

Abstract
Addition of brefeldin A (BFA) to mammalian cells rapidly results in the removal of coatomer from membranes and subsequent delivery of Golgi enzymes to the endoplasmic reticulum (ER). Microinjected anti-EAGE (intact IgG or Fab-fragments), antibodies against the “EAGE”-peptide of β-COP, inhibit BFA-induced redistribution of β-COP in vivo and block transfer of resident proteins of the Golgi complex to the ER; tubulo-vesicular clusters accumulate and Golgi membrane proteins concentrate in cytoplasmic patches containing β-COP. These patches are devoid of marker proteins of the ER, the intermediate compartment (IC), and do not contain KDEL receptor. Interestingly, relocation of KDEL receptor to the IC, where it colocalizes with ERGIC53 and ts-O45-G, is not inhibited under these conditions. While no stacked Golgi cisternae remain in these injected cells, reassembly of stacks of Golgi cisternae following BFA wash-out is inhibited to only ∼50%. Mono- or divalent anti-EAGE stabilize binding of coatomer to membranes in vitro, at least as efficiently as GTPγS. Taken together these results suggest that enhanced binding of coatomer to membranes completely inhibits the BFA-induced retrograde transport of Golgi resident proteins to the ER, probably by inhibiting fusion of Golgi with ER membranes, but does not interfere with the disassembly of the stacked Golgi cisternae and recycling of KDEL receptor to the IC. These results confirm our previous results suggesting that COPI is involved in anterograde membrane transport from the ER/IC to the Golgi complex (Pepperkok et al., 1993), and corroborate that COPI regulates retrograde membrane transport between the Golgi complex and ER in mammalian cells.
Vectorial protein and membrane transport in the secretory pathway is mediated by vesicular carriers. Despite the enormous membrane traffic through this pathway, each compartment maintains a unique and defined membrane composition; in addition a balance between forward membrane transport and recycling must be kept. Two basic mechanisms operating in the regulation of this membrane flow are sorting and recycling. Coat proteins (COPs)1 have been implicated in sorting of cargo into coated buds and formation of vesicular transport intermediates (Robinson, 1994; Rothman, 1994; Kreis et al., 1995; Schekman and Orci, 1996): COPII at the ER (Barlowe et al., 1994; Aridor et al., 1995), COPI at the ER/IC (Pepperkok et al., 1993; Griffiths et al., 1995) and Golgi complex (Malhotra et al., 1989), clathrin and AP1 (Robinson, 1994), as well as so far poorly characterized COPs at the trans-Golgi network (TGN) the exit site of the Golgi complex (Griffiths et al., 1985; Ladinsky et al., 1994; Narula and Stow, 1995). Immunolocalization of COPs in mammalian cells is consistent with their sites of action (Oprins et al., 1993; Griffiths et al., 1995; Shaywitz et al., 1995). In yeast COPI and COPII appear both involved in the formation of distinct anterogradely directed transport intermediates from ER membranes (Bednarek et al., 1995). Missorted ER proteins and components of the targeting and fusion machinery bearing specific signals are retrieved from the IC and the Golgi complex by specific receptors, like the KDEL receptor for example (Pelham, 1995; see also Miesenbock and Rothman, 1995), and genetic evidence in yeast indicates that retrograde transport of proteins with an ER retrieval signal KKXX depends on COPI (Letourneur et al., 1994; Cosson et al., 1996; see however also Duden et al., 1994; Wuestehube et al., 1996).
The organization of membrane traffic in tissue culture cells is disrupted by BFA. BFA interferes with the function of a membrane bound GTP exchange factor and thus prevents association of ARFs, small GTPases (Boman and Kahn, 1995), with membranes (Donaldson et al., 1992; Helms and Rothman, 1992; Stamnes and Rothman, 1993). As a consequence, binding of COPI (and most likely some other related COPs), as well as the TGN, but not the cell surface, clathrin adaptors, to membranes is inhibited (Donaldson et al., 1990; Robinson and Kreis, 1992). The final result is a dramatic reorganization of endocellular membranes; membranes of compartments connected in the secretory and endocytic pathways appear to fuse with one another in a nonregulated manner and sorting of cargo is impaired (Hunziker et al., 1991; Lippincott-Schwartz et al., 1991; Orci et al., 1991; Wood et al., 1991). Most significantly, membranes of the Golgi complex relocate into the ER leading to the rapid disappearance of morphologically distinct stacks of Golgi cisternae (Misumi et al., 1986; Oda et al., 1987; Lippincott-Schwartz et al., 1989; see also Ulmer and Palade, 1991). This BFA induced relocation has given valuable insights into components and mechanisms that might be involved in the regulation of transport in the early secretory pathway (Klausner et al., 1992). Yet, it remains to be shown how closely BFA-induced relocation of Golgi components into the ER resembles the normal retrograde pathway(s) involved in recycling material from the Golgi complex to the IC/ER, and the role of coatomer in these processes is not yet fully understood (Pelham, 1991). The BFA-induced transfer of Golgi components to the ER may be due to uncovering a fusion machinery following dissociation of coatomer (Orci et al., 1991). This process could then lead to a nonregulated fusion of related membranes that normally do not fuse, a consequence of uncoupling the sequential events of budding, uncoating, and fusion of transport intermediates (Elazar et al., 1994; Rothman and Warren, 1994). Alternatively, and not mutually exclusive, exposure of receptors for microtubule-based molecular motors may induce the movement of cis-Golgi membranes to sites adjacent to ER membranes and somehow facilitate their fusion (Pelham, 1991; Lippincott-Schwartz et al., 1995).
Microinjection of antibodies into living cells has proven a powerful alternative approach to studies with cell-free systems and genetic analyses in yeast for characterizing functions of COPs in membrane traffic in mammalian cells in vivo. In this study we have used specific polyclonal antibodies raised against peptides of β-COP, a subunit of coatomer (Duden et al., 1991), for further characterizing the role of COPI in membrane traffic between the ER/IC and Golgi complex. Microinjected antibodies against the EAGE peptide inhibit transport of newly synthesized temperature sensitive membrane glycoprotein of vesicular stomatitis virus (ts-O45-G) between the ER/IC and the Golgi complex (Pepperkok et al., 1993). We show here that these antibodies also interfere with the BFA-induced transport of Golgi proteins into the ER. Biochemical and morphological analyses of these inhibitory effects suggest that anti-EAGE interfere with dissociation of coatomer from membranes leading to the inhibition of the subsequent fusion of membranes of the Golgi complex with those of the ER. The inhibitory effects of anti-EAGE on anterograde transport from the IC to the Golgi complex and on BFAinduced retrograde transport appear to be different, suggesting that the function of COPI may be regulated differently in these two pathways.
Materials and Methods
Cell Culture, Microinjection, and Microscopy
Vero cells (African green monkey kidney cells, Amer. Type Culture Collection, Rockville, MD; CCL81) were maintained and infected with tsO45VSV (Indiana serotype) as described earlier (Kreis and Lodish, 1986). Microinjection was performed on a computer automated microinjection system (AIS, Zeiss, Zürich, Switzerland) as described (Pepperkok et al., 1993). Vero cells stably expressing myc-tagged NAGT1 were obtained from Dr. Brian Storrie (Blacksburg). Immunofluorescence was performed as described (Pepperkok et al., 1993) with antibodies against calnexin (Hammond and Helenius, 1994), PDI (Vaux et al., 1990), ERGIC53 (Schweizer et al., 1988), KDEL receptor (Tang et al., 1995), p58 (Bloom and Brashear, 1989), β-COP (Pepperkok et al., 1993), β′-COP (Lowe and Kreis, 1995), and VSV-G (Kreis, 1986). The Golgi complex was visualized in Vero cells expressing myc-tagged NAGT1 with a monoclonal antibody against the myc-epitope (Evan et al., 1985) as described (Pierre et al., 1994).
Images of immunolabeled cells were recorded on a Zeiss inverted fluorescence microscope (Axiovert TV135) equipped with a cooled CCD camera (Photometrics CH250, 1317X1035 pixels, Tucson, AZ), controlled by a Power Macintosh 8100/100 and the software package IPLab spectrum V2.7 (Signals Analytics Corp., Vienna, VA). Images were further processed using Adobe Photoshop V3.0 before printing with a Xanté laser press 1800 (Conware Informatik AG, Baar, Switzerland).
Quantitative electron microscopy (EM) on microinjected cells was performed as described (Pepperkok et al., 1993), and the volume density of Golgi membranes per cell determined by point counting (Weibel, 1979).
Metabolic Labeling and Biochemical Analyses of Maturation of ts-O45-G
500–1,000 cells, grown on a glass coverslip (Pepperkok et al., 1993), were infected with tsO45-VSV and incubated for 1.5 h in culture medium followed by 10 min incubation in labeling medium (culture medium lacking methionine and cysteine). Cells were metabolically labeled with 2.8 mCi [35S]methionine (Amersham Corp., Arlington Heights, IL) per ml labeling medium for 20 min. Cells were washed and placed into low carbonate culture medium containing 100 μg/ml cycloheximide for microinjection (all incubations up to this step and the microinjection were done at 39.5°C). Cells were further chased (in culture medium containing 100 μg/ml cycloheximide) as indicated in the figure legends. Preparation of cell lysates, immunoprecipitation of ts-O45-G, endoglycosidase H (endo H) digestion, gel electrophoresis, and fluorography were performed as described (Pepperkok et al., 1993).
Preparation of Rat Liver Cytosol and Enriched IC/Golgi Membranes
To prepare rat liver cytosol 30 g of frozen rat liver was thawed at 4°C, minced in 60 ml of homogenization buffer (20 mM Hepes-KOH [pH 7.0], 100 mM KCl, 2.5 mM MgCl2, 1 mM PMSF, 0.5 mM 1:10 phenanthroline, 2 mM pepstatin A, 2 mg/ml aprotinin, 0.5 μg/ml leupeptin) and homogenized with a Polytron using three 30-s bursts on setting 4. The resulting homogenate was centrifuged at 12,000 g for 15 min in a 18.50 rotor (Heraeus SA, Carouge-Genève, Switzerland). The supernatant was collected and centrifuged at 100,000 g for 1 h in a TST 41.14 rotor (Koutron Instruments, Schlieren, Switzerland).AU: Please give name of of manufacturer, city and state The resulting supernatant was collected and centrifuged for a further 2 h at 200,000 g in the TST 41.14 rotor. The 200,000-g supernatant (rat liver cytosol) was frozen in liquid nitrogen and stored at −80°C. Golgi/IC enriched membranes were prepared from rat liver using the method of Malhotra et al. (1989).
Quantifying Binding of Coatomer to Membranes In Vitro
Incubations (120 μl total volume) were carried out for 10 min at 37°C in the presence of 14 μg Golgi/IC membranes, 25 mM Hepes-KOH (pH 7.0), 25 mM KCl, 2.5 mM MgCl2, 50 μM ATP, 2 mM creatine phosphate, 12.5 U/ml creatine kinase, and 160 μg rat liver cytosol. Further additions were made as indicated in the figure legends. Reactions were layered on top of 200 μl of 15% sucrose (in 25 mM Hepes-KOH [pH 7.0], 25 mM KCl) and centrifuged at 4°C in a microcentrifuge for 30 min at 15,000 rpm. The supernatants were discarded and the membrane pellets resuspended in 15 μl SDS sample buffer. Proteins were separated by SDS-PAGE and transferred to nitrocellulose for immunoblotting. β-COP was detected using the monoclonal antibody M3A5 (Allan and Kreis, 1986) at a 1:1,000 dilution followed by horseradish peroxidase–conjugated goat anti–mouse IgG (Cappel, Malvern, PA) at a 1:2,000 dilution. Peroxidase labeling was detected using the ECL kit (Amersham) and quantified by densitometry.
Results
Microinjection of Anti-EAGE Inhibits BFA-induced Transfer of Golgi Enzymes to the ER
Anti-EAGE, when microinjected into cells, inhibit anterograde transport of ts-O45-G from the ER/IC to the Golgi complex (Pepperkok et al., 1993). These antibodies may directly inhibit this transport by interfering with the formation or function of transport vesicles, or the inhibition may be indirect by preventing recycling of essential factors for anterograde transport. In an attempt to further clarify this issue we analyzed the effect of injected anti-EAGE on the putative recycling of Golgi enzymes between the Golgi complex and the ER/IC, and on BFA-induced relocation of Golgi proteins to the ER/IC. If continuous recycling of Golgi enzymes through the ER/IC normally occurred, one should expect an accumulation of these enzymes in the ER/IC when anterograde transport between the ER/IC and Golgi complex is selectively blocked. As a consequence, Golgi specific trimming of the viral glycoprotein accumulated in the ER/IC should be detected.
To analyze a possible recycling of Golgi enzymes through the ER/IC, Vero cells were infected with tsO45-VSV and kept at nonpermissive temperature (39.5°C, predominantly ER accumulation), or 15°C (IC accumulation). Under these conditions, ts-O45-G arrested in the ER or IC may serve as a substrate for oligosaccharide-processing Golgi enzymes, and transport of these enzymes into the ER/IC can be studied by biochemical analyses of modifications of the oligosaccharide side chains of the viral glycoprotein (Doms et al., 1989). Even after accumulation of ts-O45-G in the ER or IC for up to 6 h no trace of endo H-resistant glycoprotein could be detected in cells injected with anti-EAGE at the respective temperature (as in noninjected control cells; Fig. 1 A). Moreover, when cells were injected with anti-EAGE at 39.5°C and kept for three additional hours at the nonpermissive temperature and then for 3 h at 15°C with ts-O45-G finally accumulated in the IC (anti-EAGE does not inhibit ER to IC transport) no endo H-resistant fraction of the viral glycoprotein could be detected (not shown). Thus, either the recycling of (even a small fraction of) Golgi enzymes through the ER/IC is inhibited by the injected antibodies, or these enzymes, once located at their proper site of function within the Golgi complex, are retained. To investigate directly the role of coatomer in transport from the Golgi complex to the ER/IC in vivo, we used BFA-induced transfer of Golgi enzymes into the ER combined with microinjection of specific antibodies raised against synthetic peptides of β-COP (anti-EAGE and anti-110-12; Pepperkok et al., 1993) and β′-COP (Lowe and Kreis, 1995).
Figure 1.

Biochemical analysis of transfer of Golgi enzymes to the ER. Transfer of Golgi enzymes to the ER was determined biochemically by analyzing acquisition of endo H resistance of tsO45-G arrested in the ER. 500–1000 Vero cells infected with tsO45-VSV were incubated for 1.5 h at 39.5°C, metabolically labeled for 10 min with 35S-met and microinjected at 39.5°C or 15°C with Fab-fragments of anti-EAGE (EAGE), divalent anti–110-12 (110-12), or kept as noninjected controls (n.i.). (A) Noninjected control cells were lysed directly after the pulse with 35S-met and ts-O45-G analyzed for endo H sensitivity (0 h). 39.5°C: control or injected cells were lysed and ts-O45-G analyzed for endo H sensitivity after a chase of 6 h at 39.5°C; 15°C: temperature was shifted for 2 h to 15°C before injection at 15°C, and the cells were kept for an additional 4 h at 15°C before lysis and analysis of endo H resistance of ts-O45-G. (B) Control and injected cells were chased for 0 h or 2.5 h at 39.5°C in the presence of 5 μg/ml BFA before lysis and analysis of endo H resistance of ts-O45-G. Microinjected anti-EAGE, but not anti-110-12 block BFA induced acquisition of endo H resistance of ts-O45-G.
Under normal conditions, ts-O45-G is not modified by Golgi enzymes at 39.5°C and therefore remains sensitive to digestion with endo H for at least 6 h (Fig. 1 A, n.i. 6h). Treatment of cells with BFA relocates Golgi enzymes to the ER within 15–30 min (Domes et al., 1989; LippincottSchwartz et al., 1989) and renders ER accumulated ts-O45-G endo H resistant at 39.5°C (Fig. 1 B, n.i. 2.5h). However, ts-O45-G remains completely endo H sensitive at this temperature in cells, microinjected with anti-EAGE or its Fab-fragments, which have subsequently been treated with BFA (Fig. 1 B, EAGE). Thus, microinjection of antiEAGE blocks the transfer of Golgi enzymes to the ER where ts-O45-G is accumulated. This inhibition is specific for anti-EAGE, since microinjected anti-110-12 have no effect on BFA induced acquisition of endo H resistance of ts-O45-G (Fig. 1 B, 110-12).
Distribution of Golgi, IC, and ER Proteins in BFA-treated Cells Microinjected with Antibodies against β-COP
In control noninjected cells ts-O45-G accumulates in the ER at 39.5°C and colocalizes with PDI or calnexin (data not shown). Microinjection of anti-EAGE has no effect on the localization of ts-O45-G in the ER at nonpermissive temperature (e.g., Fig. 3, a and b). The lack of BFA-induced acquisition of endo H resistance of ts-O45-G in these cells must therefore be due to inhibition of delivery of Golgi enzymes to the ER. To analyze this transport block morphologically, we colocalized microinjected antibodies and several marker proteins for the ER, IC, and Golgi complex by immunofluorescence microscopy.
Figure 3.

Effect of microinjected anti-EAGE and BFA on the distribution of ts-O45-G. ts-O45 VSV-infected Vero cells, kept for 2 h at 39.5°C, were microinjected at nonpermissive temperature with Fab-fragments of anti-EAGE (a–d) or with anti-110-12 (e and f). After injection control cells were incubated for further 6 h at 39.5°C (a and b), or for 2.5 h at 39.5°C with 5 μg/ml BFA (c–f). Cells were then fixed and labeled for injected antibodies (b, d, and f) and ts-O45-G (a, c, and e). Injected anti-EAGE has no effect on ts-O45-G in the ER in the control cells kept at 39.5°C (a and b). BFA treatment, however, induces accumulation of ts-O45-G in cytosolic patches (arrows in c and e) which are distinct from the patches where anti-EAGE accumulate (arrowheads in d). Anti-110-12 maintains its diffuse cytosolic distribution after BFA treatment (f). Bar, 15 μm.
The resident Golgi membrane proteins NAGT I (Fig. 2 e) and Man II (not shown), as well as p58, a protein associated with the cytoplasmic surface of Golgi membranes (Bloom and Brashear, 1989), accumulate in β-COP positive patches (Fig. 2 f) when virus-infected cells, microinjected with anti-EAGE, are treated with BFA while continuously kept at 39.5°C. These patches do not contain the “IC marker lectin” ERGIC53 (Schweizer et al., 1988; Schindler et al., 1993; Arar et al., 1995; Fig. 2, c and d), and the ER marker proteins, PDI and calnexin (not shown), maintain their reticular distributions and are not enriched in these structures either (Fig. 2, a and b). Furthermore, no ts-O45-G accumulates in these patches under nonpermissive conditions (Fig. 3, c and d). Interestingly, the normally predominantly cis-Golgi localized KDEL receptor accumulates in these BFA-treated cells in the patches containing ts-O45-G (Fig. 4, d and e) and ERGIC53, and not in those positive for Golgi enzymes or injected anti-EAGE (Fig. 4 c). This result indicates that injected anti-EAGE does not interfere with the BFA-induced relocation of KDEL receptor to the IC. Furthermore, the distribution of KDEL receptor (Fig. 4 a) appears not significantly changed in control cells microinjected with anti-EAGE (Fig. 4 b); occasionally, formation of tubular structures can be observed. The formation of these aggregates is specific for injected anti-EAGE, since neither microinjected anti110-12 nor anti–β′-COP (not shown; these antibodies when injected into cells bind to coatomer in vivo) lead to an accumulation of coat proteins in patches under these conditions (Fig. 3, e and f), and no significant number of the β-COP and Golgi protein containing aggregates form in the absence of BFA at nonpermissive temperature (Fig. 3, a and b). Furthermore, the effect of injected anti-EAGE is not dominant over the effect of BFA, since if the antibodies were injected after BFA treatment of infected cells, no patches could be seen (not shown). It is also an early effect of BFA action, since patches containing Golgi proteins and β-COP can already be observed ∼5 min after addition of the drug (data not shown). These results corroborate our biochemical data which suggest that ts-O45-G does not meet Golgi enzymes in BFA-treated cells which have been injected with anti-EAGE. Thus, injected anti-EAGE inhibits the BFA-induced transfer of resident Golgi proteins to the ER and leads to a rapid accumulation of Golgi proteins in COPI containing cytoplasmic aggregates distinct from the ER/IC.
Figure 2.
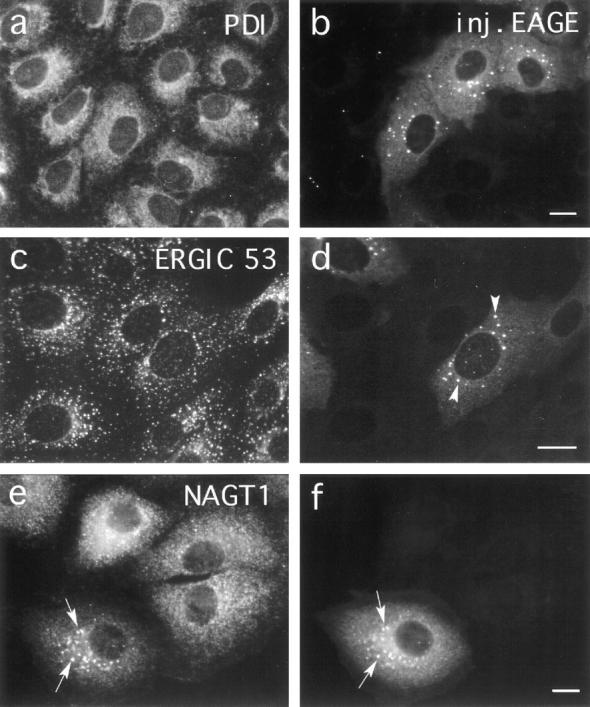
Effect of microinjected anti-EAGE on BFA induced relocation of Golgi enzymes. Vero cells were microinjected with Fabfragments of anti-EAGE. 30 min after injection 5 μg/ml BFA was added to the medium and cells were incubated for further 2.5 h at 37°C. Cells were then fixed and double stained for injected antibodies (b, d, and f) and marker proteins for (a) the ER (PDI), (c) the intermediate compartment (ERGIC 53), and (e) the Golgi complex (NAGT1). Microinjected antibodies accumulate in patches (arrowheads in d; arrows in f) which colocalize with NAGT1 (arrows in e) but not ERGIC 53 and PDI. The injected anti-EAGE thus affect the BFA-induced relocalization of Golgi proteins to the ER. Bar, 10 μm.
Figure 4.

Effect of microinjected anti-EAGE on BFA induced relocation of KDEL receptor. ts-O45VSV–infected Vero cells were microinjected at 39.5°C with Fab-fragments of anti-EAGE (asterisks). 30 min after injection, cells were transferred into medium containing 5 μg/ml BFA (c–e) or kept in normal medium (a and b). Cells were fixed after 2 h and double labeled with a murine monoclonal antibody recognizing KDEL receptor (a) and rabbit anti-EAGE (b), or triple labeled for injected anti-EAGE (c), KDEL receptor (d), and ts-O45-G (e). Anti-KDEL receptor was visualized with cy3-labeled antibodies against mouse IgG and anti-EAGE Fab-fragments with fluorescein antibodies against rabbit IgG. Cells were then incubated with 5 mg/ml mouse IgG, after this first incubation with secondary antibodies, to saturate free binding sites for mouse IgG on cy3-anti–mouse. Finally, ts-O45-G was labeled with cy5-conjugated P5D4. Arrows indicate aggregates containing antiEAGE; note that these patches do not contain KDEL receptor or ts-O45-G. Arrowheads indicate patches containing KDEL receptor and ts-O45-G note that these patches do not contain antiEAGE. Bar, 10 μm.
Interestingly, upon addition of BFA to noninjected virusinfected control cells kept at nonpermissive temperature, a significant fraction of ts-O45-G also accumulates in patchy structures (Fig. 5; see also Fig. 3, c and e). These patches show no enhanced staining with antibodies against PDI or calnexin (not shown) which retain their typical ER distribution (Fig. 5 b). However, they are enriched in ERGIC53 (Fig. 5 d), p58 (Fig. 5 h), and contain β-COP (Fig. 5 f). These patches are usually not observed in noninfected cells. These results suggest that in these BFA-treated cells newly synthesized membrane proteins destined to the cell surface, as well as Golgi proteins and proteins recycling between the ER/IC and the Golgi complex, either accumulate in a subdomain of the ER which probably corresponds to the IC (see also Hammond and Helenius, 1994), or are transported in a BFA/COPI independent way from the ER to the IC.
Figure 5.
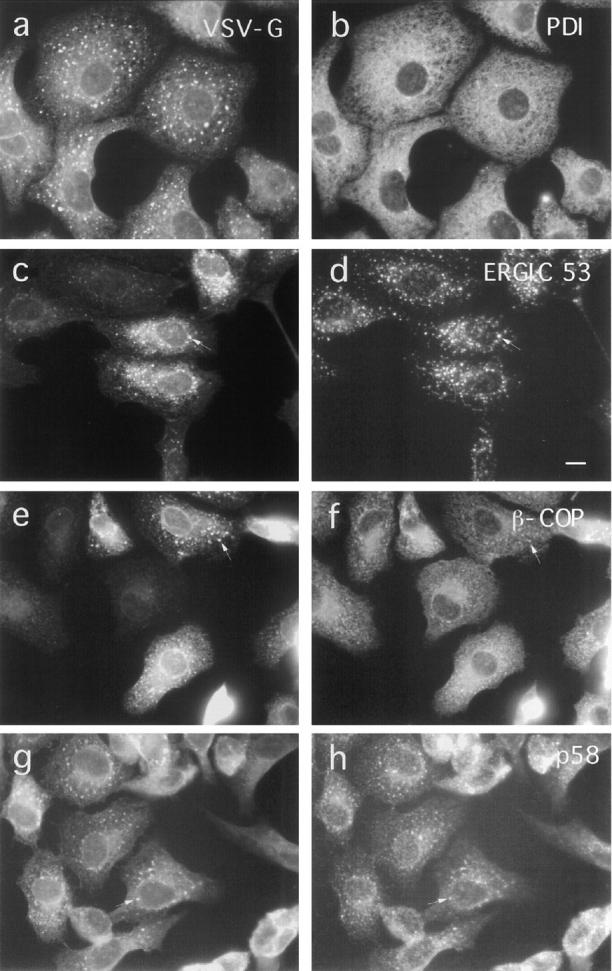
Effect of BFA on the distribution of ER, IC, and Golgi complex-associated proteins in VSV-infected cells. ts-O45 VSV-infected Vero cells were incubated with 5 μg/ml BFA for 2.5 h at 39.5°C and then fixed and double stained for ts-O45-G (a, c, e, and g) and the ER protein PDI (b), the IC marker protein ERGIC 53 (d), β-COP (f), and Golgi membrane associated p58 (h). ts-O45-G accumulates in patches enriched in ERGIC53, β−COP, and p58; PDI maintains its typical ER distribution and is not affected by BFA. Bar, 10 μm.
Quantitative EM analysis of thin sections revealed that virtually no identifiable Golgi stacks remained after a 1-h treatment with BFA at 39.5°C in control (anti-110-12) or anti–EAGE-injected cells (Fig. 6 a). Thus, the scattered patches of Golgi membranes positive for β-COP seen by immunofluorescence microscopy in cells injected with antiEAGE and after BFA treatment are unlikely to represent Golgi mini-stacks. In contrast, numerous dense spherical aggregates (Fig. 7 c) were found in the BFA-treated cells microinjected with anti-EAGE but not anti-110-12 (Fig. 6 c). The number of these spherical aggregates appears to correspond roughly with the number of β-COP–positive patches expected from light microscopy observations. Similar structures shown to contain β-COP and membranes have previously been observed in different cell types under various conditions of treatment (Oprins et al., 1993; Orci et al., 1993; Pepperkok et al., 1993; Hendricks et al., 1993). Furthermore, a significant fraction of tubulo-vesicular structures (Fig. 7, a, d, and e) appear in the injected cells; although more of these clusters are found in cells injected with anti-EAGE, a significant number is also present in control injected cells (Fig. 6 b). Thus, it is possible that in cells injected with anti-EAGE, BFA-induced relocation of resident Golgi proteins is arrested in these spherical aggregates. Since the tubulo-vesicular clusters are present in cells injected with either antibodies, they probably represent the structures where ts-O45-G, ERGIC53, and Golgi markers accumulate in BFA-treated cells, i.e., the IC.
Figure 6.

Quantitative EM analysis of the effect of injected antibodies against β-COP on the BFA-induced reorganization of the Golgi complex. ts-O45 VSV-infected (2 h) Vero cells were microinjected with antibodies against β-COP before or after treatment with 5 μg/ml BFA for 1 h. Thin sections of these cells were analyzed by EM and the volume density of Golgi stacks, tubulo- vesicular clusters, and dense spherical aggregates determined (Weibel, 1979) in at least 10 different fields (see Fig. 6 for examples of these structures). Injected antibodies have no effect on the disappearance of distinct Golgi stacks. Injected Fab-fragments of anti-EAGE, but not anti-110-12, inhibit, however, the reassembly of Golgi stacks by ∼50% after BFA wash-out for 1 h and numerous tubulo-vesicular clusters remain which are abundant after treatment of cells with BFA. Also, electron dense spherical aggregates form in anti–EAGE-injected BFA-treated cells, and they readily disappear upon removal of the drug. Error bars represent the standard mean error.
Figure 7.
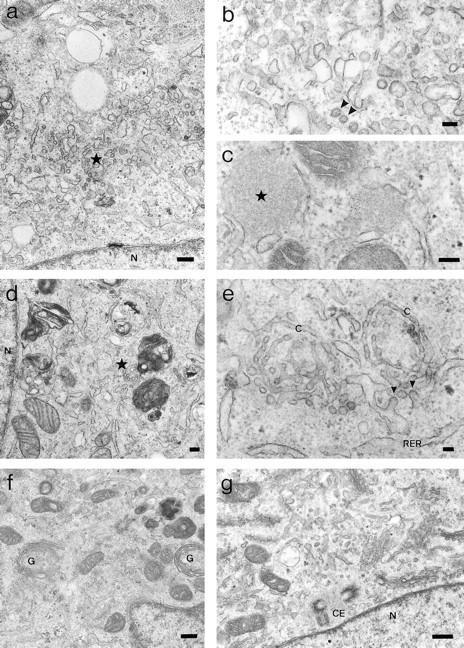
Morphological EM analysis of the effect of injected antibodies against β-COP on the BFA-induced reorganization of the Golgi complex. Typical examples for the structures identified and quantified in Fig. 5 in BFA-treated ts-O45 VSV-infected cells injected with Fab-fragments of anti-EAGE (a–c and f) or with anti-110-12 (d, e, and g) are shown. Cells in a–e have been treated for 1 h with 5 μg/ml BFA 30 min after injection with antibodies, while BFA has been washed-out for 1 h from cells shown in f and g. a shows a low magnification and b a higher magnification micrograph of tubulo-vesicular clusters that typically accumulate in BFA-treated cells injected with anti-EAGE; electron dense spherical aggregates are illustrated in c. In d and e tubulo-vesicular clusters are shown at low and high magnification in control cells injected with anti-110-12. Reassembled stacks of Golgi cisternae are shown in cells injected with anti-EAGE (f) or anti-110-12 (g). C, cisterna; CE, centriole pair; G, stacks of Golgi cisternae; N, nucleus; RER, rough ER; asterisks in a and d indicate tubulo-vesicular clusters, and in c, dense spherical aggregates. Arrowheads indicate possible COP-coated buds or vesicles. Bars: (b, c, and e) 100 nm; (a, d, f, and g) 200 nm.
Microinjected Anti-EAGE Inhibits Reformation of Golgi Stacks after BFA Wash-Out
Anti-EAGE inhibit protein transport in the anterograde (Pepperkok et al., 1993), as well as in the BFA-induced retrograde (this report) direction in vivo. While mixing of Golgi and ER membranes is induced by BFA, sorting of Golgi- from ER-specific components and subsequent transfer of these components to the location where the new Golgi complex will reassemble must occur upon removal of the drug. These processes may depend on the machinery regulating anterograde transport or reflect an intrinsic property of Golgi specific components to self assemble. If coatomer is essential for anterograde membrane traffic, and Golgi reformation after wash-out of BFA followed that pathway, then microinjected anti-EAGE should inhibit Golgi reassembly.
Virus-infected Vero cells were treated with BFA for 1 h, microinjected with antibodies in the presence of BFA at 39.5°C, and subsequently transferred to medium at permissive temperature lacking the drug. Initially, ts-O45-G is accumulated in ERGIC53 positive patches (Fig. 8, a and b). In uninjected cells or cells injected with anti-110-12, tsO45-G is transported normally to the plasma membrane through the reforming Golgi apparatus (data not shown). However, in cells microinjected with anti-EAGE, ts-O45-G is arrested in tubular structures (Fig. 8 c) positive for ERGIC53 (Fig. 8 d), suggesting that under these conditions anterograde protein transport is blocked in the IC, apparently in identical fashion to injected cells shifted to permissive temperature that had not been treated with BFA (Pepperkok et al., 1993). Similarly to the anti–EAGE– injected cells not treated with BFA, β-COP accumulates in patches which were often found on or at the ends of tubules containing ERGIC53 and ts-O45-G (Fig. 8, e and f). Interestingly, NAGT1 (myc-tagged and stably expressed in Vero cells) and p58 (not shown) resumed an apparently normal Golgi distribution in about half of the cells injected with anti-EAGE within 1 h after removal of BFA (Fig. 9, c and d); in the other cells, NAGT1 remained in perinuclear clusters (e.g., lower injected cell in Fig. 9 c). No significant effect on Golgi reassembly was found in cells injected with anti-110-12 (Fig. 9, a and b). It appears that coatomer does not accumulate in a vesicular pattern at the periphery of the Golgi complex demarcated by a medial-Golgi resident protein (NAGT1) in cells injected with anti-EAGE (Fig. 9, c and d), as is typically observed in control cells or cells injected with anti-110-12 (see arrows in Fig. 9, a, b, e, and f).
Figure 8.

Effect of microinjected antiEAGE on transport of ts-O45-G following BFA wash-out. ts-O45 VSVinfected Vero cells were treated for 1 h at 39.5°C with 5 μg/ml BFA. These cells were then microinjected at nonpermissive temperature with Fab-fragments of anti-EAGE and coumarin conjugated BSA as an injection indicator. Cells were incubated for further 30 min at 39.5°C in the presence of BFA after microinjection and then fixed (a and b) or incubated in medium without BFA for one more hour at 31°C to wash-out the drug before fixation (c–f). Cells were immunolabeled for ERGIC 53 (b and d), ts-O45-G (a, c, and e), or injected anti-EAGE (f). Injected cells were identified in a–d by the co-injected coumarine BSA (not shown). ts-O45-G is arrested in tubular structures colocalizing with ERGIC 53 (arrowheads in c–e). Arrows in e and f indicate ts-O45-G positive dots which are labeled with the injected antibodies against β-COP. Bar, 10 μm.
Figure 9.
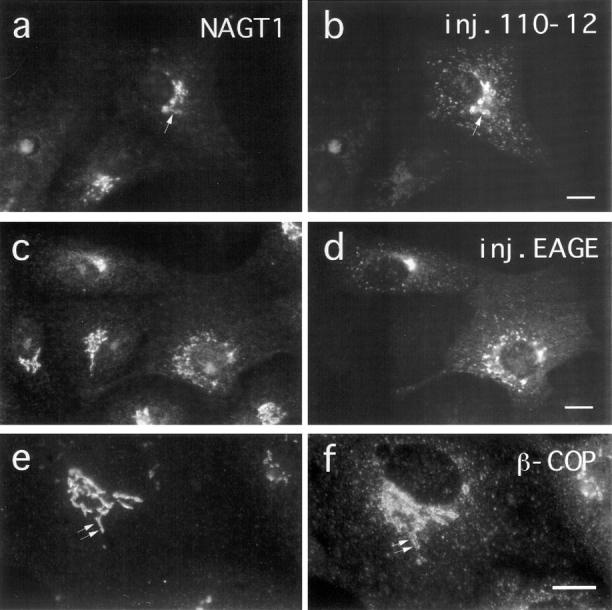
Effect of injected antibodies against β−COP on the localization of NAGT1 after BFA wash-out in VSV-infected cells. ts-O45 VSV-infected Vero cells were incubated for 1 h at 39.5°C with 5 μg/ml BFA and subsequently microinjected at 39.5°C with Fab-fragments of anti-EAGE or with anti-110-12 in the presence of BFA. Cells were transferred into normal medium immediately after injection and BFA washed-out at 31°C for 1 h. Cells were then fixed and stained for injected antibodies (b and d) and NAGT1 (a and c). Control cells were double labeled for NAGT1 (e) and β-COP (f) for comparison. In many cells anti-EAGE interfere with the proper reassembly of a compact Golgi complex in a juxtanuclear region (e.g., lower injected cells in d). Injected anti-EAGE also appears to interfere with the distribution of coatomer; β-COP and NAGT1 are more tightly colocalized in these injected cells (c and d) than in cells injected with anti110-12 or in control cells where β-COP is “wrapped-around” the NAGT1 positive membranes (arrows in a, b, e, and f). Bar, 10 μm.
Golgi complex reassembly during BFA wash-out was further studied by quantitative EM on thin sections of microinjected cells and determination of the volume density of Golgi membranes was performed. Golgi stacks were absent in BFA-treated cells before the wash-out of the drug (not shown). In uninjected control cells or cells injected with anti-110-12, Golgi stacks reappear rapidly after washout of BFA (Fig. 6 a). The percent cell volume of reformed Golgi stacks was however inhibited by ∼50% in cells microinjected with anti-EAGE (Fig. 6 a) and in contrast to the control cells, many of the BFA-induced tubulo-vesicular clusters remained (Fig. 6 b). No dense spherical aggregates have, however, been detected in cells injected with either of the two antibodies after BFA wash-out (Fig. 6 c). We thus conclude that reassembly of the Golgi complex is only partly inhibited by the injected anti-EAGE. Interestingly, biochemical analyses also revealed only ∼50% inhibition of acquisition of endo H resistance of ts-O45-G, although virtually no viral glycoprotein reaches the cell surface under these conditions (Pepperkok et al., 1993).
Anti-EAGE Stabilizes Membrane Binding of β-COP
Relocation of coatomer is a rapid and early event of BFA action (Donaldson et al., 1990). In cells injected with antiEAGE, however, β-COP remains associated with patches containing Golgi membrane proteins for at least 2 h after addition of the drug (Fig. 3, e and f). This is an effect specific for microinjected anti-EAGE. Analyses of early time points (1–20 min) after addition of the drug to cells microinjected with these antibodies gave no evidence for an intermittent dissociation of β-COP from membranes, indicating that β-COP remains associated with the membranes during their redistribution into patches. Thus anti-EAGE appears to inhibit the dissociation of coatomer from membranes in vivo. Since interference with the dynamics of membrane interactions of β-COP could be the mechanism by which anti-EAGE affects membrane traffic, we characterized the membrane binding of β-COP in a cell-free system.
A small fraction of β-COP remains associated in vitro with membranes enriched in Golgi and IC after incubation with rat liver cytosol (Fig. 10). This binding is significantly increased (up to threefold) in the presence of GTPγS, in agreement with previous reports (Donaldson et al., 1991). A 2–3-fold enhanced binding of coatomer to Golgi membranes, similar to the levels obtained with GTPγS, can also be observed in the presence of anti-EAGE (Fig. 10). This increased binding occurs also with monovalent Fab-fragments, can be completely competed by pre-incubation of anti-EAGE with the EAGE peptide, and is specific since it does not occur with control antibodies anti-110-12 (Fig. 10). Thus, anti-EAGE significantly increases the fraction of membrane-bound coatomer. These results and the block of BFA's activity to remove β-COP from membranes in living cells microinjected with anti-EAGE suggest that anti-EAGE interferes with the regulation of dissociation of β-COP from membranes, most likely by locking it together with the other subunits of coatomer, in its membrane-bound form. We assume that this inhibition of membrane dissociation of coatomer by anti-EAGE interferes with membrane traffic between the ER/IC and the Golgi complex.
Figure 10.
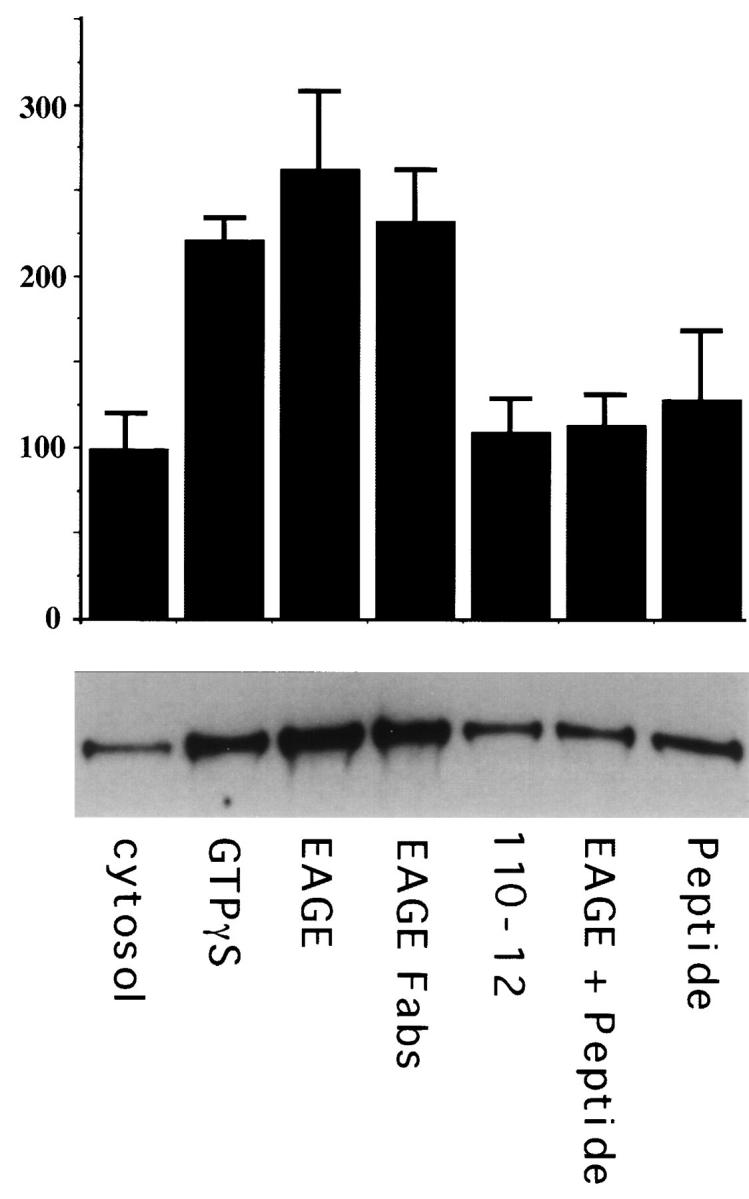
Anti-EAGE increases binding of β−COP to membranes in vitro. 14 μg of membranes enriched in IC and Golgi were incubated for 10 min at 37°C with 160 μg rat liver cytosol in a reaction mixture containing either no additions (cytosol), 25 μM GTPγS (GTPγS), 10 μg of anti-EAGE (EAGE), 10 μg Fab-fragments of anti-EAGE (EAGE-Fabs), 10 μg anti-110-12 (110-12), anti-EAGE preincubated with the EAGE peptide (EAGE + Peptide), or 30 μg EAGE peptide (Peptide). To block anti-EAGE antibodies 10 μg IgG was incubated with 30 μg EAGE peptide in PBS for 30 min on ice immediately before addition to the reaction. Membrane bound β−COP was detected by immunoblotting with M3A5 as described in Materials and Methods. Shown is a quantitation of the immunoblots from two duplicate experiments (top) and a corresponding autoradiogram (bottom). Error bars represent the standard mean error.
Discussion
We microinjected specific antibodies against β-COP to further dissect the role of coatomer in membrane traffic in living mammalian cells. We show here that microinjected antibodies directed against the EAGE-peptide of β-COP block BFA-induced transfer of resident Golgi enzymes to the ER, presumably by inhibiting the fusion of Golgi with ER membranes. Interestingly, relocation of KDEL receptor, a membrane protein cycling between the cis-Golgi and the IC, is not affected in these cells. Together with our previous results, which show inhibition of anterograde protein transport from the ER/IC to the Golgi complex by the same antibodies (Pepperkok et al., 1993), these results indicate that COPI is involved in the regulation of both anterograde (IC to Golgi) and retrograde (Golgi to ER) membrane traffic in the early exocytic pathway.
Anti-EAGE stabilizes in vitro binding of coatomer to membranes at least as efficiently as GTPγS, and in vivo, a significant fraction of coatomer remains bound to aggregates of membranes in cells treated with BFA. Out of two dozen antibodies raised against peptides along the sequence of β-COP only two (anti-EAGE, anti-110-12) bound to coatomer when injected into cells and only anti-EAGE interferes with coatomer function (Pepperkok et al., 1993). The other epitopes must thus be buried inside a protein fold, or are inaccessible within the stable hetero-oligomeric protein complex. Indeed, upon reversible disassembly of coatomer into its monomeric subunits these “hidden” epitopes of β-COP become accessible to the respective peptide antibodies (Lowe and Kreis, 1995). Thus, the domain around the EAGE-epitope appears to be a major site for heterologous liaisons involved in regulating membrane binding of coatomer. Anti-EAGE may either mask the site where a factor binds β-COP that regulates dissociation of coatomer from its membrane receptor(s), or interfere with a conformational change of β-COP essential for dissociation of coatomer from membranes. While strong evidence suggests that a subcomplex of coatomer composed of α-, β′-, and ε-COP binds to membrane proteins with an ER-retrieval motif (Cosson and Letourneur, 1994; Lowe and Kreis, 1995), and that another coatomer subcomplex (containing ζ- and γ-COP) may interact with members of the p24 membrane proteins with a putative “phenylalanine” anterograde transport motif (Fiedler et al., 1996; Harter et al., 1996), our data indicate that β-COP must be intimately involved in the regulation of membrane binding of the coatomer complex. Interestingly, binding of the α-, β′-, and ε-COP subcomplex to membranes in vitro is insensitive to GTPγS (Lowe and Kreis, 1995). It is possible that β-COP, perhaps together with δ-COP with which it interacts directly in the complex (Lowe and Kreis, 1995), may be involved in conferring ARF-dependent, GTP sensitivity to coatomer–membrane interaction.
In normal cells at steady state, membrane-bound coatomer visualized with antibodies against β-COP is on vesicular structures scattered throughout the cytoplasm or closely attached to, and surrounding, the Golgi complex (Duden et al., 1991; Kreis et al., 1995). Interestingly, while about half of the β-COP coated vesicular structures that can be identified immediately upon shifting tsO45-VSV–infected cells to the permissive temperature (after accumulation of ts-O45-G in the IC at 15°C) colocalize with ts-O45-G, the other half appears not to contain viral glycoprotein and may thus be recycling vesicles (Griffiths et al., 1995). This observation is consistent with the hypothesis that coatomer is involved in both anterograde and retrograde membrane transport. When injected anti-EAGE interfere with transport from the IC to the Golgi complex, cargo (e.g., ts-O45-G) is found in tubular structures containing ERGIC53 and β-COP (Pepperkok et al., 1993). However, when anti-EAGE interferes with BFA induced relocation of resident Golgi membrane proteins to the ER, virtually no overlap of β-COP with ERGIC-53 and KDEL receptor is observed, yet coatomer closely colocalizes with “Golgiderived cargo.” In both situations no obvious vesicular structures accumulate, but numerous aggregates can be seen with which β-COP is associated (see also Pepperkok et al., 1993; Oprins et al., 1993). Since these aggregates are significantly more abundant in injected, BFA-treated cells and contain Golgi resident membrane proteins, but not recycling KDEL receptor, ERGIC53, or ER proteins, they are most likely Golgi derived. These results are thus consistent with genetic data from yeast suggesting that coatomer is involved in regulating membrane transport from the Golgi complex to the ER.
Interestingly, injected anti-EAGE neither leads to accumulation of KDEL receptor in the Golgi complex in normal cells, nor inhibit BFA-dependent relocation of KDEL receptor to the IC. It has been reported that at steady state most of the KDEL receptor is localized to the cis-Golgi (Lewis and Pelham, 1992; Tang et al., 1993). Since the BFA-induced relocation of Golgi resident membrane proteins to the ER/IC aggregates is inhibited by injected antiEAGE, direct retrograde transport of KDEL receptor to the IC and BFA-induced transfer of Golgi resident proteins to the ER must follow independent routes. The simplest explanation for this finding is that the bulk of the KDEL receptor cycles at, or between, the interfaces between the ER (IC) and the Golgi complex (cis-Golgi network; see also Griffiths et al., 1994; Tang et al., 1995), and that in this cycle, recycling is independent of COPI that is recognized by anti-EAGE. Anti-EAGE will only affect relocation to the ER of resident Golgi proteins. This observation in fact further supports a role for COPI in anterograde early secretory membrane traffic. If recycling of an essential factor were inhibited by injected anti-EAGE, then KDEL receptor retrieval should also be affected. It is possible that in normal cells anti-EAGE affects anterograde COPI-dependent membrane traffic more effectively, because it binds to this form of coatomer with higher affinity. This possibility is fully consistent with the hypothesis that coatomer is involved in more than one transport step, and that for each distinct transport step, coatomer has different conformation or composition (i.e., different posttranslational modification of subunits, different subunit isoforms, etc.; see Whitney et al., 1995; Scheff et al., 1996; Fiedler et al., 1996; Lowe and Kreis, 1996).
We consider it likely that anti-EAGE inhibits the budding of COPI-coated vesicles in vivo. Indeed, the number of COPI-coated vesicles decreases significantly in microinjected cells (Pepperkok et al., 1993). This is in contrast to the action of GTPγS, which also stabilizes binding of coatomer to membranes and inhibits BFA-induced relocation of resident Golgi proteins to the ER in permeabilized cells (Donaldson et al., 1991), but does not interfere with the formation of COPI-coated vesicles in vitro (Melançon et al., 1987). The nature of the accumulating tubulo-vesicular clusters in BFA-treated cells injected with anti-EAGE (many of which remain after wash-out of the drug) is unclear; they probably represent ER/IC membranes that accumulate when early exocytic membrane traffic is inhibited by the injected antibodies and membranes pile up as COPI-coated vesicles cannot bud (see also Pepperkok et al., 1993). On the other hand, the aggregates (containing resident Golgi transmembrane proteins) that form in the BFA-treated cells injected with anti-EAGE are most likely remnants of Golgi complex derived membranes which cannot fuse with ER membranes as a consequence of the antibody induced stabilization of membrane bound coatomer. It is tempting to speculate that components of the membrane fusion machinery (e.g., v-SNAREs) are present in these aggregates and that they are inactive while covered by coatomer.
Anti-EAGE inhibits reformation of Golgi stacks upon BFA wash-out to only ∼50%. This indicates that two mechanisms may lead to Golgi complex reassembly, one COPI dependent and one COPI independent. This is consistent with previous observations that not all Golgi membranes partition into the ER upon treatment of cells with BFA (Oprins et al., 1993; Orci et al., 1993; Hendricks et al., 1993). In addition, two functionally different domains have been predicted within Golgi cisternae based on two different pathways leading to mitotic Golgi disassembly (Misteli and Warren, 1995), and postmitotic reassembly of the Golgi complex is regulated by two distinct fusion events depending on NSF-SNAPs-p115 and p97 (Rabouille et al., 1995; Acharya et al., 1995). Interestingly however, although coatomer is stably bound to membranes of the Golgi complex in cells injected with anti-EAGE, and coatomerdependent budding of vesicles appears inhibited, BFA still leads to the complete disappearance of stacked Golgi cisternae. This result suggests that the BFA induced morphological changes of the Golgi complex cannot be attributed alone to dissociation of COPI from Golgi membranes. Other factors (e.g., ARF), closer in the cascade of events to the action of BFA, may be more directly responsible for this process.
Microinjected anti-EAGE appear to inhibit membrane traffic from the IC to the cis-Golgi by a different mechanism from that by which they inhibit BFA-induced relocation of Golgi enzymes to the ER. Only ∼50% of the normal transport of newly synthesized ts-O45-G to the Golgi is inhibited by the injected antibodies (Pepperkok et al., 1993), whereas virtually no BFA-induced transfer of Golgi glycosidases to the ER can be measured in cells injected with anti-EAGE. While the precise reason for this major difference remains unclear, several possibilities can be discussed. It could for example be argued that coatomer alone is essential for all retrograde transport, but to some extent redundant (with COPII) for transport in the anterograde direction. It could also be speculated that significantly more binding sites for coatomer reside on membranes of the Golgi complex (consistent with the predominant localization of COPI to the region of the Golgi complex in normal cells) and that as a consequence inhibition of BFA- induced Golgi to ER transport by injected anti-EAGE is more efficient than is transport from the IC to the cisGolgi. Anti-EAGE may also more efficiently stabilize Golgi membrane bound coatomer due to different conformations of coatomer subcomplexes (Lowe and Kreis, 1995, 1996; Fiedler et al., 1996) primed to produce retrograde or anterograde directed transport vesicles. Clearly, further experiments will be required to resolve this conundrum.
Given the recent evidence for a role of a coat immunologically related to COPI in the endocytic pathway (Whitney et al., 1995; Aniento et al., 1996) and the presence of COPI subunit isoforms generated by differential phosphorylation (Sheff et al., 1996), it would not be surprising if different forms of coatomer mediated anterograde and retrograde transport between the ER/IC and the Golgi complex. Alternatively, COPI-coated vesicles may provide a “paternoster” continuously recycling immature cargo, its receptors, as well as membrane proteins of the vesicle docking and fusion machinery between the ER/IC and the Golgi complex. One signal for release of cargo into the Golgi complex would then be its dissociation from chaperones which are part of the sequential quality control machinery (Hammond and Helenius, 1995). Simultaneous visualization of movement of fluorescently modified cargo and coat proteins in living cells may provide further insight into the mechanisms of regulation of early secretory membrane traffic.
Acknowledgments
We thank Jean Gruenberg and Andy Whitney for helpful comments on the manuscript, Brian Storrie for the NAGT1-Vero cells, and George Bloom, Steve Fuller, Hans-Peter Hauri, Ari Helenius, Wanjin Hong, and Bor Luen Tang for antibodies; we appreciated Heinz Horstmann's help with electron microscopy.
Abbreviations used in this paper
- BFA
brefeldin A
- COP
coat protein
- EM
electron microscopy
- endo H
endoglycosidase H
- IC
intermediate compartment
Footnotes
R. Pepperkok was a recipient of a European Molecular Biology Organization (EMBO) longterm postdoctoral fellowship and M. Lowe was supported by a Traveling Research Fellowship from the Welcome Trust. T.E. Kreis was supported by grants from the Fonds Nationale Suisse, the Canton de Genève, and the International Human Frontier Science Program.
J. Scheel and R. Pepperkok contributed equally to this work and are first co-authors.
References
- Acharya U, McCaffery JM, Jacobs R, Malhotra V. Reconstitution of vesiculated Golgi membranes into stacks of cisternae: requirement of NSF in stack formation. J Cell Biol. 1995;129:577–589. doi: 10.1083/jcb.129.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan VJ, Kreis TE. A microtubule-binding protein associated with membranes of the Golgi apparatus. J Cell Biol. 1986;103:2229–2239. doi: 10.1083/jcb.103.6.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aniento F, Gu F, Parton RG, Gruenberg J. An endosomal βCOP is involved in the pH-dependent formation of transport vesicles destined for late endosomes. J Cell Biol. 1996;133:29–41. doi: 10.1083/jcb.133.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arar C, Carpentier V, Le Caer JP, Monsigny M, Legrand A, Roche AC. ERGIC-53, a membrane protein of the endoplasmic reticulumGolgi intermediate compartment, is identical to MR60, an intracellular mannose-specific lectin of myelomonocytic cells. J Biol Chem. 1995;270:3551–3553. doi: 10.1074/jbc.270.8.3551. [DOI] [PubMed] [Google Scholar]
- Aridor M, Bannykh SI, Rowe T, Balch WE. Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J Cell Biol. 1995;131:875–893. doi: 10.1083/jcb.131.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R. COPII: a membrane coat formed by sec proteins that drive vesicle budding from the Endoplasmic Reticulum. Cell. 1994;77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Bednarek SY, Ravazzola M, Hosobuchi M, Amherdt M, Perrelet A, Schekman R, Orci L. COPI- and COPII-coated vesicles bud directly from the endoplasmic reticulum in yeast. Cell. 1995;83:1183–1196. doi: 10.1016/0092-8674(95)90144-2. [DOI] [PubMed] [Google Scholar]
- Bloom GS, Brashear TA. A novel 58-kDa protein associates with the Golgi apparatus and microtubules. J Biol Chem. 1989;264:16083–16092. [PubMed] [Google Scholar]
- Boman AL, Kahn RA. Arf proteins: the membrane traffic police? . Trends Biochem Sci. 1995;20:147–150. doi: 10.1016/s0968-0004(00)88991-4. [DOI] [PubMed] [Google Scholar]
- Cosson P, Démollière C, Hennecke S, Duden R, Letourneur F. δ- and ζ-COP, two coatomer subunits homologous to clathrin-associated proteins, are involved in ER retrieval. EMBO (Eur Mol Biol Organ) J. 1996;15:1792–1798. [PMC free article] [PubMed] [Google Scholar]
- Cosson P, Letourneur F. Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science (Wash DC) 1994;263:1629–1631. doi: 10.1126/science.8128252. [DOI] [PubMed] [Google Scholar]
- Doms RW, Russ G, Yewdell J. Brefeldin A redistributes resident and itinerant Golgi proteins to the endoplasmic reticulum. J Cell Biol. 1989;109:61–72. doi: 10.1083/jcb.109.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG, Finazzi D, Klausner RD. Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature (Lond) 1992;360:350–352. doi: 10.1038/360350a0. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Lippincott-Schwartz J, Klausner RD. Guanine nucleotides modulate the effects of brefeldin A in semipermeable cells: regulation of the association of a 110-kD peripheral membrane protein with the Golgi complex. J Cell Biol. 1991;112:579–588. doi: 10.1083/jcb.112.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG, Lippincott-Schwartz J, Bloom GS, Kreis TE, Klausner RD. Binding of ARF and β-COP to Golgi membranes: possible regulation by trimeric G protein. Science (Wash DC) 1991;254:1197–1199. doi: 10.1126/science.1957170. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Lippincott-Schwartz J, Bloom GS, Kreis TE, Klausner RD. Dissociation of a 110-kD peripheral membrane protein from the Golgi apparatus is an early event in brefeldin A action. J Cell Biol. 1990;111:2295–2306. doi: 10.1083/jcb.111.6.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duden R, Hosobuchi M, Hamamoto S, Winey M, Byers B, Schekman R. Yeast β-andβ′-coat proteins (COP). Two coatomer subunits essential for endoplasmic reticulum-to-Golgi protein traffic. J Biol Chem. 1994;269:24486–24495. [PubMed] [Google Scholar]
- Duden R, Griffiths G, Frank R, Argos P, Kreis TE. β-COP, a 110 kD protein associated with non-clathrin-coated vesicles and the Golgi complex, shows homology to β-adaptin. Cell. 1991;64:649–665. doi: 10.1016/0092-8674(91)90248-w. [DOI] [PubMed] [Google Scholar]
- Elazar Z, Orci L, Ostermann J, Amherdt M, Tanigawa G, Rothman JE. ADP-ribosylation factor and coatomer couple fusion to vesicle budding. J Cell Biol. 1994;124:415–424. doi: 10.1083/jcb.124.4.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler K, Veit M, Stamnes MA, Rothman JE. Bimodal interaction of coatomer with the p24 family of putative cargo receptors. Science (Wash DC) 1996;273:1396–1399. doi: 10.1126/science.273.5280.1396. [DOI] [PubMed] [Google Scholar]
- Griffiths G, Pepperkok R, Locker JK, Kreis TE. Immunocytochemical localization of β-COP to the ER-Golgi boundary and the TGN. J Cell Sci. 1995;108:2839–2856. doi: 10.1242/jcs.108.8.2839. [DOI] [PubMed] [Google Scholar]
- Griffiths G, Pfeiffer S, Simons K, Matlin K. Exit of newly synthesized membrane proteins from the trans cisterna of the Golgi complex to the plasma membrane. J Cell Biol. 1985;101:949–964. doi: 10.1083/jcb.101.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths G, Ericsson M, Krijnse-Locker J, Nilsson T, Goud B, Soling HD, Tang SH, Wong SH, Hong W. Localization of the Lys, Asp, Glu, Leu tetrapeptide receptor to the Golgi complex and the intermediate compartment in mammalian cells. J Cell Biol. 1994;127:1557–1574. doi: 10.1083/jcb.127.6.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harter C, Pavel J, Draken E, Wehehingel S, Tschochner H, Wieland F. Nonclathrin coat protein gamma, a subunit of coatomer, binds to the cytoplasmic dilysine motif of membrane proteins of the early secretory pathway. Proc Natl Acad Sci USA. 1996;93:1902–1906. doi: 10.1073/pnas.93.5.1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C, Helenius A. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J Cell Biol. 1994;126:41–52. doi: 10.1083/jcb.126.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C, Helenius A. Quality control in the secretory pathway. Curr Opin Cell Biol. 1995;7:523–529. doi: 10.1016/0955-0674(95)80009-3. [DOI] [PubMed] [Google Scholar]
- Helms JB, Rothman JE. Inhibition by brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature (Lond) 1992;360:352–354. doi: 10.1038/360352a0. [DOI] [PubMed] [Google Scholar]
- Hendricks LC, McCaffery M, Palade GE, Farquhar MG. Disruption of endoplasmic reticulum to Golgi transport leads to the accumulation of large aggregates containing β-COP in pancreatic acinar cells. Mol Biol Cell. 1993;4:413–424. doi: 10.1091/mbc.4.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker W, Whitney JA, Mellman I. Selective inhibition of transcytosis by brefeldin A in MDCK cells. Cell. 1991;67:617–627. doi: 10.1016/0092-8674(91)90535-7. [DOI] [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG, Lippincott-Schwartz J. Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis TE, Lowe M, Pepperkok R. COPs regulating membrane traffic. Annu Rev Cell Dev Biol. 1995;11:677–706. doi: 10.1146/annurev.cb.11.110195.003333. [DOI] [PubMed] [Google Scholar]
- Kreis TE. Microinjected antibodies against the cytoplasmic domain of vesicular stomatitis virus glycoprotein block its transport to the cell surface. EMBO (Eur Mol Biol Organ) J. 1986;5:931–941. doi: 10.1002/j.1460-2075.1986.tb04306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis TE, Lodish HF. Oligomerization is essential for transport of vesicular stomatitis viral glycoprotein to the cell surface. Cell. 1986;46:929–937. doi: 10.1016/0092-8674(86)90075-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladinsky MS, Kremer JR, Furcinitti PS, McIntosh JR, Howell KE. HVEM tomography of the trans-Golgi network: structural insights and identification of a lace-like vesicle coat. J Cell Biol. 1994;127:29–38. doi: 10.1083/jcb.127.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneur F, Gaynor EC, Hennecke S, Demolliere C, Duden R, Emr SD, Riezman H, Cosson P. Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell. 1994;79:1199–1207. doi: 10.1016/0092-8674(94)90011-6. [DOI] [PubMed] [Google Scholar]
- Lewis MJ, Pelham HR. Ligand-induced redistribution of the human KDEL receptor from the Golgi complex to the endoplasmic reticulum. Cell. 1992;68:353–364. doi: 10.1016/0092-8674(92)90476-s. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Yuan LC, Bonifacino JS, Klausner RD. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from the Golgi to ER. Cell. 1989;56:801–813. doi: 10.1016/0092-8674(89)90685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Yuan L, Tipper C, Amherdt M, Orci L, Klausner RD. Brefeldin A's effects on endosomes, lysosomes, and the TGN suggest a general mechanism for regulating organelle structure and membrane traffic. Cell. 1991;67:601–616. doi: 10.1016/0092-8674(91)90534-6. [DOI] [PubMed] [Google Scholar]
- Lippincott Schwartz, J., N.B. Cole, A. Marotta, P.A. Conrad, and G.S. Bloom. Kinesin is the motor for microtubule-mediated Golgi-to-ER membrane traffic. J Cell Biol. 1995;128:293–306. doi: 10.1083/jcb.128.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe M, Kreis TE. In vitro assembly and disassembly of coatomer. J Biol Chem. 1995;270:31364–31371. doi: 10.1074/jbc.270.52.31364. [DOI] [PubMed] [Google Scholar]
- Lowe M, Kreis TE. In vivo assembly of coatomer, the COP-I coat precursor. J Biol Chem. 1996;271:30725–30730. doi: 10.1074/jbc.271.48.30725. [DOI] [PubMed] [Google Scholar]
- Malhotra V, Serafini T, Orci L, Shepherd JC, Rothman JE. Purification of a novel class of coated vesicles mediating biosynthetic protein transport through the Golgi stack. Cell. 1989;58:329–336. doi: 10.1016/0092-8674(89)90847-7. [DOI] [PubMed] [Google Scholar]
- Melançon P, Glick BS, Malhotra V, Weidmann PJ, Serafini T, Gleason ML, Orci L, Rothman JE. Involvement of GTP-binding “G” proteins in transport through the Golgi stack. Cell. 1987;51:1053–1062. doi: 10.1016/0092-8674(87)90591-5. [DOI] [PubMed] [Google Scholar]
- Miesenbock G, Rothman JE. The capacity to retrieve escaped ER proteins extends to the trans-most cisterna of the Golgi stack. J Cell Biol. 1995;129:309–319. doi: 10.1083/jcb.129.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T, Warren G. A role for tubular networks and a COP I-independent pathway in the mitotic fragmentation of Golgi stacks in a cell-free system. J Cell Biol. 1995;130:1027–1039. doi: 10.1083/jcb.130.5.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misumi Y, Misumi Y, Miki K, Takatsuki A, Tamura G, Ikehara Y. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J Biol Chem. 1986;261:11398–11403. [PubMed] [Google Scholar]
- Narula N, Stow JL. Distinct coated vesicles labeled for p200 bud from trans-Golgi network membranes. Proc Natl Acad Sci USA. 1995;92:2874–2878. doi: 10.1073/pnas.92.7.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda K, Hirose S, Takami N, Misumi Y, Takatsuki A, Ikehara Y. Brefeldin A arrests the intracellular transport of a precursor of complement C3 before its conversion site in rat hepatocytes. FEBS Lett. 1987;214:135–138. doi: 10.1016/0014-5793(87)80028-5. [DOI] [PubMed] [Google Scholar]
- Oprins A, Duden R, Kreis TE, Geuze HJ, Slot JW. β-COP localizes mainly to the cis-Golgi side in exocrine pancreas. . J Cell Biol. 1993;121:49–59. doi: 10.1083/jcb.121.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L, Perrelet A, Ravazzola M, Wieland FT, Schekman R, Rothman JE. “BFA bodies”: a subcompartment of the endoplasmic reticulum. Proc Natl Acad Sci USA. 1993;90:11089–11093. doi: 10.1073/pnas.90.23.11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L, Tagaya M, Amherdt M, Perrelet A, Donaldson JG, Lippincott J, Schwartz, Klausner RD, Rothman JE. Brefeldin A, a drug that blocks secretion, prevents the assembly of non-clathrin-coated buds on Golgi cisternae. Cell. 1991;64:1183–1195. doi: 10.1016/0092-8674(91)90273-2. [DOI] [PubMed] [Google Scholar]
- Pelham HR. Sorting and retrieval between the endoplasmic reticulum and Golgi apparatus. Curr Opin Cell Biol. 1995;7:530–535. doi: 10.1016/0955-0674(95)80010-7. [DOI] [PubMed] [Google Scholar]
- Pelham HRB. Multiple targets for brefeldin A. Cell. 1991;67:449–451. doi: 10.1016/0092-8674(91)90517-3. [DOI] [PubMed] [Google Scholar]
- Pepperkok R, Scheel J, Horstmann H, Hauri HP, Griffiths G, Kreis TE. β-COP is essential for biosynthetic membrane transport from the endoplasmic reticulum to the Golgi complex in vivo. Cell. 1993;74:71–82. doi: 10.1016/0092-8674(93)90295-2. [DOI] [PubMed] [Google Scholar]
- Pierre P, Pepperkok R, Kreis TE. Molecular characterization of two functional domains of CLIP-170 in vivo. J Cell Sci. 1994;107:1909–1920. doi: 10.1242/jcs.107.7.1909. [DOI] [PubMed] [Google Scholar]
- Rabouille C, Levine TP, Peters JM, Warren G. An NSF-like ATPase, p97, and NSF mediate cisternal regrowth from mitotic Golgi fragments. Cell. 1995;82:905–914. doi: 10.1016/0092-8674(95)90270-8. [DOI] [PubMed] [Google Scholar]
- Robinson MS. The role of clathrin, adaptors and dynamin in endocytosis. Curr Opin Cell Biol. 1994;6:538–544. doi: 10.1016/0955-0674(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Robinson MS, Kreis TE. Recruitment of coat proteins onto Golgi membranes in intact and permeabilized cells: effects of brefeldin A and G protein activators. Cell. 1992;69:129–138. doi: 10.1016/0092-8674(92)90124-u. [DOI] [PubMed] [Google Scholar]
- Rothman JE. Mechanisms of intracellular protein transport. Nature (Lond) 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Rothman JE, Warren G. Implications of the SNARE hypothesis for intracellular membrane topology and dynamics. Curr Biol. 1994;4:220–233. doi: 10.1016/s0960-9822(00)00051-8. [DOI] [PubMed] [Google Scholar]
- Schekman R, Orci L. Coat proteins and vesicle budding. Science (Wash DC) 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Schindler R, Itin C, Zerial M, Lottspeich F, Hauri HP. ERGIC53, a membrane protein of the ER-Golgi intermediate compartment, carries an ER retention motif. Eur J Cell Biol. 1993;61:1–9. [PubMed] [Google Scholar]
- Schweizer A, Fransen JAM, Bächi T, Ginsel L, Hauri H-P. Identification, by a monoclonal antibody, of a 53-kD protein associated with a tubulo-vesicular compartment at the cis-side of the Golgi complex. J Cell Biol. 1988;107:1643–1653. doi: 10.1083/jcb.107.5.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaywitz DA, Orci L, Ravazzola M, Swaroop A, Kaiser CA. Human SEC13Rp functions in yeast and is located on transport vesicles budding from the endoplasmic reticulum. J Cell Biol. 1995;128:769–777. doi: 10.1083/jcb.128.5.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheff D, Lowe M, Kreis TE, Mellman I. Biochemical heterogeneity and phosphorylation of coatomer subunits. J Biol Chem. 1996;271:7230–7236. doi: 10.1074/jbc.271.12.7230. [DOI] [PubMed] [Google Scholar]
- Stamnes MA, Rothman JE. The binding of AP-1 clathrin adaptor particles to Golgi membranes requires ADP-ribosylation factor, a small GTP-binding protein. Cell. 1993;73:999–1005. doi: 10.1016/0092-8674(93)90277-w. [DOI] [PubMed] [Google Scholar]
- Tang BL, Low SH, Hauri H-P, Hong W. Segregation of ERGIC53 and the mammalian KDEL receptor upon exit from the 15°C compartment. Eur J Cell Biol. 1995;68:398–410. [PubMed] [Google Scholar]
- Tang BL, Wong SH, Qi XL, Low SH, Hong W. Molecular cloning, characterization, subcellular localization and dynamics of p23, the mammalian KDEL receptor. J Cell Biol. 1993;120:325–338. doi: 10.1083/jcb.120.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmer JB, Palade GE. Effects of Brefeldin A on the Golgi complex, endoplasmic reticulum and viral envelope glycoproteins in murine erythroleukemia cells. Eur J Cell Biol. 1991;54:38–54. [PubMed] [Google Scholar]
- Vaux D, Tooze J, Fuller S. Identification by anti-idiotype antibodies of an intracellular membrane protein that recognizes a mammalian endoplasmic reticulum retention signal. Nature (Lond) 1990;345:495–502. doi: 10.1038/345495a0. [DOI] [PubMed] [Google Scholar]
- Weibel, E.R. 1979. Stereological Methods: Practical Methods for Biological Morphometry. Vol. 1. Academic Press, NY.
- Whitney JA, Gomez M, Sheff D, Kreis TE, Mellman I. Cytoplasmic coat proteins involved in endosome function. Cell. 1995;83:703–713. doi: 10.1016/0092-8674(95)90183-3. [DOI] [PubMed] [Google Scholar]
- Wood SA, Park JE, Brown WJ. Brefeldin A causes a microtubule-mediated fusion of the trans-Golgi network and early endosomes. Cell. 1991;67:591–600. doi: 10.1016/0092-8674(91)90533-5. [DOI] [PubMed] [Google Scholar]
- Wuestehube L, Duden R, Eun A, Hamamoto S, Korn P, Ram R, Schekman R. New mutants of Saccharomyces cerevisiaeaffected in the transport of proteins from the endoplasmic reticulum to the Golgi complex. Genetics. 1996;142:393–406. doi: 10.1093/genetics/142.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]