

Abstract
The transcription regulators, PDR1 and PDR3, have been shown to activate the transcription of numerous genes involved in a wide range of functions, including resistance to physical and chemical stress, membrane transport, and organelle function in Saccharomyces cerevisiae. We report here that PDR1 and PDR3 also regulate the transcription of one or more undetermined genes that translocate endogenous and fluorescent-labeled (M-C6-NBD-PE) phosphatidylethanolamine across the plasma membrane. A combination of fluorescence microscopy, fluorometry, and quantitative analysis demonstrated that M-C6-NBD-PE can be translocated both inward and outward across the plasma membrane of yeast cells. Mutants, defective in the accumulation of M-C6-NBD-PE, were isolated by selectively photokilling normal cells that accumulated the fluorescent phospholipid. This led to the isolation of numerous trafficking in phosphatidylethanolamine (tpe) mutants that were defective in intracellular accumulation of M-C6-NBD-PE. Complementation cloning and linkage analysis led to the identification of the dominant mutation TPE1-1 as a new allele of PDR1 and the semidominant mutation tpe2-1 as a new allele of PDR3. The amount of endogenous phosphatidylethanolamine exposed to the outer leaflet of the plasma membrane was measured by covalent labeling with the impermeant amino reagent, trinitrobenzenesulfonic acid. The amount of outer leaflet phosphatidylethanolamine in both mutant strains increased four- to fivefold relative to the parent Tpe+ strain, indicating that the net inward flux of endogenous phosphatidylethanolamine as well as M-C6-NBD-PE was decreased. Targeted deletions of PDR1 in the new allele, PDR1-11, and PDR3 in the new allele, pdr3-11, resulted in normal M-C6-NBD-PE accumulation, confirming that PDR1-11 and pdr3-11 were gain-of-function mutations in PDR1 and PDR3, respectively. Both mutant alleles resulted in resistance to the drugs cycloheximide, oligomycin, and 4-nitroquinoline N-oxide (4-NQO). However, a previously identified drug-resistant allele, pdr3-2, accumulated normal amounts of M-C6-NBD-PE, indicating allele specificity for the loss of M-C6-NBD-PE accumulation. These data demonstrated that PDR1 and PDR3 regulate the net rate of M-C6-NBD-PE translocation (flip-flop) and the steady-state distribution of endogenous phosphatidylethanolamine across the plasma membrane.
An asymmetric distribution of phospholipids across the plasma membrane of cells has been documented for numerous cell types (15, 16, 54). In all cells where reliable measurements have been made, the majority of phosphatidylserine and phosphatidylethanolamine are located on the inner leaflet, whereas phosphatidylcholine, sphingomyelin, and glycolipids are located on the outer leaflet. Regulation of the loss of this asymmetric distribution has been shown to function as a signal for several important physiological processes, including platelet activation (39), clearance of senescent red cells (11), and phagocytosis of apoptotic cells (17, 18). Apart from these intercellular signaling functions, the role for the establishment and regulation of an asymmetric distribution of phospholipids across the plasma membrane of cells is not understood. Although Mg2+, ATP-dependent flip-flop of the aminophospholipids, phosphatidylserine and phosphatidylethanolamine, by an aminophospholipid translocase is thought to be responsible for establishing their asymmetric distribution, it is not known how many phospholipid translocases are required or how they are regulated to establish the appropriate phospholipid distribution.
In this paper, we developed a genetic approach using the yeast Saccharomyces cerevisiae to address these questions. First, we characterized the influx and efflux pathways for 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD)1-labeled phosphatidylethanolamine (M-C6-NBD-PE) and found that influx occurred by a mechanism that has similar characteristics to the flippase-mediated uptake of phosphatidylethanolamine in mammalian cells. Second, we developed a novel method of mutant enrichment by dye-sensitized photokilling and isolated new mutant alleles in two genes (PDR1 and PDR3) that result in the loss of M-C6-NBD-PE accumulation in cells and an increase in the amount of endogenous phosphatidylethanolamine in the outer leaflet of the plasma membrane. Both PDR1 and PDR3 encode transcription factors that activate the transcription of numerous genes that confer resistance to a wide range of drugs (5). The results indicated that PDR1 and PDR3 regulate phosphatidylethanolamine distribution across the plasma membrane, presumably by increasing the transcription of one or more undetermined genes that regulate the net translocation (flip-flop) of phosphatidylethanolamine across the plasma membrane.
Materials and Methods
Materials
Yeast media were obtained from Difco Laboratories (Detroit, MI). Restriction enzymes and molecular biology reagents were purchased from Life Technologies (Gaithersburg, MD). Unless otherwise noted, all other reagents were purchased from Sigma Chemical Co. (St. Louis, MO).
Yeast Strains and Culture
The Saccharomyces cerevisiae strains used are shown in Table I. For all experiments, early to mid-log phase cultures (OD600 = 0.2–1) were grown from either overnight cultures or fresh plates in the indicated media as described in Sherman et al. (44). The media used were: YPD, medium containing 1% yeast extract, 2% glucose, 2% peptone; YEP-GE, medium containing 1% yeast extract, 2% peptone, 2% glycerol, and 2% ethanol; YP-Gal, medium containing 1% yeast extract, 2% galactose, 2% peptone; SDC, synthetic complete media: 0.67% yeast nitrogen base, 2% glucose, complete amino acid supplements; SCNaN3, SDC lacking glucose but containing 2% sorbitol and 20 mM sodium azide. Temperature-sensitive strains were grown at a permissive temperature of 23°C, and internalization assays were performed at the nonpermissive temperature of 37°C. Yeast transformations were performed as described by Gietz et al. (19).
Table I.
Yeast Strains Used in This Study
| Strain | Genotype | Source | ||
|---|---|---|---|---|
| CRY3* | MAT a Sec+ can1-100 ade2-1 his 3-11,15 leu2-3, 112 trpl-1 ura3-1 | R.S. Fuller lab collection | ||
| MATα Sec+ can1-100 ade2-1 his3-11,15 leu2-3, 112 trpl-1 ura3-1 | ||||
| KRY78-4A | MAT a Tpe+ can1-100 ade2-1 his3-11,15 leu2-3, 112 trp1-1 ura3-1 lys2 | R.S. Fuller lab collection | ||
| CRY2* | MATα Tpe+ can1-100 ade2-1 his3-11,15 leu2-3, 112 trpl-1 ura3-1 LYS2 | R.S. Fuller lab collection | ||
| LKY6-2B | MAT a Tpe+ ade2 HIS3 ura3-1 lys2 | This study | ||
| LKY118 | MATα Tpe+ can1-100 ade2-1 HIS3- leu2-3, 112 trp1-1 ura3-1 lys2 | This study | ||
| LKY115 | MAT a Tpe+ can1-100 ade2-1 his3-11,15 leu2-3, 112 trp1-1 ura3-1 LYS2 | This study | ||
| LKY155 | MATα TPE1-1 ade2-1 his3-11-15 ura3-1 LYS2 | This study | ||
| LKY149 | MATα TPE1-1 ade2-1 HIS3 ura3-1 lys2 | This study | ||
| LKY141 | MAT a TPE1-2 ade2-1 HIS3 ura3-1 lys2 | This study | ||
| LKY157 | MAT a tpe2-1 ade2-1 his3-11-15 ura3-1 LYS2 | This study | ||
| LKY156 | MAT a tpe2-1 ade2-1 HIS3 ura3-1 lys2 | This study | ||
| LKY161 | MATα tpe2-1 ade2-1 HIS3 ura3-1 lys2 | This study | ||
| AGY10 | MAT a ade8 | J.W. Nichols lab collection | ||
| AGY124 | pAG3 integrated into PDR3 locus in LKY161 | This study | ||
| YYMI4-O3 | MAT a PDR3 trp1 his3 ura3 leu2 ade2 (parental strain of YYMI4-A4) | K. Kuchler lab collection | ||
| YYMI4-A4‡ | MAT a pdr3-2 trp1 his3 ura3 leu2 ade2 | K. Kuchler lab collection | ||
| YALA-G4§ | MAT a PDRI-3 ura3-52 leu2-3,112 his3-11,115 trp1-1 | K. Kuchler lab collection | ||
| YALA-B1 | MAT a ura3-52 leu2-3,112 his 3-11,115 trp1-1 PDR1 | K. Kuchler lab collection | ||
| YPH500 | MATα ura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 ade2-101 lys2-801 (parental strain of YKKB-13, YYM3 and YYM5) | K. Kuchler lab collection | ||
| YKKB-13 | MATα ura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 ade2-101 lys2-801 Δpdr5::TRP1 | K. Kuchler lab collection | ||
| YYM3 | MATα ura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 ade2-101 lys2-801 Δsnq2::hisG Δpdr5::TRP1 | K. Kuchler lab collection | ||
| YYM5 | MATα ura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 ade2-101 lys2-801 Δsnq2::hisG | K. Kuchler lab collection | ||
| NY13 | MATa Sec+ ura3-52 | P. Novick, Yale University | ||
| NY17 | MATa sec6-4 ura3-52 | P. Novick, Yale University | ||
| KRY30-2C* | MATa sec1-1 can1-100 ade2-1 his3-11,15 leu2-3, 112 trp1-1 | R.S. Fuller lab collection | ||
| KRY31-3C* | MATa sec7-1 can1-100 ade2-1 his3-11,15 leu2-3, 112 trp1-1 | R.S. Fuller lab collection | ||
| CWS11-2* | MATa sec11 ade2-1 his2-11,15 leu2-3-112 trpl-1 ura3-1 | R.S. Fuller lab collection | ||
| KRY33-2C* | MATα sec14 can1-100 ade2-1 his3-11,15 leu2-3, 112 trp1-1 ura3-1 | R.S. Fuller lab collection | ||
| CWS18-1 | MATα sec18 can1-100 ade2-1 his3-11,15 leu2-3, 112 trp1-1 ura3-1 | R.S. Fuller lab collection | ||
| X2180-1A | MATa Sec+ SUC2 mal gal2 CUP1 | Yeast Genetic Stock Center | ||
| SF266-1C | MATa sec12-4 SUC2 mal gal2 CUP1 | Yeast Genetic Stock Center |
Strains were derived from a nominally isogenic pair of strains, W303-1A (Mat a can1-100 ade 2-1 his3-11,15 leu2-3, 112 trp1-1) and W3031B (W303-1A MATα) originally obtained from R. Rothstein, Columbia University (50).
The pdr3-2 mutant allele was initially isolated by Guerineau et al. (20) and subsequently identified as an allele of PDR3 by Subik et al., 1986 (48).
Vesicle Preparation
M-C6-NBD-PE, M-C6-NBD-PC, dioleoylphosphatidylcholine (DOPC), and N-rhodamine-dioleoylphosphatidylethanolamine (N-Rh-DOPE) were from Avanti Polar Lipids Inc. (Alabaster, AL). FM4-64 was purchased from Molecular Probes, Inc. (Eugene, OR). Phospholipids were stored at −20°C, periodically monitored for purity by thin-layer chromatography (TLC), and repurified when necessary. Phospholipid concentrations were determined by lipid phosphorus assay (2). To prepare vesicles, lipids were first mixed in desired proportions, and the chloroform was removed by evaporation under nitrogen followed by overnight vacuum desiccation. Desiccated lipids were then solubilized either in SDC medium or SDC medium lacking glucose (for ATP-depletion experiments), and the mixture was passed eight times through a Lipex Extruder (Lipex Biomembranes, Inc., Vancouver, BC, Canada) equipped with 0.1-mm filters to produce vesicles. Total lipid concentration in the stock vesicle preparation was 1 mM. Typically, proportions were 40 mol% NBD-labeled phospholipid, 2 mol% N-Rh-DOPE, and 58 mol% DOPC.
Internalization of Fluorescent Lipids into Yeast Cells
Cells were grown to mid-log phase in YPD or SDC at 30°C. Cells were washed three times in SDC and resuspended at ∼107 cells/ml. Donor vesicles containing 40% NBD-labeled phospholipid, 58% DOPC, and 2% N-Rh-DOPE (50 μM total lipid concentration) were added to the cells and incubated for 30–120 min. Cells were washed three times with ice-cold SCNaN3 before microscopy. Specific incubation conditions for the different fluorescent lipids are as follows: Internalization experiments with M-C6-NBD-PE typically entailed incubation with donor vesicles for 30 min before microscopy. Experiments with M-C6-NBD-PC typically entailed incubation for 2 h with the fluorescent lipid before microscopy. FM4-64 internalization experiments were performed by adding 1 μl of this dye from a 1 mg/ml ethanolic stock solution to 1 ml of cells at ∼1 × 107/ml and continuing incubation for 2 h before microscopy.
For M-C6-NBD-PE internalization experiments with sec mutants, growth to mid-log phase was carried out at 23°C. Cultures were then split, and half of the cells were warmed to the restrictive temperature of 37°C, while the other half remained at 23°C. Warming to 37°C was accomplished by gradually increasing the temperature over a period of ∼10 min. After shifting the temperature for 30 min, vesicles were added, and incubation at 37°C was continued for 1 h. Cells were then transferred to an ice-water bath and washed rapidly with ice-cold SCNaN3 medium in preparation for microscopy. Controls were processed identically.
To quantitate the results with the sec mutants and their isogenic Sec+ strains, 15 to 30 cells were analyzed for each of the strains at their permissive (23°C) and nonpermissive (37°C) temperatures for growth. Although there were strain to strain variations in M-C6-NBD-PE accumulation at the permissive temperature for each isogenic pair, the difference in accumulation at 23°C did not exceed 27%. The temperature-sensitive accumulation of M-C6-NBD-PE for each strain was expressed as a ratio of the average pixel brightness at 37°C relative to 23°C. Inhibition of growth of the sec mutants at 37°C was used to confirm the temperature-sensitive growth phenotype of all of the sec strains used in these studies.
To deplete intracellular ATP, cells were incubated with shaking in SCNaN3 for 45–50 min at 30°C before labeling as described above. We have previously shown that this treatment results in 80% reduction in cellular ATP levels (26). To test the effect of N-ethylmaleimide (NEM) treatment on M-C6-NBD-PE accumulation, mid-log phase cells were cooled to 2°C, harvested by centrifugation, and resuspended in buffered media (SDC containing 50 mM sodium phosphate buffer, pH 7.5) containing 2 mM NEM. Cells were incubated on ice for 30 min and were shaken after the first 15 min. Cells recovered by centrifugation were resuspended in buffered medium containing 2 mM DTT and incubated on ice for 5 min. Cells were pelleted again, washed once with buffered medium, and resuspended in buffered medium before labeling with M-C6-NBD-PE as described above. After labeling, cells were incubated on ice for 1.5 h and washed three times with ice-cold SCNaN3 buffered with 50 mM sodium phosphate, pH 7.5, before fluorescence microscopy.
Fluorescence Microscopy
Fluorescence microscopy was performed on a microscope (model Axiovert; Carl Zeiss, Inc., Thornwood, NY) equipped with barrier filters that allowed no detectable crossover of NBD and rhodamine fluorescence. For localization of 4′,6-diamidino-2-phenylindole (DAPI) fluorescence, barrier filters were used that did not allow detectable crossover of NBD and DAPI fluorescence. The fluorescence image was enhanced with an image-intensifying camera (model SIT 66 or VE1000-SIT; DAGE-MTI, Inc., Michigan City, IN), digitized, and stored. Contrast enhancement and pixel brightness analysis of the digital images were performed with Image-1/AT or Metamorph software (Universal Imaging Corp., West Chester, PA). Average pixel brightness was measured from a rectangle (1 × 3 pixels) in the middle of vacuoles (for vacuolar labeling with M-C6-NBD-PC) or a rectangle surrounding the entire cell (for all other labeling experiments). Pixel brightness analysis of DOPC vesicles containing known amounts of M-C6-NBD-PC confirmed that the measurements fell within the linear range of the SIT camera. To confirm that the pixel brightness measurements accurately reflected the amount of internalized M-C6-NBD-PE, labeled cells were extracted (22) and analyzed by TLC and fluorometry (26). Fluorescence readings of the total extract as well as the TLC-purified M-C6-NBD-PE were proportional to the pixel brightness measurements.
Trinitrobenzenesulfonic Acid Labeling
Cells were grown 18 h at 30°C to mid-log phase (OD600 = 0.4–0.7) in SDC containing 500 μCi of [32P]KH2PO4 (specific activity 1 Ci/mmol; New England Nuclear, Boston, MA). Plasma membrane, outer leaflet phosphatidylethanolamine was labeled with TNBS as described previously (30). Cells were harvested by centrifugation, washed twice in ice-cold 40 mM NaCl, 120 mM NaHCO3, pH 8.4, resuspended in the same buffer containing 5 mM trinitrobenzenesulfonic acid (TNBS) (Sigma Chemical Co.), and immediately placed on ice for 1 h with periodic vortex mixing. After TNBS labeling, cells were washed by centrifugation three times in fresh buffer, pelleted, and disrupted by vortexing with glass beads. Cellular lipids were extracted with chloroform/methanol (2:1) and separated by two- dimensional TLC (first solvent: chloroform/methanol/ammonium hydroxide, 65:35:5; second solvent: chloroform/methanol/acetone/acetic acid/water; 50:10:20:10:5). Spots were identified by comparison with known standards. The percentages of phosphatidylethanolamine and TNP-phosphatidylethanolamine were quantified by phosphorimaging with a PhosphorImager SI scanning instrument (Molecular Dynamics, Sunnyvale, CA). Percent viability was determined by counting the number of cells labeled after dilution into 0.02% methylene blue with a hemocytometer.
Isolation of Trafficking in Phosphatidylethanolamine (tpe) Mutants
Isogenic strains LKY6-2B (MAT a) and CRY2 (MATα) were mutagenized at 23°C with ethylmethanesulfonate to 80% killing (27). For dye-sensitized photokilling, mutagenized cells were grown to mid-log phase in SDC at 23°C before shifting to 37°C for 30 min. Donor vesicles (50 μM total lipid concentration) containing 40 mol% M-C6-NBD-PE were then added as described above, and incubation continued for 30 min. After the initial 30-min incubation, cells (7 ml at 1 × 107 cells/ml) were transferred to a 60-mm Petri dish, which was placed in a chamber equilibrated to 37°C by a circulating water bath, and incubation was continued for an additional 2 h (in the continuous presence of vesicles). During this incubation, cells were irradiated with the light from a 150-W xenon lamp (Second Source Inc., LaVerne, CA) that was passed through a narrow band-pass filter at 470–480 nm, producing an intense blue light. After irradiation, cells were washed three times in SDC and either plated for single colonies or incubated overnight at 23°C in preparation for successive rounds of photokilling. The temperature shift from 37°C (incubation with vesicles and irradiation) to 23°C (growth overnight) allowed the potential isolation of temperature-sensitive mutants. The tpe mutants were isolated after five rounds of dye-sensitized photokilling.
Drug Sensitivity Assays
1 mg/ml stock solutions of drugs were made by dissolving drugs in DMSO or ethanol. Resistance to drugs such as cycloheximide, oligomycin, and 4-nitroquinoline-N-oxide was determined using the spot test assay. For the spot test assay, 5-μl aliquots of yeast mid-log phase cultures (OD600 = 0.25) were spotted onto YPD plates containing the indicated concentrations of cycloheximide or 4-nitroquinoline-N-oxide, or YPG plates containing the indicated concentrations of oligomycin. Plates were incubated for 3 d at 23, 30, or 37°C.
Cloning of TPE2
The TPE2 gene was identified by its ability to complement the temperature-sensitive growth defect of tpe2-1 on nonfermentable carbon sources. The tpe2-1 strain LKY161 was transformed with the yeast genomic library, YPH1, which contains the selectable marker LEU2. Candidate TPE2 clones were identified based on their ability to restore growth of tpe2-1 on SC-glycerol-ethanol plates lacking leucine at 37°C. Only one plasmid was isolated that was able to complement both the M-C6-NBD-PE accumulation defect as well as the temperature-sensitive growth defect on nonfermentable carbon sources of tpe2-1. This plasmid was designated p201. p201 contained a 15-kb insert that sequencing revealed was a portion of chromosome II spanning half of the SLA1 gene through approximately one third of the ORF YBL004. Only two full-length ORFs are contained within this region, PDR3 and an uncharacterized ORF YBL006.
To identify the portion of p201 responsible for complementing tpe2-1, LKY161 was transformed with subclones of p201 containing either YBL006 or PDR3. Integrative transformation with YBL006 did not complement tpe2-1; however, transformation with PDR3 resulted in full complementation of both the M-C6-NBD-PE accumulation defect as well as the temperature-sensitive growth defect on nonfermentable carbon sources. The strain transformed with PDR3 was designated AGY124.
Linkage analysis was used to determine whether the PDR3 was TPE2 (38). AGY124 was crossed to AGY10, a strain that is wild-type at the TPE2 and LEU2 loci. All spores from this cross were wild-type for the PE accumulation phenotype. This confirmed pAG3 integrated at the PDR3 locus and that TPE2 was linked to PDR3.
Disruption of PDR1 and PDR3
For disruption of the PDR3 gene, a 2.3-kb portion of the PDR3 coding region was removed by digestion with MunI and EcoRI and replaced with the HisG-URA3-HisG cassette of the vector pNKY51 (1). To disrupt PDR1, 2.2 kb of the coding region was removed by digestion with MunI and replaced by the HisG-URA3-HisG cassette. Proper integration of the disrupted alleles was confirmed by PCR or Southern blotting analysis.
Cloning and Sequencing of Mutant and Wild-Type PDR3 Alleles
PDR3 alleles were rescued using the method described by Winston et al. (55). The alleles were obtained by integrative transformation of a YIp vector near the PDR3 locus and subsequent eviction of a plasmid containing the vector along with the full-length PDR3 allele of interest. Plasmid DNA was isolated from Escherichia coli transformants and the presence of PDR3 alleles was confirmed by restriction digest analysis.
The nucleotide sequence of the entire open reading frame along with an additional 400 bp upstream of the translation initiation codon and 250 bp downstream of the termination codon of each PDR3 allele was determined. Sequencing reactions were performed using the PRISM Dye Terminator Cycle Sequencing Kit (Applied Biosystems, Inc., Foster City, CA), an automated sequencer (model 373; Applied Biosystems, Inc.), and a set of 12 16-mer primers designed to anneal to sequences ∼300 bp apart in the PDR3 gene.
Statistical Analysis
Statistical analysis was performed with the InStat statistics software from GraphPad Inc. (San Diego, CA). χ2 analysis and two-tailed P values were obtained from 3 × 2 contingency tables, or the Student's t test was used to compare the means and calculate a two-tailed P value from analysis of variance.
Results
Analysis of M-C6-NBD-PE Influx and Efflux
When the diploid strain, CRY3, was incubated at 30°C in the presence of donor vesicles containing N-Rh-DOPE, DOPC, and either M-C6-NBD-PE or M-C6-NBD-PC, the rapidly exchangeable lipids, M-C6-NBD-PE and M-C6-NBD-PC (33), were internalized (Fig. 1). The absence of rhodamine fluorescence in the cells (not shown) indicated that the nonexchangeable fluorescent lipid, N-Rh-DOPE (35), was not internalized. Thus, the accumulation of intracellular NBD fluorescence was not due to fusion of donor vesicles or internalization of intact vesicles, but rather internalization of the NBD-phospholipid from the outer leaflet of the plasma membrane after transfer from the donor vesicles (26). Fig. 1 shows that M-C6-NBD-PC and M-C6-NBD-PE are sorted to different organelles in yeast. M-C6-NBD-PC was transported primarily to the vacuole (the yeast lysosome) and, to a much smaller extent, to the mitochondria and nuclear envelope (as previously documented, [26]), while M-C6-NBD-PE was excluded from the vacuole and instead accumulated in the mitochondria and nuclear envelope, as determined by colocalization with DAPI (Fig. 2), and perhaps other unidentified organelles.
Figure 1.

M-C6-NBD-PC and M-C6-NBD-PE are sorted to different organelles in yeast. The diploid strain CRY3 was grown to mid-log phase in SDC medium at 30°C and incubated with donor vesicles containing 58 mol% DOPC, 2 mol% N-Rh-DOPE, and either 40 mol% M-C6-NBD-PC or 40 mol% M-C6-NBD-PE at 30°C for 2 h (M-C6-NBD-PC) or 1 h (M-C6-NBD-PE). Microscopy was performed as described in Materials and Methods. (A and C) Cells incubated with M-C6-NBD-PE. (B and D) Cells incubated with M-C6-NBD-PC. (A and B) NBD fluorescence. (C and D) Differential interference contrast (DIC) optics. The identification of organelles colocalizing with M-C6-NBD-PE fluorescence is shown in Fig. 2. Bar: 10 μm.
Figure 2.

M-C6-NBD-PE is sorted to the mitochondria and nuclear envelope from the yeast plasma membrane. CRY3 was grown to mid-log phase in SDC medium at 30°C and incubated with M-C6-NBD-PE–containing vesicles for 1 h as described in Materials and Methods. Colocalization of NBD fluorescence with DAPI fluorescence of nuclear and mitochondrial DNA was performed by including 2.5 μg/ml DAPI with the M-C6-NBD-PE–containing vesicles. Top row, NBD fluorescence; middle row, DAPI fluorescence; bottom row, DIC optics. Solid arrows point to nuclear envelope (NBD fluorescence) and nuclear DNA (DAPI fluorescence); open arrows point to mitochondria (NBD fluorescence) and mitochondrial DNA (DAPI fluorescence). Bar, 10 μm.
Fig. 3 shows a time course of M-C6-NBD-PE internalization and efflux by a combination of fluorescence microscopy, spectrofluorometry, and quantitation after extraction and separation by TLC. The spectrofluorometric internalization assay (Fig. 3 A) measured the increase in NBD-fluorescence as M-C6-NBD-PE transferred from the quenching environment of the donor vesicles to the nonquenching environment of the yeast cellular membranes. The accuracy of the spectrofluorometric assay, as well as fluorescence microscopy digital imaging, to measure the accumulation of M-C6-NBD-PE were confirmed by concomitant extraction and TLC separation. No degradation products of M-C6-NBD-PE were detected in the cell extracts, indicating that only intact M-C6-NBD-PE was internalized and that either intracellular degradation was insignificant or that degraded products were rapidly effluxed from the cell. All three techniques illustrated that the net amount of internalized M-C6-NBD-PE steadily increased during an hour of incubation with the donor vesicles (Fig. 3, A and D).
Figure 3.
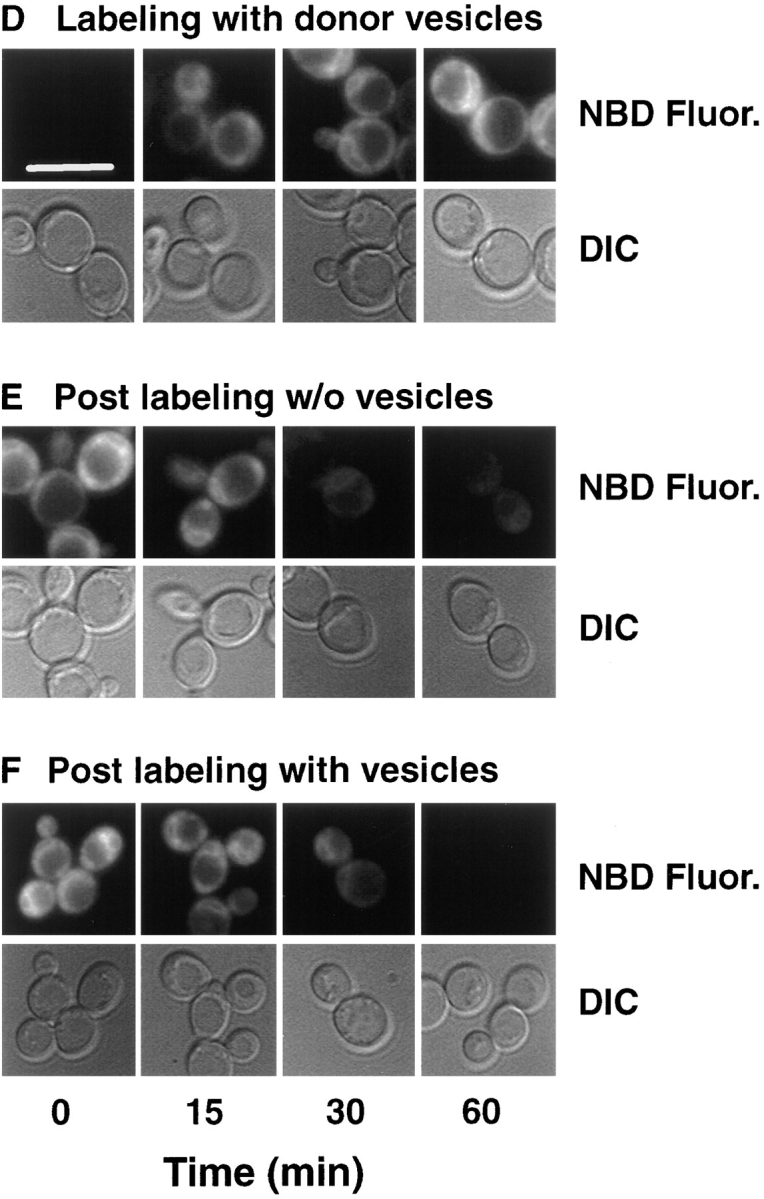

Cellular M-C6-NBD-PE influx and efflux measured by fluorescence microscopy, fluorometry, and quantitative TLC. CRY2 was grown to mid-log phase in SDC medium at 30°C. NY17 (sec6-4) was grown to mid-log in SDC at room temperature (∼23°C). The sec6-4 cells were warmed to 37°C for 30 min before labeling with M-C6-NBD-PE and were treated identically to the CRY2 cells as described below with the exception that they were maintained at 37°C during influx and efflux instead of 30°C. CRY2 cells were pelleted and resuspended in SDC + 2% sorbitol at OD600 = 0.16. Labeling of cells was initiated by the addition of M-C6-NBD-PE–containing donor vesicles (vesicle phospholipid concentration, 50 μM). A 2-ml aliquot was immediately placed in a stirred fluorometer cuvette at 30°C and NBD fluorescence (excitation, 475 nm; emission, 530 nm) was recorded continuously. The remainder were placed in a shaker incubator at 30°C. Aliquots were removed at the indicated times and washed three times in SCNaN3. A small aliquot was removed for fluorescence microscopy and the remainder were extracted, separated by TLC, and quantified by digital imaging of the M-C6-NBD-PE fluorescent spots. After 1 h, the remaining labeled cells were washed three times in ice-cold SDC + 2% sorbitol. Measurement of M-C6-NBD-PE in the cells and medium post labeling was initiated by returning the cells to 30°C in the shaker incubator in the presence or absence of unlabeled DOPC vesicles (50 μM). 2-ml aliquots were removed and placed in the fluorometer for continuous recording as above. Aliquots were removed at the indicated times for fluorescence microscopy, TLC separation and quantification as described above. (A and D) Labeling in the presence of M-C6-NBD-PE donor vesicles. (B and E) Post labeling in the absence of acceptor vesicles. (C and F) Post labeling in the presence of acceptor vesicles. Images in D–F are of CRY2 cells only. The appearance of the sec6-4 cells were similar and are not shown. Solid lines are fluorometer traces adjusted to 100% of maximum signal. Filled symbols refer to extracted, cell-associated M-C6-NBD-PE; open symbols refer to the M-C6-NBD-PE extracted from the supernatant. Circles, CRY2; squares, sec6-4. Bar, 10 μm.
After the net internalization of M-C6-NBD-PE, cells were washed free of the donor vesicles and incubated at 30°C with (Fig. 3, C and F) or without (Fig. 3, B and E) excess unlabeled acceptor vesicles, and the NBD fluorescence was monitored by the three techniques described above. Since NBD fluorescence is highly sensitive to the polarity of its immediate environment (32, 34), degradation of M-C6-NBD-PE to water-soluble products resulted in almost complete loss of fluorescence. Thus, the data presented in Fig. 3, B and E, illustrated a time-dependent degradation of M-C6-NBD-PE. To determine whether the degradation occurred intracellularly or was subsequent to efflux from the cells, the rate of loss of cellular fluorescence and degradation were monitored in the presence of excess unlabeled acceptor vesicles. Removal of the intact probe from the cell surface by its transfer to the acceptor vesicles would be expected to prevent its degradation and therefore inhibit the overall rate of loss of the fluorescence signal. The result (Fig. 3, C and F) illustrated that concomitant with its loss from the cells, intact M-C6-NBD-PE appeared in the acceptor vesicle–containing medium. This result was confirmed by the slower rate of degradation detected by fluorometry (Fig. 3 C). These data indicated that intact M-C6-NBD-PE was effluxed from the cells and degraded. The existence of an extracellular phospholipase B capable of hydrolyzing NBD-labeled lipids has been described previously (28, 56). These data are consistent with the interpretation that in the presence of excess M-C6-NBD-PE–containing donor vesicles, the concurrent influx and efflux resulted in net intracellular accumulation. Upon removal of the donor vesicles, the net flux reversed, resulting in degradation of M-C6-NBD-PE by extracellular phospholipases unless it was removed from the cell surface by transferring to acceptor vesicles in the medium.
Similar results were obtained when the above experiment was performed with the temperature-sensitive sec6-4 strain. At the nonpermissive temperature (37°C), the transport of secretory vesicles to the plasma membrane in this strain is blocked, resulting in the accumulation of excess intracellular secretory vesicles (53). Numerous sec mutants, including sec6-4, that inhibit vesicular traffic (36) also inhibit the endocytic uptake of M-C6-NBD-PC (26) and the lipophillic membrane probe, FM4-64 (52). Thus, the observation that M-C6-NBD-PE internalization and efflux occurred at rates similar to that observed in the wild-type Sec+ strain (Fig. 3, A and C) indicated that M-C6-NBD-PE influx was not dependent on the same pathway as M-C6-NBD-PC and FM4-64 endocytosis nor was its efflux dependent on secretion.
Additional sec mutant strains were tested for their ability to accumulate M-C6-NBD-PE by measuring the ratio of M-C6-NBD-PE accumulation (measured by average pixel brightness) at the nonpermissive temperature (37°C) to that at the permissive temperature (23°C). This ratio for sec1, 7, 11, 12, 14, and 18 ranged from 70 to 97% of their isogenic Sec+ strains. Thus, M-C6-NBD-PE internalization occurs even when the endocytosis of M-C6-NBD-PC and FM4-64 are inhibited. In addition, the observations that M-C6-NBD-PE is not transported to the vacuole (Figs. 1 and 2) and is not degraded intracellularly further support the conclusion that M-C6-NBD-PE is internalized primarily by a mechanism that does not require endocytosis.
M-C6-NBD-PE transport in yeast possessed many of the same characteristics as NBD-labeled phosphatidylethanolamine sorting in mammalian cells. In addition to targeting to the same intracellular organelles, M-C6-NBD-PE transport in yeast was inhibited by ATP depletion, low temperature, and NEM treatment (Fig. 4). Preincubation of the CRY2 strain with SCNaN3 medium depleted cellular ATP by 80% (26) and reduced the accumulation of M-C6-NBD-PE to 7% of the pixel brightness of the untreated control cells. Incubation at 2°C reduced accumulation to 10% of controls. Pretreatment and labeling of CRY2 cells at 2°C in the presence of 2 mM NEM further reduced the pixel brightness to 4%. The similarity of these characteristics of M-C6-NBD-PE traffic in yeast to NBD-labeled phosphatidylethanolamine traffic in mammalian cells suggests that it is translocated across the yeast plasma membrane by a protein analogous to the mammalian aminophospholipid translocase (31, 45, 49).
Figure 4.

M-C6-NBD-PE transport in yeast is ATP-, temperature- and NEM-sensitive. Cells were treated with SCNaN3 medium (to deplete ATP stores), low temperature, or NEM as described in Materials and Methods. M-C6-NBD-PE internalization assays, fluorescence microscopy, and pixel brightness analysis were then performed as described in Materials and Methods. At least 20 cells were analyzed to determine the mean pixel brightness for each condition. These numeric values are shown above each column.
Isolation of Mutants That Do Not Accumulate M-C6-NBD-PE
We developed a novel scheme for isolating yeast mutants (referred to as tpe mutants) that were defective in the accumulation of M-C6-NBD-PE. As shown above (Figs. 1–3), wild-type yeast cells became highly fluorescent when they internalized M-C6-NBD-PE. Exposure of the labeled cell cultures to intense light between 470–480 nm (the excitation maximum for NBD) for 2 h resulted in the death of >95% of the cells. Photokilling required both incubation with vesicles and illumination. Elimination of either incubation condition eliminated the photokilling. Mutagenized cultures were subjected to five “rounds” of photokilling enrichment (described in detail in Materials and Methods) and then plated for single colonies. Individual colonies were examined microscopically for defects in M-C6-NBD-PE accumulation. Testing multiple individual colonies that grew on rich medium containing glucose (YPD medium) after five rounds of dye-sensitized photokilling revealed a >25:1 ratio of tpe mutants/wild-type survivors. 8–10 Tpe− survivors from each independently mutagenized culture were saved for further analysis.
Fig. 5 shows the phenotype of tpe mutants representing two different complementation groups (see analysis below) obtained from the photokilling enrichment. After incubation with donor vesicles containing M-C6-NBD-PE, tpe mutants showed no fluorescence when microscopy was performed with a neutral density filter that allowed 5% of the light from a 100-W lamp to reach the sample, although in the Tpe+ parent strain, highly fluorescent mitochondria and nuclear envelopes were apparent (Fig. 5, compare A and D). When the neutral density filter was removed, low levels of intracellular and cell surface fluorescence could be observed in both tpe mutants, whereas fluorescent emission from the Tpe+ cells was oversaturated (Fig. 5, compare B, E, and H). Inhibition of the M-C6-NBD-PE accumulation into the tpe mutants detected by fluorescence microscopy was confirmed by extraction of total cellular lipids and recording of total NBD fluorescence in the extract as well as the fluorescence of M-C6-NBD-PE purified from the extract by TLC. Thus, the lack of fluorescence detected by microscopy reflected a quantitative decrease in M-C6-NBD-PE cellular accumulation, and the “ring staining” pattern in the tpe mutants presumably reflected accumulation of M-C6-NBD-PE in the plasma membrane because of inhibition of its inward or activation of its outward translocation across the plasma membrane.
Figure 5.

tpe mutants are inhibited in the accumulation of M-C6-NBD-PE. TPE1-1 and its Tpe+ parent strain, CRY2, were grown to mid-log phase at 23°C. The incubation temperature was then shifted to 37°C for 30 min, and M-C6-NBD-PE internalization assays were performed for 1 h before microscopic analysis as described in Materials and Methods. (A–C) CRY2. (D–F) TPE1-1. (G–I) tpe2-1. (A, B, D, E, G, and H) NBD fluorescence. (C, F, and I) DIC optics. In A and D, a neutral density filter was used that attenuated the excitatory light by 95%. In B and E, the filter was removed to allow 100% of the excitatory light to impinge upon the sample.
Genetic Analysis of the tpe Mutants
The Tpe− clones were placed into two complementation groups by partial complementation of the M-C6-NBD-PE accumulation defect. Two independent isolates from the first complementation group and one from the second were selected for further characterization. These three mutant strains were back-crossed three times against the Tpe+ parent strains to produce congenic Tpe− strains. Tpe+/Tpe− phenotypes segregated 2:2 in each of the nine tetrads analyzed per back-cross for all three Tpe− strains, indicating that for each tpe allele, a single gene was responsible for the Tpe− phenotype. Diploids containing one Tpe+ and one Tpe− allele were not capable of accumulating M-C6-NBD-PE to wild-type levels. Diploids resulting from crosses of the two isolates from the first complementation group to their isogenic parent strain remained defective in M-C6-NBD-PE accumulation, and thus, these two mutant alleles appeared to be dominant. The isolate from the second complementation group when crossed to its parent strain showed a partial defect in M-C6-NBD-PE accumulation and was considered to be semidominant. The two dominant Tpe− strains were assigned to the TPE1-1 and TPE1-2 (capitals to denote dominance) complementation group and the semidominant strain was assigned to tpe2-1.
tpe Mutants Are Temperature-sensitive for Oxidative Phosphorylation
Growth of TPE1-1 and tpe2-1 mutants was temperature-sensitive on nonfermentable carbon sources (Fig. 6), indicating a temperature-sensitive loss of respiratory function in the mutants. After the third back-cross of TPE1-1, TPE1-2, and tpe2-1, the segregation pattern of the respiratory deficiency phenotype (Respiratory−) was determined. In each of the nine tetrads dissected for each mutant allele, Respiratory+/Respiratory− segregated 2:2, indicating that a single gene was responsible for this phenotype. Furthermore, the Respiratory− and M-C6-NBD-PE-accumulation defect cosegregated in 100% of the tetrads analyzed, indicating that the same gene or a tightly linked gene was responsible for both phenotypes. tpe2-1 exhibited a more severe respiration defect than the TPE1-1 mutant, being unable to grow on glycerol/ethanol medium (YEP-GE medium) (Fig. 6) or lactate (not shown) at 30°C, whereas the TPE1-1 mutant exhibited this growth defect only at 37°C. Both TPE1-1 and tpe2-1 mutants grew on YEP-GE medium at 23°C and on the fermentable carbon sources glucose (YPD medium) (Fig. 6) and galactose (YP-Gal medium) (not shown) at 23, 30, and 37°C. TPE1-1/tpe2-1 double mutants exhibited synthetic lethality in that growth on YPD at 37°C and growth on YP-glycerol at 23°C was inhibited (not shown).
Figure 6.
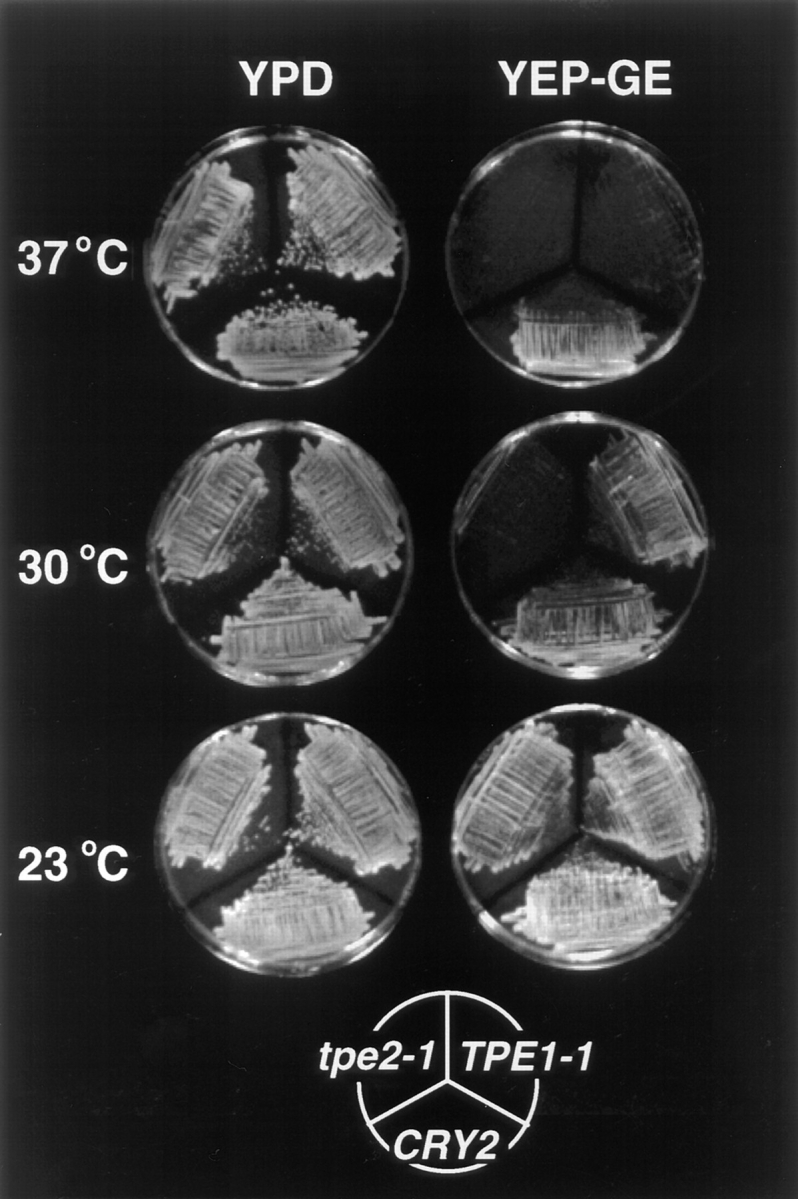
TPE1-1 and tpe2-1 display temperature-sensitive growth defects on nonfermentable carbon sources. TPE1-1, tpe2-1, and CRY2 were plated on rich media containing both a fermentable carbon source (2% glucose) (YPD), or the nonfermentable carbon sources glycerol (2%) and ethanol (2%) (YEP-GE). Plates were incubated for 2 d (30° and 37°C plates) or 4 d (23°C) before photography.
Although the tpe mutants did not arrest when grown in fermentable carbon sources at 37°C, they did exhibit growth defects under these conditions. Growth of TPE1-1 and tpe2-1 in YP-Gal was slowed twofold compared to the isogenic Tpe+ strains, and growth on glucose was reduced by 1.6-fold in rich media (YPD) and fivefold in synthetic media (SDC). Thus, although the growth defects at 37°C in the tpe mutants were predominantly manifested on nonfermentable carbon sources, to some extent growth defects were observed on all media tested.
The Percentage of Endogenous Phosphatidylethanolamine Exposed to the Outer Leaflet of the Plasma Membrane Is Increased in the tpe Mutants
To determine whether the transport of M-C6-NBD-PE across the plasma membrane was indicative of the behavior of endogenous phosphatidylethanolamine, the amount of phosphatidylethanolamine in the outer leaflets of TPE1-1, tpe2-1, and their parent Tpe+ strain was measured by labeling with TNBS using membrane impermeant conditions. The percentage of TNP-labeled phosphatidylethanolamine increased four to fivefold in both mutants relative to the Tpe+ strain (Table II). Thus, the amount of phosphatidylethanolamine in the outer leaflet of the plasma membrane increased in the mutant strains consistent with a decrease in the net influx rate of endogenous phosphatidylethanolamine as well as M-C6-NBD-PE.
Table II.
TNBS Labeling of Phosphatidylethanolamine in the Outer Leaflet of the Plasma Membrane
| Strains | ||||||
|---|---|---|---|---|---|---|
| CRY2 | TPE1-1/ PDR1-11 | tpe2-1/ pdr3-11 | ||||
| Phosphatidyl- ethanolamine | 98.2 ± 0.4 | 91.8 ± 1.5 | 90.3 ± 1.5 | |||
| TPN-phosphatidyl- ethanolamine | 1.8 ± 0.4 | 8.2 ± 1.5 | 9.7 ± 1.4 | |||
32P-labeled cells were reacted with TNBS on ice, washed, extracted, and separated by TLC as described in Materials and Methods. The percentages of phosphatidylethanolamine and TNP-phosphatidylethanolamine were determined by phosphorimaging analysis. The percentage of viable cells was 96.1 ± 0.4, 98.2 ± 0.5, and 95. 1 ± 0.4 for CRY2, TPE1-1, and tpe2-1, respectively. Data are reported as the mean and standard error of four independent analyses from one experiment that was representative of three.
tpe Mutants Are Defective in Membrane-Lipid Endocytosis
The effect of the tpe mutations on endocytosis was tested for two different membrane markers. M-C6-NBD-PC (26) and FM4-64 (52) have been shown previously to be endocytosed to the vacuole in S. cerevisiae. As shown in Table III, rows B and C, accumulation of both probes was inhibited to similar extents in the tpe mutants relative to the parent strain. Although the extent of accumulation of both probes was attenuated, FM4-64 and M-C6-NBD-PC fluorescence was localized to the vacuolar membrane and lumen, respectively (data not shown). Thus, there was a generalized defect in endocytosis of membrane-lipids in the tpe mutants. It is important to note that although endocytosis was inhibited in the tpe mutants, it was not reduced as severely as was the nonendocytic traffic of M-C6-NBD-PE. Estimates of the extent of M-C6-NBD-PC uptake compared to M-C6-NBD-PE by pixel brightness analysis showed that there was at least a 10-fold greater accumulation of M-C6-NBD-PC than M-C6-NBD-PE in the tpe mutants at 37°C (Table III, rows A and B).
Table III.
Internalization of NBD-labeled Lipids and FM4-64 into Mutant and Wild-Type Yeast Strains
| Strains | ||||||
|---|---|---|---|---|---|---|
| CRY2 | TPE1-1/PDR1-11 | tpe2-1/pdr3-11 | ||||
| A M-C6-NBD-PE | 100 ± 16 | 1 ± 1 | 0.3 ± 0.3 | |||
| B M-C6-NBD-PC | 100 ± 38 | 12 ± 3 | 18 ± 8 | |||
| C FM4-64 | 100 ± 58 | 35 ± 23 | 36 ± 24 | |||
Yeast strains were grown to mid-log phase, and lipid internalization assays, microscopy, and pixel brightness analysis were perfomed as described in Materials and Methods. For each determination, the mean pixel brightness of 10–38 cells and standard deviation were determined. For each internalization condition, the pixel brightness of CRY2 was set to 100%, and the pixel brightness in the other strains is shown as a percentage of this value. The pixel brightness of CRY2 under the various incubation conditions was: (A) 177 ± 28; (B) 132 ± 50; (C) 12 ± 7.
The nonspecific internalization in the TPE1-1 and tpe2-1 strains of other amphipathic fluorescent probes, including DAPI and 3,3′-dihexyloxacarbocyanine iodide (DiOC6) (data not shown), to the same level as their isogenic parent strains indicated that inhibition of NBD-lipid accumulation was not due to a nonspecific block of internalization of all lipophillic molecules.
tpe Mutants are Drug Resistant
Given the identification of MDR1 and MDR2 as phospholipid transporters (40, 46, 51), we tested the possibility that the sorting defects in the tpe mutants were due to the upregulation of an MDR-like efflux pump. Activation of a variety of MDR homologues in yeast leads to increased resistance to a large array of drugs because of enhanced efflux of these drugs from the cells (5, 7). If the tpe phenotype resulted from an activated MDR-like pump, mutants would also be expected to display increased drug-resistant phenotypes. We therefore tested the sensitivity of TPE1-1, tpe2-1, and their parent Tpe+ strains to three structurally unrelated drugs, oligomycin, 4-nitroquinoline N-oxide (4-NQO), and cycloheximide, using a spot test assay (see Materials and Methods). Resistance to cycloheximide and 4-NQO has been shown to result primarily from the amplification of the ABC transporters, Pdr5p and Snq2p, respectively (12, 24, 29). Resistance to oligomycin is at least partly due to the amplification of Yor1p (25).
Table IV shows that both TPE1-1 and tpe2-1 are resistant to 4-NQO and cycloheximide at 30°C and are resistant to oligomycin at 23°C, compared to the isogenic Tpe+ strain. The isogenic Tpe+ strain does not grow on YPD plates containing 0.2 μg/ml cycloheximide or greater; however, both tpe mutants grow on plates containing 1 μg/ml cycloheximide (Table IV). TPE1-1 shows a slightly greater resistance to oligomycin and 4-NQO than tpe2-1; however, they both are significantly more resistant than the Tpe+ strain (Table IV). The relative resistance of TPE1-1 and tpe2-1 to cycloheximide and 4-NQO was similar at 23, 30, and 37°C. Tests of oligomycin resistance required growth on a nonfermentable carbon source. Therefore, because of the temperature-sensitive growth defect of the mutants, tests were limited to 23°C.
Table IV.
Drug Resistance Phenotypes of tpe and pdr Strains
| CYH | Oligomycin | 4-NQO | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| μg/ml | μg/ml | μg/ml | ||||||||||||||||
| Strains | 0.2 | 0.5 | 1.0 | 0.3 | 0.5 | 1.0 | 0.5 | 0.8 | 1.0 | |||||||||
| CRY2 (TPE2 TPE1) | − | − | − | − | − | − | +/− | − | − | |||||||||
| LKY161 (tpe2-1/pdr3-11) | + | + | +/− | + | + | − | + | + | +/− | |||||||||
| LKY155 (TPE1-1/PDR1-11) | + | + | +/− | + | + | + | + | + | + | |||||||||
| YYMI4-O3 (PDR3) | − | − | − | − | − | − | +/− | − | − | |||||||||
| YYMI4-A4 (pdr3-2) | + | + | +/− | − | − | − | +/− | +/− | − | |||||||||
| YALA-B1 (PDR1) | − | − | − | − | − | − | +/− | − | − | |||||||||
| YALA-G4 (PDR1-3) | + | + | +/− | + | + | − | + | + | + | |||||||||
The growth of strains spotted on YPD plates containing 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, and 1.0 μg/ml of cycloheximide (CYH) or 4-nitroquinoline-N-oxide (4-NQO) or YPG plates containing the same concentrations of oligomycin was scored after 3–4 d of growth at 30°C for YPD plates and 23°C for YPG plates. +, normal growth; −, no growth; +/−, slow but visible growth.
Identification of TPE2 and TPE1
Complementation of the temperature-sensitive growth defect on nonfermentable carbon sources of tpe2-1 was used to isolate the TPE2 gene (see Materials and Methods). Transformation with PDR3 resulted in full complementation of the M-C6-NBD-PE accumulation defect (Fig. 7 A), cycloheximide resistance (Fig. 7 B), and the temperature-sensitive growth defect on nonfermentable carbon sources of tpe2-1 (data not shown). Linkage analysis determined that tpe2-1 was allelic with PDR3 (38). PDR3 has been identified as a transcriptional activator of the pleiotropic drug resistance network (14).
Figure 7.

Complementation of tpe2-1 by PDR3. The tpe2-1 strain LKY161 was transformed with either the plasmid pRS405 alone or the pRS405 plasmid containing the PDR3, pRS405-PDR3. Four transformants from each transformation were grown to mid-log phase in SDC medium lacking leucine at 23°C. (A) 1 ml of each culture was incubated at 30°C for 30 min, and M-C6-NBD-PE internalization assays and fluorescence microscopy were performed as described in Materials and Methods. (a–c) tpe2-1 transformed with vector pRS405 alone. (d–f) tpe2-1 transformed with pRS405-PDR3. (a, b, d, and e) NBD fluorescence. (c and f) DIC optics. In a and d, a neutral density filter was used that attenuated the excitatory light by 95%. In b and e, the neutral density filter was removed to allow 100% of the excitatory light to reach the samples. The M-C6-NBD-PE accumulation of the cells shown in this figure are representative of that observed for all four transformants analyzed. (B) 5 μl of each culture was spotted onto SDC plates lacking leucine and containing 0 (not shown), 0.25, 0.50, or 0.75 μg/ml cycloheximide and incubated at 30°C for 3 d.
We hypothesized that TPE1 was identical to PDR1 and therefore tested the linkage of these two genes based on the following three observations: (a) TPE2 and PDR3 were determined to be identical; (b) PDR1 is a transcriptional activator of the drug resistance network and is homologous to PDR3 (5); and (c) TPE1-1 is drug resistant (Table IV). Tetrad analysis of TPE1-1 and PDR1-3 for cycloheximide resistance indicated that TPE1 and PDR1 were tightly linked.
TPE1-1 and tpe2-1 Are Respective Gain-of-Function Alleles of PDR1 and PDR3
To test the predicted identity of TPE1 with PDR1 and TPE2 with PDR3, the nonessential genes PDR1 and PDR3 were deleted in both tpe mutant strains. Deletion of PDR1 in the TPE1-1 mutant strain restored wild-type levels of M-C6-NBD-PE accumulation (Fig. 8) and cycloheximide sensitivity (data not shown). Similarly, PDR3 deletion in the tpe2-1 strain also restored wild-type levels of M-C6-NBD-PE accumulation and cycloheximide sensitivity. Conversely, deletion of PDR1 in tpe2-1 and deletion of PDR3 in TPE1-1 had no effect on either mutant phenotype. These data indicated that TPE1-1 is a gain-of-function mutant allele of PDR1, and tpe2-1 is a gain-of-function mutant allele of PDR3. Consistent with this interpretation, deletion of PDR1 and PDR3 in the parental Tpe+ strain resulted in wild-type levels of M-C6-NBD-PE accumulation and cycloheximide sensitivity (data not shown). From this point on, TPE1-1 will be referred to as PDR1-11, and tpe2-1 will be referred to as pdr3-11.
Figure 8.

Deletion of PDR1 in TPE1-1 and PDR3 in tpe2-1 suppresses their M-C6-NBD-PE accumulation defect. Strains were grown to mid-log phase in SDC medium at 23°C. 1 ml of each culture was incubated at 37°C for 30 min and M-C6-NBD-PE internalization assays and fluorescence microscopy were performed as described in Materials and Methods. (A–C) tpe2-1 Δpdr3. (D–F) TPE1-1 Δpdr3. (G–I) tpe2-1 Δpdr1. (J–L) TPE1-1 Δpdr1. (A, B, D, E, G, H, J, and K) NBD fluorescence. (C, F, I, and L) DIC optics. In A, D, G, and J, a neutral density filter was used that attenuated the excitatory light by 95%. In B, E, H, and K, the neutral density filter was removed to allow 100% of the excitatory light to reach the samples.
The Defect in M-C6-NBD-PE Accumulation in PDR3 Is Allele Specific
The strain carrying the mutant allele pdr3-2 was originally isolated based on its resistance to chloramphenicol and cycloheximide (20) and subsequently identified as an allele of PDR3 (48). This strain was compared to the tpe2-1/ pdr3-11 allele in relation to M-C6-NBD-PE accumulation and drug resistance (Fig. 9, Table IV). Allele specificities were observed in both. Unlike tpe2-1/pdr3-11, pdr3-2 did not exhibit a defect in M-C6-NBD-PE accumulation; however, both alleles exhibited similar resistance to cycloheximide (Table IV). pdr3-2 was also less resistant to 4-NQO and oligomycin than tpe2-1.
Figure 9.

The inhibition of M-C6-NBD-PE accumulation by mutations in PDR3 is allele specific. pdr3-2 and tpe2-1/pdr3-11 strains were grown to mid-log phase in SDC medium at 23°C. 1 ml of each culture was incubated at 37°C for 30 min and M-C6-NBD-PE internalization assays and fluorescence microscopy were performed as described in Materials and Methods. (A–C) tpe2-1/pdr3-11. (D–F) pdr3-2. (A, B, D, and E) NBD fluorescence. (C and F) DIC optics. In A and D, a neutral density filter was used that attenuated the excitatory light by 95%. In B and E, the neutral density filter was removed to allow 100% of the excitatory light to reach the samples.
The PDR3 mutant allele, tpe2-1/pdr3-11, was rescued by integrative transformation followed by plasmid eviction of the mutant allele. Sequencing revealed a single point mutation. Glycine957, which is predicted to be in the activation domain, was replaced by an aspartate.
Discussion
Model for M-C6-NBD-PE Translocation
The data presented here provide evidence that M-C6-NBD-PE is internalized into yeast by a mechanism that is distinct from the major pathway for M-C6-NBD-PC internalization. This mechanism is detailed in the model shown in Fig. 10. In step 1, M-C6-NBD-PE molecules dissociate from donor vesicles and insert in the outer leaflet of the yeast plasma membrane. Even though M-C6-NBD-PE molecules are predominantly membrane bound (their molar concentration in the membrane is 107 times greater than their concentration in water [33]), they rapidly exchange through the medium between membranes (t 1/2 = ∼2 s at 25°C [33]). Furthermore, by incorporating a “nonexchangeable” fluorescent lipid (N-Rh-DOPE) into the donor vesicles, we determined that the cell-associated fluorescence was not due to donor vesicle fusion or internalization, but rather due to the movement of monomers of M-C6-NBD-PE from the donor vesicles into the plasma membrane.
Figure 10.

Schematic model for the regulation of M-C6-NBD-PE flip-flop and sorting.
Translocation across the plasma membrane (step 2) is modeled as protein mediated based on the inhibition of M-C6-NBD-PE accumulation by treatment with NEM at low temperature. Inhibition of M-C6-NBD-PE accumulation by depletion of intracellular ATP indicates a yet to be defined requirement for cellular ATP or another nucleotide in the translocation process. Given the relatively high partition coefficient of M-C6-NBD-PE (33), the flippase model, in which an amphiphile partitions into one leaflet of a membrane and its polar group is flipped to the other side (21), is the most likely mechanism to explain M-C6-NBD-PE translocation across the plasma membrane. These characteristics of M-C6-NBD-PE internalization in yeast are similar to the characteristics of the flippase-mediated phosphatidylethanolamine uptake that has been described in mammalian cells (10, 31, 42) and yeast (3).
Our results show that the predominant pathway for M-C6-NBD-PE internalization occurs even when lipid endocytosis is inhibited. M-C6-NBD-PE is not delivered to the vacuole like other lipid markers that are internalized by endocytosis, nor is its internalization inhibited in sec mutants shown previously to inhibit lipid-endocytosis (26, 52). However, although M-C6-NBD-PE accumulation occurs when endocytosis is inhibited, our results do not exclude the possibility that under normal conditions, topological translocation of M-C6-NBD-PE to the cytoplasmic surface also occurs subsequently to the early steps of endocytosis.
After translocation to the cytoplasmic leaflet of the plasma membrane or endocytic vesicle, M-C6-NBD-PE is transported to intracellular organelles including, but not necessarily limited to, the nuclear envelope and mitochondria (step 4). Given the rapid rate of exchange of M-C6-NBD-PE between membranes measured in vitro (33), it is likely that transfer from the inner leaflet of the plasma membrane to other intracellular organelles occurs spontaneously. However, the different organelle membranes are not labeled to the same extent. For example, M-C6-NBD-PE labeling was clearly seen in the mitochondria and nuclear envelope but never in the vacuolar membrane. Thus, the rapidly exchanging M-C6-NBD-PE molecules appeared to be sorted into distinct organelle membranes. The observed sorting may result from directed transport to specific organelles or it may reflect an equilibrium distribution determined by the properties of the organelle membranes. At this time, the mechanisms responsible for the apparent nonuniform labeling of the intracellular membranes is unknown.
In addition to its endocytosis-independent internalization, our results also provide evidence (Fig. 3) for the efflux of intact M-C6-NBD-PE molecules from yeast cells (step 3). It is important to realize that although we monitor this efflux only after the cells are loaded with M-C6-NBD-PE, influx and efflux are likely occurring concurrently. In the presence of medium containing excess M-C6-NBD-PE donor vesicles (Fig. 3, A and D), the balance leads to intracellular accumulation of fluorescent lipid. When these donor vesicles are removed, the balance shifts to favor a depletion of intracellular fluorescence. Our data do not distinguish whether a single protein is responsible for both influx and efflux, or whether influx and efflux are mediated in different directions by different proteins.
Relevance of Short Acyl Chain, NBD-labeled Phospholipid Trafficking to Endogenous Phospholipid Trafficking
Although NBD-labeled lipids have been used for many years to study lipid sorting and trafficking in cultured cells (37), one must consider the extent to which the presence of the NBD group and/or the truncation of the sn-2 acyl chain alters the behavior of the phospholipid analogue relative to its endogenous counterpart. The differential pattern of accumulation of M-C6-NBD-PE relative to M-C6-NBD-PC (Fig. 1) demonstrated unequivocally that the yeast cells distinguish between the head group structures of these two molecules and import them to different locations. Since the diacyl glycerol portion of these two molecules is identical, one can conclude that the NBD group does not determine the uptake pathway nor does it interfere with the discrimination of different head groups by the yeast plasma membrane.
It is clear that truncation of one or both acyl chains of a phospholipid molecule has a profound effect on its stability in membranes. Based on the relative rates of transfer through the water phase between liposomes, 1-palmitoyl, 2-oleoyl phosphatidylcholine is 1.4 × 105 times (23) and dimyristoyl phosphatidylcholine is 3,000 times (23) less likely to dissociate from a membrane into the water than M-C6-NBD-PC (33). Thus, the usefulness of truncated NBD-labeled lipids for studying the effect of acyl chain length and structure on intermembrane transfer is limited. However, as stated above, even though the truncated NBD-labeled lipids are significantly more water soluble, their concentration in membranes is still 107 times greater than in water (33). Thus, even though they are readily exchangeable, they exist predominantly in the membrane.
PDR1 and PDR3 Regulate M-C6-NBD-PE and Endogenous Phosphatidylethanolamine Net Translocation Across the Plasma Membrane
A novel mutant enrichment procedure based on photokilling of cells that accumulated normal amounts of M-C6-NBD-PE was developed to identify genes required for the accumulation of intracellular M-C6-NBD-PE. Two genes, originally named TPE1 and TPE2, were identified and found to be identical to two previously identified pleiotropic drug resistance genes, PDR1 (6) and PDR3 (14), respectively. PDR1 and PDR3 are transcription regulators that control the transcription of numerous genes involved in a wide range of functions, encompassing resistance to chemical and physical stresses, membrane transport, and organelle function (5). Gain-of-function alleles of PDR3 mediate resistance to a wide variety of drugs by activating transcription of the corresponding ABC transporters that efflux the drugs out of the cells. ABC transporters shown to be regulated by PDR3 include YOR1 (25), SNQ2 (12, 29), and PDR5 (8, 24), which mediate the efflux of oligomycin, 4-NQO, and cycloheximide, respectively. PDR3 autoregulates itself (13) and mediates resistance to other drugs as well and is consequently believed to regulate other genes (4, 5). PDR3 is most homologous (36% identical) and functionally analogous to another transcriptional activator of this PDR family, PDR1, which also mediates drug resistance through the aforementioned ABC transporters. Preliminary evidence suggests that PDR1 may be involved in the regulation of a host of other genes such as PDR10, PDR15, GAS1, glycerol-3-phosphate dehydrogenase (G3PD), and others as well as PDR3.
The observation that M-C6-NBD-PE appears to flip-flop in both the inward and outward directions suggests that the steady-state amount of M-C6-NBD-PE accumulation resulting from our standard assay is determined by the relative rates of each process. In wild-type cells, M-C6-NBD-PE is accumulated because the rate of inward- directed flippase activity exceeds the outward-directed activity. Given that drug resistance is conferred by increasing the rate of drug efflux from yeast cells and the demonstration that a mechanism for M-C6-NBD-PE efflux exists in wild-type cells (Fig. 3), it is likely that TPE1-1/PDR1-11 and tpe2-1/pdr3-11 activate the transcription of one or more flippases that actively transport M-C6-NBD-PE in the outward direction. However, one cannot rule out the possibility that these mutant alleles decrease the activity of an inward-directed flippase either by inhibiting its transcription or by activating the transcription of an inhibitor.
In addition to a decreased net inward flux of M-C6-NBD-PE, TPE1-1/PDR1-11 and tpe2-1/pdr3-11 strains also exhibited a four to fivefold increase in the amount of endogenous phosphatidylethanolamine exposed in the outer leaflet of the plasma membrane (Table II). This result supports the assertion that the observed changes in M-C6-NBD-PE accumulation in the tpe mutants reflect alterations in the distribution of endogenous phosphatidylethanolamine.
Identification of Flippases in Yeast
Several recent reports have identified proteins with flippase activity in mammalian cells. The ABC transporters, human MDR1 and MDR3, as well as mouse mdr2, have been shown to have outward-directed phospholipid flippase activity (40, 47, 51). This suggests the likely possibility that the flippase responsible for the outward-directed flip-flop of M-C6-NBD-PE will be included within a limited class of yeast homologues of ABC transporters that contain the consensus sequence for the DNA binding domain of PDR1 in their promoter regions (5). Inward-directed phosphatidylserine flippase activity has been reported to be encoded by the DRS2 gene—a yeast homologue of a bovine chromaffin granule, P-type ATPase. Deletion of DRS2 results in the inability to internalize NBD-labeled phosphatidylserine across the plasma membrane (49). Using our standard labeling protocol, a DRS2 deletion strain accumulated M-C6-NBD-PE to the same extent as its isogenic Drs2+ parent strain (Grant, A.M., and J.W. Nichols, unpublished observation). Thus, DRS2 is not solely responsible for the internalization of M-C6-NBD-PE. Several genetic approaches are available that should allow the identification of the inward- and outward- directed flippases and the mechanisms involved in the regulation of phosphatidylethanolamine transport and its steady-state distribution across the plasma membrane.
Significance of Flippases in Unicellular Organisms
Numerous mammalian cell types (for reviews see references 15, 16, 54), as well as the yeast Saccharomyces carlbergensis (9), have been reported to have an asymmetric distribution of phospholipids across their plasma membranes. In general, the aminophospholipids, phosphatidylserine and phosphatidylethanolamine, are sequestered to the inner leaflet while the choline lipids, phosphatidylcholine and sphingomyelin, and glycolipids are concentrated in the outer leaflet. Establishment of this asymmetric distribution is thought to result from the activity of one or more aminophospholipid translocases that flip-flop phosphatidylserine and phosphatidylethanolamine from the outer to inner leaflet. Although the loss of the asymmetric distribution of phosphatidylserine acts as a signal for several intercellular or systemic signaling events, including blood coagulation (39), clearance of senescent red cells (11), and phagocytosis of apoptotic cells (17, 18), the role of the establishment and maintenance of an asymmetric phospholipid distribution for the function of single cells has yet to be determined. It has been proposed that inward-directed phosphatidylserine and/or phosphatidylethanolamine translocation is required for the inward budding of the plasma membrane during endocytosis (15). Active inward translocation of these lipids is hypothesized to increase the phospholipid surface density in the inner leaflet, inducing inward bending of the bilayer as predicted by the bilayer-couple hypothesis (43). The observation that mutant strains defective in M-C6-NBD-PE accumulation (PDR1-11 and pdr3-11) are also defective in the endocytosis of lipid probes is consistent with this hypothesis. Regardless of whether M-C6-NBD-PE accumulation in the mutants is inhibited by an increased efflux or a decreased influx, the net result would be a loss of the ability to establish the excess surface density in the inner leaflet predicted to be required for inward vesicle budding.
The data presented here establish that yeast cells have the ability to translocate M-C6-NBD-PE in the inward and outward direction and that the net direction of translocation is under transcriptional regulation by PDR1 and PDR3. This suggests a fundamental cellular role for phospholipid flippases unrelated to intercellular signaling and interactions. The presence of flippases that pump in opposite directions suggests that yeast cells have the ability to coordinately regulate their activities to produce dynamic membrane perturbations or to produce an appropriate transbilayer phospholipid distribution for their survival in a wide range of environments during different stages of growth.
Acknowledgments
The authors wish to thank Don Wigston for the use of his microscope and digital image analysis system.
This work was supported by National Institutes of Health grants GM39697 and GM50915 to R.S. Fuller and by National Institutes of Health grant GM52410 to J.W. Nichols. L.S. Kean is a recipient of a Howard Hughes Medical Institute Predoctoral Fellowship, and A.M. Grant is a recipient of a National Institutes of Health Minority Predoctoral Fellowship. Karl Kuchler was supported by a grant from the Austrian Science Foundation (FWF-MOB-1023).
Abbreviations used in this paper
- 4-NQO
4-nitroquinoline N-oxide
- DAPI
4′,6-diamidino-2-phenylindol
- DIC
differential interference contrast
- DOPC
dioleoylphosphatidylcholine
- M-C6-NBD-PC
1-myristoyl-2-[6-(NBD) aminocaproyl]-phosphatidylcholine
- M-C6-NBD-PE
1-myristoyl-2-[6-(NBD) aminocaproyl]-phosphatidylethanolamine
- NBD
7-nitrobenz-2-oxa-1,3- diazol-4-yl
- NBD-PE
1-acyl-2-[6-(NBD) aminocaproyl]-phosphatidylethanolamine
- N-Rh-DOPE
N-rhodamine-dioleoylphosphatidylethanolamine
- NEM
N-ethlymaleimide
- TLC
thin-layer chromatography
- TNBS
trinitrobenzenesulfonic acid
- tpe
trafficking of phosphatidylethanolamine
Footnotes
Please address all correspondence to J. Wylie Nichols, Department of Physiology, Emory University School of Medicine, Atlanta, GA 30322. Tel.: (404) 727-7422. Fax: (404) 727-2648. E-mail: wnichols@physio.emory.edu
Leslie S. Kean and Althea M. Grant made equivalent contributions to the work presented and should be considered co-first authors.
References
- 1.Alani E, Cao L, Kleckner N. A method for gene disruption that allows repeated use of URA3selection in the construction of multiply disrupted yeast strains. Genetics. 1987;116:541–545. doi: 10.1534/genetics.112.541.test. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ames BN, Dubin DT. The role of polyamines in the neutralization of bacteriophage deoxyribonucleic acid. J Biol Chem. 1960;235:769–775. [PubMed] [Google Scholar]
- 3.Balasubramanian K, Gupta CM. Transbilayer phosphatidylethanolamine movements in the yeast plasma membrane. Evidence for a protein-mediated, energy-dependent mechanism. Eur J Biochem. 1996;240:798–806. doi: 10.1111/j.1432-1033.1996.0798h.x. [DOI] [PubMed] [Google Scholar]
- 4.Balzi E, Goffeau A. Genetics and biochemistry of yeast multidrug resistance. Biochim Biophys Acta. 1994;1187:152–162. doi: 10.1016/0005-2728(94)90102-3. [DOI] [PubMed] [Google Scholar]
- 5.Balzi E, Goffeau A. Yeast multidrug resistance: the PDR network. J Bioenerg Biomembr. 1995;27:71–76. doi: 10.1007/BF02110333. [DOI] [PubMed] [Google Scholar]
- 6.Balzi E, Chen W, Ulazewski S, Capieaux E, Goffeau A. The multidrug resistance gene PDR1 from Saccharomyces cerevisiae. . J Biol Sci. 1987;262:16871–16879. [PubMed] [Google Scholar]
- 7.Balzi E, Wang M, Leterme S, Dyck LV, Goffeau A. PDR5, a novel yeast multidrug resistance conferring transporter controlled by the transcriptional regulator PDR1. . J Biol Chem. 1994;269:2206–2214. [PubMed] [Google Scholar]
- 8.Bissinger PH, Kuchler K. Molecular cloning and expression of the Saccharomyces cerevisiae STS1gene product. A yeast ABC transporter conferring mycotoxin resistance. J Biol Chem. 1994;269:4180–4186. [PubMed] [Google Scholar]
- 9.Cerbon J, Calderon V. Changes of the compositional asymmetry of phospholipids associated to the increment in the membrane surface potential. Biochim Biophys Acta. 1991;1067:139–144. doi: 10.1016/0005-2736(91)90035-7. [DOI] [PubMed] [Google Scholar]
- 10.Connor J, Pak CH, Zwaal RF, Schroit AJ. Bidirectional transbilayer movement of phospholipid analogs in human red blood cells. Evidence for an ATP-dependent and protein-mediated process. J Biol Chem. 1992;267:19412–19417. [PubMed] [Google Scholar]
- 11.Connor J, Pak CC, Schroit AJ. Exposure of phosphatidylserine in the outer leaflet of human red blood cells. Relationship to cell density, cell age and clearance by mononuclear cells. J Biol Chem. 1994;269:2399–2404. [PubMed] [Google Scholar]
- 12.Decottignies A, Lambert L, Catty P, Degand H, Epping EA, Moye-Rowley WS, Balzi E, Goffeau A. Identification and characterization of SNQ2, a new multidrug ATP binding cassette transporter of the yeast plasma membrane. J Biol Chem. 1995;270:18150–18157. doi: 10.1074/jbc.270.30.18150. [DOI] [PubMed] [Google Scholar]
- 13.Delahodde A, Delaveau T, Jacq C. Positive autoregulation of the yeast transcription factor Pdr3p, which is involved in control of drug resistance. Mol Cell Biol. 1995;15:4043–4051. doi: 10.1128/mcb.15.8.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deleveau T, Delahodde A, Carvajal E, Subik J, Jacq C. PDR3, a new yeast regulatory gene, is homologous to PDR1and controls the multidrug resistance phenomenon. Mol Gen Genet. 1994;244:501–511. doi: 10.1007/BF00583901. [DOI] [PubMed] [Google Scholar]
- 15.Devaux PF, Zachowski A. Maintenance and consequences of membrane phospholipid asymmetry. Chem Phys Lipids. 1994;73:107–120. [Google Scholar]
- 16.Diaz C, Schroit AJ. Role of translocases in the generation of phosphatidylserine asymmetry. J Membr Biol. 1996;151:1–9. doi: 10.1007/s002329900051. [DOI] [PubMed] [Google Scholar]
- 17.Fadok VA, Savill JS, Haslett C, Bratton DL, Doherty DE, Campbell PA, Henson PM. Different populations of macrophages use either the vitronectin receptor or the phosphatidylserine receptor to recognize and remove apoptotic cells. J Immunol. 1992;149:4029–4035. [PubMed] [Google Scholar]
- 18.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 19.Gietz D, John AS, Woods RA, Schiestl RH. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guerineau M, Slonimski PP, Avner PR. Yeast episome: oligomycin resistance associated with a small covalently closed non-mitochondrial circular DNA. Biochem Biophys Res Commun. 1974;61:462–469. doi: 10.1016/0006-291x(74)90979-6. [DOI] [PubMed] [Google Scholar]
- 21.Higgins CF. Flip-Flop: the transmembrane translocation of lipids. Cell. 1994;79:393–395. doi: 10.1016/0092-8674(94)90248-8. [DOI] [PubMed] [Google Scholar]
- 22.Hjelmstad RH, Bell RM. Mutants of Saccharomyces cerevisiae defective in sn-1, 2-diacylglycerol cholinephosphotransferase. J Biol Chem. 1987;262:3909–3917. [PubMed] [Google Scholar]
- 23.Jones JD, Thompson TE. Mechanism of spontaneous, concentration-dependent phospholipid transfer between bilayers. Biochemistry. 1990;29:1593–1600. doi: 10.1021/bi00458a034. [DOI] [PubMed] [Google Scholar]
- 24.Katzmann D, Burnett P, Golin J, Mahe Y, Moye-Rowley WS. Transcriptional control of the yeast PDR5 gene by the PDR3gene product. Mol Cell Biol. 1994;14:4653–4661. doi: 10.1128/mcb.14.7.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katzmann D, Hallstrom TC, Voet M, Wysock W, Golin J, Volckaert G, Moye-Rowley WS. Expression of an ATP-binding cassette transporter gene (YOR1) is required for oligomycin resistance in Saccharomyces cerevisiae. . Mol Cell Biol. 1995;15:6875–6883. doi: 10.1128/mcb.15.12.6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kean LS, Fuller RS, Nichols JW. Retrograde lipid traffic in yeast: identification of two distinct pathways for internalization of fluorescent-labeled phosphatidylcholine from the plasma membrane. J Cell Biol. 1993;123:1403–1419. doi: 10.1083/jcb.123.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawrence CW. Classical mutagenesis techniques. Methods Enzymol. 1991;194:273–280. doi: 10.1016/0076-6879(91)94021-4. [DOI] [PubMed] [Google Scholar]
- 28.Lee KS, Patton JL, Fido M, Hines LK, Kohlwein SD, Paltauf F, Henry SA, Levin DE. The Saccharomyces cerevisiae PLB1gene encodes a protein required for lysophospholipase and phospholipase B activity. J Biol Chem. 1994;269:19725–19730. [PubMed] [Google Scholar]
- 29.Mahe Y, Parle-McDermott A, Nourani A, Delahodde A, Lamprecht A, Kuchler K. The ATP binding cassette multidrug transporter Snq2 of Saccharomyces cerevisiae: a novel target for the transcription factors Pdr1 and Pdr3. . Mol Microbiol. 1996;20:109–117. doi: 10.1111/j.1365-2958.1996.tb02493.x. [DOI] [PubMed] [Google Scholar]
- 30.Marinetti GV, Love R. Differential reaction of cell membrane phospholipids and proteins with chemical probes. Chem Phys Lipids. 1976;16:239–254. doi: 10.1016/0009-3084(76)90019-0. [DOI] [PubMed] [Google Scholar]
- 31.Martin OC, Pagano RE. Transbilayer movement of fluorescent analogs of phosphatidylserine and phosphatidylethanolamine at the plasma membrane of cultured cells. J Biol Chem. 1987;262:5890–5898. [PubMed] [Google Scholar]
- 32.Monti JA, Christian ST, Shaw WA, Finley WH. Synthesis and properties of a fluorescent derivative of phosphatidylcholine. Life Sci. 1977;21:345–356. doi: 10.1016/0024-3205(77)90515-x. [DOI] [PubMed] [Google Scholar]
- 33.Nichols JW. Thermodynamics and kinetics of phospholipid monomer-vesicle interaction. Biochemistry. 1985;24:6390–6398. doi: 10.1021/bi00344a011. [DOI] [PubMed] [Google Scholar]
- 34.Nichols JW. Binding of fluorescent-labeled phosphatidylcholine to rat liver nonspecific lipid transfer protein. J Biol Chem. 1987;262:14172–14177. [PubMed] [Google Scholar]
- 35.Nichols JW, Pagano RE. Resonance energy transfer assay of protein-mediated lipid transfer between vesicles. J Biol Chem. 1983;258:5368–5371. [PubMed] [Google Scholar]
- 36.Novick P, Field C, Schekman R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 1980;21:205–215. doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- 37.Pagano RE, Sleight RG. Defining lipid transport pathways in animal cells. Science (Wash DC) 1985;229:1051–1057. doi: 10.1126/science.4035344. [DOI] [PubMed] [Google Scholar]
- 38.Rose, M., and J. Broach. 1991. Cloning genes by complementation in yeast. In Guide to Yeast Genetics and Molecular Biology. Vol. 194. C. Guthrie and G.R. Fink, editors. Academic Press, Inc., San Diego. 195–230. [DOI] [PubMed]
- 39.Rosing R, Tans G, Govers-Riemslag JWP, Zwaal RFA, Hemker HC. The role of phospholipids and factor Va in the prothrombinase complex. J Biol Chem. 1980;255:274–283. [PubMed] [Google Scholar]
- 40.Ruetz S, Gros P. Phosphatidylcholine translocase: a physiological role for the mdr2gene. Cell. 1994;77:1071–1081. doi: 10.1016/0092-8674(94)90446-4. [DOI] [PubMed] [Google Scholar]
- 41.Saunders GW, Rank GH. Allelism of pleitropic drug resistance in Saccharomyces cerevisiae. . Can J Genet Cytol. 1982;24:493–503. doi: 10.1139/g82-053. [DOI] [PubMed] [Google Scholar]
- 42.Schroit AJ, Zwaal RFA. Transbilayer movement of phospholipids in red cell and platelet membranes. Biochim Biophys Acta. 1991;1071:313–329. doi: 10.1016/0304-4157(91)90019-s. [DOI] [PubMed] [Google Scholar]
- 43.Sheetz MP, Singer SJ. Biological membranes as bilayer couples: a mechanism of drug-erythrocyte interactions. Proc Natl Acad Sci USA. 1974;71:4457–4461. doi: 10.1073/pnas.71.11.4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sherman, F., G.R. Fink, and J.B. Hicks. 1986. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 164– 165.
- 45.Sleight RG, Pagano RE. Transbilayer movement of a fluorescent phosphatidylethanolamine analog across the plasma membranes of cultured mammalian cells. J Biol Chem. 1985;260:1146–1154. [PubMed] [Google Scholar]
- 46.Smit JJM, Schinkel AH, Oude RPJ, Elferink, Groen AK, Wagenaar E, van Deemter L, Mol CAAM, Ottenhoff R, van der Lugt NMT, van Roon MA, et al. Homozygous disruption of the murine mdr2P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451–462. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 47.Smith AJ, Timmermans-Hereijgers JL, Roelofsen B, Wirtz KW, van Blitterswijk WJ, Smit JJ, Schinkel AH, Borst P. The human MDR3 P-glycoprotein promotes translocation of phosphatidylcholine through the plasma membrane of fibroblasts from transgenic mice. FEBS Lett. 1994;354:263–266. doi: 10.1016/0014-5793(94)01135-4. [DOI] [PubMed] [Google Scholar]
- 48.Subik J, Ulaszewski S, Goffeau A. Genetic mapping of nuclear mucidin resistance mutations in Saccharomyces cerevisiae.A new pdr locus on chromosome II. Curr Genet. 1986;10:665–670. doi: 10.1007/BF00410914. [DOI] [PubMed] [Google Scholar]
- 49.Tang X, Halleck MS, Schlegel RA, Williamson P. A subfamily of P-Type ATPases with aminophospholipid transporting activity. Science (Wash DC) 1996;272:1495–1497. doi: 10.1126/science.272.5267.1495. [DOI] [PubMed] [Google Scholar]
- 50.Thomas BJ, Rothstein R. Elevated recombination rates in transcriptionally active DNA. Cell. 1989;56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- 51.van Helvoort A, Smith AJ, Sprong H, Fritzsche I, Schinkel AH, Borst P, van Meer G. MDR1 p-glycoprotein is a lipid translocase of broad specificity, while MDR3 p-glycoprotein specifically translocates phosphatidylcholine. Cell. 1996;87:507–517. doi: 10.1016/s0092-8674(00)81370-7. [DOI] [PubMed] [Google Scholar]
- 52.Vida TA, Emr SD. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol. 1995;128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walworth NC, Novick PJ. Purification and characterization of constitutive secretory vesicles from yeast. J Cell Biol. 1987;105:163–174. doi: 10.1083/jcb.105.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williamson P, Schlegel RA. Back and forth: the regulation and function of transbilayer phospholipid movement in eukaryotic cells (Review) Mol Membr Biol. 1994;11:199–216. doi: 10.3109/09687689409160430. [DOI] [PubMed] [Google Scholar]
- 55.Winston F, Chumley F, Fink GR. Eviction and transplacement of mutant genes in yeast. Methods Enzymol. 1993;101:211–227. doi: 10.1016/0076-6879(83)01016-2. [DOI] [PubMed] [Google Scholar]
- 56.Witt W, Mertsching A, Konig E. Secretion of phospholipase B from Saccharomyces cerevisiae. . Biochim Biophys Acta. 1984;795:117–124. doi: 10.1016/0005-2760(84)90111-5. [DOI] [PubMed] [Google Scholar]