

Abstract
We have identified a human Bcl-2–interacting protein, p28 Bap31. It is a 28-kD (p28) polytopic integral protein of the endoplasmic reticulum whose COOH-terminal cytosolic region contains overlapping predicted leucine zipper and weak death effector homology domains, flanked on either side by identical caspase recognition sites. In cotransfected 293T cells, p28 is part of a complex that includes Bcl-2/Bcl-XL and procaspase-8 (pro-FLICE). Bax, a pro-apoptotic member of the Bcl-2 family, does not associate with the complex; however, it prevents Bcl-2 from doing so. In the absence (but not presence) of elevated Bcl-2 levels, apoptotic signaling by adenovirus E1A oncoproteins promote cleavage of p28 at the two caspase recognition sites. Purified caspase-8 (FLICE/MACH/Mch5) and caspase-1(ICE), but not caspase-3 (CPP32/apopain/ Yama), efficiently catalyze this reaction in vitro. The resulting NH2-terminal p20 fragment induces apoptosis when expressed ectopically in otherwise normal cells. Taken together, the results suggest that p28 Bap31 is part of a complex in the endoplasmic reticulum that mechanically bridges an apoptosis-initiating caspase, like procaspase-8, with the anti-apoptotic regulator Bcl-2 or Bcl-XL. This raises the possibility that the p28 complex contributes to the regulation of procaspase-8 or a related caspase in response to E1A, dependent on the status of the Bcl-2 setpoint within the complex.
Despite the complexity of signals that can induce apoptotic programmed cell death, many appear to converge on a common execution pathway that is initiated upon pro-enzyme activation of the Ced-3/ICE (caspase) family of cysteine proteases (Kumar and Lavin, 1996; Alnemri et al., 1996). There are at least 10 known members of the family whose activities lead to site-specific cleavage and consequent activation/inactivation of various target molecules. Additionally, these enzymes may operate as part of a cascade in which initiator caspases such as caspase-8 (FLICE/MACH/Mch5) and caspase-10 (Mch4) activate downstream effector caspases such as caspase-3 (CPP32/apopain/Yama; Fernandes-Alnemri et al., 1996; Srinivasula et al., 1996; Muzio et al., 1997). Insight into the likely mechanism of activation of initiator caspases has come with the finding that the large pro-regions of initiator caspases contain a death effector domain that physically links these pro-enzymes to an apoptotic signaling complex. In the case of the Fas (CD95/Apo-1)/TNFR-1 complexes, recruitment of procaspase-8 involves the adaptor molecule FADD via interactions between the death effector domains within the two molecules (Boldin et al., 1996; Muzio et al., 1996). Fas and TNFR-1, however, are highly restricted in the death signals to which they respond. An important question, therefore, is the extent to which the Fas/TNFR-1 paradigm extends to other apoptotic signaling pathways.
For many of these other death pathways, the decision to induce apoptosis in response to specific death signals depends on the status of various cellular regulators, including p53 and the Bcl-2Bax family set point (White, 1996; Yang and Korsmeyer, 1996). The latter arises through heterodimerization between the Bcl-2/Bcl-XL (Hockenbery et al., 1990; Strasser et al., 1991; Boise et al., 1993) and Bax/Bak (Oltvai et al., 1993; Chittenden et al., 1995; Farrow et al., 1995; Kiefer et al., 1995) family of suppressors and promoters, respectively, in which the ratio of the heterodimerizing partners determines the outcome (cell death or cell survival) in response to various death signals. Bad, a more distantly related family member, is a regulator of the set point (Yang et al., 1995) by a mechanism that is governed by phosphorylation (Zha et al., 1996). This may involve Bcl-2–dependent recruitment of Raf-1 kinase (Wang et al., 1996a ), albeit indirectly.
Although it is now known that Bcl-2/Bcl-XL controls the apoptotic execution pathway at a point that is either at or upstream of pro-enzyme activation of CPP32 (Armstrong et al., 1996; Boulakia et al., 1996; Chinnaiyan et al., 1996), how this is achieved remains to be elucidated. It is undoubtedly relevant, however, that Bcl-2/Bcl-XL family members are integral membrane proteins with a restricted subcellular distribution. They are anchored in the mitochondrial outer membrane and ER/nuclear envelope via a single COOH-terminal transmembrane segment, leaving a protease-sensitive domain exposed to the cytosol (Krajewski et al., 1993; Nguyen et al., 1993; Gonzalez-Garcia et al., 1994). Recent studies have revealed intriguing pore-forming properties for the cytosolic domain of Bcl-XL in synthetic lipid bilayers (Minn et al., 1997), raising the possibility that the cytosolic domain may have the potential to influence channel activities in these organelles. Mitochondria and ER, either in parallel or in cooperation, are well documented to control a number of electrochemical events that may be linked to the induction of apoptosis and to come under the control of Bcl-2 (Hockenbery et al., 1993; Gottlieb et al., 1996; Lam et al., 1994). Moreover, mitochondria may export pro-apoptotic molecules by a Bcl-2–regulated mechanism (Kluck et al., 1997; Kroemer et al., 1997; Yang et al., 1997); one of these, cytochrome c, activates the caspase-3 zymogen when added to cytosolic extracts. Whether or not mitochondria and ER contribute to proximal events during the initiation of the caspase cascade, however, remains to be elucidated. If so, Bcl-2/Bcl-XL may be strategically positioned within the cell to influence both upstream and downstream events in this pathway, perhaps involving coordinated signaling between the two organelles.
In addition to dimerizing members of the Bcl-2 family itself, several proteins have been suggested as candidate targets for Bcl-2 interaction (Fernandez-Sarabia and Bischoff, 1993; Boyd et al., 1994; Wang et al., 1994; Naumovski and Cleary, 1996). With the exception of Raf-1 and its Bcl- 2–interacting effector, Bag-1 (Wang et al., 1996a ,b), however, the majority of these have not been linked functionally to apoptosis. Here, we describe a polytopic integral membrane protein of the ER, p28 Bap31, that is part of a complex that contains Bcl-2 proteins and procaspase-8. In the absence of Bcl-2, p28 itself becomes a target of a FLICE/ICE-related caspase upon induction of apoptosis, resulting in removal of a cytosolic segment that contains overlapping death effector and leucine zipper homology domains. The resulting product, p20, induces apoptosis upon ectopic expression in transfected cells. It may be that p28 is a Bcl-2–regulated component of an apoptosis signaling pathway, possibly involving coordinated ER–mitochondrial events.
Materials and Methods
Cells and Viruses
Human KB cells expressing the neomycin resistance gene (neo) either alone or together with BCL-2 (Nguyen et al., 1994) were cultured in α-MEM supplemented with 10% FBS and 100 U/ml streptomycin and penicillin. After reaching 80% confluency, the medium was replaced with fresh medium containing either no virus or 25–35 PFU/cell adenovirus type 5 lacking expression of E1B 19K (pm1716/2072; McLorie et al., 1991) or adenovirus type 5 expressing only the 243R form (12S) of E1A and no E1B products (dl520E1B−; Shepherd et al., 1993). After incubation for 1 h at 37°C, fresh medium was added and cells were collected at various times for analysis. Both forms of the virus elicit a cytotoxic response in infected cells that exhibit all of the hallmark features of apoptosis (Nguyen et al., 1994; Teodoro et al., 1995).
Bacterial Expression and Purification of 32P-labeled Bcl-2 Cytosolic Domain for Ligang Blot (Far Western) Analyses
cDNA encoding the cytosolic domain of human Bcl-2 (i.e., lacking the COOH-terminal 21 amino acids) was inserted into the pTrchis vector (Invitrogen, Carlsbad, CA), and standard PCR methodology was used to extend hexahistidine at the COOH terminus to include a heart muscle kinase recognition sequence using the oligonucleotides 5′-CTAGCGCCCGCCGCGCCTCTGTGGAATTCTGAA and 5′-AGCTTTCAGAATTCCACAGAGGCGCGGCGGGCG-3′. The final construct encoded Bcl-2 (amino acids 1–218), hexahis, Arg, Arg, Ala, Ser-COOH, and the protein designated Bcl-2Δc21/his6/HMK. The Bcl-2 portion contained three additional mutations that were introduced for reasons not related to this project (Met 16 to Leu; Lys 17 to Arg; Lys 22 to Arg). Stable epithelial cell lines that express full length Bcl-2 harboring these mutations were found to be as effective as cells expressing wild-type Bcl-2 in countering apoptotic death stimuli (not shown).
Escherichia coli MC1061 was transformed with pBcl-2Δc21/his6/HMK. When 500-ml cultures reached an A600 of 0.6, they were treated with 1.0 mM IPTG, and cells were recovered by centrifugation 4 h later. Packed cells (2.5–3.0 ml) were suspended in 15 ml extraction medium (20 mM Na phosphate, pH 7.4, 0.5 M NaCl, 0.05% vol/vol Triton X-100, 10 mM β-mercaptoethanol, 1.0 mM PMSF, and 1.0 mM benzamidine) and were sonicated eight times with a Vibra Cell probe sonicator (Sonics & Materials, Inc., Danbury, CT) operating at setting 7.5 for 15 s at 4°C. The sonicate was adjusted to 10% (vol/vol) glycerol and centrifuged at 25,000 rpm for 30 min at 4°C in a Ti 50.2 rotor (Beckman Instruments, Inc., Fullerton, CA). The supernatant was added to 1.2 ml Ni2+-NTA agarose (QIAGEN Inc., Chatsworth, CA; a 1:1 [vol/vol] mixture with extraction medium) and incubated for 1.5 h at 4°C. The beads were washed extensively in extraction medium containing 20% (vol/vol) glycerol and 22 mM imidazole, and Bcl-2Δc21/his6/HMK was eluted in extraction medium containing 20% glycerol and 0.3M imidazole. 1 liter of induced culture yielded 0.8–1.0 mg protein that was >95% pure. The purified protein was labeled with 32P after incubation with heart muscle kinase (HMK)1 and 32P[γ-ATP], yielding 2.0–2.5 × 106 cpm/μg protein, and it was used for ligand blotting as described in Blanar and Rutter (1992).
Purification and Identification of p20 Fragment
KB cells were cultured in 20 15-cm plates until 80% confluency was reached, and infected with 25 pfu/cell adenovirus type 5 lacking expression of E1B 19K (pm1716/2072). After 60 h, total cells (65–70% nonviable, as judged by exclusion of trypan blue) were collected and rinsed, and the packed cells (∼1.5 ml) were suspended in 6 ml ice-cold lysis medium containing 10 mM Tris HCl, pH 7.5, 140 mM NaCl, 1.5 mM MgCl2, 0.5% Triton X-100, and 1 mM PMSF) and separated into three equal portions. Each was subjected to sonication for 4 X 10 s using an Artek probe sonicator (Artek Systems Corp., Farmingdale, NY) operating at setting 6.0. The combined sonicates were centrifuged at 11,000 g for 20 min, and the supernatant was mixed with 0.25 vol of 5× SDS sample buffer (250 mM Tris HCl, pH 6.8, 50% glycerol, 0.5% bromophenol blue, 10% SDS, and 1 M DTT). The total volume was subjected to preparative 14% SDS-PAGE using a Prep Cell 491 system (Bio Rad Laboratories, Hercules, CA) fitted with a 37-mm diam resolving gel chamber. Fractions were collected at a flow rate of 1 ml/min, assayed for the presence of p20 by ligand blotting using 32P-Bcl-2Δc21/his6/HMK as a probe, and the reactive peak fractions combined and concentrated fivefold in a Centriprep-10 concentrator (Amicon, Inc., Beverly, MA). The concentrated sample was mixed with an equal volume of 0.12% trifluoroacetic acid and subjected to reverse-phase HPLC in a system (1090; Hewlett-Packard Co., Palo Alto, CA) outfitted with a Vydac C4 column (The Nest Group, Inc., Southboro, MA) (0.21 × 20 cm) prefixed with two SDS removal cartridges (2.1 × 20 mm). The column was developed with a linear gradient of 0 to 80% n-propyl alcohol containing 0.12% trifluoroacetic acid at a flow rate of 0.1 ml/min and was monitored at A280. Fractions (0.1-ml) were collected, and those containing Bcl-2–reactive p20, as judged by ligand blotting, were individually subjected to NH2-terminal peptide sequence analysis at Harvard Microchem (Harvard University, Cambridge, MA).
Cloning of p28 Bap31/CDM cDNA
The coding region of p28 was cloned by reverse transcription PCR using human fibroblast RNA together with primers derived from the sequence of human BAP31 (these sequence data are available from GenBank/ EMBL/DDBJ under accession number X81817). Conditions were exactly as described by Goping et al. (1995a), and we used 5′-TCTCTAGAACAAACAGAAGTACTGGA-3′ as the antisense primer, and 5′-GATCTAGACATCTTCCTGTGGGAA-3′ as the sense primer. Authenticity was confirmed by DNA sequence analysis.
Glutathione S-Transferase Fusion Proteins
PCR was used to generate cDNA fragments corresponding to p28 amino acids 1–246 (full length), 1–164, 122–164, and 165–246, using primers that contained either 5′-BamH1 or 3′-EcoR1 overhangs, respectively. The primers were 5′-GCGGATCCATGAGTCTGCAGTGGACT-3′ and 5′-GCGAATTCTTACTCTTCCTTCTTGTC-3′ for p28 amino acids 1–246; 5′-GCGGATCCATGAGTCTGCAGTGGACT-3′ and 5′-GCGAATTCAGTCAACAGCAGCTCCCTT-3′ for p28 amino acids 1–164; 5′-GCGCGGATCCCTCATTTCGCAGCAGGCC-3′ and 5′-GCGAATTCAGTCAACAGCAGCTCCCTT-3′ for p28 amino acids 122–164; and 5′-GCGGATCCGGAGGCAAGTTGGATGTC-3′ and 5′-GCGAATTCTTACTCTTCCTTCTTGTC-3′ for p28 amino acids 165–246. Fragments generated by PCR were digested with BamH1 and EcoR1, inserted between the BamH1 and EcoR1 sites of pGEX-2t (Pharmacia Fine Chemicals, Piscataway, NJ), and the recombinant plasmids introduced into E. coli MC1061. Packed cells from 500 ml of induced culture were recovered, suspended in 25 ml PBS and 0.1% Triton X-100, and sonicated using a Vibra Cell probe sonicator operating at setting 7.5 for 4 × 15 s at 4°C. After centrifugation at 25,000 rpm in a Beckman Ti 50.2 rotor for 25 min, the supernatant was recovered, mixed with 750 μl of a 1:1 suspension of glutathione-Sepharose 4B beads, and the mixture rotated at 4°C for 45 min. After extensive washing of the beads in PBS and 0.1% Triton X-100, glutathione S-transferase (GST) fusion protein was eluted with 3 ml of 50 mM Tris HCl, pH 8.0, and 12 mM reduced glutathione.
Antibodies
GST fusion proteins were injected into chickens, and the resulting IgY antibodies were recovered from the eggs, exactly as described by Goping et al. (1995a). After adsorption of IgY that reacted with immobilized GST, antibodies specific for p28 sequences were purified by affinity binding to immobilized GST-p28(165-246) or GST-p28(122-164) fusion protein, using the methods described in the Amino Link Plus kit (Pierce Chemical Co., Rockford, IL).
Transient Transfections
CHO LR73 cells were seeded at a density of 5 × 105 cells/well in six-well plates. 24 h later, the cells in each well were transfected by calcium phosphate precipitation with 0.5 μg luciferase reporter plasmid, 10 μg RcRSV-p28 or RcRSV-p20, and 10 μg sheared salmon sperm DNA (Goping et al., 1995b ). After 24 h, cells were shocked with 15% glycerol and collected 24 h later. Cells from each well were lysed in 0.4 ml 0.5% NP-40 and 50 mM Tris HCl, pH 7.8, and aliquots were assayed for luciferase activity as described previously (Goping et al., 1995b ). 293T cells in 10-cm culture plates were similarly transfected with 15 μg total plasmid DNA when cells reached 50–60% confluency.
Coimmunoprecipitations
Approximately 30 h after transfection, 293T cells were washed in PBS and homogenized in 1.0 ml lysis medium/10-cm culture plate (50 mM Hepes, pH 7.4, 150 mM NaCl, 1 mM ethylenediamine tetraacetate, 0.5% vol/vol NP-40, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mM PMSF). After centrifugation at 11,000 g, the supernatant was incubated with 50 μl of a 1:1 slurry of protein G Sepharose for 1 h at 4°C. The Sepharose was removed and the supernatant incubated with mouse M2 anti-Flag antibody (IBI-A Kodak Co., New Haven, CT) at 4°C for 6 to 8 h, at which time 20 μl of a 1:1 slurry of protein G Sepharose was added. After 1 h at 4°C, the beads were recovered, washed, and boiled in SDS sample buffer. After SDS-PAGE and transfer to nitrocellulose, blots were developed with either mouse anti-Myc 9E1D antibody or mouse anti-HA 12CA5 antibody (both from Babco, Berkeley, CA) or rabbit anti-Bax sc-526 antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
Results
Appearance and Identification of a Bcl-2–interacting Polypeptide after Induction of Apoptosis by E1A
To detect potential Bcl-2–interacting polypeptides by ligand blot (Far Western) analysis, a 32P-labeled probe was constructed by expressing a modified version of the cytosolic domain of Bcl-2 in E. coli. To that end, the last 21 amino acids of Bcl-2 were deleted and substituted with hexahistidine (his6) plus an HMK recognition peptide to facilitate purification and 32P labeling, respectively. Isolation conditions were developed in which the recombinant protein was purified as a soluble product comprised of mixed monomers and dimers at pH 7.4, as judged by FPLC molecular sieve chromatography. The 32P-labeled probe (32P-Bcl-2Δc21/his6/HMK) readily detected either recombinant Bcl-2 or Bax as the only radioactive products on a ligand blot of total bacterial lysate (not shown). When used as a probe to analyze potential Bcl-2–interacting polypeptides in cells induced to undergo apoptosis in response to various stimuli (including adenovirus E1A expression or treatment with puromycin), a product of ∼20 kD in size (p20) as judged by SDS-PAGE was observed consistently.
Fig. 1 shows one such example after infection of neo- and BCL-2–expressing human KB cells with adenovirus type 5 producing either 12S E1A mRNA (which encodes 243R E1A protein) or both 12S and 13S E1A mRNAs (which encode 243R and 289R E1A proteins, respectively), but lacking expression of the dominant suppressor of apoptotic cell death, E1B 19-kD protein (19K). Induction of apoptotic cell death by either virus (Nguyen et al., 1994; Teodoro et al., 1995) was accompanied by the appearance of p20 Bcl-2–binding activity, whereas apparent binding to Bax did not change significantly during the time course of infection. Of note, however, is the observation that stable expression of Bcl-2 in these cells countered cell death and prevented the appearance of p20 Bcl-2–binding activity after viral infection.
Figure 1.

Appearance of a Bcl-2–interacting polypeptide during E1A-induced apoptosis. KB cells expressing neomycin resistance, either alone (Neo) or together with Bcl-2, were infected with either adenovirus dl520E1B− (expressing 12S E1A and no E1B products) or pm1760/ 2072 (expressing 12S and 13S E1A but not E1B 19K). At the indicated times after infection, samples of cells were either assessed for viability by exclusion of trypan blue (graph) or prepared for ligand blot (Far Western) analysis, as described in Materials and Methods, using 32P-Bcl-2Δc21/his6/HMK as a probe (upper panel, Bcl- 2–expressing cells; lower panel, Neo control cells). Ligand blots were visualized by phosphorimaging. The radioactive band associated with a polypeptide of M r 20 kD is labeled p20, whereas that which comigrates with Bax is designated p21 Bax. The latter was determined using a blot cut along the vertical midline of a protein lane and developing one half by immunoblot analysis with anti–human Bax (Chen et al., 1996) and the other by ligand blotting with 32P-Bcl-2Δc21/ his6/HMK (results not shown).
Identification of the p20 Bcl-2–binding polypeptide was obtained by NH2-terminal peptide sequence analysis of p20 after its purification by a combination of differential solubilization in detergent, preparative SDS-PAGE, and reverse-phase HPLC (Fig. 2, A and B). Several individual HPLC fractions were subjected to peptide sequence analysis to detect a polypeptide sequence whose appearance correlated with the appearance of p20 Bcl-2–binding activity (Fig. 2). One candidate sequence emerged, and it was the only sequence that was detected in the peak fraction of Bcl-2–binding activity (Fig. 2 B, fraction 54). It showed a perfect match with amino acids 2–11 of human Bap31 (these sequence data available from GenBank/EMBL/DDBJ under accession number X81817)/CDM (these sequence data available from GenBank/EMBL/DDBJ under accession number Z31696), suggesting that p20 derives from the NH2 terminus of this 27,991-kD (p28) protein. Reverse transcription-PCR analysis of the p28 coding region, using total RNA obtained from KB cells after induction of apoptosis, showed no evidence that p20 arose by differential splicing of p28 mRNA (data not shown). As demonstrated below, Bcl-2 also associates with full length p28 in vitro and in vivo; failure to observe this interaction in the original ligand blot analyses (Fig. 1) was likely the result of relatively inefficient transfer of p28 to nitrocellulose blots.
Figure 2.




Identification of p20. (A) Preparative SDS-PAGE of differentially solubilized protein from KB cells 60 h after infection with adenovirus pm1760/2072. Aliquots of fractions eluted from the gel were assayed by 32P-Bcl-2Δc21/his6/HMK Far Western, and the radioactive bands corresponding to p20 and p21 Bax were detected and quantified by phosphorimaging. The levels relative to the maximal signal detected (set at 100) were plotted as a bar graph (upper panel). Equal aliquots from the same fractions were also subjected to analytical 12% SDS-PAGE, and the gels were stained with Coomassie brilliant blue (lower panel). The positions of molecular mass marker proteins are indicated. black, p20; gray, Bax. (B) Proteins eluting from preparative SDS-PAGE between 190 and 220 ml were concentrated and resolved by reverse-phase HPLC. The upper panel shows the A280 profile. Equal aliquots from all fractions were assayed for 32P-Bcl-2Δc21/his6/HMK–interacting protein by Far Western, for which only p20 was detected. Amounts relative to the maximal signal detected (set at 100) were plotted as a bar graph (lower panel). Fractions 52, 53, 54 (peak activity), and 55 were individually subjected to NH2-terminal peptide sequence analysis. (C) Polypeptide sequence of p28 Bap31/CDM (single-letter code). Peptide sequencing of p20 revealed a perfect match with amino acids 2–11 of human Bap31(underlined; these sequence data available from GenBank/EMBL/ DDBJ under accession number X81817). This was the only detectable sequence in fraction 54, was detectable together with other sequences in fraction 53, and was not detected in fractions 52 and 55. Predicted TM segments are boxed and contain charged amino acids in TM1 and TM3 (asterisks). The predicted caspase recognition sites, AAVD·G, are highlighted, and cleavage is denoted by arrows following Asp at positions 164 and 238. A potential leucine zipper located between the caspase recognition sites is denoted by bold letters, as is the KKXX ER retention signal at the COOH terminus. (D) Comparison of putative death effector domain sequences for the indicated proteins. The sequences, given in the single-letter amino acid code, were obtained from GenBank/EMBL/DDBJ, and their relative positions in the molecule are shown in parentheses. Sequences were aligned using the PILEUP program of the GCG software package and were optimized by spacing (shown as dashes). Identical residues and conserved substitutions that were recorded for at least half of the sequences analyzed are shaded in gray.
p28 is identical to a protein previously described as Bap31 and CDM, which is ubiquitously expressed (Adachi et al., 1996). CDM was discovered because of its proximity to the adrenoleukodystrophy locus (Mosser et al., 1994), and Bap31 because it was one of several polypeptides that were found in immunoprecipitates of the B cell receptor complex obtained from detergent-solubilized cells (Kim et al., 1994; Adachi et al., 1996). Another protein present in these precipitates, Bap32, is a homologue of the rat protein prohibitin (McClung et al., 1989). It is noteworthy, however, that prohibitin has recently been localized to the mitochondrial periphery in transfected BHK cells (Ikonen et al., 1995) and that Bap31 (p28) is largely restricted to the ER in rat liver hepatocytes (see below), whereas the B cell receptor is located in the plasma membrane.
p28 Bap31
Fig. 2 C highlights several predicted motifs in the human p28 sequence. There are three potential transmembrane (TM) segments (Kyte and Doolittle, 1982) located in the NH2-terminal half of the molecule. TM1 and TM3 each contain charged residues. Additionally, two potential caspase cleavage sites comprised of identical P1–P4 tetrapeptide recognition sequences (Ala-Ala-Val-Asp) plus a preferred small amino acid (Gly) in the P1′ position are located at positions 164 and 238 in the polypeptide, on either side of a predicted leucine zipper domain (Fig. 2 C) and overlapping weak homology to death effector domains found in such proteins as procaspase-8/10 and FADD (Fig. 2 D). Cleavage at the proximal caspase recognition site would generate a product (calculated Mr of 18.8 kD) similar in size to p20. Interestingly, the distal caspase recognition site is lacking in the mouse sequence (Fig. 2 D). Finally, the molecule terminates in Lys-Lys-Glu-Glu, which conforms to a canonical KKXX COOH-terminal signal that retains integral ER proteins containing COOH termini exposed to the cytosol within this organelle, preventing their exit into the distal secretory pathway (Jackson et al., 1993).
As shown in Fig. 3, p28 was efficiently inserted cotranslationally into dog pancreas microsomes. In contrast to β-lactamase, which was translocated across the ER membrane and deposited in the lumen as a soluble protein, p28 was recovered as an integral protein after release from the ribosome. Whereas the processed form of β-lactamase was protected from external protease (Fig. 3, lane 4) and liberated from microsomes by alkaline extraction (Fig. 3, lane 3), p28 was resistant to alkaline extraction (Fig. 3, lane 7), and exhibited sensitivity to external protease (Fig. 3, lane 8), resulting in the generation of proteolytic fragments that would be expected for a multispanning integral protein with an exposed cytosolic domain. Unlike β-lactamase, whose NH2-terminal signal sequence was removed during translocation (Fig. 3, compare lanes 1 and 4), processing of p28 was not observed (Fig. 3, compare lanes 5 and 6), suggesting that insertion into the microsomal membrane is initiated by an uncleaved signal anchor. Though not studied in detail, the observed properties of p28 (Fig. 3), together with predictions for the orientation of transmembrane segments in the ER based on charge difference rules (von Heijne, 1986; Hartmann et al., 1989), suggest a topology for p28 in the ER membrane in which the NH2 terminus of this triple-spanning polypeptide faces the lumen, leaving an ∼13-kD COOH-terminal fragment containing predicted caspase cleavage sites, leucine zipper/death effector-like domain, and ER retention motif exposed to the cytosol (see Fig. 3). Biochemical fractionation and cryoimmunocytochemical electron microscopy confirmed that p28 is predominantly located in the ER in rat hepatocytes (not shown).
Figure 3.

Insertion of p28 into ER microsomes. Pre–β-lactamase (lanes 1–4) and p28 (lanes 5–8) mRNA was translated in a rabbit reticulocyte lysate system in the presence of [35S]methionine, in the presence (lanes 2–4 and 6–8) or absence (lanes 1 and 5) of ribosome-stripped canine pancreas microsomes (Walter and Blobel, 1983). At the end of the reaction, microsomes were recovered and analyzed by SDS-PAGE and fluorography either directly (lanes 2 and 6) or after isolation of alkali-insoluble (NaCO3, pH 11.5) product (lanes 3 and 7; Nguyen et al., 1993), or after treatment with proteinase K (lanes 4 and 8; McBride et al., 1992). The positions of p28, pre–β-lactamase (pre-β-L), and processed β-lactamase (β-L) are indicated, as is the gel front. c, marker translation product. The schematic shown below the fluorogram depicts the deduced topology of p28 in the ER membrane (see text).
Recombinant p28 and p20 Interact with Bcl-2
Various p28 fusion proteins were constructed in which GST was linked to p28 amino acids 1–246 (full length p28), 1–164 (p20), 122–164, and 165–246. These constructs, together with GST itself, were purified, and equal amounts were examined for their ability to bind to the cytosolic domain of Bcl-2 in a ligand blot assay. As shown in Fig. 4 A, reactivity was observed for both GST-p28 (lane 5) and GST-p20 (lane 4), with weak activity possibly registering with the COOH-terminal 165–246 amino acid domain (lane 2), and none detected for the middle 122–164 amino acid domain (lane 3) or for GST alone (lane 1).
Figure 4.



Associations of p28 with Bcl-2, Bcl-XL, and procaspase-8 (pro-FLICE). (A) GST (lane 1) or GST fused to p28 amino acids 165–246 (lane 2), 122–164 (lane 3), 1–164 (lane 4), and 1–246 (lane 5) were expressed in bacteria, purified, and transferred to nitrocellulose in duplicate after SDS-PAGE. One blot was stained with Ponceau S and the other probed by ligand blotting (Far Western) with 32P-Bcl-2Δc22/his6/HMK, as indicated. Constructs and results are summarized below the blots. (B) Standard recombinant DNA manipulations were used to create cDNAs encoding Bcl-XL tagged at the COOH terminus with the Myc epitope EQKLISEEDL (Chinnayan et al., 1997); pro-FLICE tagged at the COOH terminus with the hemagglutinin (HA) epitope, YPYDVPDYA (Chinnayan et al., 1997); Bcl-2 tagged at the NH2 terminus with the HA epitope (Nguyen et al., 1994); and p28 tagged with the Flag epitope, in which the Flag sequence MDYKDDDDKA was inserted between Pro240 and Met241 of p28. The recombinant cDNAs, or Flag DNA alone (Control-Flag), were inserted into pcDNA 3 (Invitrogen) or RcRSV (Pharmacia Fine Chemicals) (HA-Bcl-2) and transfected into 293T cells, as indicated (pluses and minuses). After incubation of cell lysates with anti-Flag antibody, immunoprecipitates (ip) and lysates were resolved by SDS-PAGE, transferred to nitrocellulose, and the blot developed with the indicated antibody and visualized by enhanced chemiluminescence. Ig HC, immunoglobulin heavy chain. (C) Same as in B, except that Bax was included in cotransfections, as indicated.
Bcl-2 Proteins and Procaspase-8 (pro-FLICE) Associate with p28 In Vivo
Epitope-tagged versions of Bcl-XL, Bcl-2, and wild-type procaspase-8 were expressed separately or in combination in 293T cells, together with either p28-Flag or control Flag. The presence of p28-associated proteins present in immunoprecipitates recovered with anti-Flag was then assessed by immunoblot (Western) analysis (Fig. 4 B). To avoid interference with the function of the ER retention signal in p28, the Flag epitope was inserted just upstream of the KKEE motif at the COOH terminus of the protein. As judged by coimmunoprecipitation, Bcl-XL, procaspase-8, and Bcl-2 each demonstrated a specific association with p28 (Fig. 4 B, lanes 3, 8, and 14, respectively). Procaspase-8 was observed as a doublet band that migrated immediately below the Ig heavy chain; transcription–translation of the cDNA in vitro likewise generated a doublet of similar size (not shown). Interestingly, Bcl-XL and procaspase-8, when expressed in combination, did not mutually antagonize each other's ability to associate with p28 (Fig. 4 B, lanes 5 and 10; note the lower input levels of Bcl-XL in the cell lysate in lane 5). Although some activation of wild-type procaspase-8 might be expected, the full length proenzyme was readily detectable at similar levels in both the presence and absence of Bcl-XL (Fig. 4 B, lanes 8 and 10).
Finally, the pro-apoptotic member of the Bcl-2 family, Bax (Oltvai et al., 1993), did not coimmunoprecipitate with p28 after their coexpression in 293T cells (Fig. 4 C, lane 5), despite the fact that significant expression levels of transfected Bax were recorded (lanes 2 and 3). However, Bax prevented Bcl-2 from associating with p28 (Fig. 4 B, compare lanes 11 and 12). Although the level of Bcl-2 in cell lysates was somewhat lower in transfectants containing Bax (Fig. 4 B, compare lanes 8 and 9), such a level of Bcl-2 would otherwise have been sufficient to readily detect coimmunoprecipitation of Bcl-2 and p28.
p28 is Cleaved to p20 by FLICE-related Caspase
To test the possibility that the predicted caspase recognition sequences in p28, AAVD·G, can in fact be recognized by one or more of these enzymes, 35S-labeled p28 trans– cription–translation product was incubated with increasing concentrations of either caspase-1 (ICE) or caspase-3 (CPP32) in vitro (Nicholson et al., 1995), and the products were examined after SDS-PAGE (Fig. 5 A). Little reactivity was observed for caspase-3 over a wide range of enzyme concentration. caspase-1, on the other hand, generated two products (denoted a and b in Fig. 5 A), whose apparent sizes in SDS gels (∼27 and ∼20 kD, respectively) are consistent with cleavage occurring at both AAVD/G sites. Of note, p28 was more sensitive to cleavage by caspase-8 (FLICE) than by caspase-1 (ICE; Fig. 5 B).
Figure 5.
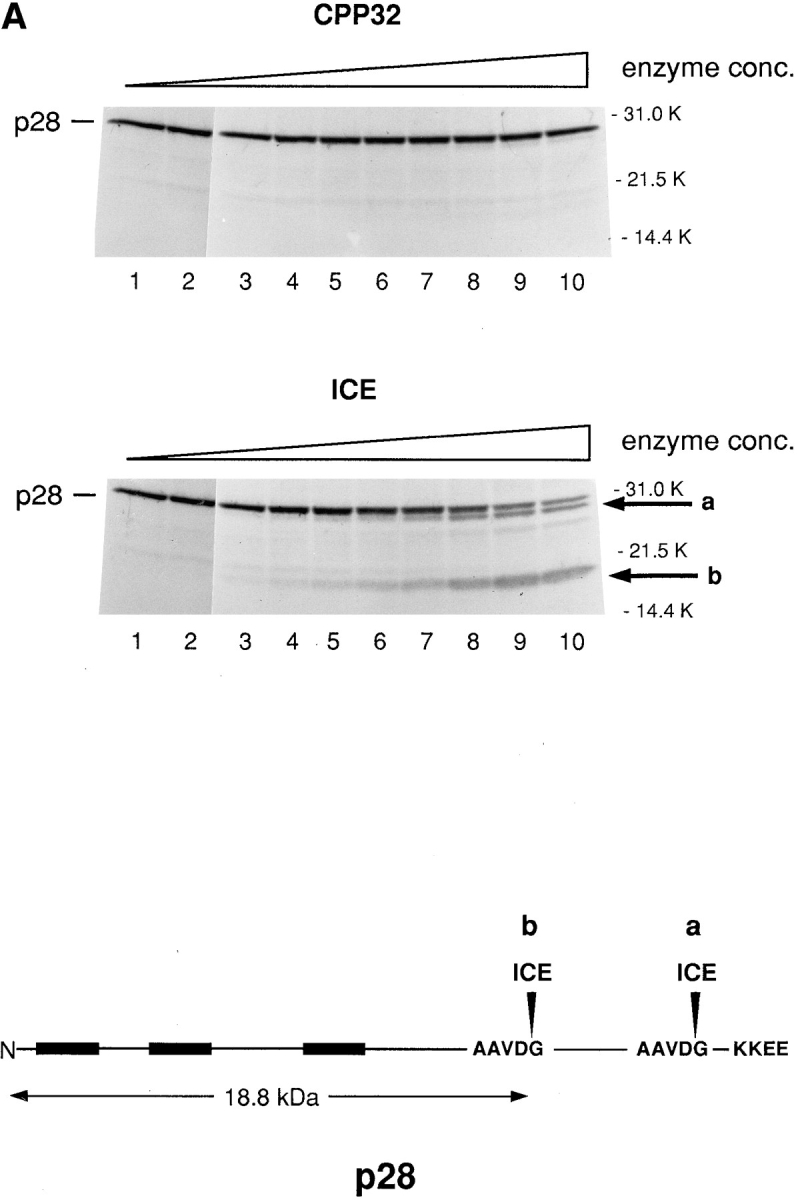

In vitro cleavage of p28 by caspase-1 (ICE) and caspase-8 (FLICE) but not by caspase-3 (CPP23). (A) 35S-labeled transcription–translation product of p28 cDNA was incubated with increasing concentrations of CPP32 or ICE, and the products were resolved by SDS-PAGE (Nicholson et al., 1995). Units of enzyme added per 25 μl reaction mixture were: none (lane 1), 0.0056 (lane 2), 0.98 (lane 3), 1.95 (lane 4), 3.9 (lane 5), 7.8 (lane 6), 15.6 (lane 7), 31.2 (lane 8), 62.5 (lane 9), and 125 (lane 10). The positions of polypeptide molecular mass markers are shown. The arrows designated a and b denote cleavage products whose sizes are consistent with cleavage of p28 at the sites indicated by a and b in the schematic at the bottom of the figure. (B) Same analysis as in A, except that p28 was incubated with purified ICE or FLICE, and the resulting p20 cleavage product was quantified using a Phosphorimager. 1 U of caspase enzyme activity is equivalent to 1 pmol aminomethylcoumarin liberated from fluorogenic tetrapeptide-AMC per min at 25°C at saturating substrate concentrations (Nicholson et al., 1995).
Two cleavage products of p28, similar in size to those seen in vitro, were also observed in cells that had been induced to undergo apoptotic cell death in response to infection by 19K-defective adenovirus (Fig. 6). In Fig. 6 A, p28 cleavage during apoptosis in vivo was analyzed using antibodies raised in chicken to either of two regions of the protein: p28 amino acids 122–164 (α p28-M) and 165–246 (α p28-C). Cleavage products were detected with α p28-M but not with α p28-C, a finding consistent with the suggestion from peptide sequence analysis that the p20 cleavage product derives from the NH2 terminus of p28 (Fig. 2). α p28-C also failed to detect the larger of the two cleavage products (designated a in Fig. 6 A) despite the predicted overlap of this product with the sequence injected into chickens. Presumably, this means that the extreme eight amino acids of p28 are critically important for epitope recognition by this antibody. Finally, protein electrophoretic blots were developed from apoptotic cell extracts, cut in half along the vertical midline of a protein lane, and one half probed with α p28-M and the other with 32P-Bcl-2Δc21/his6/HMK. p20 detected by the Bcl-2 probe migrated exactly with p20 detected by α p28-M immunoblotting (not shown).
Figure 6.


Induction of p28 cleavage and procaspase-3 (pro-CPP32) processing during apoptosis in vivo. (A) Cell extracts were obtained from KB cells that had either been infected for 60 h with adenovirus pm1716/2072 lacking expression of E1B 19K or had been mock infected (+ or − Apoptosis, respectively). After 12% SDS-PAGE and transfer to nitrocellulose, blots were incubated with affinity-purified chicken antibody against p28 amino acids 165–246 (α p28-C) or p28 amino acids 122–164 (α p28-M) and were developed with secondary antibody conjugated either to HRP and visualized by electrochemiluminescence (Amersham Intl., Arlington Heights, IL) (α p28-M) or to alkaline phosphatase and visualized with NBT/BCIP (Boehringer Mannheim Biochemicals, Indianapolis, IN) (α p28-C), according to the manufacturer's instructions. Bands corresponding to p28 are indicated. Arrows labeled a and b denote products whose sizes are consistent with cleavage of p28 at the sites designated a and b in the schematic. (B) KB cells expressing neomycin resistance either alone (minus Bcl-2, lanes 6–10) or together with Bcl-2 (plus Bcl-2, lanes 1–5) were infected with adenovirus pm1716/2072 lacking expression of E1B 19K and cell extracts prepared at 0, 24, 36, 48, and 60 h postinfection (p.i.; lanes 1 and 6, 2 and 7, 3 and 8, 4 and 9, and 5 and 10, respectively). Aliquots (15 μg protein) were subjected to 12% SDS-PAGE, transferred to nitrocellulose, and blots were probed with antibody against p28-M or against the 17-kD subunit of CPP32 (Boulakia et al., 1996), and the products were developed as described in A. The positions of p28 and the cleavage products a and b are indicated in the upper panels. The arrow denotes a cross-reacting product whose appearance is variable (e.g., it did not appear in A). The positions of full length pro-CPP32 and the processed 17-kD subunit (p17) and putative 29-kD processing intermediate (asterisk) are indicated in the lower panels.
In Fig. 6 B, the effect of Bcl-2 on the appearance of p28 cleavage products after cell infection with 19K-deficient adenovirus was examined using the α p28-M antibody. In the absence of Bcl-2 expression, the time course of appearance of these products closely followed the time course for activation of procaspase-3 (CPP32), as judged by processing of the pro-enzyme to the p17 catalytic subunit (Fig. 6 B, lanes 6–10). However, both p28 cleavage and procaspase-3 processing were blocked in virus-infected cells that express Bcl-2 (Fig. 6 B, lanes 1–5).
Ectopic Expression of p20 Induces Apoptosis
CHO (neo) cells were transiently cotransfected with a luciferase reporter gene together with RcRSV expressing either p28 or p20. Subsequent measurements revealed that coexpression of p20 with the reporter severely depressed the amount of luciferase activity obtained relative to coexpression with p28 (Fig. 7 A). p28, on the other hand, had no deleterious effect on the recovery of luciferase activity compared to a control RcRSV plasmid that did not encode protein (not shown). If these same transfections were conducted in cells stably expressing Bcl-2, however, Bcl-2 largely overcame the dominant negative influence of p20 on luciferase activity (Fig. 7 A), presumably because p20 could no longer interfere with normal p28 function. Because of this protective effect by Bcl-2, we conclude that the negative influence of p20 on luciferase activity was the result of induction of apoptosis. This was confirmed by microscopic analysis, which revealed apoptotic nuclei in p20- but not p28-transfected cells (Fig. 7 B). The findings described in Fig. 7 have been consistently observed many times and in different cell types.
Figure 7.


Ectopic expression of p20 induces apoptosis. (A) CHO LR73 cells expressing neomycin resistance, either alone (− Bcl-2) or together with Bcl-2 (+ Bcl-2), were cotransfected with a luciferase reporter plasmid and Rc/RSV expressing either full length p28 or p28 amino acids 1–164 (i.e., p20). After 2 d, cells were recovered, analyzed for luciferase activity, and the enzyme activity expressed relative to the values obtained in the presence of p28 (arbitrarily set at 100). The results shown are the average of two separate experiments. (B) CHO cells were transfected with the p28 and p20 expression plasmids together with pHook (Invitrogen). 24 h later, transfected cells were recovered with Capture-Tec beads, cultured on coverslips, stained with 4′,6′-diamidino-2-phenyl indole (DAPI), and visualized under a microscope.
Discussion
We have identified p28 Bap31 as a component of a putative apoptotic signaling complex in the ER that also includes Bcl-2/Bcl-XL and procaspase-8 (pro-FLICE). At least with respect to recruitment of procaspase-8, the function of the p28 complex may be analogous to Fas and TNFR-1, apoptotic receptors in the plasma membrane that respond to FasL and TNF, respectively, by activating receptor-associated procaspase-8 and initiating the caspase cascade (for review see Nagata, 1997). Association of procaspase-8 with these receptor complexes is achieved through interactions of its death effector domain with an analogous domain in the adaptor molecule FADD (Boldin et al., 1996; Muzio et al., 1996). Despite the fact that p28 contains a weak death effector homologous domain within its cytosolic region, however, we find no evidence that p28 and procaspase-8 interact directly, at least based on binding assays in vitro (not shown). Thus, evidence for an autonomously functional death effector domain in p28 is lacking. The observed association of procaspase-8 with p28 in vivo, therefore, likely depends on an as yet unidentified adaptor component within the p28 complex. Expression of the adenovirus E1A oncoprotein presumably results in activation of procaspase-8, as judged by the ensuing rapid and specific cleavage of p28 in vivo at sites that exhibit a marked substrate preference by purified caspase-8 in vitro. Such a claim is consistent with the finding that CrmA, an inhibitor of FLICE-related caspases but not of caspase-3 (Srinivasula et al., 1996; Zhou et al., 1997), significantly retards the onset of E1A-induced apoptosis in viral infected cells (our unpublished data). It remains to be determined, however, if E1A results in activation of the procaspase-8 (or related caspase) members that are directly associated with the p28 complex. Whichever the case, p28 is cleaved to p20 after E1A expression, and the p20 molecule itself can induce cellular apoptotis. p20 may act either to ampilify a caspase-8–initiated protease cascade (Srinivasula et al., 1996; Muzio et al., 1997) or to contribute to a parallel pathway. Significantly, Bcl-2 blocks the appearance of p20 after E1A expression. It remains to be determined whether this is because Bcl-2 subverts the E1A signal that results in activation of procaspase-8 or because it protects p28 against activated caspase-8. Bax, a pro-apoptotic member of the Bcl-2 family, does not appear to associate with the p28 complex. However, it blocks Bcl-2 from doing so, presumably because competing interactions between Bcl-2 and Bax (Oltvai et al., 1993) prevent interactions between Bcl-2 and p28.
There is a growing body of evidence that mitochondria may play an important role in apoptotic signaling leading to downstream activation of caspase-3 and other effector proteases (Kroemer et al., 1997; Kluck et al., 1997; Yang et al., 1997). Moreover, these or other mitochondrial events may be controlled by phosphorylation (Wang et al., 1996a ; Zha et al., 1996) which in turn may be influenced by Bcl-2 (Wang et al., 1996a ). Under normal conditions, Bcl-2 and Bcl-XL are localized to both the mitochondrial outer membrane and ER, suggesting that events influencing these proteins may apply to both membrane sites. Nevertheless, replacing the COOH-terminal Bcl-2 signal anchor with either a heterologous mitochondrial outer membrane signal anchor (Mas70p; McBride et al., 1992) or a posttranslational ER-specific insertion sequence (aldehyde dehydrogenase; Masaki et al., 1994) can restore anti-apoptotic activity to the protein (Nguyen et al., 1994; our unpublished data). Similar results have been obtained with other mitochondrial- and ER-specific targeting sequences (Zhu et al., 1996). These findings are consistent with the idea that ER and mitochondria may cooperate to elicit apoptotic signals, and that regulation of these signals by Bcl-2 at either compartment may be effective under certain conditions. Alternatively, apoptotic regulatory systems at the two membrane sites may respond differentially to individual inducers. Although the potential role for Ca2+ in apoptosis remains unresolved, the ability of mitochondria to decode ER-transmitted oscillating Ca2+ signals (Hojnóczky et al., 1995; Camacho and Lechleiter, 1995; Jouaville et al., 1995) is one example in which the two organelles clearly cooperate to regulate metabolic events. Consistent with a role for calcium signaling in apoptosis is the recent finding that inhibition of the type I inositol 1,4,5-triphosphate receptor calcium release channel in the ER confers resistance to diverse apoptotic stimuli. It remains to be determined whether or not p28 is involved in either this or another ER–mitochondrial program. If so, however, the fact that predicted transmembrane segments 1 and 3 contain positively charged residues implies that p28 may be part of a larger complex that thermodynamically stabilizes these residues within the membrane lipid bilayer. Such a structure might also be compatible with the predicted pore-forming properties of the Bcl-XL (Minn et al., 1997) and Bcl-2 (Schendel et al., 1997) cytosolic domains and contribute to p28–Bcl-2 interactions.
Finally, genetic studies in Caenorhabditis elegans have identified and ordered a core machinery for regulation of cell death in which the Bcl-2 homologue, Ced-9, prevents Ced-4 from activating the ICE-like caspase Ced-3 (Hengartner et al., 1996; Shaham and Horvitz, 1996). Reconstitution of these events in mammalian cells has recently been achieved (Chinnaiyan et al., 1997; Wu et al., 1997) and has revealed that Ced-4 can mechanically link the initiator procaspase-8 and -1 pro-enzymes with Bcl-XL (Chinnaiyan et al., 1997). Moreover, Ced-4 can compete for interaction of procaspase-8 with the predicted endogenous molecule(s) in mammalian cells that bridges this Bcl-2 protein with procaspase-8. Ced-4, like the cytosolic tail of p28, may contain a weak death effector homologous domain (Bauer et al., 1997). These findings all conform with the predicted function of the putative p28 apoptotic signaling complex described here. Moreover, they raise the possibility that the mammalian homologue of Ced-4, if one exists, may be a constituent of this complex.
Acknowledgments
We are grateful to Robert MacKenzie, John Bergeron, Jerry Pelletier, Anne Claude Gingras, Josée Dostie, and Josée Lavoie for discussions and technical advice.
Abbreviations used in this paper
- GST
glutathione S-transferase
- HMK
heart muscle kinase
- TM
transmembrane
Footnotes
Address all correspondence to Gordon C. Shore, Department of Biochemistry, McIntyre Medical Sciences Building, McGill University, Montreal, QC, Canada H3G 1Y6. Tel.: (514) 398-7282; Fax: (514) 398-7384; E-mail: shore@medcor.mcgill.ca
The present address for James A. Cromlish is Institut de Recherches Cliniques de Montréal, Montréal, QC, Canada H2W 1R7.
This work was financed by operating grants from the National Cancer Institute of Canada and the Medical Research Council.
References
- Adachi T, Schamel WW, Kim KM, Watanabe T, Becker B, Nielsen PJ, Reth M. The specificity of association of the IgD molecule with the accessory proteins BAP31/BAP29 lies in the IgD transmembrane sequence. EMBO J. 1996;15:1534–1541. [PMC free article] [PubMed] [Google Scholar]
- Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan JY. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- Armstrong RC, Aja T, Xiang J, Gaur S, Krebs JF, Hoang K, Bai X, Korsmeyer SJ, Karanewsky DS, Fritz LC, Tomaselli KJ. Fas-induced activation of the cell death-related protease CPP32 is inhibited by Bcl-2 and by ICE family protease inhibitors. J Biol Chem. 1996;271:16850–16855. doi: 10.1074/jbc.271.28.16850. [DOI] [PubMed] [Google Scholar]
- Bauer MKA, Wesselborg S, Schulze-Osthoff K. The Caenorhabditis elegansdeath protein Ced-4 contains a motif with similarity to the mammalian death effector domain. FEBS Lett. 1997;402:256–258. doi: 10.1016/s0014-5793(96)01497-4. [DOI] [PubMed] [Google Scholar]
- Blanar MA, Rutter WJ. Interaction cloning: identification of a helix-loop-helix zipper protein that interacts with c-Fos. Science (Wash DC) 1992;256:1014–1018. doi: 10.1126/science.1589769. [DOI] [PubMed] [Google Scholar]
- Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- Boulakia CA, Chen G, Ng FWH, Teodoro JG, Branton PE, Nicholson DW, Poirier GG, Shore GC. Bcl-2 and adenovirus E1B 19-kDA protein prevent E1A-induced processing of CPP32 and cleavage of poly(ADP-ribose) polymerase. Oncogene. 1996;12:529–535. [PubMed] [Google Scholar]
- Boyd JM, Malstrom S, Subramanian T, Venkatesh LK, Schaeper U, Elangovan B, D'Sa EC, Chinnadurai G. Adenovirus E1B 19-kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell. 1994;79:341–351. doi: 10.1016/0092-8674(94)90202-x. [DOI] [PubMed] [Google Scholar]
- Camacho P, Lechleiter JD. Calreticulin inhibits repetitive intracellular Ca2+waves. Cell. 1995;82:765–771. doi: 10.1016/0092-8674(95)90473-5. [DOI] [PubMed] [Google Scholar]
- Chen G, Branton PE, Yang E, Korsmeyer SJ, Shore GC. Adenovirus E1B 19-kDa death suppressor protein interacts with Bax but not with Bad. J Biol Chem. 1996;271:24221–24225. doi: 10.1074/jbc.271.39.24221. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, Orth K, O'Rourke K, Duan H, Poirier GG, Dixit VM. Molecular ordering of the cell death pathway. Bcl-2 and Bcl-XLfunction upstream of the CED-3-like apoptotic proteases. J Biol Chem. 1996;271:4573–4576. doi: 10.1074/jbc.271.9.4573. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, O'Rourke K, Lane BR, Dixit VM. Interaction of Ced-4 with Ced-3 and Ced-9: a molecular frame work for cell death. Science (Wash DC) 1997;275:1122–1126. doi: 10.1126/science.275.5303.1122. [DOI] [PubMed] [Google Scholar]
- Chittenden T, Harrington EA, O'Connor R, Flemington C, Lutz RJ, Evan GI, Guild BC. Induction of apoptosis by the Bcl-2 homologue Bak. Nature (Lond) 1995;374:733–736. doi: 10.1038/374733a0. [DOI] [PubMed] [Google Scholar]
- Farrow SN, White JH, Martinou I, Raven T, Pun KT, Grinham CJ, Martinou JC, Brown R. Cloning of a Bcl-2 homologue by interaction with adenovirus E1B 19K. Nature (Lond) 1995;374:731–733. doi: 10.1038/374731a0. [DOI] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Armstrong RC, Krebs J, Srinivasula SM, Wang L, Bullrich F, Fritz LC, Trapani JA, Tomaselli KJ, Litwack G, Alnemri A. In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc Natl Acad Sci USA. 1996;93:7464–7469. doi: 10.1073/pnas.93.15.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Sarabia M, Bischoff JR. Bcl-2 associates with the ras- related protein R-ras p23. Nature (Lond) 1993;366:274–275. doi: 10.1038/366274a0. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Garcia M, Parez-Ballestero R, Ding L, Boise LH, Thompson CB, Nunez G. Bcl-XLis the major Bcl-X mRNA form expressed during murine development and its product localizes to mitochondria. Development (Camb) 1994;120:3033–3042. doi: 10.1242/dev.120.10.3033. [DOI] [PubMed] [Google Scholar]
- Goping IS, Millar DG, Shore GC. Identification of the human mitochondrial protein import receptor, huMas20p. Complementation of delta mas20 in yeast. FEBS Lett. 1995a;373:45–50. doi: 10.1016/0014-5793(95)01010-c. [DOI] [PubMed] [Google Scholar]
- Goping IS, Lamontagne S, Shore GC, Nguyen M. A gene-type-specific enhancer regulates the carbamyl phosphate synthetase I promoter by cooperating with the proximal GAG activating element. Nucleic Acids Res. 1995b;23:1717–1721. doi: 10.1093/nar/23.10.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb RA, Nordberg J, Skowronski E, Babior BM. Apoptosis induced in Jurkat cells by several agents is preceded by intracellular acidification. Proc Natl Acad Sci USA. 1996;93:654–658. doi: 10.1073/pnas.93.2.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann E, Rapoport TA, Lodish HF. Predicting the orientation of eukaryotic membrane-spanning proteins. Proc Natl Acad Sci USA. 1989;86:5786–5790. doi: 10.1073/pnas.86.15.5786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengartner MO. Programmed cell death in invertabrates. Curr Opin Genet Dev. 1996;6:34–38. doi: 10.1016/s0959-437x(96)90007-6. [DOI] [PubMed] [Google Scholar]
- Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature (Lond) 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- Hojnóczky G, Robb-Gaspers L, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Ikonen E, Fiedler K, Parton RG, Simons K. Prohibitin, an antiproliferative protein, is localized to mitochondria. FEBS Lett. 1995;358:273–277. doi: 10.1016/0014-5793(94)01444-6. [DOI] [PubMed] [Google Scholar]
- Jackson MR, Nilsson T, Peterson PA. Retrieval of transmembrane proteins to the endoplasmic reticulum. J Cell Biol. 1993;121:317–333. doi: 10.1083/jcb.121.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman T, Marks AR. T cells deficient in inositol 1,4,5-triphosphate receptor are resistant to apoptosis. Mol Cell Biol. 1997;17:3005–3012. doi: 10.1128/mcb.17.6.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouaville LS, Ichas F, Holmuhamedov EL, Camacho P, Lechleiter JD. Synchronization of calcium waves by mitochondrial substrates in Xenopus laevisoocytes. Nature (Lond) 1995;377:438–441. doi: 10.1038/377438a0. [DOI] [PubMed] [Google Scholar]
- Kiefer MC, Brauer MJ, Powers VC, Wu JJ, Umansky SR, Tomei LD, Barr PJ. Modulation of apoptosis by the widely distributed Bcl-2 homologue Bak. Nature (Lond) 1995;374:736–739. doi: 10.1038/374736a0. [DOI] [PubMed] [Google Scholar]
- Kim KM, Adachi T, Nielsen PJ, Terashima M, Lamers MC, Kohler G, Reth M. Two new proteins preferentially associated with membrane immunoglobulin D. EMBO J. 1994;13:3793–3800. doi: 10.1002/j.1460-2075.1994.tb06690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science (Wash DC) 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Krajewski S, Tanaka S, Takayama S, Schibler M, Fenton W, Reed JC. Investigation of the subcellular distribution of the Bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- Kroemer G, Zamzami N, Susin S. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- Kumar S, Lavin MF. The ICE family of cysteine proteases as effectors of cell death. Cell Death & Differentiation. 1996;3:255–267. [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Lam M, Dubyak G, Chen L, Nunez G, Miesfeld RL, Distelhorst CW. Evidence that Bcl-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+fluxes. Proc Natl Acad Sci USA. 1994;91:6569–6573. doi: 10.1073/pnas.91.14.6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki R, Yamamoto A, Tashiro Y. Microsomal aldehyde dehydrogenase is localized to the endoplasmic reticulum via its carboxyl-terminal 35 amino acids. J Cell Biol. 1994;126:1407–1420. doi: 10.1083/jcb.126.6.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride HM, Millar DG, Li JM, Shore GC. A signal-anchor sequence selective for the mitochondrial outer membrane. J Cell Biol. 1992;119:1451–1457. doi: 10.1083/jcb.119.6.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung JK, Danner DB, Stewart DA, Smith JR, Schneider EL, Lumpkin CK, Dell'Orco RT, Nuell MJ. Isolation of a cDNA that hybrid selects antiproliferative mRNA from rat liver. Biochem Biophys Res Commun. 1989;164:1316–1322. doi: 10.1016/0006-291x(89)91813-5. [DOI] [PubMed] [Google Scholar]
- McLorie W, McGlade CJ, Takayesu D, Branton PE. Individual adenovirus E1B proteins induce transformation independently but by additive pathways. J Gen Virol. 1991;72:1467–1471. doi: 10.1099/0022-1317-72-6-1467. [DOI] [PubMed] [Google Scholar]
- Minn AJ, Vélez P, Schendel SL, Liang H, Muchmore SW, Fesik SW, Fill M, Thompson CB. Bcl-XLforms an ion channel in synthetic lipid membranes. Nature (Lond) 1997;385:353–357. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- Mosser J, Sarde CO, Vicaire S, Yates JR, Mandel JL. A new human gene (DXS1357E) with ubiquitous expression, located in Xq28 adjacent to the adrenoleukodystrophy gene. Genomics. 1994;22:469–471. doi: 10.1006/geno.1994.1413. [DOI] [PubMed] [Google Scholar]
- Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Muzio M, Salvensen GS, Dixit VM. FLICE induced apoptosis in a cell free system–cleavage of caspase zymogens. J Biol Chem. 1997;272:2952–2956. doi: 10.1074/jbc.272.5.2952. [DOI] [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Naumovski L, Cleary ML. The p53-binding protein 53BP2 also interacts with Bc12 and impedes cell cycle progression at G2/M. Mol Cell Biol. 1996;16:3884–3892. doi: 10.1128/mcb.16.7.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M, Millar DG, Yong VW, Korsmeyer SJ, Shore GC. Targeting of Bcl-2 to the mitochondrial outer membrane by a COOH-terminal signal anchor sequence. J Biol Chem. 1993;268:25265–25268. [PubMed] [Google Scholar]
- Nguyen M, Branton PE, Walton PA, Oltvai ZN, Korsmeyer SJ, Shore GC. Role of membrane anchor domain of Bcl-2 in suppression of apoptosis caused by E1B-defective adenovirus. J Biol Chem. 1994;269:16521–16524. [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature (Lond) 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M, Reed JC. Channel formation by anti-apoptotic protein Bcl-2. Proc Natl Acad Sci USA. 1997;94:5113–5118. doi: 10.1073/pnas.94.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham S, Horvitz HR. Developing Caenorhabditis elegansneurons may contain both cell-death protective and killer activities. Genes Dev. 1996;10:578–591. doi: 10.1101/gad.10.5.578. [DOI] [PubMed] [Google Scholar]
- Shepherd SE, Howe JA, Mymryk JS, Bayley ST. Induction of the cell cycle in baby rat kidney cells by adenovirus type 5 E1A in the absence of E1B and a possible influence of p53. J Virol. 1993;67:2944–2949. doi: 10.1128/jvi.67.5.2944-2949.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasula SM, Ahmad M, Fernandes-Almeri T, Litwack G, Alnemri ES. Molecular ordering of the Fas-apoptotic pathway: the Fas/apo-1 protease Mch5 is a CrmA-inhibitable protease that activates multiple Ced-3/ ICE-like cysteine proteases. Proc Natl Acad Sci USA. 1996;93:14486–14491. doi: 10.1073/pnas.93.25.14486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A, Harris AW, Cory S. bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell. 1991;67:889–899. doi: 10.1016/0092-8674(91)90362-3. [DOI] [PubMed] [Google Scholar]
- Teodoro JG, Shore GC, Branton PE. Adenovirus E1A proteins induce apoptosis by both p53-dependent and p53-independent mechanisms. Oncogene. 1995;11:467–474. [PubMed] [Google Scholar]
- von Heijne G. Net N-C charge imbalance may be important for signal sequence function in bacteria. J Mol Biol. 1986;192:287–290. doi: 10.1016/0022-2836(86)90365-7. [DOI] [PubMed] [Google Scholar]
- Walter P, Blobel G. Preparation of microsomal membranes for cotranslational protein translocation. Methods Enzymol. 1983;96:84–93. doi: 10.1016/s0076-6879(83)96010-x. [DOI] [PubMed] [Google Scholar]
- Wang H-G, Miyashita T, Takayama S, Sato T, Torigoe T, Krajewski S, Tanaka S, Hovey L, III, Troppmair J, Rapp UR, Reed JC. Apoptosis regulation by interaction of Bcl-2 protein and Raf-1 kinase. Oncogene. 1994;9:2751–2756. [PubMed] [Google Scholar]
- Wang H-G, Rapp UR, Reed JC. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell. 1996a;87:629–638. doi: 10.1016/s0092-8674(00)81383-5. [DOI] [PubMed] [Google Scholar]
- Wang H-G, Takayama S, Rapp UR, Reed JC. Bcl-2 interacting protein, Bag-1, binds to and activates the kinase Raf-1. Proc Natl Acad Sci USA. 1996b;93:7063–7068. doi: 10.1073/pnas.93.14.7063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. Life, death, and the pursuit of apoptosis. Genes Dev. 1996;10:1–15. doi: 10.1101/gad.10.1.1. [DOI] [PubMed] [Google Scholar]
- Wu D, Wallen HD, Nuñez G. Interaction and regulation of subcellular localization of Ced-4 by Ced-9. Science (Wash DC) 1997;275:126–1129. doi: 10.1126/science.275.5303.1126. [DOI] [PubMed] [Google Scholar]
- Yang E, Korsmeyer SJ. Molecular thanatopsis: a discourse on the Bcl-2 family and cell death. Blood. 1996;88:386–401. [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XLand Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng T-I, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science (Lond) 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist Bad in response to survival factor results in binding to 14-3-3 not Bcl-XL . Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvensen GS. Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J Biol Chem. 1997;272:7797–7800. doi: 10.1074/jbc.272.12.7797. [DOI] [PubMed] [Google Scholar]
- Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DW. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J. 1996;15:4130–4141. [PMC free article] [PubMed] [Google Scholar]