

Abstract
We have cloned and characterized Xlrbpa, a double-stranded RNA-binding protein from Xenopus laevis. Xlrbpa is a protein of 33 kD and contains three tandemly arranged, double-stranded RNA-binding domains (dsRBDs) that bind exclusively to double-stranded RNA in vitro, but fail to bind either single-stranded RNA or DNA. Sequence data and the overall organization of the protein suggest that Xlrbpa is the Xenopus homologue of human TAR-RNA binding protein (TRBP), a protein isolated by its ability to bind to human immunodeficiency virus (HIV) TAR-RNA. In transfection assays, TRBP has also been shown to inhibit the interferon-induced protein kinase PKR possibly by direct physical interaction. To determine the function of Xlrbpa and its human homologue we studied the expression and intracellular distribution of the two proteins. Xlrbpa is ubiquitously expressed with marked quantitative differences amongst all tissues. Xlrbpa and human TRBP can be detected in the cytoplasm and nucleus by immunofluorescence staining and Western blotting.
Sedimentation gradient analyses and immunoprecipitation experiments suggest an association of cytoplasmic Xlrbpa with ribosomes. In contrast, a control construct containing two dsRBDs fails to associate with ribosomes in microinjected Xenopus oocytes. Nuclear staining of Xenopus lampbrush chromosome preparations showed the association of the protein with nucleoli, again indicating an association of the protein with ribosomal RNAs. Additionally, Xlrbpa could be located on lampbrush chromosomes and in snurposomes. Immunoprecipitations of nuclear extracts demonstrated the presence of the protein in heterogeneous nuclear (hn) RNP particles, but not in small nuclear RNPs, explaining the chromosomal localization of the protein. It thus appears that Xlrbpa is a general double-stranded RNA-binding protein which is associated with the majority of cellular RNAs, ribosomal RNAs, and hnRNAs either alone or as part of an hnRNP complex.
Most cellular RNAs associate with a variety of proteins that are required for several important cellular functions. Heterogeneous nuclear (hn) RNP1 proteins, for instance, package newly transcribed hnRNA into hnRNP particles (Piñol-Roma et al., 1988). Splicing factors, in combination with splicing small nuclear (sn) RNPs, on the other hand, mediate the removal of introns from pre-mRNAs (reviewed by Nilsen, 1994). Other modifying proteins are required for capping, polyadenylation, or editing of mRNA (Adam et al., 1986; Christofori and Keller, 1989; Hamm and Mattaj, 1990; Wahle, 1991; Bass, 1993; Polson and Bass, 1994).
Structural RNAs such as ribosomal RNAs or snRNAs also have to be processed, modified, and complexed with proteins to become functionally active (Reddy and Bush, 1988; Tyc and Steitz, 1989; Savino and Gerbi, 1990; Wool et al., 1990; Peculis and Steitz, 1993; Mougey et al., 1993).
RNA-binding proteins can even be involved in the control of gene expression, such as the iron regulatory factor that controls the stability of human transferrin receptor mRNA and the translatability of ferritin mRNA by binding to these RNAs (Leibold and Munro, 1988; Müllner et al., 1989). Other RNA-binding proteins are required for the proper localization of certain RNAs thereby controlling cell polarity or body axis and pattern formation during development (for review see St Johnston, 1995).
Some RNA-interacting proteins bind RNA directly while others contact RNA only as part of a larger complex (Piñol-Roma et al., 1988). RNA–protein interaction is frequently mediated through specific RNA-binding domains (RBDs). These domains can be quite similar to each other in very different proteins, both in their primary amino acid sequence and at the structural level, and can thus be defined by specific consensus sequences. So far, several conserved RBDs could be defined by their homologies to each other. Among these, the 90–100-amino acid RNA recognition motif is the most abundant and probably best-characterized RBD (Bandziulis et al., 1989; Nagai et al., 1990; Wittekind et al., 1992). Other well-characterized RBDs include the zinc-finger motif, the arginine-rich motif, the RGG box, and the KH box (for review see Burd and Dreyfuss, 1994).
A new RNA-binding motif that binds exclusively to double-stranded RNA or RNA–DNA hybrids is the so-called double-stranded RNA-binding domain (dsRBD) (St Johnston et al., 1992; Green and Mathews, 1992; Bass et al., 1994). This protein domain is ∼70 amino acids in length with several basic amino acids clustered at its carboxy terminus. While most dsRBDs fit a consensus sequence quite well over their entire length, some dsRBDs appear less conserved at their amino terminus than at their basic carboxy-terminal end (St Johnston et al., 1992; Krovat and Jantsch, 1996). Like other RBDs, the dsRBD can sometimes be found in multiple copies within a single protein. So far, dsRBDs could be identified in almost 30 putative or known RNA-binding proteins from Escherichia coli to man (Kharrat et al., 1995; Krovat and Jantsch, 1996). Recently the in-solution structure of two different dsRBDs have been determined by nuclear magnetic resonance (NMR) analysis (Bycroft et al., 1995; Kharrat et al., 1995). Both studies determined virtually identical structures for both dsRBDs, suggesting a conserved structural organization of all dsRBDs. While some dsRBD containing proteins associate only with specific RNAs in vivo, no sequence specific binding of dsRBDs could so far be demonstrated in vitro.
From a screen for RNA-binding proteins we have isolated at least four different dsRBD-containing proteins from a Xenopus ovary-specific cDNA library. One of these proteins, named Xenopus laevis RNA-binding protein A (Xlrbpa), shows significant homology to human trans-activation–responsive (TAR)–RNA binding protein (TRBP) (Gatignol et al., 1991) and seems to be its Xenopus homologue. Human TRBP has been isolated by its ability to bind human immunodeficiency virus 1 (HIV-1) TAR-RNA, and has been postulated to be involved in the transcriptional activation of several viral promoters (Gatignol et al., 1991). TRBP has also been shown to act as an inhibitor of the cellular interferon-induced kinase PKR when overexpressed in COS-1 cells (Park et al., 1994). PKR is activated by double-stranded RNAs resulting in autophosphorylation and consecutive phosphorylation of translation initiation factor eIF2α, which leads to inhibition of protein synthesis. Inhibition of PKR by TRBP has thus been attributed to competitive binding of double-stranded RNAs by TRBP, therefore preventing their binding to PKR and subsequent activation of kinase activity (Park et al., 1994). A possible function of TRBP could therefore be the complexation of structured cellular RNAs to prevent inadvertent activation of PKR. TRBP has also been shown to dimerize with PKR through RNA bridges (Cosentino et al., 1995). In contrast, a recent study demonstrates physical interaction of the two proteins in an RNA-independent manner, suggesting a regulation of PKR by TRBP in a more direct way (Benkirane et al., 1997).
However, association of TRBP with cellular RNAs has not been investigated to this point. We therefore studied the expression and cellular distribution of Xlrbpa and its human homologue, TRBP, both cytologically and at the biochemical level. We also determined the association of these proteins with RNA. Both proteins seem ubiquitously expressed and are localized in the cytoplasm and nucleus. In the cytoplasm, most of these proteins seem associated with ribosomes. Additionally, lampbrush chromosome preparations of Xenopus germinal vesicles revealed the association of Xlrbpa with nascent hnRNAs on transcribing chromosome loops. Both observations, the association with ribosomes and hnRNPs were confirmed by biochemical experiments.
Materials and Methods
Isolation of Xlrbpa cDNA
Xlrbpa cDNA was isolated from a Xenopus laevis ovary cDNA library by Northwestern screening with 32P-labeled U1 or U2 snRNA. The screen was performed according to the procedure of Vinson et al. (1988), except that 8 M urea replaced 6 M guanidine hydrochloride as the chaotropic agent. The binding buffer contained 50 mM NaCl, 10 mM MgCl2, 10 mM Hepes, pH 7.5, 0.1 mM EDTA, and 1 mM DTT. The probe was a mixture of 32P-labeled transcripts produced by T7 polymerase from cloned U1 and U2 snRNA genes. The Xlrbpa insert was sequenced after conversion of the λZAP bacteriophage into the corresponding pBluescript phagemid (Stratagene, LaJolla, CA). These sequence data are available from EMBL/GenBank/DDBJ under accession No. M96370.
Antibody Production
A BamHI fragment encoding the carboxy-terminal two thirds of the Xlrbpa cDNA was cloned into the GST fusion vector pGEX (Pharmacia Diagnostics AB, Uppsala, Sweden). Plasmids containing the proper insert were transformed into Escherichia coli BL21. 3 ml starting cultures were grown overnight and used to inoculate larger 300-ml cultures the next morning. When the cultures reached an OD600 of 0.7, protein production was induced by the addition of isopropyl β-d-thiogalactoside (IPTG) to 1 mM final concentration. After an additional 4 h of incubation, cells were harvested by centrifugation and lysed by sonication in PBS, 1% Triton X-100.
Cell lysate was cleared by centrifugation and the supernatant was loaded on a glutathione Sepharose 4B column (Pharmacia Diagnostics AB). The column was washed extensively with PBS, 1% Triton X-100 to remove unbound protein. Bound glutathione-S-transferase (GST) fusion protein was eluted by washing the column with 5 mM glutathione in 50 mM Tris-HCl, pH 8.0. Typical yields were 3–5 mg of pure protein per 500 ml culture.
Peptide Coupling
Peptide MJ2 (CDYVKMLKDVAEELDF) was coupled to keyhole limpet hemocyanin (KLH) by γ-maleimidobutyric acid N-hydroxysuccinimide ester cross-linking according to Harlow and Lane (1988).
Antibodies
Two rabbits (Rb3 and Rb5) were immunized with KLH-coupled peptide MJ2 while two rabbits (Rb6 and Rb7) were immunized with GST fusion protein by subcutaneous injections after the immunization schemes given by Harlow and Lane (1988). 2 wk after the first booster injection, test sera were collected from the ear vein and tested for their ability to detect the GST fusion protein in Western blots. Since all animals had developed antibodies against Xlrbpa, two further booster injections were administered before animals were sacrificed, at which stage all blood was collected. Sera were prepared according to Harlow and Lane (1988) and stored frozen at −70°C.
mAb 4F4 recognizes human hnRNPs C1 and C2 (Choi and Dreyfuss, 1984). Human autoimmune sera 13751 and 92751 recognize ribosomal phosphoproteins P0, P1, and P2 as determined by comparison with reference serum SD1 obtained from CDC (Atlanta, GA) (Steiner, G., personal communication). mAb Y12 recognizes the Sm epitope of snRNPs (Lerner et al., 1981).
Western Blotting
Sample preparation and SDS gel electrophoresis was performed according to standard procedures. After electrophoresis, proteins were transferred to Immobilon–PVDF (polyvinylidenedifluoride) membranes (Millipore Corp., Waters Chromatography, Bedford, MA). For Western blotting, peptide antisera were used at 1:300 dilutions while antisera against GST fusion proteins were used at a dilution of 1:500. Bound antibodies were detected with 125I-labeled protein A and autoradiographed.
Oocyte Sections
Small pieces of Xenopus ovary containing oocytes of different developmental stages were quick-frozen by immersion in isopentane at −180°C, quickly transferred to ethanol at −70°C, and stored for 2–3 d at that temperature. Dehydrated tissue was subsequently warmed to room temperature (3 h at −20°C, 3 h at 4°C, 3 h at 20°C), and finally embedded in histosec at 65°C after a passage through paramylalcohol.
4-μm sections were cut with a glass knife on a Reichert ultramicrotome and mounted on coated slides. Sections were deparaffinized in xylene, rehydrated in a descending alcohol series, and used for immunostaining after a final wash in PBS. Primary antibodies were detected with secondary, alkaline phosphatase–labeled antibodies (Harlow and Lane, 1988).
Immunoprecipitation
For precipitation of hnRNP particles, HeLa nucleoplasm was prepared according to Choi and Dreyfuss (1984). For precipitation of ribosomal proteins, small pieces of ovary were homogenized by sonication in ribosomal buffer D (0.1 M KCl, 5 mM MgCl2, 1 mM β-mercaptoethanol, 0.1 mM EDTA, and 50 mM Tris-HCl, pH 7.8) containing 0.1% deoxycholic acid, 1 mM vanadyl–ribonucleoside complex, 100 μg/ml PMSF, 2 μg aprotinin, and 2 μg/ml pepstatin (Brown et al., 1974). The lysate was cleared from insoluble material by two rounds of centrifugation at 4°C in a table top centrifuge and the remaining supernatant was used for immunoprecipitation. For each immunoprecipitation 40 μl of serum (or preimmuneserum), 5 μl of ascites fluid or 500 μl of mAb tissue culture supernatant were coupled to 5 mg of protein A–Sepharose beads CL4B (Pharmacia Diagnostics AB, Uppsala, Sweden) according to Steitz (1989). After incubation of the coupled beads with the cell lysates, the beads were washed several times with the appropriate buffer and boiled directly in 2× SDS sample buffer before analysis by Western blotting.
Isolation of TRBP cDNA
A TRBP cDNA was isolated by PCR amplification from a HeLa cDNA library using specific primers located at the 5′ and 3′ end of the open-reading frame. Approximately 106 plaque forming units (pfu) were directly used in the PCR. The 5′ and 3′ primers carried BamHI and KpnI sites, respectively. After amplification, the PCR product was gel purified, cut with BamHI and KpnI, and cloned directly between the BamHI and KpnI sites of a pBluescript KS vector.
Immunoprecipitation of TRBP
Capped TRBP mRNA was in vitro transcribed by T7 RNA polymerase after linearization of the template with KpnI. In vitro–transcribed RNA was checked for integrity by gel electrophoresis and used for in vitro translation in a rabbit reticulocyte extract (Promega Corp., Madison, WI) in the presence of 35S-labeled methionine. The translation product was checked by SDS gel electrophoresis followed by autoradiography. To determine whether antibodies directed against Xenopus Xlrbpa would also recognize human TRBP, the in vitro–translated protein was used for immunoprecipitations with antisera Rb5, Rb6, and Rb7, coupled to protein A–Sepharose beads. Preimmune sera or beads alone were used as a control. All immune sera were able to precipitate 35S-labeled human TRBP while corresponding controls failed to do so.
Isolation of Ribosomes
Xenopus ribosomes were isolated from pieces of ovary containing ∼100 mature oocytes. The ovaries were homogenized in ribosomal buffer D containing 0.1% deoxycholic acid, 1 mM vanadyl–ribonucleoside complex, 100 μg/ml PMSF, 2 μg aprotinin, and 2 μg/ml pepstatin by ultrasonication on ice. The crude extract was centrifuged twice for 15 min in a cooled table top centrifuge. Avoiding the fatty surface layer, the supernatant was layered over a linear 10% to 30% (wt/vol) sucrose gradient prepared in ribosomal buffer E (50 mM KCl, 2 mM MgCl2, 1 mM β-mercaptoethanol, 50 mM Tris-HCl, pH 7.8) and centrifuged for 4.5 h at 26,000 rpm in a Beckman SW28 rotor (Beckman Instruments, Inc., Fullerton, CA) at 4°C (Brown et al., 1974). Gradients were fractionated in a cold room. 1-ml fractions were taken from the bottom of the gradient and absorbance at 254 nm was monitored with an UV M-II optical unit (Pharmacia Diagnostics AB).
Immunofluorescence Staining
Cells grown on acid-etched coverslips were fixed and permeabilized according to Fu and Maniatis (1990), with the exception that methanol replaced acetone in the permeabilization step. After fixation, cells were dehydrated through an ethanol series, permeabilized with methanol at −20°C for 10 min, washed in 70% ethanol, and transferred to PBS. Lampbrush chromosome preparations were performed as described (Gall et al., 1991). Immunofluorescence staining was performed as described by Wu et al. (1991).
Construction of Control Construct MP-2
Control construct MP-2 was constructed by PCR amplification of the two dsRBDs of a clone related to 4f1 (Bass et al., 1994). Primers were designed in a way to allow in-frame cloning of the amplified fragment upstream of six myc tags, followed by the polyadenylation (polyA) tail of NO38 cDNA. The resulting clone encodes a protein with a calculated molecular weight of 32 kD consisting of two dsRBDs followed by six COOH-terminal myc tags.
Protein Expression and Northwestern RNA-binding Assay
To determine the RNA-binding ability of construct MP-2, the coding region of this construct was cloned in-frame into the protein expression vector pET17b. For a comparison of RNA-binding of MP-2 and wild-type Xlrbpa, the entire coding region of Xlrbpa was cloned into the protein expression vector pET 3b, resulting in construct pET3.5. For protein expression, both clones were transformed into E. coli BL21(DE3). Small cultures were grown to mid log phase at which stage protein expression was induced by the addition of 1 mM isopropyl β-d-thiogalactoside (IPTG). After 2 h of induction, cells were harvested by centrifugation and cell lysates were prepared by sonication in SDS sample buffer. RNA-binding assays were performed as described, using radiolabeled poly rI/rC as a double-stranded RNA substrate (St Johnston et al., 1992).
Multimerization Assay
To determine a possible multimerization of Xlrbpa, the full-length protein was expressed from construct pET3.5 (see above) in E. coli BL21(DE3). Overexpressed protein was purified by two rounds of cation-exchange chromatography resulting in at least 90% pure protein as judged by SDS gel electrophoresis. Purified Xlrbpa was RNA-binding as determined by Northwestern assay.
The multimerization potential of Xlrbpa was tested by chromatography of purified protein on a Sepharose CL-4B sizing column (Pharmacia Diagnostics AB) next to suitable sizing standards. On these columns Xlrbpa eluted right at its calculated molecular weight of 32 kD, giving no indication for a potential multimerization of the protein. As a second approach to determine potential multimerization of Xlrbpa in solution, glutaraldehyde cross-linking of the protein was performed (Thompson et al., 1991). Transcription factor GCN-4 and carbonic anhydrase were used as positive and negative controls in the same assay. Cross-linking products were analyzed by SDS electrophoresis and silver staining of gels. No cross-linking of Xlrbpa could be detected in this assay. Finally, purified Xlrbpa was subjected to sedimentation gradient analysis. Again, there was no indication for a possible multimerization of Xlrbpa, as the protein always migrated at the top of the gradient.
Tissue Culture Transfection Assays
To determine the cellular localization of construct MP-2, the coding region, including the six myc tags and the polyA tail of MP2 were cloned into the eukaryotic expression vector pcDNA (Invitrogen, San Diego, CA). The resulting construct was used for transient transfection of coverslip-grown HeLa cells. Immunofluorescence staining of transfected cells was performed as described (Jantsch and Gall, 1992).
Oocyte Injection
Construct MP-2 was linearized at a unique BamHI site downstream of the NO38 polyA tail. Capped in vitro–transcribed RNA was produced using T3 RNA polymerase as described (Jantsch and Gall, 1992). Oocytes were injected with ∼50 ng RNA per oocyte and incubated overnight at 16°C to allow protein synthesis. For Western blotting, ∼10 germinal vesicles (GV) or three cytoplasms were used, per lane. For sedimentation gradient analysis, the homogenate of ∼100 microinjected oocytes were used for each gradient. The MP-2 myc fusion protein was detected by Western blotting using mAb 9E10 directed against the myc tag, and a secondary anti– mouse antibody labeled with alkaline phosphatase for enzymatic detection (Evan et al., 1985).
Results
Isolation of Xlrbpa cDNA
By Northwestern screening of a Xenopus laevis oocyte cDNA library with radiolabeled U1 and U2 snRNAs we have previously isolated several clones that encode proteins which bind to these RNAs. Sequence analysis and deletion mapping of the RNA-binding region within these clones led to the identification of the dsRBD in several cDNAs (St Johnston et al., 1992).
One group of the isolated cDNAs encodes a protein showing strong homology to human TRBP (Gatignol et al., 1991), both at the primary amino acid level and in its overall organization (Fig. 1). We called the protein encoded by these cDNAs Xenopus laevis RNA-binding protein A (Xlrbpa).
Figure 1.
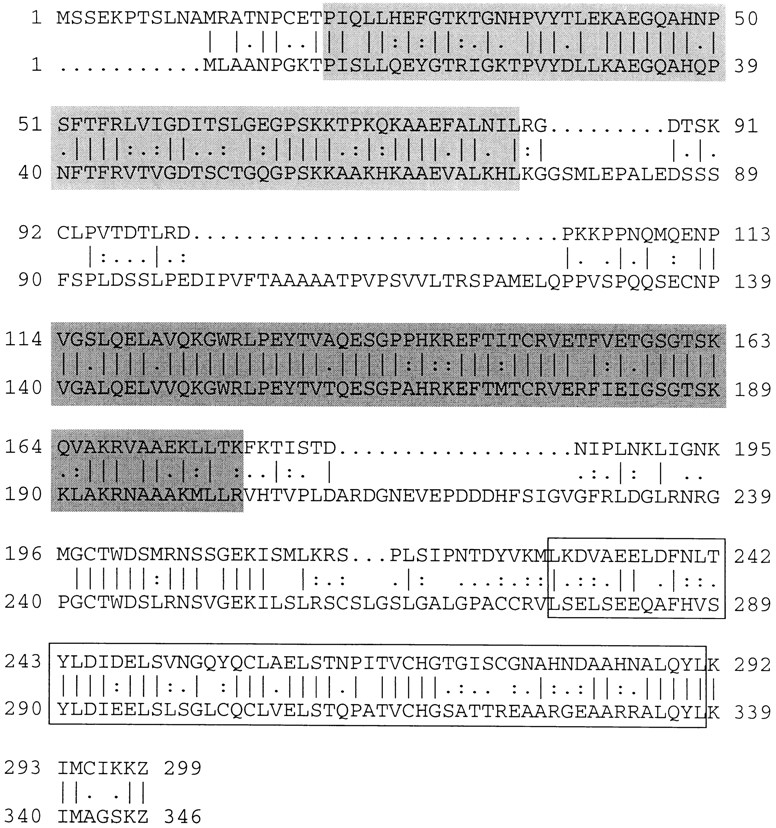
Protein sequence of Xlrbpa (top) aligned with that of human TRBP (bottom). The alignment was generated with program “Gap” of the GCG package (GCG, Madison, WI). ∣, identical amino acids; :, similar amino acids. Double-stranded RNA-binding regions are boxed. dsRBD-1 (light gray); dsRBD-2 (dark gray); dsRBD-3 (open box).
At the amino acid level Xlrbpa shows >70% homology and >55% identity to human TRBP. The two proteins are of similar size with calculated molecular weights of 32.8 and 36.9 kD for Xlrbpa and human TRBP, respectively. Additionally, both proteins are very similar in their general organization. Both contain three tandemly arranged dsRBDs, each separated by short spacers of variable length (Fig. 1). In both proteins, the third dsRBD shares a lower degree of homology with the overall dsRBD consensus sequence while the first and second dsRBDs are highly homologous to it (St Johnston et al., 1992; Krovat and Jantsch, 1996).
Taken together, the data suggest that Xlrbpa is the Xenopus homologue of human TRBP. This view is supported by two additional facts. First, in pairwise alignments of 20 dsRBDs from different proteins, the corresponding dsRBDs in Xlrbpa and human TRBP always turn out to be more closely related to each other than to any other dsRBDs in the entire alignment (St Johnston et al., 1992; Krovat and Jantsch, 1996). Second, antibodies directed against Xlrbpa recognize in vitro–translated human TRBP and a protein of identical molecular weight to TRBP in HeLa cells (see below).
Expression of Xlrbpa
To study the distribution of Xlrbpa both at the level of the entire organism and at the cellular level, we produced antibodies against an Xlrbpa fusion protein and a peptide located in the third dsRBD of Xlrbpa. All antisera recognized Xlrbpa expressed from E. coli and the endogenous protein in Xenopus oocytes; however, the antisera directed against the fusion protein (Rb6 and Rb7) gave stronger signals than peptide sera and were thus used for most parts of this study.
On Western blots, all antisera recognized a single band of 33 kD both in oocytes and XlA6, a Xenopus cell line (Fig. 2 a), while preimmune sera showed no signals on these blots (data not shown). The observed band of 33 kD corresponds well with a calculated molecular weight of 32.8 kD for Xlrbpa. In addition, we tested whether any of our antisera would recognize human TRBP, the putative homologue of Xlrbpa. All antisera directed against Xlrbpa fusion protein (Rb6 and Rb7) were able to specifically immunoprecipitate 35S-labeled, in vitro–translated human TRBP while the corresponding preimmune sera failed to do so (data not shown). Additionally, antisera were tested on Western blots for their ability to recognize human TRBP in HeLa cell extracts. Of all sera tested, Rb6 directed against the Xlrbpa fusion protein gave the strongest signal on these blots. The band observed in HeLa extracts migrates a little slower than the corresponding protein in Xenopus oocyte extracts (Fig. 2 b), which is in good agreement with the slightly larger molecular weight of 36 kD of human TRBP. The corresponding preimmune serum showed no signal in HeLa cells (data not shown). Also, the antiserum could be blocked by preincubation with E. coli–produced Xlrbpa, suggesting that serum Rb6 recognizes authentic human TRBP in HeLa cells (data not shown).
Figure 2.

Distribution of Xlrbpa and TRBP in various frog tissues and HeLa cells. (a) Equal amounts of protein extract from Xenopus oocytes (Oo) and XlA6 tissue culture cells (XLA) were run on a SDS gel, blotted onto Immobilon membrane and detected with anti-Xlrbpa antiserum Rb6. In both tissues, a single band of 33 kD is detectable. (b) Detection of putative human TRBP by polyclonal serum Rb6. Oocyte extracts (Oo) and HeLa tissue culture cell extracts (HeLa) were probed by Western blotting with polyclonal serum Rb6. Antiserum Rb6 detects Xlrbpa in Xenopus oocytes and a band of slightly larger molecular weight in HeLa cells representing the putative homologue, human TRBP. (c) Distribution of Xlrbpa in various frog tissues. Equal amounts of protein from spleen, liver, kidney, heart, nerve, and brain were probed for the presence of Xlrbpa by Western blotting with serum Rb6. Xlrbpa can be detected in all tissues but in different concentrations. Spleen and liver have the highest, heart and kidney intermediate, and nerve and brain lowest concentration of Xlrbpa. In some tissues (liver, kidney) prominent degradation products are visible. The exposure time for nerve and brain lanes was twice that of the other tissues. (d) Intracellular distribution of Xlrbpa. A single enucleated oocyte (Cytoplasm) and 20 isolated nuclei (GV) were separated on an SDS gel and probed by Western blotting with serum Rb6. About equal amounts of Xlrbpa can be detected in both lanes. Since a GV occupies <10% of the total oocyte volume, the proteins in the cytoplasmic lanes are derived from half the volume than in the GV lanes. The concentration of Xlrbpa in the nucleus is, therefore, about half of that in the cytoplasm. Arrows indicate position of Xlrbpa in all gels.
To determine the expression of Xlrbpa in adult Xenopus we performed Western blots of various tissues. Xlrbpa could be detected in all tissues investigated, including oocyte, spleen, liver, kidney, heart, nerve, brain, and XlA6, indicating that Xlrbpa is a ubiquitously expressed protein (Fig. 2, a and c). There was, however, a marked quantitative difference of Xlrbpa among these tissues. The protein seemed most abundant in oocytes and liver, while only smaller amounts could be detected in spleen, kidney, and heart. In nervous tissue Xlrbpa was only barely detectable. Besides full-length Xlrbpa, smaller bands were detectable in some tissues. The occurrence and intensity of these additional bands varied with experiments and depended on the speed of preparation, source of organs, and use of protease inhibitors. We therefore believe that these additional bands of exclusively lower molecular weight than Xlrbpa represent breakdown products of Xlrbpa. It should be noted, however, that it was almost impossible to obtain protein extracts completely free of degradation products, indicating that Xlrbpa is very sensitive to proteolytic degradation.
Intracellular localization of Xlrbpa
Human TRBP has been suggested to be involved in the transcriptional activation of HIV TAR-RNA and also has been shown to increase the expression of reporter genes when expressed under the control of other viral promoters (Gatignol et al., 1991). However, an RNA-independent interaction of TRBP with the double-stranded RNA-activated kinase PKR has also been reported recently (Benkirane et al., 1997). Direct transcriptional activation could only take place in the nucleus while interaction of TRBP with the cytoplasmic kinase PKR would most likely occur in the cytoplasm. We therefore wanted to determine the intracellular localization of Xlrbpa and its human homologue TRBP. To do this, immunostainings of oocyte sections and coverslip-grown HeLa tissue culture cells were performed. Additionally, we made Western blots of hand-enucleated oocytes and isolated germinal vesicles.
Surprisingly, most of the protein could be detected in the cytoplasm of stained oocyte sections (Fig. 3). Upon close investigation, however, minor staining could also be detected in the nucleus where staining appeared in small dots, possibly representing nucleoli or chromatin. The same result was obtained on Western blots of hand-isolated GVs and enucleated oocytes (Fig. 2 d).
Figure 3.

Distribution of Xlrbpa in Oocyte sections. Oocyte sections were stained with Rb6 preimmune serum (a), or immune serum Rb6 (b and c). (a) No staining can be observed in sections stained with preimmuneserum. (b) Staining with immuneserum Rb6 shows strong cytoplasmic and also nuclear signals, indicating that most but not all of Xlrbpa is located in the cytoplasm. An arrowhead marks the oocyte enlarged in c. (c) Enlargement of the small oocyte marked by an arrowhead in b. Besides the strong cytoplasmic signal, nuclear staining can also be observed. The observed nuclear signals (marked by arrows) could represent nucleoli or chromatin. Bars: (a and b) 200 μm; (c) 20 μm.
For comparison, we stained coverslip-grown HeLa tissue culture cells with the anti-Xlrbpa antiserum Rb6. As mentioned, this antiserum recognizes human TRBP (Fig. 2 b). Similar to the situation in oocytes, human TRBP in HeLa cells seemed to be located in both the cytoplasm and the nucleus, resulting in an almost homogeneous staining pattern (Fig. 4). However, when compared to oocytes, the concentration of TRBP in HeLa cells seemed relatively high in the nucleus. In the cytoplasm, the staining appeared in minute dots possibly colocalized with the ER. Colocalization of TRBP with the ER could be indicative of an association of the protein with ribosomes. Therefore, to compare the cytoplasmic distribution of TRBP with that of ribosomes in HeLa cells we performed double immunofluorescence staining with Rb 6 and serum 13751, an anti-ribosomal autoimmune serum that recognizes ribosomal proteins P0, P1, and P2 (Steiner, G., unpublished results), which are the putative eukaryotic homologues of bacterial ribosomal proteins L10, L7, and L12, respectively (Santos and Ballesta, 1994). In these experiments both sera showed similar staining patterns, consistent with an association of TRBP with the ER (Fig. 4 g).
Figure 4.

Localization of Xlrbpa and ribosomes in HeLa cells. Coverslip-grown HeLa cells were stained with preimmune serum Rb6 (a– c), immune serum Rb6 (d–f), and anti-ribosomal serum 13751 (g). (a and d) Phase contrast images, (b and e) DAPI images, (c and f) FITC images, and (g) rhodamine image. Preimmune serum Rb6 shows no signal in HeLa cells (c), while staining with immuneserum Rb6 reveals a homogenous, slightly punctate pattern in the entire cell (f). (g) Cells in f were double-labeled with an anti-ribosomal serum 13751 which was visualized in the rhodamine channel. Staining with both anti-Xlrbpa and anti-ribosomal serum 13751 resulted in very similar images, revealing a homogeneous, slightly punctate staining pattern over the entire cell. Bar, 10 μm.
Xlrbpa in Ribosomes
To determine whether the cytoplasmic localization of Xlrbpa and its human homologue TRBP could be due to a physical association of these proteins with ribosomes, we performed sedimentation gradient analyses of Xenopus oocyte extracts. In sucrose gradients, the majority of Xlrbpa cosedimented with the peak of 80S ribosomes. Only minor amounts could be found in the pellet and supernatant corresponding to polysomes and hnRNAs, as well as free protein, respectively (Fig. 5 A). To confirm these results and to exclude the possibility that Xlrbpa was only trapped in the ribosomal fraction, the peaks of 80S ribosomes from several gradients were concentrated by microfiltration and rerun on a second sucrose gradient. Again, the majority of Xlrbpa was cosedimenting with 80S ribosomes, confirming the association of Xlrbpa with ribosomes (Fig. 5 A, right). A ribosomal association of Xlrbpa is in agreement with our recent study which demonstrated binding of Xlrbpa to rRNA in vitro (Krovat and Jantsch, 1996). The observed association of Xlrbpa with 80S ribosomes was unchanged by puromycin treatment of oocyte extracts, but was sensitive to RNase A treatment (data not shown). It should be noted, however, that the association of Xlrbpa with ribosomes was sensitive to high salt washes. When ribosomal pellets were exposed to salt concentrations higher than 300 mM, a large fraction of Xlrbpa dissociated from the ribosomes resulting in reduced amounts of Xlrbpa cosedimenting with ribosomes and larger amounts of Xlrbpa migrating as free protein at the top of the gradient (data not shown).
Figure 5.

Sedimentation gradient analysis of Xlrbpa and control construct MP2. (A) Oocyte lysates were fractionated on sucrose gradients and tested for the distribution of Xlrbpa (left). UV absorbance at 260 nm was recorded for all fractions (top) indicating the peak of 80S ribosomes in fractions 13–23. Representative fractions were tested by Western blotting with serum Rb7 for the presence of Xlrbpa (A). The majority of Xlrbpa could be found in the ribosomal peak fractions (fractions 16 and 18). In these fractions, degradation bands of lower molecular weight could also be detected. Minor amounts of Xlrbpa could be found at the top of the gradient (fraction 34), corresponding to free protein. However, the free protein was mostly degraded. Unfractionated oocyte extract was loaded in the first two lanes (Oo). Position of full-length Xlrbpa is indicated by an arrow. Ribosomal peaks from several primary gradients were concentrated in a Centricon microconcentration device (exclusion limit = 50 kD) and rerun on a second sucrose gradient (right). Supernatant (cen) and flowthrough (ft) from the microconcentration step was also monitored for the presence of Xlrbpa. Xlrbpa was exclusively present in the top chamber, indicating the presence of the protein in a particle larger than 50 kD. UV absorbance of the gradient rerun was monitored (top) and representative fractions were tested for the presence of Xlrbpa (A). Again, the majority of Xlrbpa could be found in the ribosomal peak fractions. No free protein could be found in the supernatant fraction (34). (B) Oocyte lysates of MP2-injected oocytes were fractionated on sucrose gradients. Fractions were monitored for the presence of MP2 protein by Western blotting with mAb 9E10 directed against the myc epitope. No MP2 protein could be found in the ribosomal peak fractions. Instead all MP2 remained at the top of the gradient. Unfractionated extracts of injected oocytes (Oo) were loaded as a control.
Xlrbpa has a molecular weight of 33 kD. The free protein should therefore be able to pass through a microfiltration membrane with a molecular weight cutoff of 50,000. However, if Xlrbpa was part of a larger particle, such as a ribosome, it should remain in the top chamber of the microconcentration device. We therefore tested the material from the top and bottom chamber of the Centricon device for the presence of Xlrbpa. As can be seen in Fig. 5 A, all Xlrbpa remained in the top chamber during microconcentration, indicating the association of Xlrbpa with a physical particle >50 kD.
Purified full-length Xlrbpa does not form protein multimers as judged by sizing columns, sedimentation gradients, and glutaraldehyde cross-linking experiments (data not shown). The possibility that Xlrbpa does not pass through the microfiltration column due to the formation of protein multimers could therefore be excluded. However, RNA-dependent multimerization of TRBP and the dsRBD-containing fragment of PKR kinase has been observed in the yeast two-hybrid system and in vitro (Cosentino et al., 1995). Even direct interaction of PKR and TRBP has been reported recently (Benkirane et al., 1997). An association of Xlrbpa with ribosomes could therefore be mediated by an interaction of the protein with the ribosome-associated kinase PKR.
Nonetheless, to exclude the possibility that Xlrbpa would only stick to ribosomes in a nonspecific manner due to the protein's intrinsic RNA-binding ability or, alternatively, would form multimers through a common RNA linker, we determined whether an unrelated control construct containing two dsRBDs would also associate or cosediment with ribosomes when expressed in Xenopus oocytes. The Xenopus 4f1 protein seemed suitable for this purpose. Xenopus 4f1 contains a nuclear localization signal (NLS) and two dsRBDs, and is also very abundant in Xenopus oocytes (Fig. 6 A) (Bass et al., 1994). The 4f1 protein is homologous to the human NF90 transcription factor (Kao et al., 1994). A clone encoding an isoform of this protein was previously isolated by us (Jantsch, M., unpublished results). Since full-length 4f1 protein contains an NLS and is ∼98 kD in size, we wanted to express a part of the protein encoding the two dsRBDs only. An ∼600-bp region, including the two dsRBDs, was therefore amplified by PCR from our 4f1-related cDNA and cloned in-frame upstream of six myc tags which are followed by the 3′ UTR and polyA tail of Xenopus NO38 in a pBluescript SK vector (Fig. 6 B) (Jantsch and Gall, 1992). The resulting construct, designated as MP2, was sequenced to confirm the presence of the coding region for the two dsRBDs and the six myc tags (Fig. 6 C). The putative translation product of MP2 has a calculated molecular weight of ∼31 kD and should thus, like Xlrbpa, be able to diffuse into the nucleus. To determine whether MP2 was indeed able to bind dsRNA, the MP2 myc fusion and full-length Xlrbpa were expressed in E. coli and tested for their RNA-binding ability in a Northwestern assay. As expected, both proteins showed strong in vitro RNA-binding ability (Fig. 7, A and B).
Figure 6.

Schematic representation of control construct MP2. (A) Clone 4f1 contains two dsRBDs (gray boxes) and a NLS (hatched box). PCR primers used to amplify the sequence encoding the two dsRBDs are indicated by arrows. (B) The PCR amplified fragment was cloned upstream of six tandemly arranged myc tags (open box) and the NO38 3′ UTR and poly(A)+ tail in pBluescript KS to give construct MP2. T3 transcripts of the construct could be made after linearization at a unique restriction site downstream of the NO38 A+ tail. (C) Conceptual translation product of MP2. Gray boxes, the two dsRBDs. Underlining, the myc epitopes.
Figure 7.
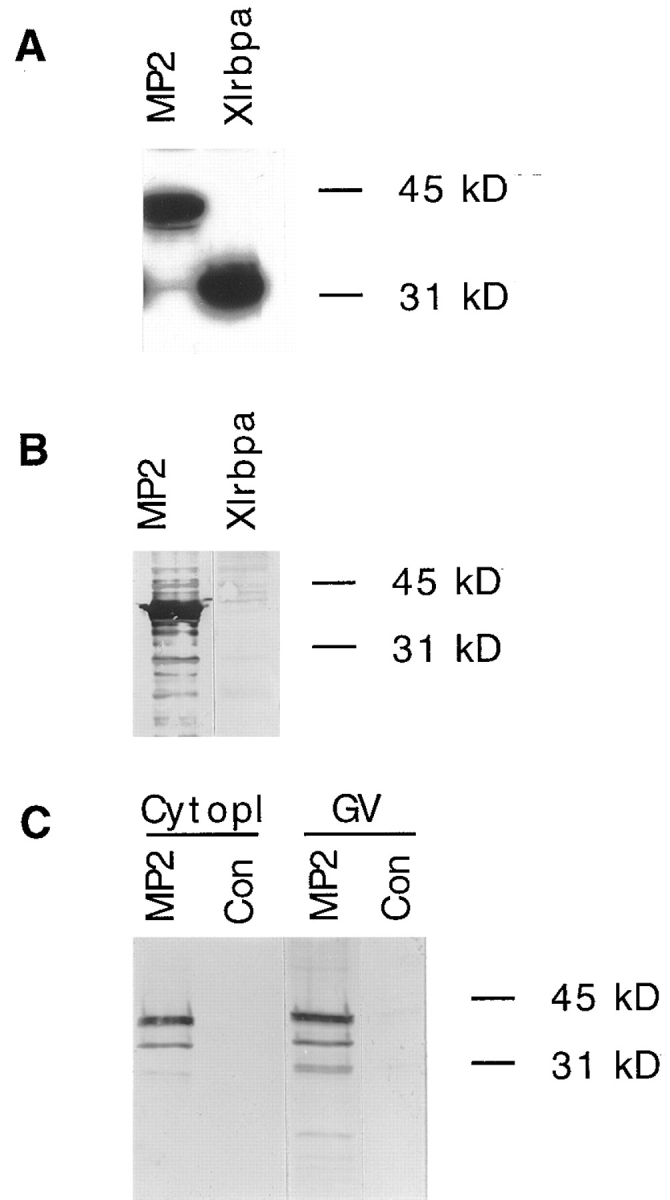
RNA-binding ability of construct MP2 and detection of the protein by myc antibodies in oocytes. (A) Control construct MP2 and full-length Xlrbpa were expressed in Escherichia coli. RNA-binding abilities of both constructs were determined by Northwestern probing of E. coli lysates. Both proteins show strong RNA-binding activity. (B) Detection of epitope-tagged MP2 protein by mAb 9E10 directed against the myc tag. E. coli lysates of cells expressing either MP2 or Xlrbpa protein were probed by Western blotting with mAb 9E10. MP2 protein is readily detectable by mAb 9E10. (C) Detection of MP2 protein in microinjected oocytes. 1 cytoplasm (Cytopl) and 10 germinal vesicles (GV) of injected (MP2) and uninjected (Con) oocytes were separated by SDS-PAGE and tested for the presence of MP2 protein by Western blotting with mAb 9E10. MP2 protein could be detected in both cellular compartments indicating that MP2 can freely diffuse into the nucleus. No protein could be detected in uninjected oocytes.
For expression of MP2 in Xenopus oocytes, in vitro transcripts of the construct were injected into oocytes. Western blots of nuclear and cytoplasmic extracts were detected with mAb 9E10 directed against the myc tag (Evan et al., 1985). Although the protein migrated at a slightly aberrant molecular weight of ∼38 kD, MP2 could be detected in the cytoplasm and the nucleus, indicating that MP2 carried no cryptic NLS but could freely diffuse into the nucleus (Fig. 7 C).
To determine whether MP2 would associate with ribosomes, we performed sedimentation gradient analyses of oocytes expressing MP2. 100 oocytes were injected with MP2 RNA. After overnight incubation, to allow protein synthesis to occur, the oocytes were homogenized and loaded on a sedimentation gradient. Fractions of these gradients were then analyzed for the presence of MP2 protein (Fig. 5 B). As expected, no MP2 protein could be found cosedimenting with ribosomes. Instead all MP2 protein remained at the top of the gradient indicating that MP2 protein, although RNA-binding, failed to form multimers or to associate with ribosomes.
Colocalization in situ and cosedimentation of Xlrbpa with ribosomes is indicative, but does not provide absolute proof for the physical association of the protein with ribosomes. Therefore, to test the association of Xlrbpa and TRBP with ribosomes in a more direct manner, we performed coimmunoprecipitation experiments. Two different anti-ribosomal autoimmune sera were used to precipitate ribosomes from oocyte extracts. The precipitated material was then tested for the presence of Xlrbpa by Western blotting. Both antisera, serum 9275 and serum 13751 were able to coprecipitate Xlrbpa with ribosomes, while all Xlrbpa remained in the supernatant when no antibody was added to the beads (Fig. 8 A). In contrast, when MP2-injected oocytes were used in the same experiment, no coprecipitation of MP2 protein with ribosomes could be detected (Fig. 8 B, and data not shown). These data suggest that Xlrbpa and possibly its human homologue, TRBP, are closely associated with ribosomes. Our control experiments also indicate that ribosomal association of these two proteins is specific and not merely an effect of the intrinsic RNA-binding ability of any dsRBD-containing protein.
Figure 8.

Coprecipitation of Xlrbpa with ribosomal proteins. (A) Immunoprecipitations of oocyte extracts were made with anti- ribosomal sera 13751, 92751, or beads alone. The immunoprecipitated material (p) and an aliquot of the corresponding supernatant (sn) were tested for the presence of Xlrbpa. Both sera 13751 and 92751 were able to coprecipitate Xlrbpa, while no protein could be precipitated when no antibody had been added to the beads. As a control, a small aliquot of total oocyte extract was loaded (total). An arrow indicates position of Xlrbpa. (B) In contrast, MP2 protein could not be coprecipitated by either anti- ribosomal serum. When oocytes injected with MP2 RNA were used for the same experiment all MP2 protein remained in the supernatant. An arrow indicates position of MP2 protein.
Xlrbpa in the Nucleus
Xlrbpa and TRBP could not only be detected in the cytoplasm, but were also present in the nucleus as shown by in situ immunostaining and Western blotting of hand-enucleated oocytes and germinal vesicles. Xlrbpa does not contain an obvious consensus nuclear localization signal (Dingwall and Laskey, 1991). Its presence in the nucleus and the relatively small molecular weight of <33 kD therefore suggest that the protein can enter the nucleus by diffusion.
To investigate the nuclear localization of Xlrbpa and TRBP in more detail we performed immunofluorescence stainings of spread Xenopus germinal vesicles. Because of its size, the amphibian germinal vesicle allows a detailed observation of nuclear organelles like lampbrush chromosomes, amplified nucleoli and snurposomes, spherical organelles enriched in snRNPs, and several accessory splicing factors (Wu et al., 1991). Nascent transcripts on the chromosomes can easily be detected as well as pre-ribosomal RNAs on nucleoli.
In these spreads Xlrbpa was detectable in essentially all RNA-containing structures; nucleoli, lampbrush chromosomes, and snurposomes showed moderate to strong staining with anti-Xlrbpa antibodies, indicating the association of the protein with most, if not all nuclear RNAs. This general picture was independent of the antibody used for immunofluorescence staining (Fig. 9).
Figure 9.

Localization of Xlrbpa in nuclear spreads of Xenopus GVs. (a and b) Lampbrush chromosome preparations were stained with preimmune serum Rb6 or (c and d) immune serum Rb6. Antibodies were detected with a secondary FITC-labeled antibody. (a and c) Phase contrast; (b and d) fluorescence image. (a) Lampbrush chromosomes, C snurposomes (C), B snurposomes (B), and nucleoli (N) can be seen in the phase contrast image. The large C snurposome (C) has a smaller B snurposome attached to its surface. (b) No staining can be observed with preimmune serum Rb6. (c) Nuclear organelles are labeled as in (a). The large C snurposome (C) has an attached B snurposome (B). (d) Essentially all nuclear structures are labeled after staining with serum Rb6. Lampbrush chromosomes, nucleoli (N), C snurposomes (C), and B snurposomes (B) are easily detectable. C snurposomes label most brilliantly while the other structures show only moderate staining with this antibody. A comparison of B and C snurposome staining can easily be made on the large, intensely labeled C snurposome (C) which has a moderately stained, smaller B snurposome on its surface. Bar, 10 μm.
The staining on the amplified nucleoli suggests that ribosomal transcripts already associate with Xlrbpa before their export from the nucleus and assembly into mature ribosomes. This observation is in good agreement with our finding that Xlrbpa is present in ribosomes. Xlrbpa is a double-stranded RNA-binding protein; ribosomal RNAs are known to contain long, double-stranded stem-loop regions and hairpins (Noller et al., 1981), and could thus provide enough binding sites for Xlrbpa.
In addition to nucleoli, the RNP matrix of lampbrush chromosomes stained homogeneously along the length of the entire chromosome. This was somewhat surprising since it seemed unlikely that all transcripts contain sufficient double-stranded regions to allow binding of significant amounts of Xlrbpa. However, the homogeneous and relatively intense staining of all transcripts could be explained if Xlrbpa associated with nascent transcripts as part of a larger hnRNP complex (Piñol-Roma et al., 1989). In this case, not Xlrbpa itself, but other proteins within the hnRNP complex would make the contact with RNA where Xlrbpa would only bind directly to RNA if sufficient double-stranded regions were present. To test this hypothesis, we wanted to determine whether Xlrbpa was associated with hnRNP particles. We therefore made immunoprecipitations with anti-Xlrbpa antibodies and tested the precipitated material for the presence of known hnRNPs. As a precipitating antibody we used antisera Rb6 or Rb7 and the corresponding preimmune sera as controls. Both antisera are able to precipitate in vitro–translated human TRBP (data not shown) and recognize human TRBP on Western blots (Fig. 2 b). For the detection of hnRNP proteins we used mAb 4F4 directed against human hnRNPs C1 and C2 (Choi and Dreyfuss, 1984). Since mAb 4F4 only recognizes human hnRNPs, and not the corresponding proteins from Xenopus, we used HeLa nucleoplasmic extracts as starting material. Both antisera Rb 6 (Fig. 10 A) and Rb 7 (not shown) were able to coprecipitate hnRNPs C1 and C2 very efficiently while no signals were seen when either preimmuneserum was used. Coprecipitation of hnRNPs C1 and C2 by anti-Xlrbpa antisera was resistant to predigestion of the nucleoplasmic extracts with RNases, indicating a physical association of Xlrbpa with these hnRNPs. We thus believe that Xlrbpa is part of hnRNP particles, which would also explain its presence on nascent transcripts.
Figure 10.

Association of Xlrbpa with hnRNPs. (A) HeLa nuclear extracts were used for immunoprecipitations with anti-Xlrbpa serum Rb6 (#6), or the corresponding preimmuneserum (PI #6). Some of the material was digested with RNases (+RNAse) or left undigested (−RNAse) before incubation with the antibody-coupled beads. Before washing, the beads were pelleted by centrifugation and an aliquot of the supernatant was saved (sn). After several washes, beads were boiled in SDS sample buffer, and corresponding supernatants (sn) and pellets (p) were assayed for the presence of hnRNPs C1 and C2 by Western blotting with mAb 4F4. As a reference, total HeLa nucleoplasmic extracts were loaded (total). Antiserum Rb6 (#6) could coprecipitate hnRNPs C1 and C2 while the corresponding preimmuneserum did not. Coprecipitation of hnRNPs was resistant to RNase digestion (+RNAse), indicating a physical association of Xlrbpa with these proteins. An arrow indicates the position of hnRNPs C1 and C2. (B) Immunoprecipitations of nuclear extracts from HeLa cells stably expressing MP2 protein performed with anti-hnRNP antibody 4F4 (4F4), antiserum Rb6 (#6) or beads alone (beads). The precipitated material (p) and corresponding supernatants (sn) were tested for the presence of MP2 protein by Western blotting with mAb 9E10. No MP2 protein could be coprecipitated with either antibody, instead all MP2 protein remained in the supernatant. As a control total extracts of HeLa cells (total) expressing MP2 (+) or untransfected cells (−) were loaded. An arrow indicates position of MP2 protein. The strong background bands in the 4F4 pellet lanes derive from IgG heavy and light chains, which are detected by the secondary anti–mouse antibody used to detect mAb 9E10.
To determine whether the dsRBD-containing control construct MP2 would also associate with hnRNP particles, we performed immunoprecipitation experiments with HeLa cells which were stably transfected with a eukaryotic vector expressing MP2 protein. Immunofluorescence staining of these cell lines with mAb 9E10 showed a homogeneous distribution of MP2 protein throughout the entire cell (data not shown). Immunoprecipitations of stably expressing cell lines were performed with either mAb 4F4 directed against hnRNPs C1 and C2, or with polyclonal serum Rb6 directed against Xlrbpa (TRBP). Immunoprecipitates were then probed with mAb 9E10 for the presence of myc-tagged MP2 protein (Fig. 10 B). In these experiments, MP2 protein could not be coprecipitated with either antibody, indicating that MP2 protein does not associate with hnRNP particles.
The positive staining of snurposomes indicated the presence of Xlrbpa in these structures. Two types of snurposomes (B and C) can be distinguished in Xenopus GVs. The smaller B snurposomes, which can sometimes be found associated with C snurposomes, are highly enriched in the five splicing snRNPs, U1, U2, U5, and U4/U6 (Wu et al., 1991). Besides these snRNPs, accessory splicing factors such as SC-35 or SR proteins can be found in these structures (Roth et al., 1991; Zahler et al., 1992). However, also hnRNPs can be detected in B snurposomes (Wu et al., 1991). In contrast, no hnRNPs could so far be detected in the generally larger C snurposomes. Instead, C snurposomes seem to contain U7 snRNPs as the only snRNP component (Wu and Gall, 1993). The presence of p80 coilin in these structures suggests a homology of C snurposomes and coiled bodies found in somatic cells (Wu et al., 1994; Gall et al., 1995).
In our immunofluorescence stainings, C snurposomes labeled generally more intensely than B snurposomes, suggesting a higher concentration of Xlrbpa in the former structures (Fig. 9). This was somewhat surprising since no hnRNPs could so far be detected in C snurposomes. However, since all snurposomes contain snRNPs, we wanted to determine whether the presence of Xlrbpa in snurposomes could be due to an association of the protein with snRNPs. We performed immunoprecipitation experiments with anti-Xlrbpa and anti-snRNP antibodies and tested the precipitated material for the presence of snRNP proteins or Xlrbpa, respectively. In these experiments, no association of Xlrbpa with snRNPs could be detected (data not shown). It thus seems unlikely that Xlrbpa is associated with snRNAs in snurposomes. Instead, Xlrbpa could be associated with other hnRNPs, at least in B snurposomes.
Discussion
We have cloned and characterized Xlrbpa, a Xenopus double-stranded RNA-binding, 33-kD protein. Our data indicate that Xlrbpa is the putative homologue of human TRBP (Gatignol et al., 1991). Both proteins show a high degree of homology at the primary amino acid level and in their overall organization. They contain three tandemly arranged dsRBDs. In both proteins, the first two dsRBDs show a high degree of homology to a dsRBD consensus sequence while the third dsRBD fits the consensus only at its carboxy-terminal end (St Johnston et al., 1992). The dsRBD has been shown to exclusively interact with double-stranded RNAs or RNA–DNA hybrids (St Johnston et al., 1992; Bass et al., 1994). A preference for these two substrates could be explained by the similar structures exhibited by these double-stranded molecules; both RNA– DNA and RNA–RNA helices assume A form helices, which differ considerably from the B form helices adopted by most double-stranded DNAs. Although both substrates, RNA–RNA and RNA–DNA duplexes have been shown to be recognized by the dsRBD in vitro, no example of an RNA–DNA hybrid substrate has been shown to be the natural substrate for any known dsRBD-containing protein. In contrast, in all cases where the substrate for a dsRBD containing protein has been identified, it represents an RNA duplex molecule. Consistent with this idea, we could demonstrate that Xlrbpa and human TRBP are associated with ribosomes and hnRNAs. In both cases RNA–RNA hybrids rather than RNA–DNA hybrids seem the likely substrates of these proteins.
Xlrbpa, a Ribosomal Protein?
Xlrbpa is ubiquitously expressed in essentially all tissues tested and it seems likely that human TRBP is also ubiquitously expressed in humans (Gatignol et al., 1991). However, we observed marked differences among the tissues tested. Especially nerve and brain tissue showed a very low level of Xlrbpa, while oocytes had the highest concentrations. These differences in concentration could possibly be explained by our finding that Xlrbpa and TRBP associate with ribosomes. It is known that amphibian oocytes contain extremely high levels of ribosomes. In contrast, although neurons contain considerable amounts of ribosomes, nervous tissue as a whole has a relatively low ribosome content. Thus, it appears that Xlrbpa concentration resembles the concentration of ribosomes in a given tissue. Since ribosomal RNAs represent the majority of all RNAs in a cell, it is also obvious that the low amounts of Xlrbpa (or TRBP) assembled with hnRNPs do not fall into account for this comparison.
We could demonstrate the association of Xlrbpa and TRBP with ribosomes by immunofluorescence, cofractionation on density gradients, and coimmunoprecipitation experiments. Additionally, we could purify ribosomal RNAs in immunoprecipitation experiments with anti-Xlrbpa antibodies (data not shown). Sticking, and therefore non-specific association of Xlrbpa (or human TRBP) with ribosomes, could be excluded by our control experiments in which a dsRNA-binding control construct, containing two dsRBDs failed to associate with ribosomes. Taken together, we are confident that Xlrbpa and TRBP are closely associated with ribosomes. This finding is also supported by our previous observation that Xlrbpa can bind rRNA in vitro (Krovat and Jantsch, 1996). A ribosomal association of Xlrbpa or TRBP is also consistent with the recent finding that TRBP can interact with the ribosome-associated kinase PKR (Benkirane et al., 1997). Ribosomes and their components have been intensely studied over the last decades. It is thus surprising that neither Xlrbpa, nor human TRBP have been described as ribosomal proteins, especially when considering their relative abundance. However, most ribosomal preparations include a high salt wash to remove associated proteins. dsRBDs lose their in vitro–RNA-binding ability at salt concentrations >300 mM (Jantsch, M., unpublished data). Consistently, we observed a dissociation of Xlrbpa from ribosomes in sedimentation gradients at high salt concentrations, which could explain its absence from standard ribosomal preparations. Thus, we believe that Xlrbpa is merely a ribosome-associated factor, but not a necessary component of translationally active ribosomes.
Xlrbpa on Nascent Transcripts
Xlrbpa and human TRBP could also be found in the nucleus. Although both proteins lack a clear nuclear localization signal, they seem small enough to enter the nucleus by simple diffusion through nuclear pores. In nuclear spreads of Xenopus GVs, we could localize the protein in essentially all RNA-containing structures: amplified nucleoli, lampbrush chromosomes, and snurposomes. The nucleolar localization is in agreement with our finding that Xlrbpa and TRBP are associated with ribosomes. It also suggests that the proteins can associate with ribosomal RNAs, possibly even nascent pre-ribosomal RNAs before they are processed, exported to the cytoplasm, and assembled into mature ribosomes.
The localization of Xlrbpa on lampbrush chromosomes indicated an association of the protein with essentially all nascent transcripts. Xlrbpa binds exclusively to double-stranded RNAs in vitro (St Johnston et al., 1992; Bass et al., 1994). This raises the question as to how the protein can associate with the many transcripts lacking double-stranded regions. However, the homogeneous distribution of Xlrbpa on essentially all hnRNAs could be explained by our finding that Xlrbpa is associated with hnRNP complexes. In this case, not Xlrbpa itself but the entire hnRNP complex, including Xlrbpa, would associate with the nascent transcripts. Thus, Xlrbpa would only bind RNA when sufficient double-stranded regions are available on the corresponding transcript. In the absence of extensive double-stranded structures, however, other hnRNP proteins within the complex would bind to the RNA. The decision of which RNA-binding protein within an hnRNP complex binds directly to RNA would thereby be governed by competition of the proteins for a suitable binding substrate. The fact that our control construct, MP2, fails to associate with hnRNP particles indicates that the association of Xlrbpa or human TRBP with hnRNPs is specific, and not caused by the intrinsic RNA-binding ability of the two proteins.
An analogous situation has been found for the distribution of snRNPs and splicing components in amphibian GVs. Surprisingly, all components of the splicing machinery can be found on most nascent transcripts irrespective of whether those transcripts contain suitable splice sites or not (Wu et al., 1991). Thus, it seems as if splicing components associate with nascent transcripts, possibly as part of larger RNP complexes, independent of the availability of processing sites. This situation is similar to the one found by us, where Xlrbpa is associated with essentially all nascent transcripts seemingly independent of the availability of secondary structures required for the direct binding of the protein to RNA. Instead, Xlrbpa seems to be associated with transcripts as part of a larger hnRNP complex.
Besides an association of Xlrbpa with nucleoli and nascent transcripts we could find the protein in snurposomes within the Xenopus GV. Snurposomes are spherical structures of 1–10 μm diam, which can be distinguished based on their structure and molecular composition in B and C snurposomes. In Xenopus, the smaller B snurposomes are highly enriched in the five splicing snRNPs, U1, U2, U5, and U4/U6 as well as several accessory splicing factors (Wu et al., 1991). Additionally, staining with anti-hnRNP antibodies indicates the presence of hnRNPs in B snurposomes (Wu et al., 1991). The generally larger C snurposomes, in contrast, seem to lack splicing snRNPs but contain the U7 snRNP (Wu and Gall, 1993). C snurposomes frequently have B snurposomes attached to their surface or internalized and are thus also called sphere organelles. These structures also contain p80 coilin, suggesting that sphere organelles are homologous to coiled bodies found in several somatic cells (Wu et al., 1994; Gall et al., 1995). No hnRNPs are detectable in C snurposomes (Wu et al., 1991). The presence of Xlrbpa in B snurposomes and its homogeneous distribution on nascent transcripts resembles that of splicing snRNPs and splicing factors, as well as that of hnRNP proteins in the amphibian GV closely. However, in immunoprecipitations with the anti-Sm antibody, Y12 (Lerner et al., 1981), no association of Xlrbpa with splicing snRNPs could be detected. It is thus likely that Xlrbpa is either present as free protein in B snurposomes or, alternatively, associated with the other hnRNP proteins found in these structures. Since Xlrbpa can be found associated with hnRNP particles, the latter hypothesis appears more likely.
The presence of Xlrbpa in C snurposomes is more problematic. In view of the fact that anti-Sm antibodies were unable to precipitate Xlrbpa, an association of the protein with the U7 snRNP seems very unlikely. Also, no other known hnRNPs could so far be detected in these organelles. Additionally, in HeLa cells we observed no indication for the presence of the Xlrbpa homologue, TRBP, in coiled bodies, which are believed to be homologous to sphere organelles. All these facts make a reasonable explanation for the presence of Xlrbpa in C snurposomes very difficult at this point. However, future studies will be aimed to explain the mechanism that is responsible for the localization of Xlrbpa in these structures.
Possible Functions of Xlrbpa
Human TRBP has been isolated by its ability to bind HIV TAR-RNA in vitro, and has been suggested to be a human-specific factor specifically required for the replication of HIV in human cells (Gatignol et al., 1991). Binding specificity of TRBP for TAR-RNA has been determined by the introduction of mutations in this RNA which abolished binding of the protein to this sequence (Gatignol et al., 1991). However, several points argue against the notion that human TRBP is a human specific factor necessary for the replication of HIV.
First, TRBP does not seem to be exclusively present in human cells. Our data strongly suggests that Xlrbpa is the Xenopus homologue of TRBP, thus demonstrating the presence of a TRBP homologue in an organism incompatible with the replication of HIV.
Second, Xlrbpa and human TRBP contain three tandemly arranged dsRBDs. So far, no sequence-specific binding of dsRBDs could be determined in vitro. In contrast, Xlrbpa has been isolated by its ability to bind U1 or U2 snRNAs in vitro, but can also bind poly rI/rC very efficiently (St Johnston et al., 1992; Krovat and Jantsch, 1996). Also, the third dsRBD in the Drosophila Staufen protein binds both U1 snRNA and poly rI/rC in vitro (St Johnston et al., 1992). None of these substrates used for in vitro assays seem to be the natural substrates of these proteins. Thus it is also possible that TAR-RNA is not the in vivo substrate for human TRBP. TAR-RNA forms a stable double-stranded hairpin region. To prove the binding specificity of TRBP for TAR-RNA, several mutations were introduced in this RNA, some of which abolished binding of TRBP. However, all mutations affecting TRBP binding were located in the base-paired region of the TAR-RNA hairpin, thus destabilizing the double-stranded region (Gatignol et al., 1991). Therefore, it seems likely that structural changes in the RNA, rather than changes in the primary nucleic acid sequence were responsible for the observed loss of binding of TRBP to TAR-RNA.
It has been shown that chloramphenicol transferase (CAT)-reporter genes under the control of viral promoters are more strongly expressed when TRBP was cotransfected in these cells (Gatignol et al., 1991). This effect has been proposed to be caused by a stimulation of these promoters by TRBP. However, it has not been shown that transcription from the promoter itself was increased by TRBP. Recently, another study reported increased levels of expression of transfected genes upon cotransfection of either TRBP or the vaccinia virus E3L gene, another dsRBD-containing protein (Park et al., 1994). In this case, increased expression of the transfected genes correlated in part with increased mRNA amounts of the corresponding genes. Both effects, increased reporter gene activity and increased mRNA amounts of certain transfected genes, could be explained in part by mRNA stabilization mediated by Xlrbpa or TRBP. We could show that TRBP is part of hnRNP particles. It is thus possible that Xlrbpa or TRBP can stabilize certain mRNAs with extensive secondary structures by binding to them.
It has also been shown that human TRBP acts as an inhibitor of the cellular, double-stranded–activated, interferon-dependent protein kinase PKR (Park et al., 1994). This kinase phosphorylates translation initiation factor, eIF2α, when activated by double-stranded RNAs upon viral infection, resulting in an inhibition of translation (Kaufmann et al., 1989; Manche et al., 1992). Inhibition of PKR by TRBP and other dsRBD-containing proteins has been attributed to the masking of double-stranded RNAs, thereby preventing binding and subsequent activation of PKR by these substrate RNAs (Watson et al., 1991; Langland et al., 1994; Park et al., 1994). Additionally, TRBP has been suggested to be involved in the masking of cellular mRNAs containing extensive double-stranded regions, thus precluding inadvertent activation of PKR by cellular RNAs. This view is consistent with our finding that Xlrbpa or TRBP associate with essentially all cellular double-stranded RNAs. Binding of Xlrbpa or TRBP to double-stranded RNAs might therefore help to prevent the activation of PKR by these RNAs.
However, our finding that Xlrbpa is associated with ribosomes can also be interpreted in an alternative way. Rather than masking ribosomal RNAs to avoid activation of PKR, Xlrbpa or TRBP might compete with the ribosome-associated kinase PKR for suitable substrates directly at the ribosome. Although no substrate specificities of dsRBDs could so far be determined in vitro, biological data suggests that dsRBD-containing proteins can bind in a sequence-specific manner (St Johnston et al., 1991). It is therefore possible that activation of PKR in vivo depends on the availability of high affinity substrates for this kinase. Xlrbpa would thereby act as a competitor to PKR and prevent the binding of low affinity substrates to the kinase by binding to them at the ribosome.
RNA-dependent dimerization of TRBP and PKR has recently been demonstrated in the yeast two hybrid system and in vitro (Cosentino et al., 1995). Direct (RNA-independent) interaction of TRBP and PKR has also been reported recently (Benkirane et al., 1997). The same study shows that TRBP can down-regulate PKR activity in an RNA-independent manner, possibly through direct protein–protein contact (Benkirane et al., 1997). Association of TRBP or Xlrbpa with ribosomes could therefore be mediated by an association of these proteins with PKR, a kinase which has also been shown to be ribosome associated (Levin and London, 1978). However, to demonstrate RNA-independent down-regulation of PKR by TRBP, a mutant version of TRBP (defective in its second dsRBD and thus unable to bind dsRNA) was used (Benkirane et al., 1997). A similar mutation in Xlrbpa, in which the second dsRBD has either been inactivated by mutation or completely deleted, still shows significant RNA-binding activity in our hands (Krovat and Jantsch, 1996). This raises the question of whether down-regulation of PKR by a mutated TRBP is indeed mediated by a completely RNA-independent mechanism or, alternatively, whether residual RNA-binding activity of the mutated TRBP might lead to the observed effect, possibly by competition of TRBP for dsRNAs that activate PKR.
Nonetheless, no PKR homologous kinase has so far been described in Xenopus. Therefore, several other functions can also be envisaged for Xlrbpa. On the one hand, Xlrbpa might act as a general hnRNP whose main function is the complexation and packaging of RNA. Complexation of RNA with hnRNPs has been shown to be important for the proper processing and export of RNA (Bennet et al., 1992; Mayeda and Krainer, 1992; Piñol-Roma and Dreyfuss, 1993). At the same time, hnRNPs might stabilize and protect RNA from degradation. On the other hand, Xlrbpa might itself influence the folding state of RNA. Since Xlrbpa binds exclusively to double-stranded regions it might either help in the formation of such structured regions, stabilize such regions or, alternatively, destabilize double-stranded regions. Double-strand annealing and unwinding activities have also been reported for other RNA-binding proteins (Cobianchi et al., 1993; Portman and Dreyfuss, 1994; Xiao et al., 1994). The structure of two dsRBDs from different proteins has recently been solved and showed a conserved structure for those two dsRBDs (Bycroft et al., 1995; Kharrat et al., 1995). Mutational analyses of Xlrbpa suggest that the dsRBDs found in Xlrbpa assume the same conserved tertiary structure (Krovat and Jantsch, 1996). However, little is known about the way double-stranded RNA-binding domains interact with RNA which makes an assessment about other functions of Xlrbpa purely speculative at this point. Nonetheless, studies to determine the mode of interaction of dsRBDs with RNA are under way and will hopefully help to shed some light on these unsolved questions. It will also be interesting to determine whether Xlrbpa and its human counterpart, TRBP, play a role in mRNA turnover and translation of cellular RNAs.
Acknowledgments
We thank J. Steitz for mAb Y12, and G. Dreyfuss for mAb 4F4. We also thank G. Steiner for human sera 13751 and 92751. A. Wolffe kindly supplied the Xenopus ovary cDNA library, and I. Mattaj the clones of Xenopus U snRNAs. The authors are thankful to A. Barta, G. Steiner, J.G. Gall, and members of the department for a critical review of the manuscript. A. Neunteufl provided excellent technical assistance.
This work was supported by Austrian Science Foundation research grant P 09665. C.R. Eckmann is the recipient of a Ph.D. fellowship from the Austrian Academy of Sciences.
Footnotes
1. Abbreviations used in this paper: ds, double-stranded; GST, glutathione-S-transferase; GV, germinal vesicle; HIV, human immunodeficiency virus; hn, heterogeneous nuclear; NLS, nuclear localization signal; polyA, polyadenylation; RBD, RNA-binding domain; sn, small nuclear; TAR, trans-activation–responsive; TRBP, TAR-RNA binding protein.
Please address all correspondence to Michael Jantsch, Department of Cytology and Genetics, Institute of Botany, University of Vienna, Rennweg 14, A-1030 Vienna, Austria. Tel.: 43-1-79-794-177. Fax: 43-1-79-794-131.
References
- Adam SA, Nakagawa T, Swanson MS, Woodruff TK, Dreyfuss G. mRNA polyadenylate-binding protein: gene isolation and sequencing and identification of a ribonucleoprotein consensus sequence. Mol Cell Biol. 1986;6:2932–2943. doi: 10.1128/mcb.6.8.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandziulis RJ, Swanson MS, Dreyfuss G. RNA-binding proteins as developmental regulators. Genes Dev. 1989;3:431–437. doi: 10.1101/gad.3.4.431. [DOI] [PubMed] [Google Scholar]
- Bass, B.L. 1993. RNA editing: new uses for old players in the RNA world. In The RNA World. R.F. Gesteland and J.F. Atkins, editors. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 383–418.
- Bass BL, Hurst SR, Singer JD. Binding properties of newly identified Xenopusproteins containing dsRNA-binding motifs. Curr Biol. 1994;4:301–314. doi: 10.1016/s0960-9822(00)00069-5. [DOI] [PubMed] [Google Scholar]
- Benkirane M, Neuveut C, Chun RF, Smith SM, Samuel CE, Gatignol A, Jeang K-T. Oncogenic potential of TAR RNA binding protein TRBP and its regulatory interaction with RNA-dependent protein kinase PKR. EMBO (Eur Mol Biol Organ) J. 1997;16:611–624. doi: 10.1093/emboj/16.3.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennet M, Piñol-Roma S, Staknis D, Dreyfuss G, Reed R. Transcript-dependent packaging of pre-mRNA into hnRNP complexes prior to spliceosome assembly in vitro. Mol Cell Biol. 1992;12:3165–3175. doi: 10.1128/mcb.12.7.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GE, Kolb AJ, Stanley WM., Jr A general procedure for the preparation of highly active eukaryotic ribosomes and ribosomal subunits. Meth Enzymol. 1974;30:368–387. doi: 10.1016/0076-6879(74)30039-0. [DOI] [PubMed] [Google Scholar]
- Burd CG, Dreyfuss G. Conserved structures and diversity of functions of RNA-binding proteins. Science (Wash DC) 1994;265:615–621. doi: 10.1126/science.8036511. [DOI] [PubMed] [Google Scholar]
- Bycroft M, Grünert S, Murzin AG, Proctor M, St Johnston D. NMR solution structure of a dsRNA binding domain from Drosophilastaufen protein reveals homology to the N-terminal domain of ribosomal protein S5. EMBO (Eur Mol Biol Organ) J. 1995;14:3563–3571. doi: 10.1002/j.1460-2075.1995.tb07362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YD, Dreyfuss G. Monoclonal antibody characterization of the C proteins of heterogeneous nuclear ribonucleoprotein complexes in vertebrate cells. J Cell Biol. 1984;99:1997–2004. doi: 10.1083/jcb.99.6.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofori G, Keller W. Poly(A) polymerase purified from HeLa cell nuclear extract is required for both cleavage and polyadenylation of pre-mRNA in vitro. Mol Cell Biol. 1989;9:193–203. doi: 10.1128/mcb.9.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobianchi F, Calvio C, Stoppini M, Buvoli M, Riva S. Phosphorylation of human hnRNP protein A1 abrogates in vitro strand annealing activity. Nucleic Acids Res. 1993;21:949–955. doi: 10.1093/nar/21.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosentino GP, Venkatesan S, Serluca FC, Green S, Mathews MB, Sonenberg N. Double-stranded-RNA-dependent protein kinase and TAR RNA-binding protein form homo- and heterodimers in vivo. Proc Natl Acad Sci USA. 1995;92:9445–9449. doi: 10.1073/pnas.92.21.9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingwall C, Laskey RA. Nuclear targeting sequences—a consensus? . Trends Biochem Sci. 1991;16:478–481. doi: 10.1016/0968-0004(91)90184-w. [DOI] [PubMed] [Google Scholar]
- Evan G, Lewis G, Ramsay G, Bishop M. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–3636. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X-D, Maniatis T. Factor required for mammalian spliceosome assembly is localized to discrete regions in the nucleus. Nature (Lond) 1990;343:437–441. doi: 10.1038/343437a0. [DOI] [PubMed] [Google Scholar]
- Gall JG, Murphy C, Callan HG, Wu Z. Lampbrush chromosomes. Methods Cell Biol. 1991;36:149–166. [PubMed] [Google Scholar]
- Gall JG, Tsvetkov A, Wu Z, Murphy C. Is the sphere organelle/ coiled body a universal nuclear component? . Dev Genet. 1995;16:25–35. doi: 10.1002/dvg.1020160107. [DOI] [PubMed] [Google Scholar]
- Gatignol A, Buckler-White A, Berkhout B, Jeang K-T. Characterization of a human TAR-RNA–binding protein that activates the HIV-1 LTR. Science (Wash DC) 1991;251:1597–1600. doi: 10.1126/science.2011739. [DOI] [PubMed] [Google Scholar]
- Green SR, Mathews MB. Two RNA-binding motifs in the double-stranded RNA-activated protein kinase, DAI. Genes Dev. 1992;6:2478–2490. doi: 10.1101/gad.6.12b.2478. [DOI] [PubMed] [Google Scholar]
- Hamm J, Mattaj IW. Monomethylated cap structures facilitate RNA export from the nucleus. Cell. 1990;63:109–118. doi: 10.1016/0092-8674(90)90292-m. [DOI] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1988. Antibodies—A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 53–137.
- Jantsch MF, Gall JG. Assembly and localization of the U1-specific snRNP C protein in the amphibian oocyte. J Cell Biol. 1992;119:1037–1046. doi: 10.1083/jcb.119.5.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao PN, Chen L, Brock G, Ng J, Kenny J, Smith AJ, Corthesy B. Cloning and expression of cyclosporin A- and FK506-sensitive nuclear factor of activated T-cells: NF45 and NF90. J Biol Chem. 1994;269:20691–20699. [PubMed] [Google Scholar]
- Kaufman RJ, Davies MV, Pathak VK, Hershey JWB. The phosphorylation of eukaryotic initiation factor 2 alters translational efficiency of specific mRNAs. Mol Cell Biol. 1989;9:946–958. doi: 10.1128/mcb.9.3.946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharrat A, Macias MJ, Gibson TJ, Nigels M, Pastore A. Structure of the dsRNA binding domain of E. coliRnase III. EMBO (Eur Mol Biol Organ) J. 1995;14:3572–3584. doi: 10.1002/j.1460-2075.1995.tb07363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krovat BC, Jantsch MF. Comparative mutational analysis of the double-stranded RNA binding domains of Xenopus laevisRNA-binding protein A. J Biol Chem. 1996;271:28112–28119. doi: 10.1074/jbc.271.45.28112. [DOI] [PubMed] [Google Scholar]
- Langland JO, Pettiford S, Jiang B, Jacobs BL. Products of the porcine group C rotavirus NSP3 gene bind specifically to double-stranded RNA and inhibit activation of the interferon-induced protein kinase PKR. J Virol. 1994;68:3821–3829. doi: 10.1128/jvi.68.6.3821-3829.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibold EA, Munro HN. Cytoplasmic protein binds in vitro to a highly conserved sequence in the 5′ untranslated region of ferritin heavy- and light-subunit mRNAs. Proc Natl Acad Sci USA. 1988;85:2171–2175. doi: 10.1073/pnas.85.7.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner EA, Lerner MR, Janeway CA, Steitz JA. Monoclonal antibodies to nucleic acid-containing cellular constituents: probes for molecular biology and autoimmune disease. Proc Natl Acad Sci USA. 1981;78:2737–2741. doi: 10.1073/pnas.78.5.2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin D, London IM. Regulation of protein synthesis: activation by double-stranded RNA of a protein kinase that phosphorylates eukaryotic initiation factor 2. Proc Natl Acad Sci USA. 1978;75:1121–1125. doi: 10.1073/pnas.75.3.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manche L, Green SR, Schmedt C, Mathews MB. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol Cell Biol. 1992;12:5238–5248. doi: 10.1128/mcb.12.11.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeda A, Krainer AR. Regulation of alternative pre-mRNA splicing by hnRNP A1 and splicing factor SF2. Cell. 1992;68:365–375. doi: 10.1016/0092-8674(92)90477-t. [DOI] [PubMed] [Google Scholar]
- Mougey EB, Pape LK, Sollner-Webb B. A U3 small nuclear ribonucleoprotein-requiring processing event in the 5′ external transcribed spacer of Xenopusprecursor rRNA. Mol Cell Biol. 1993;13:5990–5998. doi: 10.1128/mcb.13.10.5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müllner EW, Neupert B, Kühn LC. A specific mRNA binding factor regulates the iron-dependent stability of cytoplasmic transferrin receptor mRNA. Cell. 1989;58:373–382. doi: 10.1016/0092-8674(89)90851-9. [DOI] [PubMed] [Google Scholar]
- Nagai K, Oubridge C, Jessen TH, Li J, Evans PR. Crystal structure of the RNA-binding domain of the U1 small nuclear ribonucleoprotein A. Nature (Lond) 1990;348:515–520. doi: 10.1038/348515a0. [DOI] [PubMed] [Google Scholar]
- Nilsen TW. RNA–RNA interactions in the spliceosome: unraveling the ties that bind. Cell. 1994;78:1–4. doi: 10.1016/0092-8674(94)90563-0. [DOI] [PubMed] [Google Scholar]
- Noller HF, Kop J, Wheaton V, Brosius J, Gutell R, Koplov AM, Dohme F, Herr W, Stahl DA, Gupta R, et al. Secondary structure model for 23S ribosomal RNA. Nucleic Acids Res. 1981;9:6167–6189. doi: 10.1093/nar/9.22.6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Davies MV, Langland JO, Chang H-W, Nam YS, Tartaglia J, Paoletti E, Jacobs BL, Kaufman RJ, Venkatesan S. TAR RNA-binding protein is an inhibitor of the interferon-induced protein kinase PKR. Proc Natl Acad Sci USA. 1994;91:4713–4717. doi: 10.1073/pnas.91.11.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peculis BA, Steitz JA. Disruption of U8 nucleolar snRNA inhibits 5.8S and 28S rRNA processing in the Xenopusoocyte. Cell. 1993;73:1233–1245. doi: 10.1016/0092-8674(93)90651-6. [DOI] [PubMed] [Google Scholar]
- Piñol-Roma S, Dreyfuss G. hnRNP proteins: localization and transport between the nucleus and the cytoplasm. Trends Cell Biol. 1993;3:151–155. doi: 10.1016/0962-8924(93)90135-n. [DOI] [PubMed] [Google Scholar]
- Piñol-Roma S, Choi YD, Matunis MJ, Dreyfuss G. Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev. 1988;2:215–227. doi: 10.1101/gad.2.2.215. [DOI] [PubMed] [Google Scholar]
- Piñol-Roma S, Swanson MS, Gall JG, Dreyfuss G. A novel heterogeneous nuclear RNP protein with a unique distribution on nascent transcripts. J Cell Biol. 1989;109:2575–2587. doi: 10.1083/jcb.109.6.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polson AG, Bass BL. Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO (Eur Mol Biol Organ) J. 1994;13:5701–5711. doi: 10.1002/j.1460-2075.1994.tb06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portman DS, Dreyfuss G. RNA annealing activities in HeLa nuclei. EMBO (Eur Mol Biol Organ) J. 1994;13:213–221. doi: 10.1002/j.1460-2075.1994.tb06251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, R., and H. Busch. 1988. Small nuclear RNAs: RNA sequences, structure, and modifications. In Small Nuclear Ribonucleoprotein Particles. M.L. Birnstiel, editor. Springer-Verlag. Berlin/Heidelberg/New York. 1–37.
- Roth MB, Zahler AM, Stolk JA. A conserved family of nuclear phosphoproteins localized to sites of polymerase II transcription. J Cell Biol. 1991;115:587–596. doi: 10.1083/jcb.115.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Johnston D. The intracellular localization of messenger RNAs. Cell. 1995;81:161–170. doi: 10.1016/0092-8674(95)90324-0. [DOI] [PubMed] [Google Scholar]
- St Johnston D, Beuchle D, Nüsslein-Volhard C. staufen, a gene required to localize maternal RNAs in the Drosophilaegg. Cell. 1991;66:51–63. doi: 10.1016/0092-8674(91)90138-o. [DOI] [PubMed] [Google Scholar]
- St Johnston D, Brown NH, Gall JG, Jantsch M. A conserved double-stranded RNA-binding domain. Proc Natl Acad Sci USA. 1992;89:10979–10983. doi: 10.1073/pnas.89.22.10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos C, Ballesta JPG. Ribosomal protein P0, contrary to phosphoproteins P1 and P2, is required for ribosome activity and Saccharomyces cerevisiaeviability. J Biol Chem. 1994;269:15689–15696. [PubMed] [Google Scholar]
- Savino R, Gerbi SA. In vivo disruption of XenopusU3 affects ribosomal RNA processing. EMBO (Eur Mol Biol Organ) J. 1990;9:2299–2308. doi: 10.1002/j.1460-2075.1990.tb07401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz JA. Immunoprecipitation of riboproteins using autoantibodies. Methods Enzym. 1989;180:468–481. doi: 10.1016/0076-6879(89)80118-1. [DOI] [PubMed] [Google Scholar]
- Thompson CC, Brown TA, McKnight SL. Convergence of Ets- and Notch-related structural motifs in a heteromeric DNA binding complex. Science (Wash DC) 1991;253:762–768. doi: 10.1126/science.1876833. [DOI] [PubMed] [Google Scholar]
- Tyc K, Steitz JA. U3, U8, and U13 comprise a new class of mammalian snRNPs localized in the cell nucleolus. EMBO (Eur Mol Biol Organ) J. 1989;8:3113–3119. doi: 10.1002/j.1460-2075.1989.tb08463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson CR, LaMarco KL, Johnson PF, Landschulz WH, McKnight SL. In situ detection of sequence-specific DNA binding activity specified by a recombinant bacteriophage. Genes Dev. 1988;2:801–806. doi: 10.1101/gad.2.7.801. [DOI] [PubMed] [Google Scholar]
- Wahle E. A novel poly(A)-binding protein acts as a specificity factor in the second phase of messenger RNA polyadenylation. Cell. 1991;66:759–768. doi: 10.1016/0092-8674(91)90119-j. [DOI] [PubMed] [Google Scholar]
- Watson JC, Chang HW, Jacobs BL. Characterization of a vaccinia virus-encoded double-stranded RNA-binding protein that may be involved in inhibition of the double-stranded RNA-dependent protein kinase. Virology. 1991;185:206–216. doi: 10.1016/0042-6822(91)90768-7. [DOI] [PubMed] [Google Scholar]
- Wittekind M, Görlach M, Friedrichs M, Dreyfuss G, Mueller L. 1H,13C, and 15N NMR assignments and global folding pattern of the RNA-binding domain of the human hnRNP C protein. Biochemistry. 1992;31:6254–6265. doi: 10.1021/bi00142a013. [DOI] [PubMed] [Google Scholar]
- Wool, I.G., Y. Endo, Y-L. Chan, and A. Glück. 1990. Structure function, and evolution of mammalian ribosomes. In The Ribosome: Structure, Function & Evolution. W.E. Hill, A. Dahlberg, R.A. Garret, P.B. Boore, D. Schlessinger, and J.R. Warner, editors. American Society for Microbiology, Washington, DC. 203–216.
- Wu C-HH, Gall JG. U7 small nuclear RNA in C snurposomes of the Xenopusgerminal vesicle. Proc Natl Acad Sci USA. 1993;90:6257–6259. doi: 10.1073/pnas.90.13.6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Murphy C, Callan HG, Gall JG. Small nuclear ribonucleoproteins and heterogeneous nuclear ribonucleoproteins in the amphibian germinal vesicle: loops, spheres, and snurposomes. J Cell Biol. 1991;113:465–483. doi: 10.1083/jcb.113.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Murphy C, Gall JG. Human p80-coilin is targeted to sphere organelles in the amphibian germinal vesicle. Mol Cell Biol. 1994;5:1119–1127. doi: 10.1091/mbc.5.10.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Sharp TV, Jeffrey IW, James MC, Pruijn GJM, van Venrooij WJ, Clemens MJ. The La antigen inhibits the activation of the interferon-inducible protein kinase PKR by sequestering and unwinding double-stranded RNA. Nucleic Acids Res. 1994;22:2512–2518. doi: 10.1093/nar/22.13.2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler AM, Lane WS, Stolk JA, Roth MB. SR proteins: a conserved family of pre-mRNA splicing factors. Genes Dev. 1992;6:837–847. doi: 10.1101/gad.6.5.837. [DOI] [PubMed] [Google Scholar]