

Abstract
While the protrusive event of cell locomotion is thought to be driven by actin polymerization, the mechanism of forward translocation of the cell body is unclear. To elucidate the mechanism of cell body translocation, we analyzed the supramolecular organization of the actin–myosin II system and the dynamics of myosin II in fish epidermal keratocytes. In lamellipodia, long actin filaments formed dense networks with numerous free ends in a brushlike manner near the leading edge. Shorter actin filaments often formed T junctions with longer filaments in the brushlike area, suggesting that new filaments could be nucleated at sides of preexisting filaments or linked to them immediately after nucleation. The polarity of actin filaments was almost uniform, with barbed ends forward throughout most of the lamellipodia but mixed in arc-shaped filament bundles at the lamellipodial/cell body boundary. Myosin II formed discrete clusters of bipolar minifilaments in lamellipodia that increased in size and density towards the cell body boundary and colocalized with actin in boundary bundles. Time-lapse observation demonstrated that myosin clusters appeared in the lamellipodia and remained stationary with respect to the substratum in locomoting cells, but they exhibited retrograde flow in cells tethered in epithelioid colonies. Consequently, both in locomoting and stationary cells, myosin clusters approached the cell body boundary, where they became compressed and aligned, resulting in the formation of boundary bundles. In locomoting cells, the compression was associated with forward displacement of myosin features. These data are not consistent with either sarcomeric or polarized transport mechanisms of cell body translocation. We propose that the forward translocation of the cell body and retrograde flow in the lamellipodia are both driven by contraction of an actin–myosin network in the lamellipodial/cell body transition zone.
The crawling motion of animal cells involves three basic steps: formation of a lamellipodial protrusion at the front of the cell, adhesion of the lamellipodium to the substratum, and translocation forward of the cell body. Numerous studies indicate that protrusion is driven by polymerization of actin at the leading edge of the lamellipodium (for reviews see Condeelis, 1993; Mitchison and Cramer, 1996; Small et al., 1993; Mogilner and Oster, 1996). While existing models of this process differ in the details of the actin polymerization mechanism and network structure (e.g., treadmilling [Small et al., 1993] and nucleation release [Theriot and Mitchison, 1992]), it is generally accepted that actin filaments grow at their barbed, forward-facing ends, thus providing the force for protrusion, but polymerized domains do not move forward relative to the substratum. In contrast, major components of the cell body, such as the nucleus and other organelles, actually move forward. Consequently, translocation of the cell body requires elements in addition to lamellar protrusion.
A minimalistic scheme for cell body translocation could rely on a passive means of maintaining cell integrity, such as the mechanical continuity and elasticity of the plasma membrane and/or cortical cytoskeleton. In this model, filament polymerization inside a closed container propels the container forward with its contents following passively. Such a treadmilling mechanism has been proposed to explain translocation of Ascaris sperm (Roberts and King, 1991), although in this novel example of cell motility, the filament-forming protein is unrelated to actin, and both actin and myosin are absent from the sperm.
In general, however, cell motility is based on actin and myosin, and it is reasonable to consider that actin-dependent motor proteins actively contribute to the translocation of the cell body. Knockout of myosin II in Dictyostelium resulted in a dramatic decrease in the rate of cell locomotion (Wessels et al., 1988) or in a block of locomotion in an environment of increased resistance (Doolittle et al., 1995; Jay et al., 1995). Thus, myosin II, the only member of the myosin superfamily with the ability to form polymeric supramolecular assemblies (Cheney et al., 1993; Goodson, 1994), has been clearly shown to participate in the overall process of cell motility. However, in contrast to a conceptually clear paradigm for protrusion of the leading edge, it is not obvious how myosin II is involved in cell motility and, more specifically, how it brings about translocation of the cell body. Among the mechanisms discussed in the literature are contraction of microfilament bundles organized in a sarcomeric-like manner (Huxley, 1973; Sanger and Sanger, 1980; Byers et al., 1984; Langanger et al., 1986) and myosin-driven transport of cell body components along uniformly polarized actin arrays (Maciver, 1996; Mitchison and Cramer, 1996; Cramer et al., 1997). Analysis of the organization and dynamics of myosin with respect to actin is required to evaluate these hypotheses and to determine the mechanism of cell body translocation.
Most studies of cytoskeletal organization and dynamics have been conducted on a classic model of cell motility, the mammalian or avian fibroblast. However, fibroblasts may not be an ideal system for analysis of cell motility because they exhibit a relatively slow and uncoordinated locomotion, lack persistent polarization, and contain a complex system of actin–myosin II fibers oriented at various angles to the direction of movement. To more clearly establish a link between myosin organization and cell motility, one would prefer to study a cell of a simpler shape and pattern of movement.
Fish epidermal keratocytes, with their fast locomotion, persistent polarization, and simple, stable shape seem an excellent model (Cooper and Schliwa, 1986; Lee et al., 1993a ). Free locomoting keratocytes are characteristically wing shaped with a large lamellipodium filled with actin. They move with a velocity of a few tens of micrometer per minute in a direction perpendicular to the long axis of the cell. Keratocytes can also be cultivated as an epithelioid colony where locomotion of cells is restrained by firm attachments to each other. Experiments with photoactivation of a microinjected actin probe demonstrated that the actin cytoskeleton in lamellipodia of locomoting keratocytes remains stationary relative to the substratum, indicating that the rate of actin polymerization equals the rate of protrusion (Theriot and Mitchison, 1991). Ultrastructural study of keratocyte lamellipodia demonstrated that it contained an extremely dense organization of actin filaments (Small et al., 1995). Polarity was only determined on filament portions elongated with exogenous actin beyond the leading edge of permeabilized cells because the density of filaments precluded determination of actin polarity within the lamellipodium (Small et al., 1995). As in other cell types (Begg et al., 1978; Small et al., 1978), actin filament polarity was uniform with fast growing barbed ends forward, which is consistent with the idea of incorporation of new actin subunits into the network at the extreme leading edge, as it has been directly shown for cultured fibroblasts (Symons and Mitchison, 1991). These ultrastructural results provided morphological support for the treadmilling model of leading edge protrusion.
Normally, protrusion of the leading edge in freely locomoting keratocytes is tightly coupled to the translocation of the cell body, resulting in a remarkable conservation of the cell's shape, which was kinematically described in terms of a graded radial extension model (Lee et al., 1993b ). However, cell body translocation could also proceed in the absence of front protrusion (Anderson et al., 1996), indicating the existence of an active mechanism independent of actin polymerization. As in other cells, it was suggested that this mechanism operates through the interaction of actin with myosin II, and consistent with this view, myosin II was localized by immunofluorescence to the rear part of the lamellipodia and to the lamellipodia–cell body transition zone (Anderson et al., 1996; Strohmeier and Bereiter-Hahn, 1984). A more specific model concerning spatial coordination of actomyosin activity in keratocytes was also developed. It was proposed that the tension required to move the cell body forward was generated by actin–myosin II bundles at the sides of the body and that the cell body rolled around these bundles as axles (Anderson et al., 1996). However, this hypothesis was based only on the characterization of the overall myosin distribution and on the observation of cell body rotation. It did not speak to the local mechanism of force generation in terms of contraction or transport because neither myosin supramolecular organization nor dynamics and mode of interaction with actin were known. A detailed study of organization and dynamics of myosin II was required.
In this study, we establish the supramolecular organization of the actin–myosin II system and dynamics of myosin II in fish keratocytes in a manner similar to that previously accomplished for the fibroblast model (Verkhovsky and Borisy, 1993; Verkhovsky et al., 1995). The unique regularity of keratocyte motility and the simplicity of its overall cytoskeletal arrangement allowed us to relate actin and myosin arrangement to cell motility and to put forward a novel model for the role of myosin II in cell body translocation. The general principles of this model may be applicable to other cells.
Materials and Methods
Keratocyte Culture
Black tetra (Gymnocorymbus ternetzi) keratocytes were cultured in DMEM (Hepes modification; Sigma Immunochemicals, St. Louis, MO) supplemented with 20% FBS (Hyclone Laboratories, Inc., Logan, UT) and antibiotics. Fish scales were extracted with tweezers, placed external side up on dry coverslips, and allowed to adhere for 30–60 s (until almost dried) to prevent floating. Culture medium was then added and the scales were kept at 30°C overnight to allow for migration of keratocytes onto the coverslips. Colonies of migrated cells were treated with 0.2% trypsin and 0.02% EDTA in PBS for ∼30–60 s. The extent of treatment was monitored with phase contrast optics, and the trypsin/EDTA solution was replaced with culture medium when the cells were mostly separated from each other but before a significant number of them detached from the coverslip. Cells were allowed to recover for 1–3 h in fresh medium before observation by light or electron microscopy. Cell cultures prepared this way typically contained sufficient numbers of both freely locomoting cells and cells tethered to a colony.
Microscopy
Procedures for detergent extraction, immunostaining, S1 decoration, light and electron microscopy were described previously (Svitkina et al., 1995, 1996; Verkhovsky et al., 1995). Briefly, cells were washed in PBS or serum-free media and extracted for 5 min at room temperature with a cytoskeleton-stabilizing solution (50 mM imidazole, 50 mM KCl, 0.5 mM MgCl2, 0.1 mM EDTA, 1 mM EGTA, and either 0.5 μM TRITC-phalloidin [for light microscopy] or 10 μM phalloidin [for EM]; Sigma) containing 1% Triton X-100 and 4% polyethylene glycol, M r 40,000. Extracted cells were briefly washed with the cytoskeleton-stabilizing solution and fixed with 2% glutaraldehyde. Polyclonal myosin antibody or S1 in the cytoskeleton-stabilizing solution were applied to extracted cells before glutaraldehyde fixation. Wet cleavage of cells was performed as described (Brands and Feltkamp, 1988) with slight modifications. Briefly, coverslips with attached cells were rinsed in the cytoskeleton-stabilizing solution buffer and overlaid with 0.22-μm nitrocellulose membrane filters soaked in the same buffer and blotted. After ∼1 min, filters were peeled up, and the cells were rinsed and fixed with glutaraldehyde.
Determination of Actin Filament Polarity
The “double rope” appearance of S1-decorated actin filaments in platinum replicas (Heuser and Cooke, 1983) differs from the well-known arrowhead pattern observed by negative contrast (Huxley, 1963) or thin sectioning (Ishikawa et al., 1969). We determined the polarity of actin filaments in replicas based on the asymmetry of individual turns of the “rope” as described (Heuser and Cooke, 1983; Verkhovsky et al., 1997). The thin tapered end of an individual “rope” element points toward the barbed end of the actin filament, while the thick, rounded end is directed toward the pointed end.
For quantitation of actin filament polarity, a narrow (4–5 μm wide) sector of the central lamellipodia of each keratocyte spanning its entire depth from the leading edge to the cell body was divided into 2-μm zones parallel to the leading edge. Polarity was determined in all zones and was expressed as an angle between the direction of the filament-pointed end and the leading edge. According to the angle, all filaments with determined polarity were put into three categories: (a) “parallel to the edge” category included filaments that were oriented at an angle of <20° to the leading edge; (b) “barbed end forward” or (c) “pointed end forward” filaments included those with angles of >20° and oriented with the respective end to the leading edge.
Microinjection of Myosin II and Time-Lapse Observation
Preparation of tetramethylrhodamine-myosin and microinjection was performed as described (Verkhovsky et al., 1995). Tethered keratocytes were injected into the cell body. For locomoting cells, the sides of the cell body were the most convenient sites for injection. After injection, cells were allowed to recover for 5–10 min and relocated using their position relative to scales or epithelioid colonies. Time-lapse images were collected as described (Verkhovsky et al., 1995) in intervals of 8–20 s.
Observation of Cell Body Rolling
Following (with modifications) the procedure of Anderson et al. (1996), fluorescent latex beads (Fluoresbrite Carboxylate Microspheres, 0.2 μm; Polysciences Inc., Warrington, PA), diluted 1:10 in culture media, were placed on top of locomoting keratocytes with a blunt microinjection tip. Unbound beads were removed by a flush of culture media. Some of the bound beads presumably remained at the cell surface and accumulated at the lamellipodia–cell body boundary, while others were endocytosed and underwent circular motion within the cell body. Serendipitously, we found that in the cells to which microbeads were applied, mitochondria were also brightly fluorescent, presumably because of a leak of fluorescent dye from beads. Thus, we monitored cell body rolling by following the dynamics of both beads and mitochondria.
Results
Distribution of Actin and Myosin II in Keratocytes
To provide orientation for detailed analysis of the supramolecular organization of the actin–myosin II system of keratocytes, it is necessary first to establish the overall distribution of these components with fluorescence microscopy. Important information (which we mostly confirm) is contained in previous studies (Strohmeier and Bereiter-Hahn, 1984; Small et al., 1995; Anderson et al., 1996). Here, we briefly describe the images that were obtained using the same extraction/fixation procedures that were used as for further ultrastructural analysis, and we pay special attention to details of the relative distribution of actin and myosin II to identify possible regions of cytoskeletal rearrangement and development of tension.
Three cellular domains distinct in cytoskeletal organization were identified in keratocytes: lamellipodia, the lamellipodia–cell body transition zone, and the cell body proper.
Lamellipodia.
In lamellipodia, actin was organized as a continuous network often exhibiting a fine criss-cross pattern, while myosin II formed distinct spotlike accumulations (Fig. 1 a). The intensity of actin staining in lamellipodia was maximal at the leading edge and gradually decreased toward the cell body. Although it has been reported that extraction before fixation selectively depleted actin from the front of lamellipodia, resulting in an apparently uniform distribution (Small et al., 1995), cells extracted by our protocol exhibited a graded actin distribution similar to cells fixed before permeabilization. Quantitative analysis of fluorescent phalloidin binding provided a measure of the actin filament concentration and a means of evaluating extraction/fixation protocols. The ratio of actin intensity at the front of lamellipodia to that at the rear was 1.80 ± 0.40, n = 9, for cells that were first extracted and then fixed, and 1.78 ± 0.32, n = 7, for cells that were first fixed and then extracted, indicating that no significant redistribution of actin occurred upon extraction.
Figure 1.
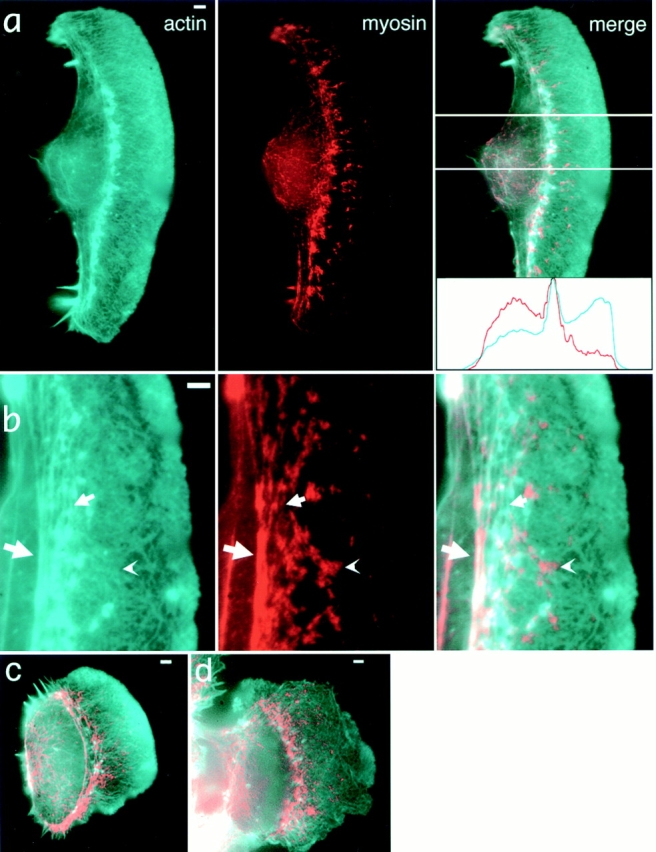
Localization of actin and myosin II in keratocytes by fluorescence microscopy. Actin (cyan) and myosin (red) distributions are revealed by TRITC-phalloidin and indirect immunofluorescence staining, respectively. Overall actin and myosin II organization in a typical wing-shaped locomoting cell (a), enlarged portion of another cell exhibiting various patterns of actin and myosin mutual arrangement (b), locomoting cell of a symmetrical shape (c), and a tethered cell (d) are shown. All cells exhibit discrete myosin spots among continuous actin network in lamellipodia, as well as accumulation of both actin and myosin at the lamellipodia/cell body boundary. Intensity profiles of actin (cyan) and myosin (red) within the cell area indicated in the “merge” panel of a are shown in the inset, and they illustrate reverse gradients of actin and myosin in lamellipodia. (b) Examples of a myosin spot in the lamellipodia that does not coincide with any discrete actin structure (arrowhead), myosin spots coinciding with small actin bundles merging to boundary bundles (small arrow), and colocalization of actin and myosin in the boundary bundle (large arrow). Bars, 2 μm.
Spots of myosin II in lamellipodia increased in size and density in the direction from front to rear. Thus, myosin exhibited a gradient of reverse orientation compared to the gradient of actin (see intensity profiles, Fig. 1 a, inset). In most cases, no detectable accumulations of actin colocalized with myosin spots in the bulk of the lamellipodia (Fig. 1, a and b, arrowhead). Lack of overall correlation between the distributions of actin and myosin II could be indicative of the independent assembly of the two components and the absence of their strong interaction in this region.
Lamellipodia–Cell Body Transition Zone.
In contrast to lamellipodia, actin and myosin II displayed highly correlated distribution in the transition zone between the lamellipodium and the cell body (Fig. 1, a and b), where the two proteins concentrated in distinct arc-shaped fibers (Fig. 1 b, big arrow). In some cells, small actin fibers associated with myosin spots were also found in the posterior region of lamellipodia (Fig. 1 b, small arrow). These fibers apparently merged to the main fiber in the transition zone, suggestive of possible reorganization of the lamellipodial network into arc-shaped fibers via an intermediate stage of small fibers.
Cell Body.
The cell body proper exhibited less intense staining for both actin and myosin than the transition zone. Here, both proteins were distributed in a diffuse and fine particulate manner, and they colocalized in retraction fibers and in occasional internal fibers within the cell body.
The three cytoskeletal domains were also distinct in terms of the distribution of microtubules and intermediate filaments. Both of these fibril systems were mostly localized to the cell body. Very few fibrils of either kind extended beyond the accumulation of actin and myosin at the transition zone.
The organization of the three cytoskeletal domains was similar in all locomoting keratocytes. Faster locomoting cells were wing shaped with width significantly exceeding length (Fig. 1, a and b). Less elongated cells were fibroblast-like in shape and were characterized by slower locomotion (Fig. 1 c). The cells at the border of an epithelioid colony also exhibited cytoskeletal organization similar to freely locomoting cells (Fig. 1 d), but showed less pronounced gradients of actin and myosin concentration in the lamellipodia, less well-defined actin–myosin II fibers at the cell body boundary, and more extensive penetration of microtubules and intermediate filaments into lamellipodia. Overall, fluorescence images were indicative of a possible rearrangement of actin–myosin II system at the lamellipodia/cell body transition zone. This putative rearrangement was more dramatic in locomoting cells, suggestive of a role in cell body translocation.
Supramolecular Organization of the Keratocyte Cytoskeleton
For detailed study of the structural organization of the keratocyte actin–myosin II system, we used a previously developed procedure of EM of detergent-extracted and critical point dried cells (Svitkina et al., 1995). In combination with wet cleavage, S1 decoration, gelsolin treatment, and immunogold staining, this technique allowed determination of important features of the actomyosin machinery, such as actin filament organization and polarity throughout the cell and arrangement of myosin filaments and patterns of their interactions with actin filaments.
Organization of Actin.
Negative staining of whole-mount keratocyte preparations (Small et al., 1995) previously revealed orthogonal networks of actin filaments in the main body of lamellipodia, but was not satisfactory for the determination of actin organization in regions with higher actin density, such as the leading edge and transition zone. Using the platinum replica technique, we were able to visualize clearly actin filament organization in all parts of the locomoting keratocyte.
In the lamellipodia of locomoting keratocytes, actin filaments were organized into networks with the highest density at the leading edge and a gradual decrease towards the nucleus (Fig. 2). Although determination of filament length distribution was not possible because of high filament density, numerous long filaments (with length comparable to the entire width of lamellipodia) were apparent. The actin network at the leading edge was characterized by an abundance of free ends in a characteristic brushlike appearance (Fig. 2, b and c) in contrast to the more smooth network in deeper parts of the lamellipodia (Fig. 2, b and d). The abundance of actin filament ends near the leading edge was suggestive of intensive actin polymerization in this area. In accord with this suggestion, the width of the brushlike zone correlated with the magnitude of lamellipodial protrusion at the respective site. As a rule, it was maximal (1.2 ± 0.3 μm) in central, forward-facing domains of the leading edge and was less at the lateral edges. The extent of the brushlike zone was greater in cells whose shape was indicative of rapid locomotion as compared to relatively stationary cells, e.g., tethered cells in keratocyte colonies.
Figure 2.

Organization of actin filaments in keratocyte lamellipodia. EM of detergent-extracted cells. (a) Overview of a locomoting cell; (b) actin network in lamellipodia from the leading edge (top) to the transitional zone (bottom); (c) brushlike zone at the leading edge with numerous filament ends; (d) smooth actin filament network in the middle part of lamellipodia; (e–h), T junctions (arrowheads) between filaments at the extreme leading edge (e), within the brushlike zone (f), in the central lamellipodia (g), and close to the lateral edge of the lamellipodia (h). The cell's leading edge is oriented upward in all panels. Boxed region in a is enlarged in b; upper and lower boxed regions in b are enlarged in c and d, respectively. Bars: (b) 1 μm; (e–h) 50 nm.
Proximal ends of actin filaments often terminated on other actin filaments at approximately a right angle, resulting in the formation of T junctions (Fig. 2, e–h). Near the leading edge of the lamellipodia, many actin filaments were rather short, so it was often possible to see both ends of a filament, one free and the other linked to a longer filament (Fig. 2, e and f). Behind the marginal brushlike zone and throughout almost the entire lamellipodia, actin filaments formed multilayer arrays consisting of long diagonally oriented filaments (Fig. 2, b and d) similar to that described by Small et al. (1995). In contrast to the leading edge, where free ends were common, no free filament ends were observed in the deeper regions of the lamellipodia. T junctions between actin filaments were also seen here (Fig. 2, g and h).
At the transition zone with the cell body, the criss-cross actin network gradually transformed into arc-shaped actin bundle(s) oriented parallel to the leading edge. The transitional zone had an irregular actin arrangement and, along with an actin filament network, contained small bundles and asters (Fig. 2 b). In many cases, continuity of actin filaments between the lamellipodial network and transition zone bundles was apparent. The actin network in the middle parts of lamellipodia was often thin enough to allow for the surface of the substratum to be visible beneath the filaments, suggesting that the whole depth of the cytoskeleton was visualized in this region.
In the cell body, the nucleus and dense cytoskeletal network, including abundant intermediate filaments, interfered with the visualization of actin organization at the substratum plane. To expose the cytoskeletal organization in internal cell regions (e.g., at the bottom of the cell body), we used a wet cleavage procedure (see Materials and Methods). We were particularly interested in cells where the cleavage plane passed between the nucleus and the ventral plasma membrane (Fig. 3). Most of these cells retained a dense actin network in the lamellipodial region, but had very few membrane-associated actin filaments within the cell body (Fig. 3 a), suggesting that either the actin network is not prominent here or it is more strongly attached to the nucleus. In some cells, however, the actin filament network underlying the nucleus was retained. It was usually limited to areas adjacent to transition bundles (Fig. 3 b), but sometimes more elaborated ventral actin arrays were found (Fig. 3 c). Variations in the density of the actin filament network under cell nuclei correlated with the variability of diffuse fluorescence in the cell body after staining with rhodamine-phalloidin (see above). No preferential orientation of actin filaments was observed in either case. Thus, actin filaments formed a physically continuous and gradually transforming network spanning the entire cytoplasmic region from the leading edge to the transition bundles, which, in turn, were loosely connected to a sparse network at the ventral surface of the cell body.
Figure 3.
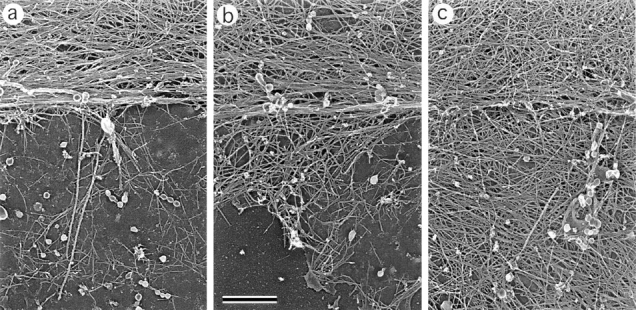
Organization of actin filaments in the lamellipodia–cell body transition zone. EM of wet-cleaved cells with nuclei removed shows actin bundles in the transition zone (top of each panel) and a network of actin filaments at the bottom of the cell body (bottom of each panel). Different amounts of actin filaments remain associated with bottom plasma membrane (a–c). Orientation of actin filaments is random (a and b) or approximately parallel to the transition zone bundle (c). The cell's leading edge is oriented upward in all panels. Bar, 0.1 μm.
Actin Filament Polarity.
Within the cytoskeleton, actin filament polarity is an important characteristic that determines the possible direction of myosin movement. For the determination of actin filament polarity, we used decoration with myosin S1 (Fig. 4). Free filament ends within the brushlike zone at the leading edge were identified as barbed ends. When polarity of actin filaments making T junctions was analyzed, filaments were found oriented with pointed ends toward the base of a fork (Fig. 4 e). Throughout the lamellipodia, the predominant orientation of actin filaments was with barbed ends forward (Fig. 4, a–c). Although polarity could be estimated only in a limited fraction of filaments (20–40%) because of their high density, a quantitative assay revealed a strong bias in filament polarity in lamellipodia not only at the leading edge, but in deeper parts of lamellipodia as well (Fig. 5). The fraction of filaments oriented with the pointed end forward was extremely low (∼5%) and approximately constant throughout the lamellipodia, including transitional zone. The fraction of filaments oriented with the barbed end forward was high (∼80%) and did not change appreciably with distance from the leading edge until the transitional zone was reached. Here, the fraction of filaments with the barbed end facing forward decreased with a concomitant increase in the fraction of filaments oriented approximately parallel to the leading edge, changes related to the formation of bundles. In arc-shaped bundles, the polarity of actin filaments was mixed in the center (Fig. 4 d), while the terminal parts of the bundles contained more filaments with the barbed ends facing the nearest cell edge (not shown). Retraction fibers at the rear had uniformly oriented filaments with barbed ends directed outward (Fig. 4 f). Thus, polarity of actin filaments suggests that filaments arising with the barbed ends forward at the leading edge undergo no significant reorganization throughout most of the lamellipodia; however, reorientation of filaments occurs at the lamellipodia/cell body transition.
Figure 4.

Polarity of actin filaments in keratocyte cytoskeleton. EM of detergent-extracted cells after myosin S1 decoration. (a) Leading edge; (b) middle portion of a lamellipodium; (c) transitional zone; (d) boundary bundle; (e) T junctions between actin filaments at the leading edge; (f) retraction fiber at the cell rear. Directions of pointed ends of some filaments are shown by arrowheads located next to a filament. Filaments are oriented primarily with barbed end forward throughout the lamellipodia (a–c), while the boundary bundle has mixed filament polarity (d). At T junctions, filaments are oriented with pointed end toward the junction (e). The retraction fiber (f) contains uniformly oriented filaments with their barbed ends to the tip of the fiber. Unlabeled intermediate filaments can be seen in some panels. The cell's leading edge is oriented upward in all panels. Bars, 0.1 μm.
Figure 5.

Quantitation of actin filament polarity in keratocyte lamellipodia. Polarity of filament orientation was determined with respect to the leading edge as being in one of three categories (see Materials and Methods): barbed end forward, pointed end forward, or parallel to the edge. Determinations were made in cells of similar size and morphology, covering the whole width of the lamellipodia, within 2-μm zones parallel to the leading edge, and for a depth of 12 μm behind the leading edge. A total of 3,761 filaments were scored in five cells, converted to percentage per cell, and the mean percentages were plotted against distance from the leading edge. The percentage of filaments in each category remained constant throughout the lamellipodia (0–8 μm) until the transitional zone was reached (8–12 μm).
Organization of Myosin.
Although immunofluorescence microscopy demonstrates prominent myosin arrays in keratocytes (see above), they are not readily seen in EM images because of abundant actin. To reveal myosin II filaments in keratocytes, we applied an actin-severing protein, gelsolin, to detergent-extracted cells as previously described for fibroblasts (Svitkina et al., 1989; Verkhovsky and Borisy, 1993; Verkhovsky et al., 1995). Gelsolin treatment removed actin and revealed not only myosin filaments, but also intermediate filaments and, if taxol was added, microtubules. Intermediate filaments were particularly abundant in the cell body, where they partially obscured myosin filament arrangement. In lamellipodia, intermediate filaments were sparse, but because of their similarity in thickness to myosin filament rods, they also interfered with clear imaging of myosin distribution, especially at low magnification. To facilitate the visualization of myosin arrangement in keratocytes and to prove the molecular nature of bipolar filaments, we combined gelsolin treatment with myosin immunogold decoration, and we examined both labeled and unlabeled specimens for organization of myosin II (Fig. 6).
Figure 6.
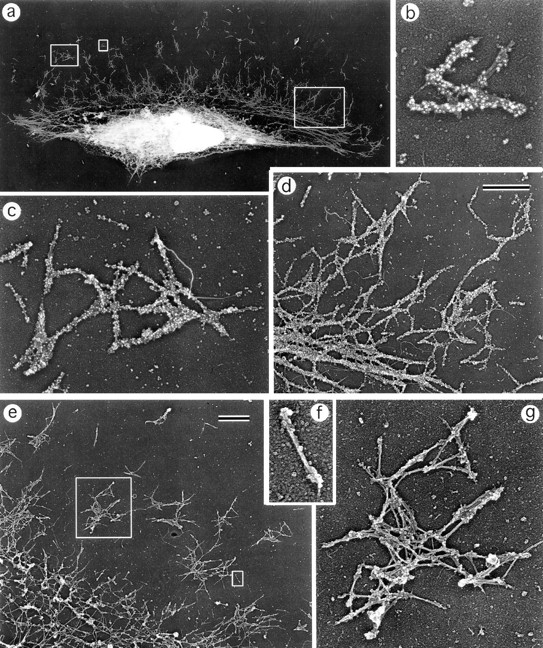
Organization of myosin II filaments in keratocytes. EM of detergent-extracted and gelsolin-treated cells with (a–d) or without (e–g) myosin immunogold labeling. (a) Overview of a cell; (b and c) clusters of gold-labeled myosin filaments; (d) gold-labeled myosin filament network (upper right) that gradually transforms into boundary bundle (lower left); (e) part of a cell without labeling; (f) individual myosin bipolar filament; (g) cluster of myosin bipolar filaments associated predominantly at their heads. The cell's leading edge is oriented upward in all panels. Left, middle, and right boxed regions in a are enlarged in b–d, respectively. Small and large boxed regions in e are enlarged in f and g, respectively. Bars, 1 μm.
The general distribution of myosin II revealed after gelsolin treatment and immunogold decoration was similar to that observed by immunofluorescence (Fig. 6 a). In lamellipodia, myosin II formed clusters of variable size (Fig. 6, b–g) that apparently corresponded to immunofluorescent spots revealed by light microscopy. The clusters were composed of rod-shaped units of length 0.38 ± 0.04 μm, correlating well with the length of myosin filaments in fibroblasts (Verkhovsky and Borisy, 1993). In unlabeled specimens, a dumbbell shape of these units characteristic for myosin bipolar filaments was easily recognized (Fig. 6, f and g). Myosin filaments in clusters had no preferential orientation with respect to each other or to the cell, in contrast to REF-52 fibroblasts, where zigzag and ladder-like arrangements occurred (Verkhovsky et al., 1995). As in fibroblasts, individual filaments associated mostly by ends, although contacts involving myosin filament rods were also found (Fig. 6 g). The size of myosin clusters usually increased from the periphery, where individual myosin bipolar filaments were common, towards the cell body, where clusters tended to associate into extensive networks with filaments still oriented randomly (Fig. 6, d and e). In the transition zone, this network gradually transformed into a bundle-like assembly of aligned myosin filaments oriented along the lamellipodia–cell body boundary (Fig. 6, a and d). Thus, myosin II in the keratocyte cytoskeleton is present in the form of bipolar minifilaments that tend to associate with each other by forming isolated clusters in lamellipodia and a consolidated network close to the cell body. Randomly oriented myosin filaments in clusters and network become aligned at the lamellipodia–cell body boundary, suggesting the structural reorganization of myosin in this region.
Correlation of Actin and Myosin Organization
Actin and myosin filament organization, when studied separately, displayed similar patterns of rearrangement, suggesting that these events may be interdependent. To correlate the organization of actin and myosin in the same cells, we performed myosin immunogold labeling of intact cytoskeletons not treated with gelsolin (Figs. 7 and 8). Myosin staining was usually absent from the peripheral brushlike zone of lamellipodia. Individual myosin filaments that looked like rod-shaped groups of gold particles with a characteristic length of 0.4 μm were found in distal parts of lamellipodia behind the brushlike zone (Fig. 7). Clusters of myosin filaments were scattered within the actin network in the central lamellar region (Fig. 8). In the vicinity of small myosin clusters in lamellipodia, several actin filaments often seemed to converge to each myosin filament and align with it (Figs. 7 and 8), suggesting a role for myosin in the reorientation of actin filaments. A more pronounced reorientation of actin filaments at sites of myosin localization was observed in the transitional zone. Myosin filament clusters here were often found at sites where many actin filaments changed their course and converged into small bundles and asters. Myosin was highly concentrated in arc-shaped actin bundles, and individual myosin filaments that sometimes could be resolved there were mostly oriented along the bundle. Numerous gold particles were also found in the cell body (not shown). Thus, simultaneous analysis of actin and myosin organization in the same cells suggests that myosin assemblies drive the reorientation of actin filaments. Large myosin assemblies close to the cell body boundary seem to cause significant changes in the adjacent actin filament network, while individual filaments and clusters scattered in lamellipodia at most are able to align a few nearby actin filaments.
Figure 7.

Relative distribution of actin and myosin II filaments in keratocyte lamellipodia. EM of detergent-extracted cells after myosin immunogold labeling shows a few myosin filaments (revealed as rod-shaped groups of gold particles) among actin filaments. Actin filaments contacting myosin tend to be arranged into small bundles. b Same image as a, but with gold particles digitally colorized in yellow. The cell's leading edge is oriented upward. Bars, 0.1 μm.
Figure 8.
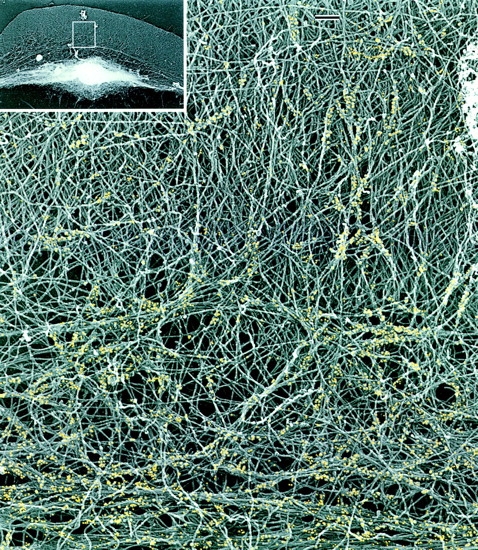
Relative distribution of actin and myosin II filaments in the keratocyte lamellipodia–cell body transition zone. EM of a detergent-extracted cell (overview in inset) after myosin immunogold labeling shows myosin filament clusters and the boundary bundle (bottom) within an actin filament network. Actin filaments forming small bundles and changing their course can be seen at sites of myosin localization. For better visualization, gold particles are digitally colorized in yellow. Bars, 0.2 μm.
Myosin Dynamics
While static snapshots at both light and electron microscopic levels were suggestive of reorganization of the actin–myosin II system at the cell body boundary, a dynamic study was necessary to determine the actual sequence of events and to correlate it to the cell locomotion. Study of myosin dynamics includes two aspects: determination of the morphogenetic pathway of individual myosin features to elucidate the assembly/disassembly cycle of myosin within the cell, and characterization of motility of myosin features relative to the substratum, cell margin, and to each other, which is of indispensable diagnostic value for the locomotion mechanism. We were especially interested to see if, when, and where the myosin features move forward in locomoting cells.
Fluorescently labeled smooth muscle myosin was injected into both freely locomoting keratocytes and into the cells at the border of an epithelioid colony. Fluorescence microscopy of living keratocytes revealed that the distribution of injected myosin II was similar to the distribution of endogenous myosin II in extracted cells: distinct myosin spots in lamellipodia increasing in size and density towards the cell body, as well as bundles at the lamellipodia–cell body border were clearly observed (Figs. 9 and 10). The only difference was that living, microinjected cells exhibited a much brighter diffuse fluorescence in the cell body than extracted cells. This could be explained by the contribution of a soluble and extractable pool of myosin II, which (if uniformly distributed throughout the cell volume) should be more apparent in the cell body because of its thickness. Dim (compared to the cell body) diffuse fluorescence was also detected in the thin lamellipodia of living cells. This feature was used to determine the position of the cell's leading edge in fluorescence images. We conclude that, similar to fibroblasts, labeled myosin II was a faithful reporter of the distribution of endogenous myosin II.
Figure 9.
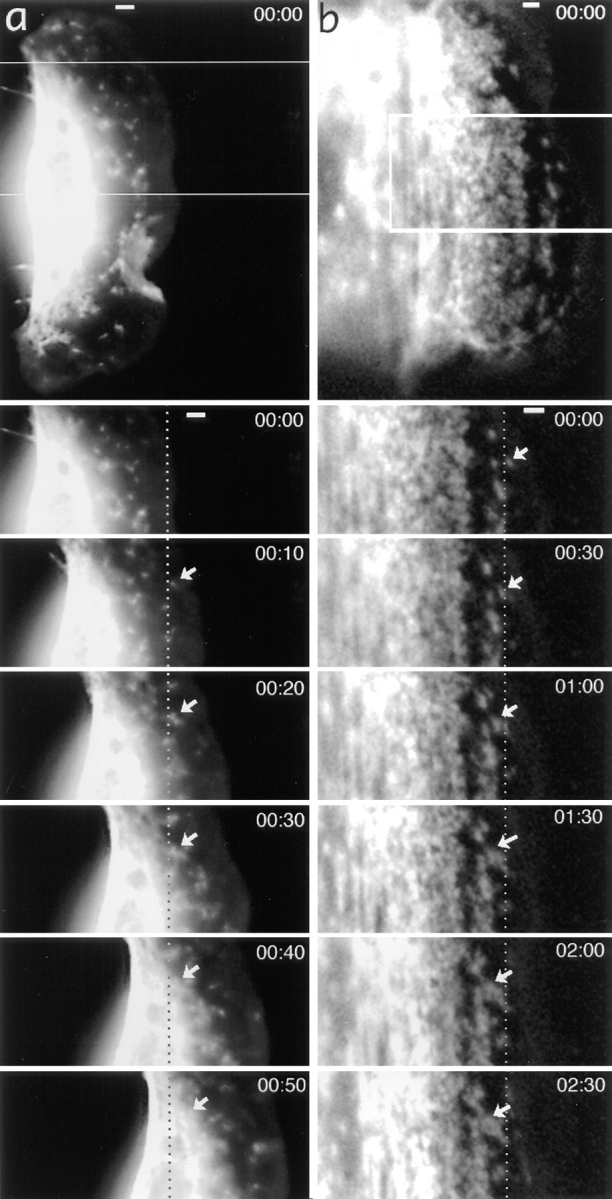
Myosin spots are stationary in the lamellipodia of a locomoting keratocyte (a), but they exhibit retrograde flow in the lamellipodia of a tethered cell (b). General views of tetramethylrhodamine-myosin–injected cells and time-lapse sequences for boxed areas are shown with time indicated in minute and seconds. Dotted lines indicate fixed positions with respect to the substratum. Selected myosin spots are shown with arrows. In a, the marked myosin spot is stationary while in the lamellipodia, but exhibits forward displacement at 50 s when it reaches the cell body. Bars, 2 μm.
Figure 10.
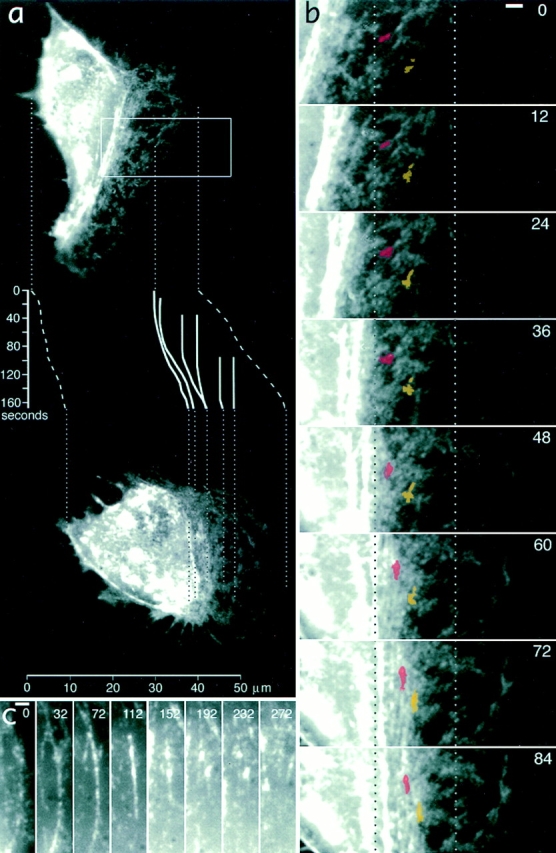
Formation of myosin bundles in the lamellipodia–cell body transition zone of a locomoting keratocyte is associated with forward translocation of myosin features. (a) An overview of a keratocyte at the start of observation (top) and after 168 s (bottom). Positions of two images reflect actual displacement of the cell in the horizontal direction. Traces of the cell's leading and rear edges (dashed lines) and selected myosin features (solid lines) are shown in the inset with time indicated in seconds on the vertical scale. One of the myosin spots traced is visible on the image at time 0, others have arisen at later time points and ended up, depending on time of their appearance and initial position, in the lamellipodium, in the lamellipodia–cell body transition zone, and as part of contracted myosin aggregates in the cell body at 168 s. Traces illustrate that myosin spots are initially stationary but become sequentially involved in forward translocation as the cell advances. (b) Details of bundle formation in the cell region indicated with box in a. Two myosin spots are highlighted with red and yellow. Dotted lines indicate positions fixed with respect to the substratum. Myosin spots are compressed in a horizontal direction (direction of locomotion), resulting in bundle formation and displacement to the right (forward). (c) The fate of a small myosin bundle as it forms at the cell body boundary (time 0) and contracts (112 s), fragments (152–232 s), and disappears (272 s) within the cell body. Bars, 2 μm.
Origination and Growth of Myosin Spots.
Time-lapse observation showed that myosin spots (clusters of myosin filaments as shown above by EM) arose continuously in the lamellipodia of both locomoting and tethered cells, as previously demonstrated for fibroblasts (McKenna et al., 1989; Verkhovsky et al., 1995). Typically, 5–20 new myosin spots per min were observed to form in every cell. In smoothly locomoting cells (majority of the cells), myosin spots usually arose at some distance from the leading edge, but in a few cells that exhibited ruffling activity at the edge, myosin spots arose at the edge in association with ruffle withdrawal similarly to what was reported for fibroblasts (Verkhovsky et al., 1995). Each individual myosin spot exhibited consistent growth (increase in size and brightness) over time, indicative of progressive enlargement of myosin filament clusters.
Motile Behavior of Myosin Spots in the Lamellipodia.
Motility of myosin spots differed in locomoting and tethered cells. In all cells analyzed that locomoted rapidly and consistently during observation (n = 21), myosin spots in the bulk of the lamellipodia remained stationary with respect to the substratum, but moved back with respect to the cell margin and, consequently, rapidly approached the cell body boundary (Fig. 9 a). Forward translocation of myosin spots that might be expected, based on actin polarity in lamellipodia, was not observed. In slowly or irregularly locomoting isolated cells (n = 6) and in all cells tethered at the border of epithelioid colony (n = 6), myosin spots in lamellipodia uniformly moved back with respect to the substratum, thus approaching the cell body border, as in locomoting cells (Fig. 9 b). However, the rates of backward myosin flow in tethered cells with respect to the substratum (1–2 μm/min) were typically 5–10 times lower than the rates of locomotion of free cells (3.5–15 μm/min).
Reorganization and Forward Translocation at the Cell Body Boundary.
The behavior of myosin features close to the cell body was of special interest because it might be related to the mechanism of translocation of the cell body, and because the structural data (see above) were suggestive of the reorganization of actin–myosin II system at the cell body boundary. In rapidly locomoting cells, characterized by a thick, spindle-shaped cell body, bright diffuse fluorescence in the advancing cell body interfered with the visualization of myosin dynamics in the transitional zone. In these cells, it was only possible to observe that myosin spots exhibited a brief period of forward translocation immediately before they were consumed by the cell body (see highlighted spot in Fig. 9 a, at 50 s), but the details of the process were not resolved. Better observation conditions were offered by cells having a flatter cell body, presumably because of relatively strong attachment to the substratum (Fig. 10 a). These cells were typically locomoting at moderate rates (3.5–8 μm/min), and they exhibited alternating phases of elongation and shortening of the cell body. In favorable cases, it was possible to observe that conglomerates of myosin spots next to the cell body boundary compressed to form arc-shaped bundles (Fig. 10 b). This process was associated with forward translocation of rear myosin features, while at the onset of compression, front features of the forming bundle remained stationary with respect to the substratum (compare the spots highlighted with red and yellow in Fig. 10 b). As compression of bundles continued, they became thinner and brighter and moved forward as a whole, perpendicular to their long axis. The rate of translocation of mature bundles was lower than the rate of cell body translocation, and in some cases, bundles could even stall after the initial translocation period. As a result, previously formed bundles entered the cell body while the new bundles continued to form in front of them at the cell body boundary. Thus, over time, new myosin features from lamellipodia became sequentially involved into forward translocation in this region (see traces of myosin spots in Fig. 10 a).
To verify if black tetra keratocytes exhibited cell body rolling as described for trout cells (Anderson et al., 1996) and to estimate its contribution to forward translocation, we examined the motility of endocytosed fluorescent beads and endogenous mitochondria. Both kinds of intracellular particles clearly rotated, as evidenced by their faster forward movement along the upper surface than at the lower surface of the cell (not shown). From measurement of the cell body diameter and the distance travelled by the cell during one or one-half revolution of the cell body, we estimated that the rotation was responsible for only 46 ± 6% of the total distance travelled (mean ± SD, n = 10), confirming that forward sliding occurred concomitantly with rolling.
Similar processes of condensation of myosin spots into bundles were also observed in tethered cells. Myosin spots at the cell body boundary ceased or slowed their retrograde flow and, as in locomoting cells, compressed to form bundles that remained stationary or (in case of cells advancing with an epithelial sheet) slowly translocated forward and frequently contracted (not shown). Thus, both in locomoting and tethered cells, condensation of myosin spots into bundles resulted in the displacement of myosin features at the cell body boundary relative to the lamellipodium. This displacement could represent a source of forward translocation of the body relative to stationary lamellipodia of locomoting cells, as well as a source of backward flow of the lamellipodia relative to the stationary body of tethered cells. In addition, the displacement of myosin spots near the lateral edges of the transition zone showed a component perpendicular to the direction of locomotion, moving toward the center concomitant with bundle formation (not shown).
Dynamics in the Cell Body.
Within the cell body, bundles were frequently observed to contract, fragment, and eventually disappear (Fig. 10 c). Small bundles merged with myosin background fluorescence, while bigger bundles transformed into bright amorphous aggregates that moved along with the cell body.
Discussion
The fish keratocyte, because of its rapid, predictable motility and thin, extended lamellipodium, is a model system for investigating cell locomotion. One might expect that the design of a moving machine should be revealed by the manner in which the elements of its mechanism are connected and how they move during action. This expectation guided the structural and dynamics approach of this study. Our focus here was on the organization of myosin II and its role in cell body translocation. In addition, novel features of the actin network that may have implications for the mechanism of lamellipodial protrusion were revealed.
Organization of Actin in Lamellipodia
Examination of platinum replicas of keratocyte lamellipodia confirmed general conclusions that were obtained using negatively contrasted preparations (Small et al., 1995): actin filaments in lamellipodia were long, abundant, and their density decreased with distance from the leading edge. In addition, the greater clarity of replica preparations allowed us to determine actin filament polarity throughout the lamellipodia and to visualize the pattern of actin filament termination. As a result, a comprehensive picture of actin filament arrangement has emerged, characterized by the following features: (a) free barbed ends of actin filaments are abundant in the distal 1-μm zone, but they are almost absent farther away from the leading edge; (b) filaments are oriented over a range of angles, but the barbed ends are generally directed toward the leading edge; and (c) the pointed ends of filaments are rarely observed free; throughout the lamellipodium, they usually terminate on other filaments, making T junctions. These observations are consistent with the idea that actin filaments in lamellipodia are connected to each other like branches of a bush, with the many “twigs” of the “bush” located at the leading edge and the less numerous “stems” close to cell body.
Implications for the Mechanism of Lamellipodia Protrusion
Several problems are usually considered in connection with the protrusion mechanism (Condeelis, 1993; Theriot, 1997; Zigmond, 1993): (a) how actin polymerization is spatially controlled at the leading edge; (b) the identity of nucleation sites for actin polymerization; (c) how actin filaments are cross-linked; and (d) how actin depolymerization is controlled to provide subunits for repolymerization.
Spatial Control of Polymerization.
Our data are consistent with the treadmilling mechanism of protrusion, where actin filaments have a broad-length distribution, with their elongating barbed ends being localized at the leading edge and their pointed ends distributed throughout the lamellipodia (Small et al., 1993, 1995). While it has been suggested that actin monomer–binding proteins and membrane phospholipids spatially control actin polymerization by providing high concentrations of polymerization competent actin near the plasma membrane (Condeelis, 1993), treadmilling offers an additional possibility of control by spatially segregating barbed and pointed filament ends.
Nucleation and Cross-Linking.
Although the treadmilling mechanism of lamellar protrusion depends primarily on steady-state elongation of preexisting filaments, the formation of new filaments is also necessary to compensate for the loss of diagonally oriented filaments that are predicted to treadmill to the sides of lamellipodia and eventually be removed from the protruding region (Anderson et al., 1996; Small et al., 1995). Our observation of numerous T junctions between short and long filaments at the leading edge suggests that new actin filaments either are nucleated at the membrane and become anchored to preexisting filaments immediately after nucleation, or that nuclei are anchored to a preexisting actin lattice. An advantage of tight coupling between nucleation and T junction formation would be that nascent polymerizing filaments, being anchored to an extensive actin network, could immediately push against the membrane. Tight coupling also offers a potential mechanism for exponential increase of actin filament number analogous to the dichotomic branching of a bush. Such a mechanism may be important for the expansion or turning of lamellipodia.
What protein(s) may mediate the coupling between actin filament nucleation and cross-linking? Nearly perpendicular branching of actin filaments in vitro occurs in the presence of actin-binding protein, with the pointed filament ends directed toward a branch point, and this pattern has been speculated to account for the blunt shape of cell protrusions (Hartwig et al., 1980). The molecule responsible for T junctions was identified as filamin or ABP-280 (Hartwig and Shelvin, 1986; Gorlin et al., 1990), and it is essential for the formation of cellular protrusions (Cunningham et al., 1992). Another candidate is an Arp2/3 complex, which contains actin-related proteins 2 and 3 and five other polypeptides (reviewed in Machesky, 1997), and has recently been shown to be sufficient to promote actin assembly around Listeria (Welch et al., 1997). The three-dimensional structure of the Arp2/Arp3 heterodimer suggests that it may imitate the barbed end of an actin filament and thus form a nucleation site for actin polymerization (Kelleher et al., 1995). In vitro, the Arp2/3 complex binds to the sides of actin filaments (Mullins et al., 1997), thus possibly mediating association of nucleation sites with preexisting filaments. It remains to be determined if ABP-280, Arp2/3 complex, or other protein(s) mediate actin branching in locomoting keratocytes. However, our study provides the first clear observation of T junctions between short and long actin filaments at known sites of intensive actin polymerization in situ.
Depolymerization.
In agreement with a previous report (Small et al., 1995), we found that actin concentration at the rear of lamellipodia was ∼1.8 times lower than at the front. This indicates that actin filaments undergo net depolymerization on their way from the cell edge to the center. How does depolymerization proceed despite the apparent absence of free pointed ends? One possibility is dissociation of T junctions followed by rapid depolymerization of the pointed ends until a halt at the next junction. Free pointed ends in this mechanism exist only transiently and thus could escape identification. Fast depolymerization at the pointed ends is consistent with the recent data showing that facilitated disassembly of actin filaments in vivo is mediated by an actin-depolymerizing factor (ADF1/cofilin; Carlier et al., 1997; Rosenblatt et al., 1997; Theriot, 1997). Because members of ADF/cofilin family seem to be less effective for ATP- or ADP.Pi-containing actin subunits than for ADP-actin (Maciver and Weeds, 1994; Carlier et al., 1997), their depolymerizing activity is predicted to be low at the leading edge, where nascent ATP-bound filaments dominate, and higher farther away, where the proportion of ADP-containing subunits increases.
Despite net depolymerization, the density of the actin network remains high, even in the rear of the lamellipodium. We propose that actin depolymerization continues as filaments are reorganized into bundles in the lamellipodial–cell body transition zone. This is consistent with the proposed increase of ADF/cofilin activity with filament lifetime. Also, the bending of actin filaments, which occurs here under the action of myosin (see below), may facilitate the severing action of ADF/cofilin, since this protein preferentially severs filaments at preexistent bends (for review see Moon and Drubin, 1995).
Organization of Myosin II and the Evaluation of Proposed Mechanisms of Cell Body Translocation
Myosin II in locomoting fish keratocytes was organized mostly in the form of stationary clusters of bipolar filaments in lamellipodia and filament bundles in the transition zone, which translocated forward. Existing models of cell body translocation make distinct predictions about the organization and behavior of myosin II. We evaluate these models based on how their predictions are fitted by our observations.
Sarcomeric Contraction.
This model implies the existence of actin–myosin II bundles organized in a semisarcomeric fashion and contracting during locomotion. In keratocytes, the only prominent actin filament bundles possibly organized this way were arc-shaped bundles at the cell body boundary. However, they were oriented perpendicular to the direction of cell locomotion and, therefore, not likely to provide the driving force. Moreover, since these bundles were arc shaped, with their convex side forward and presumably attached strongest at their trailing lateral edges (as suggested by wrinkling of elastic substrata [Lee et al., 1994]), their contraction would create a force component in the direction opposite to the direction of locomotion (Fig. 11 I).
Figure 11.

Models of cell body translocation. Actin filaments are shown as gray lines, and myosin filaments are shown as black lines or dumbbell figures. (I) The sarcomeric mechanism cannot drive the cell body forward because arc-shaped actin– myosin II bundles, upon contraction, would produce a backward-directed net force on the cell body. (II) The transport model does not fit the data because myosin clusters in the lamellipodia, the only part of a cell where transport tracks are present, do not move forward. (III) Forward rolling driven by actin–myosin axles seems problematic because of the predominantly backward orientation of flanking bundles (a, top view). These bundles may participate in rolling by exerting a rearward-directed force at the bottom (b, vertical section); however, the origin of forward-directed force remains unclear (question mark). (IV) According to the dynamic network model, contraction of an actin–myosin network in the lamellipodia– cell body transition zone is coupled to forward translocation. (a) In the lamellipodium, the network of divergent actin filaments interacts with clusters of myosin bipolar filaments. Whereas small myosin clusters situated in the dense network close to cell front cannot move, bigger clusters in the sparser network in the transition zone are capable of approaching the barbed ends of diverging filaments and moving forward. (b) As myosin clusters move forward, they align actin filaments parallel to the leading edge. (c) Overall, network contraction at the lamellipodia–cell body transition zone results in formation of actin–myosin bundles and forward translocation of the cell body. (d) Vertical section view shows that forward rolling would result from a combination of network contraction in front of the cell body with rearward drag resulting from actin–myosin bundles at the bottom of the body.
Transport Along Uniform Actin Arrays.
In this model, one would expect myosin II to be associated with uniform actin arrays and translocated forward during locomotion. Actin filaments in lamellipodia could serve as transport tracks because they are almost uniformly oriented with barbed ends forward (this study) and are stationary with respect to the substratum (Theriot and Mitchison, 1991). However, we observed no forward movement of myosin features in the bulk of the lamellipodia. In the transition zone and cell body, where actual forward movement did occur, no apparent transport tracks were found, neither by the whole-mount nor by the wet cleavage technique. More specifically, in the transition zone, actin filaments changed their course from approximately diagonal to perpendicular to the direction of locomotion, inconsistent with the idea of actin being a stationary track for a transport vehicle. Thus, polarized transport is unlikely to account for cell body translocation (Fig. 11 II).
Rolling of the Cell Body.
The rolling model (Anderson et al., 1996) holds that the cell body translocates forward via rotation. Actin–myosin bundles splaying forward into the flanks of the lamellipodium were proposed to drive the rotation. However, the forward splaying reported was slight (7.8°), and Fig. 8 of Anderson et al. (1996), which presented to demonstrate forward splaying, showed as much or more backward splaying as well. We confirmed the rolling of the cell body, but also note that forward splaying bundles were observed only in a small fraction of the cell population, and they were usually less pronounced than bundles splaying backward in the same cells. Consequently, the actin–myosin bundles seem poorly positioned to generate a forward component of force, which was also discussed earlier in relation to the sarcomeric model (Fig. 11, I and III). In addition, translocation of the cell body cannot be fully accounted for by rolling. In a pure rolling mechanism, the bottom surface of the cell should be stationary with respect to the substratum. In contrast to this expectation, the myosin bundles at the substratum plane translocated forward, indicating that the cell body slid forward in addition to rolling. Comparison of the rates of rotation and translocation (Anderson et al., 1996; our data) is also indicative of forward sliding, which cannot be explained by rolling about an actin–myosin axle.
Dynamic Network Model
The above models seem to be inconsistent with our results. We present an alternative model, which holds that forces for forward movement are generated by contraction of an actin–myosin II network. The network undergoes continuous assembly in the lamellipodia, contraction (with formation of bundles) in the transition zone, and disassembly in the cell body. This scheme represents a modification and development of our network model that had been proposed earlier for fibroblasts (Verkhovsky and Borisy, 1993; Verkhovsky et al., 1995, 1997).
Assembly of the Actin–Myosin II Network.
Clusters of interconnected myosin II bipolar minifilaments arise and grow spontaneously in keratocyte lamellipodia, similar to what has been shown earlier for fibroblasts (McKenna et al., 1989; Verkhovsky et al., 1995), and they eventually merge into an extended network. The nascent myosin clusters are not likely to contribute directly to cell locomotion for several reasons: (a) the clusters do not move forward themselves; (b) actin filaments are also stationary with respect to the substratum (Theriot and Mitchison, 1991), indicating that there is no relative translocation of actin and myosin in the lamellipodia; and (c) the myosin clusters are unlikely to exert any forward-directed force on the cell body through actin filaments because the polarity of actin in lamellipodia is consistent with backward rather than forward translocation of actin.
The lack of relative translocation of myosin and actin in the lamellipodium may be explained by differential regulation of myosin activity in the lamellipodia and in the transition zone. Another explanation is that myosin is active throughout the lamellipodium, but the actin–myosin network is too interconnected and rigid to allow for relative translocation of filaments (Taylor and Fechheimer, 1982; Kolega et al., 1991; Verkhovsky et al., 1997). More specifically, one may speculate that in keratocyte lamellipodia, each myosin cluster interacts with many divergent actin filaments and can neither follow simultaneously all these tracks nor, because of the rigidity of the actin network, converge them into one track.
Contraction and Formation of Actin–Myosin Bundles.
The potential contractile properties of the network may be selectively expressed in the vicinity of the cell body. Actin and myosin exhibit inversely related, graded distributions in lamellipodia: while actin density (and therefore network rigidity) decrease towards the cell body, myosin clusters increase in number and size. At some critical distance from the leading edge, the myosin clusters are expected to acquire the critical strength to overcome actin rigidity and start the contraction process. The following observations suggest that contraction leads to the formation of boundary bundles: (a) continuity between filaments in lamellipodia and bundles; (b) intermediates in the form of accumulations of actin filaments with associated myosin; and (c) direct observation of bundle formation in living cells. The idea of myosin II–driven formation of actin bundles from networks in the lamellum has been also proposed for fibroblasts (Verkhovsky et al., 1995). Formation of transverse bundles in keratocytes may be analogous to the formation of arc-shaped bundles in fibroblasts (Heath, 1983). Small et al. (1996) also discussed derivation of arclike actin bundles from the lamellipodia, although actin filament flux rather than the action of myosin was deemed the driving force.
The bundles in keratocytes are always oriented parallel to the leading edge, suggesting that compression of the network occurred along the anterior/posterior axis. Such orientation could be explained based on network geometry. Given that the contracting area is located within the sparser network next to the cell body, which is physically continuous with the denser network at the leading edge, the contracting portion of the network would compress to the border of the rigid area, forming a bundle parallel to the leading edge. At the supramolecular level, the only geometrical possibility for a cluster of interconnected myosin filaments to travel towards the barbed ends of many divergent actin filaments simultaneously would be by bending them and aligning them into a transverse bundle (Fig. 11 IV).
How Network Contraction and Bundle Formation Is Related to Forward Translocation and Retrograde Flow.
In the process of bundle formation, actin and myosin features in the transition zone move forward relative to the lamellipodia. The cell body would also move relative to lamellipodia, being attached to the contracting network in the transition zone either directly or through other cytoskeletal structures; e.g., intermediate filaments. The force along the anterior–posterior axis is transiently generated by each of the numerous portions of the compressing network continuously replacing one another and altogether producing a permanent pulling action on the cell body. Dynamic network elements operate along the entire cell body boundary, allowing the whole system to adapt to different configurations of the lamella, including the split lamellum, as described by Anderson et al. (1996). Our model is consistent with the transient connection of the cell body to the central part of lamellipodia, proposed by Anderson et al. (1996), to reconcile cell body rolling with lack of rotation in lamellipodia. In contrast to these authors, we consider these transient connections to be sufficient to drag the cell body. We propose that the pulling force is generated by the network along the entire cell body boundary, instead of flank bundles, as suggested by Anderson et al. (1996). Bundles represent the result of network contraction, but because of their orientation, they have little or no role in the generation of forward force.
The outcome of cell body translocation relative to the lamellipodia would depend upon attachment to the substratum. In locomoting keratocytes, the lamellipodial network is stationary because of relatively strong substrate attachment, but the cell body is weakly attached and therefore rides forward on a wave of network contraction at the cell body boundary. The rolling of the cell body may be explained by the assumption that the bottom surface of the body experiences more resistance and thus moves slower than the top surface. Arc-shaped bundles at the bottom of the cell would contribute to resistance by contracting and producing the net backward-directed force. Thus, the role of flank bundles in rotation may be opposite to that proposed by Anderson et al. (1996); i.e., we suggest they provide a backward-directed component of force along the cell bottom instead of a forward component along the top surface (Fig. 11, III and IV).
In tethered keratocytes, the cell body cannot move forward. Thus, contraction of the lamellipodial network would result in the lamellipodia as a whole moving backward rather than the cell body moving forward. Slower backward flow of lamellipodia in tethered cells compared to the movement of cell body in locomoting ones could be explained by greater substrate resistance experienced by lamellipodia. Thus, both the force for cell body translocation and retrograde flow can be explained by the process of contraction of an actin–myosin network in the transition zone.
Disassembly of Myosin Structures.
Since the formation and growth of new myosin features was continuously observed in the lamellipodia, the disassembly of myosin structures to replenish the soluble pool for new assembly must proceed elsewhere. We have actually observed that myosin fibers enter the cell body, where they contract, fragment, and finally merge with the background. A similar cycle of assembly at the periphery and disassembly in the perinuclear region has been proposed for fibroblasts, based on observations of stress fibers in serum-starved cells (Giuliano and Taylor, 1990) and estimation of local polymer/monomer ratio in locomoting cells (Kolega and Taylor, 1993).
Despite a simpler overall pattern of myosin distribution, the local organization and behavior of myosin features in keratocytes revealed remarkable similarity to fibroblasts. In particular, radially spreading fibroblasts exhibit the same essential elements of organization as tethered keratocytes (broad lamellipodia, retrograde flow, and actin– myosin bundles transverse to the direction of spreading; i.e., arc-shaped, circumferential bundles). Other cells might be different from keratocytes in the geometry of the leading edge and relative strength of attachment at front, sides, and rear of the cell. Consequently, contraction of the lamellipodial network might result in bundles parallel as well as perpendicular to the direction of locomotion. Nevertheless, myosin-dependent network contraction may be the primary event determining forces acting on the cell body. We propose that a model of assembly, contraction-driven translocation and disassembly of the actin–myosin network provides a conceptual framework that might be applicable, with modifications, to the motility of vertebrate cells in general.
Acknowledgments
We are grateful to John Peloquin for the preparation of fluorescent derivative of myosin II and to Steve Limbach for excellent light and electron microscope support. This work was supported by grants from the American Cancer Society CB-95 and the National Institutes of Health GM 25062.
Footnotes
Address all correspondence to Tatyana M. Svitkina, Laboratory of Molecular Biology, Bock Laboratories, University of Wisconsin, 1525 Linden Avenue, Madison, WI 53706. Tel.: (608) 262-1365; Fax: (608) 262-4570; E-mail: tsvitkin@facstaff.wisc.edu
1. Abbreviation used in this paper: ADF, actin depolymerizing factor.
References
- Anderson KI, Wang Y-L, Small JV. Coordination of protrusion and translocation of the keratocyte involves rolling of the cell body. J Cell Biol. 1996;134:1209–1218. doi: 10.1083/jcb.134.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begg DA, Rodewald R, Rebhun LI. The visualization of actin filament polarity in thin sections: evidence for the uniform polarity of membrane associated filaments. J Cell Biol. 1978;79:846–852. doi: 10.1083/jcb.79.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brands R, Feltkamp CA. Wet cleaving of cells: a method to introduce macromolecules into the cytoplasm. Application for immunolocalization of cytosol-exposed antigens. Exp Cell Res. 1988;176:309–318. doi: 10.1016/0014-4827(88)90333-3. [DOI] [PubMed] [Google Scholar]
- Byers, H.R., G.E. White, and K. Fujiwara. 1984. Organization and function of stress fibers in cells in vitro and in situ. In Cell and Muscle Motility. Vol. 5. J.W. Shay, editor. Plenum Publishing Corp., New York. 83–137. [DOI] [PubMed]
- Carlier M-F, Laurent V, Santolini J, Melki R, Didry D, Xia G-X, Hong Y, Chua N-H, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol. 1997;136:1307–1323. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheney RE, Riley MA, Mooseker MS. Phylogenetic analysis of the myosin superfamily. Cell Motil Cytoskel. 1993;24:215–223. doi: 10.1002/cm.970240402. [DOI] [PubMed] [Google Scholar]
- Condeelis J. Life at the leading edge: the formation of cell protrusions. Annu Rev Cell Biol. 1993;9:411–444. doi: 10.1146/annurev.cb.09.110193.002211. [DOI] [PubMed] [Google Scholar]
- Cooper MS, Schliwa M. Motility of cultured fish epidermal cells in the presence and absence of direct current electric fields. J Cell Biol. 1986;102:1384–1399. doi: 10.1083/jcb.102.4.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer LP, Siebert M, Mitchison TJ. Identification of novel graded polarity actin filament bundles in locomoting heart fibroblasts: implications for the generation of motile force. J Cell Biol. 1997;136:1287–1305. doi: 10.1083/jcb.136.6.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C, Gorlin J, Kwiatkowski D, Hartwig J, Janmey P, Stossel T. Requirement for actin binding protein for cortical stability and efficient locomotion. Science (Wash DC) 1992;255:325–327. doi: 10.1126/science.1549777. [DOI] [PubMed] [Google Scholar]
- Doolittle KW, Reddy I, McNally JG. 3D analysis of cell movement during normal and myosin-II-null cell morphogenesis in Dictyostelium. . Dev Biol. 1995;167:118–129. doi: 10.1006/dbio.1995.1011. [DOI] [PubMed] [Google Scholar]
- Giuliano KA, Taylor DL. Formation, transport, contraction and disassembly of stress fibers in fibroblasts. Cell Motil Cytoskel. 1990;16:14–21. doi: 10.1002/cm.970160104. [DOI] [PubMed] [Google Scholar]
- Goodson HV. Molecular evolution of the myosin superfamily: application of phylogenetic techniques to cell biological questions. Soc Gen Physiol Ser. 1994;49:141–157. [PubMed] [Google Scholar]
- Gorlin J, Yamin R, Egan S, Stewart M, Stossel T. Human endothelial actin binding protein (ABP-280, non-muscle filamin): a molecular leaf spring. J Cell Biol. 1990;111:1089–1105. doi: 10.1083/jcb.111.3.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig J, Shevlin P. The architecture of actin filaments and the ultrastructural location of actin-binding protein in the periphery of lung macrophages. J Cell Biol. 1986;103:1007–1020. doi: 10.1083/jcb.103.3.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig J H, Tyler J, Stossel T P. Actin-binding protein promotes the bipolar and perpendicular branching of actin filaments. J Cell Biol. 1980;87:841–848. doi: 10.1083/jcb.87.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath J. Behavior and structure of the leading lamella in moving fibroblasts. J Cell Sci. 1983;60:331–354. doi: 10.1242/jcs.60.1.331. [DOI] [PubMed] [Google Scholar]
- Heuser JE, Cooke R. Actin-myosin interactions visualized by the quick-freeze, deep-etch replica technique. J Mol Biol. 1983;169:97–122. doi: 10.1016/s0022-2836(83)80177-6. [DOI] [PubMed] [Google Scholar]
- Huxley HE. Electron microscope studies on the structure of natural and synthetic protein filaments from striated muscle. J Mol Biol. 1963;7:281–308. doi: 10.1016/s0022-2836(63)80008-x. [DOI] [PubMed] [Google Scholar]
- Huxley HE. Muscular contraction and cell motility. Nature (Lond) 1973;243:445–449. doi: 10.1038/243445a0. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Bischoff R, Holtzer H. Mitosis and intermediate-sized filaments in developing skeletal muscle. J Cell Biol. 1969;38:538–555. doi: 10.1083/jcb.38.3.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay PY, Pham PA, Wong SA, Elson EL. A mechanical function of myosin II in cell motility. J Cell Sci. 1995;108:387–393. doi: 10.1242/jcs.108.1.387. [DOI] [PubMed] [Google Scholar]
- Kelleher JF, Atkinson SJ, Pollard TD. Sequences, structural models, and cellular localization of the actin-related proteins Arp2 and Arp3 from Acanthamoeba. . J Cell Biol. 1995;131:385–397. doi: 10.1083/jcb.131.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolega J, Janson LW, Taylor DL. The role of solation-contraction coupling in regulating stress-fiber dynamics in nonmuscle cells. J Cell Biol. 1991;114:993–1003. doi: 10.1083/jcb.114.5.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolega J, Taylor DL. Gradients in the concentration and assembly of myosin II in living fibroblasts during locomotion and fiber transport. Mol Biol Cell. 1993;4:819–836. doi: 10.1091/mbc.4.8.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langanger G, Moeremans M, Daneels G, Sobieszek A, De Brabander M, De Mey J. The molecular organization of myosin in stress fibers of cultured cells. J Cell Biol. 1986;102:200–209. doi: 10.1083/jcb.102.1.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J., A. Ishihara, and K. Jacobson. 1993a. The fish epidermal keratocyte as a model system for the study of cell locomotion. In Cell Behavior: Adhesion and Motility. G. Jones, C. Wigley, and R. Warn, editors. The Company of Biologists Ltd., Cambridge, UK. 73–89. [PubMed]
- Lee J, Ishihara A, Theriot JA, Jacobson K. Principles of locomotion for simple shaped cells. Nature (Lond) 1993b;362:167–171. doi: 10.1038/362167a0. [DOI] [PubMed] [Google Scholar]
- Lee J, Leonard M, Oliver T, Ishihara A, Jacobson K. Traction forces generated by locomoting keratocytes. J Cell Biol. 1994;127:1957–1964. doi: 10.1083/jcb.127.6.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky LM. Cell motility: complex dynamics at the leading edge. Curr Biol. 1997;7:R164–R167. doi: 10.1016/s0960-9822(97)70079-4. [DOI] [PubMed] [Google Scholar]
- Maciver SK. Myosin II function in non-muscle cells. BioEssays. 1996;18:179–182. doi: 10.1002/bies.950180304. [DOI] [PubMed] [Google Scholar]
- Maciver SK, Weeds AG. Actophorin preferentially binds monomeric ADP-actin over ATP-bound actin: consequences for cell locomotion. FEBS Lett. 1994;347:251–256. doi: 10.1016/0014-5793(94)00552-4. [DOI] [PubMed] [Google Scholar]
- McKenna NM, Wang Y-L, Konkel ME. Formation and movement of myosin-containing structures in living fibroblasts. J Cell Biol. 1989;109:1163–1172. doi: 10.1083/jcb.109.3.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- Mogilner A, Oster G. Cell motility driven by actin polymerization. Biophys J. 1996;71:3030–3045. doi: 10.1016/S0006-3495(96)79496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon A, Drubin DG. The ADF/cofilin proteins: stimulus-responsive modulators of actin dynamics. Mol Biol Cell. 1995;6:1423–1431. doi: 10.1091/mbc.6.11.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RD, Stafford WF, Pollard TD. Structure, subunit topology, and actin-binding activity of the Arp2/3 complex from Acanthamoeba. . J Cell Biol. 1997;136:331–343. doi: 10.1083/jcb.136.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TM, King KL. Centripetal flow and directed reassembly of the major sperm protein (MSP) cytoskeleton in the amoeboid sperm of the nematode, Ascaris suum. . Cell Motil Cytoskel. 1991;20:228–241. doi: 10.1002/cm.970200306. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J, Agnew BJ, Abe H, Bamburg JR, Mitchison TJ. Xenopus actin depolymerizing factor/cofilin (XAC) is responsible for the turnover of actin filaments in Listeriamonocytogenes tails. J Cell Biol. 1997;136:1323–1332. doi: 10.1083/jcb.136.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger JM, Sanger JW. Banding and polarity of actin filaments in interphase and cleaving cells. J Cell Biol. 1980;86:568–575. doi: 10.1083/jcb.86.2.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small JV, Herzog M, Anderson K. Actin filament organization in fish keratocyte lamellipodium. J Cell Biol. 1995;129:1275–1286. doi: 10.1083/jcb.129.5.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small JV, Isenberg G, Celis JE. Polarity of actin at the leading edge of cultured cells. Nature (Lond) 1978;272:638–639. doi: 10.1038/272638a0. [DOI] [PubMed] [Google Scholar]
- Small, J. V., A. Rohlfs, and M. Herzog. 1993. Actin and cell movement. In Cell Behavior: Adhesion and Motility. G. Jones, C. Wigley, and R. Warn, editors. The Company of Biologists Ltd., Cambridge, UK. 57–71.
- Strohmeier R, Bereiter-Hahn J. Control of cell shape and locomotion by external calcium. Exp Cell Res. 1984;154:412–420. doi: 10.1016/0014-4827(84)90165-4. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Surgucheva IG, Verkhovsky AB, Gelfand VI, Moeremans M, DeMay J. Direct visualization of myosin bipolar filaments in stress fibers of cultured fibroblasts. Cell Motil Cytoskel. 1989;12:150–156. doi: 10.1002/cm.970120304. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Verkhovsky AB, Borisy GG. Improved procedures for electron microscopic visualization of the cytoskeleton of cultured cells. J Struct Biol. 1995;115:290–303. doi: 10.1006/jsbi.1995.1054. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Verkhovsky AB, Borisy GG. Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J Cell Biol. 1996;135:991–1007. doi: 10.1083/jcb.135.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons MH, Mitchison TJ. Control of actin polymerization in live and permeabilized fibroblasts. J Cell Biol. 1991;114:503–513. doi: 10.1083/jcb.114.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DL, Fechheimer M. Cytoplasmic structure and contractility: the solation contraction hypothesis. Philos Trans R Soc Lond B Biol Sci. 1982;299:185–197. doi: 10.1098/rstb.1982.0125. [DOI] [PubMed] [Google Scholar]
- Theriot JA, Mitchison TJ. Actin microfilament dynamics in locomoting cells. Nature (Lond) 1991;352:126–131. doi: 10.1038/352126a0. [DOI] [PubMed] [Google Scholar]
- Theriot JA, Mitchison TJ. The nucleation-release model of actin filament dynamics in cell motility. Trends Cell Biol. 1992;2:219–222. doi: 10.1016/0962-8924(92)90298-2. [DOI] [PubMed] [Google Scholar]
- Theriot JA. Accelerating on a treadmill: ADF/cofilin promotes rapid actin filament turnover in the dynamic cytoskeleton. J Cell Biol. 1997;136:1165–1168. doi: 10.1083/jcb.136.6.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhovsky AB, Borisy GG. Non-sarcomeric mode of myosin II organization in the fibroblast lamellum. J Cell Biol. 1993;123:637–652. doi: 10.1083/jcb.123.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhovsky AB, Svitkina TM, Borisy GG. Myosin II filament assemblies in the active lamella of fibroblasts: their morphogenesis and role in the formation of actin filament bundles. J Cell Biol. 1995;131:989–1002. doi: 10.1083/jcb.131.4.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhovsky AB, Svitkina TM, Borisy GG. Polarity sorting of actin filaments in cytochalasin-treated fibroblasts. J Cell Sci. 1997;110:1693–1704. doi: 10.1242/jcs.110.15.1693. [DOI] [PubMed] [Google Scholar]
- Welch MD, Iwamatsu A, Mitchison TJ. Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. . Nature (Lond) 1997;385:265–269. doi: 10.1038/385265a0. [DOI] [PubMed] [Google Scholar]
- Wessels D, Soll DR, Knecht D, Loomis WF, De Lozanne A, Spudich J. Cell motility and chemotaxis in Dictyosteliumamebae lacking myosin heavy chain. Dev Biol. 1988;128:164–177. doi: 10.1016/0012-1606(88)90279-5. [DOI] [PubMed] [Google Scholar]
- Zigmond SH. Recent quantitative studies of actin filament turnover during cell locomotion. Cell Motil Cytoskel. 1993;25:309–316. doi: 10.1002/cm.970250402. [DOI] [PubMed] [Google Scholar]