

Abstract
While the localization of chemoattractant receptors on randomly oriented cells has been previously studied by immunohistochemistry, the instantaneous distribution of receptors on living cells undergoing directed migration has not been determined. To do this, we replaced cAR1, the primary cAMP receptor of Dictyostelium, with a cAR1-green fluorescence protein fusion construct. We found that this chimeric protein is functionally indistinguishable from wild-type cAR1. By time-lapse imaging of single cells, we observed that the receptors remained evenly distributed on the cell surface and all of its projections during chemotaxis involving turns and reversals of polarity directed by repositioning of a chemoattractant-filled micropipet. Thus, cell polarization cannot result from a gradient-induced asymmetric distribution of chemoattractant receptors. Some newly extended pseudopods at migration fronts showed a transient drop in fluorescence signals, suggesting that the flow of receptors into these zones may slightly lag behind the protrusion process. Challenge with a uniform increase in chemoattractant, sufficient to cause a dramatic decrease in the affinity of surface binding sites and cell desensitization, also did not significantly alter the distribution profile. Hence, the induced reduction in binding activity and cellular sensitivity cannot be due to receptor relocalization. The chimeric receptors were able to “cap” rapidly during treatment with Con A, suggesting that they are mobile in the plane of the cell membrane. This capping was not influenced by pretreatment with chemoattractant.
Chemotaxis is a fascinating phenomenon whereby motile cells sense and respond directionally to chemical gradients. This important biological response plays a fundamental role in inflammation, neurogenesis, angiogenesis, and other morphogenetic processes (Behar et al., 1994; Szekanecz et al., 1994; Hogan and Foster, 1996). With the identification of cAR1, the cAMP chemoattractant receptor of Dictyostelium discoideum (Klein et al., 1985), it became clear that receptors mediating chemotaxis belong to the G protein-coupled receptor (GPCR)1 superfamily (Strader et al., 1995). Since then ∼20 functionally related “chemokine” receptors have been identified that mediate the chemotactic responses of neutrophils, macrophages, and lymphocytes. The features of chemotaxis and the spectrum of the biochemical responses triggered by chemoattractants are remarkably conserved between these evolutionarily distant cell types (Chen et al., 1996). The recent discovery that certain chemokine receptors act as the coreceptor for HIV virus infection has raised new interest in this class of receptors (Wells et al., 1996; Baiocchi et al., 1997).
A critical, unanswered question concerns the distribution profile of these receptors. Previous immunohistochemical studies of randomly oriented cells or cells taken from shaken suspension have shown chemoattractant receptors to be uniformly distributed around the cell periphery (Raposo et al., 1987; Wang et al., 1988; Trogadis et al., 1995). However, since chemotaxis is a dynamic process, the receptors must be instantaneously visualized in cells undergoing directional migration. Within a few minutes of being placed in a gradient, cells become polarized; and often, they will turn when the gradient direction is reversed. While the trailing and lateral edges of the cells do remain sensitive to chemoattractant, higher concentrations are required to elicit new pseudopods in these regions (Swanson and Taylor, 1982; Fisher et al., 1989). This gradient-induced polarization might be mediated by a redistribution of receptors. That is, the altered sensitivities may be due to a reversible accumulation of receptors at the anterior end. Recent studies have shown that coronin, a cytoplasmic actin-associated protein that is enriched at the cortical sites of moving D. discoideum cells, does transiently accumulate at the leading edge of chemotaxing cells (Gerisch et al., 1995; Maniak et al., 1995). We sought to determine whether cAR1, which is responsible for triggering the events that lead to actin and coronin translocation, displays a similar dynamic localization profile.
Another important question about GPCRs in general concerns their desensitization pathways. Desensitization is a series of processes that prevent continuous activation of the cell during prolonged exposure to agonist, thus protecting it from over stimulation. The commonly recognized mechanisms of desensitization are: (a) reduction in ligand binding capacity due to internalization or decreased affinity; (b) decrease in the efficiency of coupling to cognate G proteins and down-stream effectors; and (c) reduction in the receptor protein level. In many systems, these processes are promoted by agonist-induced phosphorylation of the receptors. A number of GPCRs including the β-adrenergic receptor (for review see Perkins et al., 1991), interleukin 8 receptor (Prado et al., 1996), thrombin receptor (Chen et al., 1995), muscarinic receptor (Maloteaux and Hermans, 1994), and angiotensin receptor (Hunyady et al., 1994) have been observed to be internalized in response to agonist treatment. By immunofluoresence, we have observed that ligand occupancy of cAR1 can lead to its clustering or internalization (Wang et al., 1988). However, we have also obtained conflicting biochemical evidence indicating that cAR1 remains on the plasma membrane even after prolonged cAMP stimulation (Caterina et al., 1995).
Previously it has not been possible to characterize chemotaxis and desensitization under physiological conditions, since most of the available techniques involve immunohistochemistry or immunofluorescence, which requires the prefixation of cells. In alternative methods which allow study of live cells, the receptor still has to be prelabeled with fluorecent ligands or antibodies. These reactions could cause unwanted alterations in receptor metabolism or trafficking. Green fluorescence protein (GFP), a stable fluorescent protein from a Pacific Northwest jellyfish, Aequorea victoria, has been extensively used as a visualization tag to study a variety of physiological phenomena (Cubitt et al., 1995). Only in a few cases were GFP successfully fused with integral membrane protein (Marshall et al., 1995; Hampton et al., 1996). A GFP fusion with odr-10, a presumptive seven-transmembrane domain olfactory receptor in Caenorhabditis elegans has been previously constructed (Sengupta et al., 1996). In that study, the fusion was mainly used in a fixed whole animal fluorescence assay to determine organ localization of the protein. No detailed biochemical study or study on the cellular level was carried out.
We fused GFP to the COOH terminus of cAR1 and found that this chimeric protein is indistinguishable from wild-type cAR1 in all testable biochemical and genetic properties, including agonist binding, agonist-induced phosphorylation, and phenotypic rescue of cAR1-null cells. By expressing this construct in a cAR1-null cell line, we could follow the distribution of receptors during chemotaxis and desensitization nonintrusively. This represents the first successful attempt to study a GPCR in unperturbed living cells and to instantaneously visualize a receptor during stimulus presentation. Our results show for the first time that chemoattractant receptors remain uniformly distributed on the surface of cells that have been polarized by chemotactic gradients and also in cells that have been desensitized by persistent treatment with chemoattractant. This study demonstrates that GFP fusions with GPCRs may be an effective means to study the localization of these receptors.
Materials and Methods
Construction of cAR1-GFP Fusion Protein
A mutant GFP sequence (S65T), cloned into the BamHI site of pRSETB (Invitrogen, Carlsbad, CA), was kindly provided by Dr. Roger Tsien (University of California, San Diego, CA). This mutant has been shown to give greater brightness and sustain slower photobleaching than wild-type GFP (Cubitt et al., 1995). The multiple linker site was removed by digesting the plasmid with HindIII to XhoI and blunt-end ligating the new ends after Klenow enzyme treatment. The entire coding sequence of cAR1, including the 5′ ribosome-binding site was PCR amplified and cloned into the remaining EcoRI site in front of the GFP sequence. This cAR1-GFP fusion sequence was then released by BamHI digestion and cloned into the BglII site of pJK1, a D. discoideum extra-chromosomal expression vector (Kim and Devreotes, 1994). Transformants with the correct orientation were selected by PCR. DNA was purified and transformed into RI9 cells (car1−/car3− cells), and these cells served as the cells under study (cAR1-GFP cells).
Immunoblotting
Protein samples were solubilized in SDS-sample buffer and resolved by SDS-PAGE on 10% gels. In the case of ligand-induced receptor phosphorylation, the samples were run on gels with lower cross-linker concentration (Klein et al., 1985) to effectively resolve the two forms of receptor (unmodified vs. phosphorylated). cAR1-GFP was detected by immunoblotting with anti-GFP antibody (Clontech, Palo Alto, CA) or cAR1 antiserum (R4 serum; Klein et al., 1985).
Detergent Resistance of cAR1-GFP
The procedure was essentially as described previously (Xiao and Devreotes, 1997). Briefly, cells expressing cAR1-GFP were resuspended to 1 × 108 cells/ml, and Chaps was added to a final concentration of 1.5%. Extraction was allowed to proceed for 5 min on ice. Lysates were separated into soluble and pellet (detergent-resistant membrane) fractions by centrifugation at 15,000 g for 10 min. The fractionation profile of cAR1-GFP was monitored by immunoblotting.
Ligand-induced Phosphorylation and Electrophoretic Mobility Shift of cAR1-GFP
cAR1-GFP cells were washed once with development buffer (DB; 10 mM sodium phosphate, pH 6.2, 1 mM MgCl2, 0.2 mM CaCl2) and developed in DB suspension at a cell density of 2 × 107/ml for 6 h. Cells were first shaken at 200 rpm for 30 min in the presence of 4 mM caffeine and then treated with increasing doses of cAMP in the presence of 10 mM DTT for 15 min (Kim and Devreotes, 1994). Samples were resolved on 10% low-bisacrylamide PAGE, transferred to PVDF membranes, and immunoblotted with cAR1 antiserum.
Desensitization of Fusion Receptor and Loss-of-Ligand Binding
The loss-of-ligand binding (LLB) assay was performed essentially as described (Caterina et al., 1995). Briefly, cells were washed, resuspended in DB, and divided into two equal aliquots. One received 10−5 M cAMP with 10 mM DTT while the other received DTT alone. After treatment for 20 min, cells were diluted into 10-fold excess of ice-cold phosphate buffer (PB; DB without the MgCl2 and CaCl2) and pelleted by centrifugation at 4° (Sorvall SS34, 2,000 rpm, 3 min). After three washes in cold PB, cells were resuspended to 108/ml. Binding of 16 nM [3H]cAMP was then performed by a sedimentation assay.
Confocal Fluorescence Microscopy Analysis
Chemotaxis-competent cAR1-GFP cells were plated onto glass surfaces and allowed to adhere. They were fixed with 4% paraformaldehyde/0.1% Triton X-100 in PBS for 8 min, rinsed twice with PB, submerged in PB, and then analyzed with a confocal laser scanning microscope (Noran OZ; Noran, Middleton, WI) at an excitation wavelength 488 nm from a Krypton-Argon multi-line laser. A barrier filter was used to detect 500–550-nm emissions. Cells were examined through an inverted microscope (IX-50; Olympus, New Hyde Park, NY) with a U plan-apo 100×/1.35 NA oil immersion lens. Z-axis images were processed and three-dimensional reconstructions created and rotated with Intervision 1.6 software (Noran).
Chemotactic Stimulation of Cells through Micropipettes
Cells at 2 × 107/ml were shaken in DB suspension with addition of 50 nM cAMP every 6 min for 6 h to allow the expression of the full complement of chemotactic proteins (Devreotes et al., 1987). Cells (at 106 cells/ml) were washed, dissociated by gentle vortexing, and transferred onto a glass coverslip. After cell adhesion, the coverslip was mounted on the bottom of an observation chamber (5.5 × 4.0 cm) and covered with 2 ml DB to prevent drying and allow chemotaxis to take place. Micropipettes (Gerisch and Keller, 1981) filled with 10−4 M cAMP were used to stimulate the chemotaxis of these cells. Cells were observed on an inverted microscope (Axiovert 135 TV; Zeiss, Inc., Thornwood, NY) equipped with a water condenser and a phase-contrast 40 or 100× oil-immersion plan neofluar objective lens. Their movement was recorded using a cooled CCD camera (PXL; Photometrics, Tucson, AZ) controlled by IPLab-Spectrum software (Signal Analytics, Vienna, VA). Fluorescence microscopy was performed on the same microscope with an HBO 100-W mercury lamp and a 100× oil neoflur objective lens. To avoid UV injury to the cells, the light intensity was reduced by neutral density filters, and usually 20–50 ms- exposure images were taken at 10- to 15-s intervals for a total duration of 5 to 10 min.
Ligand-induced Desensitization of Cells and Con A-stimulated Receptor Capping
For desensitization, a concentrated dose of cAMP was applied to the vicinity of the cells adherent to a glass coverslip, and time-lapse fluorescence images were taken at 20-s intervals for a total duration of 5 to 10 min. The final equilibrated concentration of cAMP inside the observation chamber was >10−5 M, sufficient to induce receptor desensitization and LLB (Caterina et al., 1995).
For Con A treatment, Con A was added to a final concentration of 20 to 50 μg/ml. Fluorescence images were taken before and after the treatment at intervals of 40 s for a total duration of 5 to 10 min.
Results
The cAR1-GFP Fusion Protein Targets to the Plasma Membrane and Is Biochemically and Functionally Indistinguishable from WT cAR1
We expressed the cAR1-GFP fusion construct in a cAR1-null cell line, thereby replacing the endogenous receptor. Multiple lines of evidence suggested that the fusion protein correctly reported the distribution of the functionally active chemoattractant receptors. First, we investigated whether the fusion protein is effectively synthesized and processed by immunoblotting the whole cell protein extract with purified GFP antibody or cAR1 antibody (Fig. 1 A). Both antibodies detected an identical 65-kD protein, which is the expected size of the fusion protein. Smaller proteins corresponding to the size of free GFP (30 kD) or cAR1 (40 kD) were not observed. There was a faint band at 85 kD which corresponds to GFP fused to a minor 55-kD form of cAR1 routinely observed in cells expressing WT cAR1. We have speculated that this minor form may derive from a low level of premature translation from an upstream initiation site or a post-translational modification. Our previous study has established that WT cAR1 resides on special detergent-resistant microdomains of plasma membrane (Xiao and Devreotes, 1997). Crude subcellular fractionation indicated that both the major and minor GFP fusion proteins, like WT cAR1, mostly associated with this detergent-resistant subdomain (Fig. 1 B). Further fractionation of cellular lysates on sucrose density gradients showed that both bands were quantitatively localized to the plasma membrane microdomains (data not shown). All these observations indicate that cAR1-GFP is a stable fusion protein that is correctly targeted to the proper location.
Figure 1.
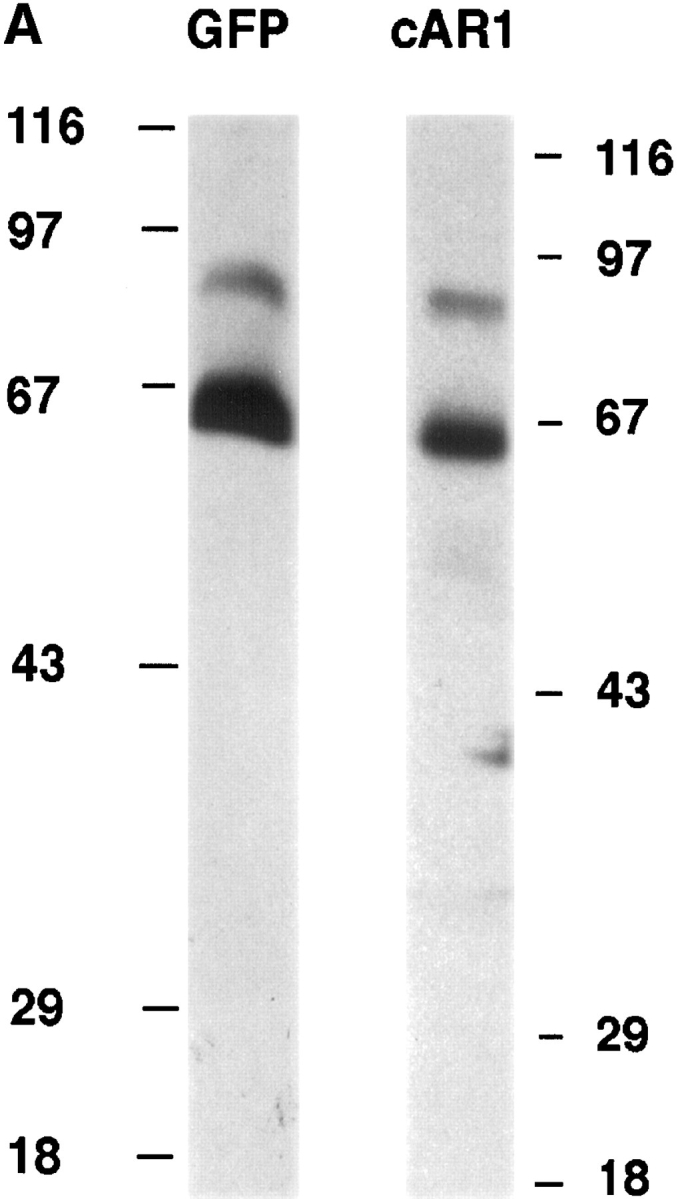

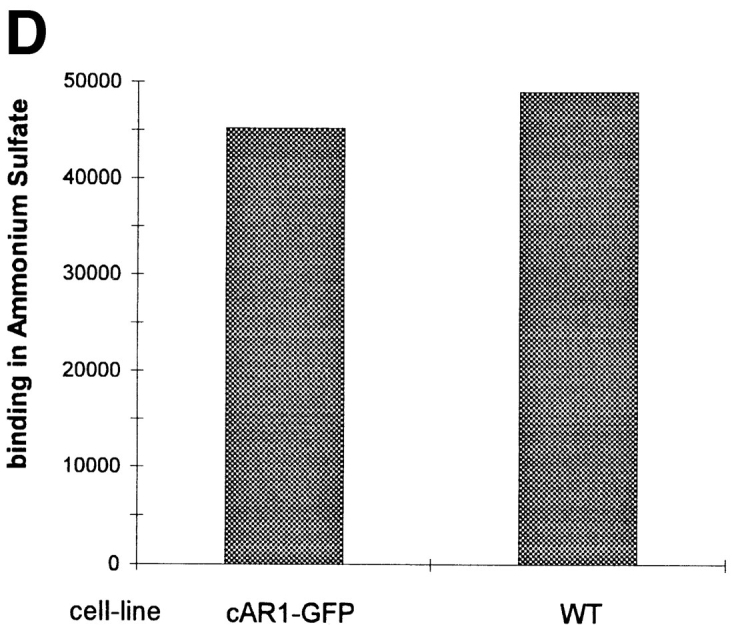

cAR1-GFP is functionally indistinguishable from wild-type cAR1. (A) cAR1-GFP fusion protein as detected by immunoblotting with GFP and cAR1 antisera. Proteins were solubilized in SDS-sample buffer, resolved on 10% SDS-PAGE, and transferred onto PVDF membranes. Duplicate samples were loaded and analyzed in parallel. In the left lane, the fusion protein was detected with affinity-purified anti-GFP antibodies. In the right lane, it was detected with anti-cAR1 antiserum. (B) Both the major and minor forms of cAR1-GFP are localized to a detergent-resistant plasma membrane subdomain. Whole cells (lane 1) were extracted with 1.5% CHAPS at 4°C and lysate separated into soluble (lane 2) and detergent-resistant membrane (lane 3) fractions by centrifugation, as described in Materials and Methods. Protein samples were resolved by SDS-PAGE, transferred to membranes, and fusion protein detected by anti-cAR1 antibody. (C) Agonist-induced phosphorylation of cAR1-GFP and gel mobility shift assay. cAR1-GFP cells were treated with increasing doses of cAMP for 15 min to induce the phosphorylation of the COOH terminus of cAR1. cAMP doses: 1, 0 nM; 2, 5 nM; 3, 20 nM; 4, 50 nM; 5, 100 nM; 6, 200 nM; 7, 500 nM; 8, 1 μM; and 9, 5 μM. The unmodified or phosphorylated forms of the fusion receptor were detected with cAR1 antiserum. (D) cAR1-GFP cells express the same total surface cAMP-binding sites as wild-type cAR1 cells. cAR1-GFP and cAR1 expressing cells were washed in PB and resuspended to 1 × 108 cells/ml density in ice-cold PB. 60 μl cells were used in the ammonium sulfate-binding assay (Materials and Methods). The specific binding for both cell lines in cpm were compared and later translated into the number of binding sites per cell (see text).
To investigate the ligand-binding affinity and functional properties of cAR1-GFP, we carried out the cAMP-induced receptor gel mobility shift assay. Cells were treated with increasing doses of cAMP to induce different extents of steady-state receptor phosphorylation. This phosphorylation can be visualized as a mobility shift on SDS-PAGE; the phosphorylated protein migrates more slowly. For wild-type receptors, the concentration of cAMP at which half of the molecules are shifted to the lower mobility form (defined as EC50) is closely correlated with the dissociation constant (Kd) for cAMP binding (Johnson et al., 1992). As shown in Fig. 1 C, the fusion protein showed a similar mobility shift, with an EC50 between 20 and 50 nM. Densitometric analysis gave a value of 27 nM, which is essentially identical to the wild-type value (30 nM; Kim and Devreotes, 1994). This observation indicates that cAR1-GFP has the same cAMP-binding properties as cAR1 and couples with equal efficiency to the receptor kinase.
To assess whether the fusion receptor is expressed to the same level as cAR1, we performed [3H]cAMP binding for both the fusion cell line and a control cell line that expresses cAR1 in the same expression vector. It has been previously shown that ammonium sulfate converts the receptors into a single high-affinity species with a Kd value of 5 nM (Johnson et al., 1991). Therefore, binding obtained at 20 nM [3H]cAMP represents the total number of surface cAMP binding sites. As shown in Fig. 1 D, the cAR1-GFP cells express about the same number of sites as the WT cAR1 cells (3.5 × 105 sites/cell). This value is ∼70% more than that of optimally differentiated AX3 cells (2 × 105 sites/cell) in which cAR1 is expressed from its endogenous promotor.
To further test the functionality of the fusion protein, we plated the cells on non-nutrient agar. Under nutrient starvation, D. discoideum cells start a developmental program which allows them to form multicellular aggregates that differentiate into fruiting bodies. Central cells initially start to secrete cAMP, which causes neighboring cells to move chemotactically towards the center; in turn these cells secrete cAMP, attracting even more distal cells. This chain of events culminates in the formation of aggregates containing up to 105 cells. cAR1 is primarily responsible for mediating this process (Devreotes, 1994). As shown in Fig. 2, cAR1 null cells cannot initiate this pathway and hence remain unaggregated (3), and wild-type cAR1 rescues the program such that normal fruiting bodies are eventually formed (2). The cAR1-GFP–expressing cell line displayed a normal developmental phenotype, reaching each stage of the program at the same time as the WT cAR1 cells (data not shown). Fig. 2, 1 shows its final fruiting body stage. These observations indicate that cAR1-GFP and WT cAR1 are functionally interchangeable.
Figure 2.

cAR1-GFP rescues the developmental defect of cAR1-null cells. The cAR1 null cell line was transformed with 1, cAR1-GFP construct; 2, wild-type cAR1; and 3, empty vector. Transformed cells were washed with DB and developed on a 35-mm agar plate (1 × 107 cells/plate). After 36 h, pictures were taken of each plate for the formation of fruiting bodies.
The Chimeric Receptor Is Uniformly Distributed on the Cell Surface and Remains So during Chemotaxis
Under conventional fluorescence microscopy, the cAR1-GFP cells displayed intense fluorescence on the cell periphery; the cell body showed a faint diffuse signal (see below). Such a profile is expected if the protein is evenly localized on the surface membrane and further suggests that GFP does not interfere with the correct targeting of cAR1 to the plasma membrane. The uniformity of the fluorescence signal was remarkably preserved during random movement of the cell. Rapidly extending and retracting pseudopods and fine filopods were clearly visible due to the bright fluorescence, despite the fact that the signals showed little preferential localization onto these structures or to the leading or trailing edges of the cell (see below). The peripheral fluorescence might have been anticipated based on previous immunohistochemical studies, but it was not possible to observe the receptors on thin cellular projections in those experiments.
We next used the micropipette assay to study distribution of the cAR1-GFP receptor during chemotaxis (Fig. 3, 1–8). cAR1-GFP cells were able to move chemotactically towards the source of cAMP in a fashion that is indistinguishable from cells expressing wild-type cAR1. We detected little change in receptor localization profiles under any conditions. The fluorescence signals remained uniform on the cell surface and projections, even though the cells were undergoing rapid morphological changes associated with chemotaxis. Even in highly polarized cells, there was no increase of signal at the migrating front. Specifically, in Fig. 3, 1–4, which represents one complete sequence, cell “a” initially extended two pseudopods (1, arrowhead). The one pointed in the wrong direction was quickly suppressed and taken over by the correct one, which persisted throughout the process (Fig. 3, 2, arrowhead). In Fig. 3, 3, the cell extended a pseudopod in the wrong direction (arrowhead), which was again competed out by the right one. Cell “b” provided another example of this behavior. Little apparent change in receptor distribution accompanied these contortions.
Figure 3.
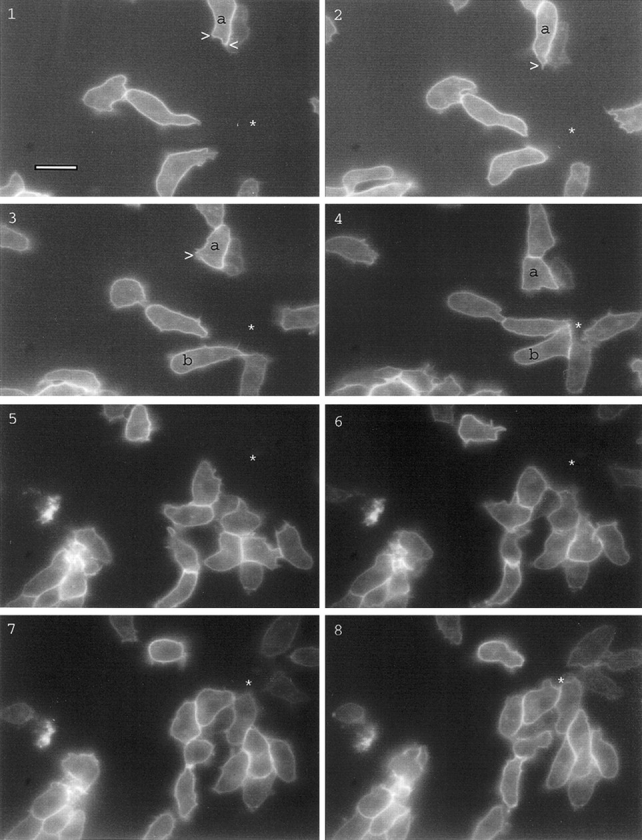
Distribution of cAR1-GFP during chemotaxis. All figures shown were fluorescence images captured in a 50–100-millisecond exposure; phase contrast images were unnecessary since cell shapes were obvious from the peripheral fluorescence of cAR1-GFP. There was a 10-15 second interval between consecutive frames. Frames 1–4 show the complete sequence of one chemotaxis event, frames 5–8 showed another. Positions of the micropipette were indicated by “*”. Arrowheads indicate pseudopods. Bar, 5 μm.
We frequently switched the position of the pipette to induce formation of novel chemotactic fronts (Fig. 4, 1–8). Cells initially moved towards the right asterisk (Fig. 4, 1 and 2). Upon repositioning of the micropipette to the bottom left asterisk (Fig. 4, 3–8), cells extended pseudopods from the old migrating front towards the new spot. This was followed by a dramatic shape change that bent the cells in the direction of the new gradient. The cells reoriented themselves, and thus the old front served as the new front. In other instances, a new front would form de novo from the side of the cell proximal to the new cAMP source, and the old front would disappear. In either case little change in the receptor localization profile was noticed.
Figure 4.
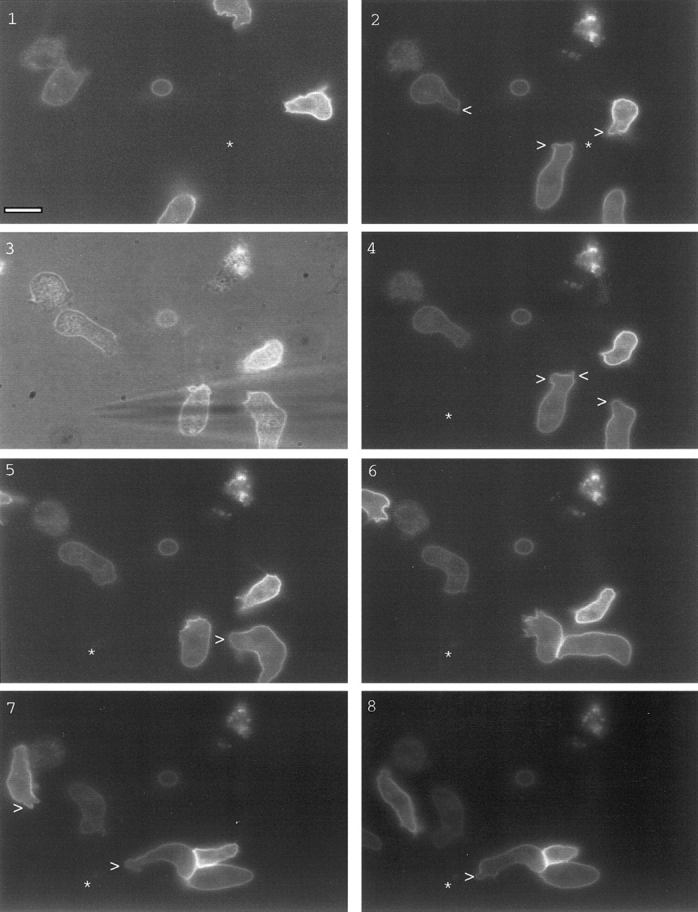
Distribution of cAR1-GFP during cell turning after chemoattractant source movement. Sequence shows cells turning in response to cAMP source movement. Frame 3 is a phase-contrast image to highlight the micropipette position shift. In frames 1 and 2, asterisks indicate the original cAMP source; in all other frames, the asterisk denotes the new position. Pseudopod positions were indicated with arrowheads. Time interval is 15 s. Bar, 5 μm.
To quantitatively assess the uniformity of the signals around the cell periphery, we directly counted the pixel numbers of fluorescent signals from various regions or converted the digitized signal to a false color image. We set the color range in such a way that signal intensity variations of >10% would cause a different color to appear. In most cases, the signal was uniform along the circumference and varied randomly by <10–20%. In some instances there was a decrease in intensity of ∼20 to 30% on the extending fronts, especially on the most recently projected pseudopods.
Confocal Image Analysis of cAR1-GFP Distribution on Cells
To further elucidate the details of receptor distribution on various regions of the cell membrane, we carried out a Z-axis confocal analysis (Fig. 5 A). It was necessary to lightly fix the cells since they are extremely mobile. Sections of 0.5 μm in thickness were taken from the bottom of the cell close to the substratum to the upper surface at a distance of 1.0 μm per section. The cell under study was a typically flat and adhesive cell with many pseudopods and fine filopods. The bottom section (Fig. 5 A, 1) showed fluorescence in the interior of the profile, which was likely due to the upward invaginations of its basal membrane. The latter frames of Fig. 5 A displayed mostly peripheral fluorescence and little internal signal, indicating that cAR1 is highly localized on the surface. Pseudopods and filopods seemed most apparent in the lower sections. The upper surface of the cell seemed to be quite uneven: one portion of the cell (near bottom of the frame) rose up higher than the other portion. After Fig. 5 A, 3, only cross-sections of this portion were clearly present. The receptors seem to be more or less evenly distributed on the plasma membrane, although some small, randomly localized regions do contain more signals than neighboring domains.
Figure 5.

(A) Z-axis two-dimensional confocal fluorescence image scanning analysis of cAR1-GFP cells. Cells adhering to a glass coverslip were fixed in 4% paraformaldehyde/0.1% Triton X-100 and subjected to confocal analysis. Section thickness: 0.5 μm; frame interval: 1.0 μm. (B) Three-dimensional reconstruction image of cAR1-GFP cell fluorescence. All three-dimensional images were created from a Z-axis scanning series with the Intervision 1.6 software program. The reconstructed image was rotated around the X-axis for 150° starting from the bottom to top image (top of cell projecting into paper plane). Images were captured every 30°. Final image shows top to bottom view. Bars, 10 μm.
A three-dimensional reconstruction was created for another cell (Fig. 5 B). Rotation of the image along the x axis provides further spatial information of the receptor. The first image is the view from bottom to top with the top projecting into the plane of the paper. After a 150° rotation, the last image displays the view from top to bottom. The receptors again appear to be uniformly distributed on the flat part of the cell surface, as well as along the numerous folds, creases, and projections found on the surface.
During Persistent Stimulation with Chemoattractant Leading to Cell Desensitization, cAR1-GFP Undergoes a Decrease in Affinity and Remains on the Cell Surface
We next compared the properties of the wild-type and fusion receptors during agonist-induced desensitization. As shown previously, agonist occupancy leads to phosphorylation of cAR1. The phosphorylated receptors then display diminished ligand binding capacity due to a decreased affinity (LLB; Caterina et al., 1995). As shown in Fig. 6 A, cells expressing either type of receptor display ∼80–90% apparent LLB after cAMP pretreatment, in good agreement with previous studies. This demonstrates, as expected from its capacity to be normally phosphorylated, that cAR1-GFP desensitizes as efficiently as the wild-type receptor and confirms the physiological relevance of using the fusion construct to study this phenomenon.
Figure 6.


(A) Cells expressing cAR1-GFP receptors display normal desensitization properties. Wild-type cAR1 cells and cAR1-GFP cells were washed in PB and treated with either buffer (control) or 10−5 M cAMP in the presence of 10 mM DTT for 15 min to induce LLB. After extensive washing, the cells were assayed for their cAMP binding (16 nM [3H]cAMP) in PB (Materials and Methods). The binding was expressed as a percentage of the binding obtained from buffer-treated control cells. (B) Distribution of desensitized cAR1-GFP analyzed through time-lapse fluorescence imaging. All frames are fluorescence images. Frame 1 showed the cell in random movement. cAMP was applied 5 s before frame 2 was taken. Time interval is 1 min between 2 and 4; 90 s between 4 and 6. (C) Z-axis confocal analysis of receptor distribution on desensitized cells. Cells were pretreated with excess cAMP for 5 to 10 min to induce desensitization and then quickly fixed with 4% paraformaldehyde/0.08% Triton X-100. Z-axis sections were taken from the bottom of the cell (substrate adhesion side) to the top surface. Section thickness: 0.5 μm; frame distance: 1.0 μm. Bars, 10 μm.
To visualize the possible changes in receptor distribution during persistent treatment with chemoattractant, we gently applied a dose of cAMP to cells adherent on a glass surface; the addition caused a uniform increase of cAMP concentration to ∼10−5 M (the effects of phosphodiesterase are negligible here since there are very few cells plated and they are in a large volume of buffer). The cells were monitored by time-lapse imaging before and after cAMP addition (Fig. 6 B). Fig. 6 B, 1 shows three randomly moving cells with different fluorescence intensities. Note the rapidly extending and retracting pseudopods. All of the green fluorescence signals were primarily localized to the cell surface. (Certain internal speckled light spots were visible, but careful study revealed that these were yellow and red autofluorescence signals, perhaps due to mitochondrially bound NADH [Arbin, 1979]. This condition seemed especially pronounced for inactive or apoptotic cells [note the two nonfluorescent and totally inert cells in the same field].) cAMP was applied 5 s before Fig. 6 B, 2. From Fig. 6 B, 2 to 6, ∼5 min elapsed. The cells displayed a general trend of rounding up throughout the process, which is a phenomenon usually associated with cell adaptation induced by prolonged application of uniform stimulus. The initial pseudopods of the cells were quickly withdrawn after the ligand application, but in the case of the dimmer cell (Fig. 6 B, upper right cell), new ones were subsequently produced at random orientations before they were eventually suppressed. Visual monitoring of this process suggested little rearragement (internalization, degradation, local depletion, or concentration) of the receptor. Quantitative measurements further proved that the fluorescence signals remained essentially unchanged on the cell surface during this response to persistent stimulation.
Although the above result showed that under a conventional microscope, receptors do not display gross rearrangement after desensitization, it was possibile that a small percentage of receptors was internalized to certain interior compartments or plasma membrane-proximal vesicle structures that may not be obvious under our test conditions. To address these issues and further investigate the desensitization phenomenon, we carried out two-dimensional confocal analysis of persistently stimulated cells (Fig. 6 C). This cell had been completely desensitized by treatment with 10−5 M cAMP for 7 min and then quickly fixed. In contrast to the previous confocal profile (Fig. 5 A), the size of the cells was smaller in the lower sections and gradually increased in the upper sections, consistent with the rounded shape of desensitized cells. Otherwise, this profile is similar to the previous one. The bottom frame displayed the most internal fluorescence, indicating upward projections of its basal membranes, and all the upper frames showed essentially uniform peripheral signals with little internal fluorescence. This result strongly corroborates our earlier observations. Desensitization does not lead to receptor rearrangement.
Con A Is Able to Cause the Patching and Capping of cAR1-GFP
The multivalent lectin Con A induces cross-linking of cell surface glycoproteins, resulting in the patching and eventual “capping” of these surface membrane proteins (Patton et al., 1990). Since cAR1-GFP maintained a uniform distribution on cell surface under all physiological conditions, we tested whether the Con A treatment would shift its location along with other surface proteins. As shown in Fig. 7 A, the distribution does change drastically. Fig. 7 A, 1 and 2, shows an elongated cell involved in random movement, with a number of pseudopods. Con A (final volume 20–50 μg/ml) was added 10 s before Fig. 7 A, 2 was taken. A detectable enrichment of signal on the bottom right corner of the cell quickly occurred (Fig. 7 A, 2, arrowhead). Massive patching and/or capping of the receptor then took place at three regions (Fig. 7 A, 3, arrowheads). After this, the cell gradually rounded up, and one major capping event proceeded to completion at the bottom edge of the cell. The patch on the opposite end persisted but did not culminate into an equally impressive cap. During this stage, certain regions of the membrane lost receptors, as was evident from the depletion of fluorescence signals (Fig. 7 A, 5 and 6, arrowheads). The rounded cells were not able to respond to micropipettes containing cAMP. Higher Con A concentration (>0.5 mg/ml) resulted in very inefficient patching and no subsequent capping (data not shown), as might be expected for a multivalent ligand.
Figure 7.
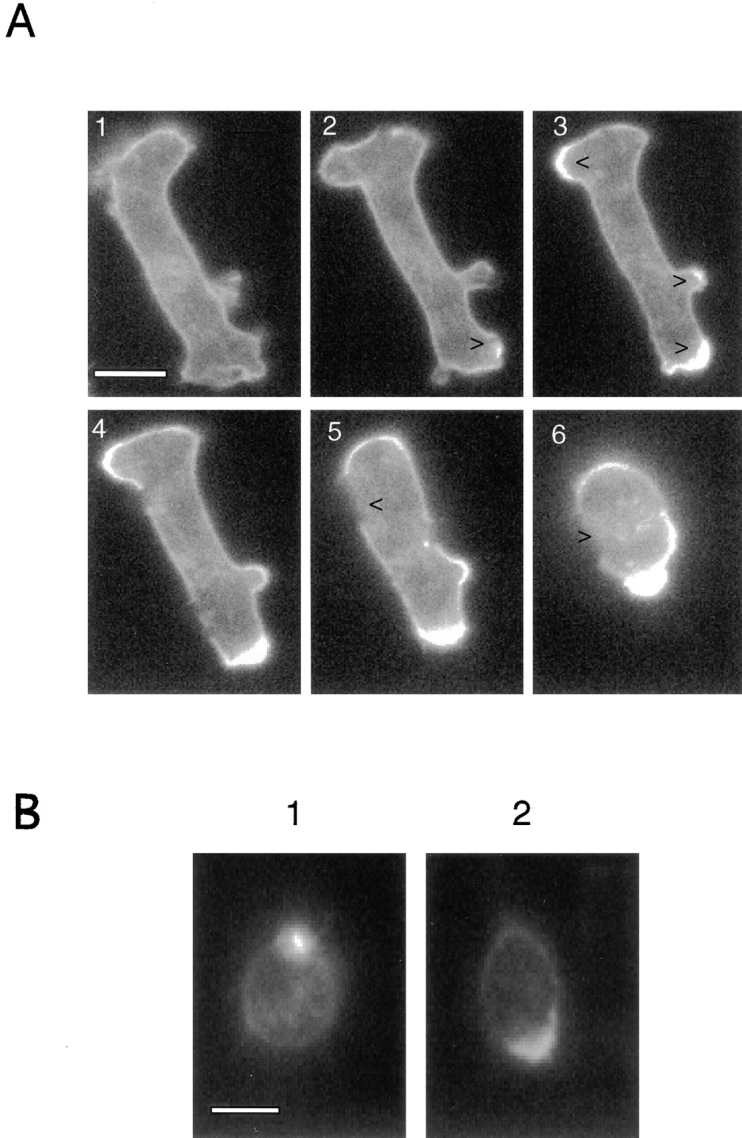
(A) Con A treatment of cAR1-GFP cells to induce receptor capping. All images are fluorescence ones. Frame 1: cell before treatment. Frames 2–6: after application of 50 μg/ml Con A. Con A was applied 15 s before frame 2 was taken. Time interval is 45 s. In frames 2 and 3, arrowheads indicate patching and capping of signals. In frames 5 and 6, they show the regions devoid of signals. (B) Desensitized cells can still carry out efficient Con A-induced receptor capping. Cells were either pretreated with buffer (1) or 10−5 M cAMP (2) for 10 min to induce desensitization. Con A was then added to induce receptor capping. Images were taken after 5 min of Con A addition.
Since the Con A capping process has been postulated to be mediated through the cytoskeletal structures (de Priester et al., 1990; Espinosa-Cantellano et al., 1994), the above result may indicate a close association between chemoattractant receptors and the cytoskeleton. We were interested in knowing whether receptor phosphorylation and cell desensitization would affect the capacity of the receptor to be capped by Con A treatment, hence reflecting a corresponding change of this presumptive association. Towards this end, we treated two identical sets of cells with either buffer (Fig. 7 B, 1) or 10−5 M cAMP (Fig. 7 B, 2) for 10 min to induce receptor desensitization. Con A was then added to both sets of cells and changes in receptor distribution were monitored. The photographs were taken after 5 min of Con A treatment. According to the previous test, this time should be sufficient for capping to take place. This experiment clearly demonstrated that the receptors on completely desensitized cells were still able to be efficiently capped. Further study also indicated that the time frames of the capping process were not significantly different between the two sets of cells (data not shown).
Discussion
We were able to study the details of the cellular distribution of a chemoattractant receptor during chemotaxis and under constant ligand stimulation in real time within motile cells by the use of a GFP fusion protein. Our study represents the first application of the GFP fusion technology towards the study of a GPCR. In randomly moving cells, the receptor is evenly distributed throughout the plasma membrane; and during chemotaxis, the distribution undergoes minimal change. If challenged with an uniform increase in cAMP, which is sufficient to induce receptors phosphorylation, a decrease in affinity, and cellular desensitization, the localization profile remains essentially unaltered. The receptor is not absolutely static on cell surface since Con A induces its dramatic redistribution into a polar cap. Thus, while both coronin, a cytoplasmic actin-associated protein, and filamentous actin (F-actin) accumulate at the migration front during chemotaxis, the chemoattractant receptor showed no overall rearrangement in distribution. This indicates that local populations of receptors are sufficient for the signal transduction processes required to redistribute the cytoskeletal proteins and that global mobilization is unnecessary. Often cells display a strong degree of polarization upon gradient reversals. Our results demonstrate that this gradient-induced polarization is not due to receptor redistribution.
We observed that the frequently extended and retracted pseudopods and filopods in the chemotactic cells are uniformly covered with chemoattractant receptors (Figs. 3 and 4, and data not shown). These observations support a “pilot pseudopod” model of chemotaxis (Gerisch et al., 1975; Devreotes and Zigmond, 1988). In this model, cells constantly extend pseudopods in various directions: those in the correct direction are more likely to persist and become part of the migrating front; those in the wrong direction are more easily suppressed. Since the receptors coat these projections, we can speculate that receptor-mediated sensing mechanisms are continuously involved in these decisions. As the cell thrusts a portion of its periphery into a chemoattractant gradient to form a pseudopod, receptors on this protrusion experience an increase in agonist occupancy leading to recruitment of further cytoskeletal and cytoplasmic components into this region. Conversely, receptors on filopods or pseudopods extending down the gradient or in other directions will experience a decrease or no increase in occupancy, so no further recruitment of cellular material into this domain would occur. These events could bias orientations and overall directions of cell migration.
In certain instances (Fig. 3), we noticed slightly reduced signals on some of the migration front pseudopods. This may be simply due to a transient thinning of the cell front caused by pseudopod protrusion. Alternatively, the protruded, streched out membrane may initially have a lower density of receptors that is quickly replenished by receptors flowing in from adjacent zones. Further confocal studies of chemotaxing cells (at the migration front) will be required to resolve this issue. It appears that generation of a pseudopod corresponds to simple protrusion of a portion of cell plasma membrane rather than deposit of new materials, and when pseudopods retract, the extended membrane directly falls back into surrounding membranes.
We fused GFP to the COOH terminus of cAR1, and this apparently did not interfere with either signal sequence recognition or correct translocation of its multiple membrane-spanning domains. Furthermore, the fusion receptor retains most of the biochemical properties and physiological functions of cAR1. This suggests that the COOH terminus of a GPCR can be highly tolerant of genetic manipulation. Besides allowing real time imaging, the GFP fusion technique also avoids fixation-induced artifacts inherent in conventional immunohistochemical techniques.
The distribution of the receptor did not depend on its expression level. Our observations were obtained in a cell line that overexpresses the fusion protein by less than onefold compared with optimally developed wild-type cells. A similar level of overexpression has been observed for wild-type cAR1 in the same expression vector. However, individual cells displayed fluorescent intensities that varied about fivefold (Fig. 4). Previous immunoperoxidase (HRP) labeling study of cAR1 also showed a wide range of labeling intensities among different cells (Xiao and Devreotes, 1997). We do not know the cause of this expression level polymorphism, but it was useful in analyzing the results. We carefully observed both bright and less intense cells. These should have been receptor populations both greater than and less than wild-type cells. Nevertheless, the behaviors of cells with high or low receptor levels were indistinguishable (Fig. 4). This indicates that our observations are not complicated by overexpression-caused artifacts.
The uniform distribution of the chemoattractant receptors on the plasma membrane may be actively maintained. D. discoideum cells are vigorous phagocytes, similar to macrophages; the entire cell membrane turns over within 45 min, mainly through endocytosis and vesicular trafficking. Several mechanisms may preserve the uniform distribution of the receptor in the face of this dynamic membrane turnover. An active sorting program may restrict cAR1 from the internalizing vesicles; or a retrieval system may quickly return cAR1 back to plasma membrane. Earlier studies showed that cAR1 can be quantitatively recovered from detergent lysates of cells as bilayered membrane fragments. These domains are highly enriched in sterol but poor in phospholipids (Xiao and Devreotes, 1997). Studies have shown that the sterol content of the plasma membrane can have a dramatic impact on sorting and trafficking of membrane proteins. Stieger et al. (1994) have been able to disrupt the asymmetric distributions of various apical and basolateral membrane markers in rat hepatocytes by altering the cholesterol content of cellular membranes. If the high sterol content or low phospholipids level inhibits the recruitment of these domains into endocytic vesicles, the persistent presence of cAR1 on plasma membranes might be expected.
The apparently static distribution of receptor on cell surface should not be confused with an absolute immobility. Rather, this only means that there is no net movement of receptor within the membrane or insertion and removal from the membrane. Individual receptor molecules may be very laterally mobile and involved in active intra-membrane protein trafficking. It will be interesting to compare the lateral mobility of the receptor with that of other membrane proteins that do display visible translocations, and to compare mobilities of receptors at front and rear ends of the same chemotacting cell. It will be equally intriguing to compare the mobility of receptors in resting cells with that of desensitized cells. We are currently attempting to address these issues through fluorescence photobleaching recovery techniques.
The Con A capping test showed that the uniformity of receptor distribution is only relative, and its gross rearrangement can take place during capping. Cytoskeletal components, such as myosin and actin, have been shown to be required for this capping process. We speculate that cAR1 is associated with cytoskeletal proteins that underlie the plasma membrane or with other surface glycoproteins. cAR1 itself does not appear to be glycosylated. The chemoattractant-induced phosphorylation of cAR1 does not interfere with its response to Con A. Hence, if the capping is due to the interaction of receptor with another component, this interaction is not perturbed during desensitization.
There have been many studies on the fate of desensitized GPCRs after persistent stimulation (Chuang et al., 1996; Freedman and Lefkowitz, 1996; Lohse et al., 1996). The most frequently discussed phenomena include internalization through endosomal-like structures or “sequestration” into coated or noncoated vesicles near the cell surface. Typically, only a small fraction (15–25%) of receptors is “sequestered.” It is not clear whether receptor phosphorylation is required for this process (von Zastrow and Kobilika, 1992; Barak et al., 1994; Roettger et al., 1995a ). Our study indicates that cAR1 does not undergo a significant rearrangement during desensitization, although we cannot rule out slight changes. Most of the receptor remains at the cell surface but with dramatically reduced affinity. It has been shown definitively that phosphorylation of two serine residues on cAR1 is absolutely required for this affinity decrease (Caterina et al., 1995).
Two previous studies have proposed that desensitized GPCR remains on the cell surface in special plasma membrane microdomains. The CCK (Cholecystokinin) receptor, a GPCR involved in the hormonal regulation of zymogen secretion from pancreatic acinar cells after ingestion of a meal, appears to desensitize through a process termed “insulation,” whereby the full complement of receptor remains on the plasma membrane but with greatly reduced lateral mobility (Roettger et al., 1995b ). Studies on the N-formyl chemotactic peptide receptors in human neutrophils also revealed that while basal state receptors reside on the regular plasma membrane fraction, desensitized receptors are segregated into different lateral microdomains of the plasma membrane enriched in cytoskeletal components and devoid of G proteins (Jesaitis et al., 1988, 1989). In both cases, the proposed major functions for these domains are to prevent the coupling between the desensitized receptor with G proteins and facilitate the resensitization processes of receptors. However, in neither case were these presumptive domains morphologically observable.
In our case, both basal and desensitized forms of cAR1 display a diffuse pattern of localization on plasma membrane through the current GFP fluorescence study and previous immunogold electron microscopy studies (data not shown). Adapted receptors remain on surface membranes; this may provide the cells with greater flexibility and sensitivity. No de novo synthesis or recruitment of internal receptors is necessary; deadaptation can be accomplished on the plasma membrane.
Acknowledgments
We wish to thank Stephen M. Mattessich for technical help during the micropipette assay and Dr. Roger Tsien for providing the GFP S65T construct.
This work is supported by the National Institutes of Health Grant GM34933 to P.N. Devreotes.
Abbreviations used in this paper
- F-actin
filamentous actin
- GFP
green fluorescent protein
- GPCR
G protein-coupled receptor
- LLB
loss-of-ligand binding
Footnotes
Address all correspondence to Peter N. Devreotes, Department of Biological Chemistry, School of Medicine, Johns Hopkins University, Baltimore, MD 21205. Tel.: (410) 955-3225. Fax: (410) 955-5757.
References
- Baiocchi M, Olivetta E, Chelucci C, Santarcangelo AC, Bona R, d'Aloja P, Testa U, Komatsu N, Verani P, Federico M. Human immunodeficiency virus (HIV)-resistant CD4+ UT-7 megakaryocytic human cell line becomes highly HIV-1 and HIV-2 susceptible upon CXCR4 transfection: induction of cell differentiation by HIV-1 infection. Blood. 1997;89:2670–2678. [PubMed] [Google Scholar]
- Barak LS, Tiberi M, Freedman NJ, Kwatra MM, Lefkowitz RJ, Caron MG. A highly conserved tyrosine residue in G protein-coupled receptors is required for agonist-mediated β 2-adrenergic receptor sequestration. J Biol Chem. 1994;269:2790–2795. [PubMed] [Google Scholar]
- Behar TN, Schaffner AE, Tran HT, Barker JL. Correlation of gp140trk expression and NGF-induced neuroblast chemotaxis in the embryonic rat spinal cord. Brain Res. 1994;664:155–166. doi: 10.1016/0006-8993(94)91966-6. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Devreotes PN, Borleis J, Hereld D. Agonist- induced loss of ligand binding is correlated with phosphorylation of cAR1, a G protein-coupled chemoattractant receptor from Dictyostelium. . J Biol Chem. 1995;270:8667–8672. doi: 10.1074/jbc.270.15.8667. [DOI] [PubMed] [Google Scholar]
- Chen X, Berrou J, Nguyen G, Sraer JD, Rondeau E. Endothelin-1 induces rapid and long lasting internalization of the thrombin receptor in human glomerular epithelial cells. Biochem Biophys Res Commun. 1995;217:445–451. doi: 10.1006/bbrc.1995.2796. [DOI] [PubMed] [Google Scholar]
- Chen MY, Insall RH, Devreotes PN. Signaling through chemoattractant receptors in Dictyostelium. . Trends Genet. 1996;12:52–57. doi: 10.1016/0168-9525(96)81400-4. [DOI] [PubMed] [Google Scholar]
- Chuang TT, Iacovelli L, Sallese M, De Blasi A. G protein-coupled receptors: heterologous regulation of homologous desensitization and its implications. Trends Pharmacol Sci. 1996;17:416–421. doi: 10.1016/s0165-6147(96)10048-1. [DOI] [PubMed] [Google Scholar]
- Cubitt AB, Heim R, Adams SR, Boyd AE, Gross LA, Tsien RY. Understanding, improving and using green fluorescent proteins. Trends Biochem Sci. 1995;20:448–455. doi: 10.1016/s0968-0004(00)89099-4. [DOI] [PubMed] [Google Scholar]
- de Priester W, Bakker A, Lamers G. Capping of Con A receptors and actin distribution are influenced by disruption of microtubules in Dictyostelium discoideum. . Eur J Cell Biol. 1990;51:23–32. [PubMed] [Google Scholar]
- Devreotes PN. G protein-linked signaling pathways control the developmental program of Dictyostelium. . Neuron. 1994;12:235–241. doi: 10.1016/0896-6273(94)90267-4. [DOI] [PubMed] [Google Scholar]
- Devreotes PN, Zigmond SH. Chemotaxis in eukaryotic cells: a focus on leukocytes and Dictyostelium. . Annu Rev Cell Biol. 1988;4:649–686. doi: 10.1146/annurev.cb.04.110188.003245. [DOI] [PubMed] [Google Scholar]
- Devreotes, P.N., D. Fontana, P. Klein, J. Sherring, and A. Theibert. 1987. Transmembrane signaling in Dictyostelium. In Methods in Cell Biology. Vol. 28. J. Spudich, editor. Academic Press, Inc., New York. 299–329. [DOI] [PubMed]
- Espinosa-Cantellano M, Martinez-Palomo A. Entamoeba histolytica: mechanism of surface receptor capping. Exp Parasitol. 1994;79:424–435. doi: 10.1006/expr.1994.1104. [DOI] [PubMed] [Google Scholar]
- Fisher PR, Merkl R, Gerisch G. Quantitative analysis of cell motility and chemotaxis in Dictyostelium discoideumby using an image processing system and a novel chemotaxis chamber providing stationary chemical gradients. J Cell Biol. 1989;108:973–984. doi: 10.1083/jcb.108.3.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman NJ, Lefkowitz RJ. Desensitization of G protein-coupled receptors. Recent Prog Horm Res. 1996;51:319–351. [PubMed] [Google Scholar]
- Gerisch G, Keller HU. Chemotactic reorientation of granulocytes stimulated with micropipettes containing fMet-Leu-Phe. J Cell Sci. 1981;52:1–10. doi: 10.1242/jcs.52.1.1. [DOI] [PubMed] [Google Scholar]
- Gerisch G, Hulser D, Malchow D, Wick U. Cell communication by periodic cyclic AMP pulses. Philos Trans R Soc Lond B Biol Sci. 1975;272:181–192. doi: 10.1098/rstb.1975.0080. [DOI] [PubMed] [Google Scholar]
- Gerisch G, Albrecht R, Heizer C, Hodgkinson S, Maniak M. Chemoattractant-controlled accumulation of coronin at the leading edge of Dictyosteliumcells monitored using a green fluorescent protein-coronin fusion protein. Curr Biol. 1995;5:1280–1285. doi: 10.1016/s0960-9822(95)00254-5. [DOI] [PubMed] [Google Scholar]
- Hampton RY, Koning A, Wright R, Rine J. In vivo examination of membrane protein localization and degradation with green fluorescent protein. Proc Natl Acad Sci USA. 1996;93:828–833. doi: 10.1073/pnas.93.2.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan SP, Foster PS. Cellular and molecular mechanisms involved in the regulation of eosinophil trafficking in vivo. Med Res Rev. 1996;16:407–432. doi: 10.1002/(SICI)1098-1128(199609)16:5<407::AID-MED2>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Hunyady L, Bor M, Balla T, Catt KJ. Identification of a cytoplasmic Ser-Thr-Leu motif that determines agonist-induced internalization of the AT1 angiotensin receptor. J Biol Chem. 1994;269:31378–31382. [PubMed] [Google Scholar]
- Jesaitis AJ, Bokoch GM, Tolley JO, Allen RA. Lateral segregation of neutrophil chemotactic receptors into actin- and fodrin-rich plasma membrane microdomains depleted in guanyl nucleotide regulatory proteins. J Cell Biol. 1988;107:921–928. doi: 10.1083/jcb.107.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesaitis AJ, Tolley JO, Bokoch GM, Allen RA. Regulation of chemoattractant receptor interaction with transducing proteins by organizational control in the plasma membrane of human neutrophils. J Cell Biol. 1989;109:2783–2790. doi: 10.1083/jcb.109.6.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RL, Vaughan RA, Caterina MJ, Van Haastert PJ, Devreotes PN. Overexpression of the cAMP receptor 1 in growing Dictyosteliumcells. Biochemistry. 1991;30:6982–6986. doi: 10.1021/bi00242a025. [DOI] [PubMed] [Google Scholar]
- Johnson RL, Van Haastert PJ, Kimmel AR, Saxe CL, III, Jastorff B, Devreotes PN. The cyclic nucleotide specificity of three cAMP receptors in Dictyostelium. . J Biol Chem. 1992;267:4600–4607. [PubMed] [Google Scholar]
- Kim JY, Devreotes PN. Random chimeragenesis of G protein-coupled receptors. Mapping the affinity of the cAMP chemoattractant receptors in Dictyostelium. . J Biol Chem. 1994;269:28724–28731. [PubMed] [Google Scholar]
- Klein P, Theibert A, Fontana D, Devreotes PN. Identification and cyclic AMP-induced modification of the cyclic AMP receptor in Dictyostelium discoideum. . J Biol Chem. 1985;260:1757–1764. [PubMed] [Google Scholar]
- Lohse, M.J., S. Engelhardt, S. Danner, and M. Bohm. 1996. Mechanisms of β-adrenergic receptor desensitization: from molecular biology to heart failure. Basic Res. Cardiol. 91(Suppl):29–34. [DOI] [PubMed]
- Maloteaux JM, Hermans E. Agonist-induced muscarinic cholinergic receptor internalization, recycling and degradation in cultured neuronal cells. Cellular mechanisms and role in desensitization. Biochem Pharmacol. 1994;47:77–88. doi: 10.1016/0006-2952(94)90439-1. [DOI] [PubMed] [Google Scholar]
- Maniak M, Rauchenberger R, Albrecht R, Murphy J, Gerisch G. Coronin involved in phagocytosis: dynamics of particle-induced relocalization visualized by a green fluorescent protein Tag. Cell. 1995;83:915–924. doi: 10.1016/0092-8674(95)90207-4. [DOI] [PubMed] [Google Scholar]
- Marshall J, Molloy R, Moss GW, Howe JR, Hughes TE. The jellyfish green fluorescent protein: a new tool for studying ion channel expression and function. Neuron. 1995;14:211–215. doi: 10.1016/0896-6273(95)90279-1. [DOI] [PubMed] [Google Scholar]
- Patton WF, Dhanak MR, Jacobson BS. Analysis of plasma membrane protein changes in Dictyostelium discoideumduring concanavalin A induced receptor redistribution using two-dimensional gel electrophoresis. Electrophoresis. 1990;11:79–85. doi: 10.1002/elps.1150110116. [DOI] [PubMed] [Google Scholar]
- Petty HR, Francis JW. Polymorphonuclear leukocyte histamine receptors: occurrence in cell surface clusters and their redistribution during locomotion. Proc Natl Acad Sci USA. 1986;83:4332–4335. doi: 10.1073/pnas.83.12.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins, J.P., W.P. Hausdorff, and R.J. Lefkowitz. 1991. The β-Adrenergic receptor. J.P. Perkins, editor. Humana Press, Clifton, NJ. 125–180.
- Prado GN, Suzuki H, Wilkinson N, Cousins B, Navarro J. Role of the C terminus of the interleukin 8 receptor in signal transduction and internalization. J Biol Chem. 1996;271:19186–19190. doi: 10.1074/jbc.271.32.19186. [DOI] [PubMed] [Google Scholar]
- Raposo G, Dunia I, Marullo S, Andre C, Guillet JG, Strosberg AD, Benedetti EL, Hoebeke J. Redistribution of muscarinic acetylcholine receptors on human fibroblasts induced by regulatory ligands. Biol Cell. 1987;60:117–123. doi: 10.1111/j.1768-322x.1987.tb00551.x. [DOI] [PubMed] [Google Scholar]
- Roettger BF, Rentsch RU, Pinon D, Holicky E, Hadac E, Larkin JM, Miller LJ. Dual pathways of internalization of the cholecystokinin receptor. J Cell Biol. 1995a;128:1029–1041. doi: 10.1083/jcb.128.6.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roettger BF, Rentsch RU, Hadac EM, Hellen EH, Burghardt TP, Miller LJ. Insulation of a G protein-coupled receptor on the plasmalemmal surface of the pancreatic acinar cell. J Cell Biol. 1995b;130:579–590. doi: 10.1083/jcb.130.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P, Chou JH, Bargmann CI. ord-10 encodes a seven transmembrane domain olfactory receptor required for responses to the odorant diacetyl. Cell. 1996;84:899–909. doi: 10.1016/s0092-8674(00)81068-5. [DOI] [PubMed] [Google Scholar]
- Stieger B, Meier PJ, Landmann L. Effect of obstructive cholestasis on membrane traffic and domain-specific expression of plasma membrane proteins in rat liver parenchymal cells. Hepatology. 1994;20:201–212. doi: 10.1016/0270-9139(94)90154-6. [DOI] [PubMed] [Google Scholar]
- Strader C, Fong T, Graziano M, Tota M. The family of G-protein-coupled receptors. FASEB J. 1995;9:745–754. [PubMed] [Google Scholar]
- Swanson JA, Taylor DL. Local and spatially coordinated movements in Dictyostelium discoideumamoebae during chemotaxis. Cell. 1982;28:225–232. doi: 10.1016/0092-8674(82)90340-3. [DOI] [PubMed] [Google Scholar]
- Szekanecz Z, Shah MR, Harlow LA, Pearce WH, Koch AE. Interleukin-8 and tumor necrosis factor-α are involved in human aortic endothelial cell migration. The possible role of these cytokines in human aortic aneurysmal blood vessel growth. Pathobiology. 1994;62:134–139. doi: 10.1159/000163891. [DOI] [PubMed] [Google Scholar]
- von Zastrow M, Kobilka BK. Ligand-regulated internalization and recycling of human β 2-adrenergic receptors between the plasma membrane and endosomes containing transferrin receptors. J Biol Chem. 1992;267:3530–3538. [PubMed] [Google Scholar]
- Wang M, Van Haastert PJ, Devreotes PN, Schaap P. Localization of chemoattractant receptors on Dictyostelium discoideumcells during aggregation and down-regulation. Dev Biol. 1988;128:72–77. doi: 10.1016/0012-1606(88)90268-0. [DOI] [PubMed] [Google Scholar]
- Wells TN, Proudfoot AE, Power CA, Marsh M. Chemokine receptors: the new frontier for AIDS research. Chem Biol. 1996;3:603–609. doi: 10.1016/s1074-5521(96)90126-x. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Devreotes PN. Identification of detergent-resistant plasma membrane microdomains in Dictyostelium: enrichment of signal transduction proteins. Mol Biol Cell. 1997;8:855–869. doi: 10.1091/mbc.8.5.855. [DOI] [PMC free article] [PubMed] [Google Scholar]