

Abstract
The assembly in living cells of heterotrimeric guanine nucleotide binding proteins from their constituent α, β, and γ subunits is a complex process, compounded by the multiplicity of the genes that encode them, and the diversity of receptors and effectors with which they interact. Monoclonal anti-β antibodies (ARC5 and ARC9), raised against immunoaffinity purified βγ complexes, recognize β subunits when not associated with γ and can thus be used to monitor assembly of βγ complexes. Complex formation starts immediately after synthesis and is complete within 30 min. Assembly occurs predominantly in the cytosol, and association of βγ complexes with the plasma membrane fraction starts between 15–30 min of chase. Three pools of β subunits can be distinguished based on their association with γ subunits, their localization, and their detergent solubility. Association of β and α subunits with detergent-insoluble domains occurs within 1 min of chase, and increases to reach a plateau of near complete detergent resistance within 30 min of chase. Brefeldin A treatment does not interfere with delivery of βγ subunits to detergent-insoluble domains, suggesting that assembly of G protein subunits with their receptors occurs distally from the BFA-imposed block of intracellular membrane trafficking and may occur directly at the plasma membrane.
Heterotrimeric guanine nucleotide binding proteins (G proteins)1 serve two major functions in eukaryotic cells. Foremost is their role in signal transduction, which comprises the interaction of a ligandbound heptahelical receptor with the intracellular machinery, such that signals delivered extracellularly result in the appropriate cellular response, which is dependent on the downstream effector system employed (19, 56). Heterotrimeric G proteins may also function as regulatory elements in membrane traffic (5). Separate from these two major functions is their possible role in the control of other enzyme systems, such as the Bruton-tyrosine kinase (BTK) (66) or the T cell receptor-specific kinase pathways (Rehm, A., and H.L. Ploegh, unpublished observation; 32), which are perhaps not linked directly to heptahelical receptors. The classical cycle starts the heterotrimeric G proteins in its inactive state as a complex of α, β, and a γ subunit in which the guanine nucleotide binding site of the α subunit is occupied by GDP (8). Upon receptor activation by an extracellular signal and interaction of the triggered receptors with G proteins, the α subunit releases GDP in exchange for GTP. The trimer then dissociates into α and βγ subunits that can now interact independently with effectors (46). The α and βγ subunits are not obligatorily derived from this activity cycle, but may also exist as preformed solitary pools (3, 4, 15).
A major challenge for maintaining the specificity with which G proteins convey signals from heptahelical receptors arises from the complexity of the component parts of this signaling system (24, 53). An increasing number of cloned heptahelical receptors must be integrated into a diverse system of effectors and G protein subunits, where 20 α, 5 β, and 8 γ subunits have been described so far (42, 46). Even if only a minor fraction of the full combinatorial complexity of receptors, G protein subunits and effectors were indeed available, how are the specificity of these interactions and specificity of signaling maintained?
Heptahelical receptors are integral membrane proteins that are synthesized on membrane-bound ribosomes and inserted into the ER. Many of these receptors carry N-linked glycans. They reach their final destination from the ER via the Golgi-complex, and their proper folding and glycosylation depend on this pathway. In contrast to the biosynthetic route followed by the receptors, the subunits of heterotrimeric G proteins are synthesized on ribosomes in the cytosol. None of the G protein subunits contain obvious sequences that would target the newly synthesized polypeptides to the ER. Modifications such as myristoylation or palmitoylation of the α subunits and the prenylation of the γ subunits, which provides the βγ complex with a membrane anchor, occur post- or cotranslationally, presumably in a cytosolic compartment (10, 39, 41). After G protein activation, myristoylation of certain G protein α subunits may be required for continued association of α with the plasma membrane (26, 27, 44). Acylation of the α subunit may further contribute to its affinity for βγ (34), and the βγ subunits themselves promote the association of α subunits with phospholipid vesicles in vitro (63). G protein synthesis clearly does not involve lumenal modifications, and the orientation of G proteins in the final complex with a receptor is cytosolic. Where and when do G protein subunits assemble with their signaling partners? The oligomerization of G proteins, receptors, and effectors comprises the assembly of proteins with a lumenal component, as well as the assembly of the multimeric G protein on the cytosolic face of membranes with the former. To our knowledge, these issues have not been addressed in living cells.
What has been established is the occurrence of heterotrimeric G proteins in specialized membrane domains that have been referred to as caveolae (38). These specializations are found in the TGN and at the plasma membrane, are enriched in glycosyl-phosphatidylinositol (GPI)–anchored membrane proteins, and are relatively resistant to extraction by non-ionic detergents (1, 50). In this study we provide insight into the kinetics of integration of βγ into detergent-resistant membranes.
Much of the effort to characterize the specificity of G protein–receptor interactions has been devoted towards identifying functional combinations of subunits in transfected cells or in cells manipulated by antisense oligonucleotide injections (29, 51). However, differential targeting to membrane subdomains may also contribute to the specificity of receptor, G protein, and effector interactions (47). As a first step towards understanding the assembly of G protein–coupled signal transduction chains, we here study the formation of the βγ complex. To trace biosynthetic intermediates in the assembly of G proteins in living cells, we made use of anti–α monoclonal antibodies and monoclonal antibodies against β subunits capable of distinguishing between free and assembled β subunits. Through combination of immunoprecipitation, subcellular fractionation, and pulse-chase analysis we define kinetically distinct pools of βγ and α subunits.
Materials and Methods
Antibodies
The β subunit specific monoclonal antibodies, ARC5 and ARC9, as well as the polyclonal rabbit anti-β serum, were raised as described (52). The anti-αo and -αi specific monoclonal antibodies 3C2 and 3E7 have been described (67, 69). The polyclonal rabbit anti-β1 antibody U-49 was prepared as previously described (43) (kindly provided by Dr. S. Mumby, Dallas, TX). The Na+/K+-ATPase, α subunit specific antibody served as plasma membrane marker (gift of Dr. W.J. Nelson, Stanford University, Stanford, CA). W6/32.HL (anti-MHC class I) has been described extensively (2).
Gel Electrophoresis
SDS-gel electrophoresis was performed as described (31), either on 12.5% or 15% acrylamide gels. Radioactively labeled samples were visualized by fluorography using DMSO-PPO and exposure to Kodak XAR-5 films.
Immunoblotting
Polypeptides were resolved on gel as indicated in the figure legends and blotted to nitrocellulose (0.45 μm pore size, Bio-Rad Labs, Hercules, CA). The blots were incubated with the first antibody, followed by HRF–coupled goat anti–mouse or –goat anti–rabbit immunoglobulin (Southern Biotechnology, Birmingham, AL) antibody. Bound antibody was visualized by chemoluminescence (ECL detection kit, Kirkegaard and Perry, Gaithersburg, MD) and exposure to Kodak XAR-5 films.
Cell Culture, Metabolic Labeling, and Immunoprecipitations
The human neuroblastoma cell line IMR-32 was routinely grown in DME medium, supplemented with 10% FCS, l-glutamine (2 mM), penicillin (1: 1,000 dilution U/ml), and streptomycin (100 μg/ml).
Adherently growing cells were grown to subconfluency, incubated for 45 min in methionine- and cysteine-free DME medium, and then pulselabeled with [35S]methionine/cysteine (80/20) (Express protein labeling mix, Du Pont-New England Nuclear, Boston, MA) as indicated in the figure legends. For overnight labeling, 50–75 μCi were added to a 10-cm-diam Petri dish of adherently growing cells or to 107 cells in suspension. In pulse-chase experiments, incorporation of label was stopped by addition of complete medium supplemented with nonradioactive methionine/ cysteine (1 mM). Brefeldin A (Sigma Chem. Co., St. Louis, MO) was added at a concentration of 10 μg/ml 2 min before pulse-labeling, and it was present in the chase medium as indicated in the figure legends. Interferon-γ (IFN-γ) was added to cell cultures at 50 U/ml for 48 h, where indicated.
For certain experiments, cells were maintained in the absence of FCS and instead cultured in DME medium supplemented with 0.4% (wt/vol) bovine serum albumin (Boehringer Mannheim, Indianapolis, IN) for 16– 18 h. Control cells were maintained in DME supplemented with 20% FCS.
Cells were lysed in 1.5 ml NP-40/Lubrol (50 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 0.5% NP-40, 0.1% Lubrol, 1 mM EDTA, 1 mM PMSF) lysis buffer. After 45 min at 4°C lysates were depleted of nuclei and cell debris by centrifugation for 10 min at 14,000 rpm in an Eppendorf centrifuge, and then precleared twice with normal rabbit serum and formalin-fixed Staphylococcus aureus bacteria (Staph. A). Immunoprecipitations for recovery of β subunits were done with 4–5-μl ascites of ARC5 or ARC9 mAb, α subunits were specifically precipitated with either mAbs 3C2 or 3E7, or a mixture of both mAbs (50 μl culture supernatant each). SDS was added as indicated in the figure legends. Immune complexes were precipitated by adsorption to Staph. A, followed by 4–5 washes in NET buffer (50 mM Tris-HCl, pH 7.5, 5 mM EDTA, 150 mM NaCl, 0.5% NP-40).
Endoglycosidase H (Endo H) (New England Biolabs, Beverly, MA) digestion was done as described (68).
Isolation of Detergent-resistant Membrane Domains
In some experiments cells were solubilized in NP-40/Lubrol, and the lysates were centrifuged at 14,000 rpm for 10 min in an Eppendorf centrifuge at 4°C for 10 min. The pellets, corresponding to the detergent-resistant membrane fraction, were solubilized in 200 μl PBS/1% SDS, heated for 3 min at 95°C, and passed through a 21-gauge needle several times (59). This procedure was repeated once, and for immunoprecipitation the solubilized pellets were brought to the same SDS concentration as the NP40/Lubrol supernatants by addition of non-ionic detergent buffer.
To study quantitative changes of detergent-resistant G protein subunit pools, cells were essentially extracted as described (37). Briefly, cells were lysed for 30 min on ice with 1.5 ml 25 mM Pipes, pH 6.5, 0.15 M NaCl, 1% Triton X-100 (Surfact-Amps X-100, Pierce, Rockford, IL). Direct lysis or re-extraction in the above buffers was done in the presence of 60 mM octyl-glucoside (Calbiochem, La Jolla, CA) (see figure legends).
To purify detergent-resistant membrane domains a modification of a method described previously was employed (38). Pulse-labeled and -chased IMR-32 cells were homogenized in 25 mM Pipes, pH 6.5, 0.15 M NaCl, 1% Triton X-100 containing 1 mM PMSF with 20 strokes of a tight-fitting Dounce homogenizer. The amount of radioactivity was normalized for individual chase points after TCA precipitation of extract aliquots and determination of counts in a β counter. The homogenate was adjusted to 40% (wt/vol) sucrose and placed at the bottom of an SW-41 centrifuge tube, and then overlaid with successive layers of 35, 25, 15, and 5% sucrose. After centrifugation at 39,000 rpm for 16 h in a SW41 rotor (Beckman Instruments, Palo Alto, CA), 1-ml fractions were collected from the top and immediately lysed in 1 ml 2× NP-40/Lubrol lysis buffer supplemented with 0.4% (wt/vol) SDS and 1 mM PMSF.
Detection of Endogenous GPI-anchored Proteins in Detergent-resistant Membrane Domains
Detergent-resistant membranes were first raised using the Triton X-100 extraction procedure described before. The resulting detergent-resistant membrane pellet was then re-extracted for 20 min at 37°C in 1 ml TNE/ Triton X-114 (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% precondensed Triton X-114 [Sigma], 1 mM PMSF, 1 mM leupeptin, 2 μg/ml aprotinin). Membrane extracts were centrifuged at 14,000 rpm for 10 min at 4°C, followed by temperature-induced phase separation of the supernatant exactly as described (6, 35). Detergent phases were re-extracted three times and then incubated in the absence and presence of phosphatidylinositol-specific phospholipase C (5 U/ml) (PI-PLC; ICN Pharmaceuticals, Costa Mesa, CA) for 1 h at 37°C. After three additional phase separations resulting aqueous phases were quantitatively precipitated by using acetone. Precipitates were solubilized in Laemmli sample buffer and analyzed on SDS-PAGE.
Subcellular Fractionation
Homogenization.
Cells (suspended in 1 or 2 ml ice-cold homogenization buffer, 10 mM Tris-HCl, pH 7.4, 250 mM sucrose, 1 mM EDTA, 1 mM PMSF) were homogenized on ice with 35 strokes of a Dounce homogenizer with a tight-fitting pestle.
Differential Centrifugation.
A postnuclear supernatant was centrifuged at 100,000 g for 1 h at 4°C to separate cytosol from particulate fractions. A different protocol was used to further separate plasma membranes from microsomal membranes (48). Briefly, each homogenate was centrifuged at 12,000 g for 15 min, followed by resuspension of the pellet (plasma membranes, mitochondria), and centrifuged at 28,000 g for 15 min. This additional centrifugation step allowed further removal of cytosolic components and yielded a pellet designated as plasma membranes in this study. The 12,000 g supernatant of the first centrifugation was centrifuged at 100,000 g for 1 h. The resulting pellet was considered as the microsomal fraction. Previous studies have shown that the 28,000 g pellet was enriched for plasma membrane markers, whereas the 100,000 g pellet was enriched for microsomal membrane enzyme activity (14, 48, 49). All fractions were then lysed in NP-40/Lubrol lysis buffer, followed by centrifugation for 10 min at 14,000 rpm in an Eppendorf centrifuge. The resulting supernatants were precleared and then immunoprecipitated with ARC9 in the presence of 0.2% SDS.
Discontinuous Sucrose Gradient.
Homogenates were separated on a sucrose step gradient with minor modifications of a protocol described (7). A postnuclear supernatant was adjusted to 40% (wt/vol) sucrose in a buffer containing 10 mM Tris-HCl, pH 7.4, and layered on a 50% (wt/vol) sucrose solution. Subsequently, a discontinuous sucrose gradient consisting of 0, 15, 25, and 35% (wt/vol) sucrose was poured on top of the 40% sucrose fraction. The flotation gradient was centrifuged in a SW 41 rotor at 4°C either at 85,000 g or at 170,000 g for 16 h. 0.5-ml fractions were taken from the top and an equal volume of 2× lysis buffer was added. Fractions were either analyzed directly or stored frozen at −70°C. The distribution of radioactivity was determined by liquid spectrometry after TCA precipitation.
Tryptic Cleavage Assay
IMR-32 cells were labeled and lysed in 1 ml 50 mM Tris-HCl, pH 7.5, 2 mM MgCl2, 0.1 mM EDTA, 0.5 mM DTT, 0.3% (vol/vol) Lubrol for 30 min at 4°C. Lysates were depleted of nuclei and debris, and the soluble supernatant was diluted to 0.075% (vol/vol) Lubrol using the same Tris-buffer without detergent. Lysates were treated with l-1-tosylamido-2-phenylchloromethyl ketone–treated trypsin (Sigma Chem. Co.) at 30°C for 30 min at the concentrations indicated in the figure legends. Reactions were stopped by the addition of trypsin inhibitor (Sigma) usually in a 10-fold excess (wt/wt) over trypsin. Lysates were further supplemented with 1% (vol/vol) Triton X-100 and 1 mM PMSF before immunoprecipitation.
This assay was also applied to the detection of βγ complexes in detergent-resistant membrane domains. Briefly, IMR-32 cells were first lysed in TX-100 containing lysis buffer and then centrifuged at 14,000 rpm for 10 min at 4°C. The resulting pellets were re-extracted with 50 mM Tris-HCl, pH 7.5, 2 mM MgCl2, 0.1 mM EDTA, 0.5 mM DTT, and 60 mM octylglucoside. After centrifugation the resulting supernatant was diluted threefold using the same Tris-buffer in the absence of detergent. Trypsin digestion was done as described above.
Results
Kinetics of Formation of Biosynthetic Gβ Intermediates
In pulse-chase experiments using the neuroblastoma cell line IMR-32, we took advantage of the observation that the presence of γ in a complex with β prevents recognition by the β-specific mAbs, ARC5 and ARC9 (52). In this case, the loss of reactivity of β subunits with ARC5 and ARC9 can be used to monitor assembly of the βγ complex.
Pulse-chased IMR-32 cells were lysed in the non-ionic detergents NP-40/Lubrol and equal aliquots of lysate were immunoprecipitated in the absence (−) or presence (+) of 0.2% SDS (Fig. 1 C). At 0 min of chase, the recovery of β from lysates prepared with and without SDS was identical, whereas at 2.5 min and later time points, we observed a progressive loss of immunoreactivity from lysates not exposed to SDS. After 30 min essentially no β subunits were detected in the absence of SDS (see also Fig. 1 A). Over the chase period examined, there is no loss of β in extracts prepared in the presence of SDS.
Figure 1.

Detection of biosynthetic intermediates, half-life, and complex formation of Gβ subunits. (A) IMR-32 cells were pulselabeled for 2.5 min with 150 μCi [35S]methionine and chased for up to 20 h. β subunits were recovered with mAb ARC9 from nonionic detergent lysates, either in the presence (+) or absence (−) of 0.2% SDS. Samples in A–C were subjected to 12.5% SDSPAGE. (B) Pulse-labeling of IMR-32 cells for 1.5 min with 150 μCi [35S]methionine (see A); chases were done for the designated intervals. Precipitations with ARC9 were performed as in A. (C) IMR-32 cells were pulse-labeled for 5 min with 150 μCi [35S]methionine and chased for the times indicated. β subunits were immunoprecipitated with mAb ARC9 in the absence (−) or presence (+) of 0.2% SDS.
We then sought to define more accurately when the loss of immunoreactivity of β with ARC9 starts. In a pulsechase experiment where we limited the pulse to 1.5 min (Fig. 1 B), equal amounts of β were again obtained at 0 min chase for lysates prepared plus and minus SDS. At 2.5 min chase, we observed a loss of the minus SDS form, with a further reduction at 10 min chase. Despite the fact that equal numbers of cells were used at each chase point, we consistently observed that the total amount of radioactivity recovered was maximal between 5 and 7.5 min of pulse or chase (note that the use of equal numbers of cells instead of normalized amounts of radioactivity per chase point allows the detection of these differences in recovery). This observation may reflect differences in detergent extractibility of β subunits due to the lipid environment in which the βγ subunits find themselves. While βγ subunits move through the cell, this lipid environment is likely to change (see below).
We conclude that the process of βγ complex formation is a rapid event starting within the first 2.5 min after completion of the polypeptide chains. The half-life of the β subunit in IMR-32 cells labeled for 2.5 min and chased for 20 h (Fig. 1 A) was in excess of 20 h.
Formation of βγ Complexes Determined by Tryptic Proteolysis
We verified βγ complex formation in a pulse-chase experiment using a trypsin-protection assay. Upon association with γ, the β subunit is partially protected from proteolysis by trypsin, and gives rise to a 27-kD COOH-terminal fragment and a 14-kD NH2-terminal fragment, as it has been shown for the combination β1γ2 (16, 18, 64).
We establish the occurrence of both characteristic fragments after tryptic digest of IMR-32 cell lysates using mAb ARC9 and the polyclonal rabbit anti-βγ serum (Fig. 2 A). Although we tested different concentration ranges of trypsin, we observed in these experiments the persistence of full-length forms of β, indicating that different subtype combinations of β and γ in IMR-32 cells may exist with different degrees of trypsin susceptibility, or that at least some fraction of βγ may not be fully accessible to trypsin, unlike purified βγ subunits. Re-immunoprecipitation of the 27-kD fragment with the peptide specific polyclonal antibody U-49 (derived against amino acids 131-145 of β1) established the origin of this fragment as β subunit-derived (Fig. 2 B).
Figure 2.


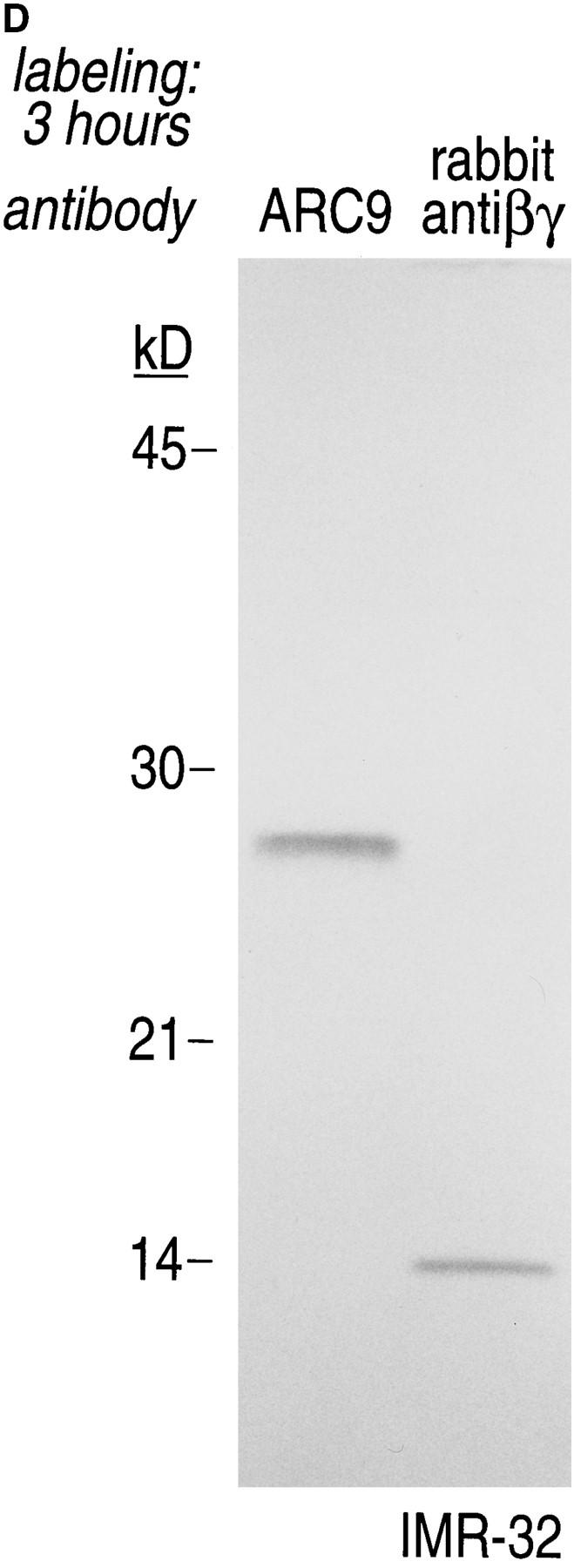
The generation of the βγ subunit derived 27-kD tryptic cleavage product in cell lysates is coincident with the appearance of newly assembled βγ complexes. (A) IMR-32 cells were labeled with 200 μCi [35S]methionine for 2 h. Cells were lysed in 0.3% Lubrol containing lysis buffer and trypsin was added at the concentrations indicated. Immunoprecipitations were done in the presence of 0.2% SDS using ARC9 or rabbit anti-βγ serum. The positions of undigested β chains and tryptic fragments are indicated on the right. (B) The origin of the 27-kD tryptic β fragment was confirmed by re-immunoprecipitation of ARC9 immunoprecipitates with the peptide-specific polyclonal antiserum U-49. (C) IMR-32 cells were pulsed for 2 min with 500 μCi [35S]methionine and chased for the times indicated. Lysates were prepared as in A, and trypsin was added at a concentration of 50 μg/ml. Immunoprecipitations were done in the presence and absence of 0.2% SDS. Note that the appearance of the 27-kD fragment is coincident with the loss of the (−) SDS form in untreated samples. (D) β and γ subunits are complexed in non-ionic detergent-resistant membrane domains. IMR-32 cells were labeled for 3 h with 150 μCi [35S]methionine and lysed in TX-100–containing lysis buffer. Detergent-resistant membranes were resolubilized in octyl-glucoside containing lysis buffer. Trypsin digestion was done as described before (A–C); immunoprecipitations with ARC9 and rabbit antiβγ serum resulted in the appearance of the 27-kD and 14-kD tryptic fragments.
In a pulse-chase experiment we observed, using ARC9, the appearance of this characteristic 27-kD band as coincident with the loss of the SDS-independent form of β, indicating that at this chase time (10 min), association of β and γ has occurred (Fig. 2 C). The NH2-terminal derived 14-kD fragment could be immunoprecipitated with the polyclonal rabbit anti-βγ antiserum (data not shown). The 27-kD fragment, which is devoid of the γ subunit after tryptic digestion, can also be immunoprecipitated with ARC9 in the absence of SDS (Fig. 2 C). The difference in recovery of this 27-kD fragment compared to immunoprecipitations in the presence of SDS is due to incomplete inhibition of trypsin activity in the absence of SDS. Accordingly, we found substantial amounts of low molecular weight degradation products present at the dye front.
A similar pattern of digestion products was observed for βγ's recovered from non-ionic detergent-insoluble domains (see below), confirming the occurrence of βγ as a complex (Fig. 2 D).
Localization of Newly Synthesized Gβ Subunits
Pulse-chase experiments in combination with subcellular fractionation were carried out to further resolve β subunit intermediates. When we fractionated cells simply into a particulate or membrane fraction (M) and a cytosolic fraction (C), and immunoprecipitated β in the absence and presence of SDS, we observed three distinct pools of β subunits (Fig. 3 A). At 0 min chase, the majority of β was detected in the C fraction as the free, uncomplexed form. After 2.5 min of chase, the ratio between cytosolic and membrane-associated β was slightly shifted, with β still found predominantly in the cytosol. Starting at 7.5 min of chase, the recovery of β in the absence of SDS was significantly reduced. The loss of free β indicates the formation of complexes that are different from those at 0 and 2.5 min of chase. At the 15-min chase interval, the distribution between cytosolic (C)- and membrane (M)-associated β appeared to be equivalent, whereas their ratio finally shifted towards the M fraction after 35 and 60 min of chase. After the 60-min chase interval, only a minor amount of β remained in the cytosolic compartment.
Figure 3.



Intracellular redistribution of newly synthesized Gβ subunits. (A) IMR-32 cells were pulse-labeled with 150 μCi [35S]methionine for 2.5 min and chased for the times indicated. A crude homogenate was separated into microsomes and cytosol by centrifugation at 100,000 g for 1 h at 4°C. The pellets recovered were considered the M fractions and contained plasma membranes and microsomal membranes, whereas the supernatant contained the cytosol (fraction C). Solubilization of subcellular fractions in NP-40/Lubrol lysis buffer was as for Fig. 1. Gβ was recovered in the absence (−) or presence (+) of 0.2% SDS. mAb ARC9 immunoprecipitates were analyzed on a 12.5% SDS-PAGE. (B) IMR-32 cells were pulse-labeled as in A and chased for up to 180 min. Membrane and cytosol fractions were prepared by differential centrifugation, yielding fractions designated plasma membranes (28,000 g pellet); microsomes (100,000 g pellet); and cytosol (100,000 g supernatant). All immunoprecipitations with ARC9 were done in the presence of 0.2% SDS. Samples were resolved by 12.5% SDS-PAGE. Although at 15-min chase some of the total cell lysate was inadvertently lost in this experiment, the ratio between the cytosolic and microsomal β as the relevant parameter can still be evaluated. (C) The plasma membrane marker α-Na+/K+-ATPase is detectable in the 28,000 g membrane fraction, but not in the 100,000 g pellet. 50-μg aliquots of membrane fractions as obtained in B were subjected to electrophoresis on a 12.5% SDS-PAGE and transferred to nitrocellulose. The blotting membrane was incubated with rabbit anti–α-Na+/K+-ATPase antiserum.
In summary, three different pools of β subunits could be identified. Pool 1 is defined as a free, uncomplexed state, and the second pool comprises cytosolic βγ subunits, whose contribution to the total pool diminishes with time. Pool 3 contains membrane-bound β subunits and requires inclusion of SDS for visualization. This fraction increases with time and is recruited from the cytosolic pool. It is reasonable to suggest that this membrane-bound fraction is in a complex with γ subunits.
To further resolve the intracellular domains into plasma membranes, microsomes, and cytosol, a postnuclear supernatant of pulse-chased IMR-32 cells was subjected to differential centrifugation (48). All immunoprecipitations were done in the presence of 0.2% SDS (Fig. 3 B). The β subunit was recovered at 0 min of chase predominantly from the cytosolic fraction and only a minor amount was associated with lower density microsomal membranes. The maximum amount of β obtained from cytosol or microsomes was found at 2.5 min of chase, followed by a decrease in both compartments of up to 45 min. The β subunits appeared after 15 min of chase in the plasma membrane pool, which contains plasma membranes, as confirmed by the occurrence of the plasma membrane marker α-Na+/ K+-ATPase (Fig. 3 C), which may also contain high density microsomes. The activity of β recovered from this compartment strongly increased at the 30-min chase interval. The microsomal and cytosolic pool of β remained at a constantly low level after 45 min of chase. We conclude that movement of β is characterized by a decrease of β in the cytosolic and microsomal fractions and a corresponding increase in the plasma membrane pool.
Distribution of Gα and Gβ Subunits Assessed by Subcellular Fractionation
The localization of G protein subunits by immunoblot on subcellular fractions does not necessarily reflect the processes or kinetics underlying the biogenesis of membrane domains in vivo. We therefore used pulse-chase labeling in conjunction with subcellular fractionation on discontinuous sucrose gradients and G protein subunit-specific immunoprecipitation to obtain a kinetic description of transport of G protein subunits in living cells (21).
A homogenate prepared from pulse-labeled IMR-32 cells was loaded on a sucrose step gradient and centrifuged at 85,000 g for 16 h. Fractions were analyzed individually by immunoprecipitation with a mixture of anti-Gα mAbs (3C2/ 3E7) or an anti-Gβ mAb (ARC9, plus 0.2% SDS). The distribution of both radiolabeled α or β in these gradients changed with time (Fig. 4, A and B). At the early time of chase (0 min), α and β were recovered from the denser region of the gradient, reflecting a status where β subunits, shortly after synthesis, are devoid of γ subunits, but they still fractionate with large dense particles, such as the cytosolic ribosomes on which they are synthesized (7). At the 20-min chase point we noted a movement of β and α subunits towards lower density fractions (B, C, and D interface). This result is consistent with the observation that after 20 min of chase most β subunits are complexed with γ subunits, and require SDS for efficient recovery in immunoprecipitation (Figs. 1 and 3). The assembly of β with the γ subunit is likely to provide the βγ complex with a membrane anchor, in the form of the COOH-terminal prenyl modification of the γ subunit. This anchor modification would allow part of the complexes to associate with lower density membranes, which are found in the B and C interface. A fraction of β subunits was still detectable in the D and E interface, where both plasma membranes (D interface), large particles and lysosomal membranes, fractionate (E interface) (7). A similar result was obtained for the α subunit, which at the 0-min chase point was found in the high density region of the gradient and after a 20-min chase moved to the low density fraction. At 60 min of chase we observed that Gα exhibited a more prominent bimodal distribution as compared to β. The peaks of this broad distribution were centered around the C and D interface. This result is consistent with the observation that the C interface accumulates light density membranes with the buoyant density of Golgi membranes, whereas plasma membranes characteristically float to the high density region (D interface) of the sucrose gradient (7). At 60 min of chase we recovered little material from the dense region of the gradient with the anti-Gβ mAb ARC9, indicating that formation of trimers in domains fractionating at the D interface might not be stoichiometric. We performed chases up to 240 min to reveal progressive changes of G protein subunit distribution as a function of time (data not shown), but no obvious alterations in comparison to the 60-min chase interval were discernible. This observation suggests that movement of G protein subunits from their site of synthesis to their final destination is essentially complete at 60 min of chase.
Figure 4.


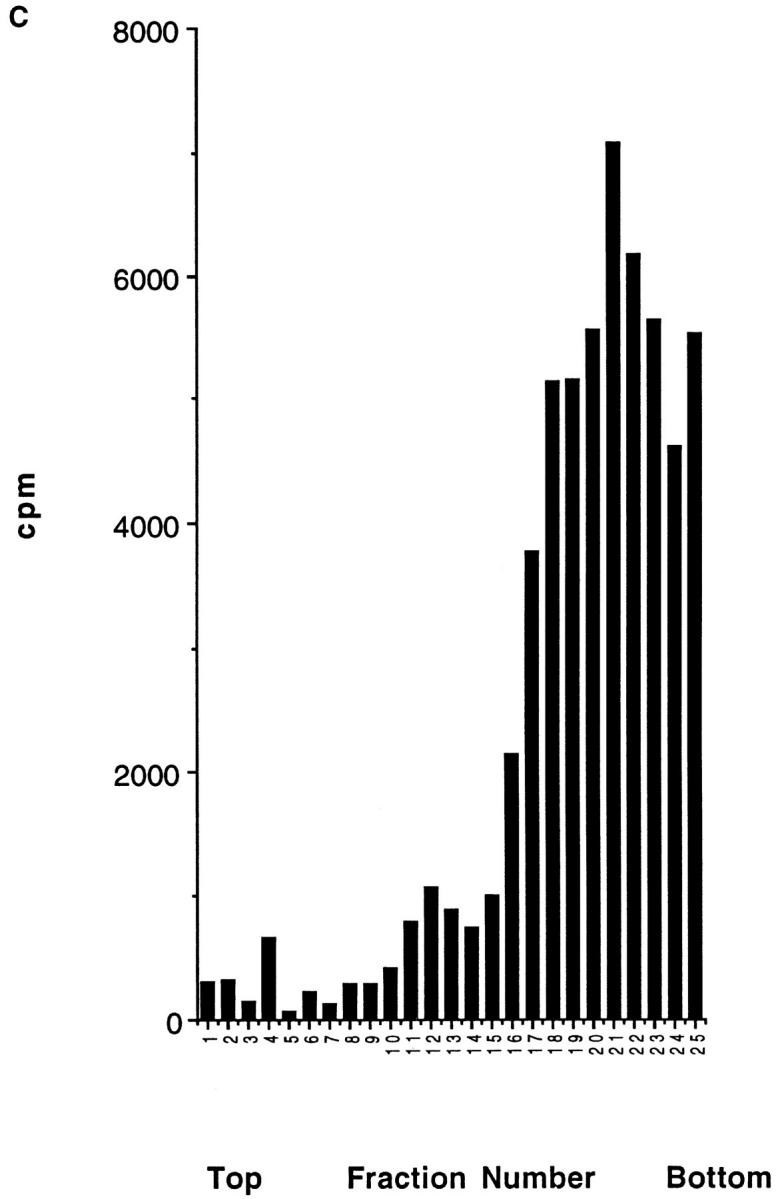
Domain localization of newly synthesized Gα and Gβ subunits. IMR-32 cells were pulse-labeled for 5 min with 500 μCi [35S]methionine and chased for the designated times. Cells were homogenized as shown (Fig. 3) and resolved on a discontinuous sucrose gradient. 0.5-ml fractions were collected from the top of each gradient, lysed in an equal volume of 2× NP-40/Lubrol lysis buffer, and aliquots from each fraction were immunoprecipitated with ARC9 (+ 0.2% SDS) (A) or with a mixture of anti-Gα antibodies (3E7, 3C2) (B). The interfaces between different sucrose densities are designated A–E, whereas the sucrose density steps are represented in percent sucrose. The distribution of radioactivity along the gradients is shown in C. Data shown are representative for at least three experiments performed in duplicate.
G Protein Subunits Rapidly Associate with Detergent-resistant Membranes
When non-ionic detergent insoluble material from labeled IMR-32 cells was analyzed for the presence of β subunits, we found substantial amounts of β subunits even after two rounds of non-ionic detergent extraction, although the majority of β subunits was still recovered from the detergent soluble fraction (Fig. 5, A and B). Whereas the recovery of β from the soluble extract required SDS, the efficiency of α recovery was independent of SDS, an indication that the epitopes recognized on α are freely accessible. To determine the time point when β associates with the insoluble pellet, IMR-32 cells were pulse-labeled for 2.5 min, chased for the times indicated, and extracted with NP-40/Lubrol. The β subunit was found in the pellet beginning at 1 min of chase (Fig. 5 C).
Figure 5.

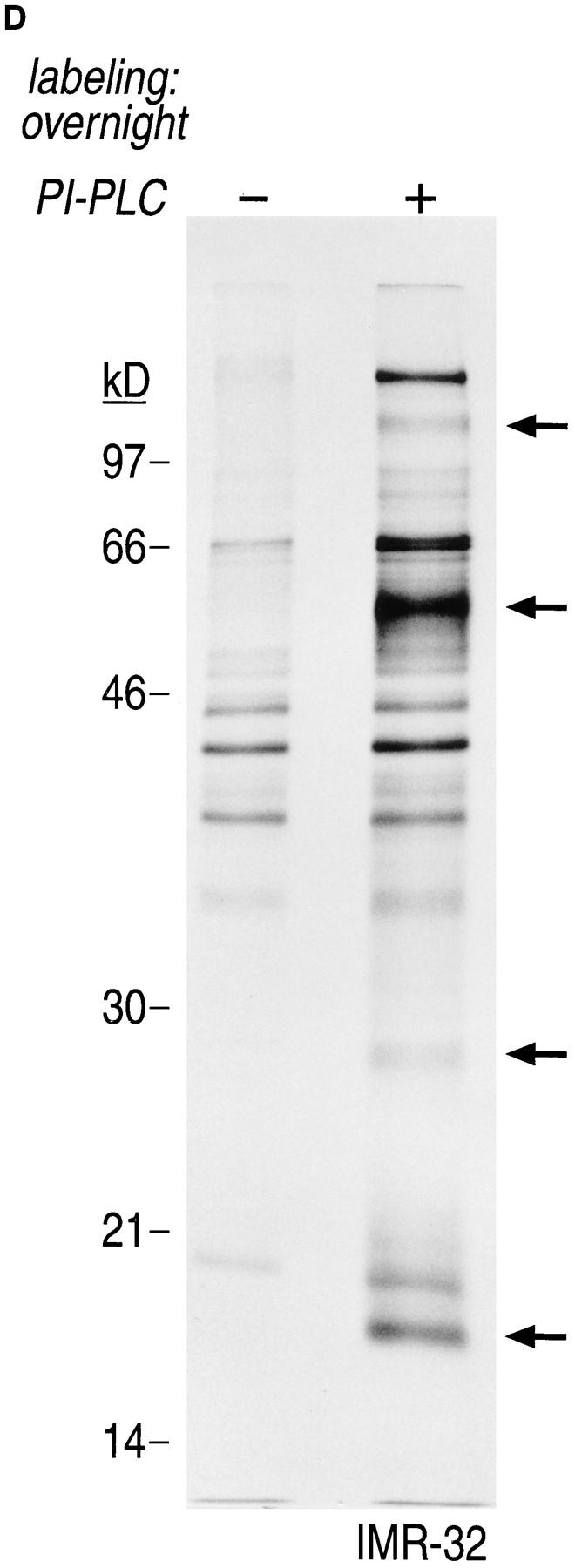
Gα and Gβ are associated with non-ionic detergent-insoluble cell pellets. (A) IMR-32 cells were labeled overnight with 100 μCi of [35S]methionine. Cells were lysed in NP-40/Lubrol lysis buffer and separated into supernatant (soluble) and insoluble pellet. The pellet was solubilized in 1% SDS, DNA was sheared by 15 passages through a 21-gauge needle and the extract was boiled twice at 100°C for 3 min. The SDS concentration in the pellet fraction was adjusted to 0.2% with NP-40/Lubrol lysis buffer. Immunoprecipitations from the supernatant were done either in the presence (+) or absence (−) of 0.2% SDS. A mixture of 3C2 and 3E7 were used to recover α subunits and β was precipitated with ARC9. Samples were analyzed on a 12.5% SDS-PAGE (A–C). Arrows indicate the positions of β and α subunits. (B) IMR-32 cells were labeled for 2 h with 250 μCi [35S]methionine followed by solubilization in NP-40/ Lubrol lysis buffer. Insoluble pellets were extracted twice with non-ionic detergent buffer and subsequently treated as shown in A. (C) Kinetics of Gβ subunit association with detergent-insoluble cell pellets. IMR-32 cells were pulsed for 2.5 min with 150 μCi [35S]methionine and chased for the time intervals indicated. Cells were separated into supernatant (soluble) and a detergent-insoluble pellet. The pellet was re-extracted as in A, followed by immunoprecipitation with ARC 9. (D) Detection of endogenous GPI-anchored proteins in non-ionic detergent-resistant membrane domains. IMR-32 cells were labeled overnight with 100 μCi [35S]methionine and lysed in buffer containing TX-100. After centrifugation, the resulting pellet was re-extracted with TX-114–containing lysis buffer for 20 min at 37°C. After centrifugation, the supernatant was subjected to temperature-induced phase separation. Samples were treated in the absence (−) and presence (+) of 5 U/ml PI-PLC. This treatment induces the transition from a hydrophobic to a hydrophilic state of GPI-anchored proteins. Polypeptides released into the aqueous phase were recovered by acetone precipitation and analyzed on a 12.5% SDS-PAGE. Arrows on the right indicate the appearance of some of the proteins specifically released by PI-PLC.
We next characterized some of the attributes of these detergent-resistant membranes, as follows. Because the neuroblastoma cell line IMR-32 used in this study does not express any of the known isoforms of caveolin (Rehm, A., and H.L. Ploegh, unpublished observations), this marker could not be used to ascertain the presence of proteins characteristically enriched in the detergent-insoluble domains. Instead, we employed a procedure in which the TX-100–resistant membrane fraction was re-extracted with TX-114, and subjected to several rounds of temperatureinduced phase separation (6, 35). This procedure results in an enrichment of GPI-anchored proteins in the detergent phase, from which they can be released by treatment with PI-PLC. The released fraction, recovered in the aqueous phase after another three cycles of phase separation, was collected by acetone precipitation and displayed by SDSPAGE (Fig. 5 D). A comparison of the profile of polypeptides released specifically by PI-PLC is indicative of the presence of GPI-anchored proteins in this detergent-resistant membrane fraction.
As was shown for βγ complexes in soluble detergent extracts (Fig. 2, A–C), we established that βγ complexes present in the detergent-resistant membranes yielded a pattern of tryptic products consistent with their proper assembly (Fig. 2 D).
The presence of βγ complexes and other proteins involved in signal transduction in detergent-resistant domains has been attributed physiological significance. It has been argued that these detergent-resistant membranes fulfill a specialized role in signaling by allowing a high local concentration of the relevant proteins. We sought to explore this issue by comparing the relative amounts of βγ in detergent-resistant membranes from cells held under serum starvation vs cells maintained in 20% FCS. For cells cultured in the presence of FCS, we observed that ∼30% of all β subunits could be retrieved in the detergent-resistant membranes, where it must be kept in mind that even non-ionic detergent may extract some βγ's (and other membrane proteins) from areas generally defined as detergent-resistant membranes (Fig. 6 A). When βγ content was analyzed for cells maintained in the presence or absence of serum (normalized for the amount of protein loaded for each sample, Fig. 6 B), we observed an approximately threefold enrichment of βγ's in detergent-resistant membranes from serum-stimulated cells.
Figure 6.


βγ complexes are enriched in detergent-resistant membrane domains. (A) IMR-32 cells (5 × 106) were lysed directly in PBS/1% SDS/2 mM DTT; alternatively an equal amount of cells was first extracted with TX-100–containing lysis buffer, followed by re-extraction of the detergent-resistant membrane fraction in PBS/1% SDS/2 mM DTT. Total cell lysate and TX-100–resistant pellet were heated twice for 3 min at 100°C in the presence of SDS, and passed through a 21-gauge needle several times. Laemmli sample buffer was added and samples were analyzed after SDSPAGE and immunoblot for the presence of G protein β subunits. (B) Equal numbers of cells were cultured for 16–18 h in the presence (+) or absence (−) of 20% FCS. Total cell lysates and detergent-resistant membrane pellets were generated as described in A. Samples were normalized for the amount of protein (100 μg/lane) using the BCA protein assay. Samples were analyzed after SDS-PAGE and immunoblot, using the anti-β mAb ARC5.
The ratios between soluble and detergent-resistant, membrane-associated β subunits were examined in a pulsechase experiment using TX-100–containing lysis buffer. Detergent-resistant pellets were then resolubilized either in 1% SDS (see above) or octylglucoside; the latter was found to solubilize α and β subunits with an efficiency comparable to SDS (Rehm, A., and H.L. Ploegh, unpublished observations). The detergent-resistant pool of β increased until 30 min, when a plateau was reached (Fig. 7, A and B). Further evidence for a kinetic shift in association of β with detergent-resistant domains is shown in Fig. 7 C, where the amount of low density protein-lipid complexes, separated by sucrose density gradient centrifugation, increased after 40 min of chase.
Figure 7.

Quantitative changes of the Triton-insoluble β subunit pool shortly after synthesis. (A) IMR-32 cells were pulsed with 500 μCi [35S]methionine and chased for the times indicated. Cells were lysed in TX-100 lysis buffer and separated as described in Fig. 5. The pellet was resolubilized in 60 mM octylglucoside, and the amounts of radioactivity for individual chase points were normalized for the soluble and pellet fraction. Immunoprecipitations were done in the presence of 0.2% SDS using mAb ARC9. (B) IMR-32 cells were pulsed and chased as indicated. Extractions and immunoprecipitations were done as described in A. (C) IMR-32 cells were pulsed for 1.5 min with 500 μCi [35S]methionine and chased for 5 or 40 min. Cells were homogenized in TX100 lysis buffer, incorporation of radioactivity was determined, and extracts containing equal amounts of counts were resolved on a discontinuous sucrose gradient. 1-ml fractions were collected from the top, lysed in an equal volume of 2× NP-40/Lubrol lysis buffer supplemented with 0.4% SDS. Immunoprecipitations were performed with ARC9.
To test for the transport pathway taken by cytosolderived βγ subunits to rapidly formed detergent-resistant domains, ER to Golgi transport was blocked by BFA. We scored for inhibition of intracellular glycoprotein transport by analyzing persistence of sensitivity of MHC class I molecules to Endo H (Fig. 8, B and C). The association of βγ subunits with detergent-resistant membranes was not affected by the BFA-imposed block of ER to Golgi transfer (Fig. 8 A). We conclude that cytosolic βγ subunits are inserted into detergent-resistant membranes at a site distal from the BFA block, probably at the plasma membrane itself.
Figure 8.

Transport and formation of detergent-resistant membranes is insensitive to BFA treatment. IMR-32 cells were stimulated for 48 h with 50 U/ml IFN-γ. Cells were pulsed in the presence and absence of 10 μg/ml brefeldin A for 10 min with 500 μCi [35S]methionine and chased (+ and −10 μg/ml BFA) for the time indicated. Cells were extracted as described in Fig. 7 and separated into a soluble and a TX-100–resistant fraction. Resolubilization of the pellet was done with octylglucoside containing lysis buffer. ARC9 was used to recover β subunits (+0.2% SDS) (A), and MHC class I heavy chains were immunoprecipitated with W6/32.HL (B and C). Endo H digestions of W6/32.HL precipitates were done to demonstrate the effects of anterograde ER to Golgi transport block. BFA renders MHC class I molecules endo H sensitive even after 60 min of chase.
Discussion
The kinetics of formation of the G protein βγ complex, and the kinetics with which these βγ subunits are targeted to distinct membrane domains were the subject of this study. Newly synthesized β and α subunits are detectable by immunoprecipitation within 1.5 min of labeling, and their halflife is in excess of 20 h (Fig. 1) (for α, see reference 33). Anti-β monoclonal antibodies, raised against immunoaffinity purified βγ, provided the tools with which to follow biosynthetic intermediates of β subunits. Formation of the βγ complex leads to masking of the epitope recognized by these mAbs on the β subunit by the association of the γ subunit (52). Coincident with this loss of immunoreactivity, we observe, upon proteolysis with trypsin, the appearance of a characteristic fragment seen only for properly assembled βγ subunits (Fig. 2) (16, 18, 64). Consequently, the pattern of immunoreactivity, seen with the anti-β mAbs, could be used to follow βγ assembly in living cells.
Purified recombinant β and γ subunits, in vitro translated βγ subunits or βγ subunits transfected into cells have been used to explore the mechanisms and structural elements required for βγ subunit assembly (10, 45, 70). Prenylation of γ is not required for βγ assembly, but this modification of the γ subunit apparently facilitates the interaction of preformed βγ complexes with the α subunit (25). The assembly of βγ apparently occurs before removal of the 3 carboxy-terminal amino acids from the prenylated γ subunit (70). The enzymatic reactivity required for this reaction is largely localized to the microsomal membranes (22, 23). Therefore, the assembly of βγ and prenylation are predominantly cytosolic processes that likely precede membrane attachment. Prevention of γ prenylation in living cells, either by site-directed mutagenesis of the prenylation target sequence or by inhibition of isoprenoid precursor synthesis, inhibits membrane targeting of the βγ complex. It is not known whether the isoprenoid moiety interacts with the lipid bilayer directly or facilitates interaction with the membrane by means of lipid-adaptor-protein interaction (13). Our data are consistent with βγ complex formation starting in the cytosol, although the process may continue for β or γ subunits that are already membrane-associated.
Our experiments did not resolve the location or timing of the isoprenoid modification of the γ subunit. The farnesyl moiety of the Ras protein facilitates targeting of the protein to detergent-resistant membranes in living cells, whereas a nonfarnesylated mutant of Ras fails to associate with this domain (62). In support of the role of the isoprenoid anchor in targeting to detergent-resistant membranes, prenylation of Gγ in yeast was shown to be a requirement for this membrane association (30).
While the timing and localization of βγ modification and assembly has been addressed, kinetics of assembly on differential membrane subdomains have not been established. Our experiments have thus far failed to identify the intracellular site at which α subunits associate with the βγ complex. As far as the formation of the βγ complex is concerned, we note that its association with the detergentresistant membrane fraction is rapid and appears not to be affected by BFA treatment (Fig. 8 A). One of the most pronounced effects of BFA is the tight block of membrane traffic out of the ER, whereas the retrograde movement of Golgi membrane components into the ER continues (28). We conclude that a sizable fraction of G protein subunits, associated with these insoluble domains, probably do not associate with the heptahelical receptors at the level of the ER but do so later. The latter presumably depend on the anterograde pathway through ER and Golgi for proper folding and maturation of their N-linked glycans.
These data suggest that cytosolically synthesized βγ subunits, once formed, can be targeted directly to the plasma membrane. Alternatively, the BFA treatment of cells could lead to a redistribution of Golgi-membrane derived lipids, causing a detergent resistance of proteins which associate with them. We consider this possibility unlikely since the induction of an enhanced retrograde trafficking pathway by BFA from the Golgi to the ER does not include the redistribution of components of the trans-Golgi network, where the occurrence of detergent-insoluble domains has been reported (28). Second, it is possible that other intracellular membranes, including the ER, may contain such detergent-insoluble domains. Insolubility may not necessarily be caused by the occurrence of βγ subunits in association with glycolipid rafts, which are expected to be largely absent from the ER (see below). A third possibility is suggested by the observation that proteins might actively recruit lipids to create detergent-insoluble domains. One example is the coclustering of a lipid into detergent-resistant membranes following aggregation of the IgE receptor (65). In this view, the newly assembled cytosol-derived βγ subunits, upon attachment to a membrane, may recruit lipids that render the βγ subunits resistant to solubilization with non-ionic detergents. The alternative interpretation would involve the creation of, and recruitment of, βγ subunits to a detergent-resistant membrane fraction in the ER itself. This possibility must be considered unlikely if such detergent resistance is caused largely by incorporation of glycolipids.
In general, detergent-resistant membranes are operationally defined by their insolubility in non-ionic detergents and by their low buoyant density in sucrose density gradients (9). They are enriched in GPI-anchored proteins, glycosphingolipids, and cholesterol (17, 40). Detergent-resistant membranes were proposed to play a role in the sorting of GPI-anchored proteins and viral proteins destined for the apical surface (57, 58). Segregation into these glycosphingolipid-enriched domains occurs in the trans-Golgi network of epithelial cells before transport to the plasma membrane (9). Other studies have suggested that detergent-resistant membranes serve other functions in addition to sorting. These include their role in signal transduction, indicated by the presence of signaling molecules like the heterotrimeric G proteins, Src family tyrosine kinases, and H-Ras (36, 38). Furthermore, these formations were shown to have a role in potocytosis, demonstrated by the internalization of folate (54, 60).
We observe an enrichment of βγ's in detergent-resistant membranes when comparing serum-exposed vs serumstarved cells. This observation might relate to the cholesterol depletion that follows serum starvation, because cholesterol renders membrane domains more resistant to extraction with non-ionic detergents. This result suggests that the formation of detergent-resistant membranes, and hence the proteins found herein, is controlled by the lipid phase of these domains (11, 61).
We show that G protein β and α subunits rapidly associate with detergent-insoluble membranes. When cellular extracts were separated into a non-ionic detergent-soluble fraction and an insoluble pellet, substantial amounts of β and α subunits were recovered in association with the insoluble pellet already after 1 min of chase (Fig. 5 C). Transient appearance in detergent-resistant membranes has also been described for human placental alkaline phosphatase and for influenza virus hemagglutinin (HA), albeit with different kinetics of association (9, 59). HA acquires some detergent insolubility, but the association of HA with the insoluble pellet is transient and found only after 15–20 min of chase. This fraction of HA was localized to the trans-Golgi network and plasma membrane. We found that α subunits associated with detergent-resistant membranes can undergo ADP-ribosylation (Rehm, A., M. Yilla, and H.L. Ploegh, unpublished observation), indicating that at least transient interactions between α and βγ subunits in this compartment must occur. The recovery of β from detergent resistant membranes after resolubilization in octylglucoside required the presence of SDS, demonstrating that β and γ are complexed in this domain. Furthermore, the tryptic digestion of detergent-resistant β results in 27- and 14-kD fragments, supporting our previous notion that β and γ find themselves associated in this domain (Fig. 2 D).
Notwithstanding the occurrence of these detergentresistant membranes, it is likely that proteins associated with them may display different degrees of detergent resistance. Obviously, the existence of membrane domains refractory to extraction with non-ionic detergents does not imply that all proteins that reside there are completely resistant to extraction. This could lead to selective loss, upon detergent extraction, of certain polypeptides from these domains. Gα's are known to undergo rapid cycles of acylation and deacylation in the course of receptor activation (55), whereas the isoprenoid moiety on the γ subunit is probably more stable. Our observation that βγ's but not α's remain associated with the detergent-resistant membranes after a second extraction step may be related to the different properties and dynamics of the lipid modifications of G protein subunits.
This result seems to be contradictory to a previous report showing that Gα subunits but not βγ complexes remain detergent resistant after TX-100 extraction. We conclude that the different protocols applied to enrich for detergent-resistant membranes might account for the differences observed. Although we have analyzed a complex cellular lysate after extraction with TX-100, Chang et al. (12) have enriched detergent-resistant caveolae with a detergent-free method and then treated those membranes with a non-ionic detergent. Therefore, it is likely that an altered protein-detergent ratio has influenced the quantitative recovery of βγ complexes from detergent-resistant domains. In addition, our data are supported by recent reports in which methods were applied for the purification of detergent-resistant membranes that are similar to the one used here (20, 62). Based on the purification criteria presented in our study we consider it unlikely that all βγ complexes are in the detergent-resistant membrane domain and are only partially removed by TX-100 extraction. Homogenization of cells in the absence of detergent results in kinetically and spatially distinguishable cytosolic, plasma membrane and microsomal pools; the latter contain endosomal and lysosomal membranes not reported to exhibit detergent resistant properties. Second, the kinetics of acquisition of detergent resistance indicate that even if this domain would constitute the final destination for βγ complexes, there still exist pools of soluble βγ complexes that could be intermediates, as revealed by immunoprecipitation and pulsechase analysis.
The topology of luminal receptors and cytosolic G protein subunits adds complexity to the problem of how G proteins, receptors, and effectors assemble. The timing and subcellular localization of assembly events may influence the composition of a signaling complex, and hence determine its specificity. Further analysis of heterotrimeric G protein assembly would therefore require the integration of a G protein–coupled receptor and downstream effectors in our experimental system.
Acknowledgments
We are grateful to Drs. Mamadi Yilla and Uta Höpken for review of the manuscript. We also thank Hisse Martien van Santen, Robert Machold, Johannes Huppa, and Matthew Bogyo for their critical evaluation of the manuscript.
Footnotes
1. Abbreviations used in this paper: Endo H, endoglycosidase H; G proteins, heterotrimeric guanine nucleotide-binding proteins; GPI, glycosylphosphatidylinositol; HA, hemagglutinin; IFN-γ, interferon-γ; Staph. A, Staphylococcus aureus bacteria.
A. Rehm was supported by a fellowship from the Deutsche Forschungsgemeinschaft (DFG). This work was supported in part by Amgen.
Please address all correspondence to H.L. Ploegh, Center for Cancer Research, Department of Biology, Massachusetts Institute of Technology, 40 Ames Street, E 17-322, Cambridge, MA 02139. Tel.: (617) 253-0520. Fax: (617) 253-9891.
References
- 1.Anderson RGW. Plasmalemmal caveolae and GPI-anchored membrane proteins. Curr Opin Cell Biol. 1993;5:647–652. doi: 10.1016/0955-0674(93)90135-d. [DOI] [PubMed] [Google Scholar]
- 2.Barnstable CJ, Bodmer WF, Brown G, Galfre G, Milstein C, Williams AF, Ziegler A. Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens. Cell. 1978;14:9–20. doi: 10.1016/0092-8674(78)90296-9. [DOI] [PubMed] [Google Scholar]
- 3.Bokoch GM. The presence of free G protein β/γ subunits in human neutrophils results in suppression of adenylate cyclase activity. J Biol Chem. 1987;262:589–594. [PubMed] [Google Scholar]
- 4.Bokoch GM, Bickford K, Bohl BP. Subcellular localization and quantitation of the major neutrophil pertussis toxin substrate, Gn . J Cell Biol. 1988;106:1927–1936. doi: 10.1083/jcb.106.6.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bomsel M, Mostov K. Role of heterotrimeric G proteins in membrane traffic. Mol Biol Cell. 1992;3:1317–1328. doi: 10.1091/mbc.3.12.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bordier C. Phase separation of integral membrane proteins in TX114 solution. J Biol Chem. 1981;256:1604–1607. [PubMed] [Google Scholar]
- 7.Bourguignon LYW, Jin H, Iida N, Brandt NR, Zhang SH. The involvement of ankyrin in the regulation of inositol 1,4,5-trisphosphate receptor mediated internal Ca2+ release from Ca2+storage vesicles in mouse T-lymphoma cells. J Biol Chem. 1993;268:7290–7297. [PubMed] [Google Scholar]
- 8.Bourne HR, Sanders DA, Mc F, Cormick The GTPase superfamily: conserved structure and molecular mechanism. Nature (Lond) 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- 9.Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- 10.Casey PJ. Lipid modifications of G proteins. Curr Opin Cell Biol. 1994;6:219–225. doi: 10.1016/0955-0674(94)90139-2. [DOI] [PubMed] [Google Scholar]
- 11.Chang W-J, Rothberg KG, Kamen BA, Anderson RGW. Lowering the cholesterol content of MA 104 cells inhibits receptor mediated transport of folate. J Cell Biol. 1992;118:63–69. doi: 10.1083/jcb.118.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang W-J, Ying Y-S, Rothberg KG, Hooper NM, Turner AJ, Gambliel HA, de Gunzburg J, Mumby SM, Gilman AG, Anderson RGW. Purification and characterization of smooth muscle cell caveolae. J Cell Biol. 1994;126:127–138. doi: 10.1083/jcb.126.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clarke S. Protein isoprenylation and methylation at carboxyl-terminal cysteine residues. Annu Rev Biochem. 1992;61:355–386. doi: 10.1146/annurev.bi.61.070192.002035. [DOI] [PubMed] [Google Scholar]
- 14.Cushman SW, Wardzala LJ. Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. J Biol Chem. 1980;255:4758–4762. [PubMed] [Google Scholar]
- 15.Denker SP, Mc JM, Caffery, Palade GE, Insel PA, Farquhar MG. Differential distribution of α subunits and βγ subunits of heterotrimeric G proteins on Golgi membranes of the exocrine pancreas. J Cell Biol. 1996;133:1027–1040. doi: 10.1083/jcb.133.5.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fung BK-K, Nash CR. Characterization of transducin from bovine retinal rod outer segments. J Biol Chem. 1983;258:10503–10510. [PubMed] [Google Scholar]
- 17.Futerman AH. Inhibition of sphingolipid synthesis: effects on glycosphingolipid-GPI-anchored protein microdomains. Trends Cell Biol. 1995;5:377–380. doi: 10.1016/s0962-8924(00)89078-9. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Higuera I, Thomas TC, Yi F, Neer EJ. Intersubunit surfaces in G protein αβγ heterotrimers. J Biol Chem. 1996;271:528–535. doi: 10.1074/jbc.271.1.528. [DOI] [PubMed] [Google Scholar]
- 19.Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 20.Gorodinsky A, Harris DA. Glycolipid-anchored proteins in neuroblastoma cells form detergent-resistant complexes without caveolin. J Cell Biol. 1995;129:619–627. doi: 10.1083/jcb.129.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Green SA, Zimmer K-P, Griffiths G, Mellman I. Kinetics of intracellular transport and sorting of lysosomal membrane and plasma membrane proteins. J Cell Biol. 1987;105:1227–1240. doi: 10.1083/jcb.105.3.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gutierrez L, Magee AI, Marshall CJ, Hancock JF. Posttranslational processing of p21 ras is two-step and involves carboxylmethylation and carboxy-terminal proteolysis. EMBO (Eur Mol Biol Organ) J. 1989;8:1093–1098. doi: 10.1002/j.1460-2075.1989.tb03478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hancock JF, Cadwallader K, Marshall CJ. Methylation and proteolysis are essential for efficient membrane binding of prenylated p21 k-ras (B) EMBO (Eur Mol Biol Organ) J. 1991;10:641–646. doi: 10.1002/j.1460-2075.1991.tb07992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hille B. G protein-coupled mechanisms and nervous signaling. Neuron. 1992;9:187–195. doi: 10.1016/0896-6273(92)90158-a. [DOI] [PubMed] [Google Scholar]
- 25.Iniguez-Lluhi J, Simon MI, Robishaw JD, Gilman AG. G protein βγ subunits synthesized in Sf 9 cells. J Biol Chem. 1992;267:23409–23417. [PubMed] [Google Scholar]
- 26.Jones TLZ, Simonds WF, Merendino JJ, Brann MR, Spiegel AM. Myristoylation of an inhibitory GTP-binding protein α subunit is essential for its membrane attachment. Proc Natl Acad Sci USA. 1990;87:568–572. doi: 10.1073/pnas.87.2.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Juhnn Y-S, Jones TLZ, Spiegel AM. Amino- and carboxyterminal deletion mutants of Gsαare localized to the particulate fraction of transfected COS cells. J Cell Biol. 1992;119:523–530. doi: 10.1083/jcb.119.3.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klausner RD, Donaldson JG, Lippincott-Schwartz J. Brefeldin A: insight into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleuss C, Scheruebl H, Hescheler J, Schultz G, Wittig B. Different β-subunits determine G-protein interaction with transmembrane receptors. Nature (Lond) 1992;358:424–426. doi: 10.1038/358424a0. [DOI] [PubMed] [Google Scholar]
- 30.Kuebler E, Dohlman HG, Lisanti MP. Identification of Triton X-100 insoluble domains in the yeast Saccharomyces cerevisiae. . J Biol Chem. 1996;271:32975–32980. doi: 10.1074/jbc.271.51.32975. [DOI] [PubMed] [Google Scholar]
- 31.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (Lond) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 32.Langhans-Rajasekaran S, Wan Y, Huang X-Y. Activation of Tsk and Btk tyrosine kinases by G protein βγ subunits. Proc Natl Acad Sci USA. 1995;92:8601–8605. doi: 10.1073/pnas.92.19.8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levis MJ, Bourne HR. Activation of the α subunit of Gs in intact cells alters its abundance, rate of degradation, and membrane avidity. J Cell Biol. 1992;119:1297–1307. doi: 10.1083/jcb.119.5.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linder ME, Pang IH, Duronio RJ, Sternweis PC, Gilman AG. Lipid modifications of G protein subunits: myristoylation of Goαincreases its affinity for βγ. J Biol Chem. 1991;266:4654–4659. [PubMed] [Google Scholar]
- 35.Lisanti MP, Sargiacomo M, Graeve L, Saltiel AR, RodriguezBoulan E. Polarized apical distribution of glycosyl-phosphatidylinositol-anchored proteins in a renal epithelial cell line. Proc Natl Acad Sci USA. 1988;85:9557–9561. doi: 10.1073/pnas.85.24.9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lisanti MP, Scherer PE, Tang ZL. Caveolae, caveolin and caveolin-rich membrane domains: a signalling hypothesis. Trends Cell Biol. 1994;4:231–235. doi: 10.1016/0962-8924(94)90114-7. [DOI] [PubMed] [Google Scholar]
- 37.Lisanti MP, Tang ZL, Sargiacomo M. Caveolin forms a hetero-oligomeric protein complex that interacts with an apical GPI-linked protein: implications for the biogenesis of caveolae. J Cell Biol. 1993;123:595–604. doi: 10.1083/jcb.123.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lisanti MP, Scherer MP, Vidugiriene J, Tang ZL, HermanowskiVosatka A, Tu Y-H, Cook RF, Sargiacomo M. Characterization of caveolin-rich membrane domains isolated from an endothelialrich source: implications for human disease. J Cell Biol. 1994;126:111–126. doi: 10.1083/jcb.126.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marshall CJ. Protein prenylation: a mediator of protein-protein interactions. Science (Wash DC) 1993;259:1865–1866. doi: 10.1126/science.8456312. [DOI] [PubMed] [Google Scholar]
- 40.Mays RW, Siemers KA, Fritz BA, Lowe AW, van Meer G, Nelson WJ. Hierarchy of mechanisms involved in generating Na/K- ATPase polarity in MDCK epithelial cells. J Cell Biol. 1995;130:1105–1115. doi: 10.1083/jcb.130.5.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milligan G, Parenti M, Magee AI. The dynamic role of palmitoylation in signal transduction. TIBS. 1995;20:181–186. doi: 10.1016/s0968-0004(00)89004-0. [DOI] [PubMed] [Google Scholar]
- 42.Morishita R, Nakayama H, Isobe T, Matsuda T, Hashimoto Y, Okano T, Fukada Y, Mizuno K, Ohno S, Kozawa O, et al. Primary structure of a γ subunit of G protein, γ12, and its phosphorylation by protein kinase C. J Biol Chem. 1995;270:29469–29475. doi: 10.1074/jbc.270.49.29469. [DOI] [PubMed] [Google Scholar]
- 43.Mumby SM, Kahn RA, Manning DR, Gilman AG. Antisera of designed specificity for subunits of guanine nucleotide-binding regulatory proteins. Proc Natl Acad Sci USA. 1986;83:265–269. doi: 10.1073/pnas.83.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mumby SM, Heukeroth RO, Gordon JI, Gilman AG. G-protein α-subunit expression, myristoylation, and membrane association in COS cells. Proc Natl Acad Sci USA. 1990;87:728–732. doi: 10.1073/pnas.87.2.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muntz KH, Sternweis PC, Gilman AG, Mumby SM. Influence of γ subunit prenylation on association of guanine nucleotide-binding regulatory proteins with membranes. Mol Biol Cell. 1992;3:49–61. doi: 10.1091/mbc.3.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neer EJ. Heterotrimeric G proteins: organizers of transmembrane signals. Cell. 1995;80:249–257. doi: 10.1016/0092-8674(95)90407-7. [DOI] [PubMed] [Google Scholar]
- 47.Neer EJ, Clapham DE. Roles of G protein subunits in transmembrane signalling. Nature (Lond) 1988;333:129–134. doi: 10.1038/333129a0. [DOI] [PubMed] [Google Scholar]
- 48.Oka Y, Czech MP. Photoaffinity labeling of insulin-sensitive hexose transporters in intact rat adipocytes. J Biol Chem. 1984;259:8125–8133. [PubMed] [Google Scholar]
- 49.Oppenheimer CL, Pessin JE, Massague J, Gitomer W, Czech MP. Insulin action rapidly modulates the apparent affinity of the insulin-like growth factor II receptor. J Biol Chem. 1983;258:4824–4830. [PubMed] [Google Scholar]
- 50.Parton RG, Simons K. Digging into caveolae. Science (Wash DC) 1995;269:1398–1399. doi: 10.1126/science.7660120. [DOI] [PubMed] [Google Scholar]
- 51.Quick MW, Simon MI, Davidson N, Lester HA, Aragay AM. Differential coupling of G protein α subunits to seven-helix receptors expressed in Xenopus oocytes. J Biol Chem. 1994;269:30164–30172. [PubMed] [Google Scholar]
- 52.Rehm A, Ploegh HL. Monoclonal antibodies that distinguish free and complexed heterotrimeric G protein β subunits. FEBS Lett. 1997;402:277–285. doi: 10.1016/s0014-5793(96)01457-3. [DOI] [PubMed] [Google Scholar]
- 53.Ross EM. Signal sorting and amplification through G protein-coupled receptors. Neuron. 1989;3:141–152. doi: 10.1016/0896-6273(89)90027-5. [DOI] [PubMed] [Google Scholar]
- 54.Rothberg KG, Ying YS, Kolhouse JF, Kamen BA, Anderson RGW. The glycophospholipid-linked folate receptor internalizes folate without entering the clathrin-coated pit endocytic pathway. J Cell Biol. 1990;110:637–649. doi: 10.1083/jcb.110.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shenoy-Scaria AM, Dietzen DJ, Kwong J, Link DC, Lublin DM. Cysteine of src family kinases determines palmitoylation and localization in caveolae. J Cell Biol. 1994;126:353–363. doi: 10.1083/jcb.126.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simon MI, Strathmann MP, Gautam N. Diversity of G proteins in signal transduction. Science (Wash DC) 1991;252:802–808. doi: 10.1126/science.1902986. [DOI] [PubMed] [Google Scholar]
- 57.Simons K, van Meer G. Lipid sorting in epithelial cells. Biochemistry. 1988;27:6197–6202. doi: 10.1021/bi00417a001. [DOI] [PubMed] [Google Scholar]
- 58.Simons K, Wandinger-Ness A. Polarized sorting in epithelia. Cell. 1990;62:207–210. doi: 10.1016/0092-8674(90)90357-k. [DOI] [PubMed] [Google Scholar]
- 59.Skibbens JE, Roth MG, Matlin KS. Differential extractibility of influenza virus hemagglutinin during intracellular transport in polarized epithelial cells and nonpolar fibroblasts. J Cell Biol. 1989;108:821–832. doi: 10.1083/jcb.108.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smart EJ, Foster DC, Ying YS, Kamen BA, Anderson RGW. Protein kinase C activators inhibit receptor-mediated potocytosis by preventing internalization of caveolae. J Cell Biol. 1994;124:307–314. doi: 10.1083/jcb.124.3.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smart JE, Ying YS, Conrad PA, Anderson RGW. Caveolin moves from caveolae to the Golgi apparatus in response to cholesterol oxidation. J Cell Biol. 1994;127:1185–1197. doi: 10.1083/jcb.127.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song KS, Li S, Okamoto T, Quilliam LA, Sargiacomo M, Lisanti MP. Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. J Biol Chem. 1996;271:9690–9697. doi: 10.1074/jbc.271.16.9690. [DOI] [PubMed] [Google Scholar]
- 63.Sternweis PC. The purified α subunits of Go and Gifrom bovine brain require βγ for association with phospholipid vesicles. J Biol Chem. 1986;261:631–637. [PubMed] [Google Scholar]
- 64.Thomas CT, Sladek T, Yi F, Smith T, Neer EJ. G protein βγ subunit: physical and chemical characterisation. Biochemistry. 1993;32:8628–8635. doi: 10.1021/bi00084a034. [DOI] [PubMed] [Google Scholar]
- 65.Thomas JL, Holowka D, Baird B, Webb WW. Large-scale co-aggregation of fluorescent lipid probes with cell surface proteins. J Cell Biol. 1994;125:795–802. doi: 10.1083/jcb.125.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsukada S, Simon MI, Witte ON, Katz A. Binding of βγ subunits of heterotrimeric G proteins to the PH domain of Bruton tyrosine kinase. Proc Natl Acad Sci USA. 1994;91:11256–11260. doi: 10.1073/pnas.91.23.11256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van der Voorn L, Tol O, Hengeveld TM, Ploegh HL. Structural and functional studies on the Goprotein. J Biol Chem. 1993;268:5131–5138. [PubMed] [Google Scholar]
- 68.Wiertz EJHJ, Jones TR, Sun L, Bogyo M, Geuze HJ, Ploegh HL. The human cytomegalovirus US 11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996;84:769–779. doi: 10.1016/s0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- 69.Yilla M, Oleinick J, Ploegh HL. A monoclonal antibody that causes the heterotrimeric G-protein Goto release its βγ subunits. FEBS Lett. 1996;387:16–22. doi: 10.1016/0014-5793(96)00441-3. [DOI] [PubMed] [Google Scholar]
- 70.Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]