

Abstract
Structural protein 4.1, first identified as a crucial 80-kD protein in the mature red cell membrane skeleton, is now known to be a diverse family of protein isoforms generated by complex alternative mRNA splicing, variable usage of translation initiation sites, and posttranslational modification. Protein 4.1 epitopes are detected at multiple intracellular sites in nucleated mammalian cells. We report here investigations of protein 4.1 in the nucleus. Reconstructions of optical sections of human diploid fibroblast nuclei using antibodies specific for 80-kD red cell 4.1 and for 4.1 peptides showed 4.1 immunofluorescent signals were intranuclear and distributed throughout the volume of the nucleus. After sequential extractions of cells in situ, 4.1 epitopes were detected in nuclear matrix both by immunofluorescence light microscopy and resinless section immunoelectron microscopy. Western blot analysis of fibroblast nuclear matrix protein fractions, isolated under identical extraction conditions as those for microscopy, revealed several polypeptide bands reactive to multiple 4.1 antibodies against different domains. Epitope-tagged protein 4.1 was detected in fibroblast nuclei after transient transfections using a construct encoding red cell 80-kD 4.1 fused to an epitope tag. Endogenous protein 4.1 epitopes were detected throughout the cell cycle but underwent dynamic spatial rearrangements during cell division. Protein 4.1 was observed in nucleoplasm and centrosomes at interphase, in the mitotic spindle during mitosis, in perichromatin during telophase, as well as in the midbody during cytokinesis. These results suggest that multiple protein 4.1 isoforms may contribute significantly to nuclear architecture and ultimately to nuclear function.
Structural proteins via diverse molecular interactions determine cell morphology, organize subcellular compartments, stabilize cell attachments, and even regulate essential cellular responses to internal or external signaling. The 80-kD structural protein, protein 4.1, was initially characterized as a crucial member of the red cell membrane skeleton where it stabilizes complexes between spectrin and actin within the skeletal network and anchors them to the overlying plasma membrane through interactions with integral membrane proteins. Deficiencies in 80-kD protein 4.1 profoundly alter red cell morphology and decrease membrane mechanical strength, leading to membrane fragmentation and hemolytic anemia.
In subsequent studies, the 80-kD 4.1 of mature red cells was identified as only one member of a large protein 4.1 family that is relatively abundant in nucleated erythroid and nonerythroid cells. In fact, Western blots of many types of mammalian and avian cells revealed 4.1 immunoreactive protein species ranging from 30–210 kD (Anderson et al., 1988; Granger and Lazarides, 1984, 1985). As in many other structural protein families, 4.1 isoform structural and functional diversity can be generated by a number of mechanisms including complex alternative splicing of 4.1 premRNA (Conboy et al., 1988, 1991; Tang et al., 1988, 1990), usage of at least two translation initiation sites, and posttranslational modifications of 4.1 proteins. These variations as well as regulated 4.1 mRNA expression can be both tissue- and differentiation-specific (for review see Conboy, 1993).
Several binding partners for specific 4.1 domains have been characterized. The amino-terminal domain of erythrocyte protein 4.1 contains binding sites for glycophorin C, calmodulin, p55 (Kelly et al., 1991; Tanaka et al., 1991; Pinder et al., 1993; Gascard and Cohen, 1994; Hemming et al., 1994, 1995; Marfatia et al., 1994, 1995), and band 3 (Jons and Drenckhahn, 1992; Lombardo et al., 1992), while a domain towards the COOH terminus contains binding sites for spectrin and actin complexes (Correas et al., 1986a ,b; Discher et al., 1993; Schischmanoff et al., 1995). Purified red cell 4.1 also interacts specifically with tubulin (Correas and Avila, 1988) and myosin (Pasternack and Racusen, 1989). The multiplicity of 4.1 isoforms combined with the diversity of possible protein–protein interactions suggests that individual 4.1 isoforms may have specific and discrete functions. However, the roles of 4.1 isoforms, other than in red cells, have not yet been defined.
In contrast to the strictly peripheral localization of 4.1 in mature red cells, 4.1 epitopes in nucleated cells have been observed by immunofluorescence throughout the cytoplasmic compartment (Cohen et al., 1982; Lue et al., 1994), including perinuclear regions such as apparent centrosomal and Golgi structures (Leto et al., 1986; Marchesi, V.T., S. Huang, T.K. Tang, and E.J. Benz. 1990. Blood. 76:12A; Chasis et al., 1993; Krauss, S.W., C.A. Larabell, C. Rogers, N. Mohandas, and J. Chasis. 1995. Blood. 86:415a; Beck, K.A., and W.J. Nelson. 1996. Mol. Cell. Biol. 302:22). Immunolabeling of 4.1 epitopes has also been observed in the nucleus (Madri et al., 1988; Tang, T.K., C.E. Mazoucco, T.L. Leto, E.J. Benz, and V.T. Marchesi. 1988. Clin. Res. 36:A405; Marchesi, V.T., S. Huang, T.K. Tang, and E.J. Benz. 1990. Blood. 76:12A; Correas, 1991; Krauss, S.W. 1994. J. Cell. Biochem. 18c:95, M208; Krauss, S.W., J.A. Chasis, S. Lockett., R. Blaschke, and N. Mohandas. 1994. Mol. Biol. Cell. 5:343a; De Carcer et al., 1995; Krauss, S.W., J.A. Chasis, C.A. Larabell, S. Lockett, R. Blaschke, and N. Mohandas. 1995. J. Cell Biochem. 21B:140, JT 309). Isoforms of protein 4.1 in the nucleus presumably could serve as structural elements. This is particularly intriguing in light of emerging evidence of the important relationship between nuclear architecture and regulation of nuclear functions.
The nucleus contains an internal nonchromatin scaffolding called the “nuclear matrix” or “nucleoskeleton.” The nuclear matrix is a three-dimensional structure, and when viewed using resinless section electron microscopy, it appears as a network of polymorphic filaments enmeshing larger masses or “dense bodies” (Capco et al., 1982; Fey et al., 1986; Nickerson et al., 1995; for review see Penman, 1995). Matrix spatial organization appears to provide functional subcompartmentalization for nuclear metabolic processes and requisite machinery (Nakamura et al., 1986; Spector, 1990, 1993; Carmo-Fonseca et al., 1991; Saunders et al., 1991; Spector et al., 1991; Wang et al., 1991). The largest nuclear domains are the nucleoli, sites of ribosomal RNA synthesis and partial assembly (Fischer et al., 1991; Scheer et al., 1993). RNA transcription and pre-mRNA splicing occur in smaller domains and along tracks within the matrix reticular network (Spector, 1990; Spector et al., 1991; Carter et al., 1993; Xing et al., 1993, 1995). DNA is attached to the nuclear skeleton, and factors involved in replication and cell cycle control are clustered into discrete replication foci (Smith and Berezney, 1982; Jackson and Cook, 1986; Cook, 1988; Leonhardt et al., 1992; Hozak et al., 1993). However, during each round of cell division, the nucleus itself undergoes dramatic morphologic remodeling: its nuclear envelope dissolves, the underlying lamin ring depolymerizes, and chromosomes condense. Thus, the nuclear scaffold must be capable of a series of ordered yet dynamic protein–protein interactions necessitated by perturbations of nuclear architecture and function during the cell cycle.
We report here that protein 4.1 isoforms are resident proteins of the nucleoskeleton during interphase and undergo dynamic reorganization in their association with nuclear components during mitosis. This suggests that by molecular interactions either previously identified or entirely novel, protein 4.1 isoforms could be integral both to nuclear structure and to modulation of nuclear activities.
Materials and Methods
Materials
WI38 cells (CCL 75: diploid human fetal lung fibroblasts), CaSki cells (CRL 1550: human cervical epidermoid carcinoma), and 3T3 cells (CCL 92: murine fibroblasts) were obtained from American Type Culture Collection (Rockville, MD). HCA cells, from human fetal foreskin, were the gift of Dr. J. Campisi (Lawrence Berkeley National Laboratory, Berkeley, CA). The plasmid pSV2NeoCMV was generously provided by Dr. P. Yaswen (Lawrence Berkeley National Laboratory). The vector contains a cytomegalovirus (CMV)1 promoter, SV-40 polyA addition and splice signals, and an EcoRI restriction site for insertion of test sequences. B4A11 monoclonal antiserum was the gift of Dr. J.A. Nickerson (Massachusetts Institute of Technology, Cambridge, MA). FITC-conjugated goat anti–rabbit IgG and 5-(and-6)-carboxy-rhodamine succinimidyl ester were purchased from Molecular Probes (Eugene, OR). Tetramethyl-rhodamine–conjugated goat anti–mouse IgG and fluorescein 5-isothiocyanate isomer 1 were from Sigma Chemical Co. (St. Louis, MO). Anti–β-tubulin and high molecular weight “Rainbow” markers were products of Amersham Corp. (Arlington Heights, IL). Pefabloc (AEBSF; 4-[2 aminoethyl]benzenesulfonyl fluoride) was obtained from Boehringer Mannheim Corp. (Indianapolis, IN). Renaissance chemiluminescence reagents were purchased from DuPont/NEN (Boston, MA). DME and LipofectAMINE were the product of GIBCO BRL (Gaithersburg, MD) and FBS was from Hyclone Labs (Logan, UT). Antibodies against lamin B (Ab-1), PCNA (Ab-1), nuclear mitotic apparatus protein (NuMA) (Ab-1), and nuclear pores (Ab-3) were either purchased or provided by Oncogene Research (Cambridge, MA).
Western Blot Analysis
Protein samples separated on 7% SDS-PAGE were transferred to nitrocellulose for 7 h at 100 V. After blocking, the blots were incubated first with 0.5 μg/ml primary IgG for 1 h at room temperature, washed, and then incubated with a secondary antibody coupled to horseradish peroxidase. Immunoreactive proteins were visualized by chemiluminescent autoradiography.
Immunofluorescence
Subconfluent WI38 fibroblasts or CaSki cells grown on coverslips were rinsed twice in PBS at pH 7.4, fixed for 10 min at 4°C in PBS containing 2% formaldehyde/0.2% Triton X-100, further permeabilized with 1% Triton X-100 for 10 min at room temperature, and then rinsed twice in PBS. Alternatively, cells were fixed in −80°C acetone or in cold MeOH for 5 min. After incubation in 10% goat serum to block nonspecific protein binding, fixed cells were incubated with 5–10 μg/ml primary antibodies for 1 h at room temperature in PBS containing 10 mg/ml BSA, washed, and then incubated with FITC- and rhodamine-conjugated secondary antibodies. Cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) at 0.5 μg/ml (Sigma Chemical Co.) to confirm nuclear location and washed again, and the coverslips were mounted on slides using Vectashield (Vector Laboratories, Burlingame, CA). Controls with equivalent amounts of nonimmune IgG or with primary antibody omitted were also included in each experiment. When photographed under the same conditions as the experimental samples, the controls did not show fluorescence.
Microscopy
Immunofluorescent cells were photographed using an epifluorescence microscope with a 63× objective and a 1.4 NA oil immersion lens. A microscope (model Axioplan; Carl Zeiss, Inc., Thornwood, NY) equipped with a 63× 1.3 Neofluor oil immersion lens, a Quantitative Image Processing System based on the MicroImager 1400 digital camera (Xillix Technologies Corp., Vancouver, BC, Canada), and the Scil-Image image analysis software package (The University of Amsterdam, The Netherlands) were used to acquire and analyze 0.2-μm optical sections of cells. A confocal scanning laser microscope (model MRC 1000; BioRad Labs, Hercules, CA) equipped with a Nikon Diaphot 200 (Melville, NY), a fluor 60× 1.4 planapo oil immersion lens, and a krypton/argon laser was also used to obtain 0.5-μm optical sections. Double-label images were collected sequentially and both signals were of similar intensity. Confocal images were constructed by means of BioRad software.
High resolution resinless section EM of immunostained cells was performed as previously described (Capco et al., 1984; Nickerson et al., 1990, Nickerson and Penman, 1992). Extracted cells were fixed with glutaraldehyde, blocked with goat serum, labeled with gold bead–conjugated goat anti–rabbit secondary antibodies (Amersham Corp.) after incubation with primary IgG, and fixed again. The samples were embedded in diethylene glycol distearate (Polysciences, Warrington, PA). Thin sections were cut, and the resin was removed before critical point drying and carbon coating. Resinless sections were examined using an electron microscope (model 1200EX; JEOL U.S.A., Inc., Peabody, MA).
Cell Culture
WI38 cells were grown in a 5% CO2 incubator at 37°C in DME and CaSki cells in RPMI-1640 medium containing 10% FCS, 4 mM glutamine, antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin) and 2.2 g/liter sodium bicarbonate. Human fibroblasts were used at passages 20–24 where >90% were capable of DNA synthesis.
Preparation of Nuclear Matrix Proteins
For isolation of nuclear matrix from nuclei, proliferating cells were trypsinized, washed three times in PBS, swollen in RSB (10 mM Tris, pH 7.4, 10 mM NaCl, 3 mM MgCl2) for 30 min on ice, and dounced in RSB containing 0.3% NP-40 for about ten strokes until nuclei were free of cytoplasm by microscopic examination. Nuclei were then pelleted through a 0.25 M sucrose-RSB cushion for 10 min at 1500 g at 4°C. Typically, >95% of nuclei were recovered. Sequential extraction procedures were performed as described previously (Fey et al., 1984, 1986; Nickerson et al., 1994) using buffers containing protease inhibitors (1 mM Pefabloc, 0.1 μg/ml aprotinin, 1 μg/ml pepstatin and leupeptin). Briefly, nuclei were successively extracted in CSK buffer (10 mM Pipes, pH 6.8, 300 mM sucrose, 100 mM NaCl, 3 mM MgCl2, 1 mM EGTA, and 0.5% Triton X-100), extraction buffer (10 mM Pipes, pH 6.8, 300 mM sucrose, 250 mM ammonium sulfate, 3 mM MgCl2, 1 mM EGTA, and 0.5% Triton X-100), digestion buffer (10 mM Pipes, pH 6.8, 300 mM sucrose, 50 mM NaCl, 3 mM MgCl2, 1 mM EGTA, and 0.5% Triton X-100) with DNase and RNase or RNase-free DNase as indicated, and reextracted with 250 mM ammonium sulfate. To remove additional proteins from nuclear matrix, the preparation was extracted with 2 M NaCl buffer. For fractionation of nuclear matrix for in situ microscopy, subconfluent cells growing on coverslips were rinsed in PBS and sequentially processed as described above.
Antibodies
Rabbits were immunized with 80-kD protein 4.1 purified from human RBC or with synthetic peptides comprised of sequences encoded by either exon 16 (peptide 10-1), exon 19 (peptide 24-2), or exon 21 (peptide 24-3) coupled to thyroglobulin (see Fig. 1). Sequences of the peptide antigens were: 10-1, KKRERLDGENIYIRHSNLMLEC; 24-2, TDDNSGDLDPGVLLTAQTITSETPSSTTTTQITKC; or 24-3, HPDMSVTKVVVH QETEIADEC. In addition, the sequence encoding peptide N-2, containing 209 amino acids following the AUG-1 start site of translation of protein 4.1 (Conboy et al., 1991) (Fig. 1), was fused to glutathione-S-transferase (GST). The construct was overexpressed, purified by glutathione agarose affinity chromatography, and used for immunization. Immune IgGs were affinity purified against the immunizing 4.1 peptide or RBC 80-kD 4.1 and analyzed to confirm no cross-reaction with either thyroglobulin or the GST moiety. Antibodies 10-1 and 24-2 have been used by us in other studies (Chasis et al., 1993; Conboy et al., 1993; Schischmanoff et al., 1995; Chasis et al., 1996). For direct labeling, IgGs (0.4 μg/ml) were conjugated to rhodamine or FITC fluorophores and then extensively dialyzed according to the instructions provided by Molecular Probes.
Figure 1.

Protein 4.1 peptide domains used to generate antibodies. (A) A schematic diagram of protein 4.1 mRNAs displaying multiple combinations of splicing pathways possible among 4.1 alternative exons. In this format, exons are coded as follows: solid bars, constitutive; shaded bars, alternative; open bars, noncoding. The arrows on top indicate the positions of alternative translation initiation sites, AUG-1 and AUG-2. The figure is derived from Conboy et al. (1991). (B) Examples of protein 4.1 isoforms derived from different translation initiation sites. The 80-kD prototypical red cell isoform is produced from AUG-2 and can be present in nucleated and nonnucleated cells. Chymotryptic fragments of this isoform include a 30-kD membrane binding domain, a 16-kD domain, a 10-kD spectrin and actin binding domain, and the 22–24-kD domain (Leto and Marchesi, 1984). Higher molecular mass 4.1 isoforms, present in nucleated cells, use AUG-1 to generate an additional 209–amino acid “NH2-terminal extension” (N-term). (C) Synthetic peptides derived from the 4.1 amino acid sequence at the positions indicated by the arrows were used to immunize rabbits. IgGs were prepared from N-2, 10-1, 24-2, and 24-3 sera by affinity purification using the homologous peptide. The sequences for the peptides are given in the Methods section.
PCR Amplification of Protein 4.1 mRNA
WI38 mRNA, prepared from early passage, proliferating cells by the guanidinium thiocyanate method (Chirgwin et al., 1979) and human reticulocyte mRNA prepared as described (Temple et al., 1977) were transcribed into cDNA and amplified by PCR under the conditions previously used by Conboy et al. (1991). For amplification of AUG-1, the oligonucleotides used were: sense strand, AACATCATGACAACAG, and antisense strand, GTGTGTTTCTGCACTGCTTA.
Transient Transfections
A DNA construct was engineered encoding the sequence for 80-kD 4.1 fused at its COOH terminus to an epitope tag (KPPTPPPEPET) derived from SV-40 large T antigen. The 80-kD 4.1 sequence began at AUG-2 and included coding sequence through exon 21 except for the alternatively spliced exons 14, 15, 17A, and 17B. The tag sequence, detected with the monoclonal antibody KT3 (MacArthur and Walter, 1984), does not contain the SV-40 nuclear localization signal. The 80-kD 4.1 epitope-tagged construct was fused to GST in pGEX-KT and expressed in Escherichia coli, and the product was isolated by glutathione chromatography (Discher et al., 1993). It was analyzed by Western blotting to verify its apparent molecular weight and reactivity both with 4.1 and KT3 antibodies.
Subconfluent murine fibroblasts (3T3 cells) growing on coverslips were transfected using LipofectAMINE and plasmid DNA according to the manufacturer's instructions. A CMV-β-gal vector was used to optimize transfection conditions. 3T3 cells were transfected for 24–48 h with pSV2NeoCMV containing an insert encoding 80-kD red cell protein 4.1 fused to an epitope tag at its COOH terminus or a control vector without an insert. Transfected 3T3 cells were fixed either with paraformaldehyde or methanol, processed for indirect immunofluorescence using Texas red–conjugated secondary antibody, and finally DAPI stained to determine subcellular distribution of expressed epitope-tagged protein. Three independent transient transfection experiments were performed.
Results
Protein 4.1 Localizes to Intranuclear Sites
To investigate nuclear localization of protein 4.1, we initially used diploid human fibroblasts (WI38 cells) since their flattened morphology is particularly amenable by microscopy to distinguish nucleus versus cytoplasm and since human fibroblasts do not spontaneously transform in culture. Multiple protein 4.1 isoforms have previously been observed in fibroblasts (Cohen et al., 1982; Conboy et al., 1993).
To detect 4.1 epitopes by immunofluorescence microscopy, we used a panel of affinity-purified IgGs. As depicted by the map of multiple alternative splicing pathways in Fig. 1 A, protein 4.1 isoforms contain varying patterns of exonic inclusion and exclusion. Using exonspecific antibodies allows detection of 4.1 isoforms having the peptide encoded by that particular exon. Our antibody panel included antibody 10-1, recognizing sequence encoded by exon 16; antibody 24-2, specific for peptide encoded by exon 19; antibody 24-3, recognizing epitope encoded by exon 21; antibody N-2, recognizing the NH2-terminal 209 amino acids translated from the AUG-1 start site (Fig 1, B and C); and anti-RBC 80-kD, against the 80-kD red cell 4.1 isoform purified from human RBC (Fig. 1 B). We established the specificity of our antibody panel for protein 4.1 by showing that IgGs 10-1, 24-2, 24-3, and anti-RBC 80-kD 4.1 each reacted with red cell 80-kD 4.1 in Western blot analysis (Fig. 2 A) and with protein 4.1 in the plasma membrane of human red cells using immunofluorescent detection (Fig. 2 B). Furthermore, immunofluorescent signals were coincident when human red blood cells were probed with 24-2 and 24-3 directly labeled with two different fluorophores (see Fig. 4).
Figure 2.

Characterization of antibodies directed against red cell 80-kD 4.1 and 4.1 peptide domains. (A) Human red cell membrane fractions (Conboy et al., 1993) were separated by SDS-PAGE, transferred to nitrocellulose, and incubated with the following IgGs: lane 1, anti-RBC 4.1; lane 2, anti–N-2; lane 3, anti–10-1; lane 4, anti–24-2; lane 5, anti–24-3; lane 6, control IgG. Protein 4.1 in red cells migrates at ∼80 kD. Although this is the predominant 4.1 isoform in red cells, a faint band at ∼135 kD detected by anti–N-2 (lane 2) is also detected by other 4.1 IgGs when increased amounts of red cell membrane preparations are applied to gels (Conboy et al., 1991). (B) Human red cells were fixed by methanol and probed by indirect immunofluorescence using primary antibodies as indicated and FITCconjugated secondary antibodies. The faint staining by anti–N-2 most likely reflects the small amount of ∼135-kD 4.1 also detected by Western blot analysis of red cell membranes. Bar, 10 μm.
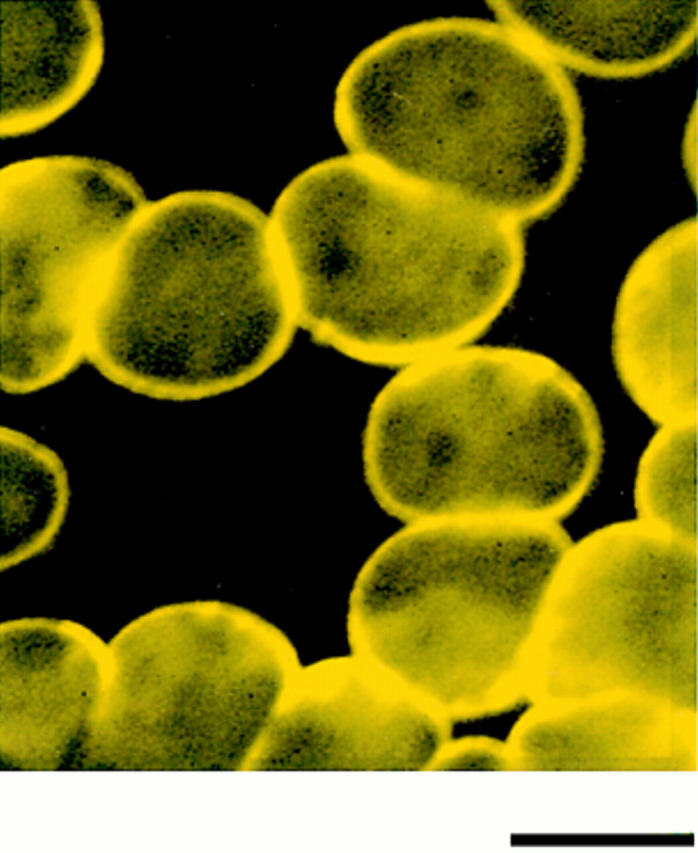
Figure 4.
Coincidence of immunofluorescent signals on human red cells probed with antibodies against two 4.1 peptide domains. Normal human red cells, which contain 80-kD 4.1 in their plasma membrane, were fixed in methanol and probed with antibody 24-2 directly conjugated to a FITC fluorophore (green) and antibody 24-3 directly conjugated to a rhodamine fluorophore (red). The yellow image indicates that the red and green signals coincide. Bar, 6 μm.
Using three different classical fixation techniques (paraformaldehyde, methanol, or acetone), prominent punctate staining in the nuclear area of WI38 fibroblasts was consistently observed by indirect immunofluorescence with each antibody. Some antibodies also generated a more diffuse nuclear staining pattern and several of the antibodies produced cytoplasmic staining (Fig. 3). Parallel samples incubated with control IgG did not produce nuclear immunofluorescence with any of these fixation methods (Fig. 3). Fibroblasts were also probed with preparations of 24-2 or 24-3 IgGs directly labeled with either of two different fluorophores; these cells displayed immunofluorescent patterns similar to those obtained by indirect methods (data not shown). Nuclear immunofluorescent staining was also observed when another human fibroblast line (HCA), a transformed epithelial human line (CaSki; see Fig. 9) and a transformed murine fibroblast line (3T3; see Fig. 8) were probed with the 4.1 antibody panel.
Figure 3.





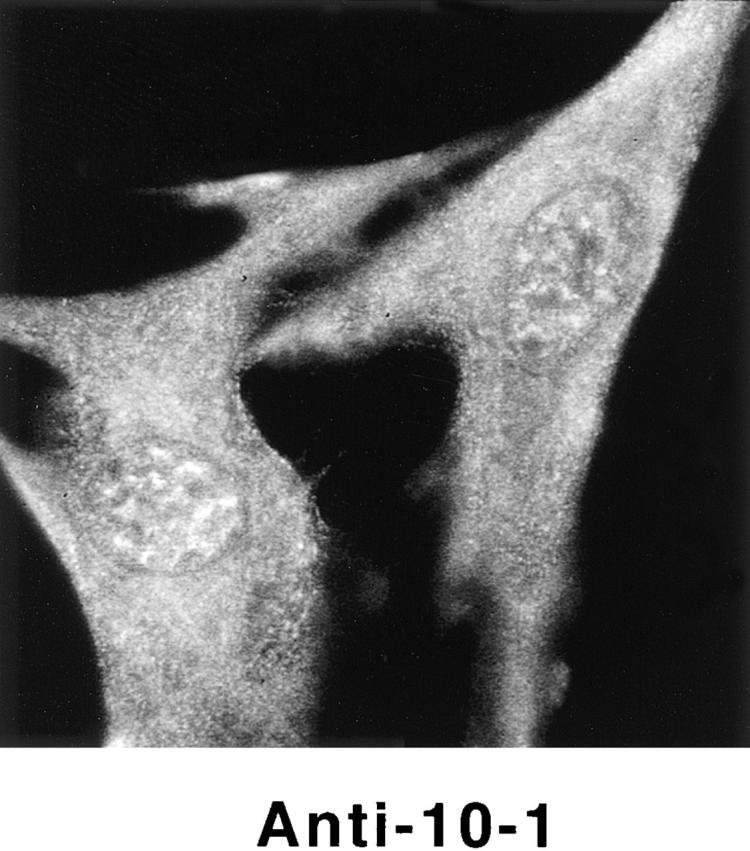
Detection of nuclear epitopes of protein 4.1 by immunofluorescent light microscopy in WI38 cells. Cultured WI38 human fibroblasts were fixed in acetone for probing with anti-RBC 80-kD 4.1 and N-2, in methanol for probing with anti–24-2, or in formaldehyde for probing with anti–10-1 and anti–24-3, followed by incubation with a FITC-labeled secondary antibody. Punctate nuclear signals, detected with all the protein 4.1 antibodies, were particularly prominent with anti-RBC 80-kD 4.1 and anti–24-2. Localization of FITC signals within the nuclei of all cells was confirmed by comparison to DAPI fluorescence of the cells with 4.1 fluorescence (not shown). Antibodies N-2, 10-1, and 24-3 also produced considerable cytoplasmic staining. Staining patterns were in large part independent of the fixation method. Controls showed no fluorescent patterns when imaged under the same conditions as experimental samples. Bar, 10 μm.
Figure 9.
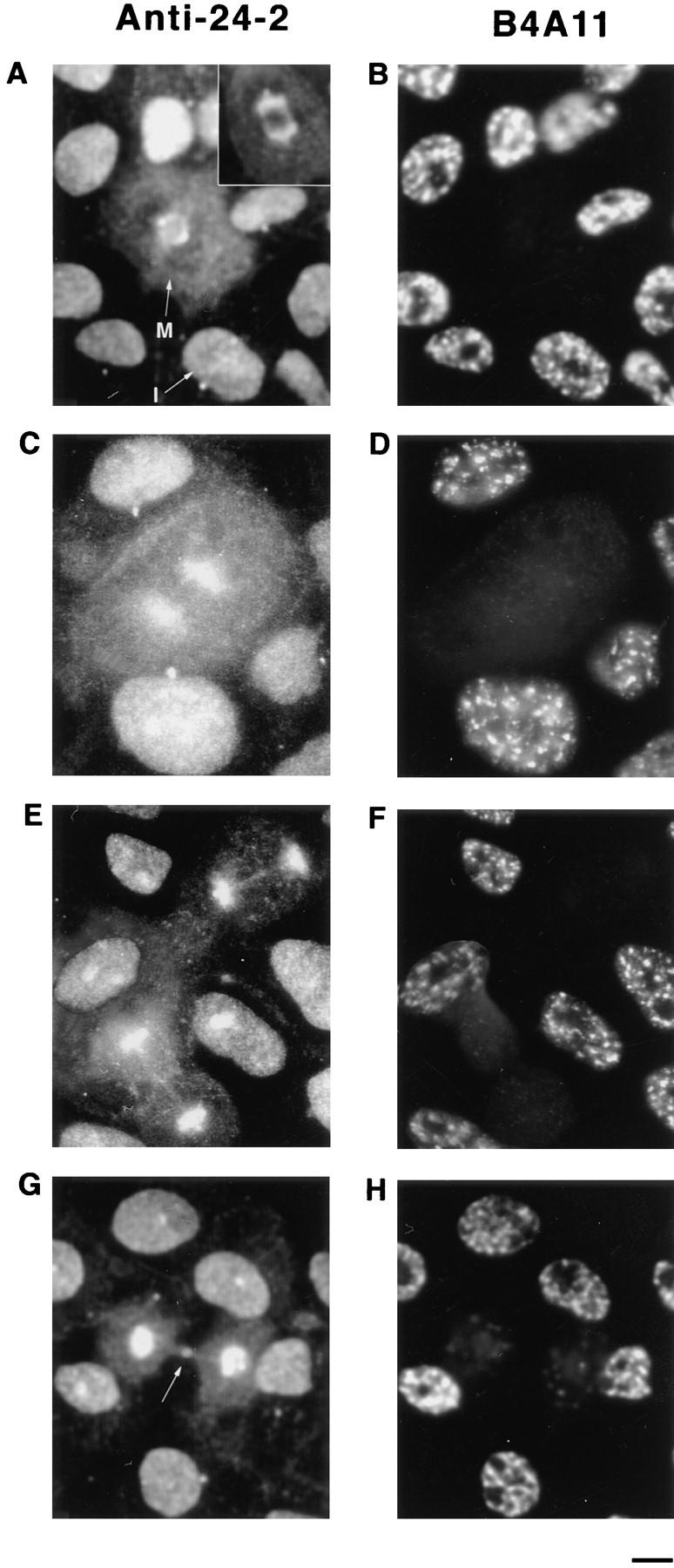
Dynamic redistribution of protein 4.1 antigens during the cell cycle. CaSki cells were permeabilized with 0.5% Triton X-100 in CSK buffer to remove membranes and soluble proteins before formaldehyde fixation, incubated with DNase I, and extracted with 0.25 M ammonium sulfate. The cell preparations were immunostained with protein 4.1 antibody 24-2 (A, C, E, and G) and with B4A11 (B, D, F, and H), a monoclonal antibody against a nuclear matrix protein that displays an intense nuclear speckle pattern at interphase but is not detectable at mitosis (Blencowe et al., 1994). Micrograph pairs show the fluorescent pattern with anti–24-2 (left) and B4A11 (right) of the same fields. Cell cycle stages were also confirmed by viewing cells using phase contrast microscopy (not shown). (A) In the center, a mitotic cell (M) showed staining of the mitotic spindle with particularly strong staining of the spindle poles. The mitotic cell is surrounded by interphase cells. Note that at opposite sides of an interphase nucleus (I), immunostained centrosomes (small spots) are visible. The inset shows the mitotic spindle of another cell intensely immunolabeled by anti–10-1. (B–F) Epitopes for B4A11 have disappeared in mitotic cells, but a strong speckled staining pattern is present in interphase nuclei. (C) In the center, a cell in anaphase retained a high degree of 4.1 staining in the area of the condensed chromosomes. (E) As the cells approached telophase and cytokinesis, the bridge between the intensely stained perichromosomal regions became visible by antibody deposition. As the daughter cells separated further apart (G), bright 4.1 staining appeared at the midbody (arrow). In the companion B4A11 fields, diffuse staining began to condense into a more focal pattern, foreshadowing the appearance of the prominent B4A11 speckles characteristic of interphase cells. Bar, 5 μm.
Figure 8.



Expression of epitope-tagged 4.1 in nuclei after transient transfection. A construct was engineered to encode the sequences for red cell 80-kD protein 4.1 fused to an epitope tag derived from SV-40 large T antigen. (A) The construct was bacterially expressed, isolated, and then analyzed by Western blotting to confirm the presence of both 4.1 and SV-40 tag epitopes. Both 24-2 IgG (lane 1) and KT3 antibody (against the epitope tag; lane 2) recognized a protein with the same apparent molecular mass. The KT3 antibody did not recognize epitopes in a whole cell lysate of 3T3 cells (lane 3). (B) Murine fibroblast 3T3 cells, probed with anti-RBC 80-kD 4.1, displayed punctate nuclear immunofluorescent signals. (C and C′) 3T3 cells, transiently transfected with pSV40NeoCMV containing sequences encoding epitope-tagged RBC 80-kD 4.1, strongly expressed epitope-tagged protein localized in nuclei, which was detected by indirect immunofluorescence using KT3 antibody. (D and D′) After parallel transient transfection of 3T3 cells with pSV40NeoCMV without a construct inserted, there was no immunofluorescent staining with KT3 antibody. Bar: (B and B′) 12 μm; (C–D′) 20 μm.
The nucleus is bounded by two concentric lipid bilayers perforated by nuclear pores and an underlying ring of lamin, a specialized intermediate filament. Nuclear-associated 4.1 immunofluorescent signals could have arisen from cytoplasmic epitopes overlying the nucleus, from nuclear membrane sites, or from interior nuclear locations. In fact, since 80-kD protein 4.1 in mature erythrocytes is a component of the plasma membrane skeleton (Fig. 4), one might predict that 4.1 would localize to the nuclear membrane or even the underlying lamin protein ring. To distinguish among these possibilities, double-label confocal microscopy was performed to localize 4.1 foci relative to epitopes for two well-characterized nuclear proteins, lamin B and the nuclear pore complex. Optical horizontal sections through the center of the nucleus revealed well-separated rhodamine (Fig. 5 A, stain for lamin; Fig. 5 B, stain for pores) and FITC (stain for 4.1) signals, leading to the conclusion that 4.1 epitopes are within the area bounded by nuclear pores embedded in the nuclear membrane and by the underlying lamin protein ring. Furthermore, the smooth circumferential staining of both pores and lamin in the preparations rules out the possibility that internal 4.1 foci are being generated by invaginations of the nuclear membrane.
Figure 5.
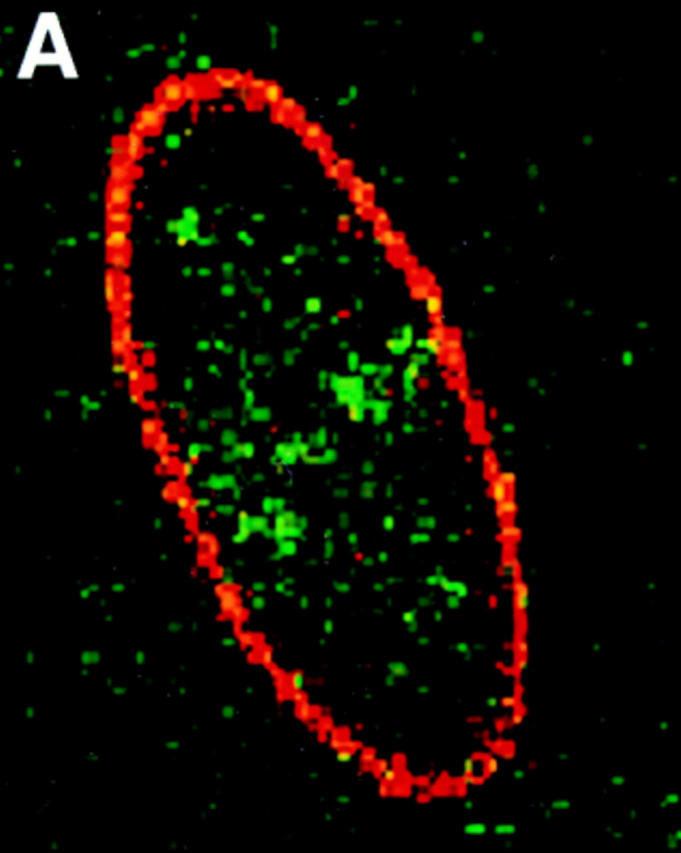
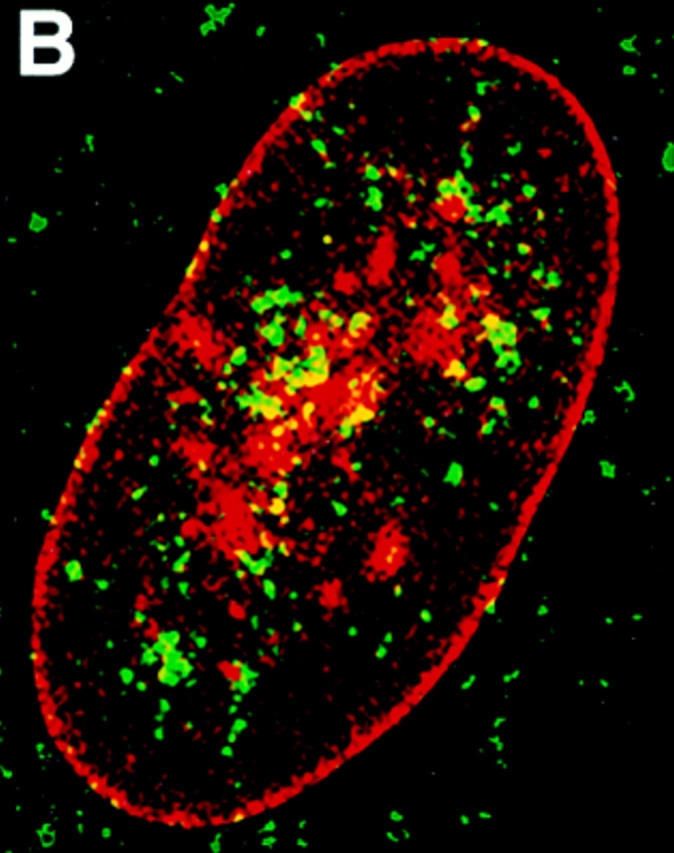





Distribution of protein 4.1 epitopes in the interphase nucleus of human fibroblasts visualized by immunofluorescence. Location of the nucleus was confirmed in all cases by DAPI staining (not shown). (A and B) Double-label confocal microscopy of cells stained by protein 4.1 antibody 24-2 (green) and (A) antibody against nuclear pores (red) or (B) lamin B (red). The majority of nuclear protein 4.1 foci are interior to the periphery of the nucleus as demarcated by immunofluorescence of pores within nuclear membrane and by its subadjacent network of lamin B fibers. (Additional lamin B sites in the nuclear interior have been reported [Bridger et al., 1993; Moir et al., 1994]). Similar results were obtained by imaging epitopes for protein 4.1 antibodies 24-3, 10-1, and N-2 relative to antipore and antilamin epitopes. (C) Three-dimensional image analysis of nuclear epitopes probed with anti-RBC 80-kD 4.1 showing a 0.3-μm horizontal optical midsection of an immunofluorescent cell with a line indicating the position of the vertical reconstruction depicted in C′. Since optical resolution in the z-axis (vertical) is less than in the x-y (horizontal) axis, the reconstructed image is less sharp; the dome shape of the top of the cell and the flat bottom plane of the coverslip are somewhat obscured by the coincidence of out of focus light. (A cartoon depicting the approximate shape of the vertical cross-section through the nucleus is presented in the bottom panel.) Thus, it is apparent that epitopes for protein 4.1 lie in numerous planes within the volume of the cell. Similar results were obtained in three-dimensional reconstructions using the 24-2, 24-3, and 10-1 antibodies as probes. (D and D′) Double-label confocal microscopy of a fibroblast using anti–24-2 (green) and anti-NuMA (red). In a horizontal plane through the midsection of the cell (D), protein 4.1 foci appeared within the NuMA-stained nuclear interior. (D′) Three-dimensional reconstructions were made of three different vertical planes from this NuMA:24-2 double-labeled cell. The flattened plane of the coverslip appears at the top of the vertical reconstructed images. The conclusion that 4.1 epitopes localized at multiple planes throughout the volume of the nucleus is entirely consistent with the observations obtained in images A, B, and C. The same conclusion was reached when cells were probed with antibodies against NuMA and RBC 80-kD protein 4.1. (E and F) Double-label confocal microscopy of cells probed with antibody 24-2 (green) and antibody against PCNA (red). Optical sections through the fibroblast nucleus show that 24-2 (green) and PCNA (red) epitopes coincided (yellow) in many areas except in the vicinities of the nucleoli, which either are relatively devoid of any signal or sometimes contained a clustering of 24-2 (green) signals. Both PCNA and 24-2 epitopes resided in multiple planes throughout the nucleus (not shown). (G and H) Double-label confocal microscopy of cells probed with antibody 24-2 (green) and antibody against SC-35 (red). Optical sections are in the plane of the cell containing SC-35 domains; most SC-35 domains in fibroblasts are located in a single plane (Carter et al., 1993). It is apparent that the vast majority of the SC-35 domains contained areas of coincident staining with 24-2 (seen as yellow coloration), most often at the periphery of the SC-35 domains. In optical sections above and below SC-35, additional 24-2 signals are observed (not shown), consistent with the conclusions from the localization data presented in A–F. Bars, 3.5 μm (8 μm in the vertical dimension [C′ and D′]).
To further characterize the distribution of 4.1 within the nucleus, fibroblast nuclei stained with IgGs against 4.1 peptide domains were optically sectioned into horizontal planes using conventional and confocal immunofluorescence microscopy, and the information was digitized. Using image analysis software, three-dimensional reconstructions were made through vertical planes of the nuclear interior. As illustrated in Fig. 5 C, 4.1 foci detected in horizontal planes are at multiple levels within the nucleus when analyzed by vertical cuts through horizontal layers (Fig. 5 C′). This observation was confirmed by a doublelabel experiment in which nucleoplasm, highlighted by rhodamine with an antibody probe against a generally distributed nuclear matrix protein NuMA (nuclear mitotic apparatus protein; Lydersen and Pettijohn, 1980; Compton et al., 1992; Yang et al., 1992), was imaged relative to 4.1 epitopes detected by FITC labeling (Fig. 5 D). The confocal optical vertical sections show 4.1 epitopes to be scattered throughout the nucleoplasm (Fig. 5 D′).
To determine whether 4.1 foci colocalize with nuclear domains involved with pre-mRNA splicing factors or with DNA replication, additional double-label immunofluorescence experiments were performed. Nuclear 4.1 epitopes were imaged relative to SC-35 splicing protein (Fu and Maniatis, 1992) and relative to proliferating cell nuclear antigen (PCNA), an accessory protein for the replicative DNA polymerase delta (Tan et al., 1986; Bravo et al., 1987; Prelich et al., 1987) (Fig. 5, E–H). Although some coincident fluorescent signals (denoted by pseudo yellow coloration) were apparent in confocal horizontal planes through fibroblast nuclei stained for 4.1 (probed with FITC) relative to either SC-35 or to PCNA (each probed with rhodaminelabeled secondary antibody), each protein was also detected alone. This is consistent with previous observations that splicing and replication centers do not coincide with each other (Leonhardt et al., 1992). Protein 4.1 foci were not detected in areas occupied by nucleoli, seen as dark round “holes” within the nucleus (Fig. 5 E). These data show 4.1 epitopes to be generally distributed throughout nonnucleolar nuclear domains involved in RNA and DNA metabolism. Taken together, it is clear from the data in Fig. 5 that 4.1 foci are intranuclear but do not localize predominantly at the nuclear membrane.
Identification of Protein 4.1 in Nuclear Matrix
Since 4.1 is an important structural protein in red cells, we hypothesized that isoforms in the nucleus might biochemically cofractionate with nonchromosomal nuclear scaffold proteins. To test this prediction, purified nuclear matrix was prepared by established techniques using sequential permeabilization, salt extraction, nuclease digestion, and salt reextraction from cells grown on coverslips and also from purified fibroblast nuclei (Capco et al., 1982; Fey et al., 1986). Fibroblasts and CaSki cells retained epitopes recognized by 80-kD RBC 4.1, 10-1, 24-2, and 24-3 antibodies when monitored by immunofluorescence during each stage of nuclear matrix preparation. When analyzed by threedimensional optical reconstructions, all extraction steps before nuclease digestion displayed nuclear foci at multiple planes. After completion of the matrix preparation, there was no detectable staining with DAPI, attesting to the near complete removal of chromatin. In contrast, immunofluorescent foci of 4.1 were retained in the matrix preparation. Microscopic optical sectioning of these samples showed a collapsed nucleus so that, in contrast to previous extraction steps, all nuclear 4.1 epitopes resided on a single plane.
Electron microscopy was used to achieve greater resolution in localizing 4.1 epitopes in the nucleus. Cells growing on coverslips were prepared for embedment-free (“resinless”) section electron microscopy, a technique developed specifically to image three-dimensional cytoplasmic and nuclear structures normally concealed in conventionally embedded samples (Capco et al., 1984; Nickerson and Penman, 1992; Nickerson et al., 1994). Samples were sequentially extracted, lightly fixed, and incubated with primary IgGs against 4.1 peptides or normal rabbit IgG. A secondary antibody coupled to gold beads was added, and the preparation was fixed again and then embedded and sectioned. After removal of the temporary embedding material, epitopes for 4.1 were detected within the residual nuclear matrix structures visualized by electron microscopy (Fig. 6), consistent with the results obtained with immunofluorescence assays run in parallel samples. At the higher resolution of electron microscopy, the 4.1 epitopes retained in nuclear matrix preparations were associated with dense bodies and not with the supporting filamentous network. Parallel samples incubated with control IgG did not show labeling of nuclear structures.
Figure 6.

Immunolocalization of protein 4.1 in nuclear matrix visualized by high-resolution resinless electron microscopy. CaSki cells were extracted with 0.5% Triton X-100 in CSK buffer, chromatin was removed by HaeIII and PstI digestion, and proteins were extracted with 0.25 M ammonium sulfate followed by extraction with 2 M NaCl. After fixation, the matrix preparation was incubated with protein 4.1 antibody 24-3 (A) and 24-2 (B) and then with colloidal gold–coupled secondary antibody. The EM micrograph of a resinless section shows a network of nuclear matrix core filaments (CF) and dense bodies (DB). The 10-nm gold beads decorated principally the periphery and some internal areas of the dense bodies (arrowheads). This localization pattern at dense bodies in the matrix was also observed in a parallel experiment using anti–10-1 IgG but not with control IgG. Bar, 100 nm.
Western Blot Analysis of Nuclear Matrix Proteins
To analyze nuclear-associated 4.1 isoforms, nuclear matrix proteins isolated from purified fibroblast nuclei were separated on SDS-PAGE and transferred to nitrocellulose, and identical gel lanes were probed with each of the 4.1 antibodies. A number of protein bands in the matrix preparation were reactive with multiple 4.1 IgGs. (Fig. 7 A, lanes 1–5). Prominent clusters of bands migrating with molecular masses in the range of ∼55 and 80–100 kD were detected. Other less abundant bands also reacted with antibodies against several different protein 4.1 epitopes: ∼65 kD (antibodies 24-2, 24-3) and ∼120–150 kD (antibodies 10-1, 24-3, 4.1). No reaction with the nuclear matrix proteins occurred when control rabbit IgG was used as a probe (Fig. 7 A, lane 6). Thus multiple isoforms of 4.1 are retained in the most insoluble fraction of nuclear proteins.
Figure 7.


Analysis of fibroblast nuclear matrix 4.1 protein by Western blot and 4.1 mRNA by reverse transcriptase–PCR. (A, lanes 1–6) Nuclear matrix proteins isolated from 3 × 106 WI38 nuclei were separated by SDSPAGE, transferred to nitrocellulose, and incubated with the following 4.1 antibodies: lane 1, RBC 80-kD 4.1; lane 2, N-2; lane 3, 10-1; lane 4, 24-2; lane 5, 24-3; lane 6, control IgG. To test the relative separation of nuclear matrix proteins from soluble cytoplasmic proteins, a cytoplasmic fraction from 2 × 105 cells was electrophoresed in parallel with a sample of nuclear matrix from 6 × 105 cells derived from the same fibroblast preparation and probed after transfer with an antibody against the cytoplasmic protein β-tubulin. While β-tubulin was abundant in the cytoplasmic fraction (lane 7), no protein band with a similar migration could be detected in the nuclear matrix fraction (lane 8) even after long exposure times. An antibody against lamin B used to probe fibroblast nuclear matrix proteins produced a band at the appropriate position, verifying the presence of a predicted nuclear matrix protein (lane 9). (B) PCR analysis of protein 4.1 mRNA containing AUG-1 in WI38 fibroblasts. The products of reverse transcriptase–PCR using primers encompassing AUG-1 were analyzed on a polyacrylamide gel. The following cDNAs were amplified: lane 1, WI38 cells; lane 2, human reticulocytes; lane 3, no DNA. The sizes of molecular weight standards are: 1353, 1078, 872, 603, 310, 234, and 194 bp.
To verify that the blotted proteins contained characteristic nuclear matrix proteins, a lane was probed with antibody against a major nuclear matrix protein, lamin B. A single band at the expected molecular mass of 66 kD was detected (Fig. 7 A, lane 9). To assess cross-contamination of nuclear matrix by cytoplasmic proteins, lanes containing cytoplasmic and nuclear matrix fractions were probed with antibody against the cytoplasmic protein β-tubulin (Fig. 7 A, lanes 7 and 8); β-tubulin could be detected only in the cytoplasmic fraction (Fig. 7 A, lane 7) and not in the lane containing nuclear matrix (Fig. 7 A, lane 8).
In both immunofluorescence and Western blotting experiments with antibody N-2, the results reported above suggest that 4.1 isoforms initiated at the upstream translation initiation site AUG-1 are expressed in fibroblasts. As an independent means of confirming the existence of such isoforms in fibroblasts, 4.1 mRNA was selectively amplified by reverse transcriptase–PCR techniques using primers designed to detect the AUG-1 sequence. As shown in Fig. 7 B, WI38 fibroblast RNA encodes AUG-1, consistent with the N-2 antibody results.
Furthermore, after Western blotting of nuclear matrix using 24-2 and 24-3 IgGs, we detected several protein bands with similar apparent molecular masses as well as other bands not in common (Fig. 5, lanes 4 and 5). This result predicted nuclear 4.1 isoforms with differing combinations of the 24-2 and 24-3 epitopes. Using preparations of 24-2 and 24-3 antibody each directly labeled with a different fluorophore, double-label immunofluorescent staining of fibroblasts revealed coincident signals in the nucleus along with some noncoincident staining for each antibody (data not shown). This result is consistent with data from Western blot analysis of nuclear matrix proteins. Taken together, immunofluorescent and Western blot data suggest that there are multiple nuclear 4.1 isoforms or homologues and that protein 4.1 fractionates with nuclear matrix proteins.
Expression of Epitope-tagged 4.1 in the Nucleus
To test for protein 4.1 in the nucleus by a second experimental strategy, a construct encoding the prototypical 80-kD protein 4.1 (Conboy et al., 1991; Tang et al., 1988, 1990) fused to an epitope tag was transiently transfected into murine fibroblast 3T3 cells. Before transfection, we determined that the protein expressed by the epitopetagged construct was recognized on Western blot analysis both by antibodies against 4.1 (24-2 IgG, Fig. 8 A, lane 1; 10-1 IgG, data not shown) and KT3 antibody directed against the epitope tag (Fig. 8 A, lane 2). As expected, the KT3 antibody did not react with proteins in extracts of the recipient host fibroblasts (Fig. 8 A, lane 3). To confirm the presence of endogenous nuclear 4.1 in murine fibroblasts, 3T3 cells were probed with anti-RBC 80-kD 4.1. The 3T3 nuclei displayed punctate immunofluorescent 4.1 signals (Fig. 8 B) similar in pattern to diploid human fibroblasts (Fig. 3). After transient transfection with the vector encoding the 4.1-tagged construct, expressed protein bearing the epitope tag produced a very strong immunofluorescent signal in the nuclei of the 3T3 cells (Fig. 8, C and C′) when probed with KT3 antibody. Of note, in parallel experiments, no immunofluorescent signal was detected when cells were transfected with the vector not containing the construct (Fig. 8, D and D′) or in mock (i.e., no vector DNA) transfected cells (data not shown).
Dynamic Rearrangement of 4.1 Epitopes during the Cell Cycle
During mitosis, the nucleoskeleton is dismantled, a mitotic spindle is assembled, and chromatin is condensed, packaged, and partitioned into daughter nuclei. Nuclear structural proteins change their subcellular localizations and some vary their roles during these events. The rearrangements of protein 4.1 epitopes as cells enter mitosis were determined in rapidly proliferating CaSki cells. Cells were labeled with antibody probes to 4.1 peptide epitopes (Fig. 9, A, C, E, and G) and with B4A11, a monoclonal antibody against a nuclear matrix protein associated with a subset of RNA splicing factor storage/assembly sites (Fig. 9, B, D, F, and H) (Blencowe et al., 1994). B4A11 epitopes produce a strong speckled staining pattern in interphase nuclei that almost completely disappears in detergentextracted cells undergoing mitosis (Blencowe et al., 1994); cells undergoing mitosis were thus identifiable by the loss of B4A11 epitopes along with altered morphology.
Antibodies to 4.1 epitopes exhibited a pattern of diffuse nuclear staining with dispersed intense fluorescent foci within interphase nuclei (Fig. 9 A, arrow I; see also C, E, and G) and stained structures resembling centrosomes in the perinuclear region (Fig. 9 A, below arrow I). This pattern dramatically altered during metaphase, when 4.1 antibodies intensely stained the spindle and spindle pole of the mitotic apparatus (Fig. 9 A, arrow M and inset) and diffusely stained the outlying areas of the dividing cell. As the spindle poles separated, 4.1 epitopes clustered around two distinct perichromosomal regions beginning to egress toward each daughter cell (Fig. 9, C, E, and G). During telophase, 4.1 antigenic material formed an intensely staining mass in each nascent nucleus and the midbody region in the vestigial connection between the daughter cells showed a distinct band of fluorescence. B4A11 epitopes, which had completely disappeared during mitosis (Fig. 9 B), began to form larger foci during telophase (Fig. 9 H) preceding movement into maturing interphase nuclei. The patterns of 4.1 epitope distribution during the cell cycle were reproducibly observed in cell populations partially synchronized by a thymidine block. Thus, throughout the cell cycle, 4.1 epitopes remain accessible to antibody binding but display a complex pattern of rearrangements, migrating from intranuclear sites after interphase to regions associated with the mitotic spindle, perichromosomal areas, and the midbody.
Discussion
While mature red cells contain predominantly an 80-kD 4.1 isoform, multiple protein 4.1 isoforms are expressed in nucleated erythroid and nonerythroid cells. Combinatorial alternative splicing of 4.1 mRNAs could in principle generate many 4.1 isoforms that participate in distinct sets of molecular interactions. This could provide mechanisms for differential cellular localization of 4.1 isoforms as well as isoform functional specificity. There are numerous precedents for a role of alternative splicing in isoform-specific localization. Among membrane skeletal proteins, for example, band 3 isoforms with different 5′ ends generated by alternative transcription initiation localize either to peripheral membranes or to perinuclear regions (Cox et al., 1995). Similarly, two β-spectrin isoforms having different carboxy termini appear to interact with different binding partners and exhibit different cellular localization (Malchiodi-Albedi et al., 1993). Alternative splicing of CaM kinase generates a nuclear localization signal that targets a specific isoform of this multifunctional protein to the nucleus (Srinivasan et al., 1994). Alternative splicing of WT1 determines if a WT1 isoform associates within the nucleus mainly with RNA splicing factors or with DNA in transcription factor domains (Larsson et al., 1995).
The focus of the present investigations has been to explore in depth the 4.1-immunoreactive epitopes in the nucleus. A currently evolving view is that the nucleus contains a highly structured internal skeletal lattice that can organize chromosomes and numerous other nuclear components into physical and functional subdomains. At a biochemical level, this internal nuclear structure could serve as a nonsoluble integrator of nuclear metabolic reactions (Nickerson et al., 1995). Isoforms of protein 4.1 may contribute to nuclear structure. The nucleus houses potential binding partners for 4.1, such as spectrin (Bachs et al., 1990; Beck et al., 1994), actin (Nakayasu and Ueda, 1986; Milankov and DeBoni, 1993; Chaly, N., and X. Chen. 1995. Mol. Biol. Cell. 6:423a), myosin (Berrios and Fisher, 1986; Hagen et al., 1986; Milankov and DeBoni, 1993), and calmodulin (Bachs et al., 1990). Moreover, protein 4.1 is known to be glycosylated and phosphorylated in vivo, posttranslational modifications used by nuclear proteins to modulate interactions during the cell cycle.
An important strategy was to use a panel of affinitypurified antipeptide IgGs against distinct areas of the 4.1 polypeptide sequence as well as traditional IgG preparations generated against red cell 80-kD 4.1. The peptides were selected in part because of their evolutionary conservation and in part because they are encoded in different functional domains of 4.1. Each antibody preparation reacted with red cell 80-kD 4.1 by Western blotting of human red cell membranes and immunofluorescent staining of human red cells. Members of the antibody panel consistently showed immunoreactivity with fibroblast nuclear structures using multiple immunofluorescent protocols, by Western blot analysis and immunoelectron microscopy. To avoid potential immunological cross-reactivity with known members of the 4.1 superfamily (ezrin, moesin, radixin, merlin/schwannonin, talin, and certain tyrosine phosphatases), antibodies used in this study were directed against domains of 4.1 not shared with these related proteins. Thus, the 4.1 epitopes located in nuclear regions should identify authentic 4.1 isoforms or very highly related homologues yet to be discovered.
However, to further verify by an independent approach that protein 4.1 can localize to the nucleus, fibroblasts were transiently transfected with a vector encoding the 80-kD red cell 4.1 sequence fused to an epitope tag. This 4.1 isoform coding sequence was selected for a number of reasons: (a) It encodes a well-characterized full-length 4.1 isoform; (b) antibody against this sequence produced strong nuclear immunofluorescent signals in human and murine fibroblasts; (c) protein bands migrating ∼80 kD were detected in nuclear matrix Western blots; and (d) this sequence contains a potential nuclear localization signal (De Carcer et al., 1995). This transfection experiment allowed the unequivocal demonstration that an individual 4.1 isoform of known structure can be localized to the nucleus.
Since 4.1 in red cells mechanically links the spectrin and actin membrane skeleton to the overlying lipid bilayer, we explored its intranuclear localization to test the idea that nuclear 4.1 might associate with the nuclear membrane– lamin region. By reconstructing three-dimensional optical sections through the nucleus, we found that 4.1 epitopes are not predominantly colocalized with nuclear membrane pores or with the underlying fibrous lamin network but are distributed throughout the interior of the nucleus.
Additional double-label experiments were performed to test for colocalization of 4.1 with known intranuclear structural or functional domains. The 4.1 distribution is complex and does not precisely colocalize with PCNA. Doublelabel confocal microscopy provided evidence for a potential association of 4.1 with SC-35–enriched splicing assemblies, which in fibroblasts are concentrated in a plane just below the midline, parallel to the growth surface (Carter et al., 1993). This potential association, however, is not exclusive: 4.1 foci are detected at lower and upper regions of the nucleus and some 4.1 signals are noncoincident even within the spliceosomal plane. Taken together, these data may represent a composite of several functionally distinct 4.1 protein isoforms or could suggest a more unified role for protein 4.1 at interfaces between the nucleoskeleton and subnuclear assemblies such as centers for splicing or DNA replication. However, many nuclear subdomains, including DNA replication foci, change in size and distribution during S phase (D'Andrea et al., 1983; Goldman et al., 1984; Hatton et al., 1988; O'Keefe et al., 1992), and it is not known whether protein 4.1 associations are static or dynamic during interphase. Therefore, further studies using tightly synchronized cell cultures and specific inhibitors will be required to quantitate the spatial associations of 4.1 with these key nuclear functional domains.
After extensive biochemical extraction, 4.1 epitopes were detected in nuclear matrix preparations by both immunofluorescence light microscopy and immunogold electron microscopy. At the higher resolution of electron microscopy, gold bead–coupled antibodies did not cluster preferentially near the nuclear pore–lamina complex, consistent with our double-label immunofluorescent confocal data. Rather, gold beads were distributed in the vicinity of dense bodies within the internal fibrous nuclear scaffold network. This observation is also entirely consistent with the double-label immunofluorescence experiments showing that 4.1 foci appeared at positions within the nuclear area stained by NuMA but were not primarily coincident. At the level of resinless electron microscopy, NuMA antibodies have been seen to decorate subsets of filaments in nuclear matrix preparations (Zeng et al., 1994); we observed 4.1 epitopes near dense bodies rather than associated with the surrounding filaments. The biochemical composition of these dense bodies within the matrix is not entirely understood. Some contain various combinations of RNA splicing factors corresponding to speckles seen in immunofluorescence but others appear to be related to centers of transcription and DNA replication, coiled bodies, RNA–protein complexes, and nucleolar remnants (for review see Nickerson et al., 1995). It remains to be established if 4.1 localization is specific for subclasses of dense bodies or is more generally associated with a variety of nuclear substructures.
Nuclear matrix proteins, isolated from purified fibroblast nuclei under extraction conditions identical to those used for immunofluorescence microscopy, revealed several polypeptides that reacted with multiple anti-4.1 IgGs in Western blots. We speculate that the complex pattern of 4.1 expression results from splicing-mediated inclusion or exclusion of various internal peptides as well as posttranslational modifications such as phosphorylation (Horne et al., 1990; Subrahmanyam et al., 1991) and/or glycosylation (Haltiwanger et al., 1990). Some additional variation may be generated by expression of alternative NH2 or COOH termini not yet characterized. For example, the lack of significant reaction of protein 4.1 antibody 24-3 with the ∼85-kD band suggests that the sequence of COOH termini may vary. Similarly, the N-2 IgG prepared against the 209–amino acid NH2-terminal extension of 80-kD 4.1 detected only a single band in the 80–100 kD region with the slowest migration (∼100 kD), indicating that apparently not all matrix polypeptides have identical NH2 termini.
One of the bands in the ∼80–100-kD cluster may be similar to the 4.1p75 polypeptide observed by De Carcer et al. (1995) using an alternative method of nuclear extraction and an independently generated 4.1 antibody. More extensive alternative splicing or usage of new unidentified promoters may be responsible for lower molecular mass bands. The broad band in the nuclear matrix preparation migrating at ∼55 kD reacted with all the 4.1 IgGs tested. This pattern could represent an authentic 4.1 isoform that is the product of an as yet unrecognized 4.1 alternative splicing pathway, or it could represent a new 4.1 homologue. For example, in the ankyrin membrane skeletal protein family, a novel small isoform with a truncated membrane binding domain has been identified in Golgi (Devarajan et al., 1996). In sum, our final matrix fraction contains multiple 4.1 isoforms, some with distinct immunoreactive patterns. This suggests that 4.1 polypeptides have differing peptide domains or modifications that may enable unique molecular interactions in the nucleus.
This speculation is particularly intriguing because during mitosis, 4.1 epitopes display dynamic rearrangements within the nuclear microarchitecture that are correlated with cell division events: at interphase they reside throughout the nucleus, then accumulate at the spindle during metaphase, coalesce around condensed chromatin during telophase, and some remain at the midbody region during cytokinesis. Many nuclear-derived components redistribute at the time of nuclear membrane dissolution to several structural zones: the chromosome scaffold, the perichromosomal region, the centrosome/kinetochore region, midbody, and spindle poles (for review see He et al., 1995). Some nuclear structural proteins, such as lamin, even migrate into the cytoplasm to be later reimported into newly formed nuclei. The 4.1 pattern appears to be a unique hybrid of several patterns reported for specific matrix-associated proteins (for review see He et al., 1995).
The recent elucidation of an isoform kindred for structural protein 4.1 suggests uncharted roles in addition to its traditionally defined function in the erythroid plasma membrane skeleton. As a structural component of the nucleus, protein 4.1 presents a new challenge to decipher mechanisms underlying its targeting to nuclear subdomains, to identify its localized molecular interactions, and to understand signals orchestrating its dynamic redistribution during the cell cycle. The versatile rearrangements of 4.1 epitopes during cell division could reflect the disposition of individual isoforms with unique structural assignments and patterns of expression. Ultimately, protein 4.1 may contribute fundamentally to modulation of the cell cycle and possibly to more specialized phenomena such as apoptosis or mammalian erythroid enucleation.
Acknowledgments
This work was supported by grant DK 32094 from the National Institutes of Health.
Footnotes
1. Abbreviations used in this paper: CMV, cytomegalovirus; DAPI, 4′6- diamidino-2-phenylindole; GST, glutathione-S-transferase; NuMA, nuclear mitotic apparatus protein; PCNA, proliferating cell nuclear antigen.
We are very grateful to Ms. Gabriela Krockmalnic (Massachusetts Institute of Technology, Cambridge, MA) for her expertise in providing the resinless section electron micrographs of nuclear matrix, to Ms. Gloria Lee for red cell experiments, and to Ms. Marilyn Parra for engineering the 4.1 epitope-tagged construct. We particularly appreciate the important suggestions of Dr. John Conboy. We thank Drs. Judith Campisi and Jeffrey A. Nickerson for many valuable discussions during the course of this work. Ms. Alicia Sheppard and Mr. Derek Clark were most helpful in the preparation of this manuscript and its figures.
Address all correspondence to Sharon Wald Krauss, University of California, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, MS 74-157, Berkeley, CA 94720. Tel.: (510) 486-6439. Fax: (510) 486-6746.
References
- Anderson RA, Correas I, Mazzucco C, Castle JD, Marchesi VT. Tissue-specific analogues of erythrocyte protein 4.1 retain functional domains. J Cell Biochem. 1988;37:269–284. doi: 10.1002/jcb.240370303. [DOI] [PubMed] [Google Scholar]
- Bachs O, Lanini L, Serratosa J, Coll MJ, Bastos K, Alique R, Ruis E, Carafoli E. Calmodulin binding proteins in the nuclei of quiescent and proliferatively activated rat liver cells. J Biol Chem. 1990;265:18595–18599. [PubMed] [Google Scholar]
- Beck KA, Buchcanan JA, Malhotra V, Nelson WJ. Golgi spectrin: identification of an erythroid β spectrin homolog associated with the Golgi complex. J Cell Biol. 1994;127:707–723. doi: 10.1083/jcb.127.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrios M, Fisher PA. A myosin heavy-chain-like polypeptide is associated with the nuclear envelope in higher eukaryotic cells. J Cell Biol. 1986;103:711–724. doi: 10.1083/jcb.103.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blencowe BJ, Nickerson JA, Issner R, Penman S, Sharp PA. Association of nuclear matrix antigens with exon-containing splicing complexes. J Cell Biol. 1994;127:593–607. doi: 10.1083/jcb.127.3.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo R, Franke R, Blundell PA, MacDonald-Bravo H. Cyclin/ PCNA is the auxiliary protein of DNA polymerase δ. Nature (Lond) 1987;326:515–517. doi: 10.1038/326515a0. [DOI] [PubMed] [Google Scholar]
- Bridger JM, Kill IR, O'Farrell M, Hutchison CJ. Internal lamin structures within G1 nuclei of human dermal fibroblasts. J Cell Sci. 1993;104:297–306. doi: 10.1242/jcs.104.2.297. [DOI] [PubMed] [Google Scholar]
- Capco DG, Wan KM, Penman S. The nuclear matrix: three-dimensional architecture and protein composition. Cell. 1982;29:847–858. doi: 10.1016/0092-8674(82)90446-9. [DOI] [PubMed] [Google Scholar]
- Capco DG, Krochmalnic G, Penman S. A new method of preparing embeddment-free sections for transmission electron microscopy: applications to the cytoskeletal framework and other three-dimensional networks. J Cell Biol. 1984;98:1878–1885. doi: 10.1083/jcb.98.5.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo-Fonseca M, Pepperkok R, Sproat ES, Ansorge W, Swanson MS, Lamond AI. In vivo detection of snRNP-rich organelles in the nuclei of mammalian cells. EMBO (Eur Mol Biol Organ) J. 1991;10:1163–1173. doi: 10.1002/j.1460-2075.1991.tb07712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter KC, Bowman D, Carrington W, Fogarty K, McNeil JA, Fay FS, Lawrence JB. A three dimensional view of precursor messenger RNA metabolism within the mammalian nucleus. Science (Wash DC) 1993;259:1330–1335. doi: 10.1126/science.8446902. [DOI] [PubMed] [Google Scholar]
- Chasis JA, Coulombel L, Conboy J, McGee S, Andrews K, Kan YW, Mohandas N. Differentiation-associated switches in protein 4.1 expression: synthesis of multiple structural isoforms during normal human erythropoiesis. J Clin Invest. 1993;91:329–338. doi: 10.1172/JCI116189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chasis JA, Coulombel L, McGee S, Lee G, Tchernia G, Conboy J, Mohandas N. Differential use of protein 4.1 translation initiation sites during erythropoiesis: implications for a maturation-induced stage-specific deficiency of protein 4.1 during erythroid development. Blood. 1996;87:5324–5331. [PubMed] [Google Scholar]
- Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- Cohen CM, Foley SF, Korsgen C. A protein immunologically related to erythrocyte band 4.1 is found on stress fibers of non-erythroid cells. Nature (Lond) 1982;299:648–650. doi: 10.1038/299648a0. [DOI] [PubMed] [Google Scholar]
- Compton DA, Szilak I, Cleveland DW. Primary structure of NuMA, an intracellular protein that defines a novel pathway for segregation of proteins at mitosis. 1992. J Cell Biol. 1992;16:1395–1408. doi: 10.1083/jcb.116.6.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy JG. Structure, function and molecular genetics of erythroid membrane skeletal protein 4.1 in normal and abnormal red blood cells. Semin Hematol. 1993;30:58–73. [PubMed] [Google Scholar]
- Conboy J G, Chan J, Mohandas N, Kan YW. Multiple protein isoforms produced by alternative splicing in human erythroid cells. Proc Natl Acad Sci USA. 1988;85:9062–9065. doi: 10.1073/pnas.85.23.9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy JG, Chan J, Chasis JA, Kan YW, Mohandas N. Tissue- and development-specific alternative RNA splicing regulates expression of multiple isoforms of erythroid membrane protein 4.1. J Biol Chem. 1991;266:8273–8280. [PubMed] [Google Scholar]
- Conboy JG, Chasis JA, Winardi R, Tchernia G, Kan YW, Mohandas N. An isoform-specific mutation in the protein 4.1 gene results in hereditary elliptocytosis and complete deficiency of protein 4.1 in erythrocytes but not in nonerythroid cells. J Clin Invest. 1993;91:77–82. doi: 10.1172/JCI116203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook P. The nucleoskeleton: artifact, passive framework or active site. J Cell Sci. 1988;90:1–6. doi: 10.1242/jcs.90.1.1. [DOI] [PubMed] [Google Scholar]
- Correas I. Characterization of isoforms of protein 4.1 present in the nucleus. Biochem J. 1991;279:581–585. doi: 10.1042/bj2790581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correas I, Avila J. Erythrocyte protein 4.1 associates with tubulin. Biochem J. 1988;255:217–221. [PMC free article] [PubMed] [Google Scholar]
- Correas I, Leto TL, Speicher DW, Marchesi VT. Identification of the functional site of erythrocyte protein 4.1 involved in spectrin-actin associations. J Biol Chem. 1986a;261:3310–3315. [PubMed] [Google Scholar]
- Correas I, Speicher DW, Marchesi VT. Structure of the spectrinactin binding site of erythrocyte protein 4.1. J Biol Chem. 1986b;261:13362–13366. [PubMed] [Google Scholar]
- Cox KH, Adair-Kirk TL, Cox JV. Four variant chicken erythroid anion exchangers. Roles of the alternative N-terminal sequences in intracellular targeting in transfected human erythroleukemia cells. J Biol Chem. 1995;270:19752–19760. doi: 10.1074/jbc.270.34.19752. [DOI] [PubMed] [Google Scholar]
- D'Andrea AD, Tantravahi U, LaLande M, Perle MA, Latt SA. High resolution analysis of the timing of replication of specific DNA sequences during S phase of mammalian cells. Nucleic Acids Res. 1983;11:4753–4774. doi: 10.1093/nar/11.14.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Carcer G, Lallena MJ, Correas I. Protein 4.1 is a component of the nuclear matrix of mammalian cells. Biochem J. 1995;312:871–877. doi: 10.1042/bj3120871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarajan P, Stabach PR, Mann AS, Ardito T, Kashgarian M, Morrow JS. Identification of a small cytoplasmic ankyrin (AnkG119) in the kidney and muscle that binds βIΣ* spectrin and associates with the Golgi apparatus. J Cell Biol. 1996;133:819–830. doi: 10.1083/jcb.133.4.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher DE, Parra M, Conboy JG, Mohandas N. Mechanochemistry of the alternatively spliced spectrin-actin binding domain in membrane skeletal protein 4.1. J Biol Chem. 1993;268:7186–7195. [PubMed] [Google Scholar]
- Fey EG, Capco DG, Krockmalnic G, Penman S. Epithelial structure revealed by chemical dissection and unembedded electron microscopy. J Cell Biol. 1984;99:203–208. doi: 10.1083/jcb.99.1.203s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fey EG, Krochmalnic G, Penman S. The non-chromatin substructures of the nucleus: the ribonucleoprotein (RNP)-containing and RNPdepleted matrices analyzed by sequential fractionation and resinless section electron microscopy. J Cell Biol. 1986;102:1654–1665. doi: 10.1083/jcb.102.5.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D, Weisenberger D, Scheer U. Assigning functions to nucleolar structures. Chromosoma (Berl) 1991;101:133–140. doi: 10.1007/BF00355363. [DOI] [PubMed] [Google Scholar]
- Fu XD, Maniatis T. Isolation of a complementary DNA that encodes the mammalian splicing factor SC35. Science (Wash DC) 1992;256:535–538. doi: 10.1126/science.1373910. [DOI] [PubMed] [Google Scholar]
- Gascard P, Cohen CM. Absence of high affinity band 4.1 sites from membranes of glycophorin C- and D-deficient (Leach phenotype) erythrocytes. Blood. 1994;83:1102–1108. [PubMed] [Google Scholar]
- Goldman MA, Holmquist GP, McGray LA, Caston LA, Nag A. Replication timing of genes and middle repetitive sequences. Science (Wash DC) 1984;224:686–692. doi: 10.1126/science.6719109. [DOI] [PubMed] [Google Scholar]
- Granger BL, Lazarides E. Membrane skeletal protein 4.1 of avian erythrocytes is composed of multiple variants that exhibit tissue-specific expression. Cell. 1984;37:595–607. doi: 10.1016/0092-8674(84)90390-8. [DOI] [PubMed] [Google Scholar]
- Granger BL, Lazarides E. Appearance of new variants of membrane skeletal protein 4.1 during terminal differential of avian erythroid and ventricular cells. Nature (Lond) 1985;313:238–241. doi: 10.1038/313238a0. [DOI] [PubMed] [Google Scholar]
- Hagen SJ, Kiehart DP, Kaiser DA, Pollard TD. Characterization of monoclonal antibodies to Acanthamoeba myosin-1 that cross-react with both myosin 11 and low molecular mass nuclear proteins. J Cell Biol. 1986;103:2121–2128. doi: 10.1083/jcb.103.6.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haltiwanger RS, Holt GD, Hart GW. Enzymatic addition of O-GlcNAc to nuclear and cytoplasmic proteins. 1990. J Biol Chem. 1990;265:2563–2568. [PubMed] [Google Scholar]
- Hatton KS, Dhar V, Gahn TA, Brown EH, Mager D, Schildkraut CL. Temporal order of replication of multigene families reflects chromosomal location and transcriptional activity. Cancer Cells. 1988;6:335–340. [Google Scholar]
- He DM, Martin T, Penman S. Localization of heterogeneous nuclear ribonucleoprotein in the interphase nuclear matrix core filaments and on perichromosomal filaments at mitosis. Proc Natl Acad Sci USA. 1991;88:7469–7473. doi: 10.1073/pnas.88.17.7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He D, Zeng C, Brinkley BR. Nuclear matrix proteins as structural and functional components of the mitotic apparatus. Int Rev Cytol. 1995;162B:1–74. doi: 10.1016/s0074-7696(08)62614-5. [DOI] [PubMed] [Google Scholar]
- Hemming NJ, Anstee DJ, Mawby WJ, Reid ME, Tanner MJA. Localization of the protein 4.1-binding site on human erythrocyte glycophorins C and D. Biochem J. 1994;299:191–196. doi: 10.1042/bj2990191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemming NJ, Anstee DJ, Staricoff MA, Tanner MJA, Mohandas N. Identification of the membrane attachment sites for protein 4.1 in the human erythrocyte. J Biol Chem. 1995;270:5360–5366. doi: 10.1074/jbc.270.10.5360. [DOI] [PubMed] [Google Scholar]
- Horne WC, Prinz WC, Tang EK. Identification of two cAMPdependent phosphorylation sites on erythrocyte protein 4.1. Biochim Biophys Acta. 1990;1055:87–92. doi: 10.1016/0167-4889(90)90095-u. [DOI] [PubMed] [Google Scholar]
- Hozak P, Hassan AB, Jackson DA, Cook PR. Visualization of replication factories attached to nucleoskeleton. Cell. 1993;73:361–373. doi: 10.1016/0092-8674(93)90235-i. [DOI] [PubMed] [Google Scholar]
- Jackson DA, Cook PR. Replication occurs at a nucleoskeleton. EMBO (Eur Mol Biol Organ) J. 1986;5:1403–1410. doi: 10.1002/j.1460-2075.1986.tb04374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jons T, Drenckhahn D. Identification of the binding interface involved in linkage of cytoskeletal protein 4.1 to the erythrocyte anion exchanger. EMBO (Eur Mol Biol Organ) J. 1992;11:2863–2867. doi: 10.1002/j.1460-2075.1992.tb05354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly GM, Zelus BD, Moon RT. Identification of a calciumdependent calmodulin-binding domain in Xenopus membrane skeleton protein 4.1. J Biol Chem. 1991;266:12469–12473. [PubMed] [Google Scholar]
- Larsson SH, Charlieu JP, Miyagawa K, Engelkamp D, Rassouizadegan M, Ross A, Cuzin F, Van Heyningen V, Hastie ND. Subnuclear localization of WT 1 in splicing or transcription factor domains is regulated by alternative splicing. Cell. 1995;81:391–401. doi: 10.1016/0092-8674(95)90392-5. [DOI] [PubMed] [Google Scholar]
- Leonhardt H, Page AW, Weier HU, Bestor TH. A targeting sequence directs DNA methyl-transferase to sites of DNA replication in mammalian nuclei. Cell. 1992;71:865–873. doi: 10.1016/0092-8674(92)90561-p. [DOI] [PubMed] [Google Scholar]
- Leto TL, Marchesi VT. A structural model of human erythrocyte protein 4.1. J Biol Chem. 1984;259:4603–4608. [PubMed] [Google Scholar]
- Leto TL, Pratt BM, Madri JA. Mechanisms of cytoskeleton regulation: modulation of aortic endothelial cell protein band 4.1 by the extracellular matrix. J Cell Physiol. 1986;127:423–431. doi: 10.1002/jcp.1041270311. [DOI] [PubMed] [Google Scholar]
- Lombardo CR, Willardson BM, Low PS. Localization of the protein 4.1-binding site on the cytoplasmic domain of erythrocyte membrane band 3. J Biol Chem. 1992;267:9540–9546. [PubMed] [Google Scholar]
- Lue RA, Marfatia SM, Branton D, Chishti AH. Cloning and characterization of hdlg: the human homologue of the Drosophila discs large tumor suppressor binds to protein 4.1. Proc Natl Acad Sci USA. 1994;91:9818–9822. doi: 10.1073/pnas.91.21.9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydersen B, Pettijohn D. Human specific nuclear protein that associates with the polar region of the mitotic apparatus: distribution in a human/ hamster hybrid cell. Cell. 1980;22:489–499. doi: 10.1016/0092-8674(80)90359-1. [DOI] [PubMed] [Google Scholar]
- MacArthur H, Walter G. Monoclonal antibodies specific for the carboxy terminus of simian virus 40 large T antigen. J Virol. 1984;52:483–491. doi: 10.1128/jvi.52.2.483-491.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madri, J.A., B.M. Pratt, and J. Yannariello-Brown. 1988. Endothelial cell-extracellular matrix interactions. In Endothelial Cell Biology in Health and Disease. N. Simionescu and M. Simionescu, editors. Plenum Press, New York. 167–188.
- Malchiodi-Albedi F, Ceccarini M, Winkelman JC, Morrow JS, Petrucci TC. The 270 kDa splice variant of erythrocyte β-spectrin (β1Σ2) segregates in vivo and in vitroto specific domains of cerebellar neurons. J Cell Sci. 1993;106:67–78. doi: 10.1242/jcs.106.1.67. [DOI] [PubMed] [Google Scholar]
- Marfatia SM, Lue R, Branton D, Chishti AH. In vitrobinding studies suggest a membrane-associated complex between erythroid p55, protein 4.1 and glycophorin C. J Biol Chem. 1994;269:8631–8634. [PubMed] [Google Scholar]
- Marfatia SM, Lue R, Branton D, Chishti AH. Identification of the protein 4.1 binding interface on glycophorin C and p55, a homologue of the Drosophila discs-large tumor suppressor protein. J Biol Chem. 1995;270:715–719. doi: 10.1074/jbc.270.2.715. [DOI] [PubMed] [Google Scholar]
- Milankov K, DeBoni U. Cytochemical localization of actin and myosin aggregates in interphase nuclei in situ. Exp Cell Res. 1993;209:189–199. doi: 10.1006/excr.1993.1301. [DOI] [PubMed] [Google Scholar]
- Moir RD, Montag-Lowy M, Goldman RD. Dynamic properties of nuclear lamins: lamin B is associated with sites of DNA replication. J Cell Biol. 1994;125:1201–1212. doi: 10.1083/jcb.125.6.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroianu J, Riordan JF. Identification of the nucleolar targeting signal of human angiogenin. Biochem Biophys Res Commun. 1994;203:1765–1772. doi: 10.1006/bbrc.1994.2391. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Morita T, Sato C. Structural organization of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp Cell Res. 1986;165:291–297. doi: 10.1016/0014-4827(86)90583-5. [DOI] [PubMed] [Google Scholar]
- Nakayasu N, Ueda K. Preferential association of acidic actin with nuclei and nuclear matrix from mouse leukemia L51784 cells. Exp Cell Res. 1986;163:327–336. doi: 10.1016/0014-4827(86)90064-9. [DOI] [PubMed] [Google Scholar]
- Nickerson JA, Penman S. Localization of nuclear matrix core filament proteins at interphase and mitosis. Cell Biol Int Rep. 1992;16:811–826. doi: 10.1016/s0309-1651(05)80024-4. [DOI] [PubMed] [Google Scholar]
- Nickerson JA, Krockmalnic G, He D, Penman S. Immunolocalization in three dimensions: immunogold staining of cytoskeletal and matrix proteins in resinless electron microscopy sections. Proc Natl Acad Sci USA. 1990;87:2259–2263. doi: 10.1073/pnas.87.6.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson, J.A., D. Krockmalnic, and S. Penman. 1994. Isolation and visualization of the nuclear matrix, the nonchromation structure of the nucleus. Cell Biology Handbook. Academic Press, Inc., Orlando, FL. 622–627.
- Nickerson JA, Blencowe BJ, Penman S. The architectural organization of nuclear metabolism. Int Rev Cytol. 1995;162A:67–123. doi: 10.1016/s0074-7696(08)61229-2. [DOI] [PubMed] [Google Scholar]
- O'Keefe RT, Henderson SC, Spector DL. Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome specific α-satellite DNA sequences. J Cell Biol. 1992;116:1095–1100. doi: 10.1083/jcb.116.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternack GR, Racusen RH. Erythrocyte protein 4.1 binds and regulates myosin. Proc Natl Acad Sci USA. 1989;86:9712–9716. doi: 10.1073/pnas.86.24.9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penman S. Rethinking cell structure. Proc Natl Acad Sci USA. 1995;92:5251–5257. doi: 10.1073/pnas.92.12.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinder JC, Chung A, Reid ME, Gratzer WB. Membrane attachment sites for the membrane cytoskeletal protein 4.1 of the red blood cell. Blood. 1993;82:3482–3488. [PubMed] [Google Scholar]
- Prelich G, Kostura M, Marshak DR, Matthews MB, Stillman B. The cell-cycle regulated proliferating cell nuclear antigen is required for SV40 DNA replication in vitro. . Nature (Lond) 1987;326:471–475. doi: 10.1038/326471a0. [DOI] [PubMed] [Google Scholar]
- Saunders WS, Cooke CA, Earnshaw WC. Compartmentalization within the nucleus: discovery of a novel subnuclear region. J Cell Biol. 1991;115:919–931. doi: 10.1083/jcb.115.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheer U, Thiry M, Goessens G. Structure, function and assembly of the nucleolus. Trends Cell Biol. 1993;3:236–241. doi: 10.1016/0962-8924(93)90123-i. [DOI] [PubMed] [Google Scholar]
- Schischmanoff PO, Winardi R, Discher DE, Parra MK, Bicknese SE, Witkowska HE, Conboy JG, Mohandas N. Defining of the minimal domain of protein 4.1 involved in spectrin-actin binding. J Biol Chem. 1995;270:21243–21250. doi: 10.1074/jbc.270.36.21243. [DOI] [PubMed] [Google Scholar]
- Smith HC, Berezney R. Nuclear matrix-bound deoxyribonucleic acid synthesis: an in vitro system. Biochemistry. 1982;21:6751–6761. doi: 10.1021/bi00269a021. [DOI] [PubMed] [Google Scholar]
- Spector DL. Higher order nuclear organization: three-dimensional distribution of small nuclear ribonucleoprotein particles. Proc Natl Acad Sci USA. 1990;87:141–151. doi: 10.1073/pnas.87.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector DL. Macromolecular domains within the cell nucleus. Annu Rev Cell Biol. 1993;9:265–315. doi: 10.1146/annurev.cb.09.110193.001405. [DOI] [PubMed] [Google Scholar]
- Spector DL, Fu XD, Maniatis T. Association between distinct pre-mRNA splicing components and the cell nucleus. EMBO (Eur Mol Biol Organ) J. 1991;10:3467–3481. doi: 10.1002/j.1460-2075.1991.tb04911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan M, Edman CF, Schulman H. Alternative splicing introduces a nuclear localization signal that targets multifunctional CaM kinase to the nucleus. J Cell Biol. 1994;126:839–852. doi: 10.1083/jcb.126.4.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subrahmanyam G, Bertics PJ, Anderson RA. Phosphorylation of protein 4.1 on tyrosine-418 modulates its function in vitro. . Proc Natl Acad Sci USA. 1991;88:5222–5226. doi: 10.1073/pnas.88.12.5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CK, Castillo C, So AG, Downey KM. An auxiliary protein for DNA polymerase delta from fetal calf thymus. J Biol Chem. 1986;261:12310–12316. [PubMed] [Google Scholar]
- Tanaka T, Kadowaki K, Lazarides E, Sobue K. Ca2+-dependent regulation of the spectrin/actin interaction by calmodulin and protein 4.1. J Biol Chem. 1991;266:1134–1140. [PubMed] [Google Scholar]
- Tang TK, Leto TL, Correas I, Alonso MA, Marchesi VT, Benz EJ., Jr Selective expression of an erythroid-specific isoform of protein generates two proteins. Proc Natl Acad Sci USA. 1988;85:3713–3717. doi: 10.1073/pnas.85.11.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TK, Quin Z, Marchesi VT, Benz EJ. Heterogeneity of mRNA and protein products arising from the protein 4.1 gene in erythroid and nonerythroid tissues. J Cell Biol. 1990;110:617–624. doi: 10.1083/jcb.110.3.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple GF, Chang JC, Kan YW. Authentic β-globin mRNA sequences in homozygous β O-thalassemia. Proc Natl Acad Sci USA. 1977;74:3047–3051. doi: 10.1073/pnas.74.7.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Cao L-G, Wang YL, Pederson T. Localization of premessenger RNA at discrete nuclear sites. Proc Natl Acad Sci USA. 1991;88:7391–7395. doi: 10.1073/pnas.88.16.7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, Johnson CV, Dobner PR, Lawrence JB. Higher level organization of individual gene transcription and RNA splicing. Science (Wash DC) 1993;259:1326–1330. doi: 10.1126/science.8446901. [DOI] [PubMed] [Google Scholar]
- Xing Y, Johnson CV, Moen PT, McNeil JA, Lawrence JB. Nonrandom gene organization: structural arrangements of specific pre-mRNA transcription and splicing SC-35 domains. J Cell Biol. 1995;131:1635–1647. doi: 10.1083/jcb.131.6.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CH, Lambie EJ, Snyder M. NuMA: an unusually long coilcoil protein in the mammalian nucleus. J Cell Biol. 1992;116:1303–1317. doi: 10.1083/jcb.116.6.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C, He D, Brinkley BR. Localization of NuMA protein isoforms in the nuclear matrix of mammalian cells. Cell Motil Cytoskel. 1994;29:167–176. doi: 10.1002/cm.970290208. [DOI] [PubMed] [Google Scholar]