CH, Jordan P, Ireland G. Evidence for two distinct mechanisms of anchorage stimulation in freshly explanted and 3T3 swiss mouse fibroblasts. Cell. 1986;44:489–496. doi: 10.1016/0092-8674(86)90470-8. [DOI] [PubMed] [Google Scholar]
- Otsuka H, Moscovitz M. Arrest of 3T3 cells in G1phase in suspension cultures. J Cell Physiol. 1975;87:213–220. doi: 10.1002/jcp.1040870209. [DOI] [PubMed] [Google Scholar]
- Pierschbacher MD, Ruoslahti E. Influence of stereochemistry of the sequence Arg-Gly-Asp-Xaa on binding specificity in cell adhesion. J Biol Chem. 1987;262:17294–17298. [PubMed] [Google Scholar]
- Pronk GJ, Ramer K, Amiri P, Williams LT. Requirement of an ICE-like protease for induction of apoptosis and ceramide generation in REAPER. Science (Wash DC) 1996;271:808–810. doi: 10.1126/science.271.5250.808. [DOI] [PubMed] [Google Scholar]
- Raff MC. Social controls on cell survival and cell death. Nature (Lond) 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- Rak J, Mitsuhashi Y, Erdos V, Huang S-N, Filmus J, Kerbel RS. Massive programmed cell death in intestinal epithelial cells induced by three-dimensional growth conditions: suppression by mutant c-H-rasoncogene expression. J Cell Biol. 1995;131:1587–1598. doi: 10.1083/jcb.131.6.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Re F, Zanetti A, Sironi M, Polentarutti N, Lanfrancone L, Dejana E, Colotta F. Inhibition of anchorage-dependant cell spreading triggers apoptosis in cultured human endothelial cells. J Cell Biol. 1994;127:537–546. doi: 10.1083/jcb.127.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinfeld B, Souza B, Albert I, Müller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P. Association of the APCgene product with β-catenin. Science (Wash DC) 1993;262:1731–1737. doi: 10.1126/science.8259518. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. Integrin signaling and matrix assembly. Tumour Biol. 1996;17:117–124. doi: 10.1159/000217975. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E, Reed JC. Anchorage dependance, integrins, and apoptosis. Cell. 1994;77:477–478. doi: 10.1016/0092-8674(94)90209-7. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Borgman CA, Cobb BC, Reynolds AB, Parsons JT. pp125FAK: a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci USA. 1992;89:5192–5196. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to FAK. Nature (Lond) 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Schwartz MA, Both G, Lechene C. Effect of cell spreading on cytoplasmic pH in normal and transformed fibroblasts. Proc Natl Acad Sci USA. 1989;86:4525–4529. doi: 10.1073/pnas.86.12.4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura A, Fisher DE. p53 in life and death. Clin Cancer Res. 1996;2:435–440. [PubMed] [Google Scholar]
- Stroker M. Anchorage and growth regulation in normal and virus-transformed cells. Int J Cancer. 1968;3:683–693. doi: 10.1002/ijc.2910030517. [DOI] [PubMed] [Google Scholar]
- Takeichi M. Cadherins in cancer: implications for invasion and metastasis. Curr Opin Cell Biol. 1993;5:806–811. doi: 10.1016/0955-0674(93)90029-p. [DOI] [PubMed] [Google Scholar]
- Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- Wagner AJ, Kokontis JM, Hay N. Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21waf/cip1 . Genes Dev. 1994;8:2817–2830. doi: 10.1101/gad.8.23.2817. [DOI] [PubMed] [Google Scholar]
- Williams GT, Smith CA. Molecular regulation of apoptosis: genetic controls on cell death. Cell. 1993;74:777–779. doi: 10.1016/0092-8674(93)90457-2. [DOI] [PubMed] [Google Scholar]
- Xu L-H, Owens LV, Sturge GC, Yang XY, Liu ET, Craven RJ, Cance WG. Attenuation of the expression of the focal adhesion kinase induces apoptosis in tumor cells. Cell Growth Differ. 1996;7:413–418. [PubMed] [Google Scholar]
- Yamada KM, Miyamoto S. Integrin transmembrane signaling and cytoskeletal control. Curr Opin Cell Biol. 1995;7:681–689. doi: 10.1016/0955-0674(95)80110-3. [DOI] [PubMed] [Google Scholar]
- Yonish-Rouach E, Resnitsky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature (Lond) 1991;352:345–347. doi: 10.1038/352345a0. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Vuori K, Reed JC, Ruoslahti E. The α5β1 integrin supports survival of cells on fibronectin and up-regulates Bcl-2 expression. Proc Natl Acad Sci USA. 1995;92:6161–6165. doi: 10.1073/pnas.92.13.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]