

Abstract
Genetic defects in a number of components of the dystrophin–glycoprotein complex (DGC) lead to distinct forms of muscular dystrophy. However, little is known about how alterations in the DGC are manifested in the pathophysiology present in dystrophic muscle tissue. One hypothesis is that the DGC protects the sarcolemma from contraction-induced damage. Using tracer molecules, we compared sarcolemmal integrity in animal models for muscular dystrophy and in muscular dystrophy patient samples. Evans blue, a low molecular weight diazo dye, does not cross into skeletal muscle fibers in normal mice. In contrast, mdx mice, a dystrophin-deficient animal model for Duchenne muscular dystrophy, showed significant Evans blue accumulation in skeletal muscle fibers. We also studied Evans blue dispersion in transgenic mice bearing different dystrophin mutations, and we demonstrated that cytoskeletal and sarcolemmal attachment of dystrophin might be a necessary requirement to prevent serious fiber damage. The extent of dye incorporation in transgenic mice correlated with the phenotypic severity of similar dystrophin mutations in humans. We furthermore assessed Evans blue incorporation in skeletal muscle of the dystrophia muscularis (dy/dy) mouse and its milder allelic variant, the dy2J/dy2J mouse, animal models for congenital muscular dystrophy. Surprisingly, these mice, which have defects in the laminin α2-chain, an extracellular ligand of the DGC, showed little Evans blue accumulation in their skeletal muscles. Taken together, these results suggest that the pathogenic mechanisms in congenital muscular dystrophy are different from those in Duchenne muscular dystrophy, although the primary defects originate in two components associated with the same protein complex.
Mutations in several components of the dystrophin–glycoprotein complex (DGC)1 are known to be involved in the pathogenesis of muscular dystrophies (Ozawa et al., 1995; Straub and Campbell, 1997). This oligomeric complex connects the subsarcolemmal cytoskeleton to the extracellular matrix (Ervasti and Campbell, 1993). The intracellular link of the DGC is the membrane-associated cytoskeletal protein dystrophin, the protein product of the Duchenne muscular dystrophy (DMD) gene (Hoffman et al., 1987). The high density of dystrophin in the subsarcolemmal cytoskeleton (Ohlendieck and Campbell, 1991a ; Ohlendieck et al., 1991b ), its homology to α-actinin and spectrin (Koenig et al., 1988; Dhermy, 1991), its costameric organization (Porter et al., 1992; Straub et al., 1992), and its expression at the myotendinous junction (Tidball, 1991) strongly suggest that it plays an important structural role in muscle fibers. Dystrophin binds with its amino-terminal and rod domain to actin (Rybakova et al., 1996) and with its carboxy terminus to the integral membrane protein β-dystroglycan (Suzuki et al., 1994; Jung et al., 1995). The peripheral membrane glycoprotein α-dystroglycan, a receptor for the heterotrimeric basement membrane protein laminin-2, binds to β-dystroglycan and so completes the connection from the inside to the outside of the cell (Henry and Campbell, 1996). Mutations in the LAMA2 gene, encoding the α2 chain of laminin-2, have been characterized in a form of congenital muscular dystrophy (CMD) linked to chromosome 6q (Tomé et al., 1994; Helbling-Leclerc et al., 1995; Nissinen et al., 1996).
It has been proposed that the initial event in muscle cell necrosis in DMD was the focal breakdown of the plasmalemma (Mokri and Engel, 1975; Schmalbruch, 1975; Carpenter and Karpati, 1979; Weller et al., 1990). Evidence for leakage of intracellular contents out of dystrophic or damaged muscle cells is provided by elevated serum levels of muscle enzymes (Rosalki, 1989) and growth factors (D'Amore et al., 1994; Kaye et al., 1996). Simultaneously, this loss of sarcolemmal integrity allows influx of molecules into muscle fibers. In particular, elevated calcium levels have been noted in dystrophin-deficient skeletal muscle (Bodensteiner and Engel, 1978; Gillis, 1996). One or both of these events may contribute to the pathogenesis of muscular dystrophy.
To study altered sarcolemmal permeability in dystrophic muscle fibers, we injected animal models for muscular dystrophy with Evans blue dye (EBD). The tetrasodium diazo salt Evans blue, also called T-1824, is a membrane-impermeant molecule that can be used in determining blood volume (Reeve, 1957). This in vivo tracer technique provides information about certain structural and dynamic features of normal and pathological skeletal muscles (Matsuda et al., 1995).
For the tracer injection, we used mdx mice, transgenic/ mdx mice, dy/dy, and dy2J/dy2J mice. The mdx mouse is a dystrophin-deficient animal model for X-linked DMD (Bulfield et al., 1984). It has been reported that the dystrophin mutation in the mdx mouse leads to an associated reduction of DGC components (Ohlendieck and Campbell, 1991b). We also injected four different transgenic/mdx mice that have been shown to result in different skeletal muscle phenotypes (Rafael et al., 1994; Phelps et al., 1995; Corrado et al., 1996; Rafael et al., 1996). These mice enabled us to analyze regions of dystrophin that are critical for maintaining sarcolemmal integrity. One of the transgenic animals expressed only the COOH-terminal isoform of dystrophin (Dp71), which is encoded by exons 63–79. These Dp71 mice display proper localization of the DGC at the sarcolemma, but still show a dystrophic phenotype (Cox et al., 1994; Greenberg et al., 1994). The other three transgenic/mdx mice we injected were generated by expressing “full length” dystrophin constructs, but with consecutive deletions within the amino terminal domain (Δ3-7; Corrado et al., 1996), the rod domain (Δ17-48; Phelps et al., 1995), and the carboxy-terminal domain (Δ71-74; Rafael et al., 1994, 1996). Mice missing exons 71–74 or exons 17–48 of the dystrophin gene display a markedly milder phenotype than mdx mice despite the expression of moderate levels of dystrophin. In contrast to deletions of exons 71–74 or in the central rod domain, proteins with a deletion in the actin-binding NH2 terminus must be expressed at high levels to prevent a dystrophic phenotype (Corrado et al., 1996).
Furthermore, we examined the dy/dy and the dy2J/dy2J mice. These murine diseases are inherited in an autosomal recessive fashion and have been studied as models for human laminin α2 chain–deficient CMD (Sunada et al., 1994; Xu et al., 1994b ), whereby the dy2J/dy2J mouse represents the milder allelic variant of the dy/dy mouse (Sunada et al., 1995). The Lama2 gene on mouse chromosome 10 was closely mapped to the dy/dy locus (Sunada et al., 1994), and a single in-frame deletion eliminating 57 amino acids was discovered in the α2 chain transcript in dy2J/dy2J mice (Sunada et al., 1995). A principal goal of our studies was to determine whether deficiency of the extracellular ligand of the DGC, laminin-2, had similar effects on sarcolemmal integrity as deficiency of its intracellular connecting link, dystrophin.
Materials and Methods
Mice
Normal control, the original mdx mutant, and dy2J/dy2J mice were bred at the University of Iowa from stocks originally obtained from the Jackson ImmunoResearch Laboratories, Inc. (Bar Harbor, ME). All mice are on a C57BL/10 background. The dy/dy mice were also obtained from the Jackson Laboratories. The transgenic/mdx mouse strains were maintained at the University of Michigan. Control and disease animals were matched by age and gender. The age range of the mice was between 4 wk and 1 yr. All animal studies were authorized by the Animal Care Use and Review Committee of the University at Iowa.
Evans Blue Injection
EBD was injected either into the tail vein of the mice or into the peritoneal cavity without anesthesia. For the assessment of EBD uptake into muscle fibers, intravenous administration of the dye was preferred. Nonspecific coloration of the diaphragm and the abdominal muscles was avoided by this route of injection. EBD was dissolved in PBS (0.15 M NaCl, 10 mM phosphate buffer, pH 7.4) and sterilized by passage through membrane filters with a 0.2-μm pore size. The concentration of the injected dye was 0.5 mg EBD/0.05 ml PBS. Animals were injected with 50 μl of this solution per 10 g body wt. 3–6 h after injection, the mice were killed by cervical dislocation. The skin of the mice was removed, and the animals were visually inspected for dye uptake into skeletal muscles, indicated by blue coloration. To better characterize dye uptake in distinct muscles of injected animals, we investigated cryosections of the femoral quadriceps, the sural triceps, the pectoral, the diaphragm, and the cardiac muscle. Additional skeletal muscle samples were taken if the muscle showed blue coloration. Muscle sections from EBD-injected animals were incubated in ice-cold acetone at −20°C for 10 min, washed 3× 10 min with PBS, and mounted with Vectashield mounting medium (Vector Laboratories, Inc., Burlingame, CA). By fluorescence microscopy analysis, EBD staining showed a bright red emission. Fiber counts of EBD-positive muscle fibers were done independently by two investigators on 7-μm cryosections of dye injected mice. All sections were examined and photographed under an Axioplan fluorescence microscope (Carl Zeiss, Inc., Thornwood, NY) or a MRC-600 laser scanning confocal microscope (Bio Rad Laboratories, Hercules, CA).
Antibodies
For immunofluorescence analysis of albumin, the AIAG3140 rabbit anti– mouse albumin antibody conjugated to FITC (Accurate Chemical & Scientific Corp., Westbury, NY) was used at a concentration of 1:75. Staining for Igs was performed with biotinylated anti–mouse IgG (H+L), biotinylated anti–mouse IgM, biotinylated anti–human IgG (H+L), and biotinylated anti–human IgM (Vector Laboratories). All anti-Ig antibodies were used at a concentration of 1:500. A monoclonal antibody VIA42A3 against dystrophin (Ervasti et al., 1990) and a polyclonal rabbit antibody against the α2 chain of laminin (Allamand et al., 1997) were used for screening purposes of mouse and patient samples. Biotinylated antibodies were detected using streptavidin conjugated to either Texas red or FITC (Vector Laboratories).
Immunofluorescence Microscopy
Muscle tissue from DMD and CMD patients was obtained from diagnostic muscle biopsies. The clinical diagnosis of DMD was confirmed by complete absence of dystrophin expression in muscle tissue. The clinical diagnosis of CMD was confirmed by white matter changes on brain magnetic resonance images and by reduction or deficiency of laminin α2 chain expression in muscle tissue. Normal human control muscle was received from patients undergoing orthopedic surgery not related to neuromuscular disorders. Immunofluorescence microscopy of 7-, 15-, and 30-μm cryosections from skeletal and cardiac muscle was performed as described previously (Ohlendieck et al., 1991a ).
Cryosections for IgG and IgM staining were preincubated for 30 min with 5% BSA in PBS, followed by a 1-h incubation with biotin-labeled goat anti–mouse IgG or IgM antibodies. The sections were washed 3× for 5 min with 1% BSA/PBS, incubated with FITC-conjugated streptavidin, and after a final wash, mounted with Vectashield mounting medium (Vector Laboratories). For albumin staining, the sections were blocked with 1% gelatin in PBS for 15 min, washed in PBS + 0.2% gelatin, and incubated for 1 h in PBS + 1% normal goat serum with a rabbit anti–mouse albumin FITC-conjugated antibody. Histochemical examination of muscle tissue was performed by hematoxylin and eosin (H&E) staining as described (Dubowitz, 1985).
Results
Loss of Sarcolemmal Integrity in mdx Mice
Within seconds after intravenous injection of EBD, discoloration of all animals was observed. A successful injection of the dye was indicated by the blue coloration of ears and paws. Control mice did not show dye uptake into their skeletal muscles by visual inspection. In EBD-injected control mice, the presence of EBD in the perivascular spaces was seen microscopically when the muscle sections were inspected without any washing steps. There were very few single fibers with a positive dye staining on sections of control animals, which were otherwise EBD negative (Table I).
Table I.
Analysis of EBD-injected Mice
| Mice | Macroscopic skeletal muscle staining | Microscopic staining | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Femoral quadriceps | Soleus | Pectoralis major | Diaphragm | |||||||
| control (n = 16) | 0% | –* | – | – | – | |||||
| mdx (n = 16) | 100% | ++§ | +‡ | + | + | |||||
| dy/dy (n = 6) | 0% | – | – | – | – | |||||
| dy2J/dy2J(n = 6) | 0% | – | – | – | – | |||||
3–6 h after intravenous EBD injection, skinned animals were visually inspected for dye uptake into skeletal muscles. Dye-positive muscles were easily detected by localized blue coloration. Only mdx mice always (100%) showed dye uptake into at least one muscle by macroscopic evaluation. This was in strong contrast to normal control, dy/dy, and dy2J/dy2J mice. The dy/dy and dy2J/dy2J mice mainly showed dye uptake into their connective tissue. Clear macroscopic EBD staining of skeletal muscles was never (0%) detected in these animals. This finding was confirmed by fluorescence microscopy analysis of 7-μm cryosections from the quadriceps femoris, soleus, pectoralis major, and diaphragm muscles of injected animals. Fiber counts of EBD-positive muscle fibers were done independently by two investigators. The number of EBD-positive fibers in a muscle was compared to the total number of fibers. Assessment of dye uptake into the diaphragm was not performed on six intraperitoneally injected mdx mice.
Less then 1% of the muscle fibers were EBD positive.
Less then 10% of the muscle fibers were EBD positive.
More then 10% of the muscle fibers were EBD positive.
In the mdx mutant, administered EBD always resulted in blue discoloration of skeletal muscles (Fig. 1). This result was independent of the age of the animals used. The accumulation of EBD tended to be more intense at certain anatomical sites, forming a characteristic topographic pattern of staining. Areas of blue staining appeared mainly within the regions of the proximal limb muscles, as well as the pelvic and the shoulder girdle (Fig. 1). The dye was particularly incorporated in gluteal, femoral quadriceps, and the ischiocrural muscles (hind limbs) and sometimes the pectoral and the triceps brachii muscles (forelimbs; Fig. 1). Affected muscles were not stained homogeneously, but they typically showed blue strands representing damaged muscle fibers. In a longitudinal alignment, these strands could be seen throughout the entire length of the muscles (Fig. 1). Although there was a general topographic distribution of the dye in skeletal muscles of mdx mice, permeability of a given muscle region varied from animal to animal and even between opposing limbs within the same animal. We found differences in both the intensity and the extent of stained regions in left and right limbs (Fig. 1 b). In addition to the proximal limb muscles, we also found discoloration of the external oblique muscle of the abdomen, the longest thoracic and lumbar muscles, the cutaneous muscle of the trunk (Fig. 1 c), and in intercostal muscles (Fig. 2 c). Anterior tibial muscles in mdx mice were often spared from dye staining. In contrast, the diaphragm of mdx mice showed areas of dye incorporation in all tested animals (Fig. 2 e). Because of the variability in dye accumulation and distribution, there was no significant difference in the staining pattern among mice that were 4–52 wk old.
Figure 1.

Macroscopic evaluation of Evans blue staining after intravenous dye injection into two 9-wk-old mdx littermates (mouse 1, a and b; mouse 2, c and d). The mice were fixed in a 8% formaldehyde solution 6 h after the dye injection. EBD staining in mouse 1 (a and b) showed staining mainly within the regions of the pelvic girdle muscles. In particular, the dye was incorporated in the gluteal and the femoral quadriceps muscles. Plasma membrane permeability for Evans blue varied from animal to animal and even from limb to limb in the same animal. In mouse 1, we found differences in both the intensity and the extent of stained regions in the left and right triceps brachii muscles (a and b, arrows). Besides dye uptake in proximal limb muscles, we also found discoloration of the external oblique muscle of the abdomen, the longest thoracic and lumbar muscles, and the cutaneous muscles of the trunk (c and d). Affected muscles were not stained homogeneously, but they typically showed blue strands according to damaged muscle fibers. Anterior tibial muscles in mdx mice were often macroscopically spared from dye staining (a and c).
Figure 2.
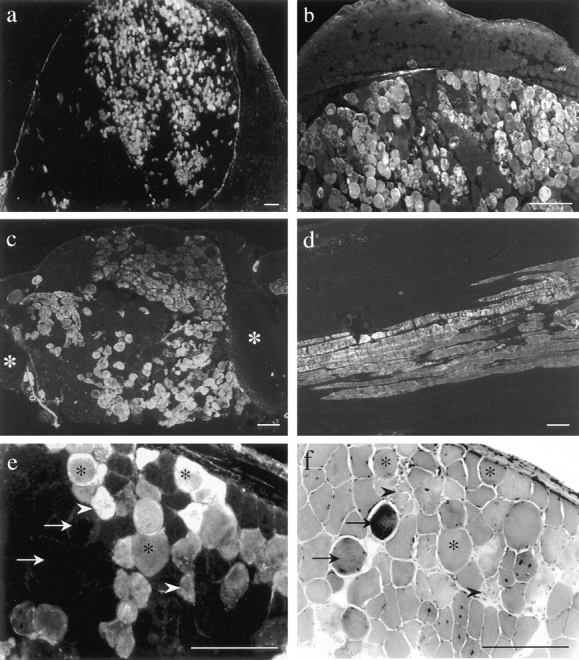
EBD (a–e) and H&E staining (f) on 7-μm (a, b, e, and f) and 15-μm (c and d) skeletal muscle cryosections from 8- (a, b, e, and f) and 16-wk-old (c and d) intravenously (a, b, e, and f) and intraperitoneally (c and d) injected mdx mice. In some animals, the number of dye-positive fibers in the femoral quadriceps muscle was >70% (a). b shows a magnification of a in which the fascia demarcates a highly damaged muscle region from the unaffected adjacent muscle. Other muscles, including the intercostal muscles (c, white asterisks indicate rips) and gluteal muscles (d, longitudinal section) took up the dye. EBD staining in mdx diaphragm (e) demonstrated variation in the dye-positive fibers. Dye positive fibers in the corresponding H&E staining (f) revealed morphological features of normal fibers (asterisk) as well as of necrosis (arrowhead). Hypercontracted fibers did not necessarily indicate membrane damage, since some of them did not take up EBD (arrows). Bars, 100 μm.
In mdx mice, skeletal muscles that visually showed dye uptake were always EBD positive by fluorescence microscopy analysis (Fig. 2 a). In most of the cases, the intracellular staining was diffusely distributed across the myofiber cytoplasm, although sometimes the fiber periphery was more strongly stained than the center of the fiber. Furthermore, the intensity of the dye signal showed differences between fibers (Fig. 2, a–e). These findings were also detected on 15- and 30-μm cross-sections and on longitudinal sections (Fig. 2). Most of the EBD-positive fibers showed intense staining, whereas in some others, the signal was faint. Transverse cryosections of blue discolored muscles never showed EBD in all fibers of a section. Instead, the frequency of EBD fibers in a given muscle varied from animal to animal and from muscle to muscle, reflecting the results of visual inspection (Fig. 1). In transverse sections of the mdx femoral quadriceps muscle, the mean frequency of dye-positive fibers was >10% of the total fiber number (Table I), with a maximum of >70% positive fibers (Fig. 2 a).
In transverse sections, the dye-positive mdx fibers occurred either singly or, more often, in groups. The grouped EBD-positive fibers were one of the most striking features of dye staining in mdx mice (Fig. 2). Although EBD-positive fibers sometimes showed signs of necrosis in the H&E staining, there was no clear correlation between dye-positive fibers and a uniform morphological feature (Fig. 2, e and f). In contrast to a previous report (Matsuda et al., 1995), we also found EBD-negative hypercontracted fibers besides positive hypercontracted fibers (Fig. 2, e and f, arrows) and therefore considered these to have an intact sarcolemma. Fibers with distinctive necrotic features in H&E seemed to be always EBD positive, no matter which animal was injected (Fig. 2, e and f, arrowheads). On longitudinal sections, we could demonstrate that dye uptake was not confined to a small part of the fiber, but appeared over long segments of the entire muscle cell (Fig. 2 d).
Evans Blue Staining in Cardiac Muscle of mdx Mice
We also examined cardiac muscle from all injected animals, since cardiac involvement is a common feature in many forms of muscular dystrophy. Control mice never showed EBD-positive fibers in cardiac muscle tissue. In contrast, 8 out of 16 mdx mice had EBD-positive lesions in the myocardium. In one 4-wk-old mouse, large areas of the cardiac section showed EBD uptake into cardiac muscle fibers (Fig. 3 a), whereas in the other mice, ranging from 6 to 52 wk of age, regions of variable size in the ventricular wall were affected (Fig. 3, b and c). On sections stained with H&E, the EBD-positive areas showed characteristic features of myocardial damage, with a strong inflammatory component (data not shown). Myocardial EBD staining in intraperitoneally injected mice (Fig. 3 c) provided evidence that the fiber damage was caused by the underlying disease and not by volume overload of intravenously administered dye, leading to a cardiac infarct.
Figure 3.
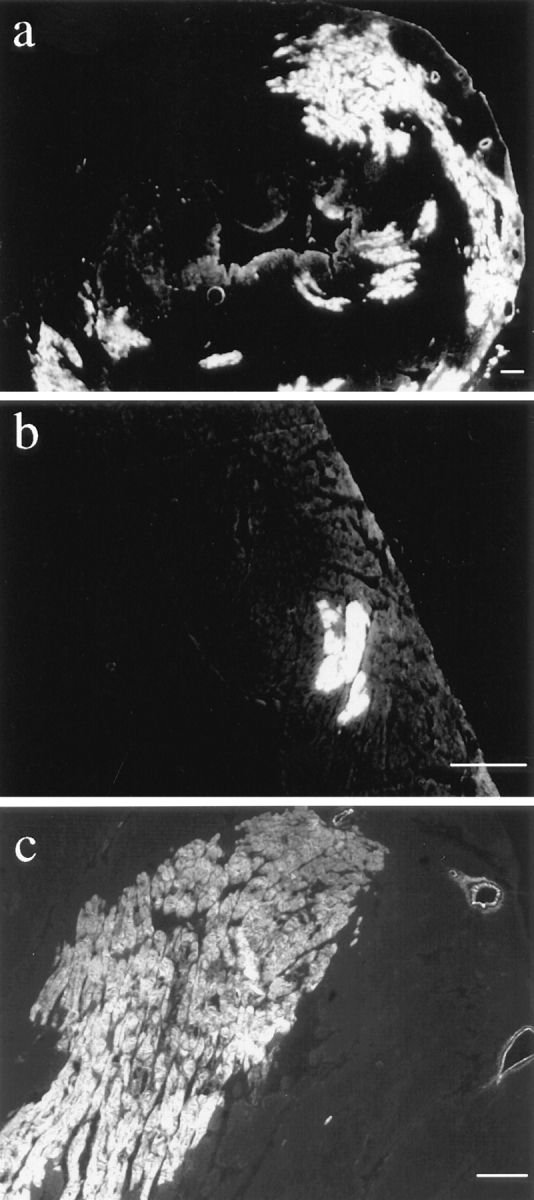
EBD staining on 7-μm cryosections of mdx cardiac muscle. EBD-positive cardiomyocytes were detected in 8 out of 16 mdx mice but never in control animals. One intravenously injected 4-wk-old animal showed large areas of dye uptake into the myocardium (a), whereas in other mice, smaller regions of EBD-positive fibers were found in the ventricular walls (b). Cardiac dye uptake in intraperitoneally injected mdx mice (c, 10-mo-old animal) demonstrated that the fiber damage was caused by the underlying disease and not by a volume overload of intravenously administered dye, leading to a cardiac infarct. Bars, 100 μm.
Presence of Serum Proteins in Damaged Skeletal Muscle Fibers
The chief characteristic of the EBD is its ability to form a tight complex with serum albumin within seconds after its injection into the bloodstream (Reeve, 1957). In view of the tight association between EBD and albumin, areas of blue macroscopic staining were taken to represent regions of albumin uptake into muscle fibers. To demonstrate that EBD-positive muscle fibers in mdx mice do take up albumin, we stained cryosections for albumin. We could show that the same fibers that took up the dye were also positive for albumin staining (Fig. 4 A).
Figure 4.

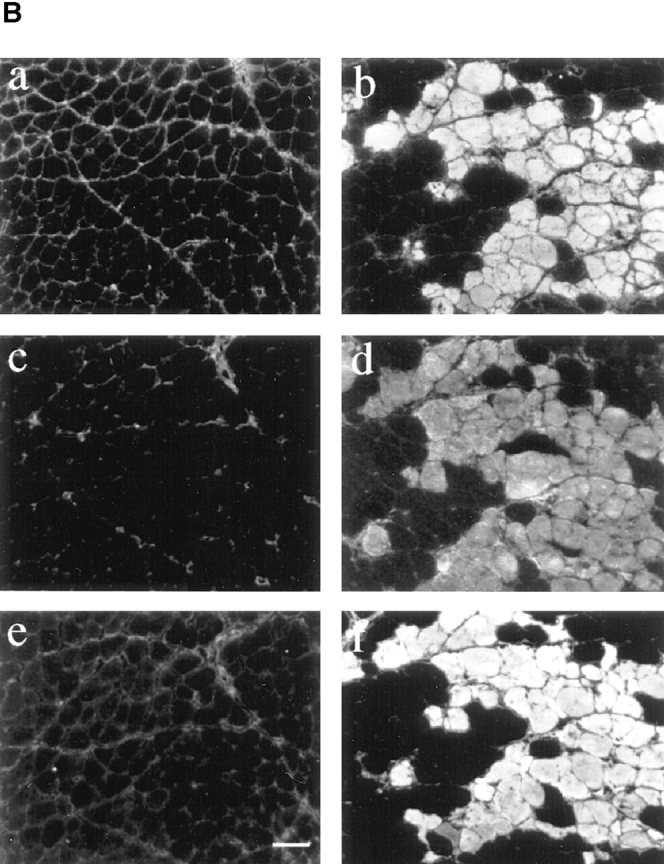
Loss of membrane integrity is indicated by intracellular staining of serum proteins. EBD-positive fibers in skeletal muscle from mdx mice also showed positive staining with antibodies against albumin on the same section. (A) Double staining for EBD (a) and albumin (b) in an 8-wk-old mdx mouse. (B) Skeletal muscle of 10-wk-old mice that were not injected with EBD were studied for uptake of serum proteins into muscle fibers. Immunohistochemical staining of 7-μm femoral quadriceps cryosection from uninjected normal and mdx mice with antibodies against mouse IgG (a and b), mouse IgM (c and d), and mouse albumin (e and f) showed accumulation of the serum markers in mdx skeletal muscle. No difference of protein uptake into muscle fibers was detected between control and dy/dy mice. Bar, 50 μm.
To further demonstrate that serum proteins cross into damaged fibers, we stained skeletal muscle sections of uninjected mice with antibodies against albumin (65 kD), IgG (150 kD), and IgM (900 kD; Fig. 4 B). In all muscles examined, positive staining with anti–mouse serum albumin antibodies, anti–mouse IgG antibodies, and anti–mouse IgM antibodies was observed in the endo- and perimysium (Fig. 4 B). Control mice did not show intracellular fiber staining with these antibodies. On the other hand, mdx mice showed intracellular staining for all antibodies within a part of their muscle fibers (Fig. 4 B). The staining patterns for the serum markers were strikingly similar to the patterns of EBD deposition. Intracellular staining was diffusely distributed across the myofiber cytoplasm, although sometimes it appeared to be localized to the fiber periphery. Examination of serial sections showed that the pattern of intracellular staining was similar for albumin, IgG, and IgM (Fig. 4 B).
The Role of Muscle Fiber Membrane Damage in Transgenic/mdx Mice
To test the importance of distinct dystrophin domains for maintaining sarcolemmal integrity, we injected EBD into transgenic lines of mdx mice expressing different portions of the dystrophin molecule (Fig. 5). Intravenous injection of EBD into Dp71 mice led to incorporation of the tracer into skeletal muscles by visual inspection. The intensity and the extent of stained regions was similar to that of mdx mice. In contrast, Δ17-48 transgenic/mdx mice, which mimic the dystrophin mutation in an extremely mild case of Becker muscular dystrophy (England et al., 1990; Phelps et al., 1995), and Δ71-74 transgenic/mdx mice, which have been reported to show no dystrophic phenotype up to at least 2 yr of age (Rafael et al., 1994, 1996), did not demonstrate macroscopic dye uptake after intravenous injections. The Δ3-7 transgenic mdx mice, in which the expression of a full-length dystrophin construct deleted for the amino-terminal, actin-binding domain improves the mdx pathology to a mild “Becker-like” phenotype (Corrado et al., 1996), did take up EBD into skeletal muscle fibers. However, sarcolemmal damage in Δ3-7 transgenic/mdx mice, as assessed by EBD incorporation, appeared less severe than in the mdx or the Dp71 mice (Fig. 5). The femoral quadriceps muscle and the diaphragm were the only muscles in which dye accumulation was visually observed in Δ3-7 transgenic/mdx mice.
Figure 5.
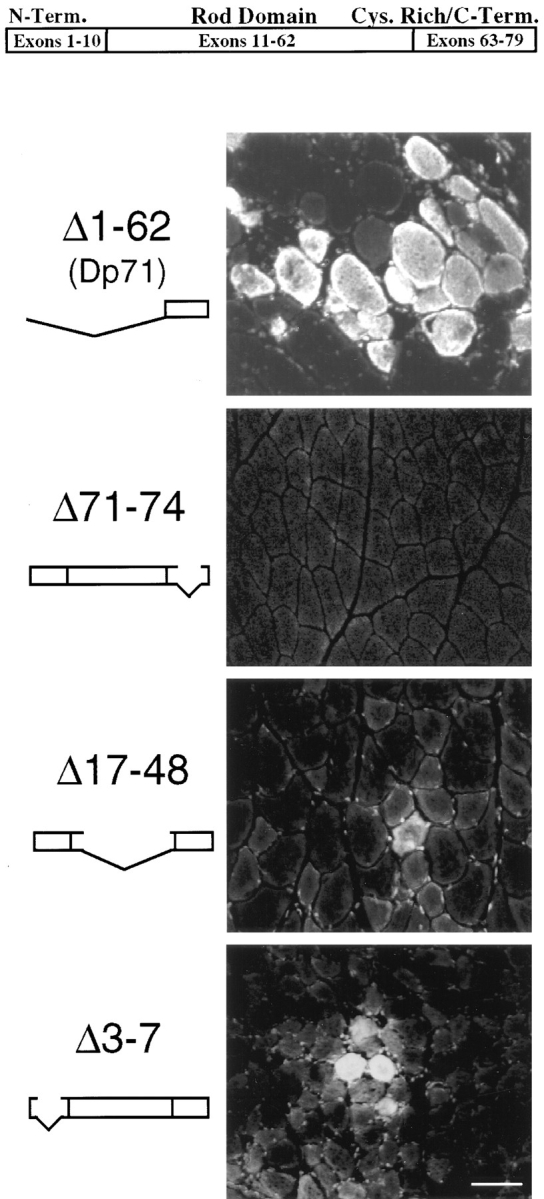
EBD staining on 7-μm cryosection of skeletal muscle from four transgenic/mdx mice. The top portion of the picture shows a model of the normal dystrophin gene. The femoral quadriceps muscle of the Dp71 mouse (20 wk old) revealed the same amount of fiber damage in the EBD assay as the original mdx mutant. Mice with a deletion of dystrophin exons 71–74 (17 wk old) did not show significantly more dye uptake into the femoral quadriceps muscle than seen in control animals. The Δ17-48 transgenic/mdx mouse (9 wk old), which has a deletion in the rod domain of dystrophin, showed few dye-positive fibers in the quadriceps femoris muscle. The Δ3-7 transgenic/mdx mouse (10 wk old), which has a deletion in the amino-terminal actin-binding domain of dystrophin, showed dye-positive fibers in the diaphragm (Δ3-7), as well as in the quadriceps femoris muscle. Bar, 50 μm.
Fluorescence microscopy analysis of Dp71 skeletal muscle cryosections revealed no difference in the amount of EBD-positive muscle fibers between Dp71 and mdx mice (Fig. 5). In the Δ17-48 transgenic/mdx mice and the Δ71-74 transgenic/mdx mice, fluorescent microscopy analysis revealed some EBD-positive fibers in the diaphragm and the femoral quadriceps muscle, although never to the extent of the mdx (Fig. 5). In Δ3-7 transgenic/mdx mice, the number of EBD-positive fibers in macroscopically blue areas of skeletal muscle was smaller, and the fibers were more loosely distributed compared to sections of injected mdx or Dp71 mice (Fig. 5).
dy/dy and dy2J/dy2J Mice Maintain Plasma Membrane Integrity
Interestingly, dy/dy and dy2J/dy2J mice did not reveal dye incorporation into skeletal muscles (Fig. 6). Because of the increase in connective tissue in dystrophic muscle, the mice showed a blue aspect resulting from dye accumulation into their connective tissue. Otherwise, we did not detect any dye accumulation in the inspected muscles (Table I).
Figure 6.

Macroscopic and microscopic evaluation of EBD staining after intravenous dye injection into 3-mo-old normal control (a and b), mdx (c and d), and dy/dy (e and f) mice. Uptake of dye into the hind legs of the mice was examined 6 h after the injection. The hind legs were fixed in a 8% formaldehyde solution. In contrast to mdx mice (c), dy/dy (e) or dy2J/dy2J mice never showed localized EBD uptake into skeletal muscles by visual inspection. Blue coloration of dy/dy and dy2J/dy2J mice observed by macroscopic evaluation was caused by dye uptake into the connective tissue, which is increased in these dystrophic animals. This finding was confirmed by fluorescence microscopy analysis of 7-μm cryosections from the quadriceps femoris muscle of injected animals. Grouped EBD-positive fibers were only detected in mdx mice (d), whereas cryosections from normal control mice (b), dy/dy (f), or dy2J/dy2J mice did not show dye uptake into groups of muscle fibers. On cryosections of dy/dy (f) mice, single necrotic fibers showed EBD staining by fluorescence microscopy. Bar, 50 μm.
The sections from injected dy/dy and dy2J/dy2J mice showed only occasional EBD-positive fibers in the diaphragm (Fig. 7) or the femoral quadriceps muscle (Fig. 6 f). The low level of intracellular fiber staining was a constant feature of the muscles we examined from dy/dy and dy2J/ dy2J mice (Table I). The dye-positive fibers always showed necrotic features according to H&E staining (Fig. 7). They appeared singly and never showed grouping, which is characteristic for mdx or Dp71 transgenic/mdx mice (Fig. 6). There were no differences in the extent or intensity of dye accumulation and distribution in skeletal muscles between dy/dy and dy2J/dy2J mice. In contrast to mdx mice, we never found EBD staining in the cardiac muscle of dy/dy and dy2J/dy2J mice.
Figure 7.
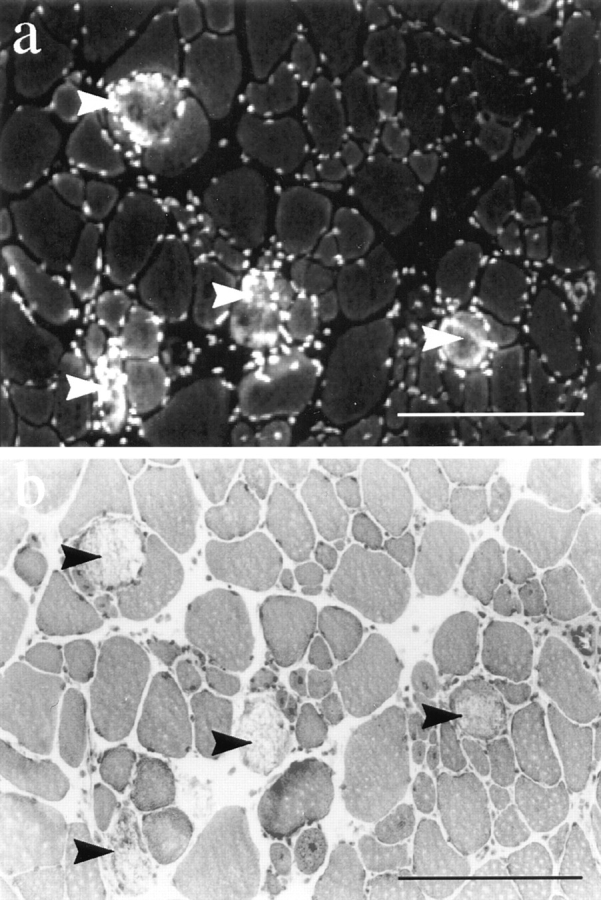
EBD staining (a) and H&E staining (b) on 7-μm cryosections from a 7-wk-old dy/dy diaphragm. The few EBD-positive fibers in dy/dy skeletal muscle always showed necrotic features in the corresponding H&E stain. The nuclei of infiltrating immune cells were also detected by the EBD. Bars, 100 μm.
Different Patterns of Sarcolemmal Disruption in Patients with DMD and Laminin α2 Chain–deficient CMD
To test whether the findings in the animal models could be reproduced in patients with DMD and laminin α2 chain– deficient CMD, we tested cryosections from diagnostic biopsies with antibodies against IgG and IgM. In three out of eight needle biopsies from DMD patients, we found IgG- and IgM-positive muscle fibers (Fig. 8). The globulin-positive fibers showed the same grouping pattern as that described for the mdx mice. On normal human muscle sections, IgG and IgM were detected only in the endo- and perimysium (Fig. 8). Interestingly, not all IgG-positive fibers in DMD patients showed IgM uptake into the cytoplasm (Fig. 8, arrow), possibly because of the different sizes of the molecules. This finding was confirmed by looking at serial sections of the biopsy samples. In the biopsies of eight patients with laminin α2 chain–deficient CMD, we never found grouped fibers with positive staining for Igs. Two of the CMD patients had a deletion in the LAMA2 gene similar to the dy2J mice and were previously characterized (Allamand et al., 1997). Single fibers with an intracellular staining signal for Igs were detected on CMD biopsies and showed morphological features of necrosis.
Figure 8.

Immunohistochemical staining of 7-μm cryosection from normal human skeletal muscle, DMD skeletal muscle, and skeletal muscle from patients with laminin α2 chain–deficient CMD with antibodies against human IgG and IgM. The plasma proteins showed intracellular fiber staining in the quadriceps femoris muscle from DMD biopsies, indicating loss of membrane integrity. Positive staining of grouped fibers was only detected in DMD patients. The antibody against human IgG did not show the same staining intensity as the antibody against human IgM. Interestingly, not all IgG-positive fibers took up IgM molecules (arrow), indicating different sizes of membrane disruptions. This observation was confirmed on serial sections throughout the damaged area. Bar 50, μm.
Discussion
Previous findings support the idea that one function of the DGC is to provide mechanical reinforcement of the sarcolemma and to maintain membrane integrity during cycles of contraction and relaxation (Weller et al., 1990; Clarke et al., 1995; Petrof et al., 1993). To test the involvement of sarcolemmal damage in the pathogenesis of muscle fiber degeneration and necrosis, we injected animal models for muscular dystrophy with EBD. Intracellular accumulation of the dye in skeletal muscle fibers indicated loss of sarcolemmal integrity due to plasma membrane disruptions. As a further test of altered membrane permeability, we examined the intracellular deposition of serum proteins in skeletal muscle fibers in the murine models and in patients with DMD and CMD.
Our results demonstrated that focal plasma membrane defects have actually occurred in vivo and not, as argued, during biopsy (Bradley and Fulthorpe, 1978). The diffuse intracellular distribution of the tracer implied a break in the structural integrity of the surface membrane. Interestingly, sarcolemmal permeability in certain muscles, including the heart, not only appeared to vary from animal to animal, but also showed alterations between the same muscle groups in the limbs of one animal. These findings suggest that in addition to the genetic disposition, sarcolemmal damage is also a function of environmental influences, such as activity in the time period between dye injection and death. By intravenous and intraperitoneal injection of EBD into mdx mice, we also demonstrated loss of membrane integrity in cardiomyocytes. These results show that the tracer technique may be helpful in evaluating the distribution and pattern of pathologic lesions in cardiac muscle, and that it might be a useful tool in studies of cardiac infarctions.
According to our findings and previous studies (Mokri and Engel, 1975; Karpati and Carpenter, 1982), we suggest that EBD-positive fibers in mdx mice do not inevitably lead to necrosis of the whole fiber, but often reflect degenerating fibers with a potential for regeneration. Segmental loss of sarcolemmal integrity or plasma membrane disruptions confined to a small region of the cell will allow the dye to cross into a muscle fiber and to diffuse along the longitudinal axis (Fig. 2 d). In the mdx or Dp71 mice, most of the EBD-positive fibers presumably undergo a stage of segmental necrosis and regeneration, and just a small group of dye positive fibers will undergo full-length fiber necrosis, depending on the amount and size of membrane disruptions. With the tracer technique, we demonstrated that in the femoral quadriceps muscle of mdx mice, >70% of the myofibers can show EBD uptake (Fig. 2 a). In addition, we could demonstrate that a number of EBD-positive fibers showed normal morphology by H&E staining (Fig. 2 f). It has been suggested that membrane defects allowed the influx of calcium-rich extracellular fluid into the muscle cells that caused hypercontraction and also initiated fiber necrosis (Mokri and Engel, 1975; Schmalbruch, 1975). Our results, on the other hand, indicate that hypercontracted fibers did not generally show primary plasma membrane defects, and they did not in and of themselves cause secondary rupture of the muscle fiber plasma membrane (Fig. 2, e and f). The interesting observation of grouped EBD fibers in cross-sections of mdx and Dp71 mice may reflect the fact that the fibers branch extensively (Bell and Conen, 1968; Schmalbruch, 1984; Head et al., 1992), and that the dye may diffuse into all branches of a fiber if one branch loses sarcolemmal integrity. Some of the faintly fluorescent muscle fibers may be segments adjacent to a damaged site from where the EBD diffused into the surviving stumps.
Furthermore, we were able to demonstrate that EBD-positive fibers were rendered transiently or permanently permeable to extracellular serum albumin and other serum proteins. These findings were confirmed in uninjected animals and in patient samples with muscular dystrophy (Figs. 3 and 6). The different molecular weights and sizes of these serum markers allowed us to draw conclusions about the size of sarcolemmal disruptions in affected fibers. The 900 kD IgM molecules were able to cross into the same fibers as 65 kD albumin proteins. The accumulation of serum proteins in the sarcoplasm also indicates that they may play a role in the pathogenic mechanism that finally leads to cell death.
As assessed by intracellular uptake of EBD, the mdx and Dp71 mice showed the most severe sarcolemmal disruptions. According to our results in the transgenic mice, the rod domain of dystrophin and the COOH-terminal domain encoded by exons 71–74 do not seem to be as important for membrane integrity. Expression of dystrophin deleted for the actin-binding sites encoded by exons 3–7 at or above normal dystrophin levels in the Δ3-7 transgenic/mdx mice resulted in a mild phenotype (Corrado et al., 1996) with relatively few EBD-positive fibers. The findings in the transgenic mice were particularly interesting with respect to the Dp71 mouse. Recent studies on the interaction of dystrophin with F-actin identified a novel F-actin– binding site near the middle of the dystrophin rod domain, and a model was proposed in which dystrophin binds F-actin in a manner analogous to actin side-binding proteins (Rybakova et al., 1996). In contrast to the Δ3-7 transgenic/mdx mouse, the Dp71 mouse lacks all known actin-binding domains of the dystrophin molecule, but is sufficient to restore the DGC at the sarcolemma. Our data indicate that the preservation of the DGC at the sarcolemma is not sufficient to prevent membrane disruptions. Taken together, these results indicate that cytoskeletal and sarcolemmal attachment of dystrophin might be a necessary requirement to prevent serious fiber damage. If these connections are altered, physiologic mechanical forces might already overstress the fragile membrane structure and cause focal plasma membrane disruptions. Pasternak et al. (1995) have demonstrated that the lack of dystrophin causes a substantial reduction in the stiffness of living muscle cells. In their model, dystrophin acts as a molecular spring that could redistribute stresses imposed locally on the sarcolemma over a wide area of the cell.
Most interestingly, the dy/dy and dy2J/dy2J mice showed no signs of sarcolemmal damage despite their severe clinical phenotype. The fact that we did not find a more significant dye accumulation in skeletal muscles of dy or dy2J mice as compared to normal controls indicated that plasma membrane disruptions do not seem to play a role in muscle cell degeneration of these animals. In the dy2J mouse, the deletion in the lama2 locus is located in the NH2-terminal domain IV, which is presumed to be involved in self-aggregation of laminin-2 heterotrimers, as in the case of laminin 1 (Schittny and Yurchenco, 1990). This mutation could disrupt the formation of the laminin-2 network in the muscle basal lamina, which could weaken or eliminate the linkage between the extracellular matrix and the muscle fiber surface. Our studies demonstrate that the laminin alteration does not effect the plasma membrane integrity. This finding is supported by electron microscopic studies suggesting that the muscular basement membrane in dy/dy mice is fragmented but plasma membrane disruptions do not occur (Xu et al., 1994a ). Similar findings have also been reported in patients with congenital muscular dystrophy with laminin α2 chain deficiency (Minetti et al., 1996).
The small size of the dy/dy and the dy2J/dy2J mice, their severe clinical phenotype, and the fact that they do not show dye accumulation in their skeletal muscles suggests that laminin-2 may transmit a specific signal to the muscle cell required for muscle function rather than play a mechanical role in maintaining membrane integrity. Laminin molecules are known to be multifunctional, performing key roles in development, differentiation, and migration through their ability to interact with cells via cell surface receptors, including α-dystroglycan. In their association with the DGC, these molecules do not seem to influence the mechanical functions of the sarcolemma.
Plasma membrane disruptions might be an alternative way into and out of the sarcoplasm and subsequently serve as a route for the release of growth factors (McNeil and Khakee, 1992) and other myocyte-derived autocrine molecules. It was proposed that such cell-mediated processes are one way to stimulate the adjustment of muscle fibers to exercise and to repair membrane damage (Clarke et al., 1995; Kaye et al., 1996). Release of growth factors in skeletal muscle might be directly coupled to plasma membrane disruptions, whether induced by exercise or caused by reduced sarcolemmal stiffness (McNeil et al., 1989; Muthukrishnan et al., 1991). In this respect, plasma membrane disruptions might be beneficial for skeletal muscle regeneration and initiate not only damage, but also the cellular processes necessary for its repair. The EBD-injected mice strains that incorporated the dye into skeletal muscle fibers and hence had sarcolemmal disruptions all showed a benign phenotype, hypertrophic muscle fibers, and a normal to increased body weight. Consistent with this hypothesis, the dy/dy and dy2J/dy2J mice, which show severe clinical symptoms, did not reveal major dye uptake, do not show hypertrophic fibers, are small in size, and weigh less than their wild-type littermates.
In conclusion, we demonstrate that loss of plasma membrane integrity in skeletal muscle fibers may play a primary role in the course of the Xp21 muscular dystrophies. Membrane disruptions induced by mechanical forces may provide a route into and out of the sarcoplasm distinct from the conventional, membrane-bound routes of endo- and exocytosis (McNeil and Khakee, 1992). In diseased muscle with a higher muscle membrane vulnerability, this route may be of technical importance for introducing foreign therapeutic substances or even genes into damaged muscle fibers. Sarcolemmal permeability is of principal interest with regard to its role in the genesis of muscle cell degeneration and necrosis, and as this role becomes better defined, in the prevention and treatment of disease.
Acknowledgments
We gratefully acknowledge Rachelle H. Crosbie and Michael Henry for their comments on this manuscript and the helpful discussion. We thank Jane Lee for her support with the dy2J/dy2J mice.
This work was supported by the Muscular Dystrophy Association. Volker Straub is supported by a grant from the Deutsche Forschungsgemeinschaft. Kevin P. Campbell is an Investigator of the Howard Hughes Medical Institute. J.S. Chamberlain was supported by National Institutes of Health grant AR40864.
Abbreviations used in this paper
- CMD
congenital muscular dystrophy
- DGC
dystrophin–glycoprotein complex
- DMD
Duchenne muscular dystrophy
- EBD
Evans blue dye
- H&E
hematoxylin and eosin
Footnotes
Address all correspondence to Kevin P. Campbell, Howard Hughes Medical Institute, 400 Eckstein Medical Research Building, The University of Iowa College of Medicine, Iowa City, Iowa 52442. Tel: (319) 335-7867. Fax: (319) 335-6957. e-mail: kevin-campbell@uiowa.edu
References
- Allamand V, Sunada Y, Salih MAM, Straub V, Ozo CO, Al-Turaike MHS, Akbar M, Kolo T, Collognato H, Zhang X, et al. Mild congenital muscular dystrophy in two patients with an internally deleted laminin α2-chain. Hum Mol Genet. 1997;6:747–752. doi: 10.1093/hmg/6.5.747. [DOI] [PubMed] [Google Scholar]
- Bell CD, Conen PE. Histopathological changes in Duchenne muscular dystrophy. J Neurol Sci. 1968;7:529–544. doi: 10.1016/0022-510x(68)90058-0. [DOI] [PubMed] [Google Scholar]
- Bodensteiner JB, Engel AG. Intracellular calcium accumulation in Duchenne dystrophy and other myopathies: a study of 567,000 muscle fibers in 114 biopsies. Neurology. 1978;28:439–446. doi: 10.1212/wnl.28.5.439. [DOI] [PubMed] [Google Scholar]
- Bradley WG, Fulthorpe JJ. Studies of sarcolemmal integrity in myopathic muscle. Neurology. 1978;28:670–677. doi: 10.1212/wnl.28.7.670. [DOI] [PubMed] [Google Scholar]
- Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S, Karpati G. Duchenne muscular dystrophy: plasma membrane loss initiates muscle cell necrosis unless it is repaired. Brain. 1979;102:147–161. doi: 10.1093/brain/102.1.147. [DOI] [PubMed] [Google Scholar]
- Clarke MS, Caldwell RW, Chiao H, Miyake K, McNeil PL. Contraction-induced cell wounding and release of fibroblast growth factor in heart. Circ Res. 1995;76:927–934. doi: 10.1161/01.res.76.6.927. [DOI] [PubMed] [Google Scholar]
- Corrado K, Rafael JA, Mills PL, Cole NM, Faulkner JA, Wang K, Chamberlain JS. Transgenic mdx mice expressing dystrophin with a deletion in the actin-binding domain display a “mild Becker” phenotype. J Cell Biol. 1996;134:873–884. doi: 10.1083/jcb.134.4.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox GA, Sunada Y, Campbell KP, Chamberlain JS. Dp71 can restore the dystrophin-associated glycoprotein complex in muscle but fails to prevent dystrophy. Nature Genet. 1994;8:333–339. doi: 10.1038/ng1294-333. [DOI] [PubMed] [Google Scholar]
- D'Amore PA, Brown RH, Jr, Ku PT, Hoffman EP, Watanabe H, Arahata K, Ishihara T, Folkman J. Elevated basic fibroblast growth factor in the serum of patients with Duchenne muscular dystrophy. Ann Neurol. 1994;35:362–365. doi: 10.1002/ana.410350320. [DOI] [PubMed] [Google Scholar]
- Dhermy D. The spectrin super-family. Biol Cell. 1991;71:249–254. [PubMed] [Google Scholar]
- Dubowitz, V. 1985. Muscle Biopsy: A practical approach. Bailliere Tindall Ltd., London. 720 pp.
- England SB, Nicholson LV, Johnson MA, Forrest SM, Love DR, Zubrzycka-Gaarn EE, Bulman DE, Harris JB, Davies KE. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature (Lond) 1990;343:180–182. doi: 10.1038/343180a0. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature (Lond) 1990;345:315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis JM. Membrane abnormalities and Ca homeostasis in muscles of the mdx mouse, an animal model of the Duchenne muscular dystrophy: a review. Acta Physiol Scand. 1996;156:397–406. doi: 10.1046/j.1365-201X.1996.201000.x. [DOI] [PubMed] [Google Scholar]
- Greenberg DS, Sunada Y, Campbell KP, Yaffe D, Nudel U. Exogenous Dp71 restores the levels of dystrophin associated proteins but does not alleviate muscle damage in mdx mice. Nature Genet. 1994;8:340–344. doi: 10.1038/ng1294-340. [DOI] [PubMed] [Google Scholar]
- Head SI, Williams DA, Stephenson DG. Abnormalities in structure and function of limb skeletal muscle fibres of dystrophic mdx mice. Proc R Soc Lond B Biol Sci. 1992;248:163–169. doi: 10.1098/rspb.1992.0058. [DOI] [PubMed] [Google Scholar]
- Helbling-Leclerc A, Topaloglu H, Tome FM, Sewry C, Gyapay G, Naom I, Muntoni F, Dubowitz V, Barois A, Estournet B, et al. Readjusting the localization of merosin (laminin alpha 2-chain) deficient congenital muscular dystrophy locus on chromosome 6q2. C R Acad Sci III. 1995;318:1245–1252. [PubMed] [Google Scholar]
- Henry MD, Campbell KP. Dystroglycan: an extracellular matrix receptor linked to the cytoskeleton. Curr Opin Cell Biol. 1996;8:625–631. doi: 10.1016/s0955-0674(96)80103-7. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Jung D, Yang B, Meyer J, Chamberlain JS, Campbell KP. Identification and characterization of the dystrophin anchoring site on beta-dystroglycan. J Biol Chem. 1995;270:27305–27310. doi: 10.1074/jbc.270.45.27305. [DOI] [PubMed] [Google Scholar]
- Karpati, G., and S. Carpenter. 1982. Micropuncture lesions of skeletal muscle cells: a new experimental model for the study of muscle cell damage, repair and regeneration. In Disorders of the Motor Unit. D. L. Schotland, editor. John Wiley, New York. 517.
- Kaye D, Pimental D, Prasad S, Maki T, Berger HJ, McNeil PL, Smith TW, Kelly R A. Role of transiently altered sarcolemmal membrane permeability and basic fibroblast growth factor release in the hypertrophic response of adult rat ventricular myocytes to increased mechanical activity in vitro. J Clin Invest. 1996;97:281–291. doi: 10.1172/JCI118414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53:219–226. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- Matsuda R, Nishikawa A, Tanaka H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: evidence of apoptosis in dystrophin-deficient muscle. J Biochem (Tokyo) 1995;118:959–964. doi: 10.1093/jb/118.5.959. [DOI] [PubMed] [Google Scholar]
- McNeil PL, Muthukrishnan L, Warder E, D'Amore PA. Growth factors are released by mechanically wounded endothelial cells. J Cell Biol. 1989;109:811–822. doi: 10.1083/jcb.109.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil PL, Khakee R. Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am J Pathol. 1992;140:1097–1109. [PMC free article] [PubMed] [Google Scholar]
- Minetti C, Bado M, Morreale G, Pedemonte M, Cordone G. Disruption of muscle basal lamina in congenital muscular dystrophy with merosin deficiency. Neurology. 1996;46:1354–1358. doi: 10.1212/wnl.46.5.1354. [DOI] [PubMed] [Google Scholar]
- Mokri B, Engel AG. Duchenne dystrophy: electron microscopic findings pointing to a basic or early abnormality in the plasma membrane of the muscle fiber. Neurology. 1975;25:1111–1120. doi: 10.1212/wnl.25.12.1111. [DOI] [PubMed] [Google Scholar]
- Muthukrishnan L, Warder E, McNeil PL. Basic fibroblast growth factor is efficiently released from a cytolsolic storage site through plasma membrane disruptions of endothelial cells. J Cell Physiol. 1991;148:1–16. doi: 10.1002/jcp.1041480102. [DOI] [PubMed] [Google Scholar]
- Nissinen M, Helbling-Leclerc A, Zhang X, Evangelista T, Topaloglu H, Cruaud C, Weissenbach J, Fardeau M, Tome F M, Schwartz K, et al. Substitution of a conserved cysteine-996 in a cysteine-rich motif of the laminin alpha2-chain in congenital muscular dystrophy with partial deficiency of the protein. Am J Hum Genet. 1996;58:1177–1184. [PMC free article] [PubMed] [Google Scholar]
- Ohlendieck K, Ervasti JM, Matsumura K, Kahl SD, Leveille CJ, Campbell KP. Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron. 1991a;7:499–508. doi: 10.1016/0896-6273(91)90301-f. [DOI] [PubMed] [Google Scholar]
- Ohlendieck K, Ervasti JM, Snook JB, Campbell KP. Dystrophin-glycoprotein complex is highly enriched in isolated skeletal muscle sarcolemma. J Cell Biol. 1991b;112:135–148. doi: 10.1083/jcb.112.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlendieck K, Campbell KP. Dystrophin constitutes 5% of membrane cytoskeleton in skeletal muscle. FEBS Lett. 1991a;283:230–234. doi: 10.1016/0014-5793(91)80595-t. [DOI] [PubMed] [Google Scholar]
- Ohlendieck K, Campbell KP. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J Cell Biol. 1991b;115:1685–1694. doi: 10.1083/jcb.115.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa, E., M. Yoshida, A. Suzuki, Y. Mizuno, Y. Hagiwara, and S. Noguchi. 1995. Dystrophin-associated proteins in muscular dystrophy. Hum. Mol. Genet. 4(Spec. No.):1711–1716. [DOI] [PubMed]
- Pasternak C, Wong S, Elson EL. Mechanical function of dystrophin in muscle cells. J Cell Biol. 1995;128:355–361. doi: 10.1083/jcb.128.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps SF, Hauser MA, Cole NM, Rafael JA, Hinkle RT, Faulkner JA, Chamberlain JS. Expression of full-length and truncated dystrophin mini-genes in transgenic mdx mice. Hum Mol Genet. 1995;4:1251–1258. doi: 10.1093/hmg/4.8.1251. [DOI] [PubMed] [Google Scholar]
- Porter GA, Dmytrenko GM, Winkelmann JC, Bloch RJ. Dystrophin colocalizes with beta-spectrin in distinct subsarcolemmal domains in mammalian skeletal muscle. J Cell Biol. 1992;117:997–1005. doi: 10.1083/jcb.117.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafael JA, Sunada Y, Cole NM, Campbell KP, Faulkner JA, Chamberlain JS. Prevention of dystrophic pathology in mdx mice by a truncated dystrophin isoform. Hum Mol Genet. 1994;3:1725–1733. doi: 10.1093/hmg/3.10.1725. [DOI] [PubMed] [Google Scholar]
- Rafael JA, Cox GA, Corrado K, Jung D, Campbell KP, Chamberlain JS. Forced expression of dystrophin deletion constructs reveals structure-function correlations. J Cell Biol. 1996;134:93–102. doi: 10.1083/jcb.134.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve EB. The contribution of I131-labeled proteins to measurements of blood volume. Ann NY Acad Sci. 1957;70:137. doi: 10.1111/j.1749-6632.1957.tb35384.x. [DOI] [PubMed] [Google Scholar]
- Rosalki SB. Serum enzymes in disease of skeletal muscle. Clin Lab Med. 1989;9:767–781. [PubMed] [Google Scholar]
- Rybakova IN, Amann KJ, Ervasti JM. A new model for the interaction of dystrophin with F-actin. J Cell Biol. 1996;135:661–672. doi: 10.1083/jcb.135.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schittny JC, Yurchenco PD. Terminal short arm domains of basement membrane laminin are critical for its self-assembly. J Cell Biol. 1990;110:825–832. doi: 10.1083/jcb.110.3.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalbruch H. Segmental fibre breakdown and defects of the plasmalemma in diseased human muscles. Acta Neuropathol (Berlin) 1975;33:129–141. doi: 10.1007/BF00687539. [DOI] [PubMed] [Google Scholar]
- Schmalbruch H. Regenerated muscle fibers in Duchenne muscular dystrophy: a serial section study. Neurology. 1984;34:60–65. doi: 10.1212/wnl.34.1.60. [DOI] [PubMed] [Google Scholar]
- Straub V, Bittner RE, Leger JJ, Voit T. Direct visualization of the dystrophin network on skeletal muscle fiber membrane. J Cell Biol. 1992;119:1183–1191. doi: 10.1083/jcb.119.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub V, Campbell KP. Muscular dystrophies and the dystrophin-glycoprotein complex. Curr Opin Neurol. 1997;10:168–175. doi: 10.1097/00019052-199704000-00016. [DOI] [PubMed] [Google Scholar]
- Sunada Y, Bernier SM, Kozak CA, Yamada Y, Campbell KP. Deficiency of merosin in dystrophic dy mice and genetic linkage of laminin M chain gene to dy locus. J Biol Chem. 1994;269:13729–13732. [PubMed] [Google Scholar]
- Sunada Y, Bernier SM, Utani A, Yamada Y, Campbell KP. Identification of a novel mutant transcript of laminin alpha 2 chain gene responsible for muscular dystrophy and dysmyelination in dy2J mice. Hum Mol Genet. 1995;4:1055–1061. doi: 10.1093/hmg/4.6.1055. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Yoshida M, Hayashi K, Mizuno Y, Hagiwara Y, Ozawa E. Molecular organization at the glycoprotein-complex-binding site of dystrophin. Three dystrophin-associated proteins bind directly to the carboxy-terminal portion of dystrophin. Eur J Biochem. 1994;220:283–292. doi: 10.1111/j.1432-1033.1994.tb18624.x. [DOI] [PubMed] [Google Scholar]
- Tidball, J.G. 1991. Force transmission across muscle cell membranes. J. Biomech. 24(Suppl. 1):43–52. [DOI] [PubMed]
- Tomé FM, Evangelista T, Leclerc A, Sunada Y, Manole E, Estournet B, Barois A, Campbell KP, Fardeau M. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III. 1994;317:351–357. [PubMed] [Google Scholar]
- Weller B, Karpati G, Carpenter S. Dystrophin-deficient mdx muscle fibers are preferentially vulnerable to necrosis induced by experimental lengthening contractions. J Neurol Sci. 1990;100:9–13. doi: 10.1016/0022-510x(90)90005-8. [DOI] [PubMed] [Google Scholar]
- Xu H, Christmas P, Wu XR, Wewer UM, Engvall E. Defective muscle basement membrane and lack of M-laminin in the dystrophic dy/dy mouse. Proc Natl Acad Sci USA. 1994a;91:5572–5576. doi: 10.1073/pnas.91.12.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Wu XR, Wewer UM, Engvall E. Murine muscular dystrophy caused by a mutation in the laminin alpha 2 (Lama2) gene. Nat Genet. 1994b;8:297–302. doi: 10.1038/ng1194-297. [DOI] [PubMed] [Google Scholar]