

Abstract
ADP-ribosylation factor (ARF) 6 localizes to the plasma membrane (PM) in its GTP state and to a tubulovesicular compartment in its GDP state in HeLa cells that express wild-type or mutant forms of this GTPase. Aluminum fluoride (AlF) treatment of ARF6-transfected cells redistributes ARF6 to the PM and stimulates the formation of actin-rich surface protrusions. Here we show that cytochalasin D (CD) treatment inhibited formation of the AlF-induced protrusions and shifted the distribution of ARF6 to a tubular membrane compartment emanating from the juxtanuclear region of cells, which resembled the compartment where the GTP-binding defective mutant of ARF6 localized. This membrane compartment was distinct from transferrin-positive endosomes, could be detected in the absence of ARF6 overexpression or CD treatment, and was accessible to loading by PM proteins lacking clathrin/AP-2 cytoplasmic targeting sequences, such as the IL-2 receptor α subunit Tac. ARF6 and surface Tac moved into this compartment and back out to the PM in the absence of pharmacologic treatment. Whereas AlF treatment blocked internalization, CD treatment blocked the recycling of wild-type ARF6 and Tac back to the PM; these blocks were mimicked by expression of ARF6 mutants Q67L and T27N, which were predicted to be in either the GTP- or GDP-bound state, respectively. Thus, the ARF6 GTP cycle regulates this membrane traffic pathway. The delivery of ARF6 and membrane to defined sites along the PM may provide components necessary for remodeling the cell surface and the underlying actin cytoskeleton.
Eukaryotic cells internalize material from the external environment through a variety of distinct endocytic pathways (Steinman et al., 1983). These pathways include clathrin-dependent endocytosis (Mellman, 1996) and a variety of clathrin-independent endocytic processes including pinocytosis (Sandvig and van Deurs, 1994; Lamaze and Schmid, 1995), macropinocytosis (Swanson and Watts, 1995), and phagocytosis (Swanson and Baer, 1995). A common feature shared by these pathways is that once cargo is delivered to its cellular destination, much of the internalized membrane is recycled back to the plasma membrane (PM).1 Studies of endocytosis using fluorescent lipid analogues and human transferrin (Koval and Pagano, 1989; Mayor et al., 1993) have shown that most of the membrane taken up by cells is returned to the cell surface. Although much of our knowledge about endocytic membrane recycling has come from studies of the clathrin-mediated transferrin receptor cycle (Gruenberg and Maxfield, 1995), it is not clear whether all recycling membrane returns to the cell surface along the same pathway as the transferrin receptor.
Small ras-related GTPases have been implicated in the regulation of endocytic membrane recycling (Gruenberg and Maxfield, 1995; Mellman, 1996). In particular, the rab family GTPases, rab4 and rab11, have been implicated in the recycling of transferrin receptors. After the release of iron, transferrin bound to transferrin receptor recycles back to the PM either rapidly from “sorting” endosomes or more slowly from a perinuclear compartment termed the “recycling” endosome (Hopkins and Trowbridge, 1983; Yamashiro et al., 1984; Hopkins et al., 1994). Rab4 is thought to regulate rapid recycling from sorting endosomes (van der Sluijs et al., 1992), and rab11 has been implicated in traffic between the sorting and recycling endosomes (Ullrich et al., 1996). It is not known whether rab proteins are also involved in the recycling of membrane internalized by other endocytic pathways or whether other regulators are involved.
The ADP-ribosylation factor (ARF) family of proteins represent another group of small GTPases that are thought to function as regulators of membrane traffic (Donaldson and Klausner, 1994; Moss and Vaughan, 1995). ARF proteins, originally identified as cofactors in the cholera toxin– catalyzed ADP ribosylation of Gs α (Kahn and Gilman, 1986), have been identified in all eukaryotes tested so far (Kahn et al., 1991) and are widely expressed in most mammalian tissues (Tsuchiya et al., 1991). ARFs also stimulate phospholipase D activity in vitro (Brown et al., 1993; Cockroft et al., 1994; Massenburg et al., 1994; Hammond et al., 1995), and a recent study suggests that this interaction may be important for ARF1 function at the Golgi complex (Ktistakis et al., 1996). Among the five known human ARF proteins, ARF1 is the most thoroughly studied and plays a critical role in the secretory pathway. Both in vivo and in vitro studies have demonstrated that ARF1 cycles between the cytosol (GDP form) and the Golgi complex (GTP form), where it mediates the binding of soluble coat complexes to Golgi membranes (Donaldson et al., 1992a ; Robinson and Kreis, 1992; Palmer et al., 1993; Traub et al., 1993). The regulated cycle of coat assembly/ disassembly is necessary to maintain both the structural integrity of the Golgi complex and transport along the secretory pathway (Melançon et al., 1987; Donaldson et al., 1991; Tanigawa et al., 1993; Dascher and Balch, 1994; Teal et al., 1994; Zhang et al., 1994). In contrast to ARF1, less is known about the functions of the other ARF proteins.
ARF6 is the most divergent member of the ARF family and, unlike ARF1, appears to function in the peripheral plasma membrane/endosomal system (D'Souza-Schorey et al., 1995; Peters et al., 1995; Radhakrishna et al., 1996). We have been studying the function of ARF6 by examining its localization and the cellular phenotypes conferred by transient expression of either the wild-type or mutant forms in mammalian cells (Peters et al., 1995; Radhakrishna et al., 1996). Peters et al. (1995) showed that wild-type ARF6 containing an HA epitope tag localizes along the cytoplasmic face of the plasma membrane and to an internal, tubulovesicular compartment. The morphology of cells expressing the HA-tagged, wild-type ARF6 appears normal. A mutant of ARF6 (Q67L) predicted to be defective in GTP hydrolysis, and thus mainly in the GTP-bound, active form, localizes exclusively to the PM and results in the formation of elaborate and protrusive PM extensions. Another mutant of ARF6 (T27N), predicted to be defective in GTP binding, and thus predominantly in the GDP-bound, inactive state, is mostly localized to the internal, tubulovesicular compartment. By immunoelectron microscopy, these tubulovesicular structures appear to be abundant within the cells (Peters et al., 1995).
More recently, we have studied the effector function of ARF6 by identifying pharmacologic reagents that shift the distribution, and presumably the GTP status, of wild-type ARF6 after transient expression in HeLa cells (Radhakrishna et al., 1996). We reasoned that whereas the mutant forms of ARF6 would be locked into either the GTP or GDP states, the wild-type protein should be capable of cycling between these two nucleotide states. We found that the G protein activator, aluminum fluoride (AlF) (Sternweis and Gilman, 1982), shifts hemagglutinin (HA)-tagged, wild-type ARF6 to the plasma membrane in transiently transfected HeLa cells and stimulates the formation of actin-rich surface protrusions (Radhakrishna et al., 1996) that are distinct from the actin rearrangements stimulated by the rho-related GTPases rac1 or rhoA. Interestingly, whereas there is an apparent increase in macropinocytosis at these protrusive sites, no effect on transferrin endocytosis is observed (Radhakrishna et al., 1996). The AlF-stimulated protrusions seen in cells expressing wild-type ARF6 resemble, in part, those observed in cells expressing ARF6/Q67L alone. This suggests that AlF treatment may result in the accumulation of ARF6-GTP at the PM, consistent with the observation that AlF can protect ARF1-GTP from GTP hydrolysis (Finazzi et al., 1994).
We have now sought to identify reagents that would shift wild-type ARF6 into the inactive, GDP-bound state. Although brefeldin A (BFA) inhibits the activation of ARF1 (Donaldson et al., 1992b ; Helms and Rothman, 1992; Randazzo et al., 1993), it has no effect on ARF6 (Peters et al., 1995; Cavenagh et al., 1996; Radhakrishna et al., 1996). An inhibitor of ARF6 activation would be expected to block AlF-induced protrusions and to result in the accumulation of ARF6 in the internal, tubulovesicular compartment. We previously showed that treatment of cells with inhibitors of actin polymerization, such as cytochalasin D (CD), prevented the AlF-induced response (Radhakrishna et al., 1996). In this study, we use CD treatment and mutants of ARF6 to demonstrate that ARF6 regulates through its GDP/GTP cycle the movement of PM into and out of a novel recycling compartment.
MATERIALS AND METHODS
Cells, Reagents, and Antibodies
HeLa cells were grown in DME supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 500 μg/ml gentamycin sulfate at 37°C with 5% CO2. Rabbit polyclonal antibodies were raised against a COOH-terminal peptide of ARF6, residues 164–175. The antiserum specifically recognizes ARF6 in transfected cells and on immunoblots; ARF6-specific labeling during immunofluorescence or immunoblotting was blocked by the immunizing peptide. A mouse mAb (16B12) against the influenza HA epitope was purchased from BabCo (Berkeley, CA). The human Tac antigen (IL-2 receptor α subunit) was detected with mouse monoclonal anti-Tac antibodies (7G7; Rubin et al., 1985). Rabbit antibodies to human transferrin were purchased from Boehringer Mannheim Biochemicals (Indianapolis, IN). Mouse antibodies against human MHC class I, W6/32 were kindly provided by Dr. Paul Roche (National Institutes of Health, Bethesda, MD). Fluorescein-conjugated WGA and Oregon green–labeled phalloidin were obtained from Molecular Probes, Inc. (Eugene, OR). Fluorescein- and rhodamine-conjugated donkey anti–mouse and donkey anti–rabbit IgG were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). All other reagents, including iron-saturated human transferrin, CD, and latrunculin B, were purchased from Sigma Chemical Co. (St. Louis, MO).
DNA Manipulations and Transient Transfections
PCR (Perkin-Elmer Cetus Instruments, Norwalk, CT) was used to remove DNA sequences encoding a COOH-terminal HA epitope tag from the cDNAs encoding human ARF6/T27N and ARF6/Q67L generated previously (Peters et al., 1995). A DNA fragment from the ClaI site at 286 bp to the end of ARF6, containing a stop codon and a BglII site, was amplified by PCR and digested with ClaI/BglII. This fragment was used to replace the corresponding fragment in the epitope-tagged T27N and Q67L mutants of ARF6. The sequences of all constructs, including regions generated by PCR, were confirmed by DNA sequencing (SeqWright DNA Sequencing, Houston, TX). A modified pCDL-SRα expression vector (termed pXS; Takebe et al., 1988) was used to express wild-type and mutant ARF6, as well as Tac cDNAs in HeLa cells.
HeLa cells were grown on glass coverslips and transfected using the calcium phosphate method as described previously (Bonifacino et al., 1989). Cotransfections of ARF6 and Tac were performed using 10 μg of each plasmid in 10-cm dishes. Under these conditions, the level of ARF6 overexpression was estimated to be 10–40-fold over the endogenous protein.
Immunofluorescence Microscopy
30–36 h after transfection, transfected cells were treated as indicated, fixed with 2% formaldehyde in PBS for 10 min at room temperature, and rinsed with 10% FBS and 0.02% azide in PBS (PBS/serum). Cells were incubated with primary antibodies diluted in PBS/serum containing 0.2% saponin for 1 h at room temperature and were washed (three times, 5 min each) with PBS/serum. The cells were then incubated with fluorescently labeled secondary antibodies diluted in PBS/serum plus 0.2% saponin for 1 h, washed again, and mounted on glass slides.
Internalization of Transferrin
Transfected cells were rinsed briefly three times with 0.5% BSA in DME, and then incubated in the same medium for 30 min at 37°C in the absence or presence of the indicated drugs. Cells were then incubated with 30 μg/ ml of iron-saturated human transferrin in the continued presence of drugs for an additional 30 min at 37°C, rinsed quickly three times with DME containing 10% FBS, fixed, and processed for immunofluorescence as above.
Internalization and Recycling of Anti-Tac Antibodies
For Tac antibody internalization, cells cotransfected with Tac and ARF6 constructs were chilled to 4°C and incubated with mouse anti-Tac antibodies for 30 min in an ice/water bath. The cells were rinsed briefly with ice-cold DME containing 10% FBS (complete medium), and they were incubated with prewarmed (37°C) complete medium in the absence or presence of the indicated drugs for 30 min at 37°C. Cells were then fixed either immediately or after washing with low pH buffer (Klausner et al., 1983). To remove anti-Tac antibody remaining at the surface, the cells were chilled to 4°C, rinsed three times quickly with 0.5% acetic acid, 0.5 M NaCl, pH 3.0 (low pH buffer), and then three times with ice-cold complete medium before fixation.
To detect ARF6 and the internalized Tac antibody, fixed cells were labeled with rabbit anti-ARF6 antiserum, washed, and then incubated with fluorescently labeled donkey anti–rabbit IgG to detect ARF6 staining, and incubated with the appropriate fluorescently labeled donkey anti– mouse IgG to detect the internalized Tac antibody. To detect Tac antibody remaining at the cell surface, some cells were incubated with fluorescently labeled donkey anti–mouse IgG in the absence of saponin; the cells were then washed and incubated in the presence of saponin with anti-ARF6 antibodies, followed by the appropriate fluorescently labeled donkey anti–rabbit IgG.
To monitor the recycling of Tac antibody, cells cotransfected with Tac and ARF6 constructs were labeled with mouse anti-Tac antibodies at 4°C as described above, and were then warmed to 37°C for 30 min either in the absence or presence of 1 μM CD. The cells were chilled to 4°C, quickly washed with low pH buffer, as described above, to remove Tac antibody remaining at the cell surface, and were warmed again to 37°C for 30 min in the absence or presence of CD before fixation. The surface reappearance of Tac antibody was detected by incubating the fixed cells with fluorescently labeled donkey anti–mouse IgG in the absence of saponin; ARF6 was then labeled with anti-ARF6 antiserum and the appropriate secondary antibodies in the presence of saponin.
RESULTS
CD Redistributes ARF6 from the PM to a Novel, Tubular Compartment That Resembles the Compartment Where the GTP Binding–defective Mutant of ARF6, T27N, Resides
We previously observed that inhibition of actin polymerization, by treatment of cells with CD, inhibited the AlF-induced protrusions in HeLa cells overexpressing ARF6, and additionally, we noticed that CD treatment shifted the distribution of ARF6 to an internal membrane compartment (Radhakrishna et al., 1996). To further investigate this response to CD, HeLa cells transfected with plasmid encoding ARF6 were treated with CD for 30 min. The cells were then fixed and immunolabeled with polyclonal anti-ARF6 antibodies, and the actin filaments were labeled with Oregon green–conjugated phalloidin. In untreated cells, ARF6 localized along the PM at the peripheral edge of cells and in internal structures near the nucleus. Fine tubular elements were observed emanating out of the juxtanuclear region towards the peripheral edge of some cells (Fig. 1, untreated). The extent to which these tubular elements were observed in untreated cells varied (for example, see tubular elements in Figs. 4 and 5); often they were difficult to discern over the cell surface labeling.
Figure 1.

CD redistributes ARF6 to juxtanuclear tubular structures that resemble the compartment where the GTP-binding defective mutant of ARF6, T27N, resides. HeLa cells transfected with wild-type ARF6 plasmid were either untreated (Untreated), or treated with 1 μM CD for 30 min (CD). Other cells were transfected with the ARF6/T27N plasmid. The cells were then fixed in 2% formaldehyde and labeled with ARF6-specific antiserum, followed by rhodamine-labeled donkey anti–rabbit IgG and Oregon green–labeled phalloidin to visualize actin filaments.
Figure 4.

Endogenous MHC-I localizes to the tubular compartment with or without ARF6 overexpression. HeLa cells were transfected with ARF6 plasmid and then incubated in the absence (Unt) or presence (CD) of 1 μM CD for 30 min at 37°C, and they were fixed and processed for immunofluorescence localization of MHC-I using mouse anti–MHC-I antibodies and for overexpression of ARF6 using antibody to ARF6. Anti–MHC-I was detected with sequential FITC-labeled goat anti–mouse and FITC-labeled donkey anti–goat antibodies, while anti-ARF6 was detected with rhodamine-conjugated donkey anti–rabbit antibody. The tubular structures (arrowheads) radiating from the perinuclear region, labeled with anti–MHC-I antibodies, become more apparent after CD treatment, and are observed in both ARF6-transfected and untransfected cells.
Figure 5.
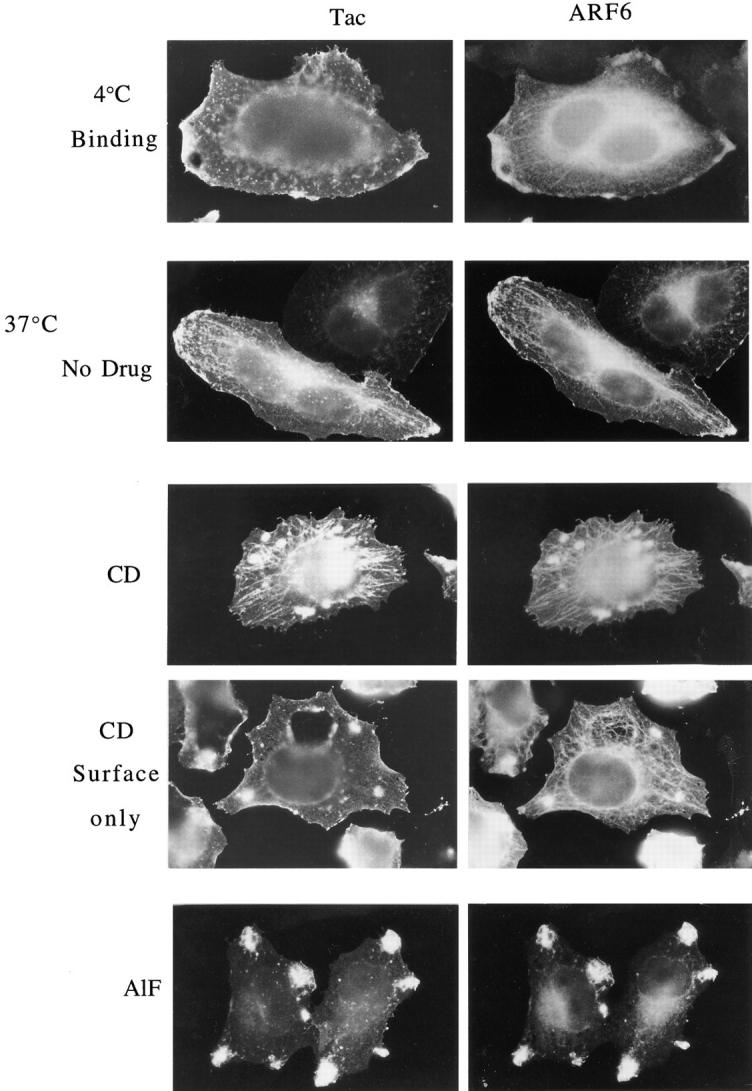
Surface Tac is internalized into and colocalizes with ARF6 in the tubular compartment, and accumulates in this compartment during CD treatment. HeLa cells expressing both ARF6 and Tac were chilled to 4°C and incubated with mouse anti-Tac antibodies (7G7) for 30 min to label surface Tac. After washing to remove excess antibodies, cells were warmed to 37°C for 30 min in the absence (No drug) or presence of 1 μM CD (CD) or AlF (AlF) to permit internalization of the bound anti-Tac antibodies, and were fixed and processed for indirect immunofluorescence. Tac antibodies were detected with fluorescent donkey anti– mouse secondary antibodies and ARF6 was localized with rabbit anti-ARF6 antiserum. In some cells, after CD treatment, Tac antibodies were localized with secondary antibodies in the absence of detergent to detect Tac antibody remaining at the surface (CD Surface only); ARF6 was subsequently localized in these cells after detergent permeabilization. Note that Tac antibodies were internalized into the tubular compartment in the absence of CD treatment (No Drug). During CD treatment, both ARF6 and the internalized Tac antibodies accumulated in the tubular compartment. In contrast, AlF treatment resulted in the sequestration of anti-Tac antibodies and ARF6 in surface protrusions and inhibited movement into the tubular compartment.
A striking enhancement of ARF6 labeling of the tubular structures was observed in cells treated with 1.0 μM CD (Fig. 1, CD). Morphologically, these tubular structures were similar to those observed in the absence of CD, but they were more extensive, suggesting that CD treatment was not creating but merely shifting more ARF6 to these structures. Other inhibitors of actin polymerization, cytochalasin B and latrunculin B (Spector et al., 1989), also resulted in extensive tubular staining for ARF6 (not shown). The effects of CD treatment on ARF6 distribution were fully reversible within 30 min after the removal of CD (not shown). Even 0.1 μM CD was sufficient to shift ARF6 to the tubular structures and inhibit the AlF response, although little change in the appearance of actin filaments was evident (data not shown).
These tubular elements were internal structures, and not invaginations of the PM, as recently described (van Deurs et al., 1996). Surface staining of fixed cells, in the absence of permeabilization, with fluorescent WGA (not shown) or antibodies to PM proteins (e.g., see Fig. 5) did not label the tubular structures. Furthermore, we could demonstrate that these were internal structures that could be loaded from the PM and thus represented an endosomal compartment (see Fig. 5). Double labeling with antibodies to ARF6 and tubulin showed that the tubular compartment was aligned along the microtubules; the ability to observe these tubular structures during CD treatment was diminished when microtubules were disrupted by pretreatment with nocodazole (data not shown).
Immunoelectron microscopy had previously shown that wild-type ARF6 localized to both the PM and internal tubulovesicular membranes (Peters et al., 1995). In contrast, the GTP-binding defective mutant, ARF6/T27N, localized exclusively to tubulovesicular structures. Since CD treatment redistributes wild-type ARF6 from the PM to tubular structures, we examined whether these structures represent the compartment where ARF6/T27N resides. ARF6/T27N localized to tubular structures originating from the perinuclear region as well as to vesicular structures scattered throughout the cell (Fig. 1). These observations suggest that CD treatment of cells expressing wild-type ARF6 causes a shift in the distribution of ARF6 to the tubular compartment where ARF6/T27N localizes.
The ability of CD to enhance the tubular morphology of the ARF6 compartment was reminiscent of the effects of BFA on increasing the tubular morphology of the transferrin receptor endosomal compartment (Hunziker et al., 1991; Lippincott-Schwartz et al., 1991; Wood et al., 1991). To investigate the relationship between these two endosomal compartments, we examined the distribution of ARF6 and transferrin in HeLa cells transfected with ARF6-HA. The cells were incubated with transferrin alone or in the presence of CD (1 μM), BFA (1 μM), or CD plus BFA for 30 min, and were then rinsed in media, fixed and processed for immunofluorescence with antibodies to HA to detect ARF6-HA-transfected cells and with antibodies to transferrin to detect cell-associated transferrin. In untreated cells, transferrin was localized to the perinuclear region and to punctate structures in the periphery, but showed little overlap with ARF6-labeled structures (Fig. 2, untreated). The distribution of transferrin in transfected cells was indistinguishable from that observed in untransfected cells. In the presence of CD, no change in the distribution of transferrin was observed, whereas the ARF6 distribution shifted towards the tubular structures emanating out of the perinuclear region (Fig. 2, CD). After treatment with BFA, the transferrin compartment exhibited tubular structures, but the ARF6 compartment was unaffected and did not colocalize with the transferrin compartment (Fig. 2, BFA). Treatment of cells with both CD and BFA resulted in tubulation of the transferrin endosome and enhanced tubular morphology of the ARF6 endosome, with little overlap between the two systems (Fig. 2, CD+BFA). The ARF6 compartment also did not colocalize with LAMP, a marker for late endosomes and lysosomes (Mellman, 1996), nor with other organelle markers tested, including those recognizing the Golgi complex and ER (data not shown).
Figure 2.
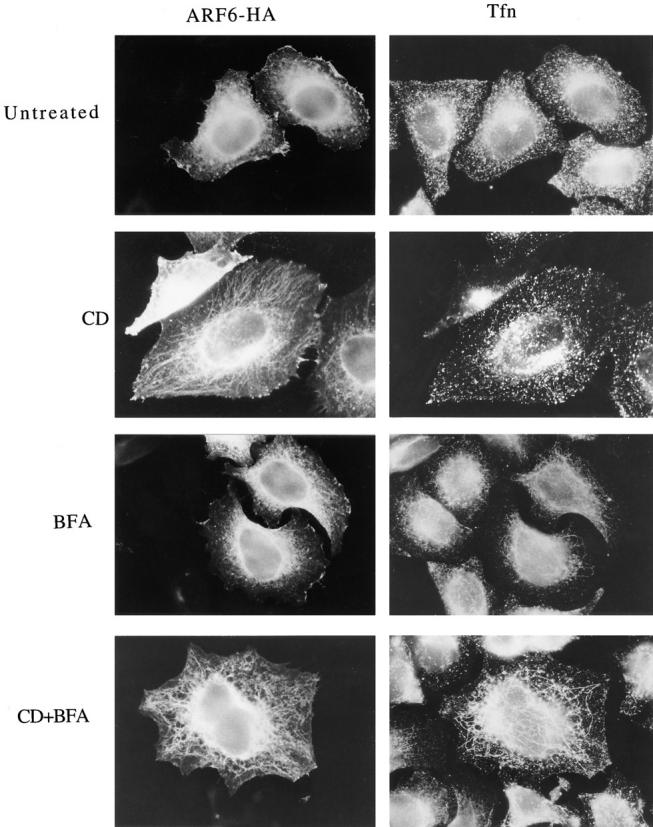
The ARF6-labeled tubular structures are distinct from transferrin-positive endosomes. Transfected HeLa cells expressing ARF6-HA were incubated with 30 μg/ ml iron-loaded human transferrin (Tfn) for 30 min at 37°C either in the absence (Untreated) or presence of 1 μM CD (CD), 1 μM BFA (BFA), or CD plus BFA (CD+BFA). Cells were washed, fixed, and processed for indirect immunofluorescence. ARF6-HA was labeled with a mouse anti-HA antibody, and transferrin was labeled with rabbit anti-Tfn antiserum; primary antibodies were visualized with appropriately labeled donkey secondary antibodies. Note that in the presence of both CD and BFA, the ARF6 tubules are distinct from the transferrin tubules.
PM Markers Colocalize with ARF6 in the Tubular Compartment
Although ARF6 redistributed from the PM to the tubular membrane compartment during CD treatment, the mechanism of transfer between these two compartments was not clear. Since ARF1 cycles between the cytosol and Golgi membranes (Donaldson and Klausner, 1994), it was possible that ARF6 might also move from the PM via the cytosol to the tubular compartment. However, given that immuno-EM localization (Peters et al., 1995) and subcellular fractionation studies (Cavenagh et al., 1996; Song, J., and J. Donaldson, unpublished observations) suggest that the majority of ARF6 in cells is membrane associated, another possibility was that ARF6 might move between the PM and the tubular compartment by a membrane-mediated process. To begin to address this, we examined whether other plasma membrane proteins might also redistribute to the tubular compartment during CD treatment.
We used Tac, the IL-2 receptor α subunit, as a PM marker. Tac has been thoroughly studied and used as a generic integral PM protein with no retention or targeting information that moves through the secretory pathway to the PM (Leonard et al., 1984; Weissman et al., 1986; Bonifacino et al., 1990; Subtil et al., 1997). We cotransfected HeLa cells with plasmids encoding Tac and wild-type ARF6, and determined the distributions of the proteins after incubation in the presence or absence of CD or AlF. Tac and ARF6 showed extensive colocalization at the plasma membrane and with an internal, juxtanuclear compartment in untreated cells (Fig. 3). Whereas CD treatment of cells shifted the localization of Tac and ARF6 to the tubular compartment, addition of AlF (obtained by adding 30 mM NaF and 50 μM AlCl3) resulted in colocalization of ARF6 and Tac in surface protrusions (Fig. 3). In HeLa cells coexpressing ARF6 and a Tac chimeric protein containing the cytoplasmic tail of HLA-DM, a sequence specifying clathrin-coated pit localization (Marks et al., 1995), the chimeric protein did not codistribute with ARF6 in either surface protrusions or the tubular compartment (not shown). These results demonstrate that PM proteins lacking known signals for clathrin-coated pit localization colocalize with ARF6 in the tubular compartment of HeLa cells during CD treatment.
Figure 3.

The PM protein Tac colocalizes with ARF6 in the tubular compartment during CD treatment and in surface protrusions during AlF treatment. HeLa cells transfected with plasmids encoding wild-type ARF6 and the PM protein Tac (IL-2 receptor α subunit) were incubated in the absence (Unt) or presence of 1 μM CD (CD) or AlF (AlF) for 30 min, and they were fixed and processed for indirect immunofluorescence localization of ARF6 with rabbit polyclonal anti-ARF6 antibodies and of Tac with monoclonal mouse anti-Tac antibodies, followed by the appropriate secondary antibodies.
The movement of Tac and ARF6 into the tubular endosomal compartment in transfected cells prompted us to examine whether an endogenous PM protein would redistribute to such a tubular compartment with or without ARF6 overexpression. MHC class I proteins (MHC-I) are found on the PM and apparently do not contain cytoplasmic tails conferring clathrin/AP-2 localization (Neefjes et al., 1990). We analyzed the distribution of endogenous MHC-I in HeLa cells, some of which were overexpressing ARF6 by transient transfection, and thus labeled with the antibody to ARF6. In untreated cells, MHC-I localized to the PM, the juxtanuclear region, and occasionally in fine tubular arrays that radiated from this region (Fig. 4, arrowheads). After CD treatment, there was an apparent increase in MHC-I labeling of these tubular membranes that extended out to the peripheral edge of the cells (Fig. 4, CD). The distribution of MHC-I was similar whether the cells were overexpressing ARF6 (detected with the ARF6 antibody) or were untransfected. In the transfected cells, ARF6 colocalized with MHC-I at the PM and in the tubular structures. These observations demonstrate that the tubular compartment is normally present in HeLa cells, becomes more elaborate in the presence of CD, and is not induced by overexpression of ARF6. Thus, we can monitor the movement of ARF6 and Tac in transfected HeLa cells to study this novel membrane system.
Surface Tac and ARF6 Move from the PM to the Tubular Compartment
Although ARF6 and Tac colocalized in the tubular compartment with CD or at surface protrusions with AlF, it was difficult to discern the ARF6- and Tac-localized structures in the juxtanuclear region. However, it is reasonable to assume that some of this labeling may represent Tac in the Golgi complex. Indeed, a tight juxtanuclear structure labeled with Tac antibody is observed in some cells. This raised the issue of whether the Tac in the tubular compartment was newly synthesized Tac en route to the PM or surface Tac redistributing from the PM.
Therefore, we assessed the movement of surface Tac from the PM into the tubular compartment in cells coexpressing ARF6 and Tac by monitoring Tac antibody internalization. Surface Tac was labeled by incubating cells at 4°C with anti-Tac antibody. The cells were then washed and incubated at 37°C in the presence or absence of CD to allow internalization of the bound antibody. After fixation, the distribution of the antibody-bound Tac was revealed by secondary antibody staining. Localization of the Tac antibody after binding at 4°C showed only staining of the cell surface, where it colocalized with PM-associated ARF6, but ARF6 was also observed in the internal perinuclear and tubular structures (Fig. 5, 4°C Binding). Subsequent incubation of cells at 37°C in the absence of CD revealed that Tac antibody was internalized into the perinuclear and tubular compartment in many cells, where, it then colocalized with ARF6; some of the Tac antibody labeling also colocalized with ARF6 staining at peripheral PM sites (Fig. 5, 37 °C No drug). This observation indicated that Tac normally moved from the PM to the tubular compartment in the absence of any perturbants. Warming the cells in the presence of CD resulted in a more dramatic redistribution of the Tac antibody and ARF6 to the tubular compartment, with little Tac antibody or ARF6 localization at the peripheral edges of the cells (Fig. 5, 37 °C CD).
To assess Tac antibody remaining on the surface after CD treatment, the fixed cells were incubated with the secondary antibodies in the absence of detergent permeabilization. After CD treatment, the Tac antibody remaining at the PM was evenly distributed along the cell surface (Fig. 5, CD, Surface only). No staining of the tubular compartment was observed, indicating that the Tac antibody taken up into the tubular compartment was inaccessible to secondary antibodies and thus not continuous with the PM. The internalization of surface Tac was not induced by antibody cross-linking during the course of the experiment, since the localization of Tac in the tubular compartment was also observed by immunofluorescence after fixation (see Fig. 3). In contrast to the internalization of PM proteins, this compartment could not be loaded with fluid phase markers (not shown). Unlike the movement of surface Tac into the tubular compartment in either the presence or absence of CD, when cells were incubated with AlF at 37°C, the Tac antibody localized to the surface protrusions along with ARF6 and did not redistribute into the tubular compartment (Fig. 5, AlF).
CD Blocks Recycling of Tac and ARF6 from the Tubular Compartment Back Out to the PM
Since surface Tac appears to move from the PM to the tubular compartment, and CD treatment causes Tac to accumulate in this compartment (see above), we wanted to determine whether internalized Tac can recycle from the tubular compartment back out to the PM. To study the fate of internalized Tac antibody, we had to remove the anti-Tac antibody that remained at the cell surface after internalization at 37°C. Rinsing cells with low pH buffers containing high salt has been shown to remove transferrin bound to its receptor at the cell surface (Klausner et al., 1983).
To determine the efficiency of removing surface-bound antibody with low pH buffer, cells expressing ARF6 and Tac were incubated with Tac antibody at 4°C to label surface Tac. Some cells were warmed to 37°C to allow internalization in the presence of CD. The cells were then either fixed immediately (Fig. 6, Total) or rinsed briefly with low pH buffer (0.5% acetic acid, 0.5 M NaCl, pH 3.0; Fig.6, Internal) before fixation and immunolabeling for ARF6 and Tac antibody. The Tac antibody labeled only the cell surface at 4°C, whereas ARF6 localized both to the PM and to internal structures (Fig. 6, 4°C Total). Rinsing with low pH buffer completely removed the surface-bound Tac antibody, but did not alter the distribution of ARF6 (Fig. 6, Internal). Incubation of cells at 37°C for 30 min in the presence of CD resulted in the internalization of Tac antibody into the tubular compartment (Fig. 6, 37 °C +CD Total). Washing with low pH buffer before fixation removed the surface Tac antibody, but did not remove the internalized Tac antibody that colocalized with ARF6 in the tubular compartment (Fig. 6, 37 °C +CD Internal). The inability of the low pH wash to remove Tac antibody lends additional support to the argument that this compartment is internal and not continuous with the PM. The tubular compartment had a beaded appearance possibly because of the low pH wash. Nevertheless, ARF6 and Tac remained colocalized in this compartment.
Figure 6.
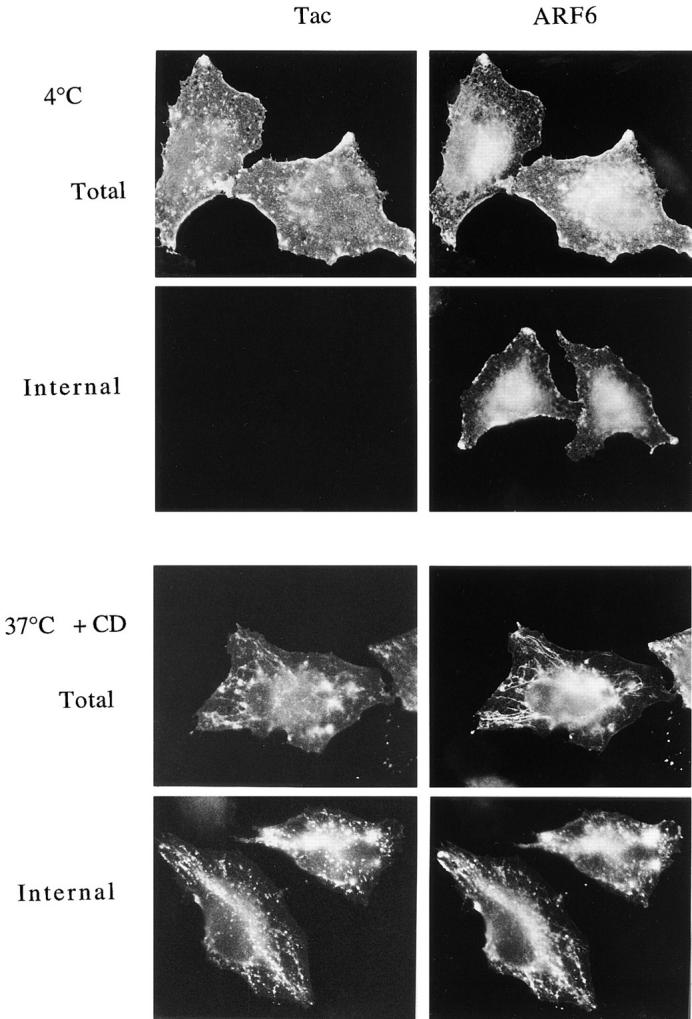
Low pH buffer removes surface-bound Tac antibodies, but not the internalized Tac antibodies in the tubular compartment. Tac- and ARF6-transfected HeLa cells were chilled to 4°C and incubated with mouse anti-Tac antibodies for 30 min. One set of cells were fixed at 4°C either before (4 °C Total) or after (4 °C Internal) rinsing with low pH buffer (0.5% acetic acid, 0.5 M NaCl, pH 3.0). Another set of cells were warmed to 37°C for 30 min in the presence of 1 μM CD and then fixed before (37 °C + CD Total) or after (37 °C +CD Internal) rinsing with low pH buffer. Fixed cells were then processed for indirect immunofluorescence.
Having demonstrated that the low pH wash could efficiently remove surface-bound Tac antibody, we examined whether internalized Tac recycled from the tubular compartment back to the PM. Cells were labeled with Tac antibody at 4°C and then warmed to 37°C in the presence of CD to accumulate Tac antibody in the tubular compartment. Antibodies remaining at the cell surface were then removed with a low pH wash at 4°C (as in Fig. 6), leaving only the internalized Tac antibody (Fig. 7, Load).
Figure 7.

CD treatment blocks the recycling of Tac from the tubular compartment back out to the PM. Anti-Tac antibodies bound to the surface of cells expressing ARF6 and Tac were loaded into the tubular compartment by incubation with 1 μM CD at 37°C for 30 min. The cells were chilled to 4°C, rinsed with low pH buffer to remove the remaining surface anti-Tac antibodies, and then either fixed immediately (Load) and assessed for the Tac antibody loaded, or warmed to 37°C for 30 min in the absence or presence of CD before fixation. Tac antibody that reappeared on the cell surface was detected by incubation with fluorescently labeled secondary antibodies in the absence of detergent permeabilization (Surface Reappearance); ARF6 was subsequently localized in these cells after permeabilization.
To determine whether the internalized Tac could recycle back to the PM, the loaded cells were subsequently incubated at 37°C in the presence or absence of CD for 30 min. Tac antibody that reappeared on the surface was detected by labeling fixed cells with fluorescent secondary antibody without detergent permeabilization; ARF6 was subsequently localized in these cells after permeabilization. The internalized Tac antibody reappeared on the surface at protrusive sites along the edges of cells, together with ARF6, in the absence of CD (Fig. 7, −CD). Phalloidin labeling of these cells indicated that the sites of Tac reappearance were enriched in actin filaments (not shown). In contrast, incubation with CD prevented the reappearance of the internalized Tac antibody back on the cell surface (Fig. 7, +CD); ARF6 remained in the tubular compartment and colocalized with the internalized Tac antibody in samples that were detergent permeabilized to visualize total Tac antibody staining (not shown).
These results demonstrate that surface Tac moves into and back out of this tubular endosomal compartment along with ARF6. In the presence of AlF, internalization into this compartment is blocked (Fig. 5), whereas in the presence of CD, recycling of Tac back out to the PM is inhibited.
ARF6 Regulates the Cycling of Membrane between the PM and the Tubular Compartment
The distribution of wild-type ARF6 in cells treated with AlF and CD mimics the localization of mutant ARF6 in cells expressing the active ARF6/Q67L and inactive ARF6/T27N mutants, respectively (Radhakrishna et al., 1996; Fig. 1 this study). Thus, we examined whether the blocks in transport observed with these drugs on the movement of Tac between the PM and the tubular endosomal compartment could be recreated in cells expressing the ARF6 mutant proteins.
We first examined the uptake of Tac antibody into cells cotransfected with plasmids encoding Tac and ARF6/ Q67L, following the protocol used in Fig. 6. After the binding at 4°C, Tac antibody was uniformly localized along the PM (Fig. 8, Binding). Upon warming to 37°C in the presence of CD, no change in the distribution of Tac antibody was observed (Fig. 8, CD Total). Washing these cells with low pH buffer before fixation removed all of the Tac antibody (Fig. 8, CD Internal), indicating that the Tac antibody remained at the cell surface during the 37°C incubation. The same results were observed when these cells were warmed in the absence of CD (not shown). This indicates that expression of the active ARF6/Q67L alone blocks the internalization of surface Tac, suggesting that GTP hydrolysis by ARF6 is required for Tac internalization.
Figure 8.

Internalization of surface Tac into the tubular compartment is blocked in cells expressing the GTPase-defective ARF6/ Q67L mutant. HeLa cells expressing Tac and ARF6/Q67L were incubated with anti-Tac antibodies at 4°C and either fixed at 4°C (4°C Binding) or warmed to 37°C for 30 min in the presence of 1 μM CD. The cells were then fixed either before (CD Total) or after rinsing with low pH buffer (CD Internal) and processed for indirect immunofluorescence.
We next examined both the internalization and recycling of Tac antibody in cells cotransfected with plasmids encoding Tac and ARF6/T27N. After binding at 4°C, Tac antibody localized to the surface of cells and did not colocalize with ARF6/T27N (Fig. 9, 4°C Binding). Cells were then incubated at 37°C in the absence of CD, followed by low pH removal of remaining surface antibody to assess the internalized Tac antibody. The Tac antibody accumulated in the tubular compartment, where it colocalized in part with ARF6/T27N (Fig. 9, Uptake). After the low pH wash, cells that had taken up the Tac antibody were then further incubated at 37°C to allow recycling, and surface reappearance of Tac antibody was determined as described above (Fig. 7). The internalized Tac antibody did not appear on the PM in these cells (Fig. 7, Surface Reappearance). This indicates that expression of the inactive ARF6/T27N mutant (ARF6-GDP) alone, in the absence of cytochalasin treatment, blocks the recycling of internalized Tac from the tubular compartment back to the PM, suggesting that activation of ARF6 is required for exit from this compartment.
Figure 9.

Recycling of Tac from the tubular compartment back out to the PM is blocked in cells expressing the GTP-binding–defective ARF6/T27N mutant. HeLa cells transfected with Tac and ARF6/T27N plasmids were incubated with anti-Tac antibodies 4°C, and were then either fixed (4 °C Binding) or warmed to 37°C for 30 min. These cells were then chilled, rinsed with low pH buffer, and either fixed (37°C Uptake) to assess the internalization of Tac antibody or warmed again to 37°C for 30 min before fixation, and surface reappearance of Tac antibody was assessed as in Fig. 7. Note that the anti-Tac antibodies accumulated in the tubular compartment in the absence of CD in cells expressing ARF6/T27N.
DISCUSSION
In this study, we describe a novel PM–endosomal recycling pathway that is regulated by the ARF6 GTP-binding protein. We previously showed that the distribution of the wild-type protein could be acutely shifted to the active PM location by treatment of the cells with AlF; dynamic, actin-rich protrusions were induced in these cells as a consequence of this treatment (Radhakrishna et al., 1996). We now demonstrate that inhibitors of actin polymerization, such as CD, shift the distribution of the protein from the PM to an internal, juxtanuclear compartment that exhibits tubular elements extending out towards the periphery. In HeLa cells, this compartment is distinct from the transferrin receptor endosome, and resembles the compartment where the GTP-binding defective mutant of ARF6, T27N, resides. Along with ARF6, “generic” PM, including membrane proteins that do not contain clathrin/AP2 localization domains such as Tac, normally moves from the PM into this compartment and back out again. Although we had difficulty discerning the morphology of the compartment in the juxtanuclear region, it was clear during the internalization experiments that Tac would appear in both the juxtanuclear and tubular portion of this compartment simultaneously, suggesting that they were connected. The two treatments that alter ARF6 distribution also result in specific blocks in this membrane traffic pathway. CD inhibits egress of ARF6 and membrane out of this system, whereas AlF prevents the internalization of ARF6 and membrane into this system. These transport blocks observed pharmacologically with CD and AlF treatment were recreated by expression of either the inactive T27N or the active Q67L mutants, respectively. This demonstrates that the membrane recycling pathway described here is regulated by the GTP cycle of ARF6.
A recent study reported that endogenous ARF6 is associated with a PM fraction isolated from CHO cells and is not associated with early endosomal membranes, identified by loading with the fluid phase marker HRP (Cavenagh et al., 1996). In transfected HeLa cells, we observe that ARF6 is associated with an internal membrane compartment in addition to the PM. This compartment is accessible to loading with surface PM proteins, is transferrin negative, and does not accumulate fluid phase endocytic tracers. Accordingly, this ARF6-associated membrane compartment that we observe would not be detected as an “early endosome,” but rather, would likely be included in the PM fraction isolated by Cavenagh et al. (1996).
We propose a tentative working model for how ARF6 regulates this membrane recycling pathway (Fig. 10). Through its GTP cycle, ARF6 moves between the PM and this recycling compartment. Nucleotide exchange onto ARF6-GDP is required for ARF6 and membrane to exit from the recycling compartment. The reappearance of ARF6-GTP and membrane at peripheral exit sites correlates with the formation of actin-based protrusions. Hydrolysis of the GTP bound to ARF6 is required to allow internalization of PM and ARF6-GDP back into this internal compartment. The specific mechanism whereby the membrane is internalized, i.e., whether by nonclathrin pinocytosis or tubular membrane invaginations that become discontinuous with the PM, is yet to be identified. The internal endosomal compartment includes ill-defined structures in the juxtanuclear region and associated tubular elements that extend out to the periphery. We speculate that treatment of cells with CD blocks ARF6–GTP exchange and membrane recycling, and that treatment with AlF blocks ARF6-GTP hydrolysis and membrane internalization. We are currently developing methods to investigate the nucleotides bound to ARF6 under these different conditions, and to further characterize the membrane intermediates involved in the movement into and out of this recycling compartment.
Figure 10.

Model for the ARF6-regulated PM–endosomal recycling pathway. We propose that activation of ARF6, through nucleotide exchange, occurs at the tubular endosome and triggers the recycling of membrane to discrete sites at the PM where ARF6-GTP stimulates the formation of protrusive structures, rich in actin. Inactivation, through GTP hydrolysis, signals the return of ARF6 to the tubular endosome via membrane intermediates that are yet to be defined. CD treatment or expression of the GTP-binding–defective mutant of ARF6, T27N, blocks recycling out to the PM. In contrast, AlF treatment or expression of the GTPase-defective mutant, Q67L, blocks membrane internalization back into the tubular endosomal compartment.
In some respects, the requirement in our model for ARF6 activation (i.e., GTP exchange) for membrane recycling to occur is similar to the observations made by D'Souza-Schorey et al. (1995) for the transferrin cycle in CHO cells. In their study, overexpression of either the wild-type ARF6 or the constitutively active Q67L mutant inhibited transferrin uptake into CHO cells, whereas expression of the T27N mutant inhibited transferrin receptor recycling back to the cell surface. Several distinctions between the two studies, however, deserve comment. First, the extent to which this endosomal recycling pathway regulated by ARF6 is separate and distinguishable from the transferrin receptor endosome, as we observed, may vary among different cell types. Indeed, in CHO cells, the transferrin receptor and the T27N mutant of ARF6 colocalize (D'Souza-Schorey et al., 1995), whereas in HeLa cells, the compartment where ARF6 localizes is clearly distinct in its morphology and behavior from the transferrin receptor compartment (Radhakrishna et al., 1996; this study). Second, we were able to confirm the existence and behavior of the ARF6-regulated compartment in HeLa cells in the absence of ARF6 overexpression by observing endogenous MHC-I in untransfected cells. Finally, in CHO cells, overexpression of the wild-type ARF6 alone causes a block in the internalization of transferrin receptors (D'Souza- Schorey et al., 1995). The inability to reverse this effect of the wild-type protein limits the use of this system for defining the role of the ARF6–GTP cycle in this process. By contrast, in HeLa cells we were able to study the full PM– endosomal recycling pathway regulated by ARF6, since overexpression of the wild-type protein, although exhibiting enhanced nascent protrusive structures at the cell surface, does not inhibit membrane movement through the pathway. This has allowed us to identify pharmacologic treatments that acutely and reversibly alter the distribution and movement of ARF6 and membrane proteins through this pathway.
In HeLa cells, the ARF6-regulated recycling compartment has a distinctive tubular morphology that emanates out of the juxtanuclear area near the microtubule organizing center. Tooze and Hollinshead (1991) were among the first to document the existence of extensive tubular endosomal networks observable by thick-section EM in a variety of cell types, including HeLa cells. Although in some cases tubular endosomes have been reported to contain the transferrin receptor (van der Sluijs et al., 1992; Hopkins et al., 1994), the ARF6-regulated tubular endosome that we observe does not. There are other reports of novel endosomal compartments distinct from the well-characterized transferrin endosomal system. The autocrine motility factor receptor localizes to a tubular compartment in MDCK and HeLa cells that does not colocalize with transferrin receptor (Benlimame et al., 1995). In NRK cells, expression of endotubin, a protein normally associated with the apical early endosomes of intestinal epithelia, localizes to a compartment that contains neither transferrin nor fluid phase markers and is not altered by treatment with BFA (Wilson and Colton, 1997). It will be interesting to determine whether either of these proteins are associated with the ARF6-regulated membrane compartment described here.
Although the tubular morphology is a hallmark of the ARF6-regulated membrane compartment in HeLa cells, this morphology may not be exhibited by ARF6-regulated compartments in other cells. For example, it was recently reported that ARF6 cofractionates with isolated chromaffin granules and may be involved in the regulated exocytosis of these granules (Galas et al., 1997). Although these granules do not exhibit tubular morphology, through its GTP cycle, ARF6 may still regulate membrane movement through such compartments.
Having demonstrated the existence of a membrane recycling pathway regulated by ARF6, one might ask what function such a pathway serves in the cell. We showed that bulk PM was capable of being internalized and recycled out of such a compartment, and that this apparently occurs on a slower time scale (loading and unloading of this compartment on the order of ∼30–60 min) than the transferrin receptor internalization and recycling pathway (loading and unloading on the order of 5–10 min; Mellman, 1996). The ARF6-regulated pathway might allow for bulk membrane to be internalized and then recycled to the cell surface at defined sites in such a way that it could be used to regulate the surface area and shape of cells. Indeed, in HeLa cells, ARF6 and membrane recycle to defined sites along the PM often associated with actin-rich protrusions. Additionally, we have demonstrated that recycling of ARF6 and membrane to the edge of cells is required during cell spreading; expression of ARF6/T27N inhibits HeLa cell spreading (Song, J., Khachikian, H. Radhakrishna, and J. Donaldson, manuscript in preparation). These observations suggest that the ARF6-regulated membrane cycle may be involved in PM remodeling events that are initiated in response to physiological stimuli (e.g., growth factors, chemotactic agents, and metastasis).
It is intriguing that the trafficking and possibly GTP status of ARF6 are influenced by actin polymerization. The observation that recycling of ARF6 and membrane back to the PM is blocked by CD treatment or expression of the T27N mutant suggests that membrane movement out of the tubular compartment requires actin polymerization and ARF6-GTP. At the present time, we cannot determine whether the requirement for actin polymerization is needed for nucleotide exchange, and thereby conversion to ARF6-GTP, or if it is required for ARF6-GTP and membrane to recycle back to the PM, for example, through an actin-myosin based transport step (Titus, 1997). It is not likely that CD is specifically inhibiting nucleotide exchange onto ARF6, analogous to the effects of BFA on ARF1 at the Golgi (Donaldson et al., 1992b ; Helms and Rothman, 1992; Randazzo et al., 1993), since other inhibitors of actin polymerization, including cytochalasin B and latrunculin, also inhibit movement of ARF6 back to the PM. Further details of the actin dependence of this step will require in vitro methods to analyze the nucleotide status of ARF6 and reconstitute the ARF6-mediated membrane recycling to the PM. Once at the PM, the accumulation of ARF6 and perhaps the delivery of membrane there results in a stimulation of actin polymerization and formation of protrusive structures. Thus, it appears that ARF6 (localization/GTP status) depends on, but is also capable of stimulating, actin polymerization. Further studies should elucidate the connection between this novel ARF6-regulated membrane traffic pathway and the actin cytoskeleton.
Acknowledgments
We thank Drs. J. Bonifacino, M. Marks, and P. Roche for generously providing reagents used in this study. We also thank Drs. O. Al-Awar, E. Korn, J. Lippincott-Schwartz, and A. Sobota for critical reading of the manuscript.
Abbreviations used in this paper
- ARF
ADP-ribosylation factor
- AlF
aluminum fluoride
- BFA
brefeldin A
- CD
cytochalasin D
- HA
hemagglutinin
- MHC-I
MHC class I proteins
- PM
plasma membrane
Footnotes
Address all correspondence to Julie Donaldson, Laboratory of Cell Biology, NHLBI, National Institues of Health, Bethesda, MD 20892. Tel.: (301) 402-2907. Fax: (301) 402-1519. e-mail: jdonalds@helix.nih.gov
REFERENCES
- Benlimame N, Simard D, Nabi IR. Autocrine motility factor receptor is a marker for a distinct membranous tubular organelle. J Cell Biol. 1995;129:459–471. doi: 10.1083/jcb.129.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Suzuki CK, Lippincott-Schwartz J, Weissman AM, Klausner RD. Pre-Golgi degradation of newly synthesized T cell antigen receptor chains: intrinsic sensitivity and the role of subunit assembly. J Cell Biol. 1989;10:73–83. doi: 10.1083/jcb.109.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Cosson P, Klausner RD. Colocalized transmembrane determinants for ER degradation and subunit assembly explain the intracellular fate of TCR chains. Cell. 1990;63:503–513. doi: 10.1016/0092-8674(90)90447-m. [DOI] [PubMed] [Google Scholar]
- Brown HA, Gutowski S, Moomaw CR, Slaughter C, Sternweis PC. ADP-ribosylation factor, a small GTP-dependent regulatory protein, stimulates phospholipase D activity. Cell. 1993;75:1137–1144. doi: 10.1016/0092-8674(93)90323-i. [DOI] [PubMed] [Google Scholar]
- Cavenagh MM, Whitney JA, Carroll K, Zhang C-j, Boman AL, Rosenwald AG, Mellman I, Kahn RA. Intracellular distribution of Arf proteins in mammalian cells. J Biol Chem. 1996;271:21767–21774. doi: 10.1074/jbc.271.36.21767. [DOI] [PubMed] [Google Scholar]
- Cockroft S, Thomas GMH, Fensome A, Geny B, Cunningham E, Gout I, Hiles I, Totty NF, Truong O, Hsuan JJ. Phospholipase D: a downstream effector of ARF in granulocytes. Science (Wash DC) 1994;263:523–526. doi: 10.1126/science.8290961. [DOI] [PubMed] [Google Scholar]
- D'Souza-Schorey C, Li G, Colombo MI, Stahl PD. A regulatory role for ARF6 in receptor-mediated endocytosis. Science (Wash DC) 1995;267:1175–1178. doi: 10.1126/science.7855600. [DOI] [PubMed] [Google Scholar]
- Dascher C, Balch WE. Dominant inhibitory mutants of ARF1 block endoplasmic reticulum to Golgi transport and trigger disassembly of the Golgi apparatus. J Biol Chem. 1994;269:1437–1448. [PubMed] [Google Scholar]
- Donaldson JG, Lippincott-Schwartz J, Klausner RD. Guanine nucleotides modulate the effects of brefeldin A in semipermeable cells: regulation of the association of a 110-kD peripheral membrane protein with the Golgi apparatus. J Cell Biol. 1991;112:579–588. doi: 10.1083/jcb.112.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG, Cassel D, Kahn RA, Klausner RD. ADP-ribosylation factor, a small GTP-binding protein, is required for binding of the coatomer protein β-COP to Golgi membranes. Proc Natl Acad Sci USA. 1992a;89:6408–6412. doi: 10.1073/pnas.89.14.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG, Finazzi D, Klausner RD. Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature (Lond) 1992b;360:350–352. doi: 10.1038/360350a0. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Klausner RD. ARF: a key regulatory switch in membrane traffic and organelle structure. Curr Opin Cell Biol. 1994;6:527–532. doi: 10.1016/0955-0674(94)90072-8. [DOI] [PubMed] [Google Scholar]
- Finazzi D, Cassel D, Donaldson JG, Klausner RD. Aluminum fluoride acts on the reversibility of ARF-dependent coat protein binding to Golgi membranes. J Biol Chem. 1994;269:13325–13330. [PubMed] [Google Scholar]
- Galas M-C, Helms JB, Vitale N, Thierse D, Aunis D, Bader M-F. Regulated exocytosis in chromaffin cells: a potential role for a secretory granule-associated ARF6 protein. J Biol Chem. 1997;272:2788–2793. doi: 10.1074/jbc.272.5.2788. [DOI] [PubMed] [Google Scholar]
- Gruenberg J, Maxfield FR. Membrane transport in the endocytic pathway. Curr Opin Cell Biol. 1995;7:552–563. doi: 10.1016/0955-0674(95)80013-1. [DOI] [PubMed] [Google Scholar]
- Hammond SM, Altshuller YM, Sung T, Rudge SA, Rose K, Engebrecht J, Morris AJ, Frohman MA. Human ADP-ribosylation factor-activated phosphatidylcholine-specific phospholipase D defines a new and highly conserved gene family. J Biol Chem. 1995;270:29640–29643. doi: 10.1074/jbc.270.50.29640. [DOI] [PubMed] [Google Scholar]
- Helms JB, Rothman JE. Inhibition by brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature (Lond) 1992;360:352–354. doi: 10.1038/360352a0. [DOI] [PubMed] [Google Scholar]
- Hopkins CR, Trowbridge IS. Internalization and processing of transferrin and transferrin receptor in human carcinoma cells. J Cell Biol. 1983;97:508–521. doi: 10.1083/jcb.97.2.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins CR, Gibson A, Shipman M, Strickland DK, Trowbridge IS. In migrating fibroblasts, recycling receptors are concentrated in narrow tubules in the pericentriolar area, and then routed to the PM of the leading lamella. J Cell Biol. 1994;125:1265–1274. doi: 10.1083/jcb.125.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker W, Whitney JA, Mellman I. Selective inhibition of transcytosis by brefeldin A in MDCK cells. Cell. 1991;67:617–627. doi: 10.1016/0092-8674(91)90535-7. [DOI] [PubMed] [Google Scholar]
- Kahn RA, Gilman AG. The protein cofactor necessary for ADP-ribosylation of Gs by cholera toxin is itself a GTP-binding protein. J Biol Chem. 1986;261:7906–7911. [PubMed] [Google Scholar]
- Kahn RA, Kern FG, Clark J, Gelmann EP, Rulka C. Human ADP-ribosylation factors. A functionally conserved family of GTP binding proteins. J Biol Chem. 1991;266:2606–2614. [PubMed] [Google Scholar]
- Klausner RD, Van Renswoude J, Ashwell G, Kempf C, Schechter AN, Dean A, Bridges KR. Receptor-mediated endocytosis of transferrin in K562 cells. J Biol Chem. 1983;258:4715–4724. [PubMed] [Google Scholar]
- Koval M, Pagano RE. Lipid recycling between the plasma membrane and intracellular compartments: transport and metabolism of fluorescent sphingomyelin analogues in cultured fibroblasts. J Cell Biol. 1989;108:2169–2181. doi: 10.1083/jcb.108.6.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ktistakis NT, Brown HA, Waters MG, Sternweis PC, Roth MG. Evidence that phospholipase D mediates ADP ribosylation factor– dependent formation of Golgi coated vesicles. J Cell Biol. 1996;134:295–306. doi: 10.1083/jcb.134.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamaze C, Schmid SL. The emergence of clathrin-independent pinocytic pathways. Curr Opin Cell Biol. 1995;7:573–580. doi: 10.1016/0955-0674(95)80015-8. [DOI] [PubMed] [Google Scholar]
- Leonard WJ, Depper JM, Crabtree GR, Rudikoff S, Pumphrey J, Robb RJ, Kronke M, Svetlik PB, Peffer NJ, Waldmann TA, Greene WC. Molecular cloning and expression of cDNAs for the human interleukin-2 receptor. Nature (Lond) 1984;311:626–631. doi: 10.1038/311626a0. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Yuan LC, Tipper C, Klausner RD. Brefeldin A's effects on endosomes, lysosomes, and TGN suggest a general mechanism for regulating organelle structure and membrane trafficking. Cell. 1991;67:601–616. doi: 10.1016/0092-8674(91)90534-6. [DOI] [PubMed] [Google Scholar]
- Marks MS, Roche PA, Van Donselaar E, Woodruff L, Peters PJ, Bonifacino JS. A lysosomal targeting signal in the cytoplasmic tail of the β chain directs HLA-DM to MHC class II compartments. J Cell Biol. 1995;131:351–369. doi: 10.1083/jcb.131.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massenburg D, Han J-S, Liyanage M, Patton WA, Rhee SG, Moss J, Vaughan M. Activation of rat brain phospholipase D by ADP-ribosylation factors 1, 5, and 6: separation of ADP-ribosylation factor-dependent and oleate-dependent enzymes. Proc Natl Acad Sci USA. 1994;91:11718–11722. doi: 10.1073/pnas.91.24.11718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor S, Presley JF, Maxfield FR. Sorting of membrane components from endosomes and subsequent recycling to the cell surface occurs by a bulk flow process. J Cell Biol. 1993;121:1257–1269. doi: 10.1083/jcb.121.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melançon P, Glick BS, Malhotra V, Weidman PJ, Serafini T, Gleason ML, Orci L, Rothman JE. Involvement of the GTP-binding “G” proteins in transport through the Golgi stack. Cell. 1987;58:329–336. doi: 10.1016/0092-8674(87)90591-5. [DOI] [PubMed] [Google Scholar]
- Mellman I. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- Moss J, Vaughan M. Structure and function of ARF proteins: activators of cholera toxin and critical components of intracellular vesicular transport processes. J Biol Chem. 1995;270:12327–12330. doi: 10.1074/jbc.270.21.12327. [DOI] [PubMed] [Google Scholar]
- Neefjes JJ, Stollorz V, Peters PJ, Geuze HJ, Ploegh HL. The biosynthetic pathway of MHC class II but not class I molecules intersects the endocytic route. Cell. 1990;61:171–183. doi: 10.1016/0092-8674(90)90224-3. [DOI] [PubMed] [Google Scholar]
- Palmer DJ, Helms JB, Beckers CJM, Orci L, Rothman JE. Binding of coatomer to Golgi membranes requires ADP-ribosylation factor. J Biol Chem. 1993;268:12083–12089. [PubMed] [Google Scholar]
- Peters PJ, Hsu VW, Ooi CE, Finazzi D, Teal SB, Oorschot V, Donaldson JG, Klausner RD. Overexpression of wild-type and mutant ARF1 and ARF6: distinct perturbations of nonoverlapping membrane compartments. J Cell Biol. 1995;128:1003–1017. doi: 10.1083/jcb.128.6.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishna H, Klausner RD, Donaldson JG. Aluminum fluoride stimulates surface protrusions in cells overexpressing the ARF6 GTPase. J Cell Biol. 1996;134:935–947. doi: 10.1083/jcb.134.4.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randazzo PA, Yang YC, Rulka C, Kahn RA. Activation of ADP-ribosylation factor by Golgi membranes: evidence for a brefeldin A and protease sensitive activating factor on Golgi membranes. J Biol Chem. 1993;268:9555–9563. [PubMed] [Google Scholar]
- Robinson MS, Kreis TE. Recruitment of coat proteins onto Golgi membranes in intact and permeabilized cells: effects of brefeldin A and G protein activators. Cell. 1992;69:129–138. doi: 10.1016/0092-8674(92)90124-u. [DOI] [PubMed] [Google Scholar]
- Rubin LA, Kurman CC, Biddison WE, Goldman ND, Nelson DL. A monoclonal antibody 7G7/B6 binds to an epitope of the human interleukin-2 (IL-2) receptor that is distinct from that recognized by IL-2 or anti-Tac. Hybridoma. 1985;4:91–102. doi: 10.1089/hyb.1985.4.91. [DOI] [PubMed] [Google Scholar]
- Sandvig K, van Deurs B. Endocytosis without clathrin. Trends Cell Biol. 1994;4:275–277. doi: 10.1016/0962-8924(94)90211-9. [DOI] [PubMed] [Google Scholar]
- Spector I, Shochet NR, Blasberger D, Kashman Y. Latrunculins—novel marine macrolides that disrupt microfilament organization and affect cell growth: I. Comparison with cytochalasin D. Cell Motil Cytoskel. 1989;13:127–144. doi: 10.1002/cm.970130302. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Mellman IS, Muller WA, Cohn ZA. Endocytosis and the recycling of plasma membrane. J Cell Biol. 1983;96:1–27. doi: 10.1083/jcb.96.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternweis PC, Gilman AG. Aluminum: a requirement for activation of the regulatory component of adenylate cyclase by fluoride. Proc Natl Acad Sci USA. 1982;79:4888–4891. doi: 10.1073/pnas.79.16.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtil A, Delepierre M, Dautry-Varsat A. An α-helical signal in the cytosolic domain of the interleukin 2 receptor β chain mediates sorting towards degradation after endocytosis. J Cell Biol. 1997;136:583–595. doi: 10.1083/jcb.136.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson JA, Watts C. Macropinocytosis. Trends Cell Biol. 1995;5:424–428. doi: 10.1016/s0962-8924(00)89101-1. [DOI] [PubMed] [Google Scholar]
- Swanson JA, Baer SC. Phagocytosis by zippers and triggers. Trends Cell Biol. 1995;5:89–93. doi: 10.1016/s0962-8924(00)88956-4. [DOI] [PubMed] [Google Scholar]
- Takebe Y, Seiki M, Fujisawa JI, Hoy P, Yokota K, Arai KI, Yoshida M, Arai N. SRα promoter: an efficient and versatile mammalian cDNA expression system composed of the simian virus 40 early promoter and the R-U5 segment of human T-cell leukemia virus type 1 long terminal repeat. Mol Cell Biol. 1988;8:466–472. doi: 10.1128/mcb.8.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanigawa G, Orci L, Amherd M, Ravazzola M, Helms JB, Rothman JE. Hydrolysis of bound GTP by ARF protein triggers uncoating of Golgi-derived COP-coated vesicles. J Cell Biol. 1993;123:1365–1371. doi: 10.1083/jcb.123.6.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teal SB, Hsu VW, Peters PJ, Klausner RD, Donaldson JG. An activating mutation in ARF1 stabilizes coatomer binding to Golgi membranes. J Biol Chem. 1994;269:3135–3138. [PubMed] [Google Scholar]
- Titus MA. Unconventional myosins: new frontiers in actin-based motors. Trends Cell Biol. 1997;7:119–123. doi: 10.1016/S0962-8924(97)01019-2. [DOI] [PubMed] [Google Scholar]
- Tooze J, Hollinshead M. Tubular early endosomal networks in AtT20 and other cells. J Cell Biol. 1991;115:635–653. doi: 10.1083/jcb.115.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub LM, Ostrom JA, Kornfeld S. Biochemical dissection of AP-1 recruitment onto Golgi membranes. J Cell Biol. 1993;123:561–573. doi: 10.1083/jcb.123.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya M, Price SR, Tsai S-C, Moss J, Vaughan M. Molecular identification of ADP-ribosylation factor mRNAs and their expression in mammalian cells. J Biol Chem. 1991;266:2772–2777. [PubMed] [Google Scholar]
- Ullrich O, Reinsch S, Urbé S, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol. 1996;135:913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sluijs P, Hull M, Webster P, Mâle P, Goud B, Mellman I. The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell. 1992;70:729–740. doi: 10.1016/0092-8674(92)90307-x. [DOI] [PubMed] [Google Scholar]
- van Deurs B, Von Bülow F, Vilhardt F, Kaae P, Holm, Sandvig K. Destabilization of plasma membrane structure by prevention of actin polymerization: microtubule-dependent tubulation of the plasma membrane. J Cell Sci. 1996;109:1655–1665. doi: 10.1242/jcs.109.7.1655. [DOI] [PubMed] [Google Scholar]
- Weissman AM, Harford JB, Svetlik PB, Leonard WJ, Depper JH, Waldmann TA, Greene WC, Klausner RD. Only high affinity receptors for interleukin-2 mediate internalization of ligand. Proc Natl Acad Sci USA. 1986;83:1463–1466. doi: 10.1073/pnas.83.5.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JM, Colton TL. Targeting of an intestinal apical endosomal protein to endosomes in nonpolarized cells. J Cell Biol. 1997;136:319–330. doi: 10.1083/jcb.136.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SA, Park JE, Brown WJ. Brefeldin A causes a microtubule-mediated fusion of the trans-Golgi network and early endosomes. Cell. 1991;67:591–600. doi: 10.1016/0092-8674(91)90533-5. [DOI] [PubMed] [Google Scholar]
- Yamashiro DJ, Tycko B, Fluss SR, Maxfield FR. Segregation of transferrin to a mildly acidic (pH 6.4) para-Golgi compartment in the recycling pathway. Cell. 1984;37:789–800. doi: 10.1016/0092-8674(84)90414-8. [DOI] [PubMed] [Google Scholar]
- Zhang C, Rosenwald AG, Willingham MC, Skuntz S, Clark J, Kahn RA. Expression of a dominant allele of human ARF1 inhibits membrane traffic in vivo. J Cell Biol. 1994;124:289–300. doi: 10.1083/jcb.124.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]