

Abstract
The small GTPases Rho and Rac regulate actin filament assembly and the formation of integrin adhesion complexes to produce stress fibers and lamellipodia, respectively, in mammalian cells. Although numerous candidate effectors that might mediate these responses have been identified using the yeast two-hybrid and affinity purification techniques, their cellular roles remain unclear. We now describe a biological assay that allows components of the Rho and Rac signaling pathways to be identified. Permeabilization of serum-starved Swiss 3T3 cells with digitonin in the presence of guanosine 5′-O-(3-thiotriphosphate) (GTPγS) induces both actin filament and focal adhesion complex assembly through activation of endogenous Rho and Rac. These responses are lost when GTPγS is added 6 min after permeabilization, but can be reconstituted using concentrated cytosolic extracts. We have achieved a 10,000-fold purification of the activity present in pig brain cytosol and protein sequence analysis shows it to contain moesin. Using recombinant proteins, we show that moesin and its close relatives ezrin and radixin can reconstitute stress fiber assembly, cortical actin polymerization and focal complex formation in response to activation of Rho and Rac.
Members of the Rho subfamily of small GTPases control the adhesion, morphology, and motility of mammalian cells, and also regulate signal transduction pathways that affect gene transcription in the nucleus (Hall, 1994; Machesky and Hall, 1996). The three best-understood members of the family, Rho, Rac, and Cdc42, share some 50% identity at the amino acid level and like the archetypal small GTPase, Ras, act as molecular switches in intracellular signaling pathways. They cycle between active (GTP-bound) and inactive (GDP-bound) forms, and a large number of potential regulators of this cycle have been identified, such as guanine nucleotide exchange factors, guanine nucleotide dissociation inhibitors (GDIs)1, and GTPase activating proteins (GAPs) (Ueda et al., 1990; Cerione and Zheng, 1996; Lamarche et al., 1996).
The biological consequences of activating Rho, Rac, and Cdc42 have been documented in some detail in Swiss 3T3 fibroblasts. Activation of Rho using extracellular stimuli such as lysophosphatidic acid, or microinjection of constitutively activated forms of recombinant Rho protein, leads to the assembly of actin stress fibers and focal adhesion complexes, whereas activation of Rac by agonists such as PDGF or insulin stimulates the formation of lamellipodia and membrane ruffles (Ridley and Hall, 1992; Ridley et al., 1992). Further analysis has revealed that the filamentous actin found in Rac-induced lamellipodia is associated with small focal complexes which, although morphologically distinct from classical focal adhesions, share many of their constituents, including integrins, vinculin, paxillin, and focal adhesion kinase (Hotchin and Hall, 1995; Nobes and Hall, 1995). Cdc42 activation in Swiss cells (by bradykinin or by microinjection of recombinant protein) triggers the formation of filopodia (microspikes, membrane protrusions containing actin filament bundles that are associated with integrin focal complexes (Kozma et al., 1995; Nobes and Hall, 1995). Activation of Cdc42 also leads to rapid activation of Rac in these cells, making Cdc42 and Rac ideal candidates for regulating cell movement in response to extracellular stimuli.
There has been a great deal of interest in the signal transduction pathways downstream of Rho, Rac, and Cdc42 and two main approaches have been used to identify proteins that associate with these GTPases: the yeast two-hybrid system using GTPases as bait, and protein purification techniques using a GTP-bound GTPase as an affinity ligand. The putative Rho effectors identified in this way to date include two families of Ser/Thr kinases, p160ROCK (p164 rho-kinase) and protein kinase N (PKN) a protein kinase C–related kinase, and several structural proteins with no obvious catalytic activity (Leung et al., 1995; Amano et al., 1996; Ishizaki et al., 1996; Matsui et al., 1996; Wanatabe et al., 1996). One of these Rho targets is the myosin-binding subunit of myosin light chain phosphatase, while myosin light chain (MLC) itself is a good substrate for p160ROCK (Kimura et al., 1996; Matsui et al., 1996). This suggests that increased phosphorylation of MLC could be an important component of the Rho-dependent assembly of actin stress fibers and focal adhesions; and recently, MLC kinase inhibitors have been used to provide evidence in support of this idea (Chrzanowska-Wodnicka and Burridge, 1996).
A growing list of targets for Rac has also emerged and these include the Ser/Thr kinases p65PAK, mixed-lineage kinase (MLK), p70S6kinase, and p160ROCK, and several structural proteins (Diekmann et al., 1994; Manser et al., 1994; Burbelo et al., 1995; Brill et al., 1996; Chou and Blenis, 1996; Hart et al., 1996; Lamarche et al., 1996; van Aelst et al., 1996). Some progress in analyzing the downstream roles of these putative targets has been achieved by introducing amino acid substitutions into the effector region of Rac that interfere selectively with target interactions (Lamarche et al., 1996). Substitutions at codon 40 in Rac, for example, prevent binding to p65PAK and MLK, but do not interfere with the induction of lamellipodia, while substitutions at codon 37, which prevent binding to p160ROCK, block lamellipodium formation.
Finally, there have been several reports that Rho and Rac control the activity of phosphatidylinositol 4-phosphate (PIP) 5-kinase thereby regulating the synthesis of phosphatidylinositol-4,5-biphosphate (PIP2) (Chong et al., 1994; Hartwig et al., 1995; Tolias et al., 1995). It is unclear whether PIP 5-kinase interacts with the GTPases directly or through an adaptor protein; however, PIP2 is known to interact with a number of actin binding proteins and has been suggested to be a key regulator of actin polymerization (Janmey and Stossel, 1989; Stossel, 1993), and it is likely, therefore, that PIP 5-kinase is an important target of Rho and Rac in mediating cytoskeletal reorganizations. A recent report suggests that PIP2 also plays an essential role in focal adhesion assembly by regulating the interaction of vinculin with talin and actin (Gilmore and Burridge, 1996).
To identify components of the Rho and Rac signal transduction pathways, we have developed a biological assay that reconstitutes the assembly of stress fibers, lamellipodia, and focal complexes in permeabilized cells. Permeabilization has been used by others to study actin filament assembly. In platelets, for example, Hartwig et al. (1995) demonstrated that thrombin triggers actin polymerization through the Rac-dependent generation of PIP2 and the consequent release of capping proteins, such as gelsolin, from actin filaments (Hartwig et al., 1995). Reorganization of actin filaments has also been observed in permeabilized mast cells and shown to be dependent on a trimeric G protein as well as on Rac and Rho, though the physiological consequences of these changes are not clear in this cell type (Norman et al., 1994). Li et al. (1995) have developed an assay for actin nucleation using permeabilized yeast cells and have shown its dependence on a functional CDC42 gene. Finally, permeabilization has been used to look at the disassembly of focal adhesions in chick fibroblasts in response to high levels of ATP; interestingly, this was blocked by peptides that inhibit actin–myosin interaction (Crowley and Horwitz, 1995).
We show here that stress fibers and focal adhesions, as well as peripheral actin structures and their associated focal complexes, are induced in quiescent Swiss 3T3 permeabilized in the presence of guanosine 5′-O-(3-thiophosphate) (GTPγS), via activation of endogenous Rho and Rac. If cells are incubated in permeabilization buffer for 6 min before nucleotide addition, the response is lost, but it can be reconstituted using concentrated cytosolic extracts. The purified bioactivity from cytosol has been identified as moesin by protein sequencing.
Materials and Methods
Cell Culture
Swiss 3T3 cells were cultured in DME with antibiotics (GIBCO BRL, Gaithersburg, MD), supplemented with 10% FCS (Sigma Chemical Co., St. Louis, MO). Cells were seeded onto 13-mm-diam round glass coverslips (Chance Propper, West Midlands, UK) at a density of 5 × 10 cells per ml and grown to quiescence over 5–8 d, and before use were serum starved overnight in DME containing 2 g/liter NaHCO3.
Permeabilization
Permeabilization buffer (DK) comprised 150 mM potassium glutamate, 10 mM Hepes/KOH, 5 mM glucose, 2 mM MgCl2, 0.4 mM EGTA, pH 7.6; this was stored as a 2× stock at −20°C. In addition, the complete buffer contained 1 mM ATP, UTP, 5 mM creatine phosphate, 10 μg/ml creatine phosphokinase, 100 μM DTT, GTP, and a protease inhibitor cocktail of 10 μg/ml chymostatin, leupeptin, aprotinin, antipain, pepstatin, and 1 mM benzamidine hydrochloride.
Each coverslip was rinsed twice in PBS and once in DK buffer, and then inverted on a 50-μl droplet of complete DK containing an additional 0.003% digitonin and a stimulus as stated (protocol 1). Alternatively (protocol 2), cells were permeabilized for 6 min at room temperature on a 60-μl droplet of DK buffer containing 0.003% digitonin, and then transferred to a 40-μl droplet of complete DK (lacking digitonin), containing a stimulus as indicated. Coverslips were incubated under humidified conditions at 37°C for 20 min and then fixed for immunofluorescence.
Immunofluorescence and Microscopy
Immunofluorescence procedures are substantially as described (Nobes and Hall, 1995). All solutions were prepared in PBS, cells were thoroughly rinsed in PBS between stages, and incubations were performed at room temperature. Cells were fixed for 10 min in 3% paraformaldehyde, and then free aldehyde groups were quenched for 10 min in 1 mg/ml sodium borohydride. The primary antibody, 50 μl of 1:200 dilution monoclonal mouse anti-vinculin (Sigma Chemical Co.), was incubated with the cells for 40 min; only in the case of unpermeabilized controls was it necessary to include 0.05% Triton X-100 as a permeabilizing agent. Two secondary antibodies were used sequentially: goat anti–mouse, and donkey anti–goat (both from Pierce, Rockford, IL, used at 1:200 dilution). The final antibody treatment also contained rhodamine-conjugated phalloidin and Hoechst dye 33342 (both at 0.1 μg/ml; Sigma Chemical Co.). Finally the coverslips were rinsed in PBS and water and mounted on slides by inversion over 5 μl Moviol mountant containing p-phenylinediamine (1 mg/ml; Sigma Chemical Co.) as an antibleaching agent. Cells were examined using a Zeiss Axiophot microscope using Zeiss 63 × 1.4 and 100 × 1.3 oil immersion objectives (Carl Zeiss, Inc., Thorwood, NY), and photographed on Kodak T-MAX 400ASA film (Eastman Kodak Co., Rochester, NY).
Preparation and Assay of Porcine Brain Extract
All procedures were carried out at 4°C. Pig brains were washed in PBS, and then homogenized in a blender in 1.5 vol of DK supplemented with 100 μM DTT, 10 μM ATP, 5 μM GTP, and 1 mM PMSF, Nα-p-Tosyl- l-lysine chloromethyl ketone (TPCK), and N-Tosyl-l-phenylalanine chloromethyl ketone (TLCK). The homogenate was ultracentrifuged for 30 min at 200,000 g in Beckman Ti50 rotors (Fullerton, CA), and the supernatant filtered through Whatman No. 1 paper (Lexington, MA). Typically the volume of supernatant obtained at this stage was equivalent to that of the starting material.
The specific activity was determined in a semi-quantitative immunofluorescence assay, by calculating the amount of protein required to generate cytoskeletal reorganization in 10–30% of permeabilized cells. For each coverslip, several fields of cells were examined at random. First, cells were counted by counting Hoechst-stained nuclei, then the cells were examined under the rhodamine and fluorescein channels, and a second count was made of cells showing focal adhesions and actin filament assembly. Fields at the edge of the coverslip were not examined, nor were fields showing evidence of damage, or fields where cells did not grow in an even monolayer. Background activity was subtracted from cells treated with stimulus or protein alone (2–5% of response in presence of stimulus + protein).
Since this was a labor-intensive assay, duplicates were rarely performed, but assays were performed three times or more; while absolute specific activities might vary within a tenfold range between assays, the relative specific activities of fractions varied within twofold.
Purification of Focal Adhesion Activity from Porcine Brain
Chromatography was carried out on a Biologic integrated chromatography system (Bio Rad Laboratories, Hercules, CA); in all cases the buffer was DKx, where x indicates the millimolar concentration of potassium glutamate. The data shown in Fig. 5 and Table I derive from a single representative purification, which was carried out without pause from homogenization of fresh brain to the assay of the second Q-Sepharose eluate, in 72 h. Activities were stable when snap frozen in small aliquots in liquid nitrogen and stored at −80°C, and also to limited freeze thawing. Gel-filtration chromatography and some biological assays were carried out on such frozen material.
Figure 5.

Purification of the active component from pig brain cytosol. (A) Cibachrome blue 3GA: a step-eluted fraction from fast-flow Q-Sepharose, diluted to 50 mM salt, was passed over a 40 ml Cibachrome blue 3GA column, washed extensively, and eluted as shown in a continuous 25-300 μS salt gradient. Protein was monitored in line by absorbence at 280 nm, and salt concentration by conductivity, as shown. The activities of column fractions were assayed and plotted (far left ordinate axis) and the two activity peaks were pooled. (B) Phenyl–Sepharose: pooled fractions from A were applied to a 15-ml phenyl–Sepharose column and proteins eluted with a 300–25 μS salt gradient. (C) Q-Sepharose: pooled active fractions from B were diluted to 20 mM salt and chromatographed on a 5-ml Q-Sepharose column. Activity was eluted in a salt gradient to 200 μS. (D) 200 μl of the peak fraction from C was applied to a Bio-Silect gel filtration column with a rated separation range of 100–5 kD. The elution positions of molecular weight standards are indicated.
Table I.
Purification of Bioactivity from Pig Brain
| Fraction | Volume | Protein | Protein | Spec. Act. | Activity | Purification | Yield | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ml | mg/ml | mg | U/mg | U | fold | % | ||||||||
| Pig brain 100k supernatant | 3,000 | 6.4 | 19,000 | 0.8 | 15,400 | 1 | 100 | |||||||
| Q-Sepharose step eluate | 150 | 10 | 1,500 | 35 | 52,500 | 44 (44) | 340 (100) | |||||||
| Blue 3GA–Sepharose | 25 | 3.8 | 95 | 200 | 19,000 | 250 (6) | 125 (36) | |||||||
| Phenyl–Sepharose | 7 | 1.3 | 9 | 1,200 | 11,000 | 1,500 (6) | 73 (21) | |||||||
| Q-Sepharose | 3 | 0.15 | 0.45 | 8,000 | 3,600 | 10,000 (6.7) | 23 (7) |
3 liters of high speed supernatant derived from pig brain homogenate was fractionated by successive passage over Q-Sepharose, Blue 3GA–agarose, phenyl–sepharose, and Q-Sepharose. In each case, column fractions were assayed for competence to restore a GTPγS-dependent biological response in permeabilized cells. The peaks of activity were pooled; the specific activities shown in this table are calculated as described in Materials and Methods and representative of at least three determinations. Parenthetical figures for purification and yield show change in those figures from the previous stage of purification. The fractions obtained after gel filtration were too dilute for accurate protein assay; therefore, since specific activities could not be determined, relative activities were assessed per volume of protein added, and these activities are listed separately in Fig. 6.
Pig brain extract was passed over a 150-ml column of Q-Sepharose Fast-Flow (Pharmacia LKB Biotechnology Inc., Piscataway, NJ). The column was washed extensively in DK10, and proteins were eluted in DK200. Pooled material was diluted to DK40 and loaded onto 40 ml of Cibachrome blue 3GA–Sepharose (Sigma Chemical Co.). Protein was eluted in a gradient of DK40–600 and two peaks of activity were detected at 250 and 410 mM. These were pooled, adjusted to 600 mM K-glutamate and passed over a 15-ml phenyl–Sepharose column; the activity was eluted at 100 mM salt in a gradient of DK600–10. The peak of activity was diluted to 20 mM, applied to a 5-ml Q-Sepharose column, and then eluted with a gradient of DK10–200. Activity was detected between 70 and 110 mM.
For gel filtration, a 200-μl aliquot of the activity peak from the final Q-Sepharose column was applied to a 6-ml Bio-Silect chromatography column (Bio Rad Laboratories: rated fractionation range 100–5 kD) and protein was eluted in DK150, sampling 0.25-ml fractions. It should be noted that the protein concentrations of these gel-filtration fractions were not assessed; therefore, the activities of the fractions (unlike all other fractions and preparations in this study) were assessed in arbitrary units, based on the volume, not the absolute quantity, of protein added.
Protein Microsequencing
The peak fraction from the final Q-Sepharose column was electrophoresed on 10% SDS-PAGE gel and the proteins transferred to PVDF (Bio Rad Laboratories) by wet-blotting in 5 mM 3-cyclohexylamino-1-propanesulfonic acid (CAPS) (Aldrich, St. Louis, MO), pH 11, 1% MeOH, 0.01% SDS at 0.75Amperes for 1.5 h. The blot was washed three times for 10 min in deionized water, and then stained with 0.1% Amido black/10% HOAc for 1 min, and destained with distilled water until the bands were clearly visible. Bands were excised, transferred to sequencing cartridges, and submitted to 20 rounds of NH2-terminal sequencing on a protein sequencer (GP1000A; Hewlett-Packard Co., Palo Alto, CA), using Routine 3.0 chemistry.
Western Blotting
The samples for Western blotting were derived from cells permeabilized under equivalent conditions to those prepared for immunofluorescence assay, except that after permeabilization the supernatant was recovered and the cell residue on the coverslip was scraped into sample buffer. (For Fig. 9, d and e, supernatant and pellet fractions were combined.) Typically, samples for electrophoresis were equivalent to 0.25–0.5 permeabilizations. Proteins were electrophoresed on 10% or 12% SDS-PAGE gels and transferred to PVDF membranes (Millipore Corp., Bedford, MA). The membranes were blocked extensively in blocking buffer (1 M glycine, 5% milk powder, 1% ovalbumin, and 5% FCS), and then immunoblotted using monoclonal antibodies against vinculin (Sigma Chemical Co.) at 1:200 dilution, and polyclonal anti-ezrin/radixin/moesin (ERM) (1:500 dilution). HRP-conjugated secondary antibodies (Pierce) was used along with Amersham ECL system (Arlington Heights, IL) for detection of immunoreactivity.
Figure 9.

F-actin binding to moesin. (a and b) F-actin binding to recombinant and purified pig brain moesin: lane 1, 35 ng recombinant moesin; lane 2, 125 ng of fraction 22 from Q-Sepharose column, estimated to contain 30 ng moesin. (A) immunoreactivity with monoclonal anti-moesin antibody; (B) F-actin binding using [32P]ATP labeled F-actin nitrocellulose overlay assay. (C) Loss of moesin from permeabilized cells. Coverslips were incubated under the following conditions: lanes 1 and 2, mock-permeabilized for 6 min in 60 μl DK without digitonin; lanes 3 and 4, permeabilized for 6 min in 60 μl DK containing 0.003% digitonin. Cell supernatants (lanes 1 and 3) and residues (lanes 2 and 4) were prepared and proteins electrophoresed on 10% SDS-PAGE gels, transferred to PVDF, and blotted with a polyclonal antibody against ERM proteins. (D and E) F-actin binding of moesin in Swiss 3T3 cells. Lane 1, 75 ng recombinant moesin; lane 2, total SDS lysate; lane 3, total SDS lysate from cells that had been first incubated for 6 min in 60 μl DK with 0.003% digitonin. Samples equivalent to half coverslips of cells were electrophoresed on 10% SDS-PAGE gels, transferred to PVDF membranes, and then probed with (D) polyclonal anti-ERM antibody (E) [32P]ATP-labeled F-actin.
Recombinant Proteins
Recombinant, constitutively activated RhoA (V14 or L63 mutants), C3 transferase, constitutively active (L61) and dominant negative (N17) Rac1, RhoGAP, and human moesin were expressed as glutathione- S-transferase (GST) fusion proteins in Escherichia coli and purified on glutathione–Sepharose beads as described by Self and Hall (1995). Cleaved proteins were obtained from the beads by addition of human thrombin (Sigma Chemical Co.; 5 U/liter of culture overnight, with an additional 5 U for 30 min before protein recovery). Contaminating thrombin was removed by incubating proteins with 10 μl p-aminobenzamidine beads (Sigma Chemical Co.) for 30 min at 4°C. GST fusion proteins were eluted from glutathione–Sepharose by addition of 5 mM GST. Recovered protein was dialyzed into buffer A (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.1 mM DTT), and then concentrated by ultrafiltration (Centricon 10; Amicon Corp., Danvers, MA). Protein concentration was assayed by Bradford assay (Bio Rad Laboratories) and purity assessed on SDS-PAGE; the activity of p21 preparations was measured as the binding of [3H]GTP in the filter-binding assay as described by Hall and Self (1986).
Lactate Dehydrogenase
Lactate dehydrogenase activity was assayed by a method based on the work of Kornberg (1955). Lysate samples equivalent to one coverslip of cells were added to an assay mixture comprising 330 μM sodium pyruvate, 10 mM Hepes/KOH, pH 7.2, 66 μM NADH, and 0.2% Triton X-100, and the decrease in absorbence at 340 nm was monitored over 1–3 min.
Rho Assay
Rho was assayed by C3-mediated ribosylation using 32P-NAD in a method based on that of Aktories and Just (1995). Samples were derived from cells permeabilized in 60-μl vol; supernatants were then removed, and cell residue harvested by briefly washing the coverslip and then scraping off residue into 60 μl 0.2% Triton X-100 in DK. 10-μl aliquots of this material were incubated for 1 h at 37°C with 4 μCi 32P-NAD and 40 ng C3 transferase. The labeled proteins were boiled in SDS-PAGE sample buffer, and 12% SDS-PAGE gels of labeled proteins were autoradiographed overnight.
Immunodepletion
Immunodepletion was carried out using polyclonal anti-moesin antiserum, a kind gift of Dr. Paul Mangeat (Université Montpelier, Montpelier, France); control antisera were rabbit anti-RhoGDI or rabbit anti–rat (Sigma Chemical Co.). Antisera were coupled to protein A–Sepharose, and the beads repeatedly washed in DK. MonoQ eluate peak activity (10-μl aliquots) was subjected to two rounds of immunodepletion on the coupled beads, by incubation for 40 min at 4°C; afterwards, the collected immunodepletion supernatants were assayed for bioactivity as described above (permeabilization protocol II). By Western blotting (not shown), the supernatant of anti-moesin antibody contained no moesin, whereas moesin was present in the supernatant of mock depletion.
Dot Blotting
Dot blots were performed essentially as described by Lamarche et al. (1996). GTPases (0.1 μg) were loaded with [γ32P]GTP by incubation in 50 mM Tris, pH 7.5, 0.5% BSA, 5 mM EDTA, and 10 μCi [γ32P]GTP for 10 min at 30°C, and then nucleotide exchange was halted by addition of MgCl2 to 10 mM. Nitrocellulose was spotted with 5-μg aliquots of GST, GST-RhoGAP, and GST-moesin in a volume of 10 μl. The nitrocellulose was dried, blocked for 1 h in Western blocking buffer, and then incubated for 5 min at 4°C with agitation in 3 ml buffer A containing 5 μM unlabeled GTP, 0.5% BSA, and the labeled GTPase. Unbound protein was removed with three brief washes in ice-cold buffer A/0.1% Tween-20, and then the nitrocellulose was dried and autoradiographed for 120 min at −80°C with intensifier screens.
F-actin Overlay
66 μM G-actin purified from rabbit muscle was a gift of Dr. Laura Machesky (MRC LMCB, University College of London, UK). G-actin was gel filtered into G buffer (2 mM Tris.HCl, pH 8.0, 50 μM CaCl2, 0.5 mM DTT, 50 μM NaN3) to reduce the concentration of free ATP, and then incubated for 120 min at room temperature with 125 μCi [α32P]ATP (800 Ci/mmol). Actin polymerization was initiated by addition of 0.1 vol of 0.5 M KCl, 20 mM MgCl2, 0.5 mM CaCl2 (10× F buffer), and continued for 20 min at room temperature, after which the F-actin was sedimented at 100,000 g for 30 min. An estimated 60% of [32P]ATP was incorporated into the F-actin pellet. The F-actin was resuspended at ∼20 μg/ml in F buffer containing 5 μM phalloidin and 1 mM DTT. For blot overlays, the protein was resuspended at 20 μg/ml in Western blocking buffer containing 5 μM phalloidin and 1 mM DTT, and incubated with preblocked Western blots for 2 h at room temperature. The blots were washed in TBS-T (150 mM NaCl, 10 mM Tris/HCl, pH 8.0, 0.2% Tween-20) four times for 5 min at room temperature, and then exposed to film at −80°C for 2–24 h.
Results
Focal Complex and Actin Filament Assembly in Permeabilized Swiss 3T3 Cells
Stress fibers and lamellipodia can be induced in quiescent, confluent serum-starved Swiss 3T3 cells by addition of extracellular agonists or by microinjection of recombinant Rho and Rac proteins, respectively. In an attempt to reconstruct these effects in vitro, we permeabilized serum-starved Swiss 3T3 cells grown on glass coverslips by exposure to isotonic buffer containing an ATP regenerating system and a low concentration (0.003%) of the nonionic detergent digitonin. After 20 min at 37°C, the permeabilized cells were fixed and F-actin visualized by immunofluorescence. Like intact serum-starved cells, permeabilized cells lack actin filament organization (Fig. 1 A, left) and focal adhesion complexes. However, permeabilization in the presence of the nonhydrolyzable GTP analogue, GTPγS, provoked cytoskeletal reorganization in up to 75% of cells, which could be detected at low magnification as an increase in F-actin staining and sharpening of cell outline owing to actin-mediated contraction of the cells (Fig. 1 A, right). Permeabilization caused no loss of cells from the substratum. Cytoskeletal reorganization was observed only within a narrow concentration range of digitonin (0.0025– 0.004%) and could not be achieved in the absence of an ATP regenerating system, or at room temperature (20°C).
Figure 1.

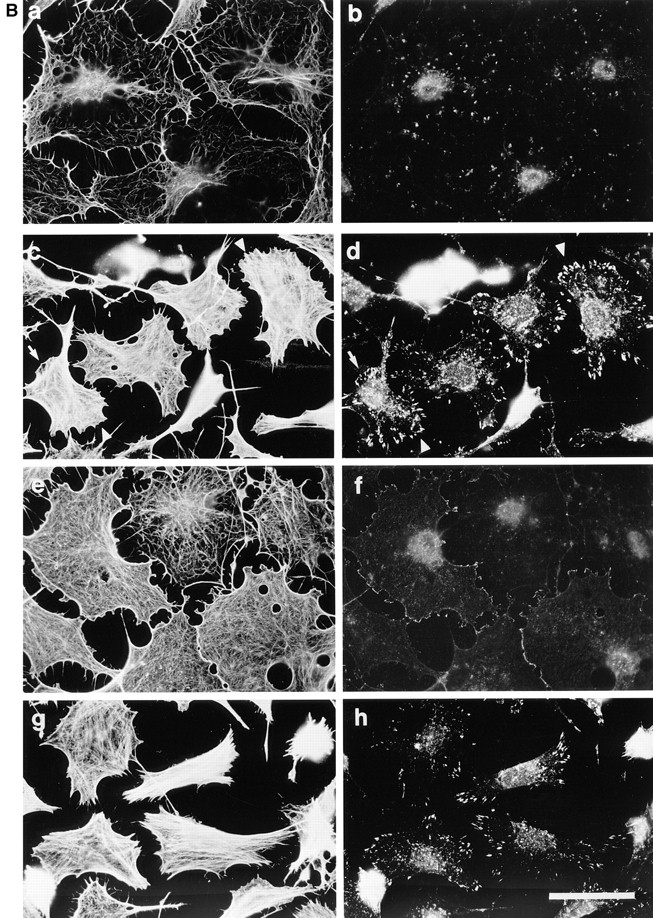
Permeabilization of quiescent Swiss 3T3 cells in the presence of GTPγS. (A) Permeabilization (protocol 1) was performed in the absence of stimulus (left) or in the presence of 50 μM GTPγS (right). Cellular F-actin was visualized using rhodamine-conjugated phalloidin. (B) Permeabilization (protocol 1) was performed in the absence of stimulus (a and b), in the presence of 50 μM GTPγS (c and d); 50 μM GTPγS with 0.1 nM C3 transferase (e and f); 50 μM GTPγS with 1 nM N17Rac (g and h). After 20 min at 37°C, cells were fixed and F-actin visualized with rhodamine-phalloidin (a, c, e, and g) and vinculin visualized with a monoclonal antibody (b, d, f, and h). In c and d, arrowheads show termini of bundled actin filaments decorated with focal adhesions, while arrows mark regions of peripheral actin polymerization decorated with linear arrays of focal complexes. Bars: (A) 150 μm; (B) 30 μm.
To visualize these effects more clearly, permeabilized cells were immunofluorescently stained for both F-actin and the focal adhesion protein vinculin, and examined at higher magnification. This showed that GTPγS treatment provoked a complex and dramatic reorganization of the actin cytoskeleton (compare Fig. 1 B, c and d of GTPγS-treated cells with untreated cells shown in Fig. 1 B, a and b). The cells displayed at least two types of actin structure: first, thick, bundled filaments that traversed the cell body, similar to actin stress fibers (Fig. 1 B, c, arrowhead); and second, dense filamentous actin localized at the cell cortex, similar to that observed in the lamellipodia of intact cells (Fig. 1 B, c, small arrow). These two types of structures were frequently found together in single cells, giving rise to the complex cytoskeletal formations revealed by F-actin staining. Moreover, cells with stress fiber–like filaments also contained typical focal adhesion complexes (Fig. 1 B, d, arrowhead), while cells showing cortical actin polymerization were decorated with peripheral, punctate focal complexes (Fig. 1 B, d, small arrow). The effects of GTPγS on the actin cytoskeleton are consistent with the activation of endogenous Rho and Rac GTPases in the permeabilized cells.
To confirm this suggestion and to dissect the morphology of the GTPγS-treated cells, we used C3 transferase and N17Rac, specific inhibitors of Rho and Rac function, respectively (Paterson et al., 1990; Ridley et al., 1992). Cells were permeabilized in the presence of GTPγS and either inhibitor. C3 transferase totally blocked the assembly of stress fibers and focal adhesions and most cells now showed clear cortical actin (Fig. 1 B, e) and peripheral focal complexes (Fig. 1 B, f). N17Rac, on the other hand, blocked cortical actin polymerization, but stress fibers (Fig. 1 B, g) and focal adhesions (Fig. 1 B, h) were unaffected. We therefore conclude that GTPγS activates endogenous Rho and Rac proteins in permeabilized fibroblasts.
We then tried to stimulate the changes in actin filament organization using recombinant, constitutively activated Rho and Rac proteins instead of GTPγS. Cells permeabilized in the presence of Rho (25 μg/ml) and incubated for 20 min showed dense actin bundles traversing the cell (Fig. 2 a); the bundles were decorated at their termini with large focal adhesions (Fig. 2 b). These effects were abrogated by inclusion of 2 ng/ml C3 transferase in the assay buffer (data not shown). We were unable to induce lamellipodial actin and peripheral integrin complexes using recombinant Rac protein.
Figure 2.
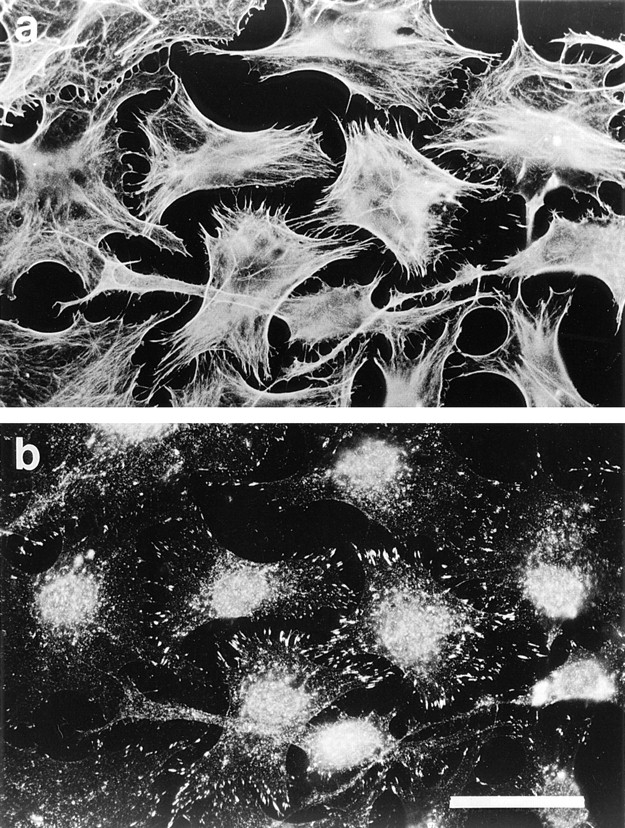
Recombinant Rho stimulates formation of focal adhesions and stress fibers in permeabilized cells. Cells were permeabilized (protocol 1) in the presence of 1 μM recombinant V14Rho. (a) Phalloidin staining for F-actin; (b) monoclonal antibody against vinculin. Bar, 30 μm.
Reconstitution of Actin Filament and Integrin Complex Assembly Using Cytosolic Extracts
Cells left in permeabilization buffer for 6 min at room temperature, before addition of either GTPγS or recombinant Rho (25 μg/ml, V14Rho), were no longer competent to assemble actin filaments or integrin complexes. (Fig. 4, a and b). It appeared, therefore, that during this incubation period one or more limiting components are either lost from cells or inactivated. Pellets and supernatants were prepared from permeabilized cells and assayed for lactate dehydrogenase (LDH; a marker for bulk cytosol), Rho and focal adhesion proteins. Although 65% of the LDH was lost from the cells during the 6-min incubation with digitonin (Fig. 3 a), ∼80% of Rho (Fig. 3 c) and essentially all cellular vinculin (Fig. 3 b), paxillin (not shown), and RhoGDI (not shown) were retained. It seemed possible, therefore, that the cells retained sufficient cytoskeletal components for actin filament and integrin complex assembly, but that a cytosolic component of the Rho and Rac signal transduction pathways had been inactivated or lost after permeabilization.
Figure 4.
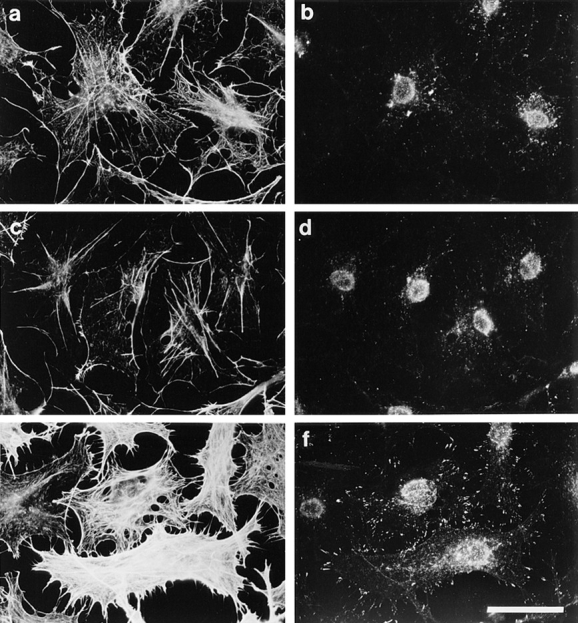
Reconstitution of Rho-induced effects after extended permeabilization using a pig brain cytosolic extract. Cells were permeabilized according to protocol 2 (i.e., permeabilized for 6 min in the presence of digitonin before addition of stimulus; see Materials and Methods) in the presence of (a and b) 25 μg/ml V14Rho, (c and d) 2 mg/ml pig brain extract, (e and f) 25 μg/ml V14Rho plus 2 mg/ml pig brain extract. F-actin in the permeabilized cells was visualized using rhodamine-conjugated phalloidin (a, c, and e) and focal adhesions with anti-vinculin antiserum (b, d, and f). Bar, 30 μm.
Figure 3.


Retention of cytosolic and cytoskeletal markers after permeabilization. (a) LDH activity, (b) vinculin visualized by Western blot, and (c) Rho visualized after ADP ribosylation with C3 transferase and 32P-NAD. (a) Coverslips were incubated for 20 min at 37°C in 50 μl vol ± 0.003% digitonin, or for 6 min at room temperature in 60 μl vol containing either 0.003% digitonin or 0.2% Triton X-100. Samples were assayed for LDH in the presence of 0.2% Triton X-100. Data are mean ± SD from four independent determinations. (b) Coverslips were incubated for 6 min at room temperature in 60 μl buffer containing no detergent, 0.003% digitonin or 0.2% (vol/vol) Triton X-100, and supernatants and pellets were recovered. Proteins were electrophoresed on 12% SDS-PAGE and transferred to PVDF, and vinculin was visualized using a monoclonal anti-vinculin antibody. (c) Cell supernatants and pellets were prepared as in b, and endogenous Rho determined as described in Materials and Methods. Positions of molecular weight standards (kD) are marked.
In an attempt to reconstitute the effects of Rho, a high speed, cytosolic supernatant was prepared from a pig brain homogenate. Swiss 3T3 cells were permeabilized at room temperature for 6 min and transferred to a fresh drop (40 μl) of buffer containing either Rho, concentrated cytosol extract (1 mg/ml), or a combination of the two, and then incubated at 37°C for another 20 min. As shown in Fig. 4, incubation with Rho plus cytosol caused cytoskeletal reorganization in up to 50% of cells (Fig. 4, e and f), while cytosol alone (Fig. 4, c and d) or Rho alone (Fig. 4, a and b) produced no effect. Similarly, pig brain extract reconstituted the morphological effects of GTPγS on permeabilized cells (data not shown). Concentrated cytosol from growing Swiss 3T3 or Rat 1 cells, mouse brain or pig liver could also reconstitute activity with GTPγS and Rho. It therefore appeared that the concentrated extracts contained a factor or factors capable of reconstituting cytoskeleton reorganization in response to Rho or Rac activation.
Purification of the Activity from Pig Brain Cytosol
To purify an active component from pig brain cytosol the permeabilization protocol was made semiquantitative. Permeabilized cells were stimulated in the presence of protein fractions, and then examined by immunofluorescence for both F-actin and vinculin. Several fields were assessed on each coverslip, and the proportion of “responding” cells was calculated; responding cells were defined operationally as those containing substantial bundled actin filaments and abundant focal adhesions. A maximum of ∼30% of cells was capable of responding to optimal stimuli, and the quantity of protein required to provoke response in 10% of the cells was defined as 0.1 units of activity. For assays during purification, GTPγS was used as a stimulus.
Pig brain extract (20 g) was first passed over a Q-Sepharose column, and the activity was eluted using a 200 mM salt step (Table I). This fraction was diluted fivefold and loaded on to a Cibachrome blue–Sepharose column and proteins eluted using a gradient of increasing ionic strength (Fig. 5 a and Table I). Two peaks of activity were obtained; these were pooled and applied to a phenyl–Sepharose column and proteins eluted using a gradient of decreasing ionic strength (Fig. 5 b and Table I). Finally, the active fractions were applied to a Q-Sepharose column and the activity eluted using a salt gradient. The specific activity of the cytosolic component increased ∼10,000-fold during the purification procedure (Table I and Fig. 5 c).
200 μl of the Q-Sepharose fraction was analyzed by gel-filtration chromatography (Figs. 5 d and 6 a) and the peak of biological activity eluted in fraction 31, although the profile of activity was broad. A prominent protein in fraction 31, whose abundance broadly correlated with biological activity across the gel-filtration profile, migrated on PAGE at just over 66 kD. To identify this protein, 22 μg of the final Q-Sepharose fraction was subjected to electrophoresis on 10% polyacrylamide gels and proteins transferred to PVDF membranes. The six major bands (marked A–F on Fig. 6 b) were excised and subjected to NH2-terminal protein microsequencing. The majority of these bands gave no peptide sequence and were presumably NH2-terminally blocked. However, two major bands around 66 kD (bands B and C) were identified as porcine moesin (Lankes et al., 1993).
Figure 6.

Moesin copurifies with GTPγS-dependent biological activity in permeabilized cells. (A) Silver-stained 10% SDS-PAGE using 10-μl aliquots from fractions 28–35 of the gel-filtration column shown in Fig. 5 D. Relative activities of the gel-filtered fractions are noted below the corresponding lanes of the blot. (B) 22 μg of fraction 22, the most active from the Q-Sepharose column shown in Fig. 5 c, was electrophoresed on 10% SDS-PAGE and electroblotted onto PVDF, and the major protein bands A–F were excised and microsequenced. The sequences obtained are shown. (C) Proteins from a parallel gel to A were transferred to PVDF and Western blotted with a polyclonal anti-ERM antibody.
Moesin Is Required for Rho and Rac Effects on the Actin Cytoskeleton
Antibodies recognizing moesin (polyclonal anti-ERM) were used to probe a Western blot of the fractions obtained after gel-filtration of the final Q-Sepharose active fraction. As seen in Fig. 6 c, moesin immunoreactivity correlated with biological activity, suggesting that this might be the active component purified from pig brain extract.
To test this, recombinant moesin was expressed in E. coli as a GST fusion protein. In digitonin-treated cells, cleaved recombinant moesin or GST–moesin fusion protein (at levels of 200–500 ng/ml), in the presence of GTPγS, induced actin stress fibers and peripheral actin polymerization, both accompanied by their attendant focal complexes (Fig. 7, a and b). No cytoskeletal reorganization was seen in cells stimulated in the presence of boiled moesin, either recombinant or purified, or in those treated with moesin alone (data not shown). In the presence of the Rho inhibitor C3 transferase, only cortical actin and peripheral focal complexes were observed (Fig. 7, c and d) whereas in the presence of N17Rac only stress fibers and focal adhesions were induced (Fig. 7, e and f). Moesin promoted stress fiber formation in the presence of recombinant V14Rho (data not shown).
Figure 7.
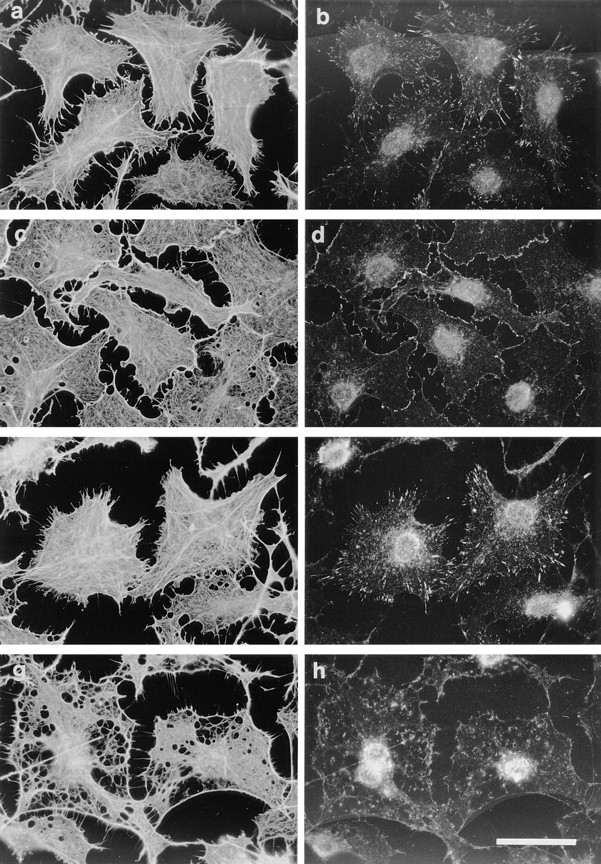
Recombinant moesin reconstitutes GTPγS- induced effects. Cells were permeabilized according to protocol 2, and incubated with 50 nM (saturating) recombinant moesin and 50 μM GTPγS (a–f); additionally, in c and d 0.1 nM C3 was present and in e and f 1 nM N17rac. The cells shown in g and h were incubated with 50 nM truncated moesin and 50 μM GTPγS. (a, c, e, and g) Phalloidin staining for F-actin; (b, d, f, and h) monoclonal antibody against vinculin. Bar, 30 μm.
As a control for the effects of full-length moesin, a construct was expressed which lacked only the COOH-terminal 22 amino acids, which constitute an F-actin binding motif (Pestonjamasp et al., 1995). This construct did not support cytoskeletal rearrangement in treated cells under any stimulus tested, at concentrations up to tenfold higher than the full-length protein (Fig. 7, g and h; and data not shown); indeed, the actin cytoskeletons of such cells were, if anything, slightly disarranged or fragmented. By titration of recombinant full-length moesin and moesin purified from pig brain (amounts judged by Western blot analysis), we estimate that the E. coli–produced protein is around 10-fold less active than the purified moesin in permeabilized cells. It is possible either that the Q-Sepharose fraction contains additional active proteins or that the E. coli–produced protein is slightly contaminated with inhibitory proteolytic fragments, or partially incorrectly folded or modified.
As an additional test of the specific activity of moesin within the semipurified pig brain activity, the purified material was subjected to immunodepletion using either a rabbit anti-moesin antiserum or an irrelevant antiserum. When tested alongside untreated pig brain activity, moesin-immunodepleted material showed an 85% loss of bioactivity, whereas mock-depleted material had suffered only a 33% loss of activity (data not shown).
Moesin belongs to a family of closely related proteins (ERM proteins) that also contains ezrin and radixin. To assess whether these family members possessed the same activity, recombinant ezrin and radixin were expressed in E. coli in the same way as moesin, and tested for re-establishment of GTPγS-dependent cytoskeletal reorganization in permeabilized cells. In parallel experiments, recombinant moesin, ezrin, and radixin exerted equivalent morphological effects at an equivalent range of concentrations (data not shown).
Moesin Does Not Interact Directly with Rho or Rac
The ability of Rho or Rac to interact directly with moesin was assessed using an in vitro bead-binding assay and a dot blot assay. GST–moesin was incubated with [α32P]GTP-bound Rho or Rac for 5 min at 4°C and glutathione– Sepharose beads added for a further 30 min. After recovery of the beads, the amount of Rho or Rac bound to moesin was measured. GST-p160ROCK was used as a positive control since this has been previously shown to bind both Rho and Rac (Lamarche et al., 1996). No significant binding of Rho or Rac to moesin was detected (data not shown). The dot blot–binding assay was also used; GST-moesin and GST-p50rhoGAP (which has also been shown to bind both Rho and Rac; Lancaster et al., 1994) were spotted on to nitrocellulose filters and incubated with [α32P]GTP-bound Rho or Rac. As seen in Fig. 8, no interaction with moesin could be detected.
Figure 8.

Moesin does not interact directly with Rho or Rac. 5 μg aliquots of GST, GST-RhoGAP, and GST-moesin were spotted onto nitrocellulose and the filter blocked with blocking buffer. The filter was then incubated with 0.1 μg of [γ32P]GTP-loaded Rho or Rac and subjected to autoradiography.
F-actin Binding Site of Moesin Is Essential for Activity
The COOH-terminal 22 residues of moesin have been shown to be required for interaction with F-actin (Pestonjamasp et al., 1995). We have compared the ability of recombinant moesin and moesin purified from pig brain cytosol to bind [α32P]ATP-labeled F-actin. As shown in Fig. 9, a and b, both preparations bind F-actin in a nitrocellulose overlay assay. To see if the F-actin binding site is required for GTPγS-stimulated actin filament assembly, a deletion construct of moesin lacking the COOH-terminal 22 residues was used in the permeabilized cell assay. This deletion mutant is unable to bind F-actin (Pestonjamasp et al., 1995) and when added to permeabilized cells along with GTPγS, it could not promote actin filament or integrin complex assembly (Fig. 7, g and h).
Next, we established the fate of endogenous moesin during digitonin permeabilization of quiescent Swiss 3T3 cells. Fig. 9 c shows that little moesin is lost during the 6-min permeabilization procedure (lane 3); most remains cell associated (lane 4). This result seemed puzzling, given that both purified and recombinant moesin were capable of reconstituting activity in these cells. To assess the ability of Swiss 3T3–derived moesin to bind F-actin, we made total SDS lysates from mock- and digitonin-treated cells. As seen in Fig. 9 d, by Western blot analysis the lysates contain equal amounts of moesin (lanes 2 and 3). Fig. 9 e shows that moesin present in quiescent Swiss cells is able to interact with F-actin (lane 2), but after digitonin permeabilization, moesin is no longer able to bind F-actin (lane 3). Quantitation (four experiments) shows a 20-fold reduction in the actin-binding capacity of moesin during a 6-min incubation with digitonin.
Discussion
Our initial challenge was to develop a protocol for permeabilizing serum-starved Swiss 3T3 fibroblasts under sufficiently mild conditions to allow subsequent morphological examination; this was achieved using low concentrations of the detergent digitonin. Permeabilization in the presence of GTPγS induced two clear types of cytoskeletal structures: (a) bundled actin filaments traversing the cells and terminating in focal adhesions; and (b) filamentous actin localized at the cell periphery and associated with small punctate focal complexes. By use of specific inhibitors, we confirmed that these effects were due to activation of endogenous Rho and Rac GTPases respectively (Ridley and Hall, 1992; Nobes and Hall, 1995). Recombinant, constitutively activated Rho was also able to stimulate focal adhesions and stress fibers when included in the permeabilization buffer, but recombinant Rac was unable to induce cortical actin polymerization or formation of integrin complexes. The reason for this difference is not clear, but one possibility is that recombinant Rac is not as efficiently posttranslationally modified in the permeabilized cells as is Rho. It is even possible that posttranslational modification does not occur under these conditions and that for Rho it is not essential. The use of baculovirus-expressed proteins may help to address this issue.
Permeabilized cells rapidly lose their ability to respond to either Rho or GTPγS, and we believed this to be caused by the loss or inactivation of a specific activity (since Rho, like numerous structural components of the cytoskeleton, was not lost under these conditions). Furthermore, the response to either GTPγS or recombinant Rho could be reconstituted using concentrated cytosol prepared from pig brain homogenates. After this observation, we undertook the purification of a factor required for GTPase-dependent cytoskeletal reorganization, not based on direct association between Rho and a putative effector, but on a biological response to GTPase activation. A 10,000-fold purification of the activity in pig brain cytosol led to the identification of moesin as its active component, and this was confirmed using E. coli–derived recombinant moesin. Moesin alone was unable to induce any cytoskeletal changes in permeabilized cells but it could cooperate with Rho to induce stress fibers and focal adhesions, and it could cooperate with Rac to induce actin polymerization at the cell periphery and the assembly of associated integrin complexes. Moesin is therefore a component of both signal transduction pathways.
Moesin is a member of the ERM family of proteins (Sato et al., 1992), which exhibit ∼70–80% amino acid homology. The ERM proteins also possess significant homology in their NH2 termini with talin, the tumor suppressor gene merlin/schwannomin (Rees et al., 1990; Trofatter et al., 1993) and Band 4.1 (Arpin et al., 1994). ERM proteins have been reported to be localized to regions of contact between actin filaments and the plasma membrane, such as microvilli and microspikes, cleavage furrows and sites of cell–cell and cell–substratum attachment (Berryman et al., 1993; Franck et al., 1993; Takeuchi et al., 1994; Amieva and Furthmayr, 1995). Their importance as membrane– cytoskeleton linkers was demonstrated by the severe disruption of cell adhesion and microvillus formation in cells treated with antisense oligonucleotides to ERM proteins (Takeuchi et al., 1994).
ERM proteins possess F-actin binding activity which, for moesin at least, is located within its COOH-terminal 22 amino acids (Pestonjamasp et al., 1995; Turunen et al., 1994). However, the regulatory mechanisms controlling this activity in cells are not yet established. Algrain et al. (1993) expressed truncations of ezrin in CV1 cells and found that the NH2-terminal portion was predominantly membrane-associated, whereas the COOH-terminal portion colocalized with actin structures (Algrain et al., 1993). In Sf9 cells, expression of the COOH-terminal portion of ezrin, but not the full-length protein, leads to the formation of long actin-rich, filopodium-like cell processes, which is reportedly inhibited by coexpression of NH2-terminal sequences (Martin et al., 1995). It is likely, therefore, that the COOH-terminal F-actin binding region is masked by NH2-terminal sequences. In addition, the F-actin binding region of all ERM proteins contains a conserved threonine (T558) and this has been shown to be phosphorylated, in moesin, during platelet activation (Nakamura et al., 1995).
More recently, further links have been established between the cytoskeleton and ERM proteins, particularly moesin, by Hirao et al. (1996). First, it was shown that PIP2 stabilized a high affinity interaction between moesin's NH2-terminal portion and CD44, via PIP2 binding to the same NH2-terminal portion of moesin; second, RhoGDI was coimmunoprecipitated with moesin from cell lysates; and third, the association between moesin and an insoluble fraction of BHK cells is modulated in vitro and in vivo by regulators of Rho family GTPases. The authors proposed that Rho activation leads to synthesis of PIP2, which then associates with moesin and alters its conformation so as to promote membrane–cytoskeleton interaction. Since phosphatidylinositol 4-phosphate kinases are among known effectors of Rho and Rac, this model provides a mechanism whereby GTPase activation may provoke local effects at cell–cell and cell–substratum adhesions, where moesin has been shown to reside. In addition, a potential link between moesin and Rho GTPases was established in the shape of RhoGDI (Ueda et al., 1990). This protein is supposed to sequester guanosinediphosphate (GDP)- bound (inactive) GTPases in the cytosol, but if the association between the Rho–RhoGDI complex and moesin is regulatable, then RhoGDI may assist the movement of the GTPase to its site of action. It should be noted, however, that GDI was not found by Western blotting in our purified bioactivity from porcine brain (data not shown).
It is clear from the results reported here that both purified porcine moesin and recombinant moesin promote GTPase-dependent cytoskeletal reorganization in permeabilized cells and both can bind F-actin. Recombinant moesin, ezrin, and radixin preparations have equivalent effects on cells, at least in this system, which presumably reflects their high homology. It is not clear whether ERM proteins are functionally redundant in vivo, though their slightly distinct localizations (Berryman et al., 1993; Franck et al., 1993) suggest that there is some specialization in their activities.
Curiously, although quiescent Swiss 3T3 cells contain moesin capable of binding F-actin, during the 6-min permeabilization this activity disappears. The reasons for this are unclear at present. One possibility is the loss of PIP2. Hirao et al. (1996) showed that the affinity of moesin for CD44 is modulated by PIP2; however, they found constitutively high affinity association between CD44 and an NH2-terminal moesin construct. Similarly, the effects of isolated NH2- and COOH-terminal ezrin constructs on cells were regulated by coexpression of the reciprocal portions of the protein (Martin et al., 1995). It is reasonable to speculate that PIP2 binding either provokes or accompanies a gross conformational change, or unclasping, in moesin, which renders the protein competent to bind both CD44 and actin. A precedent for a PIP2-regulated conformational change exists in the focal adhesion protein vinculin. Digitonin, a cholesterol-chelating detergent (Elias et al., 1978), recently has been shown to specifically extract moesin from BHK cell membranes along with a small number of other cytoskeletal proteins (Harder et al., 1997). In permeabilized cells, digitonin may rapidly extract PIP2 directly from moesin, or disrupt phospholipid turnover, thereby preventing the PIP2-dependent interaction of moesin with its normal ligands.
Another potential inactivation mechanism is an alteration in the phosphorylation of moesin on threonine 558, which in platelets is phosphorylated and dephosphorylated within seconds of stimulation; inhibitor studies suggest that these changes are additionally regulated by tyrosine kinases/phosphatases. The effects of Rho on the actin cytoskeleton are modulated by both tyrosine and serine/threonine kinase inhibitors (Ridley and Hall, 1994; Nobes and Hall, 1995), and kinases are among the putative Rho effectors (Kimura et al., 1996; Wanatabe et al., 1996); moesin may be a direct or indirect target of one of these kinases.
However, if either lipid dissociation or protein dephosphorylation causes moesin inactivation, the question of how recombinant bacterial protein resupplies this loss remains. It is doubtful that moesin is either phosphorylated or lipid associated when synthesized in bacteria; more likely, its activity is due to small amino acid changes at the NH2 terminus of the expression construct causing constitutive opening of an otherwise folded moesin structure.
In conclusion, although moesin does not interact directly with Rho or Rac, the experiments reported here show that it is required for the stress fiber formation and cortical actin polymerization. The versatility of this in vitro assay should now allow rapid progress in characterizing the role of moesin and other ERM family members in cytoskeletal reorganization, in both intact and permeabilized cells, and more general understanding of the Rho and Rac signal transduction pathways.
Abbreviations used in this paper
- GAP
GTPase activating protein
- GDI
guanine nucleotide dissociation inhibitor
- GST
glutathione-S-transferase
- GTPγS
guanosine 5′-O-(3-thiotriphosphate)
- MLC
myosin light chain
- PIP2
phosphatidylinositol-4,5-bisphosphate
- PVDF
polyvinylidene difluoride
- ROCK
Rho-kinase
Footnotes
Please address all correspondence to Alan Hall, MRC Laboratory for Molecular Cell Biology, University College London, Gower Street, London WC1E 6BT, UK. Tel.: 44-171-380-7909. Fax: 44-171-380-7909. e-mail: Alan.Hall@ucl.ac.uk
We are very grateful to B. Balch for encouragement and advice early in this project, P. Mangeat and S. Tsukita for moesin and ERM antibodies, T. Bridges for recombinant proteins and N. Tapon for GST-RhoGAP.
This work was generously supported by the Wellcome Trust (to D.J.G. Mackay and A. Hall).
References
- Aktories K, Just I. In vitro ADP-ribosylation of rho by bacterial ADP-ribosyltransferases. Methods Enzymol. 1995;256:184–195. doi: 10.1016/0076-6879(95)56023-8. [DOI] [PubMed] [Google Scholar]
- Algrain M, Turunen O, Vaheri A, Louvard D, Arpin M. Ezrin contains cytoskeleton and membrane binding domains accounting for its role as a membrane-cytoskeletal linker. J Cell Biol. 1993;120:129–139. doi: 10.1083/jcb.120.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano M, Mukai H, Ono Y, Chihara K, Matsui T, Hamajima Y, Okawa K, Iwamatsu A, Kaibuchi K. Identification of a putative target of rho as the serine-threonine kinase protein kinase N. Science (Wash DC) 1996;271:648–650. doi: 10.1126/science.271.5249.648. [DOI] [PubMed] [Google Scholar]
- Amieva MR, Furthmayr H. Subcellular localization of moesin in dynamic filopodia, retraction fibers, and other structures involved in substrate exploration, attachment and cell-cell contacts. Exp Cell Res. 1995;219:180–196. doi: 10.1006/excr.1995.1218. [DOI] [PubMed] [Google Scholar]
- Arpin M, Algrain M, Louvard D. Membrane-actin microfilament connections: an increasing diversity of players related to band 4.1. Curr Opin Cell Biol. 1994;6:136–141. doi: 10.1016/0955-0674(94)90127-9. [DOI] [PubMed] [Google Scholar]
- Berryman M, Franck Z, Bretscher A. Ezrin is concentrated in the apical microvilli of a wide variety of epithelial cells whereas moesin is found primarily in endothelial cells. J Cell Sci. 1993;105:1025–1043. doi: 10.1242/jcs.105.4.1025. [DOI] [PubMed] [Google Scholar]
- Brill S, Li S, Lyman CW, Church DM, Wasmuth JJ, Weissbach L, Bernards A, Snijders AJ. The Ras GTPase-activating-protein-related human protein IQGAP2 harbors a potential actin binding domain and interacts with calmodulin and rho family GTPases. Mol Cell Biol. 1996;16:4869–4878. doi: 10.1128/mcb.16.9.4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbelo PD, Drechsel DN, Hall A. A conserved binding motif defines numerous candidate target proteins for both Cdc42 and Rac GTPases. J Biol Chem. 1995;270:29071–29074. doi: 10.1074/jbc.270.49.29071. [DOI] [PubMed] [Google Scholar]
- Cerione RJ, Zheng Y. Dbl family of oncogenes. Curr Opin Cell Biol. 1996;8:216–222. doi: 10.1016/s0955-0674(96)80068-8. [DOI] [PubMed] [Google Scholar]
- Chong LD, Traynor-Kaplan A, Bokoch GM, Schwartz MA. The small GTP-binding protein rho regulates a phosphatidylinositol 4-phosphate 5-kinase in mammalian cells. Cell. 1994;79:507–513. doi: 10.1016/0092-8674(94)90259-3. [DOI] [PubMed] [Google Scholar]
- Chou MM, Blenis J. The 70 kDa S6 kinase complexes with and is activated by the Rho family G proteins Cdc42 and Rac1. Cell. 1996;85:753–783. doi: 10.1016/s0092-8674(00)81257-x. [DOI] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley E, Horwitz AF. Tyrosine phosphorylation and cytoskeletal tension regulate the release of fibroblast adhesions. J Cell Biol. 1995;131:179–189. doi: 10.1083/jcb.131.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekmann D, Abo A, Johnston C, Segal AW, Hall A. Interaction of Rac with p67phox and regulation of phagocytic NADPH oxidase activity. Science (Wash DC) 1994;265:531–533. doi: 10.1126/science.8036496. [DOI] [PubMed] [Google Scholar]
- Elias PM, Goerke J, Friend DS. Freeze-fracture identification of sterol-digitonin complexes in cell and liposome membranes. J Cell Biol. 1978;78:577–594. doi: 10.1083/jcb.78.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franck Z, Gary R, Bretscher A. Moesin, like ezrin, colocalizes with actin in the cortical cytoskeleton in cultured cells, but its expression is more variable. J Cell Sci. 1993;105:219–231. doi: 10.1242/jcs.105.1.219. [DOI] [PubMed] [Google Scholar]
- Gilmore AP, Burridge K. Regulation of vinculin binding to talin and actin by phosphatidylinositol-4-5-bisphosphate. Nature (Lond) 1996;381:531–535. doi: 10.1038/381531a0. [DOI] [PubMed] [Google Scholar]
- Hall A. Small GTP-binding proteins and the regulation of the actin cytoskeleton. Ann Rev Cell Biol. 1994;10:31–54. doi: 10.1146/annurev.cb.10.110194.000335. [DOI] [PubMed] [Google Scholar]
- Hall A, Self AJ. The effect of Mg++ on the guanine nucleotide exchange rate of p21N-ras . J Biol Chem. 1986;261:10963–10965. [PubMed] [Google Scholar]
- Harder T, Kellner R, Parton RG, Gruenberg J. Specific release of membrane-bound annexin II and cortical cytoskeleton elements by sequestration of membrane cholesterol. Mol Biol Cell. 1997;8:533–545. doi: 10.1091/mbc.8.3.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart MJ, Callow MG, Souza B, Polakis P. IQGAP1, a calmodulin-binding protein with a rasGAP-related domain, is a potential effector for cdc42Hs. EMBO (Eur Mol Biol Organ) J. 1996;15:2997–3005. [PMC free article] [PubMed] [Google Scholar]
- Hartwig JHH, Bokoch GM, Carpenter CL, Janmey PA, Taylor LA, Toker A, Stossel TP. Thrombin receptor ligation and activated rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell. 1995;82:643–653. doi: 10.1016/0092-8674(95)90036-5. [DOI] [PubMed] [Google Scholar]
- Hirao M, Sato N, Kondo T, Yonemura S, Monden M, Sasaki T, Takai Y, Tsukita S, Tsukita S. Regulation mechanism of ERM (Ezrin/Radixin/Moesin) protein/plasma membrane association: possible involvement of phosphatidylinositol turnover and rho-dependent signaling pathway. J Cell Biol. 1996;135:37–51. doi: 10.1083/jcb.135.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchin NA, Hall A. The assembly of integrin adhesion complexes requires both extracellular matrix and intracellular rho/rac GTPases. J Cell Biol. 1995;131:1857–1865. doi: 10.1083/jcb.131.6.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, Wanatabe N, Saito Y, Kakizuka A, Morii N, et al. The small GTP-binding protein rho binds to and activates a 160 kDa ser/thr protein kinase homologous to myotonic dystrophy kinase. EMBO (Eur Mol Biol Organ) J. 1996;15:1885–1893. [PMC free article] [PubMed] [Google Scholar]
- Janmey PA, Stossel TP. Gelsolin-polyphosphoinositide interaction: full expression of gelsolin-inhibiting function by polyphosphoinositides in vesicular form and inactivation by dilution, aggregation, or masking of the inositol head group. J Biol Chem. 1989;264:4825–4831. [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fakata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, et al. Regulation of myosin phosphatase by rho and rho-associated kinase (rho-kinase) Science (Wash DC) 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Kornberg A. Lactic dehydrogenase of muscle. Methods Enzymol. 1955;1:441–445. [Google Scholar]
- Kozma R, Ahmed S, Best A, Lim L. The ras-related protein cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol Cell Biol. 1995;15:1942–1952. doi: 10.1128/mcb.15.4.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarche N, Tapon N, Stowers L, Burbelo PD, Aspenström P, Bridges T, Chant J, Hall A. Rac and cdc42 induce actin polymerization and G1 cell cycle progression independently of p65PAKand the JNK/SAPK MAP kinase cascade. Cell. 1996;87:519–529. doi: 10.1016/s0092-8674(00)81371-9. [DOI] [PubMed] [Google Scholar]
- Lancaster CA, Taylor-Harris PM, Self AJ, Brill S, van Erp HE, Hall A. Characterization of RhoGAP. A GTPase-activating protein for rho-related small GTPases. J Biol Chem. 1994;269:1137–1142. [PubMed] [Google Scholar]
- Lankes WT, Schwartz-Albiez R, Furthmayr H. Cloning and sequencing of porcine moesin and radixin cDNA and identification of highly conserved domains. Biochem Biophys Acta. 1993;1216:479–482. doi: 10.1016/0167-4781(93)90018-9. [DOI] [PubMed] [Google Scholar]
- Leung T, Manser E, Tan L, Lim L. A novel serine/threonine kinase binding the ras-related rhoA GTPase which translocates the kinase to peripheral membranes. J Biol Chem. 1995;270:29051–29054. doi: 10.1074/jbc.270.49.29051. [DOI] [PubMed] [Google Scholar]
- Li R, Zheng Y, Drubin DG. Regulation of cortical actin cytoskeleton assembly during polarised cell growth in budding yeast. J Cell Biol. 1995;128:599–615. doi: 10.1083/jcb.128.4.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky LM, Hall A. Rho: a connection between membrane receptor signaling and the cytoskeleton. Trends Cell Biol. 1996;6:304–310. doi: 10.1016/0962-8924(96)10026-x. [DOI] [PubMed] [Google Scholar]
- Manser E, Leung T, Salihuddin H, Zhao Z, Lim L. A brain serine/ threonine protein kinase activated by cdc42 and rac1. Nature (Lond) 1994;367:40–46. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- Martin M, Andréoli C, Sahuquet A, Montcourrier P, Algrain M, Mangeat P. Ezrin NH2-terminal domain inhibits the cell extension activity of the COOH-terminal domain. J Cell Biol. 1995;128:1081–1093. doi: 10.1083/jcb.128.6.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Rho-associated kinase, a novel serine-threonine kinase, as a putative target for the small GTP-binding protein Rho. EMBO (Eur Mol Biol Organ) J. 1996;15:2208–2216. [PMC free article] [PubMed] [Google Scholar]
- Nakamura F, Amieva MR, Furthmayr H. Phosphorylation of threonine 558 in the carboxy-terminal actin-binding domain of moesin by thrombin activation of human platelets. J Biol Chem. 1995;270:31377–31385. doi: 10.1074/jbc.270.52.31377. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibres, lamellipodia and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Norman JC, Price LS, Ridley AJ, Hall A, Koffer A. Actin filament organization in activated mast cells is regulated by heterotrimeric and small GTP-binding proteins. J Cell Biol. 1994;126:1005–1015. doi: 10.1083/jcb.126.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson HF, Self AJ, Garrett MD, Just I, Aktories K, Hall A. Microinjection of recombinant p21rhoinduces rapid changes in cell morphology. J Cell Biol. 1990;111:1001–1007. doi: 10.1083/jcb.111.3.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestonjamasp K, Amieva MR, Strassel CP, Nauseef WM, Furthmayr H, Luna EJ. Moesin, ezrin and p205 are actin-binding proteins associated with neutrophil plasma membranes. Mol Biol Cell. 1995;6:247–259. doi: 10.1091/mbc.6.3.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DJ, Ades SE, Singer SJ, Hynes RO. Sequence and domain structure of talin. Nature (Lond) 1990;347:685–689. doi: 10.1038/347685a0. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibres in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. Signal transduction pathways regulating Rho-mediated stress fibre formation: requirement for a tyrosine kinase. EMBO (Eur Mol Biol Organ) J. 1994;13:2600–2610. doi: 10.1002/j.1460-2075.1994.tb06550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CJ, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Sato N, Funayama N, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S. A gene family consisting of ezrin, radixin and moesin: its specific location at actin filament/plasma membrane association sites. J Cell Sci. 1992;103:131–143. doi: 10.1242/jcs.103.1.131. [DOI] [PubMed] [Google Scholar]
- Self AJ, Hall A. Purification of recombinant rho/rac/G25K from Escherichia coli.Small GTPases and their regulation. Methods Enzymol. 1995;256:3–11. doi: 10.1016/0076-6879(95)56003-3. [DOI] [PubMed] [Google Scholar]
- Stossel TP. On the crawling of animal cells. Science (Wash DC) 1993;260:1086–1094. doi: 10.1126/science.8493552. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Sato N, Kasahara H, Funayama N, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S. Perturbation of cell adhesion and microvilli formation by antisense oligonucleotides to ERM family members. J Cell Biol. 1994;125:1371–1384. doi: 10.1083/jcb.125.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolias KF, Cantley LC, Carpenter CL. Rho family GTPases bind to phosphoinositide kinases. J Biol Chem. 1995;270:17656–17659. doi: 10.1074/jbc.270.30.17656. [DOI] [PubMed] [Google Scholar]
- Trofatter JA, MacCollin MM, Rutter JL, Murrell JR, Duyao MP, Parry DM, Eldridge R, Kley N, Menon AG, Pulaski K, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;72:791–800. doi: 10.1016/0092-8674(93)90406-g. [DOI] [PubMed] [Google Scholar]
- Turunen O, Wahlström T, Vaheri A. Ezrin has a COOH-terminal actin-binding site that is conserved in the ezrin protein family. J Cell Biol. 1994;126:1455–1453. doi: 10.1083/jcb.126.6.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda T, Kikuchi A, Ohga N, Yamamoto J, Takai Y. Purification and characterization from bovine brain cytosol of a novel regulatory protein inhibiting the dissociation of GDP from and the subsequent binding of GTP to rhoB, a ras p21-like GTP-binding protein. J Biol Chem. 1990;265:9373–9380. [PubMed] [Google Scholar]
- van Aelst L, Joneson T, Bar-Sagi D. Identification of a novel Rac-1 interacting protein involved in membrane ruffling. EMBO (Eur Mol Biol Organ) J. 1996;15:3778–3796. [PMC free article] [PubMed] [Google Scholar]
- Wanatabe G, Saito Y, Madaule P, Ishizaki T, Fujisawa K, Morii N, Mukai H, Ono Y, Kakizuka A, Narumiya S. Protein kinase N (PKN) and PKN-related protein rhophilin as targets of small GTPase rho. Science (Wash DC) 1996;271:645–648. doi: 10.1126/science.271.5249.645. [DOI] [PubMed] [Google Scholar]