

Abstract
EGF precursor (proEGF) is a member of the family of membrane-anchored EGF-like growth factors that bind with high affinity to the epidermal growth factor receptor (EGFR). In contrast to human transforming growth factor-α precursor (proTGFα), which is sorted basolaterally in Madin-Darby canine kidney (MDCK) cells (Dempsey, P., and R. Coffey, 1994. J. Biol. Chem. 269:16878–16889), we now demonstrate that human proEGF overexpressed in MDCK cells is found predominantly at the apical membrane domain under steady-state conditions. Nascent proEGF (185 kD) is not sorted but is delivered equally to the apical and basolateral membranes, where it is proteolytically cleaved within its ectodomain to release a soluble 170-kD EGF form into the medium. Unlike the fate of TGFα in MDCK cells, the soluble 170-kD EGF species accumulates in the medium, does not interact with the EGFR, and is not processed to the mature 6-kD peptide. We show that the rate of ectodomain cleavage of 185-kD proEGF is fourfold greater at the basolateral surface than at the apical surface and is sensitive to a metalloprotease inhibitor, batimastat. Batimastat dramatically inhibited the release of soluble 170-kD EGF into the apical and basal medium by 7 and 60%, respectively, and caused a concordant increase in the expression of 185-kD proEGF at the apical and basolateral cell surfaces of 150 and 280%, respectively. We propose that preferential ectodomain cleavage at the basolateral surface contributes to apical domain localization of 185-kD proEGF in MDCK cells, and this provides a novel mechanism to achieve a polarized distribution of cell surface membrane proteins under steady-state conditions. In addition, differences in disposition of EGF and TGFα in polarized epithelial cells offer a new conceptual framework to consider the actions of these polypeptide growth factors.
EGF is the prototypical member of the EGF-like family of growth factors that display high-affinity binding for the EGF receptor (EGFR).1 Other mammalian EGF-like ligands include transforming growth factor-α (TGFα), amphiregulin, heparin binding EGF-like growth factor, betacellulin and epiregulin (3, 17, 32). These growth factors are all synthesized as glycosylated, membrane-anchored precursors that contain at least one EGF-like repeat in their extracellular domains. A distinctive feature of these membrane-anchored growth factor precursors is that they are biologically active at the cell surface, although they can be proteolytically cleaved from the cell surface to release soluble, diffusible factors (3, 17, 32).
Structural and functional characteristics of the EGF precursor (proEGF) distinguish it from other EGF-like growth factors. First, human proEGF is synthesized as a very large membrane-anchored precursor of 1,207 amino acid residues, whereas the other, smaller EGF-like growth factor precursors range in length from 160 to 252 amino acid residues (3, 17, 32). Second, it is the only EGF-like growth factor that contains multiple EGF-like repeats. Nine EGF-like repeats are found in the extracellular domain of proEGF with the soluble, mature 6-kD EGF derived from the most distal EGF repeat, which is positioned near the transmembrane domain (3, 17, 32). Third, proEGF has a very restricted expression pattern in vivo compared to the other, more widely expressed EGF-like growth factors (3, 17, 32). Fourth, a polarized distribution of proEGF has been demonstrated in the kidney, where it is expressed exclusively on the luminal surface of epithelial cells in the distal convoluted tubule (5, 48, 51–53). Finally, in various epithelial cell types, differential processing of proEGF has been demonstrated to release different soluble forms of EGF. In the salivary gland, mature 6-kD EGF is secreted after intracytoplasmic proteolytic cleavage by an arginine esterase-like activity (13, 14, 28), whereas in vitro studies using NIH 3T3 cells stably transfected with proEGF have demonstrated that proEGF is proteolytically cleaved to release a high–molecular mass 160-kD form (39, 40). Recent in vivo studies indicate that the predominant EGF species released from most epithelial cells is the high–molecular mass 160-kD EGF, which is found at high concentrations in urine and milk (30, 38, 44). While the biological actions of EGF have been studied extensively (13, 14, 17, 32), these unique characteristics of proEGF suggest that it may subserve biological functions distinct from those of mature EGF and the other EGF-like growth factors.
Elucidation of the sorting, processing, and steady-state distribution of EGF-like growth factors in polarized epithelial cells, which have basolaterally restricted EGFRs, may provide insight into modes of action of this family of growth factors. We have previously used the Madin-Darby canine kidney (MDCK) cell line to investigate molecular trafficking and processing of human proTGFα (16). In polarized MDCK cells, newly synthesized human proTGFα is directly delivered to the basolateral cell surface, where it is sequentially cleaved to release mature TGFα into the basal medium. Overexpression of TGFα did not cause down-regulation of EGFRs due to the efficient recycling of EGFRs in polarized MDCK cells. The colocalization of proTGFα with EGFRs to lateral membranes of polarized MDCK cells, together with its efficient consumption by basolateral EGFRs, suggests that TGFα acts only as a basally restricted, locally acting autocrine factor.
In the present study, we have examined the biosynthesis, sorting, and processing of human proEGF constitutively expressed in polarized MDCK cells and have identified major differences from the disposition of TGFα. Under steady-state conditions, 185-kD proEGF is found predominantly on the apical cell surface. It is not sorted, but is delivered equally to the apical and basolateral membrane domains. At the cell surface, proEGF is cleaved proteolytically to release a high–molecular mass soluble 170-kD EGF that accumulates in the extracellular medium and does not appear to interact with the EGFR. We show that preferential ectodomain cleavage of proEGF at the basolateral cell surface accounts, in part, for the increased residency time of proEGF at the apical membrane and its accumulation at that surface. We suggest that this preferential proteolytic activity, which is sensitive to the metalloprotease inhibitor, batimastat, provides a novel mechanism to regulate steady-state distribution of membrane proteins in polarized MDCK cells.
Materials and Methods
Reagents and Antibodies
All chemicals were purchased from Sigma Chemical Co. (St. Louis, MO) unless otherwise stated. All cell culture reagents were purchased from Gibco Laboratories (Grand Island, NY). 35S-Translabel was purchased from ICN Biomedicals, Inc. (Costa Mesa, CA). NHS-LC-biotin, protein A–agarose, and streptavidin-agarose were purchased from Pierce (Rockford, IL). All electrophoresis reagents were purchased from BioRad Laboratories (Hercules, CA). N-glycosidase F, endoglycosidase H (endo H), O-glycosidase, and neuraminidase were obtained from Boehringer Mannheim Corp. (Indianapolis, IN). Recombinant human EGF was purchased from Collaborative Biomedical Products (Bedford, MA). Batimastat (BB94), a matrix metalloprotease inhibitor, was kindly provided by Dr. Peter Brown (British Biotech, Oxford, UK).
EGF antibodies used in this study included a monoclonal antibody to human EGF (mAb-EGF) (57) as well as Ab-1 and Ab-3 purchased from Oncogene Science (Uniondale, NY) and the rabbit polyclonal antibody 889, kindly provided by Dr. Stanley Cohen (Vanderbilt University, Nashville, TN) (40). Rabbit polyclonal antibody [Ab 1417], which recognizes the amino acid sequence 1165–1186 within the cytoplasmic domain of human proEGF (38), was kindly provided by Dr. Barbara Mroczkowski (The Agouron Institute, La Jolla, CA). Monoclonal antibody to human EGFR (mAb-528) was generously provided by Dr. Hideo Masui (Memorial Sloan-Kettering Cancer Center, New York). Affinity-purified fluorescein (DTAF)-conjugated and CY3-conjugated secondary antibodies were purchased from Jackson Immunoresearch Laboratories, Inc. (West Grove, PA).
Cells and Cell Culture
MDCK strain II cell line was obtained from Enrique Rodriguez-Boulan (Cornell University Medical College, New York). Cells were grown in routine cell culture and cultured on Transwell filter chambers (0.4 μM; Costar, Cambridge, MA) as previously described (16). Transepithelial electrical resistance was measured to assess the integrity of tight junctions (16). All experiments were performed when electrical resistance was >200 Ω·cm2. To enhance proEGF expression, cells were treated with 5 mM sodium butyrate for 16 h before any experimental procedure.
DNA Transfection and Isolation of Expressing Clones
The wild-type proEGF cDNA isolated from human kidney was kindly provided by Dr. Barbara Mroczkowski. The proEGF construct lacking the cytoplasmic domain (1,057 amino acids in length) was generated by PCR and ended in the sequence . . .LLSLWGAHYY1057.
Stable transfection of wild-type and tail minus proEGF cDNA constructs in pCB6 were performed as previously described (16). In this study, both pools and individual clones of stably transfected cells were used. All experiments were performed with transfected cell lines between passage 5 and 15, and results were verified with two to three different clonal cell lines for each construct.
Immunofluorescence
Indirect immunofluorescence and laser scanning confocal microscopy were performed as previously described with some modifications (16). Primary antibodies used for detection of human proEGF were mAb-EGF (2 μg/ml) and polyclonal rabbit antibody Ab-3 (5 μg/ml). For double staining of proEGF and EGFR, mAb-528 (15 μg/ml) was added at the same time as Ab-3. After washing, appropriate secondary antibodies were added simultaneously. Laser scanning confocal microscopy was performed using a Zeiss LSM 4 confocal microscope (Thornwood, NY). Confocal images were generated by a Personal Iris graphic workstation (Silicon Graphics, Mountain View, CA) using Vital Images software.
EGF Precursor Immunoprecipitation
Immunoprecipitations from cell lysates of metabolically labeled cells were performed at 4°C. For cells grown on Transwell filters, cells were washed, filters were cut out, and cells were lysed as previously described (16). After insoluble material was pelleted, the supernatant was incubated for various times (1–16 h) with mAb-EGF (2 μg/ml lysate), and then immunoprecipitations were performed as previously described (16). To immunoprecipitate EGF species from conditioned media, 2 mM PMSF was added, media was precleared, and then immunoprecipitations were performed as described above. All immunoprecipitates were extensively washed and either stored at −70°C or directly analyzed under reducing conditions on 7.5% SDS-PAGE. Gels were treated with Amplify (Amersham Corp., Arlington Heights, IL), fixed, dried, and fluorography was performed. Scanning densitometry was analyzed by the PhosphorImager and ImageQuant software (Molecular Dynamics, Sunnyvale, CA). The specificity of mAb-EGF for human proEGF precursor and other proEGF immunoreactive species was confirmed by preincubation of antibody with excess recombinant human EGF (10 or 100 ng per immunoprecipitate) before the immunoprecipitation procedure.
Enzymatic digestion of proEGF immunoprecipitations with N-glycosidase F, endo H, O-glycosidase, and neuraminidase were performed using the manufacturer's protocol.
Metabolic Labeling
All cell cultures were initially grown in DME/FBS. To increase proEGF expression, all cell cultures were treated with 5 mM sodium butyrate in DME/FBS for 16 h before labeling unless otherwise stated. In some labeling experiments, basolateral surface EGFRs were down-regulated by addition of 20 nM mAb-528 to the medium for 24 h before labeling cells, as well as to labeling and chase medium.
For steady-state labeling, cells grown on Transwell filters were washed with serum-free DME lacking l-methionine and l-cysteine (DME−) and then incubated for 8–24 h in DME− containing 10% FBS and 100 μCi/ml 35S-Translabel added to the basal compartment. The apical compartment received 1.5 ml of the same medium lacking 35S-Translabel. For pulse and 2-h labeling, cells grown on Transwell filters were rinsed twice with DME− and then starved for 30 min in the same medium. Cells were then labeled by placing filters on 100 μl of serum-free DME− containing 1–2 mCi/ml 35S-Translabel. 1 ml of the same medium lacking 35S-Translabel was added to the apical compartment. In pulse–chase experiments, cells were pulse labeled for the specified time at 37°C and washed once with 18°C chase medium (DME/FBS containing 10 times excess l-methionine and l-cysteine). Media was then replaced with 37°C chase medium and cells were chased for various times.
Drug Treatments and Temperature Shift
In metabolic labeling experiments, cells were exposed to either monensin (1–10 μM) or BFA (1–30 μg/ml) during starvation, labeling, and chase. For tunicamycin experiments, cells were pretreated with different concentrations of tunicamycin (0.1–5 μg/ml) for 4 h before as well as during starvation, labeling, and chase. For 18°C pulse–chase experiments, cells were pulse labeled as described above, washed with 18°C medium, and then chased at 18°C. For 18 to 37°C temperature shift experiments, cells were pulse labeled as described above, washed with 18°C chase medium, and incubated at 18°C for 1 h. Cells were then washed with 37°C chase medium and subsequently chased at 37°C.
Cell Surface Immunoprecipitation
Metabolically labeled cells were cooled on ice and then washed four times with ice-cold PBS+ (PBS-0.1 mM CaCl2 and 1.0 mM MgCl2) containing 0.2% BSA (PBS-BSA). All subsequent steps were performed on ice or at 4°C. Proteins that arrived at the cell surface were detected by addition of mAb-EGF (2 μg/ml in PBS-BSA) to either the apical (500 μl) or basolateral (1.5 ml) compartments at the indicated times and incubated for 30 min with gentle rocking. The compartments not receiving antibody were filled with PBS-BSA. Cells were then rinsed with PBS-BSA and extensively washed with four changes of PBS-BSA for 1 h. Cell surface and total lysate immunoprecipitations were performed as previously described (16). A comparison of total and cell surface immunoprecipitates was used to determine equal loading.
Protease Assay
Metabolically labeled cells were washed three times with ice-cold PBS+ and incubated with protease added to either the apical, basolateral, or both compartments. For N-tosyl-l-phenylolanine chloromethylketone (TPCK)-trypsin digestion, cells were incubated for 30 min at 37°C with 10 μg/ml TPCK-trypsin or at 4°C with 100 μg/ml TPCK-trypsin. The opposite compartment received soybean trypsin inhibitor (STI) at 200 μg/ml. After incubation, cells were washed with PBS+ containing 200 μg/ml STI. For papain digestion, cells were washed with PBS+, pH 6.8, incubated with papain at 1 mg/ml diluted in PBS+, pH 6.8, for 10 min at 37°C, and then extensively washed with PBS-BSA. Cell surface and total cell lysate immunoprecipitations as well as immunoprecipitations from equivalent TCA precipitable counts were then performed as described above. For TPCK-trypsin, cells were lysed in lysis buffer containing 200 μg/ml STI.
Batimastat Treatment
Cells grown on Transwells were metabolically labeled for 2 h, washed, and then incubated with serum-free chase medium containing different concentrations of batimastat (0.1–10 μM in DMSO) added to both compartments. Control cells received serum-free chase medium containing DMSO alone. After chase, cells and conditioned media were collected and immunoprecipitations performed as described above.
Results
Biosynthesis and Processing of ProEGF in MDCK Cells
To investigate the biosynthesis and processing of proEGF, we performed pulse–chase experiments in MDCK cells stably transfected with human proEGF. A schematic model depicting the different cellular proEGF and soluble EGF species identified is shown in Fig. 8. Cells grown on Transwell filters were metabolically labeled with 35S-Translabel for 20 min and then chased for different periods of time. Total lysates were prepared, immunoprecipitated with mAb-EGF, and then subjected to SDS-PAGE and fluorography. At 0 min of chase, a single intense band of 175 kD was detected (Fig. 1 A). The intensity of the 175-kD band slowly diminished with a half-life of ∼2 h. A second, more diffuse band of 185 kD appeared by 1 h chase (in other experiments, it was detected as early as 40 min), and it also decreased with time with a half-life of ∼2 h. Interestingly, the decrease in intensity of cellular 175-kD proEGF did not coincide with an equivalent increase in the appearance of cellular 185-kD proEGF. The detection of a soluble, high–molecular mass 170-kD EGF species in the conditioned medium followed the appearance of 185-kD proEGF form, suggesting that the 185-kD form may be processed into the soluble 170-kD EGF (Fig. 2 and Fig. 6). In contrast to the processing of proTGFα in MDCK cells (16), no mature 6-kD EGF was detected in cell lysates or conditioned medium (data not shown).
Figure 8.

A proposed model for the establishment and maintenance of an apical distribution of proEGF in polarized MDCK cells. (A) Biosynthesis and sorting of proEGF in MDCK cells. 1. 175-kD proEGF represents the endo H–sensitive, intracellular form of proEGF. 175-kD proEGF is modified during transit through the secretory pathway to form 185-kD proEGF. 2. Newly synthesized 185-kD proEGF is not sorted at the level of the TGN but is delivered equally to both apical and basolateral membrane domains. 3. 185-kD proEGF represents the endo H–resistant, cell surface form of proEGF. 4. Ectodomain cleavage of cell surface 185-kD proEGF. 5. 170-kD EGF represents a soluble high–molecular mass EGF obtained by ectodomain cleavage of cell surface 185-kD proEGF. 6. 30-kD cleavage product represents the cytoplasmic and transmembrane domains found after cell surface protease digestion. (B) In polarized MDCK cells, preferential ectodomain cleavage of cell surface proEGF at the basolateral membrane domain leads to an apical enrichment of proEGF. Enhanced basolateral processing also leads to an accumulation of soluble 170-kD EGF in the basolateral medium. A matrix metalloprotease inhibitor, batimastat, dramatically inhibits processing of 185-kD proEGF at the cell surface.
Figure 1.

Biosynthesis and processing of proEGF forms in polarized MDCK cells. (A) MDCK cells expressing proEGF were grown on Transwell filters and metabolically labeled with 35S-Translabel for 20 min. The label was then chased for different periods of time. Total cell lysates were prepared and immunoprecipitated with mAb-EGF. (B and C) Pulse–chase experiments and immunoprecipitations were performed as described above. Immunoprecipitates were washed with the appropriate buffers, divided into equal parts, and digested with either N-glycosidase F (B) or endo H (C) as described in Materials and Methods.
Figure 2.

ProEGF is cleaved in its ectodomain to release a high– molecular mass soluble 170-kD EGF into the conditioned medium. (A) MDCK cells transfected with proEGF were grown on Transwell filters and metabolically labeled with 35S-Translabel for 16 h. After labeling, conditioned medium from apical and basolateral compartments was immunoprecipitated with either the ectodomain (Ecto; mAb-EGF) or cytoplasmic domain (Cyto; Ab 1417) EGF antibodies. Total cell lysates were also immunoprecipitated with these EGF antibodies. (B) Filter-grown MDCK cells expressing proEGF were metabolically labeled with 35S-Translabel for 20 min. The label was then chased for 2 h. Cells were either untreated (control) or treated with 18°C temperature, 18°C→ 37°C temperature shift, monensin, or BFA. Monensin (3 μM) and BFA (15 μg/ml) were added during starvation and throughout the pulse-label and chase. For 18°C temperature experiments, cells were either chased at 18°C or placed at 18°C for 1 h and then shifted to 37°C and chased for 2 h. Total cell lysate immunoprecipitations were performed using mAb-EGF. Apical and basolateral conditioned media were collected and immunoprecipitated. Only results of conditioned medium from basolateral compartment are shown.
Figure 6.

185-kD proEGF is preferentially cleaved from the basolateral cell surface in MDCK cells. (A) MDCK cells transfected with proEGF were grown on Transwell filters and labeled for 2 h with 35S-Translabel. Cells were then washed and chased for time periods indicated. After the chase, cell surface (data not shown) and media (A) immunoprecipitations were performed using mAb-EGF. (B) Quantitation of soluble 170-kD EGF released into apical (open circles) and basolateral (closed circles) medium. Average of two separate experiments ± SD are shown and expressed in arbitrary units calculated from total soluble 170-kD EGF released at each time point.
In previous studies using mouse NIH 3T3 cells transfected with human proEGF, a single N-glycosylated high– molecular mass proEGF species was detected in total cell extracts (39, 40). Our results are consistent with this finding, although the observation of two cellular proEGF forms (175 and 185 kD) in MDCK cells suggests that some additional posttranslational modifications occur. To further examine the relationship between the two cellular proEGF species, pulse–chase experiments with subsequent N-glycosidase F and endo H digestion were performed. Both cellular proEGF forms were sensitive to N-glycosidase F digestion throughout the chase period with the appearance of a single, faster-migrating 165-kD band (Fig. 1 B). A similar, faster-migrating band of 165-kD was detected after tunicamycin treatment (data not shown). Only the 175-kD band was sensitive to endo H digestion; it displayed an increase in mobility similar to the 165-kD form seen with N-glycosidase F treatment (Fig. 1 C). Together, these results suggest that the predominant cellular species is the immature, endo H–sensitive, 175-kD proEGF form, which does not efficiently chase into the more mature, endo H–resistant, 185-kD proEGF form.
185-kD ProEGF Is Proteolytically Cleaved at the Cell Surface to Release a Soluble 170-kD EGF into the Medium
To further examine the relationship between processing of cellular proEGF and the soluble, high–molecular mass 170-kD EGF detected in the conditioned medium, cells grown on Transwell filters were metabolically labeled with 35S-Translabel for 2 h and then chased for 4 h to accumulate sufficient levels of soluble 170-kD EGF in the medium. Apical and basal conditioned medium were immunoprecipitated using antibodies directed to either the proEGF extracellular domain (mAb-EGF) or cytoplasmic domain (Ab 1417). Both proEGF domain antibodies readily detected the two cellular proEGF forms (175 and 185 kD) in the total lysates, indicating that both cellular forms contain the cytoplasmic domain (Fig. 2 A). In contrast, only the extracellular domain antibody was able to immunoprecipitate the soluble 170-kD EGF species from the medium (Fig. 2 A). This result argues that the cellular proEGF form(s) must be proteolytically cleaved within the ectodomain to release the soluble 170-kD EGF into the medium. This data is in agreement with previous findings of Mroczkowski and colleagues that demonstrated that the majority of proEGF was released as a high–molecular mass (∼160-kD) species (38–40).
The biochemical properties of soluble, high–molecular mass 170-kD EGF of being N-glycosylated but resistant to endo H digestion (data not shown) are consistent with it being cleaved specifically from the mature, endo H–resistant, 185-kD proEGF form. Using cell surface biotinylation, it was not possible to determine if cleavage occurred at the cell surface because of inefficient labeling of proEGF (data not shown). In an attempt to further define whether 185-kD proEGF processing occurred at the plasma membrane, we performed pulse–chase experiments in the presence of different agents that block transport in the ER–Golgi– plasma membrane pathway (monensin, BFA, and 18°C temperature shift) (Fig. 2 B). First, we examined the role of 18°C temperature block, which detains proteins in the TGN, preventing their delivery to the plasma membrane (34). In pulse–chase experiments, cells were pulse labeled for 20 min at 37°C and chased at 18°C for 2 h or placed at 18°C for 1 h and temperature shifted to 37°C and chased for 2 h. After the 2-h chase, cell lysate and conditioned medium immunoprecipitations were performed. In the 18°C chase experiment, the 175-kD proEGF form was readily detected in cell lysates throughout the chase. By contrast, the 185-kD proEGF was never detected in cell lysates, indicating that its progression in the secretory pathway had been blocked at some point before or at the TGN (Fig. 2 B). No soluble 170-kD EGF was detected in the conditioned medium under these conditions. Pulse-labeled cells that had been blocked for 1 h at 18°C and then shifted to 37°C for the chase demonstrated kinetics of appearance similar to the 185-kD form as seen in control pulse–chase experiments (data not shown, Fig. 4 B). At 2 h chase, both 185-kD proEGF and soluble 170-kD EGF were detected in the cell lysate and in the conditioned medium, respectively.
Figure 4.

Cell surface 185-kD proEGF is expressed predominantly at the apical membrane domain in polarized MDCK cells. (A) For cell surface immunoprecipitations using an EGF ectodomain antibody, MDCK cells expressing proEGF were grown on Transwell filters and labeled for 8 h with 35S-Translabel in the presence of 10% FBS. After labeling, cell surface and total cell lysate immunoprecipitations with the EGF ectodomain antibody (Ecto Ab; mAb-EGF) were performed and subjected to SDS-PAGE and fluorography. For cell surface immunoprecipitations using EGF cytodomain and EGFR antibodies, cells were metabolically labeled with 35S-Translabel for 2 h, and then cell surface and total cell lysate immunoprecipitations were performed using EGF cytodomain (Cyto Ab; Ab 1417) and EGFR (mAb-528) antibodies, respectively. Protease assay was performed using either TPCK-trypsin (B) or papain (C). MDCK cells expressing proEGF were grown on Transwell filters, metabolically labeled with 35S-Translabel for 20 min, and then chased for 2 h to allow labeled proEGF forms to accumulate on the cell surface. Cells were treated with protease added to either the apical (Ap), basolateral (Bl), or both (AB) compartments. (B) After protease treatment, cell surface and total cell lysate (not shown) immunoprecipitations were performed using the EGF ectodomain (mAb-EGF) antibody. (C) After protease treatment, total lysates were divided equally and immunoprecipitated with the EGF ectodomain (mAb-EGF) and cytodomain (1417) antibodies. Apical, basolateral or both membrane domains are represented by Ap, Bl, and AB, respectively, in this and all subsequent figures.
Similar results were obtained in pulse–chase experiments in the presence of either monensin or BFA. The 175-kD proEGF was detected in total cell lysates, but expression of both the cellular 185-kD proEGF and the soluble 170-kD EGF in the medium were completely inhibited by both drugs (Fig. 2 B). Together, these results suggest that the appearance of the endo H–resistant, 185-kD proEGF form in a post-Golgi/plasma membrane compartment is required for its cleavage to release soluble 170-kD EGF into the conditioned medium.
185-kD ProEGF Is Localized Predominantly to the Apical Membrane Domain in Polarized MDCK Cells
To determine the cell surface distribution of proEGF in polarized epithelial cells, MDCK cells expressing wild-type proEGF were grown on Transwell filters and examined by indirect immunofluorescence and confocal microscopy. ProEGF was localized primarily to the apical surface of MDCK cell monolayers (Fig. 3 A). In pooled transfectants expressing very high levels of proEGF, it was also possible to detect some proEGF staining in the lateral membranes and to a lesser extent in the basal portion of the basolateral membrane domain (Fig. 3 B). It must be emphasized, however, that under all culture conditions, proEGF was localized predominantly to the apical membrane. This is consistent with in vivo observations that showed that proEGF is localized at the luminal surface of epithelial cells in the distal convoluted tubule of the kidney (5, 48, 51–53).
Figure 3.

ProEGF is localized predominantly at the apical cell surface in polarized MDCK cells. (A) MDCK cells transfected with proEGF were cultured for 4 d on Transwell filters and stained with polyclonal rabbit EGF antibody (Ab-3). Note intense staining at the apical membrane domain. (B) A pool of transfected MDCK cells expressing proEGF was grown on Transwell filters and stained with polyclonal rabbit antibody to EGF (Ab-3) and mouse monoclonal antibody to EGFR (mAb-528). ProEGF and EGFR are visualized in red and green, respectively. Yellow represents colocalization of both proteins. Arrowheads indicate the basal surface of the monolayer.
To biochemically characterize the apical/basolateral membrane distribution of proEGF in MDCK cells under steady- state conditions, we used metabolic labeling combined with cell surface immunoprecipitation. Cells grown on Transwell filters were metabolically labeled with 35S-Translabel for either 8 or 24 h, and then cell surface immunoprecipitations were performed using an ectodomain antibody (mAb-EGF). In agreement with the immunofluorescence results, these studies suggested that the 185-kD cell surface proEGF is localized primarily on the apical surface (Fig. 4 A). In both 8- and 24-h labeling experiments, the percentage of 185-kD proEGF expressed on the apical surface was >85%.
Results from cell surface immunoprecipitation and antibody competition experiments (data not shown) also suggested that 175-kD proEGF form was expressed at the cell surface. This was a surprising finding since the endo H–sensitive, 175-kD proEGF represents an immature, intracellular form of proEGF (see Fig. 8). One possible explanation for this result is that the integrity of the plasma membrane was perturbed, allowing access of EGF antibodies to intracellular proEGF forms. To address this possibility, we first demonstrated that MDCK cells expressing proEGF were structurally and functionally polarized as determined by polarized distribution of both apical (gp135) and basolateral cell surface proteins (E-cadherin, Na+K+-ATPase and EGFR) (data not shown; Fig. 4 A). Next, we used an antibody directed against the cytoplasmic domain of proEGF (Ab 1417), which should not be able to access and therefore bind proEGF by cell surface immunoprecipitations. Using Ab 1417, no proEGF forms were detected in cell surface immunoprecipitations, but both cellular proEGF forms were readily immunoprecipitated from total cell lysates (Fig. 4 A). This result indicates that the integrity of the plasma membrane had been maintained throughout the cell surface immunoprecipitation procedure.
To confirm directly that the endo H–resistant, 185-kD proEGF, and not the endo H–sensitive, 175-kD proEGF, is the authentic cell surface form, we used a protease assay that did not rely on an antibody immunoprecipitation procedure to detect cell surface proEGF forms. In this assay, cells were metabolically labeled for 20 min and then chased for various times. Cells were then treated with protease to cleave labeled proEGF forms that had arrived at the cell surface. As shown in Fig. 4 B, 185-kD proEGF was efficiently digested from both the apical and basolateral cell surfaces (∼65–95%), whereas 175-kD proEGF was resistant (<10%) to proteolytic cleavage. To validate the cell surface protease assay, we have demonstrated that the 175-kD proEGF is sensitive to protease digestion under different experimental conditions (data not shown). To further confirm that proEGF cleavage had occurred, total lysates were immunoprecipitated with the antibody directed against the cytoplasmic domain of proEGF (Ab 1417). This antibody not only recognizes proEGF forms but can detect a 30-kD cleavage product that represents the transmembrane and cytoplasmic domains of proEGF left within the cell after cell surface proteolytic digestion (see Fig. 8). The 30-kD cleavage product was only detected when 185-kD proEGF was expressed at the cell surface (Fig. 4 C). Based on these experiments, we believe that the immature, endo H–sensitive, 175-kD proEGF is artifactually detected in the cell surface immunoprecipitation procedure because of either antibody exchange or unbound antibody binding to 175-kD proEGF after cell lysis. In all subsequent studies, the cell surface proEGF form refers only to the 185-kD form, but results obtained with the 175-kD proEGF form are included for all experiments.
Newly Synthesized ProEGF Is Delivered Directly to Both Apical and Basolateral Membrane Domains
To study direct delivery of newly synthesized proEGF to the different cell surface domains in polarized MDCK cells, we performed pulse–chase experiments combined with cell surface immunoprecipitations. As shown in Fig. 5 A, 185-kD proEGF was delivered directly to both apical and basolateral cell surfaces. The 185-kD proEGF appeared at the cell surface after 40–60 min of chase and was maximally expressed by 2 h of chase (Fig. 5 A). Quantitation of 185-kD proEGF arrival to the cell surface indicates that it is delivered in a nonpolarized fashion with an apical/basolateral ratio of 55:45.
Figure 5.

Newly synthesized 185-kD proEGF is delivered equally to both apical and basolateral membrane domains in polarized MDCK cells. MDCK cells expressing proEGF were grown on Transwell filters and pulse-labeled with 35S-Translabel for 20 min and then chased for 0, 30, 60, or 120 min as indicated. (A) After the chase, cell surface and total cell lysate immunoprecipitations were performed using mAb-EGF. (B) After the chase, cells were treated with TPCK-trypsin added to either the apical or basolateral compartments. After protease treatment, cell surface immunoprecipitations were performed using the EGF ectodomain (mAb-EGF) antibody. Total cell lysates were divided equally and immunoprecipitated with the EGF ectodomain (mAb-EGF) (not shown) and cytodomain (Ab 1417) antibodies, respectively. (C) Quantitation of the appearance of 30-kD protease cleavage product after protease digestion of either apical (open circles) or basolateral (closed circles) membrane domains during pulse–chase experiment. The results are expressed in arbitrary units calculated from total amounts of 30-kD at each time point determined after subtracting control values. Average of three separate experiments ± SD are shown.
To further verify the nonpolarized delivery of newly synthesized proEGF to the cell surface, we performed pulse–chase experiments combined with cell surface protease digestion. In this assay, 185-kD proEGF would be accessible to protease digestion upon arrival at the cell surface. If protease digestion of cell surface proEGF occurred, it would result in the appearance of the 30-kD cleavage product in total lysates. In agreement with the kinetics for the cell surface arrival of 185-kD proEGF, the 30-kD cleavage product was not observed at early chase times but was readily detected after 1 h of chase (Fig. 5 B). Quantitatively, the 30-kD cleavage product appeared equally after protease digestion of either the apical or basolateral cell surfaces (Fig. 5 C). In these protease experiments, cell integrity and polarity were maintained as measured by control cell surface proteins (data not shown). This is further supported by the fact that protease treatment of both compartments had an additive effect on both the loss of cell surface 185-kD proEGF and appearance of the 30-kD cleavage product (Fig. 4). Taken together, these results indicate that 185-kD proEGF is delivered in a nonpolarized manner in MDCK cells.
ProEGF Accumulates at the Apical Membrane Domain
The fact that newly synthesized proEGF is not sorted in MDCK cells implies that some other mechanism is involved in establishing and maintaining the apical steady-state distribution of proEGF in polarized MDCK cells. To determine the fate of proEGF at the apical and basolateral membrane domains, we performed metabolic labeling studies combined with cell surface immunoprecipitations. In these experiments, cells were labeled for 2 h and then chased for long time periods. Cell surface immunoprecipitations, as well as apical and basal conditioned medium immunoprecipitations, were performed. The 185-kD proEGF was expressed on the apical and basolateral membrane domains at 0 min of chase but was rapidly lost from the basolateral surface with a half-life of less than 30 min. In contrast, proEGF had relatively stable expression on the apical cell surface during the first 2 h of chase and then was slowly removed from the apical surface with a half-life of 4 h (data not shown). A soluble 170-kD EGF form was immunoprecipitated from both apical and basal conditioned medium (Fig. 6 A). Quantitatively, the rate of soluble 170-kD EGF release was more than fourfold greater in basal medium as compared to apical medium (Fig. 6 B) and coincided with a similar rate of loss of proEGF from the basolateral membrane domain. The steady-state, polarized distribution of proEGF at the apical membrane domain in MDCK cells observed by indirect immunofluorescence (Fig. 3) can therefore be attributed, at least in part, to the increased rate of cleavage of proEGF from the basolateral cell surface.
Metalloprotease Inhibitor Batimastat Inhibits Ectodomain Cleavage of ProEGF
If our hypothesis that preferential basolateral proteolytic activity plays a role in maintaining the polarized distribution of proEGF in MDCK cells is correct, then inhibition of ectodomain cleavage should prevent the polarized release of soluble 170-kD EGF into the basal medium and increase levels of proEGF at the cell surface. In addition, increased levels of cell surface proEGF at each domain should inversely reflect rates of cleavage at the two membrane domains. In preliminary studies, we attempted to either stimulate or inhibit proEGF ectodomain cleavage under a variety of experimental conditions. We found that endogenous proEGF proteolytic activity was not stimulated by serum or phorbol ester (PMA) and was resistant to a wide variety of protease inhibitors. However, it was sensitive to both EDTA and EGTA and was temperature dependent (data not shown). Because of the sensitivity of proEGF cleavage to EDTA/EGTA and the recent findings that metalloproteases may be involved in the processing of HB-EGF (31) and proTGFα (2), we next examined the effects of the metalloprotease inhibitor, batimastat, on proEGF cell surface cleavage. As shown in Fig. 7 A, batimastat dramatically inhibited the release of soluble 170-kD EGF from both membrane domains. Quantitatively, batimastat inhibited release of soluble 170-kD EGF into the apical and basolateral medium by 7 and 60%, respectively (Fig. 7 B). In addition, batimastat caused an increase in the cell surface expression of 185-kD proEGF at both membrane domains even though the inhibitor did not completely block release of soluble 170-kD EGF. Cell surface expression of proEGF increased at the apical and basolateral membrane domains by 150 and 280%, respectively (Fig. 7 C). Taken together, these results support our contention that preferential basolateral cleavage of proEGF directly contributes to the polarized steady-state distribution of proEGF in MDCK cells. A model for the maintenance of an apical steady-state distribution of proEGF in polarized MDCK cells is proposed in Fig. 8.
Figure 7.
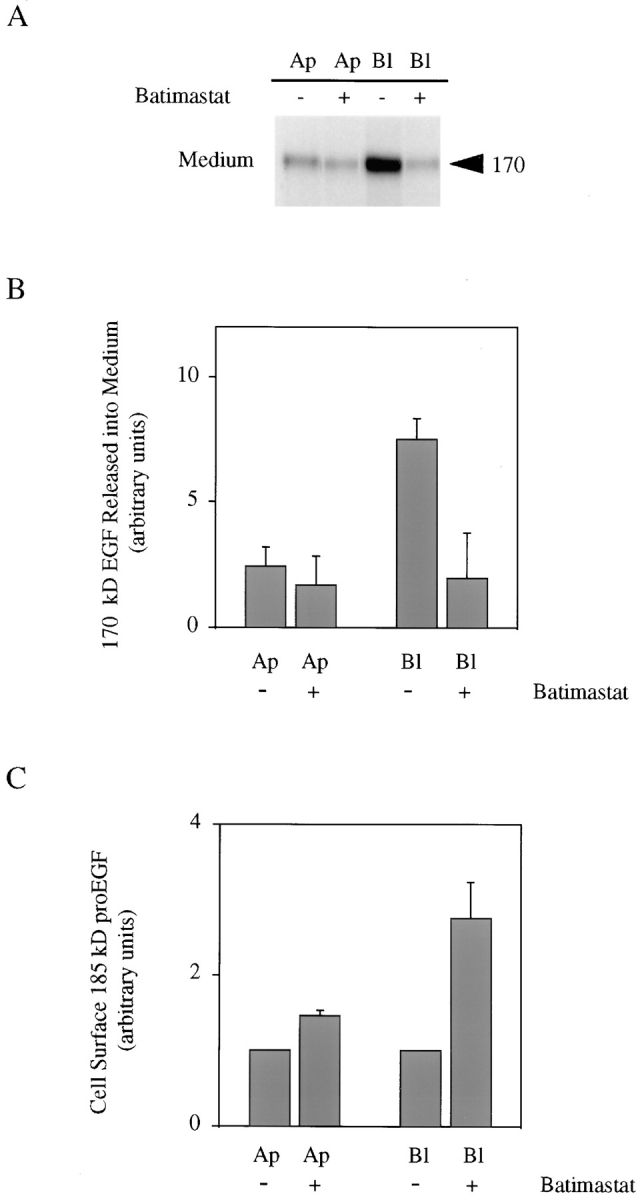
Batimastat inhibits cleavage of proEGF. MDCK cells expressing proEGF were grown on Transwell filters and labeled with 35S-Translabel for 2 h and then chased for 4 h in the absence or presence of 5 μM batimastat. (A) After the chase, media, cell surface, and total cell lysate immunoprecipitations were performed using mAb-EGF. Results for soluble 170-kD EGF released into medium are shown. (B) Quantitation of soluble 170-kD EGF released into apical and basolateral medium. Average of two separate experiments ± SD are shown and expressed in arbitrary units calculated from controls that received no batimastat. The values for apical and basolateral compartments are calculated from total soluble 170-kD released. (C) Quantitation of cell surface 185-kD proEGF expressed on the apical and basolateral membrane domains. Average of two separate experiments ± SD are shown and expressed in arbitrary units calculated from controls that received no batimastat. The values for apical and basolateral compartments are calculated separately. Cell surface values were determined from total lysate immunoprecipitates.
Discussion
Fundamental to understanding the physiological roles of the EGF family of growth factors is defining their unique biological functions. MDCK cells offer an excellent in vitro model system to study the expression of EGF-like growth factors and their interactions with basolateral EGFRs in polarized epithelial cells. In the present study, we demonstrate that the biosynthesis, sorting, and processing of proEGF in polarized MDCK cells differ significantly from proTGFα and that these differences provide a conceptual framework to understand the actions of these polypeptide growth factors. In addition, we propose that apical enrichment of human proEGF in MDCK cells involves preferential basolateral processing sensitive to a metalloprotease inhibitor, batimastat.
Ectodomain Cleavage of ProEGF Occurs at the Plasma Membrane
The membrane-anchored forms of two EGF-like ligands, proTGFα and proHB-EGF, can be cleaved from the cell surface in response to the protein kinase C activator, PMA (8, 22, 31, 41, 42, 47). In most cell types, including MDCK cells, proTGFα is sequentially cleaved at proximal and distal sites within the extracellular domain to release mature TGFα. The distal cleavage site is activated by phorbol esters to release soluble growth factor (41, 42). Within the plasma membrane compartment, proTGFα processing can be activated independent of cytosol and does not require membrane traffic (7, 8). In vitro and in vivo studies have suggested that proEGF is proteolytically cleaved to release a high–molecular mass proEGF form (27, 30, 38–40, 44). Using domain-specific antibodies in the present study, we demonstrate that mature, 185-kD proEGF is cleaved within its ectodomain to release a high–molecular mass, soluble 170-kD EGF form. Unlike TGFα, proEGF is only cleaved distally to release high–molecular mass, soluble 170-kD EGF and therefore no mature 6-kD EGF is detected in the conditioned medium. The release of high– molecular mass proEGF species from purified membranes of NIH 3T3 cells expressing proEGF and from rat kidney membranes by autolysis suggested that the proteolytic activity may be membrane associated (40, 44). To determine the cellular location of proEGF ectodomain cleavage in MDCK cells, we examined the transport and processing of proEGF in the presence of 18°C temperature block, monensin, or BFA (12, 29, 34, 45). Using these experimental procedures to block transport in the ER–Golgi–plasma membrane pathway, we show that proteolytic cleavage of 185-kD proEGF only occurs in a post-Golgi/plasma membrane compartment.
ProEGF Is Not Sorted in MDCK Cells but Accumulates at the Apical Cell Surface
Establishment and maintenance of the polarized distribution of membrane proteins in epithelial cells is regulated by several mechanisms. In MDCK cells, proteins can be sorted directly from the TGN to the apical and basolateral membrane domains (18, 37, 49, 55). For several basolateral membrane proteins, sorting signals have been identified in the cytoplasmic domain. Some but not all of these sequences overlap with endocytic motifs. Apical sorting signals are less well defined. Sorting information is conferred by a glycosyl-phosphatidyl-inositol (GPI) anchor for one specific group of apical membrane proteins; for non–GPI-anchored proteins, apical sorting information may be located in the extracellular domain and involve posttranslational modifications, such as N-glycosylation (18, 20, 37). Other membrane proteins may not be sorted preferentially but may exhibit cell surface selectivity because of differential residency times. For example, both Na+K+-ATPase and E-cadherin can be retained selectively at the basolateral membrane by interactions with the basolateral membrane cytoskeleton (23, 35, 56). Similarly, a subunit of the rat epithelial (amiloride-sensitive) Na+ channel may localize to the apical membrane because of interactions of the SH3 domain within its cytoplasmic domain and α-spectrin in the apical membrane cytoskeleton (50).
Our results provide another mechanism by which a polarized distribution of membrane proteins in epithelial cells can be achieved. Newly synthesized proEGF is delivered equally to both apical and basolateral membrane domains (Fig. 5), indicating that proEGF is not sorted at the level of the TGN. If proEGF is not sorted, how is its apical localization established and maintained in MDCK cells? The more rapid disappearance of proEGF from the basolateral cell surface combined with a concomitant accumulation of high–molecular mass, soluble 170-kD EGF in the basal medium (Fig. 6) indicates an increased level of proteolytic activity at the basolateral membrane domain. We can exclude an interaction between the proEGF cytoplasmic domain and the apical membrane cytoskeleton contributing to this effect since removal of the cytoplasmic domain of proEGF did not affect these results (data not shown). Based on these results, we submit that differences in proteolytic activity alter the residency time of proEGF at the two membrane domains. Under steady-state conditions, this would be reflected as an enrichment of proEGF at the apical cell surface. Thus, differential processing at apical and basolateral cell surfaces offers a novel mechanism to maintain a polarized distribution of proteins at the plasma membrane. A schematic model of preferential basolateral cleavage of proEGF in polarized MDCK cells is shown in Fig. 8.
Ectodomain Cleavage of ProEGF Is Sensitive to a Metalloprotease Inhibitor, Batimastat
Many membrane-anchored proteins undergo limited proteolysis to release ectodomains into the extracellular medium (1, 19). Ectodomain cleavage can also be stimulated by the protein kinase C activator, PMA, for some cell surface proteins, including membrane-anchored forms of proTGFα and proHB-EGF (8, 22, 31, 41, 42, 47). Activated proteolytic cleavage of proTGFα can also be induced by a variety of other factors, including serum and Ca2+ (8, 42), and is dependent on the COOH-terminal valine in the cytoplasmic tail (7, 8). Earlier studies on proTGFα processing indicated that certain specific serine protease inhibitors could block PMA-induced cleavage of the proTGFα ectodomain (43). However, more recent data suggests that these inhibitors block release by interfering with maturation and transport of the protein to the cell surface rather than by inhibiting proteolytic activity at the plasma membrane (2). The specificity of this proteolytic activity has only recently been established with the demonstration that a specific matrix metalloprotease inhibitor, TAPI, can inhibit PMA-induced release of proTGFα (2) and proHB-EGF (31).
Our initial studies to characterize the proEGF proteolytic activity demonstrated that proEGF proteolytic cleavage was not activated by serum or phorbol esters but was sensitive to EDTA and EGTA and was temperature dependent (data not shown). The inability to stimulate ectodomain cleavage of proEGF by various other activators meant that our analysis of potential inhibitors was dependent on the slow basal rate of proEGF cleavage with a half life of ∼2 h. This contrasts to PMA-activated release of proTGFα, which has a short half-life of 15 min (41, 42). ProEGF cleavage was not blocked by a variety of specific protease inhibitors, including pepstatin (5 μg/ml), leupeptin (5 μg/ml), PMSF (2 mM), elastatinal (100 μM), soybean trypsin inhibitor (200 U/ml), TPCK (100 μM), 3,4-DCI (100 μM), and aprotinin (200 KIU/ml) (data not shown). Interestingly, the serine protease inhibitor, aprotinin, which at very high concentrations is reported to inhibit release of EGF in an isolated perfused kidney model (27), did not block cleavage of proEGF in MDCK cells. In light of these negative results, together with the sensitivity of proEGF processing to EDTA/EGTA and the inhibition of PMA-stimulated processing of proHB-EGF and proTGFα by a matrix metalloprotease inhibitor (2, 31), we examined the effects of a specific metalloprotease inhibitor on proEGF cleavage. We found that the synthetic matrix metalloprotease inhibitor, batimastat (15, 21), effectively inhibited spontaneous cleavage of proEGF at a dose range of 2–10 μM (data not shown, Fig. 7).
Using batimastat to inhibit proEGF processing, we demonstrate that preferential basolateral ectodomain cleavage of proEGF plays an important role in establishing and maintaining a polarized apical distribution of proEGF in MDCK cells. Batimastat dramatically inhibited the polarized basolateral release of soluble 170-kD EGF and caused a concordant increase of proEGF cell surface expression at the basolateral membrane domain. The ability of batimastat to inhibit apical proEGF cleavage also suggests that some metalloprotease activity is expressed at the apical membrane domain in MDCK cells. It is also important to note that the increase in proEGF cell surface expression observed at the basolateral membrane domain after batimastat treatment was quantitatively less than would be predicted from the inhibition of release of high– molecular mass 170-kD EGF into the basolateral medium. This difference may be due, in part, to the experimental procedures used which relied on the spontaneous, basal rate of proEGF cleavage and a 4-h chase to accumulate high–molecular mass 170-kD EGF in the medium. We therefore cannot rule out the possibility that upon inhibiting the cleavage of cell surface proEGF, the cell surface fate and steady-state distribution of proEGF may be influenced by other factors such as membrane turnover, recycling, transcytosis, or membrane retention. Interestingly, a mutant proEGF lacking the cytoplasmic domain was sorted and cleaved in an identical manner to wild-type proEGF, suggesting that at least the proEGF cytoplasmic domain is not involved in any of these processes. Despite these differences, the dramatic inhibition of proEGF release by batimastat strongly suggests a role for a metalloprotease-sensitive pathway in the preferential basolateral cleavage and apical enrichment of proEGF in polarized MDCK cells.
How is differential processing of proEGF maintained at the basolateral membrane domain in MDCK cells? Matrix metalloproteases are a family of homologous enzymes that can be distinguished by their substrate specificity and can be divided structurally into secreted and membrane- anchored forms (11, 25). Disintegrin-metalloproteases or adamalysins (54) represent another family of membrane-anchored metalloproteases. A new member of the adamolysin class of metalloproteases is the tumor necrosis factor-α (TNF-α)–converting enzyme, which specifically cleaves the TNF-α precursor (6, 36). Interestingly, TNF-α–converting enzyme is inhibited by the metalloprotease inhibitor, TAPI (6), which can also inhibit both PMA-induced and spontaneous cleavage of proTGFα and proHB-EGF (2, 31). One possibility for the preferential basolateral cleavage of proEGF in polarized MDCK cells therefore is that the metalloprotease(s) involved also displays a polarized basolateral distribution. At present, the sorting, membrane distribution, or secretion of matrix metalloproteases in polarized epithelial cells has not been defined. Alternatively, most matrix metalloproteases are synthesized in a latent form and require cleavage of the amino-terminal domain to activate the enzyme (11). It is therefore possible that cofactors involved in regulating the catalytic activity of the protease may have a polarized distribution. In this context, it is interesting to note that EGFR activation can induce ectodomain cleavage of proTGFα (4). In polarized MDCK cells, EGFRs are expressed on the basolateral membrane domain (16). In this scenario, preferential basolateral cleavage of proEGF would not require the metalloprotease to have a polarized distribution because direct EGFR activation might lead to localized stimulation of proteolytic activity at the basolateral cell surface of polarized epithelial cells. Although the exact mechanism(s) involved in preferential basolateral cleavage of proEGF in polarized MDCK cells awaits identification of the specific metalloprotease(s), the present results do raise the possibility that ectodomain shedding of other membrane proteins may be regulated differently at apical and basolateral membrane domains in polarized epithelial cells.
Biological Consequences of Differential Sorting and Processing of ProEGF Compared to ProTGFα in Polarized Epithelial Cells
Dynamic recycling of EGFRs has been demonstrated in many polarized epithelial cell types. For example, in MDCK cells overexpressing proTGFα, the inability to down-regulate basolateral EGFRs concomitant with the high rate of TGFα consumption is due to efficient recycling of EGFRs to the cell surface (16). In fact, immunoreactive TGFα could only be detected in the basolateral medium upon EGFR antibody blockade (16). Hence, one distinguishing feature of MDCK cells expressing proEGF is the accumulation of soluble 170-kD EGF in the medium after its release from the cell surface (Figs. 6 and 7). Moreover, antibody blockade of EGFRs did not alter the levels of soluble 170-kD EGF found in the medium (Dempsey, P.J., unpublished observation). Species differences between EGF and its receptor cannot account for the lack of growth factor consumption because human and rodent EGF bind with high affinity to endogenous canine EGFRs on MDCK cells (9, 16, 24). Although studies using NIH 3T3 cells stably expressing proEGF demonstrated that high–molecular mass EGF released from the cell surface is biologically active (40), more recent studies using both in vivo and in vitro EGFR kinase activation and mitogenic assays have shown that soluble, high–molecular mass EGF isolated from human milk or urine is less biologically active than mature 6-kD EGF (38, 44). Therefore, it is reasonable to assume that accumulation of soluble 170-kD EGF in the basal medium of MDCK cells is a direct result of it being a low-affinity ligand for the EGFR (38) and once again underscores differences between disposition of EGF and TGFα.
What are the biological consequences of proEGF expression at and release from the apical membrane domain? In polarized epithelial cells, apical and basolateral membrane domains are separated by tight junctions that create a barrier that prevents diffusion of ions and macromolecules across the monolayer. Consequently, high– molecular mass EGF released into the apical medium cannot access basolateral EGFRs but rather bathes the apical cell surface. Similarly, membrane-anchored proEGF enriched at the apical cell surface cannot interact with EGFRs at the basolateral cell surface. In normal polarized epithelial cells, this segregation of apical ligand from basolateral EGFRs may be important to limit the capacity of EGFR signaling. However, in situations where epithelial cells depolarize, such as during cell transformation, EGFRs would have direct access to apical ligand, which may cause enhanced EGFR activation and promote cell growth.
The exclusive expression of proEGF on the luminal surface of epithelial cells in the distal convoluted tubule of the kidney (5, 48, 51–53) and on the luminal surface of alveolar cells in the mammary gland (10, 26), as well as the release of high–molecular mass proEGF into the luminal environment as seen in milk and urine (38, 44), suggests that apical proEGF does perform some relevant biological function(s). In the gastrointestinal tract, it has been proposed that mature EGF found in the lumen may act as a surveillance factor that monitors the integrity of the epithelial monolayer, and at sites of damage or injury to the monolayer, luminal EGF would be able to access and activate basolateral EGFRs (46). An analogous scenario could be envisioned for high–molecular mass 170-kD EGF. Alternatively, high–molecular mass EGF may act as a reservoir of latent growth factor in the luminal environment, which upon processing would release mature high-affinity 6-kD EGF. In urine, endoproteases and several kallikreins have been identified that could subserve this function (33). In either situation, EGF released into the luminal environment could be presented to distinct populations of EGFRs possibly far removed from their site of synthesis. Significantly, these EGFR populations would not be accessible to other basolaterally targeted EGF-like growth factors, like TGFα. It is also possible that proEGF may subserve functions within the apical compartment independent of the EGFR. For example, it has been suggested that proEGF may act as a receptor (17). Similarly, recent studies with high–molecular mass urinary proEGF indicate that it may promote cell adhesion (44). To effectively perform these functions, it may be necessary for proEGF to avoid interactions with the EGFR. Taken together, these results demonstrate marked differences in the disposition of EGF and TGFα in polarized epithelial cells and provide new insights into possible distinct actions of these growth factors.
Acknowledgments
We thank Drs. Tom Daniels and Christine Saunders, as well as members of the Coffey laboratory, for helpful discussions and critical reading of the manuscript. We would also like to thank Dr. Barbara Mroczkowski, Dr. Hideo Masui, and Dr. P. Brown for supplying reagents used in this study.
This work was supported by National Institutes of Health Grant CA46413 (to R.J. Coffey) and by the Joseph and Mary Keller Foundation. R.J. Coffey is a Veterans Administration Clinical Investigator. Analyses were performed in part through use of the Vanderbilt University Medical Center Cell Imaging Resource (supported by CA68485 and DK20593).
Abbreviations used in this paper
- BFA
brefeldin A
- EGFR
epidermal growth factor receptor
- endo H
endoglycosidase H
- MDCK
Madin-Darby canine kidney cells
- TGFα
transforming growth factor-α
- TPCK
N-tosyl-l-phenylalanine chloromethylketone
Footnotes
Address all correspondence to Dr. Peter J. Dempsey, Departments of Medicine and Cell Biology, GI Cancer Program, CC-2218 Medical Center North, Vanderbilt University School of Medicine, Nashville, TN 37232-2583. Tel.: (615) 343-0171. E-mail: peter.dempsey@mcmail.vanderbilt.edu
References
- 1.Arribas J, Massagué J. Transforming growth factor-α and β-amyloid precursor protein share a secretory mechanism. J Cell Biol. 1995;128:433–441. doi: 10.1083/jcb.128.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arribas J, Coodly L, Vollmer P, Kishimoto T, Rose-John S, Massagué J. Diverse cell surface protein ectodomains are shed by a system sensitive to metalloprotease inhibitors. J Biol Chem. 1996;271:11376–11382. doi: 10.1074/jbc.271.19.11376. [DOI] [PubMed] [Google Scholar]
- 3.Barnard J, Beauchamp R, Russell W, DuBois R, Coffey R. Epidermal growth factor-related peptides and their relevance to gastrointestinal pathophysiology. Gastroenterology. 1995;108:564–580. doi: 10.1016/0016-5085(95)90087-x. [DOI] [PubMed] [Google Scholar]
- 4.Baselga J, Mendelsohn J, Kim Y-M, Pandiella A. Autocrine regulation of membrane transforming growth factor-α cleavage. J Biol Chem. 1996;271:3279–3284. doi: 10.1074/jbc.271.6.3279. [DOI] [PubMed] [Google Scholar]
- 5.Bell G, Fong N, Stempien M, Wormsted M, Caput D, Ku L, Urdea M, Rall L, Sanchez-Pescador R. Human epidermal growth factor precursor: cDNA sequence, expression in vitro and gene organization. Nucleic Acids Res. 1986;14:8427–8446. doi: 10.1093/nar/14.21.8427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Black R, Rauch C, Kozlosky C, Peschon J, Slack J, Wolfson M, Castner B, Stocking K, Reddy P, Srinivasan S, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature (Lond) 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 7.Bosenberg M, Pandiella A, Massagué J. The cytoplasmic carboxy-terminal amino acid specifies cleavage of membrane TGFα into soluble growth factor. Cell. 1992;71:1157–1165. doi: 10.1016/s0092-8674(05)80064-9. [DOI] [PubMed] [Google Scholar]
- 8.Bosenberg M, Pandiella A, Massagué J. Activated release of membrane-anchored TGFα in the absence of cytosol. J Cell Biol. 1993;122:95–101. doi: 10.1083/jcb.122.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandli A, Adamson E, Simons K. Transcytosis of epidermal growth factor. J Biol Chem. 1991;266:8560–8566. [PubMed] [Google Scholar]
- 10.Brown C, Teng C, Pentecost B, DiAugustine R. Epidermal growth factor precursor in mouse lactating mammary gland alveolar cells. Mol Endocrinol. 1989;89:1077–1083. doi: 10.1210/mend-3-7-1077. [DOI] [PubMed] [Google Scholar]
- 11.Brown P. Matrix metalloproteinase inhibitors: a novel class of anticancer agents. Adv Enzyme Regul. 1995;35:293–301. doi: 10.1016/0065-2571(94)00022-u. [DOI] [PubMed] [Google Scholar]
- 12.Burgess T, Kelly R. Constitutive and regulated secretion of proteins. Annu Rev Cell Biol. 1987;3:243–293. doi: 10.1146/annurev.cb.03.110187.001331. [DOI] [PubMed] [Google Scholar]
- 13.Carpenter G, Cohen S. Epidermal growth factor. Annu Rev Biochem. 1979;48:193–216. doi: 10.1146/annurev.bi.48.070179.001205. [DOI] [PubMed] [Google Scholar]
- 14.Carpenter, G., and M. Wahl. 1990. The epidermal growth factor family. In Peptide Growth Factors and Their Receptors. Vol. I. M. Sporn and A. Roberts, editors. Springer-Verlag, Berlin. 69–171.
- 15.Davies B, Brown P, East N, Crimmin M, Balkwill F. A synthetic matrix metalloprotease inhibitor decreases tumor burden and prolongs survival of mice bearing human ovarian carcinoma xenografts. Cancer Res. 1993;53:2087–2091. [PubMed] [Google Scholar]
- 16.Dempsey P, Coffey R. Basolateral targeting and efficient consumption of transforming growth factor-α when expressed in Madin-Darby canine kidney cells. J Biol Chem. 1994;269:16878–16889. [PubMed] [Google Scholar]
- 17.Derynck R. The physiology of transforming growth factor-α. Cancer Res. 1992;58:27–52. doi: 10.1016/s0065-230x(08)60289-4. [DOI] [PubMed] [Google Scholar]
- 18.Drubin D, Nelson W. Origins of cell polarity. Cell. 1996;84:335–344. doi: 10.1016/s0092-8674(00)81278-7. [DOI] [PubMed] [Google Scholar]
- 19.Ehlers M, Riordan J. Membrane proteins with soluble counterparts: role of proteolysis in the release of transmembrane proteins. Biochemistry. 1991;30:10065–10074. doi: 10.1021/bi00106a001. [DOI] [PubMed] [Google Scholar]
- 20.Fiedler K, Simons K. The role of N-glycans in the secretory pathway. Cell. 1995;81:309–312. doi: 10.1016/0092-8674(95)90380-1. [DOI] [PubMed] [Google Scholar]
- 21.Gearing A, Beckett P, Christodoulou M, Churchill M, Clements J, Davidson A, Drummond A, Galloway W, Gilbert R, Gordon J, et al. Processing of tumour necrosis factor-α precursor by metalloproteinases. Nature (Lond) 1994;370:555–557. doi: 10.1038/370555a0. [DOI] [PubMed] [Google Scholar]
- 22.Goishi K, Higashiyama S, Klagsbrun M, Nakano N, Umata T, Ishikawa M, Mekada E, Taniguchi N. Phorbol ester induces the rapid processing of cell surface heparin-binding EGF-like growth factor: conversion from juxtacrine to paracrine growth factor activity. Mol Biol Cell. 1995;6:967–980. doi: 10.1091/mbc.6.8.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammerton R, Krzeminski K, Mays R, Ryan T, Wollner D, Nelson W. Mechanism for regulating cell surface distribution of Na+,K+-ATPase in polarized epithelial cells. Science (Wash DC) 1991;254:847–850. doi: 10.1126/science.1658934. [DOI] [PubMed] [Google Scholar]
- 24.Hobert M, Carlin C. Cytoplasmic juxtamembrane domain of the human EGF receptor is required for basolateral localization in MDCK cells. J Cell Physiol. 1995;162:434–446. doi: 10.1002/jcp.1041620316. [DOI] [PubMed] [Google Scholar]
- 25.Hooper N, Karran E, Turner A. Membrane protein secretases. Biochemistry. 1997;321:265–279. doi: 10.1042/bj3210265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jahnke G, Chao J, Walker M, DiAugustine R. Detection of a kallikrein in the mouse lactating mammary gland: a possible processing enzyme for the epidermal growth factor precursor. Growth Fact. 1994;11:113–123. doi: 10.1210/endo.135.5.7525260. [DOI] [PubMed] [Google Scholar]
- 27.Jorgensen P, Hilchey S, Nexo E, Poulsen S, Quilley C. Urinary epidermal growth factor is excreted from the rat isolated perfused kidney in the absence of plasma. J Endocrinol. 1993;130:227–234. doi: 10.1677/joe.0.1390227. [DOI] [PubMed] [Google Scholar]
- 28.Jorgensen P, Nexo E, Poulsen S, Almendingen M, Berg T. Processing of epidermal growth factor in the rat submandibular gland by kallikrein-like enzymes. Growth Fact. 1994;11:113–123. doi: 10.3109/08977199409001053. [DOI] [PubMed] [Google Scholar]
- 29.Klausner R, Donaldson J, Lippincott-Schwartz J. Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lakshmanan J, Salido E, Lam R, Fisher D. Epidermal growth factor prohormone is secreted in human urine. Am J Physiol Endocrinol Metab. 1992;26:E142–E150. doi: 10.1152/ajpendo.1992.263.1.E142. [DOI] [PubMed] [Google Scholar]
- 31.Lanzrein M, Garred O, Olsnes S, Sandvig K. Diphtheria toxin endocytosis and membrane translocation are dependent on the intact membrane-anchored receptor (HB-EGF precursor): studies on the cell-associated receptor cleaved by a metalloprotease in phorbol-ester-treated cells. Biochem J. 1995;310:285–289. doi: 10.1042/bj3100285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee D, Fenton S, Berkowitz E, Hissong M. Transforming growth factor-α: expression, regulation, and biological activities. Pharmacol Rev. 1995;47:51–85. [PubMed] [Google Scholar]
- 33.Margolius H. The kallikrein-kinin system and the kidney. Annu Rev Physiol. 1984;46:309–326. doi: 10.1146/annurev.ph.46.030184.001521. [DOI] [PubMed] [Google Scholar]
- 34.Matlin K, Simons K. Reduced temperature prevents transfer of a membrane glycoprotein to the cell surface but does not prevent terminal glycosylation. Cell. 1983;34:233–243. doi: 10.1016/0092-8674(83)90154-x. [DOI] [PubMed] [Google Scholar]
- 35.Mays R, Siemers K, Fritz B, Lowe A, Meer GV, Nelson W. Hierarchy of mechanisms in generating Na/K-ATPase polarity in MDCK epithelial cells. J Cell Biol. 1995;130:1105–1115. doi: 10.1083/jcb.130.5.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moss M, Jin S, Milla M, Burkhart W, Carter H, Chen W, Clay W, Didsbury J, Hassler D, Hoffman C, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature (Lond) 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- 37.Mostov K, Cardone M. Regulation of protein traffic in polarized epithelial cells. BioEssays. 1995;17:129–138. doi: 10.1002/bies.950170208. [DOI] [PubMed] [Google Scholar]
- 38.Mroczkowski B, Reich M. Identification of biologically active epidermal growth factor precursor in human fluids and secretions. Endocrinology. 1993;132:417–425. doi: 10.1210/endo.132.1.8419140. [DOI] [PubMed] [Google Scholar]
- 39.Mroczkowski B, Reich M, Whittaker J, Bell G, Cohen S. Expression of human epidermal growth factor precursor cDNA in transfected mouse NIH 3T3 cells. Proc Natl Acad Sci USA. 1988;85:126–130. doi: 10.1073/pnas.85.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mroczkowski B, Reich M, Chen K, Bell G, Cohen S. Recombinant human epidermal growth factor precursor is a glycosylated membrane protein with biological activity. Mol Cell Biol. 1989;9:2771–2778. doi: 10.1128/mcb.9.7.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pandiella A, Massagué J. Cleavage of the membrane precursor for transforming growth factor α is a regulated process. Proc Natl Acad Sci USA. 1991;88:1726–1730. doi: 10.1073/pnas.88.5.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pandiella A, Massagué J. Multiple signals activate cleavage of the membrane transforming growth factor-α precursor. J Biol Chem. 1991;266:5769–5773. [PubMed] [Google Scholar]
- 43.Pandiella A, Bosenberg M, Huang E, Besmer P, Massagué J. Cleavage of membrane-anchored growth factors involves distinct proteolytic activities regulated through common mechanisms. J Biol Chem. 1992;267:24028–24033. [PubMed] [Google Scholar]
- 44.Parries G, Chen K, Misono K, Cohen S. The human urinary epidermal growth factor (EGF) precursor. Isolation of a biologically active 160-kilodalton heparin-binding pro-EGF with a truncated carboxyl terminus. J Biol Chem. 1995;270:27954–27960. doi: 10.1074/jbc.270.46.27954. [DOI] [PubMed] [Google Scholar]
- 45.Pelham H. Multiple targets for brefeldin A. Cell. 1991;67:449–451. doi: 10.1016/0092-8674(91)90517-3. [DOI] [PubMed] [Google Scholar]
- 46.Playford R, Woodman A, Clark P, Watanapa P, Vesey D, Deprez P, Williamson R, Calam J. Effect of luminal growth factor preservation on intestinal growth [see comments] Lancet (N Am Ed) 1993;341:866–867. doi: 10.1016/0140-6736(93)93057-8. [DOI] [PubMed] [Google Scholar]
- 47.Raab G, Higashiyama S, Hetelekidis S, Abraham J, Damm D, Ono M, Klagsbrun M. Biosynthesis and processing by phorbol ester of the cell surface-associated precursor form of heparin-binding EGF-like growth factor. Biochem Biophys Res Commun. 1994;204:592–597. doi: 10.1006/bbrc.1994.2500. [DOI] [PubMed] [Google Scholar]
- 48.Rall L, Scott J, Bell G, Crawford R, Penschow J, Niall H, Coghlan J. Mouse prepro-epidermal growth factor synthesis by the kidney and other tissues. Nature (Lond) 1985;313:228–231. doi: 10.1038/313228a0. [DOI] [PubMed] [Google Scholar]
- 49.Rodriguez-Boulan E, Powell S. Polarity of epithelial and neuronal cells. Annu Rev Cell Biol. 1992;8:395–427. doi: 10.1146/annurev.cb.08.110192.002143. [DOI] [PubMed] [Google Scholar]
- 50.Rotin D, Bar-Sagi D, O'Brodovich H, Merilainen J, Lehto V, Canessa C, Rossier B, Downey G. An SH3 binding region in the epithelial Na+channel (arENaC) mediates its localization at the apical membrane. EMBO (Eur Mol Biol Organ) J. 1994;13:4440–4450. doi: 10.1002/j.1460-2075.1994.tb06766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salido E, Barajas L, Lechago J, Laborde N, Fisher D. Immunocytochemical localization of epidermal growth factor in mouse kidney. J Histochem Cytochem. 1986;34:1155–1160. doi: 10.1177/34.9.2426343. [DOI] [PubMed] [Google Scholar]
- 52.Salido E, Fisher D, Barajas L. Immunoelectron microscopy of epidermal growth factor in mouse kidney. J Ultrastruct Mol Struct Res. 1986;96:105–113. doi: 10.1016/0889-1605(86)90011-x. [DOI] [PubMed] [Google Scholar]
- 53.Salido E, Yen P, Shapiro L, Fisher D, Barajas L. In situ hybridization of prepro-epidermal growth factor mRNA in the mouse kidney. Am J Physiol. 1989;256:F632–F638. doi: 10.1152/ajprenal.1989.256.4.F632. [DOI] [PubMed] [Google Scholar]
- 54.Wolfsberg T, Primakoff P, Myles D, White J. ADAM, a novel family of membrane proteins containing a disintegrin and metalloprotease domain: multipotential functions in cell–cell and cell–matrix interactions. J Cell Biol. 1995;131:275–278. doi: 10.1083/jcb.131.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wollner D, Nelson W. Establishing and maintaining epithelial cell polarity—roles of protein sorting, delivery, and retention. J Cell Sci. 1992;102:185–190. doi: 10.1242/jcs.102.2.185. [DOI] [PubMed] [Google Scholar]
- 56.Wollner D, Krzeminski K, Nelson W. Remodeling the cell surface distribution of membrane proteins during the development of epithelial cell polarity. J Cell Biol. 1992;116:889–899. doi: 10.1083/jcb.116.4.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoshitake Y, Nishikawa K. Production of monoclonal antibodies with specificity for different epitopes on the human epidermal growth factor molecule. Arch Biochem Biophys. 1988;263:437–446. doi: 10.1016/0003-9861(88)90656-x. [DOI] [PubMed] [Google Scholar]