

Abstract
Lipoprotein transport across the blood–brain barrier (BBB) is of critical importance for the delivery of essential lipids to the brain cells. The occurrence of a low density lipoprotein (LDL) receptor on the BBB has recently been demonstrated. To examine further the function of this receptor, we have shown using an in vitro model of the BBB, that in contrast to acetylated LDL, which does not cross the BBB, LDL is specifically transcytosed across the monolayer. The C7 monoclonal antibody, known to interact with the LDL receptor-binding domain, totally blocked the transcytosis of LDL, suggesting that the transcytosis is mediated by the receptor. Furthermore, we have shown that cholesterol-depleted astrocytes upregulate the expression of the LDL receptor at the BBB. Under these conditions, we observed that the LDL transcytosis parallels the increase in the LDL receptor, indicating once more that the LDL is transcytosed by a receptor-mediated mechanism. The nondegradation of the LDL during the transcytosis indicates that the transcytotic pathway in brain capillary endothelial cells is different from the LDL receptor classical pathway. The switch between a recycling receptor to a transcytotic receptor cannot be explained by a modification of the internalization signals of the cytoplasmic domain of the receptor, since we have shown that LDL receptor messengers in growing brain capillary ECs (recycling LDL receptor) or differentiated cells (transcytotic receptor) are 100% identical, but we cannot exclude posttranslational modifications of the cytoplasmic domain, as demonstrated for the polymeric immunoglobulin receptor. Preliminary studies suggest that caveolae are likely to be involved in the potential transport of LDL from the blood to the brain.
The maintenance of the homeostasis of brain interstitial fluid, which constitutes the special microenvironment for neurons, is established by the presence of the blood–brain barrier (BBB)1 at the transition area from endothelial cells (ECs) to brain tissue. Of primary importance in the formation of a permeability barrier by these cells is the presence of continuous tight junctions that seal together the margins of the ECs and restrict the passage of substances from the blood to the brain. Furthermore, in contrast to ECs in many other organs, the brain capillary ECs contain no direct transendothelial passageways such as fenestrations or channels. But obviously, the BBB cannot be absolute. The brain is dependent upon the blood to deliver metabolic substrates and remove metabolic waste, and the BBB therefore facilitates the exchange of selected solutes. Carrier-mediated transport systems that facilitate the uptake of hexoses, amino acids, purine compounds, and mono-carboxylic acids have been revealed in the cerebral endothelium (Betz and Goldstein, 1978), but until now little information has come to light regarding the cerebral uptake of lipids.
There is growing evidence that the brain is equipped with a relatively self-sufficient transport system for maintaining cholesterol and lipid homeostasis. The presence of a low density lipoprotein (LDL) receptor has been demonstrated by immunocytochemistry in rat and monkey brains; and apolipoprotein (apo) E and apo AI-containing particles have been detected in human cerebrospinal fluid (Pitas et al., 1987). Furthermore, enzymes involved in lipid metabolism have been located within the brain: LCAT mRNA has been shown to be expressed in rat brains and cholesteryl ester transfer protein, which plays a key role in cholesterol homeostasis, has been detected in human cerebrospinal fluid and seems to be synthesized in the brain (Albers et al., 1992). The distribution of the LDL receptor-related protein, a multifunctional receptor that binds apoE, is highly restricted and limited to the gray matter, primarily associated with neuronal cell population (Wolf et al., 1992). The difference in cellular expression of ligand (apoE) and receptor (LDL receptor-related protein) may provide a pathway for intracellular transport of apoE-containing lipoproteins in the central nervous system. All these data leave little doubt that the brain is equipped with a relatively self-sufficient transport system for cholesterol.
Cholesterol could be derived from de novo synthesis within the brain and from plasma via the BBB. Malavolti et al. (1991) indicate the presence of unexpectedly close communications between extracerebral and brain cholesterol. Changes in the extracerebral cholesterol levels are readily sensed by the LDL receptor in the brain and promptly provoke appropriate modifications in its activity. Méresse et al. (1989a) provided direct evidence for the occurrence in vivo of an LDL receptor on the endothelium of brain capillaries. Furthermore, the fact that enzymes involved in the lipoprotein metabolism are present in the brain microvasculature (Brecher and Kuan, 1979) and that the entire fraction of the drug bound to lipoproteins is available for entry into the brain strongly suggest that this cerebral endothelial receptor plays a role in the interaction of plasma lipoproteins with brain capillaries. These results pinpoint the critical importance of the interactions between brain capillary ECs and lipoproteins. Owing to the fact that the neurological abnormalities that result from the inadequate absorption of dietary vitamin E can be improved by the oral administration of pharmacological doses of vitamin E, Traber and Kayden (1984) have suggested that LDL functions as a transport system for tocopherol to the brain. Furthermore, the trace amounts of apolipoprotein B that were detected by Salem et al. (1987) in cerebrospinal fluid from healthy patients using a very sensitive immunoblot technique confirm that, at most, small amounts of apolipoprotein B normally pass through the BBB. However, whether LDL is involved in the exchange is not known.
Using an in vitro model of the BBB that imitates an in vivo situation by culturing capillary ECs and astrocytes on opposite sides of a filter (Dehouck et al., 1990a , 1992), we have demonstrated that in culture, like in vivo, in contrast to peripheral endothelium and in spite of the tight apposition of ECs and their contact with physiological concentrations of lipoproteins, brain capillary ECs express an LDL receptor (Méresse et al., 1991; Dehouck et al., 1994). The capacity of ECs to bind LDLs is greater when cocultured with astrocytes than in their absence. Futhermore, we have shown that the lipid requirement of astrocytes increases the expression of the LDL receptor on brain capillary ECs. Taken together, the presence of LDL receptors on brain capillary ECs and the modulation of the expression of these receptors by the lipid composition of astrocytes suggest that cholesterol used by cells in the central nervous system may be derived, at least in part, from the periphery via transport across the BBB.
In the present study, we provide direct evidence that after binding to brain capillary ECs, there is a specific mechanism for the transport of LDL across the endothelial monolayer from the apical to the abluminal surface. This mechanism might be best explained by a process of receptor-mediated transcytosis. Preliminary results pinpoint the role of caveolae in the transcellular transport of LDL across the brain endothelium.
Materials and Methods
Cell Culture
Bovine Brain Capillary ECs.
Brain capillary ECs were isolated and characterized as described by Méresse et al. (1989b). The use of cloned endothelial cells allows the obtainment of a pure endothelial cell population without contamination by pericytes. The cells were cultured in the presence of DME supplemented with 15% (vol/vol) heat-inactivated calf serum (Hyclone Laboratories, Logan, UT), 2 mM glutamine, 50 μg/ml gentamycin, and bFGF [basic fibroblast growth factor]; 1 ng/ml, added every other day).
Bovine Adrenal Cortex Endothelial Cells (ACE).
ACE were isolated and characterized as described by Gospodarowicz et al. (1986). ACE (a gift from Dr. S. Saule, Institut Pasteur, Lille, France) were cultured like the bovine brain capillary ECs.
Rat Astrocytes.
Primary cultures of mixed astrocytes were made from newborn rat cerebral cortex. After the meninges had been removed, the brain tissue was gently forced through a nylon sieve, as described by Booher and Sensenbrenner (1972). Astrocytes were plated on six multiwell dishes (Nunclon; Nunc A/S, Roskilde, Denmark) at a concentration of 1.2 × 105 cells/ml in 2 ml of DME supplemented with 10% FCS (Hyclone Laboratories), and the medium was changed twice a week. 3 wk after seeding, cultures of astrocytes were stabilized and used for experiments. The astrocytes were characterized with glial fibrillary acidic protein (GFAP), and >95% of the population was GFAP positive (Dehouck et al., 1990b ).
Coculture of Endothelial Cells and Astrocytes.
Preparation of filters for coculture: culture plate inserts (Millicell-CM 0.4 μm; 30-mm diam; Millipore Corp., Milford, MA) were coated on the upperside with rat-tail collagen prepared by a modification of the method of Bornstein (1958).
Experimental method: cultures of astrocytes were prepared as described above. After 3 wk, coated filters were set in six multiwell dishes containing astrocytes, and ECs were plated on the upperside of the filters in 1.5 ml of medium with a concentration of 4 × 105 cells/ml for bovine brain capillary ECs and adrenal cortex ECs. The medium used for the coculture was DME supplemented with 15% CS, 2 mM glutamine, 50 μg/ml gentamycin, and 1 ng/ml bFGF. This medium was changed every other day. Under these conditions, ECs form a confluent monolayer after 7 d. Experiments were performed 5 d after confluence.
Fluorescence Microscopy
Endothelial cells grown on porous filters were fixed at room temperature for 20 min with 4% paraformaldehyde in a fibrous component-stabilizing buffer (PHEMS; 60 mM Pipes, 25 mM Hepes, 10 mM EGTA, 2 mM MgCl2, and 140 mM NaCl, pH 6.9). After being washed in PHEMS, the cells on filter fragments were permeabilized with cold acetone (−20°C) for 10 min, followed by two washes with PHEMS. Cells were then stained with filamentous actin probe, bodipy–phallacidin (Molecular Probes, Inc., Eugene, OR; 165 nM, 30 min, at room temperature). Mitotracker CMX Ros coloration was carried out as indicated by the manufacturer (Molecular Probes, Inc). For the localization of tight junction-associated protein ZO-1 to the plasma membrane, the cells were fixed with cold methanol (−20°C). Fixed cells were incubated with an affinity-purified rabbit anti– ZO-1 (Zymed Labs., S. San Francisco) diluted in Tris-HCl–buffered saline (20 mM Tris-HCl, 0.5 M NaCl, pH 7) containing 5% (wt/vol) ovalbumin and 1% heat-inactivated normal goat serum. Subsequently, the cells were incubated for 1 h at room temperature with the appropriate combination of fluorescently labeled secondary antibodies and propidium iodine (2 μg/ ml). For the luminal uptake of DiI (0,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine percholate)-LDL, the ECs were incubated for 45 min at 37°C in prewarmed DME, 5% LPDS (lipoprotein deficient serum) with DiI-LDL (35 μg/ml). The inhibition of DiI-LDL uptake was performed in the presence of filipin. The cells were pretreated with 3 μg/ml filipin for 10 min at 37°C, washed, and incubated with DiI-LDL. In the case of the study of the reversibility of filipin treatment effect, ECs were incubated for 1 h at 37°C in serum-containing medium before the addition of DiI-LDL. All incubations were terminated by three washes in ice-cold Pipes– Hepes buffer and immediately fixed with 4% paraformaldehyde.
After the final washes, the filters and their attached monolayers were mounted on glass microscope slides using Mowiol mountant (Hoechst, Frankfurt, Germany) containing p-phenylene diamine (0.1%, Sigma Chemical Co., St. Louis, MO) anti-quenching agent. All specimens were studied using a fluorescence microscope, (DMRB; Leica Mikroskopie und Systeme GmbH, Wetzlar, Germany), and pictures were taken on Kodak Tmax film.
Preparation of Low Density Lipoproteins, Acetylated LDL, and Lipoprotein-deficient Serum
LDL was isolated from human plasma by sequential ultracentrifugation at the densities of 1.03–1.053. The densities were adjusted using solid KBr. The LDL was extensively dialyzed at 4°C against 0.15 M NaCl. Acetylated LDL was prepared by treating LDL with acetic anhydride (Basu et al., 1976). LDL was radioiodinated as described by Bilheimer et al. (1972). After labeling, the LDL was chromatographed on a PD10 column (Pharmacia Fine Chemicals, Piscataway, NJ) and extensively dialyzed at 4°C against 0.15 M NaCl and 0.01% EDTA (pH 7.4). The specific activity of 125I-LDL, which was used within 10 d of preparation, ranged from 300 to 400 cpm/ng protein. LPDS was prepared from calf serum by ultracentrifugation at the density of 1.25 adjusted with solid KBr. LPDS was extensively dialyzed at 4°C against DME.
Preparation of Fluorescently Labeled Lipoprotein and Lipoprotein–Gold Complex
The fluorescent probe chosen is DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine percholate). The lipoprotein concentration should be between 1 and 2 mg lipoprotein/ml in saline–EDTA. 2 ml of lipoprotein-deficient serum is added to each 1 mg of lipoprotein to be labeled with DiI. The solution is then filtered (0.45 μm for LDL) through Millex-PF filters. The solution is gently shaken, and 50 μl of DiI in DMSO (3 mg/ml) is added per mg of lipoprotein. The mixture is incubated for 15– 20 h at 37°C. The LDLs are reisolated from the incubation mixture by ultracentrifugation at the density of 1.063. The density of 1.063 is obtained by adding 0.0834 g of KBr for each ml of incubation mixture. LDLs are reisolated using a four-rotor centrifuge (TL100; Beckman Instr., Fullerton, CA) at 100,000 rpm for 2 h at 10°C. After centrifugation, the LDLs are dialyzed against saline–EDTA.
Freshly isolated LDL with 16-nm colloidal gold was labeled according to the method of Handley et al. (1981).
125I-LDL and 125I–acLDL Binding
ECs were incubated for 2 h at 4°C with DME, 10 mM Hepes, 0.2% BSA, and 35 μg/ml of 125I-LDL and 125I–acLDL were added in the upper or lower compartment. At the end of the incubation, the cells were washed nine times at 4°C: three times with 4 ml of PBS (Ca2+/Mg2+); three times with 4 ml of PBS (Ca2+/Mg2+) containing 0.2% BSA; three times with 4 ml of PBS (Ca2+/Mg2+). EC-associated radioactivity was then determined by removing the membrane of the culture insert and counting it in a γ counter. Nonspecific binding was determined by incubating the cells with 125I-lipoproteins and a 20-fold excess of unlabeled related lipoproteins. Binding is expressed in ng/cm2, since one filter of 4.2 cm2 represents 90 μg of cellular protein.
Studies of Lipoprotein Transport Through Brain Capillary EC Monolayers
Passage of 125I-LDL or Acetylated 125I-LDL through Brain Capillary EC Monolayers.
The passage of lipoproteins through brain capillary EC monolayers was studied using the coculture model, as described above. ECs seeded on collagen-coated filters were cultured with astrocytes for 12 d. Each filter was then transferred to a six multiwell dish containing 2 ml of DME with 15% LPDS (lower compartment). At time zero, a known amount of the iodinated lipoprotein (usually 35 μg protein/ml) in DME with 15% LPDS was added on the luminal face of an EC monolayer (upper compartment; 700 μl). During the experiments, the monolayers were kept at 37°C. At several time points, filters were transferred to the next well. At the end of the experiment, all lower and upper compartments were collected. Precipitation of samples by trichloroacetic acid was performed. The determination of the total 125I-LDL passage was performed by counting, in a γ counter, the acid-precipitable fractions of the lower compartments. Nonspecific passage was determined by incubating the cells with 125I-LDL and a 20-fold excess of unlabeled LDL. Specific transport was calculated by subtracting the nonspecific from the total transport. Passage rates were expressed in ng LDL/cm2. The filter permeability to LDL was carried out with collagen-coated filters.
Degradation of 125I-LDL or Acetylated 125I-LDL during the Transcytosis.
For the determination of the degradation of LDLs during their passage (5 h) through EC monolayers, acid-soluble fractions from upper and lower compartment samples were separated by centrifugation from acid-precipitable fractions; AgNo3 (5%) precipitation was then performed to correct for free 125I. AgNo3-soluble fractions were counted in a γ counter. Nonspecific degradation was determined by incubating the cells with 125I-LDL and a 20-fold excess of unlabeled LDL. Total degradation was corrected for nonspecific flux giving the specific LDL degradation. Degradation rates are expressed in ng LDL/mg cell protein.
Degradation of Transcytosed 125I-LDL by Astrocytes.
The degradation of transcytosed 125I-LDL by astrocytes was performed using the coculture situation. 1 ml of medium containing 35 μg of labeled LDL was added in the upper compartment and 2.5 ml of medium containing 10% LPDS in the lower one. The coculture (i.e., ECs and astrocytes) was kept at 37°C for 16 h. Degradation was carried out as described above. Nonspecific degradation was determined by adding 3 μg of unlabeled LDL in the lower compartment, which represents a 10-fold excess owing to the passage of ∼100 ng for 5 h/cm2. Control experiments were carried out with ECs separated from astrocytes.
Effect of Temperature on 125I-LDL Transport.
The 125I-LDL transport experiments were performed as described above, but the monolayers were kept at 4°C.
Inhibition of 125I-LDL Passage through Brain Capillary ECs.
ECs were incubated for 15 min with different concentrations (1 and 0.1 mg/ml) of the monoclonal antibody designated immunoglobulin C-7 (American Type Culture Collection, Rockville, MD; Beisiegel et al., 1981) and with or without unlabeled LDL (upper compartment). 125I-LDL was then added to this compartment, and the rate of LDL passage was determined as described above. Irrelevant IgGs directed against apolipoprotein AI (1 mg/ ml) were used as a control.
Modulation of the Transcytosis by the Lipid Composition of Astrocytes.
The induction of LDL receptor expression (Dehouck et al., 1994) can be described in four phases: (a) coculture phase; brain capillary ECs and astrocytes were cocultured for 12 d in DME supplemented with 15% CS as described above. (b) Astrocyte LPDS phase; ECs were transferred and cocultured with other astrocytes in fresh DME supplemented with 15% calf serum. Astrocytes of the original coculture were incubated for 36 h at 37°C with DME supplemented with 10% LPDS, 2 mM glutamine, and 50 μg/ml gentamycin. As a control, astrocytes were incubated with DME supplemented with 10% FCS. (c) Induction phase; on the day of the experiment, the filter inserts with ECs, in 1.5 ml of fresh DME supplemented with 15% calf serum, were moved to a well that contained astrocytes in their 36-h LPDS conditioned medium. The cells were incubated together for different times at 37°C. (d) LDL binding phase after the “induction phase,” 125I-LDL bindings to ECs at 4°C, 125I-LDL transport, and DiI-LDL uptake experiments at 37°C were performed as described above.
Electron Microscopy Studies.
After incubation at different times with gold–LDL or horseradish peroxidase, the cells were fixed routinely with 2.5% glutaraldehyde in sodium cacodylate buffer, pH 7.2, at 4°C and postfixed in 1% OsO4 for 60 min. After being dehydrated in ethanol and embedded in araldite, sections perpendicular to the monolayer-cultured cells, contrasted with lead hydroxide, were examined with an electron microscope (420; Philips, Eindhoven, The Netherlands).
Sequencing of the LDL Receptor Cytoplasmic Domain Involved in the Basolateral Targeting
RNA Extraction.
Total cell RNA was obtained from growing brain capillary ECs, differentiated brain capillary ECs depleted or not in cholesterol, or aortic ECs, using the guanidium-thiocyanate chloride procedure according to Sambrook et al. (1989).
RT-PCR.
5 μg of total RNA was reverse transcribed by 50 U of MMLV-reverse transcriptase (Stratagene, La Jolla, CA) in a total volume of 50 μl containing 100 μM of each dNTP, 100 ng of oligod(T)12-18, 20 U Rnasin (Promega, Madison, WI), and 10 mM dithiothreitol for 1 h at 37°C. After reverse transcription, samples were heated at 95°C for 5 min to denature the MMLV-RT and then held at −20°C until PCR.
To perform PCR, specific internal primer pairs for the bovine LDL receptor were designed using the published sequence (Russel et al., 1984) and the Primer Premier version 3.1 software (Biosoft International, Palo Alto, CA). Primer sequences were as follows: BLDLR1: 5′ GAGCGTGGGTGCCCTATACA 3′ and BLDLR2: 5′ GGACTCAAGGCAGCAGCTCA 3′. PCR amplification was carried out using a thermal cycler (Maxicycler PT-100TM; MJ Research, Watertown, MA). 50 μl of reaction mixture contained 5 μl of first strand DNA, buffer (10 mM Tris-HCl, pH 8), MgCl2 (1.5 mM), dNTPs (40 μM), primers (0.4 μM), and 1.5 U Taq polymerase (Eurogentec, Seraing, Belgium). The amplification profile was 94°, 55°, and 72°C each for 1 min times 30 cycles, preceded by 5 min at 94°C and followed by 7 min at 72°C to ensure completion of extension. To check for contamination, PCR in the absence of DNA was used as a negative control.
Sequencing.
The PCR products were visualized on ethidium bromide-stained agarose gels after electrophoresis and sequenced on both strands by an Aby prism dye terminator cycle sequencing kit on an automatic sequencer (ABI-377; Applied Biosystems, Foster City, CA).
Results
The Blood–Brain Barrier Model
Fig. 1 A illustrates the typical phenotypes of confluent brain capillary ECs, cocultured for 12 d with astrocytes on an insert coated with rat-tail collagen. Mitotracker coloration shows an intense staining of the cell, indicating a large amount of mitochondriae (Fig. 1 B). However, bodipy– phallacidin staining reveals that a delicate fluorescent network of actin filaments throughout the cytoplasm formed prominent continuous bands at cell borders (Fig. 1 C). Furthermore, ZO-1 is localized at sites of cell–cell contact between ECs along the junctional complex (Fig. 1 D). This continuous network of ZO-1 suggests that it is associated with the tight junction barrier. These data, added to previously published results concerning the high electrical resistance (500–800 Ω × cm2) and the low permeability (Dehouck et al., 1990a ), support the fact that the coculture is a legitimate model of the BBB.
Figure 1.

Characterization of brain capillary EC monolayer grown on the upper face of a collagen-coated filter. (A) Phase contrast micrograph of confluent brain capillary ECs. (B) Endothelial staining with mitochondrion-selective dye, Mitotracker CMX Ros. (C) Staining with F-actin probe, bodipy–phallacidin (Molecular Probes, Inc). (D) Localization of tight junction-associated protein ZO-1 to the plasma membrane and nuclear staining by propidium iodine. Bar, 50 μm.
LDL Internalization
Since LDL internalization in most cells is mediated primarily by the LDL receptor, it was of interest to determine the extent of internalization of LDL from the luminal surface of brain capillary ECs. DiI-LDL was added to the luminal compartment and remained in contact with the cells for 45 min at 37°C. Staining obtained with an immunofluorescent microscope is shown in Fig. 2 B. DiI-LDL was found as small, individual, discrete vesicles throughout the cells. These results indicate that endocytotic pathways are functional in these cells. The cellular accumulation of gold–LDL was also examined by standard electron microscopy. Fig. 2, A and C, shows typical results demonstrating the accumulation of gold–LDL in early endosomes and in a multivesicular body. Furthermore, a coated pit could be observed only rarely, and no accumulation of gold– LDL was observed in lysosomes.
Figure 2.

Electron and fluorescence microscopy of ECs incubated with Au16nm and DiI-labeled LDL. After a 5-min incubation with gold LDL at 37°C, the tracer marks the luminal plasma membrane (A). After a 45-min incubation, the gold-labeled probe is mainly detected in multivesicular bodies (C, arrow). Arrowheads indicate caveolar structure. (B) After 45 min of incubation, the fluorescent DiI-labeled LDL is endocytosed and found as small vesicles throughout the cells. Bars: (A and C) 0.3 μm; (B) 30 μm.
To determine whether degradation of LDL occurs in the cells after internalization, degradation studies were carried out using LDL for a 5-h incubation at 37°C from the apical surface (Fig. 3). 125I-LDL degradation by endothelial cell monolayers was measured by the appearance of the iodine-free, TCA-soluble product released during incubation. No degradation of LDL occurred during the incubation period (Fig. 3 A). The functionality of the degradative pathway was checked by carrying out the same experiment using acetylated LDL (acLDL) instead of native LDL. Acetylation of the lysine and arginine residues of the LDL results in a modified LDL particle (acLDL) that is more negative and has lost its ability to bind the (B,E) receptor and the so-called scavenger receptor. When bindings of LDL and acLDL were carried out at 4°C, no difference was observed between the binding of the two lipoproteins at the luminal surface of the monolayer (4.7 ng/cm2 for LDL versus 4.2 ng/cm2 for acLDL). As shown in Fig. 3 A, acLDL is actively degraded by the brain capillary ECs. This degradation is specific, since only unlabeled acLDL can compete with acLDL (Fig. 3 B), indicating a receptor-mediated degradation (scavenger receptor ligand). These results demonstrate that in brain capillary ECs, the classic degradation pathway (via lysosomes) is functional and therefore that LDL is not directed to the lysosomes after internalization.
Figure 3.
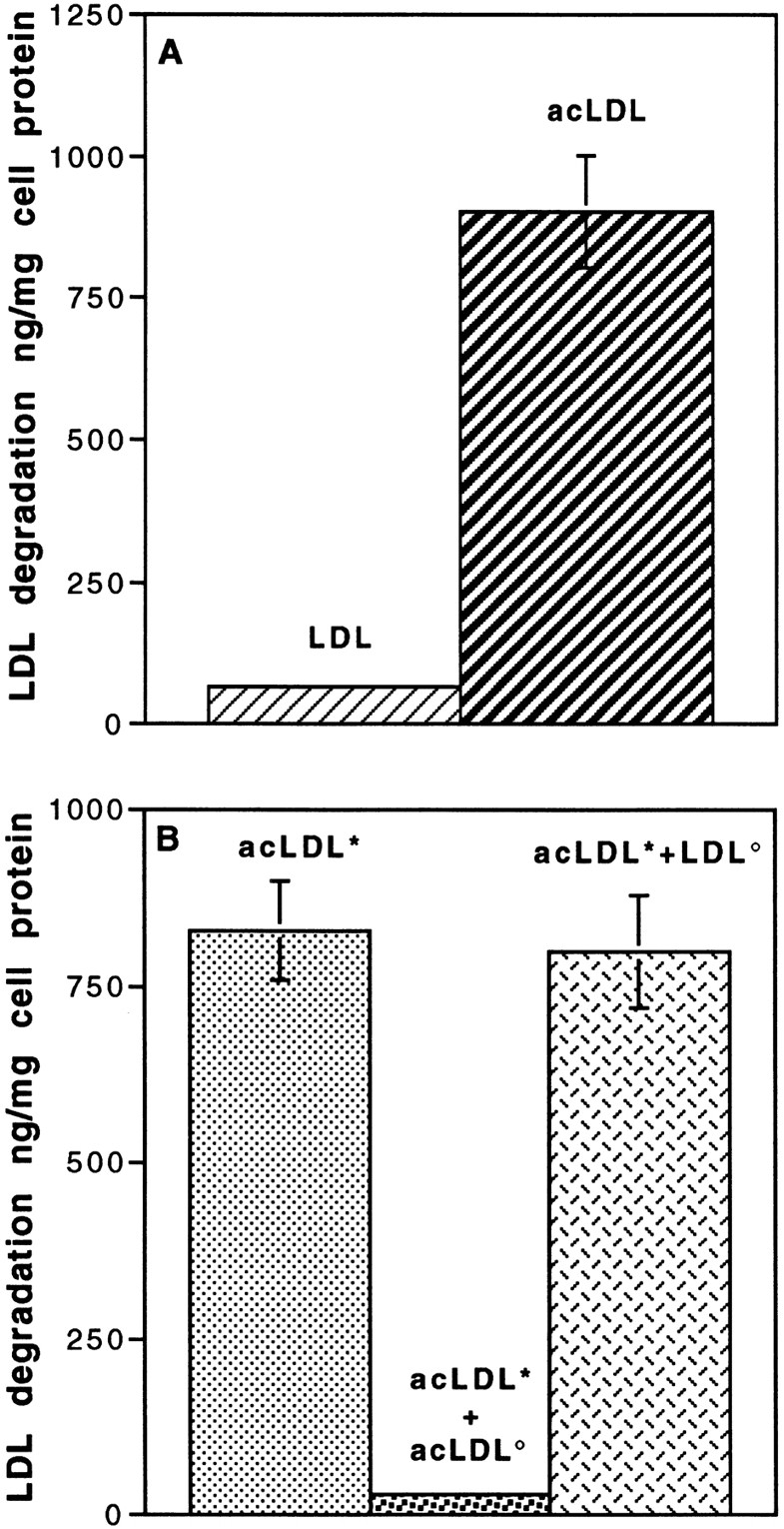
Degradation of LDL and acetylated LDL (acLDL) by brain capillary ECs. (A) Specific degradation of 125I-LDL and 125I-acLDL. Degradation was performed at the concentration of 50 μg/ml for 5 h at 37°C in the absence or the presence of a 20-fold excess of related, unlabeled lipoproteins. (B) Competition studies between labeled acLDL (*acLDL, 50 μg/ml; ░⃞ ) with unlabeled acLDL (acLDL°, 1 mg/ml; ░⃞ ) and unlabeled LDL (LDL°, 1 mg/ml; ▩ ). Determination of the degradation was carried out as described in Materials and Methods. Each point is a mean of three different filters, and the curves are representative of three series of experiments.
Transport of LDL Across the Brain Capillary EC Monolayer
From the experiments described above, it appears that brain capillary ECs bind and internalize but do not degrade LDL. Transport experiments were performed to define the role of the LDL receptor. When 125I-LDL was added to the luminal chamber of the culture, the progressive transfer of these macromolecules across the cell monolayer was observed during the 5-h incubation. The transport was examined both in the absence and in the presence of a 20-fold excess of unlabeled LDL. The toxicity of high concentrations of LDL on the integrity of the monolayer was assessed by calculating the permeability of sucrose and insulin (Dehouck et al., 1992). No effect of LDL concentrations up to 2 mg/ml was observed after an 8-h incubation (Pe = [0.52 ± 0.19] 10−3 cm/min for sucrose, n = 9). The amount of 125I-LDL (trichloro-acetic precipitable) transported is expressed in terms of ng/cm2, where the area refers to the surface area of the cells. Fig. 4 shows that the filter coated with rat-tail collagen and maintained for 12 d with rat astrocytes is not a barrier for the passage of LDL. An excess of unlabeled LDL competed with the transport of LDL from the apical to the basal membrane, although no evidence of saturation could be detected in concentrations of LDL ranging from 10 to 100 μg/ml. Limitations in specific activity and quantities of unlabeled LDL precluded examining concentrations of LDL > 100 μg/ml (125I-LDL; result not shown). Since ECs bind 4.7 ng of LDL/cm2 at 4°C at the luminal concentration of 35 μg/ml of LDL and after 5-h transcytosis ∼100 ng/cm2 cross the monolayer, during the 5-h experiment, ∼20 surface binding equivalents of LDL are transcytosed, indicating that the transcytosis time is ∼15 min. To determine if transcytosed LDL is competent for rebinding to the LDL receptor, degradation experiments of LDL by the astrocytes of the coculture were performed for 16 h. Our results showed that 3.27% of the transcytosed LDL were degraded by the astrocytes. Only 0.8% of the LDL was degraded if the astrocytes were not present during the experiment. The specificity of the astrocyte degradation was demonstrated by the fact that a 10-fold excess of unlabeled LDL in the lower compartment reduced the degradation of transcytosed LDL to 0.89%. These results indicate that transcytosed LDL are competent for binding to the LDL receptor located on astrocytes.
Figure 4.

Transport of LDL across brain capillary EC monolayers. Passage of 125I-LDL (ng/cm2) through collagen-coated filters with (solid lines) or without (dashed line) brain capillary EC monolayer was carried out at 37°C. LDL was added to the upper side of the filter (50 μg/ml 125I-LDL, 1 mg/ml unlabeled LDL). Intact 125I-LDL transport from upper to lower sides of the filter was assessed by counting in a γ counter the acid-precipitable fractions of the lower compartments. Specific transport (▪) was calculated by subtracting the radioactivity obtained in the presence of native LDL (▵) from that obtained in the absence of native LDL (○). The data are expressed as ng of 125I-LDL transported per cm2, which refers to the surface area of the cells. Each point is a mean of three different filters, and the curves are representative of seven series of independent experiments.
No specific binding on the abluminal face was observed when labeled LDL was added in the lower chamber. But, owing to the high nonspecific binding of LDL to the filter, we cannot conclude whether the LDL receptor is specifically expressed or not at the abluminal face of brain capillary ECs. There was no observed component of specific LDL transport in the basal to apical direction. These results strongly indicate the existence of a specific pathway operating in the transport of LDL from the apical to the basal surfaces, but not in the reverse direction.
When the same transport experiments were performed using radiolabeled acLDL instead of LDL in the luminal compartment, no passage of acLDL was found over a 5-h incubation. These results corroborate our conclusions with regard to a specific transport of LDL across the monolayer. Furthermore, when the same experiments were carried out with adrenal cortex ECs, a concentration-dependent, receptor-independent process was observed (results not shown). From the experiments described above, the specificity of the transport could be attributed to a receptor-mediated process in brain capillary ECs.
To confirm that the LDL receptor is involved in this receptor-mediated transcytosis, transport experiments in the presence of the C7-monoclonal antibody, known to interact with the LDL receptor-binding domain and totally block the binding of LDL to brain capillary ECs, were performed. Fig. 5 shows that coincubations of LDL with increasing concentrations of the C7 antibody, decrease the rate of passage of the LDL through the monolayer. At the concentration of 1 mg/ml, LDL transcytosis is totally abolished. Control irrelevant IgG has no effect, suggesting once more the involvement of the LDL receptor in this intracellular traffic.
Figure 5.

Inhibition of LDL transport through EC monolayers by the C7 monoclonal antibody. Specific 125I-LDL transport experiment (35 μg/ml 125I-LDL, 700 mg/ml unlabeled LDL) was performed as described in Fig. 4, legend, either in presence of C7-IgG, 1 mg/ml (•), 0.1 mg/ml (▵), or AI-IgG, 1 mg/ml (□). Each point is a mean of three different filters, and the curves are representative of three series of independent experiments.
Fig. 6 compares the effect of temperature on LDL transport from the apical to basal surfaces. A decrease in the incubation temperature from 37° to 4°C slightly affects the passage of sucrose (Fig. 6 B), whereas a dramatic decrease in the LDL transport through the monolayer is observed (Fig. 6 A), indicating that LDL is directed to the abluminal compartment by a transcellular route and that this transport system requires active mechanisms such as receptor-mediated transcytosis.
Figure 6.
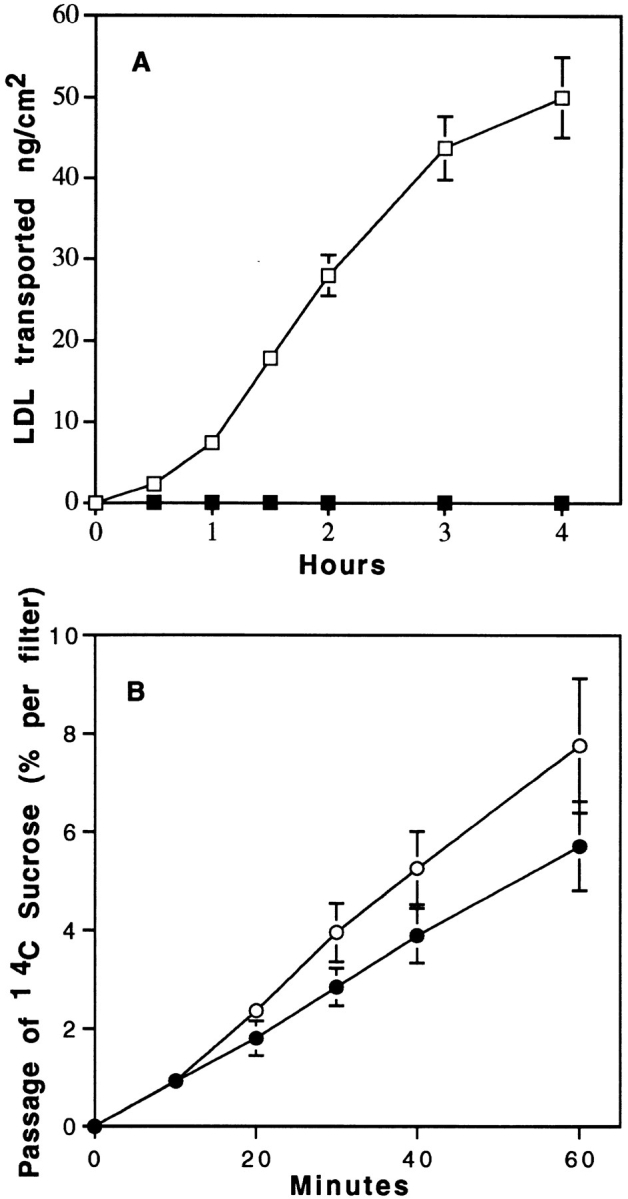
Effect of temperature on the transport of 125I-LDL (A) and sucrose (B) from apical to basal surfaces. Specific transport of LDL was performed as described in Fig. 4, legend. Transport of [14C]sucrose was expressed as the percentage of radioactivity crossing the brain capillary EC monolayer from the apical to the abluminal surfaces. 125I-LDL and sucrose transport were performed both at 37° (□, ○) and 4°C (▪, •). The data represent the means of three filters, and the curves are representative of two series of independent experiments.
We have already demonstrated that the lipid requirement of astrocytes increases the expression of endothelial cell LDL receptors (Dehouck et al., 1994). Is this upregulation of the LDL receptor at the luminal face of brain capillary ECs followed by an increase in LDL transcytosis? To answer this question the following experiments were carried out. Brain capillary ECs and astrocytes were cocultured for 12 d in a medium containing 15% CS. 36 h before the experiments, ECs were transferred onto other astrocytes, and the astrocytes of the coculture were cultured for 36 h in a medium containing 10% LPDS to modify the intracellular metabolism of these cells. After this period, brain capillary ECs and cholesterol-depleted astrocytes in their conditioned medium were again cocultured for 2 or 4 h at 37°C (induction phase), and the binding at 4°C and transcytosis experiments at 37°C were performed. Fig. 7 draws, after a 4-h induction, a parallel between the upregulation of the LDL receptor at the luminal surface of the brain capillary ECs and the increase in the LDL transcytosis across the monolayer, demonstrating once more the involvement of the LDL receptor in the transcytosis. These results were confirmed by the studies of the endocytosis of DiI-LDL. Fig. 8 B shows an increase in the fluorescent signal in ECs cocultured for 4 h with cholesterol- depleted astrocytes.
Figure 7.

LDL transport after induction of LDL receptor expression on brain capillary ECs by cholesterol-depleted astrocytes. Upregulation of LDL receptor on brain capillary ECs was performed in four phases as described in Materials and Methods. On the day of the experiment, brain capillary ECs in DME supplemented with 15% CS were cocultured for different times (1–4 h) at 37°C with cholesterol-depleted astrocytes in their 36-h incubating medium (induction phase). The increase of LDL receptor expression on ECs was studied by 125I-LDL binding at 4°C (inset). 125I-LDL transport studies were performed at 37°C, after 4 h induction and >5 h, as described in Fig. 4, legend. The induction phase was performed in the presence of either cholesterol- depleted (•) or noncholesterol-depleted (□) astrocytes. The data represent the means of three filters, and the curves are representative of three series of independent experiments.
Figure 8.
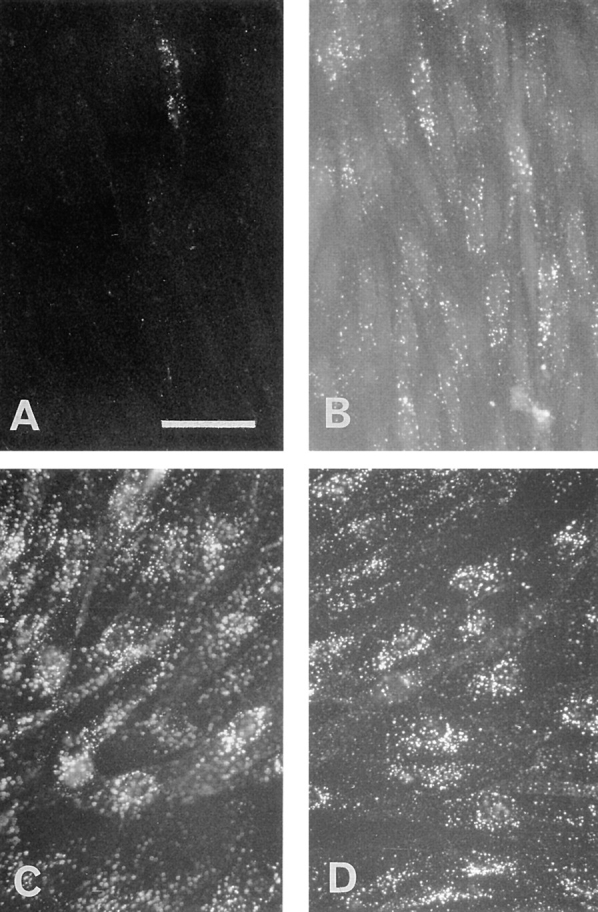
DiI-LDL endocytosis after induction of LDL receptor expression on brain capillary ECs by cholesterol-depleted astrocytes. (A) Brain capillary ECs were incubated at 37°C for 45 min with DiI-LDL. After washing, the cells were fixed and processed for fluorescent microscopy as described in Materials and Methods. (B) The same experiment was carried out with “upregulated LDL receptor” ECs as described in Fig. 7, legend. Bar, 50 μm.
One of the characteristics of brain capillary ECs is the paucity of clathrin-coated pits, known in other cell types to mediate the endocytosis of LDL. Recently, Schnitzer et al. (1994) have shown that a peripheral, continuous endothelium possesses nonclathrin-coated vesicles (caveolae) that seem to be involved in transcytosis. To study the involvement of caveolae in LDL transcytosis at the brain level, the effect of filipin on the endocytosis of LDL was studied. Indeed it has been shown that cholesterol-binding agents such as filipin cause the disassembly of caveolae. First, we checked the toxicity of filipin on the integrity of the barrier functions. No increase in sucrose permeability was observed until 20 min of incubation of the EC monolayer in the presence of concentrations of filipin ranging from 100 ng/ml to 3 μg/ml (Pe = [0.63 ± 0.24] 10−3 cm/min versus [0.52 ± 0.19] 10−3 cm/min for sucrose; n = 3). But, after 30 min incubation, a huge increase in the passage of sucrose could be observed (Pe = [2.38 ± 0.24] 10−3 cm/min for sucrose; n = 6) reflecting an alteration of the barrier properties of the monolayer. For these reasons, preincubations with filipin were carried out for 10 min only. The preincubation of endothelium for 10 min with 3 μg/ml of filipin before the addition of DiI-LDL resulted in a total inhibition of the fluorescent signal in the cells (Fig. 9 A). To rule out the possibility that filipin inhibits the binding of LDL to the cell surface, 125I-LDL binding to the cell surface of the ECs was assessed for 1 h at 4°C using confluent monolayers that were either preincubated or not with 3 μg/ml of filipin for 10 min at 37°C. We found no evidence for interference in the binding, indicating that filipin does not interact with the binding of LDL to its receptor. Since filipin is toxic for the integrity of the monolayer after 30 min and transcytosis experiments lasted at least 3 h, the quantification measurement of the effect of filipin was not possible.
Figure 9.
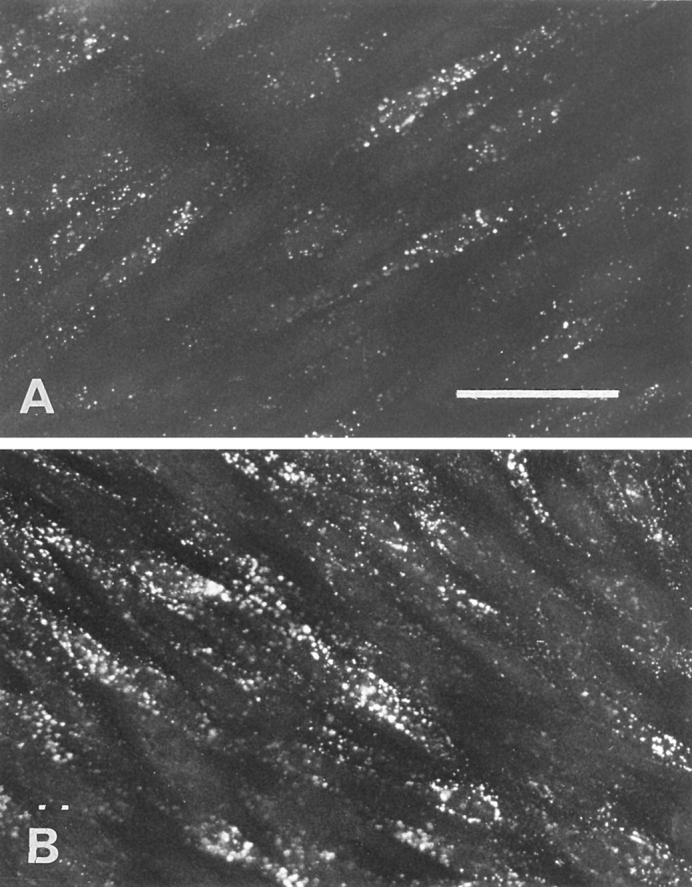
Effect of filipin on the endocytosis of LDL by brain capillary ECs. (A) The cells were pretreated with filipin (3 μg/ml) before the addition of DiI-LDL, and the endocytosis was performed as described in the Fig. 8, legend. (B) Immediately after filipin treatment, the cells were incubated in DME containing 20% CS for 30 min to reverse the effects of filipin before examining DiI-LDL endocytosis. (C and D) Endocytosis was carried out with brain capillary ECs in growing phase without (C) or with (D) pretreatment with filipin. Bar, 50 μm.
The reversibility of the effect of filipin treatment on the ECs was carried out. After filipin treatment and washings, ECs were incubated for 1 h at 37°C in serum-containing medium and were then allowed to accumulate DiI-LDL for 45 min at 37°C. As shown in Fig. 9 B, this short reversal treatment resulted in the restoration of normal endocytosis by the ECs, indicating that fundamental functions of ECs are not altered by filipin.
The effect of filipin on clathrin-coated pits was also checked using brain capillary ECs during the growing phase. Under these conditions, LDL is classically internalyzed by the clathrin pathway, where LDL is directed to lysosomes for degradation and the cholesterol is used by the cells. Fig. 9, C and D, demonstrates that in growing cells, LDL endocytosis is not inhibited by filipin treatment. The same results were obtained with differentiated ECs cocultured for 12 d and fed with LPDS for 36 h to decrease their cholesterol content. Furthermore, in these cases we can note that the accumulation of DiI-LDL creates a perinuclear punctate signal consistent with its localization in lysosomes. These results confirm that the coated vesicular degradative pathway with its associated coated pits, endosomes, and lysosomes is not altered by filipin. The compartments of this pathway are functionally intact, unlike the early stages of the noncoated vesicular pathway.
These results demonstrate that there is a shift from a recycling LDL receptor to a transcytotic LDL receptor when the cells are differentiated. Matter et al. (1993) reported that ablating one of the LDL receptor internalization signals can transform the LDL receptor from a recycling to a transcytotic trafficking pattern. To check whether such a modification may occur in differentiated brain capillary ECs, the sequences of the cytoplasmic tail of the LDL receptor required for basolateral sorting were analyzed and compared to those of the recycling receptors for growing brain capillary ECs. mRNA was extracted from these different cell lines, and the RT-PCR results show that a 245-bp fragment is obtained with each culture condition studied. Their sequences match the bovine LDL receptor perfectly (100% identity), confirming that the consensus sequence for the internalization signals is not altered in the differentiated brain capillary ECs.
Discussion
In Vitro Blood–Brain Barrier
The availability of an in vitro model to study the passage of macromolecules through the monolayers of brain capillary endothelial cells on porous membranes enabled us to investigate the characteristics of the passage of lipoproteins through endothelial monolayers. These ECs displayed: (a) tight junctions as visualized by actin and ZO-1 repartition; (b) an electrical resistance of >800 Ω × cm2; (c) a low permeability for sucrose and insulin; (d) the presence of specific transporters (amino acids, glucose, propranolol [Dehouck et al., 1995]); (e) specific enzymatic activities of the BBB (γ-glutamyl transpeptidase, monoamine oxidase). Owing to the fact that a close correlation exists between the values of the brain uptake index obtained in vitro on our monolayers and those obtained in vivo with the Oldendorf technique for a large number of drugs (Dehouck et al., 1995), the in vitro model resembles in vivo features of animal ECs. Furthermore, we have recently shown (Dehouck et al., 1994) that brain capillary ECs in coculture express, in spite of the physiological concentrations of lipoproteins in the incubation medium and the tight apposition of ECs (Méresse et al., 1991), an LDL receptor as they do in vivo. Furthermore, we have found that the fatty acid composition of cocultured ECs with astrocytes was markedly different from that of noncocultivated ECs and that these fatty acid changes might be biologically relevant, as they tended to make the fatty acid composition of brain capillary ECs more closely resemble that of freshly isolated capillaries (Bénistant et al., 1995). To summarize, our coculture enables us to apprehend, on an in vitro model that closely mimics the in vivo conditions, the cellular mechanism of LDL interaction with the BBB.
LDL Receptor Function at the Blood–Brain Barrier
What then is the function of this LDL receptor on the luminal surface, and does it represent a true LDL receptor? Since this receptor is expressed on brain capillary ECs at confluence and in the presence of LDL in the medium bathing either the basal or the apical surface, this receptor is clearly not involved in regulating cholesterol biosynthesis in ECs. Furthermore, as demonstrated (Méresse et al., 1991; Dehouck et al., 1994), this luminal surface receptor is likely to be a bona fide LDL receptor, as indicated by its saturability, its specificity, the ability of an anti-LDL receptor antibody to inhibit 125I-LDL binding, and its molecular weight of 135.
Our results suggest that there is a specific mechanism for the transport of LDL across the endothelial monolayer from the blood to the brain side. This transport is a specific transport mechanism that might be best explained by a process of receptor-mediated transcytosis. Several points lead us to this conclusion: (a) the transport is specific and unidirectional; (b) the transcytosis is inhibited by the C7 monoclonal antibody, which is known to interact with the receptor- binding domain (Beisiegel et al., 1981); (c) the transport shows a temperature dependence similar to the process involving receptor mediated internalization (Vasile et al., 1983). It is also interesting to note that the transcytosis of LDL from the luminal to the basal surface occurs in the absence of a significant route involving its degradation. The same results were obtained by Li et al. (1991) using epithelial cells. Owing to the fact that amounts of LDL and acLDL roughly bound to the luminal surface of the monolayer at 4°C, the nontranscytosis of acLDL after their internalization in the ECs and their subsequent degradation indicate that the lysosomal compartment is functional in these cells, and reinforce the hypothesis that the receptor mediated endocytotic pathway bypasses lysosomal processing.
Furthermore, the transcytosis seems highly regulated. First, it is receptor mediated and high concentration independent. Second, the brain cells are able to regulate the expression of the LDL receptor, as demonstrated by Dehouck et al. (1994). Indeed, the lipid requirement of astrocytes increases the number of LDL receptors at the luminal side of brain capillary ECs. That is why to investigate the capacity of astrocytes to modulate LDL transcytosis, the astrocytes of the coculture were preincubated in LPDS, to decrease their lipid content. Under these conditions, we observed that the LDL transcytosis parallels the increase in the LDL receptor at the luminal side. Taken together, these experiments indicate that LDL is transcytosed through the brain endothelium by a receptor-mediated mechanism and that the transcytosis could be regulated by the surrounding astrocytes.
Our in vitro results are in agreement with the in vivo clinical observations of patients with cerebrotendinuous xanthomatosis (Salem et al., 1987). Indeed, excessive amounts of cholestanol, the 5 c-dihydro derivative of cholesterol, have been detected in the brain of patients with this rare, inherited lipid storage disease. Affected patients have progressive neurological dysfunctions (dementia, spinal cord paresis, cerebellar ataxia), tendon xanthomas, and premature atherosclerosis. Cholestanol accumulates in patients because of overproduction in the liver as a consequence of a genetic defect in bile acid synthesis (Salem and Grundy, 1973). Consequently, the abnormal sterol deposits in the brain of patients with cerebrotendinuous xanthomatosis are believed to derive from plasma lipoproteins passing through the BBB. The fact that apolipoprotein B or an apolipoprotein B fragment, which increases out of proportion to the other apolipoproteins and to lecithin cholesterol acyl-transferase in the cerebrospinal fluid, strongly suggests that this apolipoprotein serves to transport sterols from the plasma to the brain in patients with cerebrotendinuous xanthomatosis. Treatment of patients with chenodeoxycholic acid not only reduced elevated sterol concentration in the cerebrospinal fluid but also decreased the abnormal levels of apolipoprotein B in the brain, suggesting that the function of the BBB returned to normal. Our results demonstrating a “basal” transport of LDL through brain capillary ECs and its upregulation by the lipid composition of glial cells are in agreement with the results observed in vivo in this disease.
In contrast, in peripheral vessels, Vasile et al. (1983), using an in situ perfusion of native LDL, showed that most of the transendothelial transport of LDL is receptor independent. Within the inherent limitations of this approach and some uncertainties about the quantitative interpretation of the morphometric data obtained, the mechanism and rate of transport of intact native LDL across the endothelium in the peripheral organ has been the subject of several in vitro studies (Navab et al., 1986; Langeler et al., 1989). Despite the absence of functionally active LDL receptors due to contact inhibitor and physiological concentrations of plasma LDL in peripheral capillaries, there is a significant transport of intact LDL across the endothelium. These in vitro results confirm the in vivo observation of Vasile et al. (1983) and describe a concentration-dependent, receptor-independent process. Our data using adrenal cortex capillary ECs and aortic ECs are in agreement with a transport of LDL independent of the simultaneous addition of an excess amount of unlabeled LDL. From our data, it is not possible to distinguish between a paracellular route for the movement of LDL or a transcellular route involving the formation of transient transendothelial channels. Thus, the normal peripheral confluent endothelium functions are not related to the direct metabolism of LDL but rather transport the particle intact, by mechanisms unique to ECs, to the subendothelial space for metabolism by other cells. This transcellular transport can deposit up to twice the plasma concentration of LDL in the interstitium of the aorta (Hoff et al., 1978; Smith and Ashall, 1983), demonstrating the nonregulation of this transport and the possible appearance of sclerotic lesions.
Transcellular Mechanism of the Passage of LDL through the Brain Capillary EC Monolayer
The existence of receptor-mediated processes that bypass lysosomes seems to be a feature of ECs. Indeed, as in continuous endothelia, ECs are linked to each other by junctional complexes that constitute a major barrier to the bidirectional exchange of macromolecules and specific receptors play a significant role in the transendothelial transport of plasma molecules to tissues. Several different albumin-binding proteins have been identified on ECs; it appears that gp30 and gp18 mediate the binding, endocytosis, and degradation of modified albumin, whereas albondin (gp60) mediates native albumin binding and subsequent transcytosis (Schnitzer and Oh, 1994). Nevertheless, there is no evidence of receptor-mediated transport of native albumin in brain capillary endothelia (Pardridge et al., 1985). In the same way, specific receptors of insulin are expressed on a variety of ECs, and it has been shown that these receptors are involved in insulin transport across ECs (King and Johnson, 1985). Specific binding of insulin to brain microvessels in vivo (Van Houtten and Posner, 1979) or in vitro (Franck and Pardridge, 1981) first suggests that the pathway for insulin entry into the CNS involved transport across the BBB. Furthermore, Franck et al. (1985) clearly show in vivo that a saturable receptor was involved in the brain uptake process. A more precise demonstration of the transcytosis of blood-borne molecules was achieved with an antibody directed against the ferrotransferrin receptor (OX-26). They have demonstrated that OX-26 selectively targets the brain relative to organs such as the kidneys, heart, or lungs. This statement is supported by the capillary depletion technique experiments of Friden et al. (1991, 1993), who have demonstrated the blood to brain entry of OX-26 conjugated to methotrexate and nerve growth factor, and by Broadwell et al. (1994), who have performed cytochemical approaches using horseradish peroxidase–OX-26. Using our model, we have provided evidence for the transcytosis of iron-loaded transferrin across the cerebral endothelium by means of a specific transferrin receptor-mediated pathway (Descamps et al., 1996). Furthermore, no intracellular degradation of transferrin was observed, indicating the occurrence of a pathway through the cultured endothelium that bypasses the lysosomal compartment.
The precise transcellular pathway for the passage of blood-borne molecules across the BBB and into the CNS has only recently begun to be elucidated (Villegas and Broadwell, 1993; Broadwell et al., 1994). Owing to the fact that early studies suggested a role for caveolae in endothelial cell transport across the monolayer (Ghitescu et al., 1986 ; Milici et al., 1987), to better appreciate possible vesicular uptake and transport of LDL in brain capillary ECs, we carried out experiments using filipin, a drug that binds sterols, thereby disrupting caveolae (Severs and Simons, 1986). But, since coated endocytic vesicles become uncoated vesicles during the transport of ligands to the lysosome, McGookey et al. (1983) found that coordinated with the dissociation of the clathrin coat from endocytic vesicles, the membranes became sensitive to the formation of filipin–sterol complexes. To rule out the possibility that the transcytotic vesicles may be derived from coated pits and affected by filipin, whereas the coated vesicles on the surface are protected, experiments were also carried out using growing cells and differentiated cells depleted in cholesterol. Under these conditions, we have shown that filipin did not alter the classic degradation pathway of the LDL receptor. In contrast, in differentiated cells still in contact with lipoproteins, a complete inhibition of the endocytosis was achieved. The dramatic increase in the sucrose permeability, after a 20-min incubation in the presence of filipin ruled out the possibility of performing quantitative studies on the effect of filipin on LDL transcytosis, since these experiments last at least 5 h. In these conditions, the huge increase in the paracellular transport would hide the expected reduction in the transcellular transport of LDL. But, added to our results demonstrating the reversibility of a short treatment with filipin on endocytosis indicating that fundamental functions of ECs are not altered by this drug, these results therefore suggest that caveolae are likely to be involved in the potential transport of LDL from the blood to the brain.
Although the capacity of caveolae to detach themselves from the cell surface has been questioned (Bundgaard, 1983; Anderson, 1993), recent results strongly support the involvement of caveolae in endocytosis (for review see Lamaze and Schmid, 1995). Since LDL receptor ligands rapidly dissociate at acid pH , tracking the pathway of the receptor using conventional antireceptor antibodies (C7 monoclonal antibody) was not possible. The use of a chimeric receptor, consisting of the extracellular and transmembrane domains of the mouse IgG Fc receptor fused to the LDL receptor cytoplasmic tail, is under investigation, since this approach will permit the utilization of a high affinity Fab fragment of the mouse IgG antibody, 2.4 G2, a well characterized probe that does not dissociate from the receptor at pH values >4.0 (Mellman et al., 1984). Furthermore, the fact that filipin stopped the fusion and/or inside internalization of caveolae in multivesicular structures argues in favor of a function of caveolae in nonclathrin-dependent endocytosis and transcytosis, as already mentioned by Montesano et al. (1982), Milici et al. (1987), and Tran et al. (1987). The work of Parton et al. (1994), showing that caveolae are dynamic structures that can be internalized into the cells leaves little doubt about the function of caveolae in endocytosis.
In peripheral vessels, the characterization of caveolin-rich membrane domains reveals known receptors for modified LDL (scavenger receptor, CD 36, and RAGE) and multiple GPI-linked proteins, but the receptor for native LDL was not mentioned (Lisanti et al., 1994). This was not surprising, owing to the fact that the LDL receptor is down regulated in physiological conditions in peripheral vessels. In brain capillaries, in spite of the high concentration of lipoproteins, the LDL receptor is not down regulated. It is not localized as in growing cells or cholesterol-depleted differentiated cells in coated pits but in caveolae. The balance between distinct endocytic pathways can be shifted in response to cellular requirements (in this case the need of cholesterol for the ECs), suggesting that these pathways could be regulated differently. As shown by Matter et al. (1993), the change from a recycling to a transcytotic receptor could have been the consequence of the ablation of one of the internalization signals of the cytoplasmic domain of the receptor. Our results demonstrated that LDL receptor messengers in growing brain capillary ECs (recycling receptor), or differentiated cells (transcytotic receptor) that are 100% identical, ruled out this hypothesis. But, as demonstrated for the polymeric IgA receptor, the shift of an endocytotic receptor to a transcytotic receptor could also be due to posttranslational modifications of the cytoplasmic domain of the receptor. Indeed, Casanova et al. (1990) and Aroeti and Mostow (1994) demonstrated that phosphorylation of Ser664 inactivates the endocytotic signal and permits the polymeric immunoglobulin receptor to be transcytosed.
Recent results have provided new tools that allow detailed study of caveolar dynamics. Parton et al. (1994) show that internalization of caveolae may be a regulated process, since increased phosphorylation due to phosphatase inhibition induces the removal of caveolae from the cell surface, whereas decreased kinase activity inhibits the internalization process. Eker et al. (1994) pinpoint the involvement of cAMP since this second messenger appears to upregulate clathrin-independent endocytosis selectively at the apical surface of polarized ECs. The hormonal regulation of caveolae internalization has been shown by Smart et al. (1995), suggesting that molecules added to the luminal surface of the endothelium could modulate the endocytosis via caveolae. Our results demonstrating an increase in transcytosis, when the astrocytes of the coculture were depleted in cholesterol, suggest that endocytosis via caveolae and transcytosis could, in part, be regulated by signal molecules secreted by astrocytes and then acting at the abluminal face of brain capillary ECs. These results provide evidence that the brain blood vessel endothelium may not be as inert a barrier as has been assumed. Appropriate stimulus can increase endocytotic activity in these cells.
In summary, taken together, these experiments indicate that LDL is transcytosed through the BBB via a receptor-mediated mechanism in specialized endocytic vesicles that avoid fusion with lysosomes. In contrast to other ECs in the body, these highly differentiated cells regulate the entry of lipoproteins into the brain. Furthermore, the influence of astrocytes on the expression of the LDL receptor and the rate of LDL transcytosis suggest the existence of focal differences in lipoprotein influx, which may depend on the lipid state of astrocytes. The BBB cannot be considered as a rigid and impermeable structure protecting the brain, but as a dynamic interface able to modify its permeability to the specific needs of certain areas of the brain.
Abbreviations used in this paper
- acLDL
acetylated LDL
- BBB
blood– brain barrier
- EC
endothelial cell
- LDL
low density lipoprotein
Footnotes
Please address all correspondence to Dr. Roméo Cecchelli, INSERM U325, Institut Pasteur, 590019 Lille Cédex, France. Tel.: (33) 3-20-87-73-81; Fax: (33) 3-20-8773-60; E-mail: romeo.cecchelli@pasteur-lille.fr
This work was partly supported by a grant from the Association Recherche et Partage, by Institut National de la Santé et de la Recherche Médicale (INSERM U 325; Professor J.C. Fruchart), and by the Centre National de la Recherche Scientifique (UMR 111; Professor A. Verbert).
References
- Albers JJ, Tollefson JH, Wolfbauer G, Albright RE. Cholesteryl ester transfer protein in human brain. Int J Clin Lab Res. 1992;21:264–266. doi: 10.1007/BF02591657. [DOI] [PubMed] [Google Scholar]
- Anderson RGW. Plasmalemmal caveolae and GPI-anchored membrane proteins. Curr Opin Cell Biol. 1993;5:647–652. doi: 10.1016/0955-0674(93)90135-d. [DOI] [PubMed] [Google Scholar]
- Aroeti B, Mostow KE. Polarized sorting of the polymeric immunoglobulin receptor in the exocytotic and endocytotic pathways is controlled by the same amino acids. EMBO (Eur Mol Biol Organ) J. 1994;13:2297–2304. doi: 10.1002/j.1460-2075.1994.tb06513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu SK, Goldstein J M, Anderson RGM, Brown MS. Degradation of cationized low density lipoprotein and regulation of cholesterol metabolism in homozygous familial hypercholesterolemia fibroblasts. Proc Natl Acad Sci USA. 1976;73:493–502. doi: 10.1073/pnas.73.9.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisiegel U, Schneider WJ, Goldstein JL, Anderson RGW, Brown MS. Monoclonal antibodies to the low-density lipoprotein receptor as a probe to study of receptor mediated endocytosis and the genetics of familial hypercholesterolemia. J Biol Chem. 1981;256:11923–11931. [PubMed] [Google Scholar]
- Bénistant C, Dehouck MP, Fruchart JC, Cecchelli R, Lagarde M. Fatty acid composition of brain capillary endothelial cells: effect of the coculture with astrocytes. J Lipid Res. 1995;36:1–9. [PubMed] [Google Scholar]
- Betz L, Goldstein GW. Polarity of the blood-brain barrier: neutral amino-acid transport into isolated brain capillaries. Science (Wash DC) 1978;202:225–227. doi: 10.1126/science.211586. [DOI] [PubMed] [Google Scholar]
- Bilheimer DM, Eisenberg S, Levy RI. The metabolism of very low density lipoproteins. Biochem Biophys Acta. 1972;260:212–221. doi: 10.1016/0005-2760(72)90034-3. [DOI] [PubMed] [Google Scholar]
- Booher J, Sensenbrenner M. Growth and cultivation of dissociated neurons and glial cells from embryonic chick, rat and human brain in flask cultures. Neurobiology. 1972;2:97–105. [PubMed] [Google Scholar]
- Bornstein MB. Reconstituted rat tail collagen used as substrate for time tissue cultures on coverslips in Maximow slides and roller tubes. Lab Invest. 1958;7:134–139. [PubMed] [Google Scholar]
- Brecher P, Kuan HT. Lipoprotein lipase and acid lipase activity in rabbit brain microvessels. J Lipid Res. 1979;20:464–471. [PubMed] [Google Scholar]
- Broadwell RD, Baker BJ, Banks WA, Friden P, Moran M, Olivier C, Villegas JC. Ferrotransferrin and antibody against the transferrin receptor as potential vehicles for drug delivery across the mammalian blood-brain barrier into the CNS. Methods Neurosci. 1994;21:93–117. [Google Scholar]
- Bundgaard M. Vesicular transport in capillary endothelium: does it occur? . FEBS (Fed Eur Biochem Soc) Proc Meet Fed Proc. 1983;42:2425–2430. [PubMed] [Google Scholar]
- Casanova JE, Breitfeld PP, Ross SA, Mostow KE. Phosphorylation of the polymeric immunoglobulin receptor required for its efficient transcytosis. Science (Wash DC) 1990;248:742–745. doi: 10.1126/science.2110383. [DOI] [PubMed] [Google Scholar]
- Dehouck MP, Méresse S, Delorme P, Fruchart JC, Cecchelli R. An easier, reproducible, and mass-production method to study the blood-brain barrier in vitro. J Neurochem. 1990a;54:1798–1801. doi: 10.1111/j.1471-4159.1990.tb01236.x. [DOI] [PubMed] [Google Scholar]
- Dehouck MP, Méresse S, Delorme P, Torpier G, Fruchart JC, Cecchelli R. The blood-brain barrier in vitro: coculture of brain capillary endothelial cells and astrocytes. Circulation et Métabolisme du Cerveau. 1990b;7:151–162. [Google Scholar]
- Dehouck MP, Jolliet-Riant P, Brée F, Fruchart JC, Cecchelli R, Tillement JP. Drug transfer across the blood-brain barrier: correlation between in vitro and in vivo models. J Neurochem. 1992;58:1790–1797. doi: 10.1111/j.1471-4159.1992.tb10055.x. [DOI] [PubMed] [Google Scholar]
- Dehouck B, Dehouck MP, Fruchart JC, Cecchelli R. Upregulation of the low-density lipoprotein receptor at the blood-brain barrier: intercommunications between brain capillary endothelial cells and astrocytes. J Cell Biol. 1994;126:465–473. doi: 10.1083/jcb.126.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehouck MP, Dehouck B, Schluep C, Fruchart JC, Lemaire M, Cecchelli R. Drug transport to the brain: comparison between in vitro and in vivo models of the blood-brain barrier. Eur J Pharm Sci. 1995;3:357–365. [Google Scholar]
- Descamps L, Dehouck MP, Torpier G, Cecchelli R. Receptor- mediated transcytosis of transferrin through blood-brain barrier endothelial cells. Am J Physiol. 1996;270:H1149–H1158. doi: 10.1152/ajpheart.1996.270.4.H1149. [DOI] [PubMed] [Google Scholar]
- Eker P, Holm PK, Van Deurs B, Sandvig K. Selective regulation of apical endocytosis in polarized Madin-Darby canine kidney cells by mastosporan and cAMP. J Biol Chem. 1994;269:18607–18615. [PubMed] [Google Scholar]
- Franck HJL, Pardridge WM. A direct in vitro demonstration of insulin binding to isolated capillaries. Diabetes. 1981;30:757–761. doi: 10.2337/diab.30.9.757. [DOI] [PubMed] [Google Scholar]
- Franck HJL, Jankovic-Vokes T, Pardridge WH, Morris WL. Enhanced insulin binding to blood-brain barrier in vivo and to brain microvessels in vitro in newborn rabbits. Diabetes. 1985;34:728–733. doi: 10.2337/diab.34.8.728. [DOI] [PubMed] [Google Scholar]
- Friden PM, Walus LR, Musso GF, Taylor MA, Malfroy B, Starzyk RM. Anti-transferrin receptor antibody and antibody-drug conjugates cross the blood-brain barrier. Proc Natl Acad Sci USA. 1991;88:4771–4775. doi: 10.1073/pnas.88.11.4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friden PM, Walus LR, Watson P, Doctrow SR, Kozarich JW, Bäckman C, Hoffer H, Bloom F, Granholm AC. Blood-brain barrier penetration and in vivo activity of an NGF conjugate. Science (Wash DC) 1993;259:373–377. doi: 10.1126/science.8420006. [DOI] [PubMed] [Google Scholar]
- Ghitescu L, Fixman A, Simionescu M, Simionescu N. Specific binding sites for albumin restricted to plasmalemmal vesicles of continuous capillary endothelium: receptor-mediated transcytosis. J Cell Biol. 1986;102:1304–1311. doi: 10.1083/jcb.102.4.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gospodarowicz D, Massoglia S, Cheng J, Fuji DK. Effect of fibroblast growth factor and lipoproteins on the proliferation of endothelial cells derived from bovine adrenal cortex, brain cortex, and corpus luteum capillaries. J Cell Physiol. 1986;127:121–136. doi: 10.1002/jcp.1041270116. [DOI] [PubMed] [Google Scholar]
- Handley DA, Arbeeny CM, Eder HA, Chien S. Hepatic binding and internalization of low density lipoprotein-gold conjugates in rats treated with 17 a-ethinyl estradiol. J Cell Biol. 1981;90:778–785. doi: 10.1083/jcb.90.3.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff HF, Gaubatz JW, Gotto AM. ApoB concentration in the normal aorta. Biochem Biophys Res Commun. 1978;85:1424–1431. doi: 10.1016/0006-291x(78)91162-2. [DOI] [PubMed] [Google Scholar]
- King GL, Johnson S. Receptor-mediated transport of insulin across endothelial cells. Science (Wash DC) 1985;227:1583–1586. doi: 10.1126/science.3883490. [DOI] [PubMed] [Google Scholar]
- Lamaze C, Schmid SL. The emergence of clathrin-independent pinocytic pathways. Curr Opin Cell Biol. 1995;7:573–580. doi: 10.1016/0955-0674(95)80015-8. [DOI] [PubMed] [Google Scholar]
- Langeler EG, Snelting-Havinga I, Van Hinsbergh VWM. Passage of low density lipoproteins through monolayers of human arterial endothelial cells. Effects of vasoactive substances in an in vitro model. Arteriosclerosis. 1989;9:550–559. doi: 10.1161/01.atv.9.4.550. [DOI] [PubMed] [Google Scholar]
- Li C, Stifanis S, Schneider WJ, Poznansky MJ. Low density lipoprotein receptors on epithelial cell (Madin-Darby Canine Kidney) monolayers. J Biol Chem. 1991;266:9263–9270. [PubMed] [Google Scholar]
- Lisanti MP, Scherer PE, Tang ZL, Hermanowski-Vosatka A Ya-Huei Tu, R.F. Cook, and M. Sargiacomo. Characterization of caveolin-rich membrane domains isolated from an endothelial rich source: implications for human disease. J Cell Biol. 1994;126:111–126. doi: 10.1083/jcb.126.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malavolti M, Fromm H, Ceryak S, Shehan KL. Cerebral low-density lipoprotein (LDL) uptake is stimulated by acute bile drainage. Biochem Biophys Acta. 1991;1081:106–108. doi: 10.1016/0005-2760(91)90257-i. [DOI] [PubMed] [Google Scholar]
- Matter K, Whitney A, Yamamoto EM, Mellman I. Common signals control low density lipoprotein receptor sorting in endosomes and the Golgi complex of MDCK cells. Cell. 1993;74:1053–1064. doi: 10.1016/0092-8674(93)90727-8. [DOI] [PubMed] [Google Scholar]
- McGookey DJ, Fagerberg K, Anderson GW. Filipin–cholesterol complexes form in uncoated vesicle membrane derived from coated vesicles during receptor-mediated endocytosis of low density lipoprotein. J Cell Biol. 1983;96:1273–1278. doi: 10.1083/jcb.96.5.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I, Plutner H, Ukkonen P. Internalization and rapid recycling of macrophages Fc receptors tagged with monovalent antireceptor antibody: possible role of prelysosomal compartment. J Cell Biol. 1984;98:1163–1169. doi: 10.1083/jcb.98.4.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méresse S, Delbart C, Fruchart JC, Cecchelli R. Low-density lipoprotein receptor on endothelium of brain capillaries. J Neurochem. 1989a;53:340–345. doi: 10.1111/j.1471-4159.1989.tb07340.x. [DOI] [PubMed] [Google Scholar]
- Méresse S, Dehouck MP, Delorme P, Bensad M, Tauber JP, Delbart C, Fruchart JC, Cecchelli R. Bovine brain endothelial cells express tight junctions and monoamine oxidase activity in long-term culture. J Neurochem. 1989b;53:1363–1371. doi: 10.1111/j.1471-4159.1989.tb08526.x. [DOI] [PubMed] [Google Scholar]
- Méresse, S., M.P. Dehouck, P. Delorme, J.C. Fruchart, and R. Cecchelli. 1991. Lipoproteins and reconstituted blood-brain barrier. In Pharmaceutical Applications of Cell and Tissue Culture to Drug Transport. G. Wilson, S.S. Davies, and L. Illum, editors. Plenum Press, New York.
- Milici AJ, Watrous NE, Stukenbrok H, Palade GE. Transcytosis of albumin in capillary endothelium. J Cell Biol. 1987;105:2603–2612. doi: 10.1083/jcb.105.6.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesano R, Roth J, Robert A, Orci L. Non-coated membrane invaginations are involved in binding and internalization of cholera and tetanus toxins. Nature (Lond) 1982;296:651–653. doi: 10.1038/296651a0. [DOI] [PubMed] [Google Scholar]
- Navab M, Hough GP, Berliner JA. Rabbit β-migrating very low density lipoprotein increases endothelial macromolecular transport without altering electrical resistance. J Clin Invest. 1986;5:135–141. doi: 10.1172/JCI112589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM, Eisenberg JB, Cephalu WT. Absence of albumin receptor on brain capillaries in vivo and vitro. Am J Physiol. 1985;249:E264–E267. doi: 10.1152/ajpendo.1985.249.3.E264. [DOI] [PubMed] [Google Scholar]
- Parton RG, Joggert B, Simons K. Regulated internalization of caveolae. J Cell Biol. 1994;127:1199–1215. doi: 10.1083/jcb.127.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitas RE, Boyles JK, Lee SH, Foss D, Maley RW. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochem Biophys Acta. 1987;917:148–161. doi: 10.1016/0005-2760(87)90295-5. [DOI] [PubMed] [Google Scholar]
- Russell DW, Schneider WJ, Yamamoto T, Juskey KL, Brown MS, Goldstein JL. Domain map of the LDL receptor: sequence homology with the epidermal growth factor precursor. Cell. 1984;37:577–585. doi: 10.1016/0092-8674(84)90388-x. [DOI] [PubMed] [Google Scholar]
- Salen G, Grundy SM. Metabolism of cholestanol, cholesterol, and bile acid in cerebrotendinous xanthomatosis. J Clin Invest. 1973;52:2822–28325. doi: 10.1172/JCI107478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salen G, Berginer V, Shore V, Horak I, Horak E, Tint GS, Shefer S. Increased concentrations of cholestanol and apolipoprotein B in the cerebrospinal fluid of patients with cerebrotendinous xanthomatosis. N Engl J Med. 1987;316:1233–1238. doi: 10.1056/NEJM198705143162002. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning, Second Edition. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York.
- Severs NJ, Simons HL. Caveolar bands and the effects of sterol-binding agents in vascular smooth muscle plasma membranes. Lab Invest. 1986;55:295–307. [PubMed] [Google Scholar]
- Schnitzer JA, Oh P. Albondin-mediated capillary permeability to albumin. J Biol Chem. 1994;269:6072–6082. [PubMed] [Google Scholar]
- Schnitzer JA, Oh P, Pinney E, Allard J. Filipin-sensitive caveolae-mediated transport in endothelium: reduced transcytosis, scavenger endocytosis, and capillary permeability of select macromolecules. J Cell Biol. 1994;127:1217–1232. doi: 10.1083/jcb.127.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart EJ, Ying YS, Anderson GW. Hormonal regulation of caveolae internalization. J Cell Biol. 1995;131:929–938. doi: 10.1083/jcb.131.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EB, Ashall C. Low density lipoprotein concentration in interstitial fluid from human atherosclerotic lesions. Biochem Biophys Acta. 1983;754:249–255. doi: 10.1016/0005-2760(83)90139-x. [DOI] [PubMed] [Google Scholar]
- Traber MG, Kayden HJ. Vitamin E is delivered to cells via the high affinity receptor for low density lipoprotein. Am J Clin Nutr. 1984;40:747–751. doi: 10.1093/ajcn/40.4.747. [DOI] [PubMed] [Google Scholar]
- Tran D, Carpentier JL, Sawano F, Gordon P, Orci L. Ligands internalized through coated or non-coated invaginations follow a common intracellular pathway. Proc Natl Acad Sci USA. 1987;84:7957–7961. doi: 10.1073/pnas.84.22.7957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasile E, Simionescu M, Simionescu N. Visualization of the binding, endocytosis of low density lipoprotein in the arterial endothelium in situ. J Cell Biol. 1983;96:1677–1689. doi: 10.1083/jcb.96.6.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Houtten M, Posner BI. Insulin binds to brain blood vessels in vivo. Nature (Lond) 1979;282:623–628. doi: 10.1038/282623a0. [DOI] [PubMed] [Google Scholar]
- Villegas JC, Broadwell RD. Transcytosis of protein through the mammalian cerebral epithelium. II. Adsorptive transcytosis of WGA-HP and the blood-brain and brain-blood barriers. J Neurocytol. 1993;22:67–80. doi: 10.1007/BF01181571. [DOI] [PubMed] [Google Scholar]
- Wolf BB, Lopez MBS, VandenBerg SR, Gonias SL. Characterization and immunohistochemical localization of α2-macroglobulin receptor (low-density lipoprotein receptor-related protein) in human brain. Am J Pathol. 1992;141:37–42. [PMC free article] [PubMed] [Google Scholar]