

Abstract
Cell interaction with adhesive proteins or growth factors in the extracellular matrix initiates Ras/ mitogen-activated protein (MAP) kinase signaling. Evidence is provided that MAP kinase (ERK1 and ERK2) influences the cells' motility machinery by phosphorylating and, thereby, enhancing myosin light chain kinase (MLCK) activity leading to phosphorylation of myosin light chains (MLC). Inhibition of MAP kinase activity causes decreased MLCK function, MLC phosphorylation, and cell migration on extracellular matrix proteins. In contrast, expression of mutationally active MAP kinase kinase causes activation of MAP kinase leading to phosphorylation of MLCK and MLC and enhanced cell migration. In vitro results support these findings since ERK-phosphorylated MLCK has an increased capacity to phosphorylate MLC and shows increased sensitivity to calmodulin. Thus, we define a signaling pathway directly downstream of MAP kinase, influencing cell migration on the extracellular matrix.
Cell adhesion/migration on the extracellular matrix plays a critical role in angiogenesis, inflammatory conditions, embryonic development, wound repair, and tumor metastasis (Yang et al., 1993; Brooks et al., 1994; Drake et al., 1995; Filardo et al., 1995; Brooks et al., 1996; Clark et al., 1996). Integrin-mediated cellular adhesion to the extracellular matrix leads to intracellular signaling, including activation of focal adhesion kinase with subsequent activation of downstream effector molecules including mitogen-activated protein (MAP)1 kinases ERK1 and ERK2 (Q. Chen et al., 1994; Schlaepfer et al., 1994; Zhu and Assoian, 1995). Activation of the MAP kinase pathway leads to transcriptional control of genes important for cell proliferation and differentiation (for reviews see Hill and Treisman, 1995; Marshall, 1995). However, both growth factor receptors and integrins promote signaling events leading to MAP kinase activity and the immediate induction of cell migration (Stoker and Gherardi, 1991; Leavesley et al., 1993; P. Chen et al., 1994; Klemke et al., 1994; Kundra et al., 1994; Yenush et al., 1994), suggesting that MAP kinase can lead to direct activation of the intracellular motility machinery independent of de novo gene transcription.
Ultimately, the motogenic signals generated by integrin or cytokine receptors impact the actin-myosin cytoskeleton and the temporal–spatial organization of cell adhesion contacts on the extracellular matrix, as these events are critical for cell movement (for reviews see Tan et al., 1992; Felsenfeld et al., 1996; Huttenlocher et al., 1996; Lauffenburger and Horwitz, 1996; Mitchison and Cramer, 1996). Myosins are actin-activated ATPases capable of generating force by promoting translational movement along actin cables (Sellers and Adelstein, 1987). While several classes of myosins have been identified, myosin II is the best characterized for its ability to promote cell contraction and migration in nonmuscle cells (McKenna et al., 1989; Giuliano and Taylor, 1990; Wilson et al., 1991; Giuliano et al., 1992; Kolega and Taylor, 1993; Jay et al., 1995). Members of this family consist of two heavy chains (200 kD) and two sets of light chains (16–20 kD) and are widely distributed in eukaryotic cells. Myosin II is concentrated at posterior regions of motile cells and along actin stress fibers in the leading lamellae, where it is thought to play a role in cell contraction and/or in breaking adhesion contacts to the extracellular matrix (Conrad et al., 1993; Gough and Taylor, 1993; Kolega and Taylor, 1993). Myosin II function is regulated by phosphorylation of the regulatory light chains by the Ca2+/calmodulin-dependent enzyme myosin light chain kinase (MLCK) (Adelstein, 1983; de Lanerolle and Paul, 1991). Phosphorylation of myosin light chains (MLC) by MLCK is a critical regulatory step in myosin function since it promotes myosin ATPase activity and polymerization of actin cables. This results in a fully functional actin-myosin motor unit involved in generating contractile force necessary for cell motility. While it is clear these events are necessary for directional cell movement (Adelstein 1983; Wilson et al., 1991; Jay et al., 1995; Smith et al., 1996), little is known about signaling components that result in the activation of MLCK and myosin-mediated cell motility.
In this report, we investigated the role that Ras/MAP kinase activation plays in regulating integrin-mediated cell migration. We show here that MAP kinase activation is required for haptotaxis cell migration on a collagen substrate based on its ability to directly phosphorylate MLCK leading to the phosphorylation of MLC. Thus, a signaling pathway can be defined that is initiated upon cell interaction with the extracellular matrix and culminates in cell migration.
Materials and Methods
Cell Culture
FG carcinoma cells were grown in RPMI 1640 (GIBCO BRL, Gaithersburg, MD) supplemented with 10% FBS, 50 μg/ml gentamicin and were free from mycoplasma during these studies. These cells have been characterized with respect to their collagen-dependent migration properties (Klemke et al., 1994). COS-7 cells were provided by Gary Bokoch (The Scripps Research Institute, La Jolla, CA) and were grown in DME with 10% FBS and 50 μg/ml gentamicin. Before testing, all cells were starved for 24 h by replacing serum containing culture media with FBS-free media.
Antibodies and Reagents
Antiactin monoclonal antibodies were purchased from Sigma Chemical Co. (St. Louis, MO). Rabbit polyclonal antibodies to MAP kinase (ERK1 and ERK2), MEK1, and RAF-1 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti–myosin IIB antibodies were kindly provided by Dr. Robert Adelstein (Molecular Cardiology, National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD). Anti-MLCK antibodies have been previously described (de Lanerolle et al., 1981). Antibodies to the influenza hemagglutinin epitope (HA) were purchased from Boehringer Mannheim Corp. (Indianapolis, IN). Antiphosphotyrosine monoclonal antibody (4G10) was from Upstate Biotechnology, Inc. (Lake Placid, NY). Goat anti–rabbit and –mouse immunoglobulin antibodies were from BioRad Labs (Hercules, CA). PD98059 (2-[2′-amino3′methoxyphenyl]-oxanaphthalen-4-one) is a compound that specifically inhibits MEK and was graciously provided by Dr. Alan Saltiel (ParkeDavis, Ann Arbor, MI). A stock concentration (10 mM) of this compound was prepared in DMSO and frozen at −70°C. The MLCK inhibitor KT5926 was purchased from Calbiochem (La Jolla, CA). Human recombinant EGF was obtained from Genzyme Corp. (Cambridge, MA). Myelin basic protein (MBP) was from Upstate Biotechnology, Inc.
Adhesive Ligands
Vitronectin was prepared as described by Yatohgo et al. (1988). Fibronectin and collagen type 1 were obtained from Upstate Biotechnology, Inc.
Transfection of COS-7 Cells
COS-7 cells (0.5 × 106 cells/10-cm plate) were cotransfected using 25 μl of lipofectamine (GIBCO BRL) and 2 μg of either the eukaryotic expression vector alone (pcDNA3; InVitrogen, San Diego, CA) or the same vector containing the cDNA encoding the mutationally active MEK1 (MEK+) along with 0.5 μg of a reporter construct encoding β-galactosidase (pSV– β-galactosidase, Promega Corp., Madison, WI). Cells were also transfected with either a myc-tagged ERK1 or HA-tagged ERK2 reporter construct (pSRα) (Minden et al., 1995). In other experiments, COS-7 cells were transfected with 0.5 μg of MEK+ cDNA and 1.5 μg of pCMV5 vector and/or the same vector containing mutationally inactive rabbit MLCK, which was generated by oligonucleotide-directed mutagenesis as previously described and shown to lack enzyme activity even though it bound calmodulin (Gallagher et al., 1991). The mutant and wild-type MLCK calmodulin binding and kinase activity assays in COS cell lysates were performed as previously described (Herring et al., 1992; Gallagher et al., 1993). Cells were washed and allowed to incubate for 40 h, which provides optimal transient expression in these cells, and then either prepared for cell migration or lysed and analyzed for expression and phosphorylation of specific proteins as described below. In typical transfection experiments, we achieved a 40% expression efficiency providing a 30–50-fold increase in the level of MEK or MLCK levels compared to endogenous COS cell enzyme levels as determined by Western blotting. Cells cotransfected with β-galactosidase were developed using X-gal as a substrate according to the manufacturer's recommendations (Promega Corp.). In some cases, cells were treated with EGF (100 ng/ml) or different doses of the MEK inhibitor PD98059 and/or the MLCK inhibitor KT5926 for various times before cell migration or protein analysis.
Cell Adhesion and Haptotaxis Migration Assays
Cell adhesion and spreading was performed on collagen-coated surfaces as previously described (Leavesley et al., 1992). Briefly, cell migration assays were performed using modified Boyden chambers (tissue culture– treated, 6.5-mm diameter, 10-μm thickness, 8-μm pores, Transwell®; Costar Corp., Cambridge, MA) containing polycarbonate membranes. The underside of the membrane of the upper chamber was coated with 10 μg/ml collagen type I (Upstate Biotechnology Inc.) for 2 h at 37°C, rinsed once with PBS, and then placed into the lower chamber containing 500 μl migration buffer (fibroblast basal medium, with 0.5% BSA, Clonetics, San Diego, CA). Serum-starved cells were removed from culture dishes with Hanks' balanced salt solution containing 5 mM EDTA and 25 mM Hepes, pH 7.2, washed twice with migration buffer, and then resuspended in fibroblast basal medium, 0.5% BSA (106 cells/ml). 50,000–100,000 cells were then added to the top of each migration chamber and allowed to migrate to the underside of the top chamber for various times. Nonmigratory cells on the upper membrane surface were removed with a cotton swab, and the migratory cells attached to the bottom surface of the membrane were stained with X-gal substrate as described above (for β-galactosidase– expressing cells) or 0.1% crystal violet in 0.1 M borate, pH 9.0, and 2% ethanol for 20 min at room temperature. The number of migratory cells per membrane were either counted with an inverted microscope using a 40× objective, or the stain was eluted with 10% acetic acid and the absorbance was determined at 600 nm, and migration was enumerated from a standard curve. Each determination represents the average of three individual wells, and error bars represent SD. All values have had background subtracted, which represents cell migration on membranes coated on the bottom with BSA (1%). In control experiments, cell migration on BSA was <0.01% of the total cell population.
Immunoprecipitation and Immunoblotting of ERK and MEK Proteins
Cells were rinsed twice with PBS and then lysed with RIPA buffer (100 mM Tris, 0.15 M NaCl, 1% deoxycholic acid, 1% Triton X-100, 0.1% SDS, 1% aprotinin, 2 mM PMSF, 10 μg/ml leupeptin, 5 mM EDTA, 1 mM sodium vanadate, and 50 mM NaF) for 1 h at 4°C. The lysate was clarified by centrifugation at 14,000 rpm for 10 min, and the amount of total protein was determined using the BCA protein assay reagent (Pierce, Rockford, IL). The protein concentration of the samples were normalized before either direct immunoblotting of total cell proteins or immunoprecipitation of equivalent amounts of proteins with ERK antibodies bound to protein A–Sepharose beads. Samples were analyzed by SDS-PAGE and then transferred to nitrocellulose and blocked with BSA (3%, Sigma Chemical Co.) before immunoblot analysis using antiphosphotyrosine monoclonal antibody (4G10) or anti-MEK and horseradish peroxidase–conjugated goat anti–mouse or rabbit secondary antibodies and the enhanced chemiluminescence system. In some cases, the immunoblots were stripped and reprobed with antibodies according to the manufacturer's recommendations (enhanced chemiluminescence, Amersham Corp., Arlington Heights, IL).
Treatment of Cells with ERK Oligonucleotides
Depletion of MAP kinase (ERK1 and ERK2) from cells was performed according to the procedure of Sale et al. (1995) with minor modifications. Briefly, FG cells grown to 60% confluency in 60-mm tissue culture dishes were incubated in culture media containing lipofectamine (40 μg/ml) and 1.5 μM ERK antisense (5′-GCC GCC GCC GCC GCC AT-3′; Genset) or control phosphorothioate oligonucleotides (5′-CGC GCG CTC GCG CAC CC-3′) for 8 h at 37°C. They were then rinsed, incubated for 16 h in fresh culture medium containing oligonucleotides, starved for 16 h in serum-free culture media containing oligonucleotides, and then tested in adhesion and migration assays as described above. An aliquot of the above cells was analyzed in parallel with cell migration assays for changes in ERK protein levels using the ERK-specific antibodies and Western blotting as described above. As a control for nonspecific depletion of proteins, the blots were stained with Ponceau dye to visualize total cellular proteins and then stripped and reprobed with antiactin, anti-MEK, and anti–Raf-1 antibodies.
In Vitro Kinase Assay for MAP Kinase
The ability of ERK to phosphorylate MBP was determined according to Boulton and Cobb (1991). Briefly, 500 μg of protein from detergent cell lysates were precleared with protein A–Sepharose for 4 h in the cold and then incubated with protein A–Sepharose coupled with anti-ERK antibodies (4 μg/100 μl stock bead suspension; Pierce) overnight in the cold. Immunoprecipitates were rinsed three times with RIPA and once with 50 mM Hepes, pH 8.0, containing 0.1 M NaCl before incubation with 100 μl of reaction mixture containing 0.5 μCi [γ-32P]ATP, 10 mM MgCl, 50 μM ATP, 1 mM DTT, 1 mM benzamidine, 0.3 mg/ml MBP, and 25 mM Hepes, pH 8.0, for 15 min at 30°C. The reaction was stopped by adding 100 μl of boiling SDS sample buffer. MBP (8 μg) was then analyzed by 15% SDSPAGE, stained with Coomassie blue, dried, and exposed to imaging film overnight.
32P Labeling and Immunoprecipitation of Myosin and MLCK from COS-7 Cells
Preparation of cell lysates and immunoprecipitation of myosin were performed according to de Lanerolle et al. (1993). Briefly, COS-7 cells cotransfected as described above with either empty vector or the MEK+ vector together with the HA-ERK2 reporter construct were cultured for 6 h in phosphate-free media and then isotopically labeled with 5 ml of DME phosphate-free media containing 250 μCi/100-mm petri dish for 2 h in the presence or absence of the MEK inhibitor PD98059 (25 μM) or the MLCK inhibitor KT5926 (10 μM). Cells were rinsed twice with ice-cold PBS, cell extracts were prepared, and the protein concentration was determined as described above. Total cell proteins (20–50 μg) were either analyzed directly for changes in phosphoproteins by SDS-PAGE and autoradiography or immunoprecipitated and Western blotted with anti–myosin IIB or MLCK-specific antibodies before autoradiography. Alternatively, myosin or MLCK was immunoprecipitated from these extracts (750 μg protein/ IP) and analyzed for changes in phosphorylation by SDS-PAGE and autoradiography.
In Vitro Phosphorylation of MLCK by Activated MAP Kinase (ERK1)
Purified chicken gizzard MLCK (4.2 μg) was incubated at 30°C in the presence or absence (control for autophosphorylation of MLCK) of 0.4 μg of purified activated ERK1 or ERK2 (a kind gift from Drs. D. Schlaepher and T. Hunter, Salk Institute, La Jolla, CA) in 50 μl of kinase reaction mixture (10 mM MgCl2, 2 mM DTT, 0.5 mM Na3VO4, 5 mM NaF, 20 mM Tris, pH 7.5, and 0.1 mM ATP (1 mCi/ml). Aliquots that contained 1 μg of MLCK were removed at 5, 10, 20, and 40 min, boiled in SDS sample buffer, separated by SDS-PAGE, and then stained with Coomassie blue and analyzed by autoradiography.
In Vitro Phosphorylation of MLC by MLCK Previously Phosphorylated by MAP Kinase (ERK1)
Phosphorylation of myosin II by MLCK was performed as previously described by de Lanerolle and Nishikawa (1988). Briefly, MLCK (50 ng) was either phosphorylated or not phosphorylated with activated MAP kinase (ERK1) as described above and then incubated with 10 μg of myosin II purified from macrophage-like J774 cells in 50 μl of reaction buffer (10 mM MgCl2, 0.5 mM ATP [1 mCi/ml], 20 mM Tris, pH 7.5) in the presence or absence of 0.5 mM EGTA or 0.4 mM calcium and 10−6 M calmodulin. The kinase reactions were performed at room temperature for various times, terminated by boiling in SDS, and analyzed by SDS-PAGE and autoradiography.
Purified MLC were phosphorylated as described above except 10 μg of MLC purified from chicken gizzard smooth muscle was used as substrate. Aliquots of the reaction mixture containing 2 μg of light chain were removed, boiled in SDS, and analyzed by SDS-PAGE and autoradiography. Importantly, the ability of activated MAP kinase to phosphorylate MLC was tested by repeating the assay under identical conditions using 5 ng of activated MAP kinase, which is the amount calculated to be present in the MLCK assays following phosphorylation by MAP kinase. The effect of calmodulin on MLC phosphorylation was determined by adding 0.4 mM calcium and various concentrations of calmodulin to the assay mixtures.
Results
MAP Kinase Is Critical for Cell Migration but Not Adhesion or Spreading on Collagen
To examine a potential role of MAP kinase in cell migration, we examined FG carcinoma cells, which readily migrate on a collagen substrate using integrin α2β1 (Leavesley et al., 1992; Klemke et al., 1994). FG cells were pretreated with antisense oligonucleotides specific for ERK1 and ERK2 (Sale et al., 1995) or a scrambled control oligonucleotide and allowed to attach, spread, and migrate on a collagen substrate. As shown in Fig. 1 A, upper panel, exposure of cells to antisense but not a control oligonucleotide reduced ERK1 and ERK2 protein levels by ∼90% yet had no effect on expression of other proteins in these cells, as determined by Western blotting with antibodies to Raf-1 and actin or a nonspecific protein recognized by the rabbit polyclonal antibodies to ERK1 and ERK2. Cells treated with the antisense oligonucleotide readily attached and spread (data not shown) on collagen, but showed 75% decreased migration (Fig. 1 A, lower panel) relative to cells treated with the control oligonucleotide. These findings suggest that MAP kinase influences cell migration yet has little apparent effect on adhesion or spreading on a collagen substrate.
Figure 1.



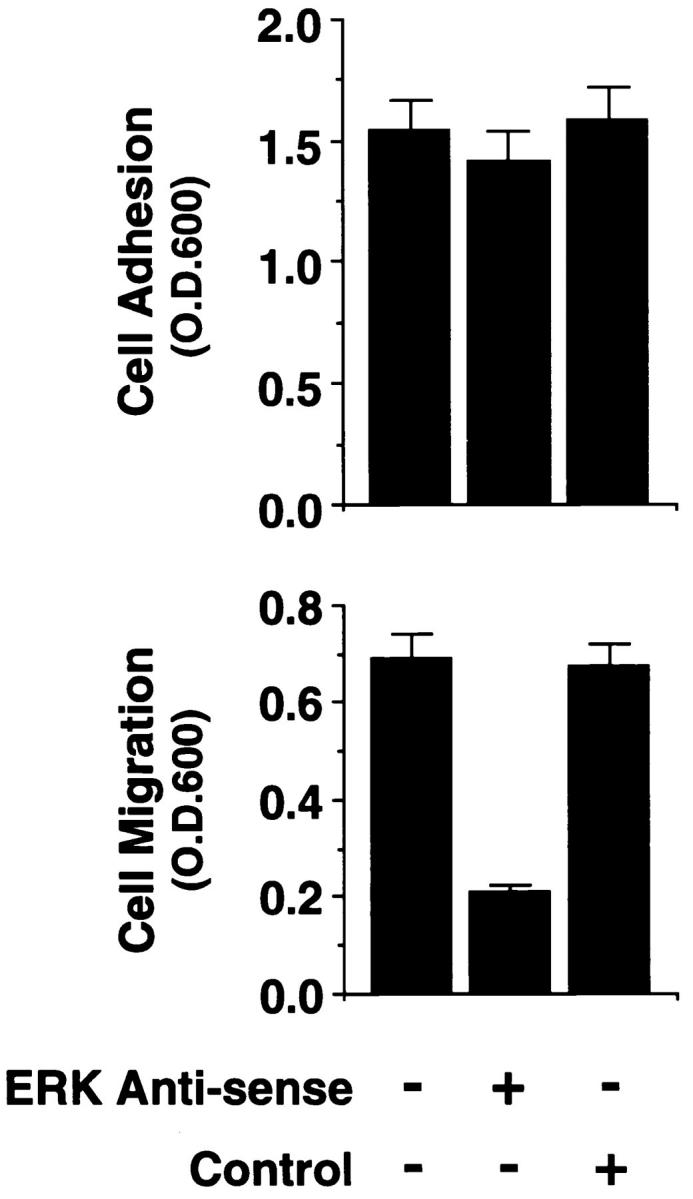

MAP kinase (ERK1 and ERK2) is required for cell migration but not adhesion or spreading on extracellular matrix proteins. (A, lower panel) Cells were allowed to adhere for 1 h or migrate for 6 h on a collagen substrate after exposure to MAP kinase–specific antisense or a scrambled control oligonucleotide. Migration studies were performed using Transwell migration chambers coated with collagen as described in Materials and Methods. (Upper panel) Total cell lysates (50 μg) prepared from an aliquot of those FG cells analyzed for migration/adhesion were immunoblotted with antibodies to Raf-1, actin, or ERK proteins. The upper band is a nonspecific protein recognized by the rabbit polyclonal ERK antisera. The result shown is a representative experiment from at least three independent experiments. (B, upper panel) Phosphotyrosine immunoblot or kinase activity of ERK1 and ERK2 proteins immunoprecipitated from cells either pretreated in the presence or absence of the MEK inhibitor PD98059 (25 μM) and allowed to attach to poly-l-lysine or collagen-coated dishes for 30 min. The ERK immunoprecipitates were also examined for their ability to phosphorylate MBP in an in vitro kinase assay as described in Materials and Methods. (Lower panel) Cells treated in the presence or absence of the MEK inhibitor as above were allowed to attach for 1 h or migrate for 6 h on a collagen substrate and quantified as described in Materials and Methods. The result shown is a representative experiment from at least three independent experiments. (C) Nomarski photomicrograph (600×) of FG cells pretreated for 6 h in the presence or absence (control) of the MEK inhibitor (100 μM) and allowed to attach and spread on collagen-coated coverslips for 10 or 60 min. The result shown is a representative experiment from at least three independent experiments. Bar, 10 μm.
ERK1 and ERK2 are specifically activated by MAP kinase kinase (MEK1) because of its ability to promote both threonine and tyrosine phosphorylation of these enzymes (Cowley et al., 1994; Mansour et al., 1994; Zheng and Guan, 1994). Therefore, cells were pretreated with an inhibitor of MEK (PD98059), which specifically prevents its ability to promote both threonine and tyrosine phosphorylation of MAP kinase (Dudley et al., 1995; Pang et al., 1995), and allowed to attach, spread, or migrate on collagen. Pretreatment of the cells with the MEK inhibitor blocked ERK1 and ERK2 phosphorylation induced by FG cell attachment to collagen in the absence of growth factors as measured by both immunoblotting with antiphosphotyrosine antibody and phosphorylation of the ERK substrate, MBP (Fig. 1 B, upper panel). Inhibition of MEK activity in these cells dramatically reduced their migration (Fig. 1 B, lower panel) yet had no effect on cell attachment or spreading (Fig. 1 C), even after 6 h of exposure to the MEK inhibitor. FG cells can be stimulated to migrate with cytokines (Klemke et al., 1994). Treatment of these cells with the MEK1 inhibitor blocked the EGF-induced migration of these cells and selectively prevented EGF-induced ERK1 and ERK2 phosphorylation without impacting other EGFdependent phosphorylation events, including the autophosphorylation of the EGF receptor (data not shown). Together, these findings implicate MAP kinase in the cell migration response induced by integrin and/or cytokine receptor ligation. Importantly, MAP kinase activity is apparently not required for cell adhesion or spreading.
MAP Kinase Activation Regulates Cell Migration
To further investigate the role of MAP kinase in cell motility, we used COS-7 cells since they efficiently express plasmids encoding members of the Ras/MAP kinase cascade (Howe et al., 1992; Cowley et al., 1994; Coso et al., 1995). Furthermore, COS-7 cells, like FG cells, migrate on collagen using integrin α2β1 (data not shown). In these experiments, we transiently expressed a mutationally active MEK1(MEK+) in COS-7 cells and examined their migratory properties on a collagen substrate. As shown in Fig. 2 A, upper panel, serum-starved MEK+ transfectant cells showed a four- to fivefold increase in migration relative to control cells transfected with the same expression vector without the MEK1 gene. However, the relative adhesive properties of these cells that were measured remained unchanged (data not shown). These cells also showed enhanced migration on fibronectin and vitronectin (data not shown), indicating that the mutationally active MEK1 could stimulate the general migratory properties of these cells. This induction of cell migration was associated with increased MAP kinase activity as measured by antiphosphotyrosine immunoblotting as well as increased phosphorylation of the substrate MBP (Fig. 2 A, lower panel). As expected, serum-starved cells exposed to EGF showed enhanced cell migration and increased MAP kinase activity (Fig. 2 A) (Howe et al., 1992). Importantly, cell migration induced by MEK+ expression was also blocked with the MEK inhibitor PD98059 (Fig. 2 B, upper panel), as was activation of ERK2 in these cells (Fig. 2 B, lower panel). These studies demonstrate that MAP kinase signaling events are critical for the induction of cell migration.
Figure 2.

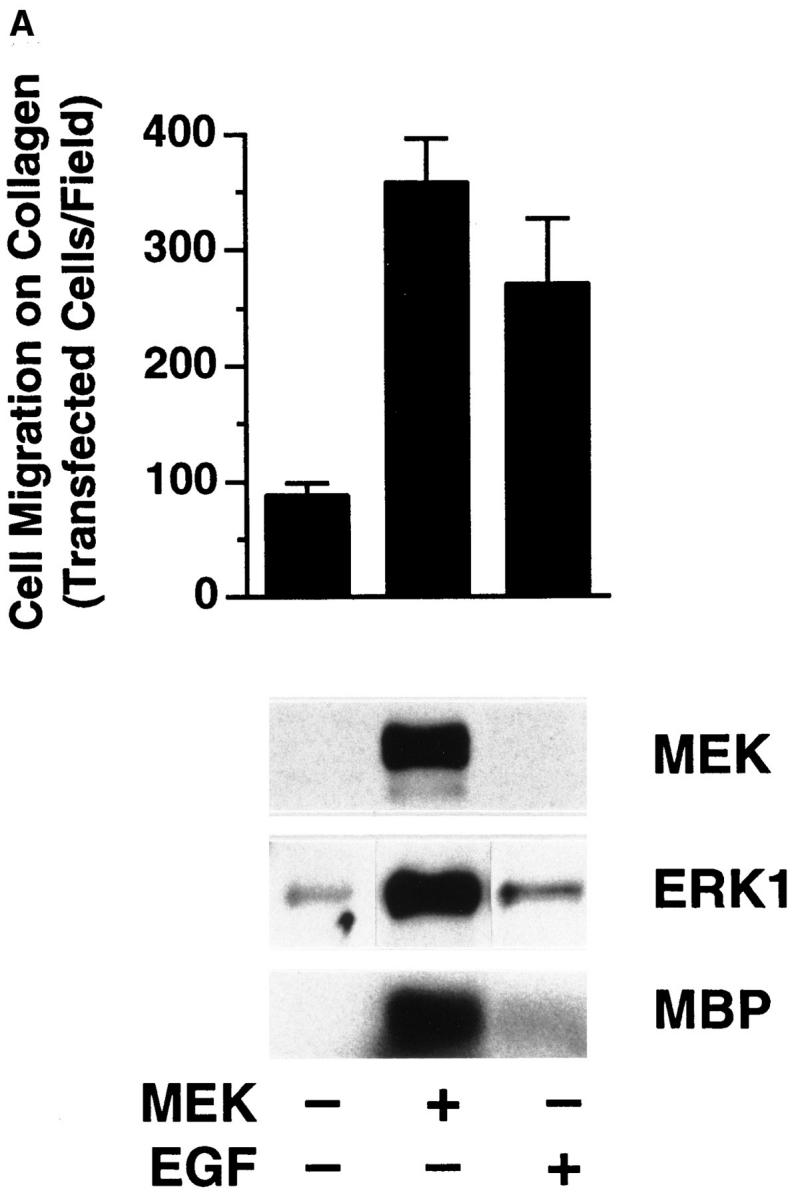

Stimulation of COS-7 cell migration after transfection with mutationally activated MEK1. (A, upper panel) COS-7 cells were serum starved for 18 h and allowed to migrate for 3 h on collagen-coated membranes after transient transfection with either the empty expression vector (pcDNA-3, control) or the expression vector containing the mutationally activated MEK1 (MEK+) as described in Materials and Methods. In both cases, cells were cotransfected with a β-galactosidase–containing vector (pSV–β-galactosidase) for in situ β-galactosidase staining with X-gal and a myc-tagged ERK1 reporter construct as described in Materials and Methods. This facilitated enumeration of only those migratory cells that had been positively transfected. In either case, transfection efficiency was routinely 30–40%. Alternatively, control transfectants were allowed to migrate in the presence of EGF (100 ng/ml) and then enumerated by counting cells on the underside of the migration chamber using an Olympus (BX60) inverted microscope. Cells per high-powered (40×) field were counted blindly by two observers. Each bar represents the mean ± SD of triplicate migration wells of an independent experiment. (Lower panel) Detergent lysates from cells treated as above were either directly immunoblotted for MEK protein expression using rabbit anti-MEK or subjected to immunoprecipitation using anti-ERK followed by immunoblotting with antiphosphotyrosine as described in Materials and Methods. In addition, these immunoprecipitates were analyzed for MAP kinase activity using an in vitro kinase assay and the substrate MBP as described in Materials and Methods. The result shown is a representative experiment from at least three independent experiments. (B, upper panel) COS-7 cells treated as described in A were allowed to migrate in the presence or absence of the MEK inhibitor (25 μM, PD98059) for 2–4 h and then stained and quantitated as described above. (Lower panel) Detergent lysates from these cells were examined for expression of MEK, ERK phosphorylation, and kinase activity using MBP as a substrate as described above.
MAP Kinase Activation Promotes MLC Phosphorylation
Potential mediators of cell migration downstream of MAP kinase were identified by examining the phosphoprotein profile from MEK+ COS-7 cells metabolically labeled with [32P]orthophosphate. We observed in MEK+ but not control transfectants the presence of labeled proteins migrating at ∼20 kD that comigrated with MLC (Fig. 3 A). We then subjected these same lysates to immunoprecipitation using an antibody directed to myosin IIB since this form of myosin is present in COS cells (Klemke, R., unpublished data). This antibody precipitated the 20-kD light chains from these cells, which showed increased phosphate incorporation in the MEK+ cells relative to vector transfected control cells Fig. 3 B. In addition, the phosphorylation of this protein was blocked by pretreatment of MEK+ cells with the MEK inhibitor PD98059 (Fig. 3).
Figure 3.


MAP kinase activity is associated with increased MLC phosphorylation. (A) COS-7 cells transfected with empty vector or MEK+ together with HA-tagged ERK2 reporter construct were labeled metabolically for 2 h with [32P]orthophosphate in the presence or absence of the MEK inhibitor (25 μM). Cells were then lysed in detergent and analyzed (20 μg total cell protein) by SDS-PAGE and autoradiography for changes in phosphorylation as described in Materials and Methods. Arrows depict expected migration of HAERK2 and MLC. (B) MLC or HA-tagged ERK2 were immunoprecipitated from the above lysates with anti–myosin IIB or anti-HA specific antibodies, respectively, and analyzed for changes in phosphorylation as described in Materials and Methods. The result shown is a representative experiment from at least three independent experiments.
Phosphorylation of MLC by the Ca2+/calmodulin-dependent enzyme, MLCK, is critical for cell migration. This reaction promotes increased myosin filament formation leading to its association with actin filaments and stimulates the actin-activated ATPase activity of myosin II. In addition, the fact that MLCK contains multiple MAP kinase consensus phosphorylation sites (P-x-S[T]-P) (Shoemaker et al., 1990) prompted us to examine the possibility that MLCK could be directly phosphorylated by MAP kinase. Therefore, MLCK was immunoprecipitated from MEK+ or control transfectants that had been metabolically labeled with [32P]orthophosphate. As shown in Fig. 4 A, anti-MLCK antibodies precipitated radiolabeled proteins of 160–200 kD, which represents isoforms of MLCK found in COS-7 cells and an additional (∼97 kD) breakdown product of MLCK as previously described (Gallagher et al., 1995). In contrast, cells transfected with a control vector express these proteins with minimal phosphate incorporation (Fig. 4 A). The phosphorylation of MLCK appeared to be caused by the activation of MAP kinase in these cells since MEK+ cells treated with the MEK inhibitor showed a loss of the MLCK phosphorylation. The enhanced phosphorylation of MLCK in MEK+ cells could not be explained by increased MLCK protein expression since immunoblotting experiments revealed that lysates from these and control cells contained the same level of MLCK (Fig. 4 B).
Figure 4.


MAP kinase activity is associated with increased phosphorylation of MLCK. (A) MLCK was immunoprecipitated from an aliquot of COS-7 cell lysates described in Fig. 3 and resolved by SDS-PAGE and autoradiography or (B) Western blotted with an MLCK-specific antibody. The phosphorylated proteins of ∼160 and 200 kD represent MLCK isoforms present in COS-7 cells, whereas the 97-kD protein may represent a break down product of MLCK. The result shown is a representative experiment from at least three independent experiments.
MAP Kinase Directly Phosphorylates MLCK Leading to MLC Phosphorylation In Vitro
To establish whether MLCK is a direct substrate for MAP kinase, we examined the ability of constitutively active MAP kinase (ERK1 or ERK2) to phosphorylate purified MLCK in an in vitro kinase assay. As shown in Fig. 5 A, purified active MAP kinase promoted a time-dependent increase in MLCK phosphorylation. This event could not be attributed to MLCK autophosphorylation since, in the absence of MAP kinase, we observed no significant phosphorylation of MLCK under the same reaction conditions (Fig. 5 A). MLCK phosphorylated with ERK2 for 40 min was then allowed to incubate with MLC in the presence or absence of calcium/calmodulin (Fig. 5 B). In this case, MLC phosphorylation was shown to depend on the presence of calcium/calmodulin. In addition, ERK2 in the absence of MLCK was incapable of phosphorylating MLC (Fig. 5 B).
Figure 5.


MAP kinase directly phosphorylates MLCK, which leads to increased MLC phosphorylation in vitro. (A) Purified MLCK was monitored for phosphorylation in the presence or absence of purified activated MAP kinase for various times in an in vitro kinase assay as described in Materials and Methods. Note that there is no detectable phosphorylation of MLCK in the absence of MAP kinase. The result shown is a representative experiment from at least three independent experiments. (B) Purified MLCK phosphorylated by MAP kinase for 40 min was tested for its ability to phosphorylate MLC in an in vitro kinase assay in the presence or absence of Ca/calmodulin. Note that MLC was not phosphorylated by purified activated MAP kinase in these experiments. The result shown is a representative experiment from at least three independent experiments. (C) MAP kinase was allowed to phosphorylate MLCK as described in vitro for 10 or 40 min as described in Materials and Methods. The MLCK treated in the presence or absence of ERK was then allowed to phosphorylate MLC for 4 min in the presence of calcium and calmodulin as described in Materials and Methods. Data are expressed as percent of control MLCK activity (i.e., MLCK incubated for 10 min in the absence of ERK). Each point represents the mean ± SE of at least three experiments. The results are significant P < 0.005.
We then examined the ability of MLCK incubated with ERK for either 10 or 40 min to phosphorylate MLC in an in vitro kinase assay. As shown in Fig. 5 C, MLCK exposed to ERK for 10 min showed little change in MLCK activity, whereas after 40 min ERK promoted increased MLCK activity. While the nonphosphorylated MLCK showed no enhanced enzymatic activity over time, MLCK phosphorylated by MAP kinase for 40 min showed an increase in MLCK activity by >50% (Fig. 5 C). This increased MLCK activity was associated with an eightfold stoichiometric increase in MLCK phosphorylation. In the absence of ERK, 0.05 mol of phosphate/mol was incorporated into MLCK, whereas 0.42 mol of phosphate/mol of MLCK was incorporated after 40 min of exposure to activated ERK.
To further examine the change in MLCK activity after phosphorylation by MAP kinase, MLCK was incubated in the presence or absence of mutationally active ERK, and the MLCK was then allowed to phosphorylate MLC in the presence of varying concentrations of calmodulin. As shown in Fig. 6, MLCK phosphorylated by ERK has increased sensitivity to calmodulin over a wide range of concentrations. In fact, half-maximal ERK-activated MLCK required two to threefold less calmodulin than MLCK not activated by ERK. Together, these findings reveal that phosphorylation of MLCK by MAP kinase leads to increased MLCK activity and a decreased requirement for calmodulin.
Figure 6.

MLCK, exposed to activated ERK or buffer (control) for 40 min, was allowed to phosphorylate MLC in the presence of varying concentrations of calmodulin as described in Materials and Methods. Data are expressed as the percent of maximal MLCK activity (i.e., MLCK phosphorylated by ERK in the presence of 1,000 nM calmodulin, which represents 1.5 μmol 32P/min/mg MLCK). Each point represents the mean ± SE of at least three experiments.
MAP Kinase–induced Cell Migration Requires MLCK Activity and MLC Phosphorylation
To establish a role for MLCK in the MAP kinase–induced cell migration response, COS cells transfected with MEK+ were allowed to migrate in the presence or absence of the MLCK inhibitor KT5926 (Nakanishi et al., 1990). As shown in Fig. 7 A, upper panel, KT5926 not only blocked the MEKinduced migration response but also prevented the phosphorylation of MLC in these cells in response to the expression of MEK+ (Fig. 7 A, lower panel). These findings suggest that MLCK plays a pivotal role in the MAP kinase–induced cell migration response.
Figure 7.




MLCK activity is required for MAP kinase-induced phosphorylation of MLC and cell migration. (A, upper panel) COS cells were transfected with empty vector or mutationally active MEK+ along with an HA-tagged ERK2 reporter construct. Cells were allowed to migrate on a collagen substrate for 3 h in the presence or absence of the MLCK inhibitor KT5926 (10 μM) as described in Materials and Methods. Cell migration was enumerated as described above. (Lower panel) Lysates prepared from these COS cells metabolically labeled with 32P were examined by SDS-PAGE and autoradiography. The position of HA-tagged ERK and MLC are denoted by arrows. (B, upper panel) COS cells were transfected with mutationally active MEK+ and/or MLCK(Mut) and allowed to migrate on a collagen substrate as described in Materials and Methods. Cell migration was quantified by counting the number of migrant cells per high powered microscopic field as described above. (Lower panel) Lysates prepared from 32P metabolically labeled COS cells transfected as above were examined for phosphorylation of HA-ERK and MLC. The result shown is a representative experiment from at least three independent experiments.
To further establish that MLCK activity is required for MAP kinase–induced cell migration, COS-7 cells expressing MEK+ were transfected with a mutationally inactivated form of rabbit smooth muscle MLCK (MLCK[Mut]) containing a single in frame deletion at K725. Residue K725 is predicted by alignment with other ser/thr kinases to have a key role in binding ATP and thereby acting as a dominant-negative enzyme (Hanks et al., 1988; Zoller et al., 1981; Knighton et al., 1991). This recombinant enzyme expressed in COS cells has no detectable catalytic activity, even at ATP concentrations that are 10-fold greater than the KmATP determined for smooth muscle MLCK (Gallagher et al., 1993). This enzyme was incapable of phosphorylating MLC in COS cells even though its calmodulin binding capacity was identical to the wild-type enzyme (Gallagher, P.J., unpublished results). Importantly, this mutant enzyme expressed in COS cells not only blocked cell migration induced by mutationally active MEK (Fig. 7 B, upper panel), but it also prevented the MEK-induced MLC phosphorylation in these cells (Fig. 7 B, lower panel). In addition, when EGF (100 ng/ml) was used to stimulate COS cell migration on collagen, expression of the mutant MLCK completely blocked the EGF-induced migration response (data not shown). These results are consistent with the notion that this mutant form of MLCK acts in a dominant-negative manner when introduced into MEK+ COS cells. It is noteworthy that the activity of MAP kinase in the MEK+ cells was not significantly impacted by either the mutationally inactive MLCK or the MLCK inhibitor KT5926 (Fig. 7), suggesting that MLCK and MLC phosphorylation are downstream of MAP kinase and necessary for the migration of these cells.
Model Depicting Role of MAP Kinase in Cell Migration
Based on the experiments described above and the work of others, it is now possible to develop a model that depicts the signaling pathway leading from integrin and/or growth factor ligation to myosin activation and cell migration (Fig. 8). This model identifies a signaling pathway involving the ability of MAP kinase to directly phosphorylate MLCK, which in turn is better able to phosphorylate MLC. Importantly, this pathway is independent of the ability of MAP kinase to translocate to the nucleus where it activates gene transcriptional events involved in cell proliferation and differentiation.
Figure 8.

Model depicting role of MAP kinase in cell migration. Proposed signaling pathway leading from integrin or growth factor receptor ligation to activation of cell migration. This model takes into account that MAP kinase can be activated by integrins and/or growth factors. As shown in this study, once activated, MAP kinase can phosphorylate MLCK, which in turn shows enhanced activity as measured by its ability to phosphorylate MLC. This model assumes that this phosphorylated MLC regulates the cell's actin-myosin motor function, thereby facilitating contraction leading to cell movement on the extracellular matrix.
Discussion
Cellular recognition of growth factors and adhesive proteins within the extracellular matrix influences whether a cell undergoes proliferation, differentiation, or migration. It is now clear that growth factor/cytokine receptors as well as adhesion receptors influence the activity of one or more members of the MAP kinase family of signaling molecules that have been implicated in a wide range of cell biological events. While MAP kinase signaling has been associated with the regulation of gene transcriptional events, we now present evidence that MAP kinase can promote cell migration on the extracellular matrix in a transcription-independent manner. We propose that MAP kinase can directly phosphorylate MLCK, thereby increasing its ability to phosphorylate MLC, which promotes cytoskeletal contraction necessary for cell movement (Sellers and Adelstein, 1987; Tan et al., 1992). While cell migration depends on cell adhesion as well as deadhesion of cell surface receptors and may be influenced by cell shape (Crowley and Horwitz, 1995), our findings that MAP kinase induction of MLCK activity has little or no influence on cell adhesion or spreading suggests these events may be differentially regulated. In fact, stress fiber formation and membrane ruffling has been associated with activation of small G proteins Rho, Cdc42, and rac (for reviews see Schwartz et al., 1995; Vojteck and Cooper, 1995; Parsons, 1996), and these, apparently, do not promote ERK1 or ERK2 activity but have been shown to activate the MAP kinases, p38 and JNK (Coso et al., 1995; Minden et al., 1995).
Growth factors/cytokines as well as integrins influence MAP kinase activity and regulate the motility of various cell types (Stoker and Gherardi, 1991; Leavesley et al., 1993; P. Chen et al., 1994; Klemke et al., 1994; Kundra et al., 1994; Yenush et al., 1994). Cellular transformation by H-Ras or c-Src is also associated with increased MAP kinase activity and enhanced cell proliferation and migration (Ochieng et al., 1991; Rodier et al., 1995). Therefore, the mitogenic and motogenic signals mediating these processes may be on a common pathway leading to MAP kinase activation, which may diverge based on the ability of MAP kinases to translocate to distinct intracellular compartments. In fact, activated MAP kinase has been shown to translocate to the nucleus (Gonzalez et al., 1993; Q. Chen et al., 1994) and/or the cytoskeleton (Gonzalez et al., 1993; Reszka et al., 1995).
In this report, we provide several lines of evidence that MAP kinase (ERK1 and ERK2) signaling can regulate cell migration by directly impacting the migratory machinery. First, blocking ERK expression with antisense oligonucleotides or ERK activity with a selective MEK inhibitor resulted in the loss of cell migration with no obvious effect on cell adhesion on a collagen substrate. Second, a mutationally active form of MEK1, when expressed in serumstarved COS-7 cells, significantly enhanced their migratory properties, which were blocked by MEK inhibition. EGF, a known inducer of cell motility, promoted collagendependent COS cell migration that was also blocked by inhibition of MEK (data not shown). Third, MLCK, a known activator of MLC phosphorylation, was found to be highly phosphorylated in cells expressing mutationally active MEK. In fact, MLCK contains multiple MAP kinase consensus phosphorylation sites (P-x-S[T]P) (Shoemaker et al., 1990; Clark-Lewis et al., 1991). Importantly, ERK1 and ERK2 were able to directly phosphorylate MLCK leading to enhanced MLC phosphorylation in vitro. This increased activity was associated with an increased sensitivity to calcium/calmodulin. In fact, MLCK activity showed a 50% increase in activity after exposure to ERK from 10–40 min in vitro. Finally, the migratory properties of MEK+ cells, containing active MAP kinase, could be blocked by selectively interfering with MLCK activity, suggesting that MLCK is downstream of MAP kinase and required for cell migration. In support of this contention, expression of a mutant form of MLCK that does not bind ATP functioned as a dominant-negative mutant, thereby inhibiting the phosphorylation of MLC and induction of cell migration by MAP kinase. Together, these findings support the notion that MAP kinase activates the cells' motility machinery by enhancing the activity of MLCK leading to increased MLC function. Therefore, we define a signaling pathway initiated from integrin and/or growth factor receptor ligation that leads to increased myosin phosphorylation (see proposed pathway depicted in Fig. 8).
While our results indicate a role for MAP kinase in cell movement, other mechanisms may regulate MLC phosphorylation and cell motility. In fact, light chain phosphorylation by Ca2+/phospholipid-dependent protein kinase II, protein kinase C, and cAMP-dependent protein kinase are important for myosin function in vivo (for review see Tan et al., 1992). Recent findings also indicate that rhoA induces MLC phosphorylation and cell contraction leading to FAK activation and focal contact formation (Chranowska-Wodnicka and Burridge, 1996). However, it is not yet clear whether any of these signaling events can directly or indirectly influence cell movement. In addition, it is possible that other MAP kinases, such as JNK or p38, once activated, impact this or other signaling pathways leading to cell migration. In any case, it is clear that in the cells examined, ERK1 and/or ERK2 are critical for collagen-dependent cell migration. Importantly, this signaling pathway appears to regulate cell migration on at least two other matrix proteins, vitronectin and fibronectin, even though it had no obvious impact on cell adhesion to each of these proteins. Thus, it is apparent that cell adhesion and migration can be differentially regulated. Perhaps this is not surprising since many adherent cell types do not migrate without prior cytokine or growth factor stimulation (Stoker and Gherardi, 1991; Leavesley et al., 1993; P. Chen et al., 1994; Klemke et al., 1994; Kundra et al., 1994; Yenush et al., 1994). In fact, recent observations indicate that inhibition of integrin-induced Ras activation of ERK2 did not affect focal contact formation, cell adhesion, or actin reorganization leading to cell spreading on fibronectin (Clark and Hynes, 1996).
Previous studies have shown that MAP kinase activity is critical for gene transcriptional events leading to cell proliferation and differentiation. Evidence presented in this report demonstrates that MLCK, a key regulator of cell motility and contraction, is a substrate for MAP kinase. This finding may explain how growth factors, cytokines, or integrins that activate MAP kinases can thereby influence cell movement on the extracellular matrix during tissue remodeling as well as tumor cell invasion.
Acknowledgments
The authors would like to thank Dr. Robert Adelstein for providing anti– myosin IIB specific antibodies and for helpful advice concerning this project. We are also grateful to Drs. Audrey Minden, Michael Karin, and Richard Ulevitch for providing the ERK reporter constructs and to Drs. D. Schlaepfer and T. Hunter for the activated MAP kinase.
Footnotes
1. Abbreviations used in this paper: HA, influenza hemagglutinin epitope; MAP, mitogen-activated protein; MBP, myelin basic protein; MLC, myosin light chain(s); MLCK, myosin light chain kinase.
D.A. Cheresh was supported by grants HL-54444, CA-50286, and CA45726 from the National Institutes of Health (NIH) and a Faculty Research Award from the American Cancer Society. R.L. Klemke was supported by National Institutes of Health fellowship grant 1F32CA67442-07. P. de Lanerolle was supported by National Science Foundation grant (MCB9631833) and the Hannet Brooks fund of the University of Illinois, Chicago, IL. S. Cai was supported by an NIH training grant HL076922. This is manuscript #10200-IMM from The Scripps Research Institute.
Address all correspondence to D.A. Cheresh, The Scripps Research Institute, (IMM-24), 10555 N. Torrey Pines Rd., La Jolla, CA 92037. Tel.: (619) 784-8281. Fax: (619) 784-8926.
References
- Adelstein RS. Regulation of contractile proteins by phosphorylation. J Clin Invest. 1983;72:1863–1866. doi: 10.1172/JCI111148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton TG, Cobb MH. Identification of multiple extracellular signal-regulated kinases (ERKs) with antipeptide antibodies. Cell Regul. 1991;2:357–371. doi: 10.1091/mbc.2.5.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PC, Clark RAF, Cheresh DA. Requirement of Integrin αVβ3 for angiogenesis. Science (Wash DC) 1994;264:569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- Brooks, P.C., S. Stromblad, L. Sanders, T. von Schalscha, R. Aimes, W. StetlerStevenson, J. Quigley, and D.A. Cheresh. 1996. Localization of matrix metalloproteinase MMP-2 to the surface of the invasive cells by interaction with integrin αVβ3. Cell. 683–693. [DOI] [PubMed]
- Chen P, Xie H, Sekar MC, Gupta K, Wells A. Epidermal growth factor receptor-mediated cell motility: phospholipase C activity is required, but mitogen-activated protein kinase activity is not sufficient for induced cell movement. J Cell Biol. 1994;127:847–857. doi: 10.1083/jcb.127.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Kinch MS, Lin TH, Burridge K, Juliano RL. Integrinmediated cell adhesion activates mitogen-activated protein kinases. J Biol Chem. 1994;6:26602–26605. [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark EA, Hynes RO. Ras activation is necessary for integrinmediated activation of extracellular signal-regulated kinase 2 and cytosolic phospholipase A2but not for cytoskeletal organization. J Biol Chem. 1996;271:14814–14818. doi: 10.1074/jbc.271.25.14814. [DOI] [PubMed] [Google Scholar]
- Clark RF, Tonnesen MG, Gailit J, Cheresh DA. Transient functional expression of αvβ3 on vascular cells during repair. Am J Pathol. 1996;148:1407–1421. [PMC free article] [PubMed] [Google Scholar]
- Clark-Lewis I, Sanghera JS, Pelech SL. Definition of a consensus sequence for peptide substrate recognition by p44 mpK, the meiosis-activated myelin basic protein kinase. J Biol Chem. 1991;266:15180–15184. [PubMed] [Google Scholar]
- Conrad PA, Giuliano KA, Fisher G, Collins K, Matsudaira PT, Taylor DL. Relative distribution of actin, myosin I, and myosin II during the wound healing response of fibroblast. J Cell Biol. 1993;120:1381–1391. doi: 10.1083/jcb.120.6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coso OA, Chiariello M, Yu J, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- Crowley E, Horwitz AF. Tyrosine phosphorylation and cytoskeletal tension regulate the release of fibroblast adhesions. J Cell Biol. 1995;131:525–537. doi: 10.1083/jcb.131.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lanerolle P, Nishikawa M. Regulation of embryonic smooth muscle myosin by protein kinase C. J Biol Chem. 1988;263:9071–9074. [PubMed] [Google Scholar]
- de Lanerolle P, Paul R. Myosin phosphorylation/dephosphorylation and regulation of airway smooth muscle contractility. Am J Physiol. 1991;261:L1–L14. doi: 10.1152/ajplung.1991.261.2.L1. [DOI] [PubMed] [Google Scholar]
- de Lanerolle P, Adelstein RS, Feramisco JR, Burridge K. Characterization of antibodies to smooth muscle myosin kinase and their use in localizing myosin kinase in nonmuscle cells. Proc Natl Acad Sci USA. 1981;78:4738–4742. doi: 10.1073/pnas.78.8.4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lanerolle P, Gorgas G, Li X, Schluns K. Myosin light chain phosphorylation does not increase during yeast phagocytosis by macrophages. J Biol Chem. 1993;268:16883–16886. [PubMed] [Google Scholar]
- Drake CJ, Cheresh DA, Little CD. An antagonist of integrin αvβ3 prevents maturation of blood vessels during embryonic neovascularization. J Cell Sci. 1995;108:2655–2661. doi: 10.1242/jcs.108.7.2655. [DOI] [PubMed] [Google Scholar]
- Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenfeld DP, Choquet D, Sheetz MP. Ligand binding regulates the directed movement of B1 integrins on fibroblasts. Nature (Lond) 1996;383:438–440. doi: 10.1038/383438a0. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Brooks PC, Deming SL, Damsky C, Cheresh DA. Requirement of the NPXY motif in the integrin β3 subunit cytoplasmic tail for melanoma cell migration in vitro and in vivo. J Cell Biol. 1995;130:441–450. doi: 10.1083/jcb.130.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher PJ, Herring BP, Griffin SA, Stull JT. Molecular characterization of a mammalian smooth muscle myosin light chain kinase. J Biol Chem. 1991;266:23936–23944. [PMC free article] [PubMed] [Google Scholar]
- Gallagher PJ, Herring BP, Tafny A, Sowadski J, Stull JT. A molecular mechanism for autoinhibition of myosin light chain kinases. J Biol Chem. 1993;268:26578–26582. [PMC free article] [PubMed] [Google Scholar]
- Gallagher PJ, Garcia JG, Herring BP. Expression of a novel myosin light chain kinase in embryonic tissues and cultured cells. J Biol Chem. 1995;270:29090–29095. doi: 10.1074/jbc.270.49.29090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliano KA, Taylor DL. Formation, transport, contraction, and disassembly of stress fibers in fibroblast. Cell Motil Cytoskel. 1990;16:14–21. doi: 10.1002/cm.970160104. [DOI] [PubMed] [Google Scholar]
- Giuliano KA, Kolega J, DeBiasro RL, Taylor DL. Myosin II phosphorylation and the dynamics of stress fibers in serum-deprived and stimulated fibroblasts. Mol Biol Cell. 1992;3:1037–1048. doi: 10.1091/mbc.3.9.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez FA, Seth A, Raden DL, Bowman DS, Fay FS, Davis RJ. Serum-induced translocation of mitogen-activated protein kinase to the cell surface ruffling membrane and the nucleus. J Cell Biol. 1993;122:1089–1101. doi: 10.1083/jcb.122.5.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough AH, Taylor DL. Fluorescence anisotropy imaging microscopy maps calmodulin binding during cellular contraction and locomotion. J Cell Biol. 1993;121:1095–1107. doi: 10.1083/jcb.121.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Quin AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science (Wash DC) 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- Herring BP, Gallagher PJ, Stull JT. Substrate specificity of myosin light chain kinases. J Biol Chem. 1992;267:25945–25950. [PubMed] [Google Scholar]
- Hill CS, Treisman R. Transcriptional regulation by extracellular signals: mechanisms and specificity. Cell. 1995;80:199–211. doi: 10.1016/0092-8674(95)90403-4. [DOI] [PubMed] [Google Scholar]
- Howe LR, Leevers SJ, Gomez N, Makielny S, Cohen P, Marshall CJ. Activation of the MAP kinase pathway by protein kinase raf. Cell. 1992;71:335–342. doi: 10.1016/0092-8674(92)90361-f. [DOI] [PubMed] [Google Scholar]
- Huttenlocher A, Ginsburg MH, Horwitz AF. Modulation of cell migration by integrin-mediated cytoskeletal linkages and ligand-binding affinity. J Cell Biol. 1996;134:1551–1562. doi: 10.1083/jcb.134.6.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay PY, Pham PA, Wong SA, Elson EL. A mechanical function of myosin II in cell motility. J Cell Sci. 1995;108:387–393. doi: 10.1242/jcs.108.1.387. [DOI] [PubMed] [Google Scholar]
- Klemke RL, Yebra M, Bayna EM, Cheresh DA. Receptor tyrosine kinase signaling required for integrin αvβ5-directed cell motility but not adhesion on vitronectin. J Cell Biol. 1994;127:859–866. doi: 10.1083/jcb.127.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knighton DR, Zeng JH, Ten LF, Eyck, Xuong NH, Taylor SS, Sowadski JM. Structure of peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science (Wash DC) 1991;253:414–420. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- Kolega J, Taylor DL. Gradients in the concentration and assembly of myosin II in living fibroblasts during locomotion and fiber transport. Mol Biol Cell. 1993;4:819–836. doi: 10.1091/mbc.4.8.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundra V, Escobedo JA, Kazlauskas A, Kim H, Rhee S, Williams LT, Zetter BR. Regulation of chemotaxis by the platelet-derived growth factor receptor-β. Nature (Lond) 1994;367:474–476. doi: 10.1038/367474a0. [DOI] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Leavesley D, Ferguson G, Wayner E, Cheresh DA. Requirement of the integrin β3 subunit for carcinoma cell spreading or migration on vitronectin and fibrinogen. J Cell Biol. 1992;117:1101–1107. doi: 10.1083/jcb.117.5.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavesley DI, Schwartz MA, Rosenfeld M, Cheresh DA. Integrin β1- and β3-mediated endothelial cell migration is triggered through distinct signaling mechanisms. J Cell Biol. 1993;121:163–170. doi: 10.1083/jcb.121.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour SJ, Matten WT, Hermann AS, Candia JM, Roug CS, Fukasawa K, Vande GF, Wonde, Ahn NG. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science (Wash DC) 1994;265:966–969. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- McKenna NM, Wang Y, Konkel ME. Formation and movement of myosin-containing structures in living fibroblasts. J Cell Biol. 1989;109:1163–1172. doi: 10.1083/jcb.109.3.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minden A, Lin A, Claret F, Abo A, Karin M. Selective activation of the JNK signaling cascade and C-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, Yamada K, Iwahashi K, Kuroda K, Kase H. KT5926, a potent and selective inhibitor of myosin light chain kinase. Mol Pharmacol. 1990;37:482–488. [PubMed] [Google Scholar]
- Ochieng J, Basolo F, Albini A, Melchiori A, Watanebe H, Elliott J, Raz A, Parodi S, Russo J. Increased, chemotactic and locomotive abilities of c-Ha-ras-transformed human breast epithelial cells. Invasion Metastasis. 1991;11:38–47. [PubMed] [Google Scholar]
- Pang L, Sawada T, Decker SJ, Saltiel AR. Inhibition of Map kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- Parsons TH. Integrin-mediated signaling: regulation by protein tyrosine kinases and small GTP-binding proteins. Curr Opin Cell Biol. 1996;8:146–152. doi: 10.1016/s0955-0674(96)80059-7. [DOI] [PubMed] [Google Scholar]
- Reszka AA, Seger R, Diltz CD, Krebs EG, Fischer EH. Association of mitogen-activated protein kinase with the microtubule cytoskeleton. Proc Natl Acad Sci USA. 1995;92:8881–8885. doi: 10.1073/pnas.92.19.8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier J, Valles A, Denoyelle M, Thiery JP, Boyer B. pp60c-srcis a positive regulator of growth factor-induced cell scattering in a rat bladder carcinoma cell line. J Cell Biol. 1995;131:761–773. doi: 10.1083/jcb.131.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale EM, Atkinson GP, Sale GJ. Requirement of MAP kinase for differentiation of fibroblasts to adipocytes for insulin activation of p90 S6 kinase and for insulin or serum stimulation of DNA synthesis. EMBO (Eur Mol Biol Organ) J. 1995;14:674–684. doi: 10.1002/j.1460-2075.1995.tb07046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrinmediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature (Lond) 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Schwartz MA, Schaller MD, Ginsberg MH. Integrins: emerging paradigms of signal transduction. Annu Rev Cell Dev Biol. 1995;11:549–599. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- Sellers JR, Adelstein RS. Regulation of contractile activity. Enzymes. 1987;18:381–418. [Google Scholar]
- Shoemaker MO, Lau W, Shattuck RL, Kwiatkowski AP, Matrisian PE, Guerra-Santos L, Wilson E, Lukas TJ, Van Eldik LJ, Watterson DM. Use of DNA sequence and mutant analyses and antisense oligodeoxynucleotides to examine the molecular basis of nonmuscle myosin light chain kinase autoinhibition, calmodulin recognition, and activity. J Cell Biol. 1990;111:1107–1125. doi: 10.1083/jcb.111.3.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JL, Silveira LA, Spudich JA. Myosin light chain kinase (MLCK) gene disruption in Dictyostelium: a role for MLCK-A in cytokinesis and evidence for multiple MLCKs. Proc Natl Acad Sci USA. 1996;93:12321–12326. doi: 10.1073/pnas.93.22.12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoker M, Gherardi E. Regulation of cell movement: the motogenic cytokines. Biochim Biophys Acta. 1991;1072:81–102. doi: 10.1016/0304-419x(91)90008-9. [DOI] [PubMed] [Google Scholar]
- Tan JL, Ravid S, Spudich JA. Control of nonmuscle myosins by phosphorylation. Annu Rev Biochem. 1992;61:721–759. doi: 10.1146/annurev.bi.61.070192.003445. [DOI] [PubMed] [Google Scholar]
- Vojtek AB, Cooper JA. Rho family members: activators of MAP kinase cascades. Cell. 1995;82:527–529. doi: 10.1016/0092-8674(95)90023-3. [DOI] [PubMed] [Google Scholar]
- Wilson AK, Gorgas G, Claypool WD, de Lanerolle P. An increase or a decrease in myosin II phosphorylation inhibits macrophage motility. J Cell Biol. 1991;114:277–283. doi: 10.1083/jcb.114.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JT, Rayburn H, Hynes RO. Embryonic mesodermal defects in α5 integrin-deficient mice. Development (Camb) 1993;119:1093–1105. doi: 10.1242/dev.119.4.1093. [DOI] [PubMed] [Google Scholar]
- Yatohgo T, Izumi M, Kashiwagi H, Hiyashi M. Novel purification of vitronectin from human plasma by heparin affinity chromatography. Cell Struct Funct. 1988;13:281–292. doi: 10.1247/csf.13.281. [DOI] [PubMed] [Google Scholar]
- Yenush L, Kundra V, White MF, Zetter BR. Functional domains of the insulin receptor responsible for chemotactic signaling. J Biol Chem. 1994;269:100–104. [PubMed] [Google Scholar]
- Zheng C, Guan K. Activation of MEK family kinases requires phosphorylation of two conserved Ser/Thr residues. EMBO (Eur Mol Biol Organ) J. 1994;13:1123–1131. doi: 10.1002/j.1460-2075.1994.tb06361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Assoian RK. Integrin-dependent activation of MAP kinase: a link to shape-dependent cell proliferation. Mol Biol Cell. 1995;6:273–282. doi: 10.1091/mbc.6.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoller MJ, Nelson NC, Taylor SS. Affinity labeling of cAMPdependent protein kinase with p-fluorosulonylbenzoyl adenosine. J Biol Chem. 1981;256:10387. [PubMed] [Google Scholar]